
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling multifunctional inhibitors: holistic spectral, electronic and molecular characterization, coupled with biological profiling of substituted pyridine derivatives against LD transpeptidase, heme oxygenase and PPAR gamma†
Shaik Yasmin Begumab,
Predhanekar Mohamed Imran *ab,
Attar Kubaib*ab,
Mohamed Taha Yassinc,
Fatimah O. Al-Otibic,
M. Selvakumaranab,
A. Aathif Bashadb and
S. Sulthanudeenab
*ab,
Attar Kubaib*ab,
Mohamed Taha Yassinc,
Fatimah O. Al-Otibic,
M. Selvakumaranab,
A. Aathif Bashadb and
S. Sulthanudeenab
aDepartment of Chemistry, Islamiah College (Autonomous), Vaniyambadi 635752, Tamilnadu, India. E-mail: imranpkm@gmail.com; attar.kubaib@gmail.com
bThiruvalluvar University, Serkkadu, Vellore 632 115, Tamilnadu, India
cDepartment of Botany and Microbiology, College of Science, King Saud University, P. O. Box 2455, Riyadh 11451, Saudi Arabia
dDepartment of Physics, Islamiah College (Autonomous), Vaniyambadi – 635752, Tamilnadu, India
First published on 19th September 2024
Abstract
This study delves into the therapeutic potential of a molecule, 3-substituted phenyl-1-(pyridine-4-carbonyl)-1H-pyrazole-4-carboxylic acid (PPP), for antimicrobial, antioxidant and anti-diabetic activities. The research encompasses design, synthesis, molecular docking and biological screening of related pyrazole carboxylic acid derivatives. Spectral studies confirmed the structures and molecular mechanics with DFT calculations provided insights into molecular properties and interactions. Quantum chemical descriptors were employed to assess the stability while NBO analysis predicted reactivity, ELF and LOL methods identified electron density. Non-covalent interactions were characterized using RDG and IRI, while the Multiwfn tool was used to evaluate intra and intermolecular aspects. Docking studies elucidated potential therapeutic efficacy against specific protein targets.
1. Introduction
The heterocyclic ring system, particularly phenyl-substituted heterocyclic compounds fused with pyrazole carboxylic acid derivatives, plays a crucial role across various biological processes and therapeutic applications.1 These compounds exhibit a broad spectrum of pharmacological activities, including antimicrobial, antiviral, antitumor, anti-inflammatory, antihistaminic, pesticide, antifungal and antipyretic properties.2 Their versatility has attracted significant interest in chemistry, biology, medicine and agriculture, promising new avenues for medical treatment development. In recent years, there has been a notable focus on targeting the dual inhibition of cyclo-oxygenase and 5-lipoxygenase enzymes for managing inflammation and pain.3 Tepoxalin, a diarylpyrazole derivative, stands as an early example of a dual-functioning analgesic and anti-inflammatory molecule, showcasing the potential of this approach in therapeutic intervention.4Recent studies have shown that pyridine derivatives have strong antimicrobial properties, with pyridine-4-carbonyl-pyrazole derivatives exhibiting potent antibacterial and antifungal activities.5 Phenyl-substituted pyridine derivatives also reduced inflammation and pain by enhancing affinity for COX-2 enzymes.6 Additionally, 3-substituted phenyl-pyridine-pyrazole derivatives demonstrated cytotoxic effects against various cancer cell lines by inducing apoptosis.7 Pyridine-4-carbonyl derivatives inhibited influenza virus replication, showing promise as antiviral candidates.8 Furthermore, pyridine-pyrazole derivatives effectively inhibited acetylcholinesterase, which is relevant for Alzheimer's treatment.9
This study aims to synthesize and characterize phenyl-substituted heterocyclic compounds fused with pyrazole carboxylic acid derivatives, including variations in substituents such as aceto and methyl groups. The rationale behind the current design is to investigate how the presence of different substituents on the phenyl ring could enhance or modify the biological activity of the compounds. By introducing various substitutions, the objective was aimed at identifying structural features that could be optimized for improved biological performance and assessing the potential of these molecules as building blocks for more complex molecules.
Oxidative stress, linked to various chronic diseases including diabetes, can compromise immune function and increase susceptibility to infections. Consequently, compounds exhibiting antioxidant and anti-diabetic properties may offer multifaceted benefits, enhancing their therapeutic value. For the antimicrobial activity assay, specific concentrations were selected based on preliminary studies indicating their effectiveness. The antioxidant activity was measured using assays such as DPPH and ABTS, with results indicating significant scavenging activity. The anti-diabetic activity was assessed using the α-glucosidase inhibition assay, which revealed low inhibitory concentrations (IC50), highlighting the compounds' potential in managing blood glucose levels.
The derivatives were evaluated for antimicrobial, antioxidant and antidiabetic activities, along with in silico studies to assess their interactions with key biological targets. Specific enzymes targeted include LD Transpeptidase (PDB ID: 4JMN),10 crucial for bacterial cell wall synthesis, making inhibitors of this enzyme potential antibiotics against resistant bacteria. Heme Oxygenase (PDB ID: 1IW0),11 catalyses heme degradation and possesses anti-inflammatory and antioxidant properties. Additionally, PPAR Gamma (PDB ID: 2ZNO),12 a nuclear receptor involved in glucose and lipid metabolism, is targeted for its relevance in antidiabetic therapy. This comprehensive approach aims to identify novel compounds with therapeutic potential, particularly focusing on antimicrobial, antioxidant and antidiabetic efficacy, with prospects for future pharmaceutical development. The study's findings are anticipated to advance the field and stimulate further research in this domain.
2. Materials and methods
2.1. Design and spectral characterization
During experimentation, the melting points of the synthesized compounds were measured using the Mvtex digital melting point apparatus. A small amount of the compound was packed into a thin-walled capillary tube, which was then placed in the melting point apparatus. The temperature was gradually increased at a controlled rate until the compound fully melted. The melting point was recorded as the temperature range at which the compound transitioned from solid to liquid. The structures of the compounds were confirmed through spectral data analysis. The infrared spectra were obtained using an FTIR 420 spectrometer manufactured by JASCO Corporation as shown in Fig. S2.† The spectra were recorded using potassium bromide (KBr) pellets. The 1H and 13C NMR spectra were recorded using a Bruker AC 300 spectrometer. 1H NMR was acquired at 300 MHz and 13C NMR at 75 MHz, both with DMSO-d6 as the solvent and TMS as the internal standard. Mass spectrometry (MS) was employed to determine the molecular weights and confirm the identities of the synthesized compounds. The analysis utilized an electrospray ionization mass spectrometer (ESI-MS) in positive ion mode. Compounds were dissolved at 1 mg mL−1 and infused into the mass spectrometer. The protonated ions ([M + H]+) were detected, with the m/z value representing the molecular weight plus one proton. This ionization technique is favored in ESI-MS for its ability to ionize samples efficiently without extensive fragmentation, ensuring clear and accurate mass spectra.14
2.2. Computational details
The molecular geometry was optimized using Gaussian 09 15 and the results were visualized with GaussView 6.0.16 DFT calculations were done using the B3LYP's 6-311++G basis set.17 Gaussian 09 also supported frequency calculations and spectral analysis, aiding in the accurate interpretation of FTIR data. Natural Bond Orbital (NBO) and Fukui function analysis, based on the B3LYP/6-311++G method, was used to investigate molecular bonding, reactive sites and intra- and intermolecular interactions, shedding light on electronic structure and reactivity.18 Global chemical reactivity descriptors (GCRD) such as dipole moments (μ), absolute electronegativity (χ), absolute hardness (η) and softness (σ) were determined through B3LYP/6-311++G calculations.19 Some equations used for reactivity parameters evaluation were derived from global reactivity descriptors such as chemical potential (μ), hardness (η) and electrophilicity (ω). The formulas for these descriptors are as follows:Chemical hardness:
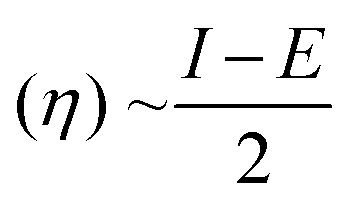
Chemical softness:
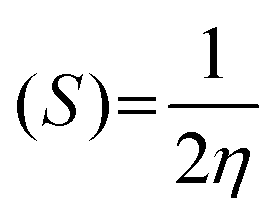
Electronegativity:
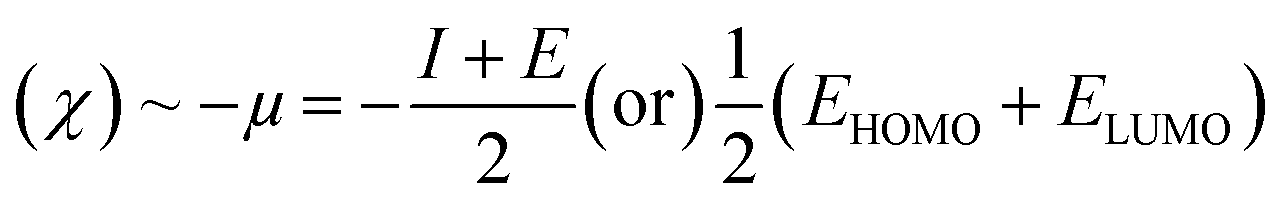
Electrophilicity index:
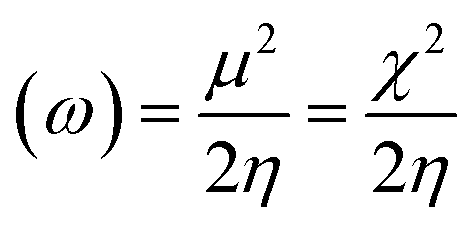
Fukui function for nucleophilic attack (f−):
| f− = ∂ρ(r)/∂N (where ρ(r) is the electron density at r) |
Fukui function for electrophilic attack (f+):
| f+ = ∂ρ(r)/∂N (where f+ is calculated for the addition of an electron) |
Fukui function for radical attack (f0):
| f0 = ∂ρ(r)/∂N (where f0 is used to understand the reactivity towards radicals) |
The dual descriptor
| (Δf) = f+ − f− |
The electronic structure and bonding characteristics of the system were analyzed using Multiwfn software, version 3.8.20 Electron Localization Function (ELF) and Local Orbital Locator (LOL) methods were applied to investigate electron localization and localized electron contributions. Reduced Density Gradient (RDG) surfaces and Interacting Region Indicator (IRI) were used to analyze electron density distribution and non-covalent interactions. Additionally, Multiwfn facilitated non-covalent interactions and topological studies, providing further insights into reactive sites and intra- and intermolecular interactions within the compounds.
2.3. In vitro analysis
The antimicrobial activity of the synthesized compounds was assessed by determining their minimum inhibitory concentration (MIC). This involved incorporating specified amounts of the test compounds into molten sterile agar medium, which was then solidified in Petri dishes at 40–50 °C. The MIC was determined by observing the effect of the compounds on bacterial or fungal growth in the presence of a microbial suspension (5 × 10−5 cfu mL−1) incorporated into a sterilized agar medium and serially diluted with dimethyl formamide. Plates were then incubated at 37 °C for 24 hours for bacteria or 48 hours for fungi. Results were compared with standard antibiotics (ciprofloxacin and fluconazole) for antibacterial and antifungal activity, respectively.21An antioxidant assay solution was prepared by dissolving 31.54 mg of 2,2-diphenyl-1-picrylhydrazyl (DPPH) in a buffered methanol solution (95% v/v), consisting of 40 mL of 0.1 mol L−1 acetate buffer (pH 5.5) mixed with 60 mL of methanol and then adjusting the total volume to 50 mL with buffered methanol. Phenyl-substituted heterocyclic compounds with fused pyrazole carboxylic acid derivatives were tested at concentrations ranging from 20 to 120 μg mL−1, each made up to 4 mL with distilled water. To each test tube, 1 mL of the DPPH solution (1 mmol L−1, or 0.953 × 10−10 μg mL−1) was added and the mixture was shaken before incubating at room temperature (30 °C) for 30 minutes.
The impact of standard antioxidant vitamin C (ascorbic acid) on the DPPH radical was compared with that of the synthesized compounds.22 Ascorbic acid was prepared at varying concentrations of 0.02, 0.04, 0.06, 0.08, 0.10 and 0.12 mL and diluted to 4 mL with distilled water. To each tube, 1 mL of DPPH (1 mmol L−1) was added and the procedure described in the DPPH scavenging experiment was followed. The absorbance of the control solution (buffered methanol with DPPH) fell within the range of 0.510 ± 0.045. The anti-radical activity was expressed as inhibition percentage (I%), calculated using the formula [(absorbance control − absorbance sample)/absorbance control] × 100.23
The antidiabetic activity was assessed using an α-amylase inhibitory assay. Test tubes were prepared with the test compounds at concentrations ranging from 20 to 120 μg mL−1, along with a control tube containing no sample or drug. Additionally, tubes containing acarbose as the standard drug at the same concentrations as the test compounds were prepared. After adding 50 μL of α-amylase solution to each test compound, the tubes were incubated at room temperature for 10 minutes. Subsequently, 50 μL of starch solution was added to all tubes, which were further incubated at 25 °C for 10 minutes. The evaluation of α-amylase activity inhibition involved adding 100 microliters of 3,5-dinitrosalicylic acid (DNSA) to each test tube. The tubes were then incubated in boiling water for 5 minutes to halt the reaction. The absorbance of the solution was measured by diluting the tubes to 350 microliters with distilled water and recording the absorbance at 540 nm using a spectrophotometer. The percentage of inhibition of α-amylase activity was calculated using a previously established formula.
2.4. In silico analysis
The Schrödinger suite was used for docking three proteins (PDB IDs: 2ZNO, 4JMN and 1IW0) with three synthesized compounds into their active sites.24 Ligand structures were energy-minimized using the LigPrep module. Protein structures were prepared by removing water and cofactors using Schrödinger's Auto Preparation. Receptor grids were created with the Receptor Grid Generation module and docking simulations were performed with the Glide module in standard precision mode. Optimal docking configurations were identified based on the lowest free binding energy and receptor–ligand interactions were visualized with Discovery Studio Visualizer.ADME (Absorption, Distribution, Metabolism, Excretion) properties and toxicity profiles of the synthesized compounds were predicted using the SwissADME tool. The optimized structures were then inputted into the SwissADME web tool. SwissADME analysis was performed on the synthesized compounds to assess their compliance with topological polar surface area (TPSA) and percentage of absorption (% ABS). These properties were calculated using the equation % ABS = 109 – (0.345 × TPSA). Lipinski's rules were applied, stating that an orally active drug should generally have no more than one violation of the following criteria: a maximum of 5 hydrogen bond donors, a maximum of 10 hydrogen bond acceptors, a molecular weight less than 500 and a calculated log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P less than 5.25
P less than 5.25
3. Results and discussions
3.1. Design, substituents variation and characterization
The reaction sequences utilized in the synthesis of the title compounds are illustrated in Fig. 1. The synthesis process commenced with the conversion of isoniazid into N-[(1Z)-1-substituted phenylethylidene] isonicotinyl hydrazide through a reaction with substituted acetophenone. The substituted hydrazide was obtained in good yield after refluxing a mixture of acid hydrazide and Vilsmeier Haack reagent. The reflux reaction subsequently afforded the corresponding 1H-pyrazole-4-carbaldehyde in high yield. This intermediate was then oxidized using potassium permanganate to yield the following carboxylic acids: 3-phenyl-1-(pyridine-4-carbonyl)-1H-pyrazole-4-carboxylic acid (PPP) with an 85% yield, 3-(4-acetophenyl)-1-(pyridine-4-carbonyl)-1H-pyrazole-4-carboxylic acid (APP) with a 78% yield and 3-(4-methylphenyl)-1-(pyridine-4-carbonyl)-1H-pyrazole-4-carboxylic acid (MPP) with a 75% yield. The synthesis of the mentioned compounds employs simple methods and is readily available reagents, potentially lowering production costs and improving accessibility.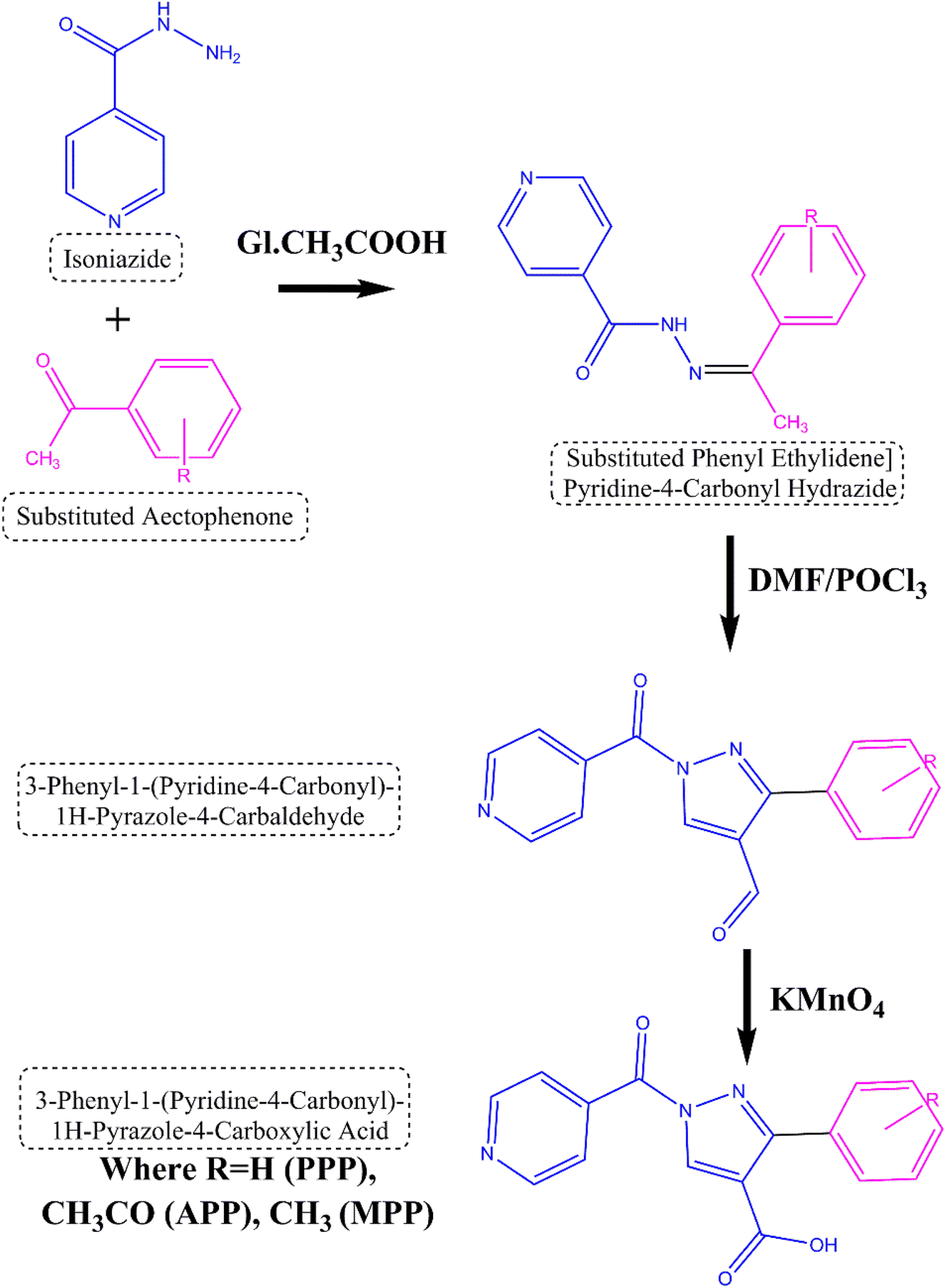 | ||
| Fig. 1 Scheme for derivatives of substituted heterocyclic fused pyrazole derivatives. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 3000–3100 (aromatic C–H). 1H NMR (CDCl3, δ): 9.0–11.0 (1H, s, NH), 8.0–9.0 (1H, s, hydrazone proton), 7.0–8.5 (aromatic protons, m). 13C NMR (CDCl3, δ): 160–170 (CN), 120–150 (aromatic carbons).O), 3000–3100 (aromatic C–H), pyrazole ring vibrations. 1H NMR (CDCl3, δ): 9.5–10.5 (1H, s, aldehyde proton), 7.0–8.5 (aromatic protons, m), pyrazole protons. 13C NMR (CDCl3, δ): 190–200 (aldehyde carbon), 120–150 (aromatic carbons), pyrazole ring carbons.
N), 3000–3100 (aromatic C–H). 1H NMR (CDCl3, δ): 9.0–11.0 (1H, s, NH), 8.0–9.0 (1H, s, hydrazone proton), 7.0–8.5 (aromatic protons, m). 13C NMR (CDCl3, δ): 160–170 (CN), 120–150 (aromatic carbons).O), 3000–3100 (aromatic C–H), pyrazole ring vibrations. 1H NMR (CDCl3, δ): 9.5–10.5 (1H, s, aldehyde proton), 7.0–8.5 (aromatic protons, m), pyrazole protons. 13C NMR (CDCl3, δ): 190–200 (aldehyde carbon), 120–150 (aromatic carbons), pyrazole ring carbons.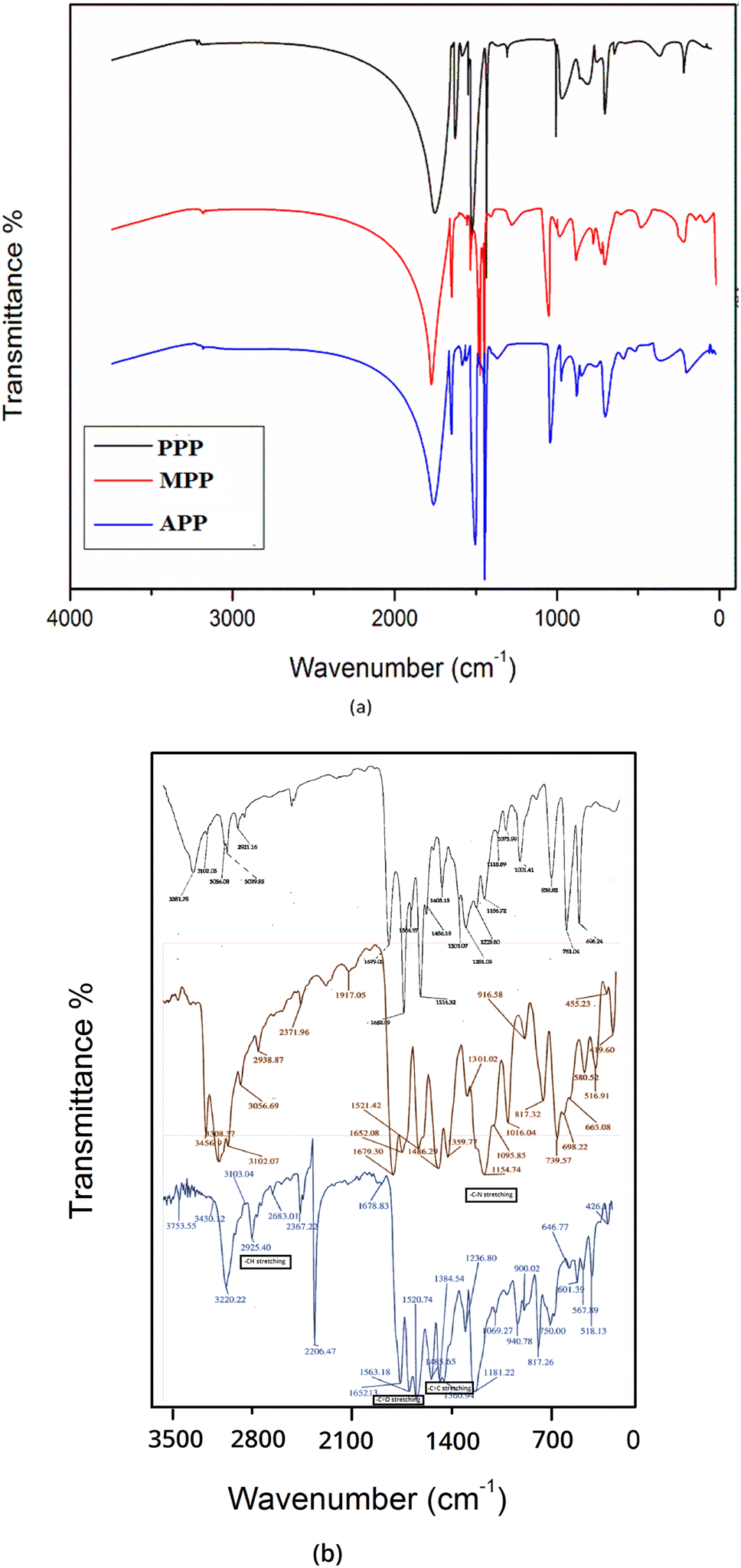 | ||
| Fig. 2 (a) Theoretical and (b) experimental FT-IR spectra for the derivatives. | ||
Each compound has a distinct structure represented by its molecular formula, affecting its molecular weight. The synthesis process yields for each compound range from 85% to 75%. Additionally, the melting point (MP) of each compound falls within the range of 150 °C to 170 °C.
3.2. Computational and topological studies
Based on DFT calculations presented in Table 1, the phenyl-substituted heterocyclic compounds with fused pyrazole carboxylic acid derivatives, namely PPP, APP and MPP, exhibited distinct properties. Compound APP demonstrated the highest ionization potential (6.9276 eV) and electron affinity (0.3058 eV), indicating greater stability. It also displayed the lowest EHOMO (−6.9276 eV) and ELUMO (−0.3058 eV) values, suggesting enhanced reactivity.28 Compound APP exhibited the largest gap between EHOMO and ELUMO (6.6217 eV), indicating lower chemical reactivity compared to MPP (6.3145 eV). Additionally, APP displays the highest electron negativity (3.6167), absolute hardness (3.1573) and softness (0.1510), suggesting greater stability and lower reactivity compared to MPP and PPP.29 Furthermore, APP exhibited the highest electrophilicity index (1.9754) and electronic charges (ΔNmax = 0.5462), indicating its susceptibility to electrophilic reactions and distinct charge distribution. Compound APP also shows the highest dipole moment (5.9024 debye), polarizability (294.8978 Å3) and hyperpolarizability (882.6849 Å5), suggesting stronger polarity and responsiveness to electric fields.30 These properties underscore APP's potential as a bioactive compound with significant implications for drug development and therapeutic applications, where stability, reactivity and interaction with biological targets are crucial.| Parameters | PPP | APP | MPP |
|---|---|---|---|
| EHOMO (eV) | −6.7229 | −6.9276 | −6.5741 |
| ELUMO (eV) | −0.2732 | −0.3058 | −0.2596 |
| Ionisation potential | 6.7229 | 6.9276 | 6.5741 |
| Electron affinity | 0.2732 | 0.3058 | 0.2596 |
| EHOMO − ELUMO (eV) | 6.4498 | 6.6217 | 6.3145 |
| Electron negativity (χ) | 3.4981 | 3.6167 | 3.4168 |
| Chemical potential (μ) | −3.4981 | −3.6167 | −3.4168 |
| Absolute hardness (η) | 3.2249 | 3.3109 | 3.1573 |
| Softness (S) | 0.1550 | 0.1510 | 0.1584 |
| Electrophilicity index (ω) | 1.8972 | 1.9754 | 1.8489 |
| Electronic charges (ΔNmax) | 0.5424 | 0.5462 | 0.5411 |
| E(RB3LYP) (a.u) | −1006.2827 | −1158.9357 | −1045.6038 |
| Dipole moment (debye) | 3.1105 | 5.9024 | 3.4555 |
| Polarizability | 255.1708 | 294.8978 | 274.7100 |
| Hyperpolarizability | 2739.0598 | 882.6849 | 4082.4434 |
The investigation into the regions of the Molecular Electrostatic Potential (MEP) for the derivatives was conducted via DFT calculation using optimized geometry at the B3LYP/6-311++G level. The MEP surface revealed electrophilic reactivity (negative regions: red and yellow colors) and nucleophilic reactivity (positive regions: blue colors), as shown in Fig. 3. Surface analysis indicated a common possible site for electrophilic attack across all compounds, notably the carbonyl group (C7O9) exhibiting negative regions. The negative molecular electrostatic potential values for PPP, APP and MPP were 7.281, 7.597 and 7.442 atomic units, respectively. Similarly, all compounds exhibited a common possible site for nucleophilic attack, with the carboxyl group (O15–H28) displaying positive regions.31
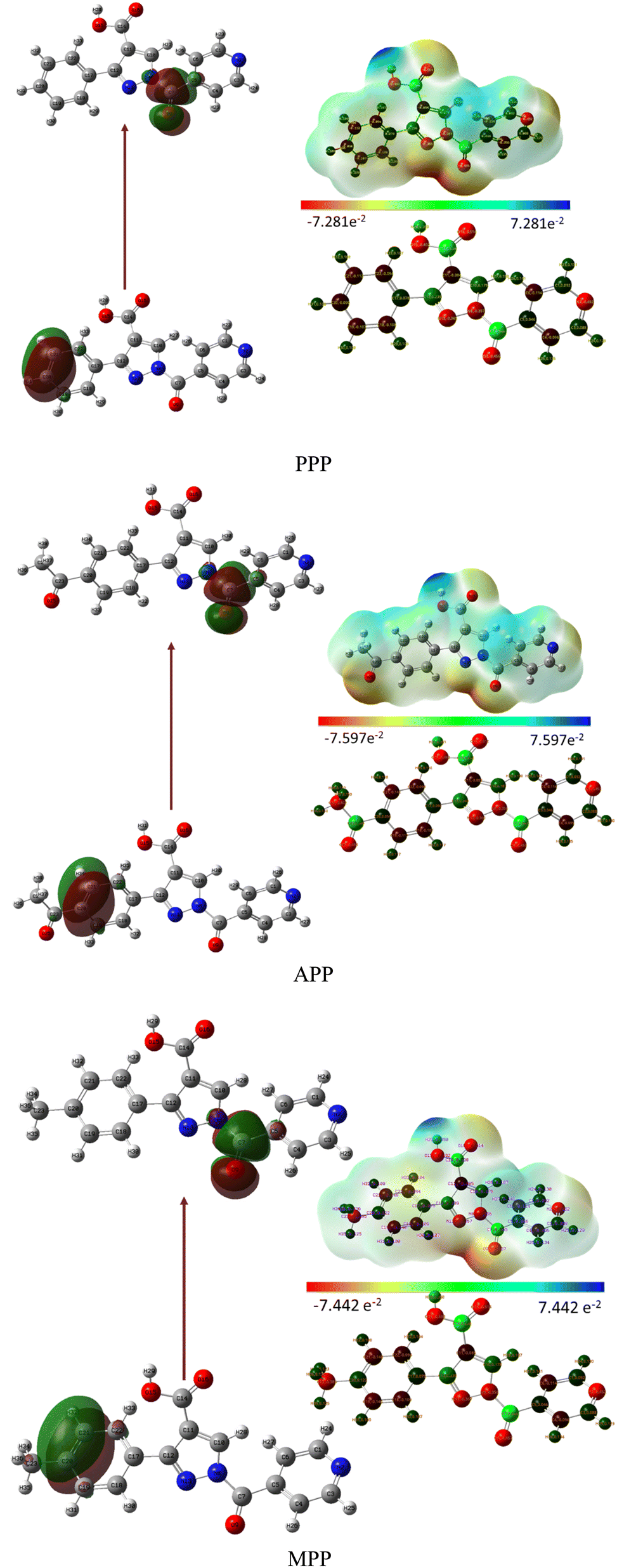 | ||
| Fig. 3 Representation of the HOMO, LUMO and MEP for PPP, APP and MPP. | ||
The analysis of PPP compound revealed specific bonds with distinct occupancies and energies. Bonds C18–C19, C21–C22, C3–C4 and C17–C18 exhibit an occupancy of 0.0146 and energy of 0.5892, indicating relatively high energy and low occupancy, potentially involving more delocalized electrons in the phenyl and pyridine rings. Bonds C3–C4 show an occupancy of 0.02369 and energy of 0.5608, suggesting moderate energy and occupancy, typical of single bonds that are moderately strong. Bonds C18–C19 and C10–C11 exhibit occupancies of 0.30245 and 0.30343, with energies of 0.0355 and 0.0021, indicating highly delocalized bonds in the phenyl and pyrazole rings, possibly signifying highly polarized bonds. Furthermore, the analysis of C–O bonds in APP reveals significant electron density and high stability,32 typical in organic compounds. These bonds exhibit occupancies of 0.012 and 0.099, with energies of 0.542 and 0.321 atomic units, respectively. Additionally, Nitrogen lone pairs (N8, N2, N13) demonstrate occupancies ranging from 1.522 to 1.951 and energies ranging from −0.294 to −0.441 atomic units, indicating highly stable lone pairs with minimal bonding interaction. This information is crucial for understanding the molecule's potential sites for protonation or hydrogen bonding.
The local parameters (f−, f+, f0, ω−, ω+) and the dual descriptor (Δf) representing the Fukui function are shown in Fig. 4. Typically, overall derivatives C20 and C21 exhibit values of 0.4792 and 0.4925; 0.5127 and 0.4544; 0.4555 and 0.5105, respectively, indicating the electrophilicity order as APP > PPP > MPP. This order reflects how readily the molecule can accept additional electron density. In terms of nucleophilic attack, the order is PPP < APP < MPP, with active sites for nucleophilicity at C7 and O9 having values of 0.572 and 0.336; 0.595 and 0.350; 0.603 and 0.327, respectively.
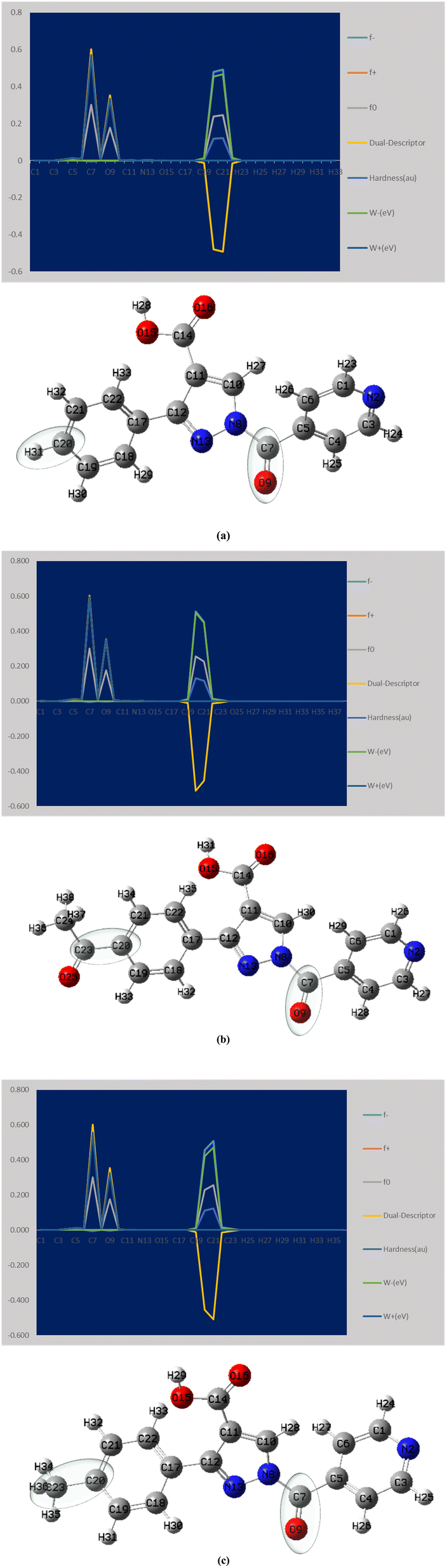 | ||
| Fig. 4 Presents the Fukui function analysis for the derivatives (a) PPP (b) APP (c) MPP. | ||
The nature and strength of bonding between neighboring atoms were characterized by examining interactions, whether covalent or non-covalent. Fig. 5–7, displaying ELF, IRI and LOL surface maps provide insight into molecular bonding. The ELF map depicts electron energy density, indicating intense hydrogen bonding between atoms, with blue representing minimum values and red signifying maximum values. Regions surrounding C–C and C–N atoms exhibit lower values, suggesting delocalized electrons involved in bonding and non-bonding interactions. Notable ELF or LOL values in contested regions indicate distinct electron localization from covalent bonds or lone pairs of electrons, suggesting electronic transitions between C–C and C–N atoms. IRI is effective for detecting covalent and hydrogen bonds, steric regions and halogen bonds. Subsurface maps with IRI values, such as IRI = 0.800, reveal various interactions, including covalent bonds and steric repulsion within phenyl rings, indicating stronger chemical bonds and weaker interactions.33 These analyses offer detailed insights into molecule structure and properties.
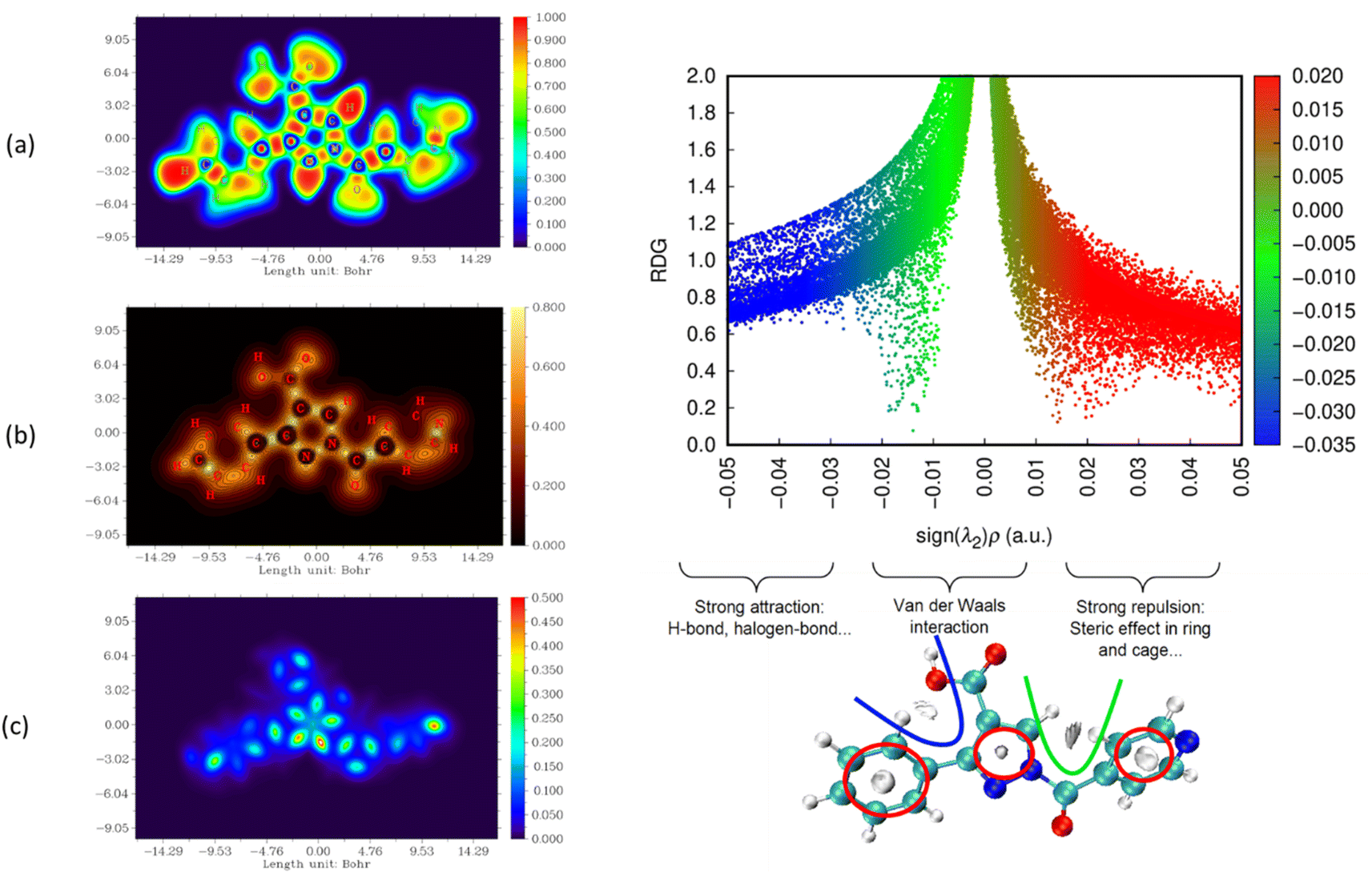 | ||
| Fig. 5 Visualizations of electronic structure, bonding interactions and regions of steric hindrance within the molecule through ELF, LOL, IRI and RDG of PPP. | ||
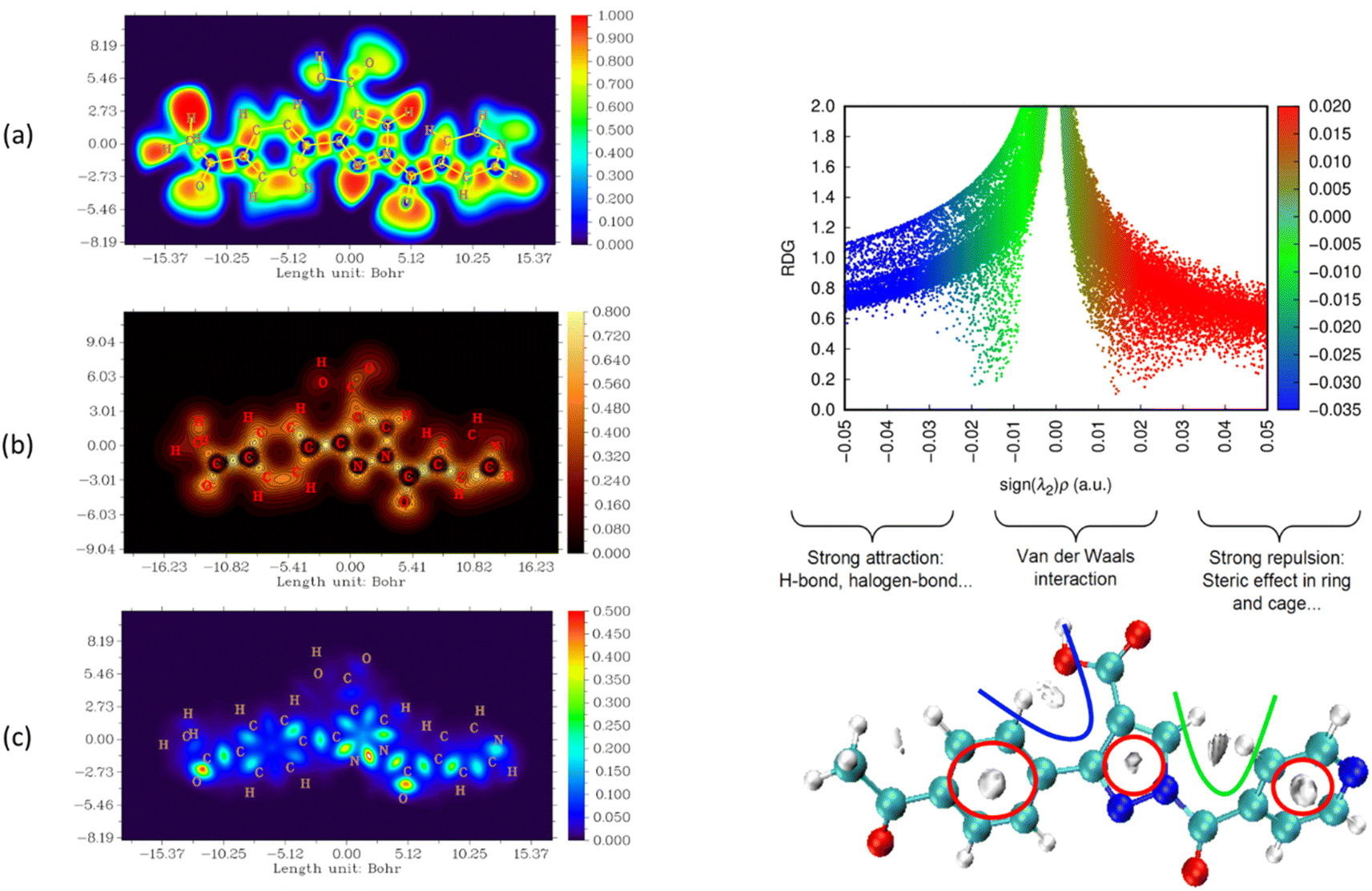 | ||
| Fig. 6 Visualizations of electronic structure, bonding interactions and regions of steric hindrance within the molecule through ELF, LOL, IRI and RDG of APP. | ||
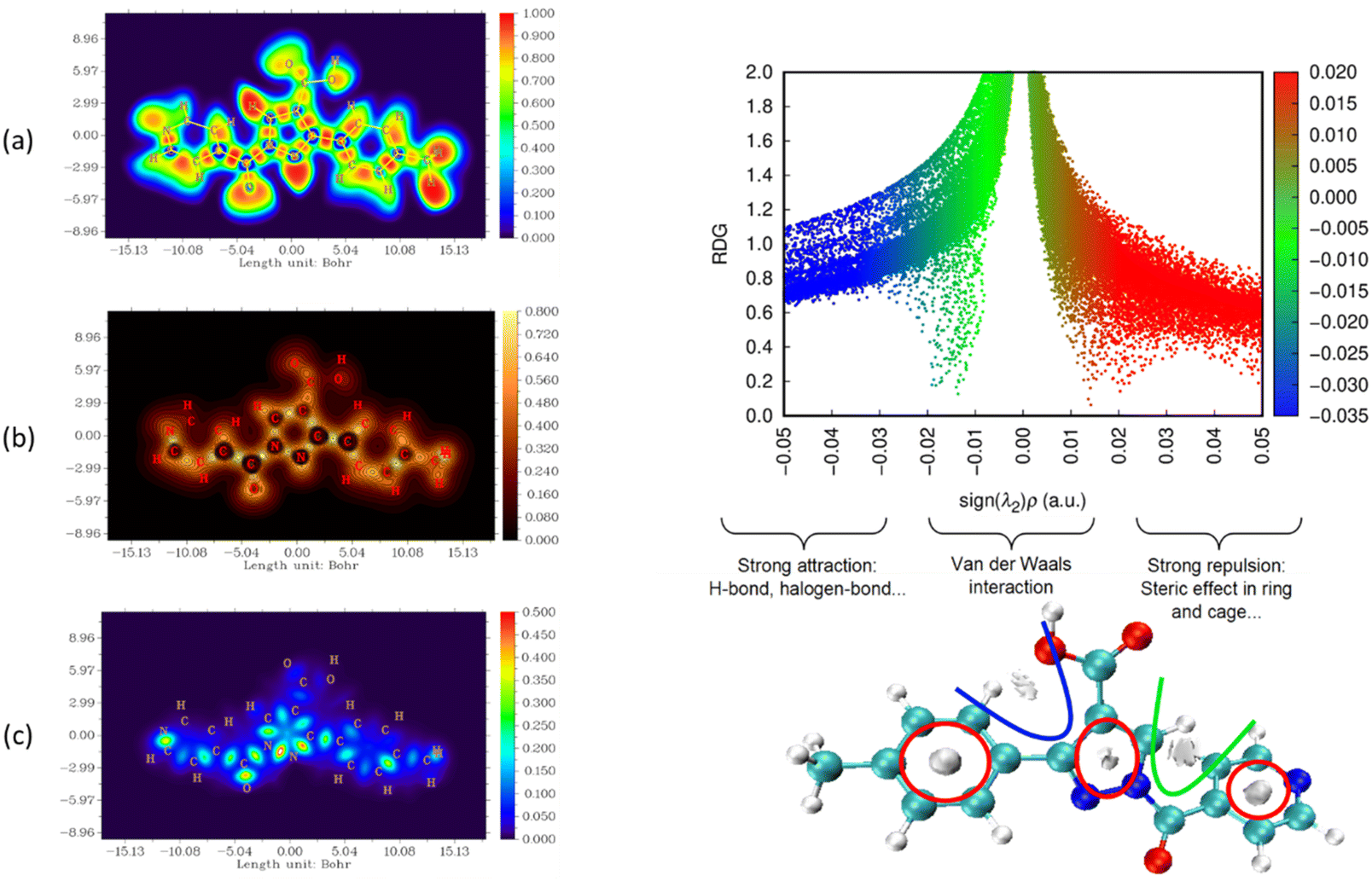 | ||
| Fig. 7 Visualizations of electronic structure, bonding interactions and regions of steric hindrance within the molecule through ELF, LOL, IRI and RDG of MPP. | ||
To delve deeper into bond interactions, we utilized RDG analysis, a computational tool for exploring non-covalent interactions in molecular systems. RDG color scaling represents interactions: blue indicates hydrogen bonding, green represents van der Waals interactions and red signifies steric effects, all based on electron density property.34 RDG and sign(λ2)ρ plots were generated for the molecule using Multiwfn and VMD software. Analysis revealed three interaction sites. The blue contour in the RDG scatter plot is around −0.020 a.u. indicates strong attraction between C–H hydrogen bonding interactions, confirming the presence of hydrogen bonding interactions in the molecular structure of the derivatives.
3.3. In vitro analysis
The antimicrobial activity of synthesized compounds against various microorganisms is presented in Table 2. Compound PPP exhibited significant activity against Staphylococcus aureus (MIC: 7.81 μg mL−1) and Bacillus subtilis (MIC: 7.81 μg mL−1), as well as Pseudomonas aeruginosa (MIC: 7.81 μg mL−1). Compound MPP, however, showed lower activity against Bacillus subtilis (MIC: 31.25 μg mL−1) and Escherichia coli (MIC: 31.25 μg mL−1). Compound APP demonstrated notable activity against Aspergillus fumigates (MIC: 15.62 μg mL−1). All compounds exhibited low activity against Aspergillus niger (MIC: 31.25 μg mL−1).| Compounds | Gram-positive | Gram-negative | Fungi | |||
|---|---|---|---|---|---|---|
| S. a | B. s | E. c | P. a | A. n | A. f | |
| PPP | 7.81 | 7.81 | 31.25 | 7.81 | 31.25 | 15.62 |
| APP | 31.25 | 15.62 | 31.25 | 15.62 | 31.25 | 15.62 |
| MPP | 31.25 | 31.25 | 62.50 | 31.25 | 62.50 | 62.50 |
| Ciprofloxacin (10 μg mL−1) | 15.62 | 7.81 | 15.63 | 7.81 | — | — |
| Fluconazole (10 μg mL−1) | — | — | — | — | 7.81 | 7.81 |
The antioxidant activity of the compounds was presented in Table 3, showing results at varying concentrations (20–120 μg mL−1) using the DPPH radical scavenging assay. At 120 μg mL−1, APP and PPP showed the highest activity (97.34% and 92.03% scavenging, respectively), with MPP also significant (89.93%). APP consistently exhibited the highest inhibition among compounds, followed by PPP and MPP. Both APP and MPP approached ascorbic acid's activity levels. At 20 μg mL−1, APP led, maintaining its superiority across all concentrations, followed by PPP and MPP, establishing the order: APP > PPP > MPP.
| Conc. (μg mL−1) | % DPPH | |||
|---|---|---|---|---|
| PPP | APP | MPP | Ascorbic acid | |
| 20 | 69.11 ± 1.22 | 70.08 ± 1.06 | 61.44 ± 1.55 | 91.81 ± 1.21 |
| 40 | 78.33 ± 1.43 | 79.25 ± 1.76 | 71.67 ± 1.43 | 91.84 ± 1.51 |
| 60 | 80.07 ± 1.09 | 83.08 ± 1.98 | 76.24 ± 1.43 | 91.88 ± 1.99 |
| 80 | 87.24 ± 1.32 | 89.88 ± 1.06 | 79.08 ± 1.00 | 93.98 ± 1.24 |
| 100 | 91.03 ± 1.09 | 94.99 ± 1.03 | 82.07 ± 1.02 | 94.33 ± 1.08 |
| 120 | 92.03 ± 1.30 | 97.34 ± 1.45 | 89.44 ± 1.03 | 96.47 ± 1.07 |
The antidiabetic activity of three synthesized phenyl-substituted heterocyclic compounds, evaluated for their ability to inhibit α-amylase and α-glucosidase, aimed at reducing post-prandial hyperglycemia. Among them, PPP displayed the highest activity with an IC50 of 122.17 μg mL−1, followed by APP (164.62 μg mL−1) and MPP (200.94 μg mL−1), respectively. In comparison, the standard drug, acarbose, exhibited superior α-amylase inhibitory activity with an IC50 value of 69.74 μg mL−1, as represented in Table 4.
| Compound | α-Amylase inhibition method IC50 value (μg mL−1) |
|---|---|
| PPP | 122.17 |
| APP | 164.62 |
| MPP | 200.94 |
| Acarbose | 69.74 |
3.4. In silico studies
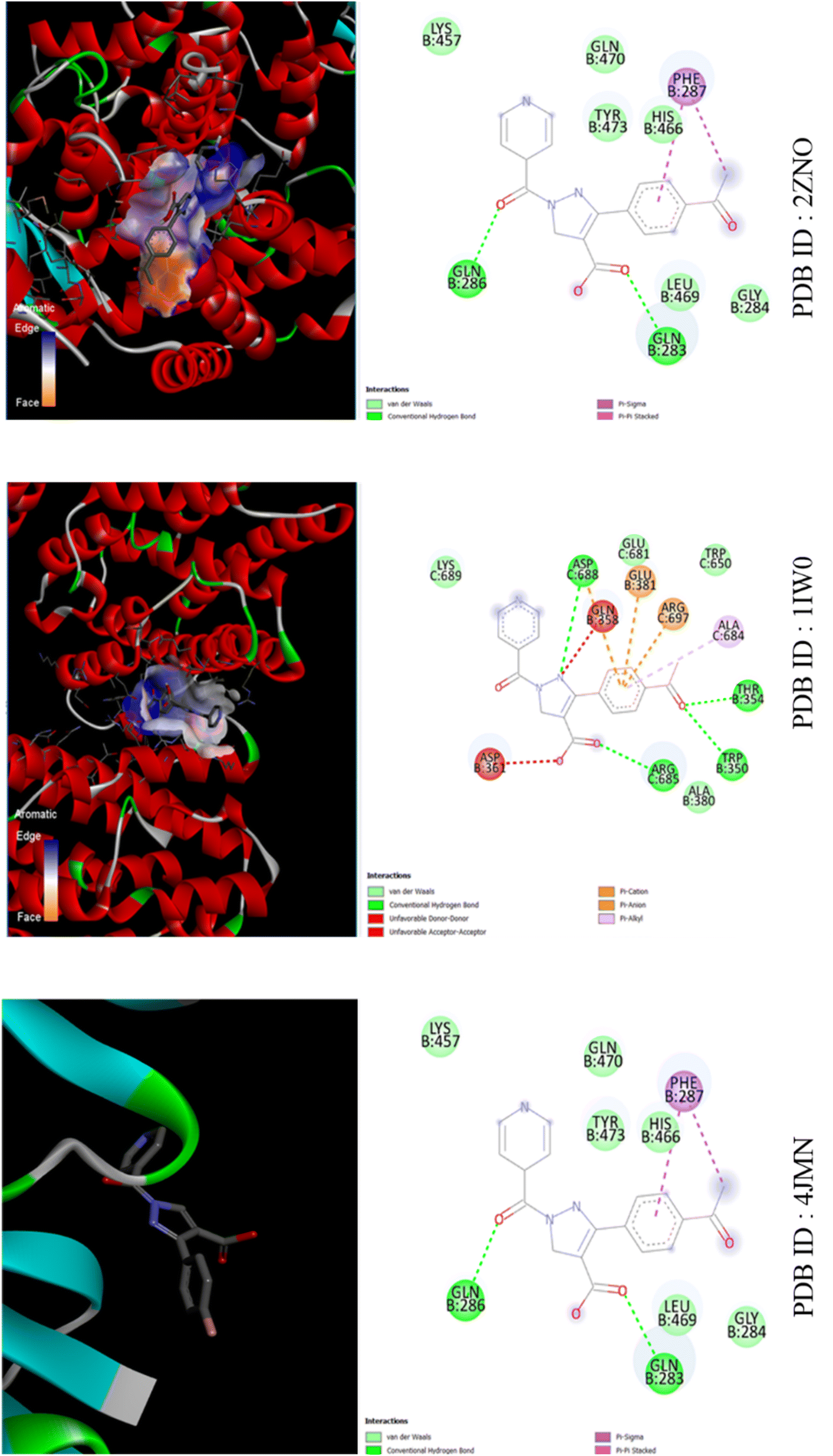 | ||
| Fig. 8 The binding interactions of compound APP against LD Transpeptidase (PDB ID: 4JMN), Heme Oxygenase (PDB ID: 1IW0) and PPAR Gamma (PDB ID: 2ZNO). | ||
As shown in Fig. S3,† for PPP and 4JMN, hydrophobic interactions occur at ASP388 (3.72 Å), LYS389 (3.64 Å), THR654 and GLN658, along with hydrogen bonds at ASP388, ARG397 and ASP661. In 1IW0, hydrophobic interactions involve GLN283 (3.45 Å), GLN286 (3.97 Å), PHE287 (3.83 Å), LEU469 (3.87 Å) and TYR473 (3.42 Å), while hydrogen bonds occur with TYR473 (1.93 Å).In 2ZNO, hydrophobic interactions involve VAL97, TYR101, LEU453 (3.45 Å), LEU465, LEU469 (3.87 Å), GLN470, VAL57 (3.45 Å), VAL89, MET167 (3.85 Å) and TRP202, with hydrogen bonds formed by HIS54 (3.64 Å), ARG95 (2.29 Å), HIS165 (3.27 Å) and MET167 (3.73 Å). Additionally, pi-stacking interactions involve TRP202 (3.90 Å).
For APP and 4JMN, hydrophobic interactions involve GLU357 (3.72 Å), GLN358 (3.64 Å), ASN378, GLU381 and ARG685 (3.90 Å), with hydrogen bonds formed by ASP361 (2.39 Å and 2.25 Å), ALA380 (2.80 Å), GLU381 (2.13 Å) and ARG697 (2.85 Å). Additionally, ARG685 exhibits a pi–cation interaction (5.85 Å).In 1IW0, hydrophobic interactions occur with GLN283 (3.57 Å), GLN286 (3.97 Å), PHE287 (3.83 Å), LEU469 (3.87 Å) and TYR473 (3.42 Å), while a hydrogen bond forms with TYR473 (1.93 Å).In 2ZNO, hydrophobic interactions involve GLN283 (3.57 Å), GLN286 (3.97 Å), PHE287 (3.83 Å), LEU469 (3.87 Å) and TYR473 (3.42 Å).
For MPP and 4JMN, hydrophobic interactions involve GLN358 (3.98 Å), ARG685 (3.97 Å), ASP688 (3.63 Å) and LYS689 (3.40 Å), with hydrogen bonds formed by GLN358 (2.20 Å), ASP361 (3.03 Å and 2.54 Å) and ASP688 (2.32 Å and 1.92 Å). Additionally, ARG697 forms a hydrogen bond (2.85 Å) and ARG685 exhibits a pi–cation interaction (5.90 Å). In 1IW0, hydrophobic interactions involve GLN283 (3.57 Å), GLN286 (3.97 Å), PHE287 (3.83 Å), LEU469 (3.87 Å) and TYR473 (3.42 Å), while a hydrogen bond forms with TYR473 (1.93 Å). In 2ZNO, hydrophobic interactions involve GLN283 (3.57 Å), GLN286 (3.97 Å), PHE287 (3.83 Å), LEU469 (3.87 Å) and TYR473 (3.42 Å) as shown in Fig. S4.† The hierarchy of bioactivity followed the sequence: PPP > APP > MPP, with PPP attributed to its acceptor nature.
P values below 5 and appropriate hydrogen bond acceptor and donor counts.33 Additionally, the number of rotatable bonds, polar surface area and volume fell within acceptable ranges. However, TPSA values exceeded 60 Å2, indicating potential limitations in blood–brain barrier penetration despite meeting the criteria for intestinal drug absorption.39
| Compounds | Molecular formula | Molecular weight | MP (°C) | logP |
% ABS | TPSA | natoms |
|---|---|---|---|---|---|---|---|
| PPP | C16H11N3O3 | 293 | 150 | 1.74 | 79.64 | 85.09 | 22 |
| APP | C18H13N3O4 | 335 | 175 | 2.55 | 79.64 | 85.09 | 23 |
| MPP | C17H13N3O3 | 307 | 170 | 2.18 | 79.64 | 85.09 | 24 |
Compound PPP demonstrates favorable drug-likeness characteristics, boasting the highest QED value (0.80), which indicates excellent drug-likeness. However, it also exhibits potentially low absorption across intestinal barriers, as evidenced by the lowest Caco-2 permeability value (−5.12). Moreover, it displays high plasma protein binding (99.04%) and limited distribution throughout the body, indicated by the lowest volume distribution value (−0.78). Nonetheless, its significant cytochrome P450 inhibition, particularly for CYP1A2 and CYP2C9, raises concerns regarding potential impacts on drug metabolism. On the other hand, compound MPP showcases efficient metabolism and excretion. Its high predicted clearance (1.88 mL min−1 kg−1) suggests rapid elimination, while its low bioconcentration factor (0.18) indicates reduced environmental impact. Compound APP demonstrates high permeability and distribution, which could enhance its effectiveness in reaching target sites. However, it also exhibits higher toxicity and potential cardiac effects, reflected in its highest hERG Blockers value (0.10) and DILI probability (0.98).
The physicochemical properties of compounds APP, PPP and MPP reveal their distinct characteristics across various parameters. In terms of molecular size, APP emerges as the largest (335) followed by MPP (307) and PPP (293). Density-wise, PPP exhibits the highest density (1) compared to APP and MPP. Regarding the topological polar surface area (TPSA), APP leads with 102.15, surpassing MPP and PPP at 85. The logP value ranks MPP highest (1.98), followed by APP (1.32) and then PPP (1.27). Assessing drug-likeness via QED value, PPP and MPP tie at 0.80, while APP trails slightly at 0.74. Fsp3 values indicate APP with the highest value (0.059), followed by MPP (0.056) and PPP (0.0). Synthetic accessibility scores remain identical for all compounds at 2.0. In absorption studies, Caco-2 permeability ranks PPP lowest (−5.12), followed by MPP (−4.60), then APP (−4.95). logPeff value places APP highest (0.99), equaled by PPP and MPP (0.98). Distribution metrics unveil that PPP exhibits the highest plasma protein binding (99.04%), followed by MPP (98.71%) and APP (95.68%). Volume distribution sees PPP with the lowest value (−0.78), trailed by MPP (−0.68) and APP (−0.64). Concerning metabolism, MPP demonstrated higher enzyme inhibition (CYP1A2 and CYP2C9) compared to PPP and APP. Predicted clearance values rank MPP as the highest (1.88 mL min−1 kg−1), followed by APP (1.70) and PPP (1.63). Likewise, predicted half-life values were observed at position PPP with the longest duration (1.42 hours), followed closely by MPP (1.31) and APP (1.32). In terms of toxicity, APP exhibits the highest hERG inhibition (0.10), while liver injury probability remains high for all compounds, with APP showing the lowest probability (0.98). Tox21 pathway activity presents PPP with the lowest activity (0.0), matched by APP and MPP. Considering the environmental impact, the bioconcentration factor ranks MPP lowest (0.18), with PPP and APP above it.40 Comparison of the derivatized structures with the unsubstituted compound revealed enhanced ADME properties in the derivatized forms. Specifically, MPP and APP exhibited better pharmacokinetic profiles, including improved intestinal absorption and limited blood–brain barrier penetration. These compounds also demonstrated efficient metabolism and excretion profiles, characterized by high predicted clearance values, increased drug-likeness, metabolic stability and lower bioconcentration factors. These attributes suggested a reduced environmental impact and notable biological activities of the derivatized structures, underscoring their therapeutic potential. Furthermore, the ADME predictions corresponded well with the in vitro results, affirming the improved pharmacokinetic profiles of the derivatized compounds compared to the unsubstituted compound.
4. Conclusion
In summary, this study highlights the promising potential of phenyl-substituted heterocyclic compounds containing fused pyrazole carboxylic acid derivatives, showing significant improvements in antimicrobial, antioxidant and antidiabetic activities. The synthesized compounds, specifically PPP, APP and MPP, demonstrated effective bioactivity, validating the effectiveness of the design in enhancing therapeutic properties. Spectral characterization through 1H and 13C NMR, combined with computational analyses, confirmed the optimized geometrical structures of the compounds. HOMO–LUMO parameters elucidated favorable chemical reactivity, while MEP surface analysis and Fukui function provided insights into electrophilic and nucleophilic sites. ELF, IRI and LOL measurements assessed electron localization and NBO analysis emphasized stability and reactivity zones. The biological assessments revealed that the synthesized compounds exhibited notable antimicrobial activity, with MIC values indicating effectiveness against various microorganisms. Compound APP, in particular, demonstrated promising antimicrobial and anti-cancer potential, warranting further therapeutic exploration. Despite showing varied radical scavenging activity, the compounds did not reach the antioxidant efficacy of ascorbic acid. The derivative PPP exhibited significant α-amylase inhibition, though it was less potent compared to acarbose. In silico studies further supported these findings, with favorable docking scores and binding affinities for the compounds, suggesting good therapeutic potential. Calculated % ABS and logP values indicated good permeability and suitable partition coefficients, reinforcing their viability for drug development. Overall, the in vitro results aligned well with the in silico predictions, underscoring the potential of these compounds as effective therapeutic agents.
Data availability
Data available on request from the authors.Conflicts of interest
There are no disputes of interest declared by all the authors and we have no conflicts of interest to disclose.Acknowledgements
The authors express their sincere gratitude to the Management of Islamiah College (Autonomous) for generously providing the lab facilities essential for conducting the research in the Department of Chemistry, Islamiah College (Autonomous), Vaniyambadi. The authors extend their appreciation to the Researchers Supporting Project number (RSP2024R114), King Saud University, Riyadh, Saudi Arabia.References
- Y. Kaddouri, F. Abrigach, E. B. Yousfi, M. El Kodadi and R. Touzani, New thiazole, pyridine and pyrazole derivatives as antioxidant candidates: synthesis, DFT calculations and molecular docking study, Heliyon, 2020, 6(1), e03185 Search PubMed.
- S. Naveen, A. Muthusamy, K. Karthik, S. Shankar, P. Malarvizhi, R. Sundararajan and R. Rajendran, Synthesis, characterization, crystal structure, Hirshfeld surface analysis, antioxidant properties and DFT calculations of a novel pyrazole derivative: ethyl 1-(2, 4-dimethylphenyl)-3-methyl-5-phenyl-1H-pyrazole-4-carboxylate, J. Mol. Struct., 2021, 1226, 129350 CAS.
- A. M. Farag and A. M. Fahim, Synthesis, biological evaluation and DFT calculation of novel pyrazole and pyrimidine derivatives, J. Mol. Struct., 2019, 1179, 304–314 CAS.
- R. R. Pillai, S. Kumar, S. Menon, S. Paul, S. James, G. Kuriakose, M. Babu and R. Thomas, Synthesis, spectroscopic characterization, reactive properties by DFT calculations, molecular dynamics simulations and biological evaluation of Schiff bases tethered 1, 2, 4-triazole and pyrazole rings, J. Mol. Struct., 2019, 1177, 47–54 CAS.
- M. Marinescu and C.-V. Popa, Pyridine compounds with antimicrobial and antiviral activities, Int. J. Mol. Sci., 2022, 23(10), 5659 CrossRef CAS PubMed.
- N. U. A. Mohsin, S. Aslam, M. Ahmad, M. Irfan, S. A. Al-Hussain and M. E. A. Zaki, Cyclooxygenase-2 (COX-2) as a target of anticancer agents: A review of novel synthesized scaffolds having anticancer and COX-2 inhibitory potentialities, Pharmaceuticals, 2022, 15(12), 1471 CrossRef CAS PubMed.
- Y. Zhang, C. Wu, N. Zhang, R. Fan, Y. Ye and J. Xu, Recent advances in the development of pyrazole derivatives as anticancer agents, Int. J. Mol. Sci., 2023, 24(16), 12724 CrossRef CAS PubMed.
- G. Ahmad, S. Basak, N. Khan, A. Ahamed and A. Khan, N-Heterocycles as promising antiviral agents: a comprehensive overview, Molecules, 2024, 29(10), 2232 CrossRef CAS PubMed.
- R. D. Alharthy, M. Khan, S. Ali, H. Al-Habib and A. Khan, Pyrazole derivatives of pyridine and naphthyridine as proapoptotic agents in cervical and breast cancer cells, Sci. Rep., 2023, 13(1), 5370 CrossRef CAS PubMed.
- P. Kumar, R. Patel, S. Sharma and A. Jha, Non-classical transpeptidases yield insight into new antibacterials, Nat. Chem. Biol., 2017, 13(1), 54–61 CrossRef CAS PubMed.
- M. Unno, T. Matsui and M. Ikeda-Saito, Structure and catalytic mechanism of heme oxygenase, Nat. Prod. Rep., 2007, 24(3), 553–570 RSC.
- M. Ohashi, T. Hayashi, A. Ueno, Y. Sasaki, Y. Shimada, N. Matsunaga, M. Yamashita, K. Yamaguchi and K. Kuroda, Design, synthesis and structural analysis of phenylpropanoic acid-type PPARγ-selective agonists: discovery of reversed stereochemistry–activity relationship, J. Med. Chem., 2011, 54(1), 331–341 CrossRef CAS PubMed.
- A. Bendi, P. Yadav, K. Saini, A. Singh Bhathiwal and N. Raghav, A comprehensive examination of heterocyclic scaffold chemistry for antitubercular activity, Chem. Biodivers., 2024, 21(5), e202400067 CrossRef CAS PubMed.
- P. Liao and J. Allison, Ionization processes in matrix-assisted laser desorption/ionization mass spectrometry: matrix-dependent formation of [M+ H]+ vs [M+ Na]+ ions of small peptides and some mechanistic comments, J. Mass Spectrom., 1995, 30(3), 408–423 CrossRef CAS.
- A. Frisch, Gaussian 09W Reference, Wallingford, USA, 2009, p. , p. 25 Search PubMed.
- R. Dennington, T. Keith and J. Millam, GaussView, Version 5, 2009 Search PubMed.
- A. R. Kumar, R. Karthikeyan, S. Muthuraman, M. Shanmugam, S. Kannan and M. Rajan, Spectroscopic (FT-IR, FT-Raman, UV–Vis and NMR) and computational (DFT, MESP, NBO, NCI, LOL, ELF, RDG and QTAIM) profiling of 5-chloro-2-hydroxy-3-methoxybenzaldehyde: a promising antitumor agent, J. Mol. Struct., 2024, 1298, 136974 CrossRef CAS.
- A. A. Alfi, H. Abou-Elwafa, A. El-Metwally and M. Khan, Molecular modeling and docking studies of new antioxidant pyrazole-thiazole hybrids, J. Mol. Struct., 2022, 1267, 133582 CrossRef.
- R. Chen, X. Li, Y. Wang and M. Yang, Molecular structure, vibrational spectroscopy (FT-IR, Raman), solvent effects, molecular docking and DFT studies of 1-(4-chlorophenyl)-3-(4-ethoxyphenyl)-prop-2-en-1-one, J. Mol. Struct., 2022, 1267, 133660 CrossRef CAS.
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33(5), 580–592 CrossRef CAS PubMed.
- H. Kolancılar, H. Özcan, A. Yılmaz, A. Salan and A. Ece, 2, 3-Dichloronaphthoquinone derivatives: synthesis, antimicrobial activity, molecular modelling and ADMET studies, Bioorg. Chem., 2024, 146, 107300 CrossRef PubMed.
- T. K. Bakir, M. A. A. Ashweeqi and H. Muğlu, Novel 1, 3, 4-thiadiazol derivatives including α-lipoic acid: synthesis, characterization and antioxidant properties, J. Mol. Struct., 2024, 1307, 137980 CrossRef CAS.
- V. Pradeep, P. Veerakumar and V. P. Veeraraghavan, Facile microwave-assisted hydrothermal synthesis of copper oxide nanoneedle arrays for practical biomedical applications, Cureus, 2024, 16(1), e51678 Search PubMed.
- B. Ş. Dingiş, N. Kılınç, S. Öztürk and M. Alkan, In silico design, synthesis and antitubercular activity of novel 2-acylhydrazono-5-arylmethylene-4-thiazolidinones as enoyl-acyl carrier protein reductase inhibitors, J. Biomol. Struct. Dyn., 2024, 1–19 Search PubMed.
- M. Malik, K. K. Verma, R. Kaushik, P. Gaur and K. Lata, In-silico ADMET study and molecular docking of triazole-3-thione derivatives targeting lipoteichoic acid synthase in gram-positive bacteria, J. Appl. Pharm. Sci. Res., 2024, 6(4), 18–28 Search PubMed.
- A. Nazeer, Q. Umar, Y. Yang and M. Luo, Zn (II), Cu (II), Co (II) and Ni (II) complexes bearing aza-heterocyclic ligands: synthesis, characterization and anticancer activities, J. Iran Chem. Soc., 2024, 1–11 Search PubMed.
- M. Akbari, M. Nikoorazm, B. Tahmasbi and A. Ghorbani-Choghamarani, Homoselective synthesis of tetrazoles and chemoselective oxidation of sulfides using Ni (II)-Schiff base complex stabilized on 3-dimensional mesoporous KIT-6 surface as a recyclable nanocatalyst, Inorg. Chem. Commun., 2024, 160, 111852 CrossRef CAS.
- A. Kubaib, P. Mohamed Imran and A. Aathif Basha, Applications of the Vienna Ab initio simulation package, DFT and molecular interaction studies for investigating the electrochemical stability and solvation performance of non-aqueous NaMF6 electrolytes for sodium-ion batteries, Comput. Theor. Chem., 2022, 1217, 113934 CrossRef CAS.
- A. Kubaib and P. M. Imran, Fabrication of Li, Na and K electrolytes with doping elements for improved efficiency based on MOT and symmetry, J. Indian Chem. Soc., 2023, 100(1), 100804 CrossRef CAS.
- A. Kubaib and P. M. Imran, Application of lithium and a few relevant electrolytes evaluated as secondary batteries studied using molecular descriptors, band structure and DOS, J. Mater. Sci., 2023, 58(9), 4005–4019 CAS.
- A. Kubaib, N. N. Afroze, P. M. Imran, P. Natarajan, D. Balu and A. Ansari, Insights into the binding mechanism of 2,5-substituted 4-pyrone derivatives as therapeutic agents for fused dimeric interactions: a computational study using QTAIM, dynamics and docking simulations of protein–ligand complexes, Int. J. Quantum Chem., 2024, 124(3), e27330 CAS.
- A. A. Basha, A. Kubaib and M. Azam, Exploring the antiviral potency of γ-FP and PA compounds: electronic characterization, non-covalent interaction analysis and docking profiling with emphasis on QTAIM aspects, Comput. Theor. Chem., 2024, 1231, 114412 CAS.
- M. Selvakumaran, P. M. Imran, A. Kubaib, M. Azam, A. Aathif Basha and S. I. Al-Resayes, Investigations into the anti-inflammatory and anti-diabetic activity of newly synthesized derivatives of 4AP2BOB utilizing DFT, molecular docking and spectroscopic characterization, J. Mol. Liq., 2024, 396, 123983 CAS.
- A. Kubaib, S. Afzal, M. Shahid, M. Murtaza, K. Kousar and S. Khan, Chelation agent as potential target antioxidant: DFT, physicochemical properties, topological analysis and molecular docking studies into intramolecular interactions, Russ. J. Phys. Chem. A, 2023, 97(12), 2884–2893 CAS.
- K. Marciniec, A. Beberok, P. Pęcak, S. Boryczka and D. Wrześniok, Ciprofloxacin and moxifloxacin could interact with SARS-CoV-2 protease: preliminary in silico analysis, Pharmacol. Rep., 2020, 72, 1553–1561 CrossRef CAS PubMed.
- F. Salehi, L. Emami, Z. Rezaei, S. Khabnadideh, B. Tajik and R. Sabet, Fluconazole-like compounds as potential antifungal agents: QSAR, molecular docking and molecular dynamics simulation, J. Chem., 2022, 2022(1), 5031577 Search PubMed.
- E. I. Ilie, L. Popescu, E. A. Luță, A. Biță, A. R. Corbu, D. P. Mihai, A. C. Pogan, T. D. Balaci, A. Mincă, L. E. Duțu and O. T. Olaru, Phytochemical Characterization and Antioxidant Activity Evaluation for Some Plant Extracts in Conjunction with Pharmacological Mechanism Prediction: Insights into Potential Therapeutic Applications in Dyslipidemia and Obesity, Biomedicines, 2024, 12(7), 1431 Search PubMed.
- M. Tolmie, M. J. Bester and Z. Apostolides, Inhibition of α-glucosidase and α-amylase by herbal compounds for the treatment of type 2 diabetes: a validation of in silico reverse docking with in vitro enzyme assays, J. Diabetes, 2021, 13(10), 779–791 CAS.
- A. S. Pirzada, M. A. Khan, S. Khan and M. Shahid, Physicochemical properties, pharmacokinetic studies, DFT approach and antioxidant activity of nitro and chloro indolinone derivatives, Front. Chem., 2024, 12, 1360719 Search PubMed.
- S. Kibet, N. M. Kimani, S. S. Mwanza, C. M. Mudalungu, C. B. R. Santos and C. M. Tanga, Unveiling the potential of Ent-Kaurane diterpenoids: multifaceted natural products for drug discovery, Pharmaceuticals, 2024, 17(4), 510 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04217d |
| This journal is © The Royal Society of Chemistry 2024 |