
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMXene/sPS nanocomposites: rheological, electrical conductivity, polymorphism, mechanical, thermal, and flammability properties†
Tongtong Zhang a,
Shuo Wangb,
Runkai Qiua,
Xiaolong Yanga and
Fangming Zhu*ac
a,
Shuo Wangb,
Runkai Qiua,
Xiaolong Yanga and
Fangming Zhu*ac
aGDHPPC Lab, School of Chemistry, Sun Yat-Sen University, Guangzhou, 510275, China
bSouth China Advanced Institute for Soft Matter Science and Technology, School of Emergent Soft Matter, South China University of Technology, Guangzhou, 510640, China
cKey Lab for Polymer Composite and Functional Materials of Ministry of Education, School of Chemistry, Sun Yat-Sen University, Guangzhou, 510275, China. E-mail: ceszfm@mail.sysu.edu.cn
First published on 16th August 2024
Abstract
MXenes as an emerging class of 2D materials have great potential in fabricating functional polymer nanocomposites. In this study, Ti3C2Tx MXene as a representative MXene filler was employed to prepare sPS nanocomposites. The abundant surface groups (mainly –OH) on MXene allowed facile modification, and thus MXene nanosheets bearing –C12H25 groups were prepared using dodecyl triethoxysiloxane (DCTES) as a modifier. Grafting –C12H25 long alkyl chains onto the surface of MXene strongly affected its affinity towards hydrophobic solvent and the sPS matrix. The rheological threshold concentration (ϕc,G′ = 0.033 vol%) and exponent (βG′ = 1.51) and the conductivity threshold concentration (ϕc,σ = 0.259 vol%) and exponent (βσ = 2.92) were determined by applying percolation scaling laws. The influences of DCTES-modified MXene on the polymorphism, mechanical, thermal and flammability properties of nanocomposites were studied. The obtained nanocomposites displayed not only enhanced mechanical properties, but also improved thermal stability and flame retardancy to some extent. Moreover, the higher the loading level of DCTES-modified MXene, the better was the improved effect.
1 Introduction
Polymer nanocomposites, defined by the particle size of the dispersed phase having at least one dimension of less than 100 nm.1,2 With even a small amount of nanofiller (≤3 wt%), polymer nanocomposites show markedly enhanced properties when compared with the neat polymers or conventional (macroscopically filled) polymer composites.3 Polymer nanocomposites are based on the formation of a very large interface between the nano-sized heterogeneities and the polymer matrix. This large interface and the corresponding interphase are supposed to produce exceptional properties that conventional polymer composites cannot achieve.4 Since the first discovery in 2011,5 MXenes, a new group of 2D nanomaterials, which are produced by selective etching of the Al layers from MAX phases, have attracted enormous attention as nanofillers due to their distinct nanostructure and excellent properties. The individual platelets of MXene are less than 1 nm thick, but their other two dimensions are usually much larger.6,7 MXenes have a general formula of Mn+1XnTx where M is an early transition metal, X is carbon and/or nitrogen, n = 1, 2, or 3, and T represents surface-terminating groups.8 However, the high hydrophilicity of MXene surface can induce the nanofillers to be easily agglomerated and hardly dispersible in polymer matrix.9 Thus, the surface functionalization is often needed to improve interfacial bonding between MXene and the polymer matrix.10Syndiotactic polystyrene (sPS) is a rapidly crystallizing engineering thermoplastic polymer with a high melting temperature. sPS possesses complex polymorphism in its crystalline region, five different unit cell forms (α, β, γ, δ and ε) can be obtained in sPS depending on the thermal histories and/or solution treatments.11 The α- and β-forms are obtained in melt-processed sPS in trans-planar (zig–zag) conformation while the γ, δ, and ε-forms are of a helical conformation that are commonly associated with solvent-induced crystallization in sPS.12 The melt-crystallized α- and β-forms can be sub-classified as two different modifications by considering their different degrees of structural order: two limited disordered modifications (α′ and β′) and two limited ordered modifications (α′′ and β′′).13,14 The addition of nanofillers has obvious effects on sPS crystallization behavior and physical properties. Clay with scale of 1–2 nm was found to greatly facilitate the formation of β-form crystals when sPS was melt- or cold-crystallized, and significantly affect the crystallinity.15,16 Carbon nanotubes have a nucleating effect on sPS crystallization, and improved thermal and conductive properties as compared with neat sPS.17,18 Chiu et al. conducted a comparative study, demonstrating that the 2D graphene nanosheets-related networks were more effective in enhancing the melt elasticity and solid conductivity of the sPS matrix than the 1D carbon nanotubes-related networks, as the sPS chains can wrap the neighbouring carbon nanotubes but can only adsorb on graphene nanosheets, which forming a stronger sPS-GNS hybrid network.19 To the best of our knowledge, there have been no reports about the effects of MXene on the crystallization of sPS.
In this study, in order to achieve the blending of hydrophilic Ti3C2Tx MXene nanosheets and hydrophobic sPS, functional alkoxysilane dodecyl triethoxysiloxane (DCTES) with a long alkyl chain (–C12H25) were grafted to the Ti3C2Tx MXene surface to enhance the hydrophobicity. The DCTES-modified Ti3C2Tx MXene/sPS nanocomposites were prepared by directly mixing the dispersions of modified MXene and the solutions of sPS and were further hot-pressed. The advantage of this process is that both modified MXene and sPS exhibit high degrees of dispersibility in organic solvents, and thus allow dispersing modified MXene individually and uniformly throughout the sPS matrix. Transmission electron microscopy (TEM), scanning electron microscopy (SEM), conventional X-ray diffraction (XRD), Haake rheometry and resistivity measurements were used to characterize modified MXene dispersibility in the sPS matrix. In situ high-temperature X-ray diffraction (HT-XRD) and isothermal differential scanning calorimetry (DSC) were used to characterize the changes in the crystalline form and crystallinity in the sPS with different loading of modified MXene. The mechanical, thermal and flammability properties of the nanocomposites were measured by dynamic mechanical analysis (DMA), thermogravimetric analysis (TGA) and microscale combustion calorimetry (MCC), respectively. Comparative studies were reported on the thermal stability and flammability performance between MXene/sPS and DCTES-modified MXene/sPS nanocomposites.
2 Experimental
2.1 Materials
Chloroform (CHCl3, ≥99.0%) and methanol (≥99.5%) were supplied by Guangzhou Chemical Reagent Factory. Ti3C2Tx MXene, DCTES-modified Ti3C2Tx MXene and syndiotactic polystyrene (sPS, Mw = 350![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, PDI = 2.65 with high syndiotacticity (see Fig. S2†)) were all self-made in our laboratory. All experimental water was deionized (DI) water, and all drugs in the experiments need no secondary treatment.
000 g mol−1, PDI = 2.65 with high syndiotacticity (see Fig. S2†)) were all self-made in our laboratory. All experimental water was deionized (DI) water, and all drugs in the experiments need no secondary treatment.
2.2 Preparation of DCTES-modified Ti3C2Tx MXene/sPS nanocomposites
The procedures to fabricate Ti3C2Tx MXene, DCTES-modified Ti3C2Tx MXene (which were abbreviated as MXene, and D-MXene, respectively) and sPS nanocomposites materials are presented in Scheme 1. The preparation and characterization details (Fig. S1†) of MXene and D-MXene are summarized in ESI.† The method of preparing nanocomposites is described as follows. Nanocomposites with various D-MXene compositions were prepared by dissolving weighed sPS powder in CHCl3 at 100 °C for 4 h, followed by adding the D-MXene dispersion (dispersions of 0.5 mg mL−1 D-MXene were prepared using ice bath ultrasound in CHCl3) and rigorously stirred for another 4 h to reach a final solid content of 1% (w/v). Subsequently, the uniform solutions were precipitated into a 20-fold excess volume of methanol. The resultant products were vacuum dried at 80 °C until the residual solvent was removed. Nanocomposites with D-MXene (including MXene and long alkyl chains contributions) weight fractions (ϕw) of 0.05, 0.1, 0.25, 0.5, 1.0, 1.5, and 2.0 wt% were prepared in this manner. The resulting D-MXene/sPS nanocomposites obtained from solution mixed were labeled as sPS-X-S, where X was referred to the weight percentage of D-MXene as compared to sPS nanocomposites. For instance, sPS-0.5-S was the sPS nanocomposite with D-MXene loading of 0.5 wt% prepared from solution mixed.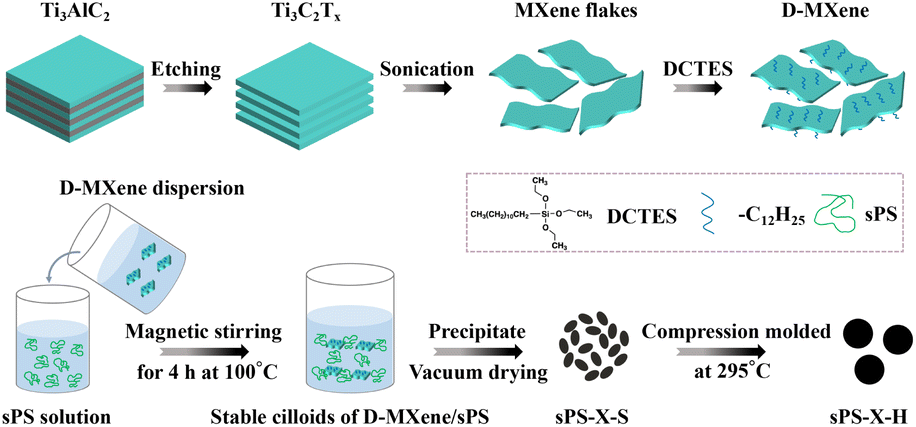 | ||
| Scheme 1 The preparation process of MXene, D-MXene and D-MXene/sPS nanocomposites. | ||
The above products were further compression molded at 295 °C under 30 MPa for 30 min to obtain the hot-pressed sPS nanocomposite materials. The resultants were labeled as sPS-X-H. For instance, sPS-0.5-H was the hot-pressed sPS nanocomposite with D-MXene loading of 0.5 wt% after solution mixed.
The control neat sPS and sPS nanocomposite with 2.0 wt% unmodified MXene prepared by the same methods (solution mixed and hot-pressed) were termed as sPS-0-S, sPS-0-H and sPS-M-S, sPS-M-H, respectively.
2.3 Characterization
Photographs of the solution mixed and the hot-pressed samples were taken using a Canon EOS 800D digital camera (Japan).Transmission electron microscopy (TEM) was performed on a JEOL JEM-2010HR TEM (Japan) at an accelerating voltage of 200 kV.
Scanning electron microscopy (SEM) images were recorded using a Hitachi S-4800 field emission scanning electron microscope (Japan) at an accelerating voltage of 10.0 kV. All the samples were fractured after immersing in liquid nitrogen for about 20 min. The fracture surface was then sputtered with a thin layer of Pt. The elementary analysis was conducted by using the energy dispersive X-ray system (EDX) attached to the S-4800 SEM (Japan) at 15.0 kV acceleration voltage to observe the distribution of Ti in the sPS nanocomposites.
Conventional X-ray diffraction (XRD) measurements were conducted on a Rigaku D/Max 2200 VPC X-ray diffractometer (Japan) at a scanning rate of 10° min−1 under Ni-filtered Cu-Kα (λ = 1.54 Å) radiation with a 2θ range of 5–30°.
The rheological dynamic frequency sweep tests were performed on a Haake MARS III rheometer (Germany) at 290 °C, with an angular frequency range from 100 to 0.1 rad s−1. The strain amplitude was selected as 0.01% in order to be in the linear viscoelastic region for all samples.
Electrical conductivity (σ) was measured by a ZC-90G resistivity meter from Shanghai Taiou Electronics (China, σ < 10−6 S cm−1) and an RTS-8 four-probe resistivity meter (China, σ > 10−6 S cm−1). The samples were prepared by compressing the solution mixed powders at 295 °C and 30 MPa into 13 mm diameter disk-shaped plates with a thickness of about 0.5 mm.
In situ high-temperature X-ray diffraction (HT-XRD) experiments were performed on a Bruker D8 Advance diffractometer (Germany) fitted with a high-temperature attachment and a nitrogen cooling unit under Ni-filtered Cu-Kα (λ = 1.54 Å) radiation. The samples were heated at a rate of 10 °C min−1. After the required temperature was reached, X-ray scans were performed in the 2θ range from 8 to 30° at a scanning rate of 10° min−1.
Differential scanning calorimetry (DSC) curves were collected using a PerkinElmer DSC 4000 instrument (USA) under nitrogen atmosphere. The follow programs were set: first, the samples were heated from 40 to 300 °C at a rate of 10 °C min−1, kept at 300 °C for 5 min to remove the thermal history. Then cooled down to 40 °C at a rate of 10 °C min−1, kept at 40 °C for 1 min followed by heating to 300 °C at a rate of 10 °C min−1 again.
Dynamic mechanical analysis (DMA) of samples with the size of 40 mm × 10 mm × 2 mm was carried out on TA DMA850 (USA) with the single cantilever beam mode and a heating rate of 5 °C min−1 in the temperature range of 40 to 200 °C. The samples were subjected to a strain of 0.01% and a frequency of 1 Hz.
Thermogravimetric analysis (TGA) were carried out on a PerkinElmer Pyris 1 TGA system (USA) from 50 °C to 800 °C at a heating rate of 10 °C min−1 under a nitrogen flow rate of 20 mL min−1.
The combustion parameters were measured with an FAA-PCFC microscale combustion calorimetry (MCC, UK), and 4–5 mg of the samples were heated from the 50 to 800 °C at a heating rate of 1 °C s−1 in a stream of nitrogen flowing at 80 mL min−1. The combustor temperature was set at 800 °C and oxygen/nitrogen flow rate was 20/80 mL mL−1.
3 Results and discussion
3.1 Appearance, morphology, rheology and electrical conductivity of sPS nanocomposites
Hydrophilic MXene and hydrophobic sPS are difficult to achieve uniform mixing, and incompatible MXene nanosheets will be phase separated and aggregated when mixed together. To solve this problem, DCTES was used in this work as a modifier to graft –C12H25 long alkyl chains onto the surface of MXene and improve the compatibility with sPS matrix. The synthetic route to nanofiller D-MXene as illustrated in Scheme 1 typically involves three steps, namely chemical etching, and ultrasonic exfoliation of MXene, then modified by DCTES to graft the –C12H25 groups onto MXene surfaces. DCTES contained three siloxy groups that can be hydrolyzed to silanol groups in the presence of water, catalyzed by base. Then the resulted silanol groups underwent condensation reaction with the hydroxyl groups on the MXene surfaces, leading to the formation of Si–O–C bonds. Following the mass conservation, compared with MXene, the extra 13.3 wt% weight loss corresponded to –C12H25 groups (Fig. S1f†). D-MXene in chloroform was homogenous and stable after 24 h (Fig. S1g†), which was crucial for the preparation of D-MXene/sPS nanocomposites. To further expand its range of practical applications, solution mixed and hot-pressed of D-MXene and sPS were used to in order to attain optimal performance. Fig. 1 shows the photographs of sPS nanocomposites with different concentrations of D-MXene after solution mixed and hot-pressed, all displaying a uniformly mixed appearance. As the content of D-MXene increased, the color of nanocomposites gradually deepened. | ||
| Fig. 1 (a) Photographs of solution mixed D-MXene/sPS nanocomposites with different D-MXene concentrations. (b–i) Photographs of hot-pressed D-MXene/sPS nanocomposites with different D-MXene concentrations after solution mixed. | ||
Good filler dispersion is generally required for better nanocomposite performance.20 SEM, TEM, and XRD may detect a micro-scale region to provide local dispersion information on the nanocomposites, whereas rheological and electrical properties coupled with the percolation law are capable of describing the global dispersion of D-MXene in macro-scale. TEM image (Fig. 2a) presents the dispersion state of D-MXene in solution mixed nanocomposites (sPS-2.0-S). The sPS-2.0-S powder was embedded in epoxy resin and then ultrathin slice was sectioned by an ultramicrotome equipped with a diamond knife and the prepared sample was used for TEM. D-MXene surrounded by sPS matrix was in the form of thin layers of sheets and there were nearly no large agglomerates observed. Therefore, most of D-MXene nanosheets are believed to be well exfoliated and well dispersed in the polymeric matrix, owing to the increased compatibility between sPS and D-MXene.
 | ||
| Fig. 2 TEM image of (a) sPS-2.0-S, the scale bar is 1 μm. SEM images of cross-section of (b) sPS-0-H, (c) sPS-M-H, (e) sPS-0.5-H, (g) sPS-1.0-H, and (i) sPS-2.0-H after brittle fracture, EDX mappings of Ti for (d) sPS-M-H, (f) sPS-0.5-H, (h) sPS-1.0-H, and (j) sPS-2.0-H, the scale bars are 10 μm. Conventional XRD patterns of (k) sPS-X-S, and (l) sPS-X-H. | ||
Fig. 2 presents the freeze-fractured microstructures of hot-pressed sPS-0-H (neat sPS), sPS-M-H (2.0 wt% MXene), sPS-0.5–2-H (0.5–2 wt% D-MXene) via SEM. In Fig. 2b, it was noted that the sPS-0-H showed a smooth surface. However, it can be seen that there were many cavities on the cross-section of sPS-M-H in Fig. 2c. It was very difficult to form good interfacial adhesion between sPS matrix and unmodified MXene. During the breaking processing, MXene was very easy to poll out from sPS matrix. For sPS-0.5-2.0-H, few defects and voids were observed on the fractured surface (Fig. 2e, g and i), indicating that the D-MXene has a good compatibility with sPS matrix. The surface of sPS nanocomposites became much rougher with the increasing content of D-MXene. The dispersion of MXene and D-MXene in the sPS matrix was also studied by EDX. The Ti element from the carbon layer of MXene was a characteristic signal in EDX. The element mapping of sPS-0.5-2.0-H indicated that D-MXene was uniformly distributed in the sPS matrix (Fig. 2f, h and j), but there was a small amount of MXene aggregation and uneven dispersion in the EDX mapping image of sPS-M-H (Fig. 2d).
Fig. 2k shows the XRD patterns of the solution mixed samples. After the D-MXene was introduced into the sPS matrix, the (002) peak of D-MXene shifted to a lower 2θ value from 5.74° (Fig. S1d†) to about 5.56°, which suggested that the blending process caused sPS segments intercalation between D-MXene layers, expanding the d-spacing of D-MXene layers. Compared to MXene/sPS nanacomposite (not display (002) peak of MXene), the dispersibility of D-MXene nanosheets in sPS matrix was improved. In Fig. 2l, it may be due to the overlap of the D-Mxene (002) peak with the low angle peak of sPS in the hot-pressed samples, and no obvious D-Mxene (002) peak was observed. The crystal structure of sPS will be discussed later.
For rheological measurements (Fig. 3a–c), the neat sPS and D-MXene/sPS nanocomposites (with 0.05, 0.1, 0.25, 0.5, 1.0, 1.5, and 2.0 wt% D-MXene) were molded at 295 °C and 30 MPa for 30 min to form disk-shaped plates about 1 mm thickness and 20 mm diameter. The volume fractions (ϕv) were obtained using the mass density of D-MXene (∼3.2 g cm−3)21 and neat sPS (∼1.05 g cm−3).22 The corresponding ϕv were 0.016, 0.033, 0.082, 0.165, 0.330, 0.495 and 0.660 vol%, respectively. The typical relationship between storage modulus (G′) and angular frequency (ω) is expected from a homogeneous polymer melt in the low frequency regime, that is, G′ is proportional to ω2. In this study, for the neat sPS melt, the exponent derived in the low frequency region for the G′ was 1.6 (see solid red line in Fig. 3a, G′–ω1.6). Our homemade sPS has a relatively large molecular weight and a wide molecular weight distribution (Fig. S2a†), some long sPS chains were not completely relaxed for frequencies down to 0.1 rad s−1 at 290 °C.19
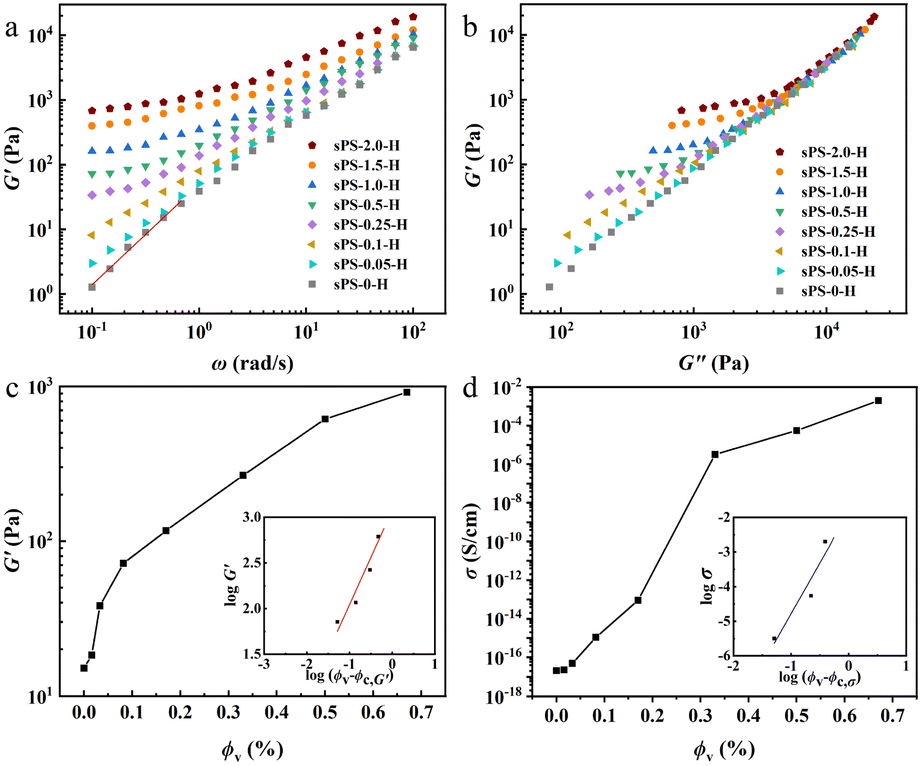 | ||
| Fig. 3 (a) G′ versus applied ω, solid red line represents the relationship between G′ and ω in the low ω; (b) Cole–Cole plots of G′ versus G′′; (c) G′ at a fixed angular frequency of 0.464 rad s−1 as a function of ϕv, solid red line in the inset is fitting the measured data to the scaling equation G′ ∼ (ϕv − ϕc,G′)βG′. The R2 coefficient for the fitting line is 0.939. (d) σ as a function of ϕv, solid blue line in the inset is fitting the measured data to the scaling equation σ ∼ (ϕv−ϕc,σ)βσ. The R2 coefficient for the fitting line is 0.918. | ||
In Fig. 3a, increasing D-MXene content to 0.1 wt% (0.033 vol%) distinctly reduced the slope in the low frequency region with the dependence of G′ on ω at low frequency weakened and increasing the content to 0.25 wt% (0.082 vol%) a small plateau was formed. Further increasing D-MXene to high loading eventually shifted the whole curve upwards. These results indicated that the gradual development of the network between D-MXene nanosheets and sPS chains above a D-MXene content of 0.05 wt% (0.016 vol%) and below 0.1 wt% (0.033 vol%), and the behavior of the nanocomposites melt rapidly transformed from liquid-like to solid-like material.19 Another approach to trace the D-MXene-related network formation is the Cole–Cole plot of storage modulus (G′) versus loss modulus (G′′) (Fig. 3b). For the neat sPS melt at 290 °C, the slope in the low G′′ region was 1.70. However, the slope for the nanocomposites melt significantly declined with the increasing D-MXene content to 0.1 wt% (0.033 vol%). The estimated transition concentration was between 0.05 wt% (0.016 vol%) and 0.1 wt% (0.033 vol%) was in fair agreement with those deduced from the G′–ω plot. To identify the accurate concentration of D-MXene for forming network between D-MXene nanosheets and sPS chains in the nanocomposite melts, percolation scaling law should be applied to determine a reliable threshold concentration. Kota et al.23 concluded that the elastic load transfer is more sensitive to the onset of percolated structures than the viscous dissipation mechanism. Thus, G′ is considered a better parameter to describe the rheological percolation than G′′. The rheological percolation values in nanocomposites were determined using the following power law equation.
| G′ = G0(ϕv − ϕc,G′)βG′ | (1) |
Fig. 3d shows the electrical conductivity (σ) of the D-MXene/sPS nanocomposites as a function of the ϕv. The measured σ of sPS-0-H was 2.1 × 10−17 S cm−1. The conductivity threshold concentration (ϕc,σ) is the minimum volume fraction of D-MXene to form a global connected filler–filler network, as the sPS matrix is an insulating material. The σ of nanocomposites exhibit a sharp transition between ϕv ranging from 0.170 to 0.330 vol%. To quantitatively determine ϕc,σ required to develop the network for electron transportation and critical exponent βσ, the percolation scaling law was applied to describe the relation between the σ and (ϕv − ϕc,σ).26
| σ = σ0(ϕv − ϕc,σ)βσ | (2) |
3.2 D-MXene effect on the polymorphism of sPS crystallization
The solution mixed D-MXene/sPS nanocomposites were subjected to HT-XRD and DSC tests to investigate the effect of D-MXene on the crystallization of nanocomposites. HT-XRD patterns (Fig. 4a–d) of sPS-0-S and sPS-0.5-2.0-S were collected at a heating rate of 10 °C min−1. Furthermore, XRD patterns of these samples after the cooling step were carried out (Fig. 4e). Fig. 4g–i show the DSC curves of the heating and the cooling scans for sPS-0-S and sPS-0.5-2.0-S. The results obtained from these DSC curves are summarized in Table 1. The degree of crystallinity (Xc) calculated by
 | (3) |
 | ||
| Fig. 4 In situ high-temperature XRD patterns during the heating stage of solution mixed samples to measure the crystalline phase transition, (a) sPS-0-S, (b) sPS-0.5-S, (c) sPS-1.0-S, (d) sPS-2.0-S. (e) In situ high-temperature XRD patterns of sPS-0-S, sPS-0.5-S, sPS-1.0-S, and sPS-2.0-S after melting and then cooling to 30 °C. (f) The calculated content of the α-form as a function of the ϕw of D-MXene after cooling. DSC thermograms of solution mixed samples, (g) first heating, (h) cooling, (i) second heating. | ||
| Samples | Process | ΔHm or ΔHc (J g−1) | Tm or Tc (°C) | Xc (%) |
|---|---|---|---|---|
| sPS-0 | First heating | 29.3 | 269.3 | — |
| Cooling | 25.8 | 235.3 | 48.5 | |
| Second heating | 24.5 | 257.6, 268.6 | — | |
| sPS-0.5 | First heating | 31.4 | 270.3 | — |
| Cooling | 26.9 | 235.5 | 50.8 | |
| Second heating | 26.2 | 257.1, 268.3 | — | |
| sPS-1.0 | First heating | 33.5 | 269.9 | — |
| Cooling | 27.4 | 235.7 | 52.0 | |
| Second heating | 26.9 | 257.6, 263.0, 268.4 | — | |
| sPS-2.0 | First heating | 35.2 | 270.3 | — |
| Cooling | 29.1 | 235.9 | 55.8 | |
| Second heating | 27.9 | 257.5, 262.8, 268.4 | — |
The XRD pattern of the blank sample consisted of a wide background peak (2θ = 7.1°), as shown in Fig. S3,† was caused by material scattering in the instrument itself. The background peak will overlap with sample peaks less than 8°. Therefore, the discussion range of HT-XRD is 8-30°. In Fig. 4a, several characteristic diffraction peaks at 2θ = 9.3°, 10.4°, 16.0°, 20.0° and 28.1° can be observed for sPS-0-S at 30 °C, implying only the presence of γ-form in the solution-cast sPS.28 For sPS-0.5-2.0-S (Fig. 4b–d), the incorporation of D-MXene into sPS matrix did not make significant change in the formation of γ-form in comparison with sPS-0-S at 30 °C, which was consistent with the conventional XRD results (Fig. 2k). As the temperature increased to 140 °C, sPS-0-S and sPS-0.5–2-S underwent an obvious reorganization of γ-form. These transitions were consistent with the DSC first heating scans (Fig. 4g) which the small exothermic peak can be attributed to reorganization of γ-form.29 At 190 °C, the intensities of the crystal characteristic peaks of γ-form gradually decreased, while the peaks of α′-form at 2θ = 11.6° and 13.4° appeared. At 210 °C, γ-form disappeared and completely transformed into α′-form (2θ = 11.6°, 13.4° and 20.4°).30 The transition was consistent with the DSC first heating scan shown in Fig. 4g which the descent of the curve can be attributed to γ-form to α′-form transition. Afterwards, α′-form became intense at 250 °C. At 270 °C, sPS-0-S exhibited addition sharp diffraction peaks at 2θ = 10.4°, 13.8°, 15.3° and 17.6° in Fig. 4a, which were typical peaks of α′′-form.30 There was a noticeable transition from α′-form to α′′-form for sPS-0-S. For sPS-0.5-S, as shown in Fig. 4b, the transition from α′-form to α′′-form was suppressed. As the content of D-MXene increased to 2.0 wt%, the content of α′′ continued to decrease at 270 °C in Fig. 4d. At 280 °C, these samples were melted and revealed amorphous broad diffraction peaks, with no obvious sharp peaks can be observed. In the DSC first heating scans (Fig. 4g), due to the high content of α′′-form in the sPS-0-S, there was a wider melting peak than sPS-0.5–2-S.
Furthermore, XRD patterns of these samples after the cooling step were carried out. As shown in Fig. 4e, a series of sharp diffraction peaks at 2θ = 12.2°, 18.8°, 21.1° and 23.7° were characteristic of β-form.28 The peaks at 2θ = 11.6° and 13.8° can be assigned to α-form. Additionally, there were three peaks at 2θ = 10.4°, 13.4° and 20.4°, where α and β-forms both had characteristic peaks at these positions. By comparing the characteristic peaks at 11.6° and 12.2° belonging to α and β-forms, sPS-0-S was almost β-form with Pα = 11.2% (the content of α-form) calculated from eqn (4). A small amount of D-MXene addition will contribute to the formation of α-form during the non-isothermal crystallization of sPS, similar to the conventional XRD results of the sPS-X-H samples (Fig. 2l). The calculated content of the α-form in Fig. 4e as a function of the ϕw of D-MXene is shown in Fig. 4f. As the content of D-MXene increased to 2.0 wt%, the Pα increased to 71.0%. The introduction of D-MXene favord α-form may be due to D-MXene can act as α-form crystal nuclei to facilitate its production. Moreover, Liao et al.31 reported it would be required to exceed a certain pressure to induce the transition from the α to β-form that the chains mobility was high enough to overcome the energy barrier. The increasing of D-MXene will limit the movement of sPS segments due to the interaction between nanofillers and matrix, making it more conducive to the formation of kinetically favourable α-form crystals.
In DSC cooling scans (Fig. 4h), the Tc and ΔHc increased with increasing D-MXene loadings. The results in Table 1 showed that the crystallinity calculated from ΔHc increased from 48.5% to 55.8% after addition only 2.0 wt% nanoscale D-MXene in sPS. Due to the high surface activity of MXene nanomaterial, sPS chains tend to adsorb onto the surface of MXene, acting as heterogeneous nucleation centers and improving the crystallinity of sPS. According to the literature,32 the relative fraction of α-form could be calculated by the equation as follows:
 | (4) |
The ratio between the intensities of the characteristic 2θ diffraction peaks located at 11.6° and 12.2° for specimens with the same thickness and crystallinity in the neat α- and β-forms is assumed to be 1.8. A(11.6) and A(12.2) are the areas of the characteristic 2θ diffraction peaks located at 11.6°(α) and 12.2°(β).
In DSC second heating scan (Fig. 4g), the sPS-0-S displayed two melting peaks (257.6 and 268.6 °C) which were all associated with β-form.33 For sPS-0.5–2-S, the minor shoulder around 263 °C in DSC second heating scan was melting of α-form. As the content of D-MXene increased in sPS nanocomposites, the content of α-form increased, leading to a more pronounced shoulder peak at around 263 °C in DSC curves. In general, during the first heating process of melting, the presence of D-MXene will inhibit the formation of α′′-form of sPS. In the process of non-isothermal cooling crystallization, the presence of D-MXene will facilitate the generation of α-form of sPS. Due to the heterogeneous nucleation of D-MXene, as the amount of D-MXene increases, the crystallinity and the crystallization temperature of sPS increase.
3.3 Mechanical, thermal and flammability properties of sPS nanocomposites
The dynamic mechanical properties of neat sPS and D-MXene/sPS nanocomposites are given in Fig. 5a, representing the variation of storage modulus (E′) and damping parameter (tanδ) with temperature. The behaviour of all samples was found to be analogous that E′ decreased systematically with an increase in temperature and a single tanδ peak (Tg) was observed. At tanδ peak (around 110 °C), the sPS chains passed from the glassy to the rubbery state, exhibiting considerable flexibility and resulting in a significant decrease in E′. Increasing the amount of D-MXene increased the E′ and Tg while reducing the peak value of tanδ. And the measured tanδ curves became broader with increasing D-MXene content. The enhancement of the E′ may be ascribed to the presence of D-MXene nanosheets in the sPS matrix can load transfer across the increased interfacial interactions. The movements of the sPS segments were restricted and a more time for relaxation were required due to confinement in between D-Mxene layers, resulting the decrease of internal friction, and therefore giving higher Tg, lower tanδ peak values and wider tanδ curves.
 | ||
| Fig. 5 (a) Temperature dependence of E′ and tanδ (inset), (b) TG, (c) DTG, and (d) MCC curves of neat sPS and sPS nanocomposites. | ||
Fig. 5b and c reveal TGA and DTG (differential thermogravimetric) curves of sPS-0-H and sPS-0.5-2.0-H. sPS is presumed to degrade in a similar manner to aPS, which is random chain scission.34 To quantitatively characterize their thermal stability, the temperature at which 5% (T5%) weight loss occurred, the temperature of the maximum decomposition rate (Tmax), and the residual mass ratio at 800 °C are extracted in Table 2. Temperatures at T5% and Tmax for sPS-0-H were 371.6 and 413.0 °C, respectively, while those were increased by 41.8 and 36.9 °C for sPS-M-H (2.0 wt% MXene). The T5% for sPS-0.5-H, sPS-1.0-H and sPS-2.0-H were 23.9, 39.7 and 71.2 °C higher than that of sPS-0-H, respectively. The Tmax for sPS-0.5-H, sPS-1.0-H and sPS-2.0-H were 9.3, 32.2 and 59.2 °C higher than that of sPS-0-H, respectively. The thermal stability of these nanocomposites increased with the increase of D-MXene content. When the nanofiller content was the same, the thermal performance of the D-MXene/sPS nanocomposite was more significantly improved than that of the MXene/sPS nanocomposite. The char residues of sPS-0-H, sPS-0.5-H, sPS-1.0-H, sPS-2.0-H, and sPS-M-H were 1.72, 2.66, 4.03, 7,85, and 3.65 wt%, respectively. This result indicated that the char residue improved with the addition of MXene and D-MXene. Moreover, the char residue of sPS-2.0-H was superior to that of sPS-M-H. Overall, D-Mxene/sPS nanocomposites materials had a much high thermostability and charring capability than neat sPS and MXene/sPS nanocomposites. This was mainly due to the well-dispersed and excellent physical barrier effect of the D-MXene.35 The improved interfacial compatibility between sPS and D-MXene can inhibit the thermal motion of sPS to some extent. Volatile decomposition products may path out of the material through a tortuous path brought by physical barriers, which restricted the permeability of volatile products during thermal decomposition and delayed the diffusion and migration of volatile products from the internal matrix to the surface. The increased char yield implied the reduction of volatile products and fuels during combustion, potentially contributing to improving flame retardancy of sPS.
| Samples | T5% (°C) | Tmax (°C) | Residues (wt%) | pHRR (W g−1) | TpHRR (°C) | THR (kJ g−1) | HRC (J g−1 K−1) |
|---|---|---|---|---|---|---|---|
| sPS-0-H | 371.6 | 413.0 | 1.72 | 939 | 443.7 | 38.4 | 938 |
| sPS-0.5-H | 395.5 | 422.3 | 2.66 | 897 | 444.5 | 37.3 | 895 |
| sPS-1.0-H | 411.3 | 445.2 | 4.03 | 723 | 452.9 | 35.9 | 768 |
| sPS-2.0-H | 442.8 | 472.2 | 7.85 | 639 | 457.7 | 31.1 | 736 |
| sPS-M-H | 413.4 | 449.9 | 3.65 | 811 | 449.3 | 38.2 | 810 |
MCC (micro combustion calorimeter) has been suggested as a method to assess the fire behavior of mg-sized sample.36 The parameters measured from this test are heat release rate (HRR) in W g−1 (calculated from the oxygen depletion measurements, the peak was pHRR), the total heat release (THR) in kJ g−1 (obtained by integrating the HRR curve), and heat release capacity (HRC) in J g−1 K−1 (obtained by dividing the sum of pHRR by the heating rate in K s−1).37 The HRR curves versus temperatures were shown in Fig. 5d and the typical data were summarized in Table 2, respectively. The essential characteristic of quantitatively describing “how big a fire is” is HRR. The value of pHRR characterizes the maximum heat release during the material combustion process. The larger the HRR and pHRR, the greater the fire hazard generated.38 After adding MXene or D-MXene to sPS, the HRR curves of sPS-M-H and sPS-0.5–2-H were significantly reduced. Compared to the pHRR value of sPS-0-H, which was 939 W g−1, the pHRR value of sPS-0.5-H, sPS-1.0-H, sPS-2.0-H and sPS-M-H decreased by 4.5%, 23.0%, 31.9%, and 13.6%, respectively. It showed that adding D-MXene to sPS can significantly suppress the combustion process. The reduced trend continued when increasing the D-MXene weight percentage in sPS nanocomposites. Compared the pHRR value of sPS-2.0-H with sPS-M-H indicated that better dispersion can form a more uniform barrier, which may lead to a “tortuous path” that played a key role in the gas barrier effect. Since the layered structure in the nanocomposite can act as a barrier to heat and mass transfer, the degradation products were retained for a more extended period and the degradation rate slow down, leading to pHRR value reduced. We also observed that the TpHRR of sPS nanocomposites was shifted to a higher temperature compared to the neat sPS. This also suggested that the flammable pyrolysis released slower.
THR is characterized as the total available energy released by the material in a fire situation. The decrease in THR of nanocomposites means that not all of the polymer has burned.39 Compared with sPS-0-H, the lowest THR value (31.1 kJ g−1) and the maximum char yield (7.85%) of sPS-2.0-H indicated that more volatile products were catalytically carbonized to participate in the charring process, rather than transferred into the MCC combustor. It was speculated that this was due to the physical barrier effect of nanosheets mentioned above, which increased the char yield and reduced the THR. HRC was accepted as the key fire parameter to predict fire risk of polymeric materials.40 As seen in Table 2, the comparison of the reduction in HRC demonstrated that D-MXene was the better candidate to improve flame retardancy of sPS than MXene. According to the data of TGA and MCC, the addition of D-MXene effectively delayed the thermal degradation of sPS and increased char formation, ultimately leading to a superior flame-retardant effect.
4 Conclusions
DCTES-modified Ti3C2Tx MXene nanosheets were used as nanofillers in the sPS matrix. Morphological observations revealed that the D-MXene nanosheets were thoroughly dispersed in the sPS matrix. The rheological and conductivity percolation thresholds were determined by percolation scaling law. A small amount of D-MXene could facilitate the nucleation of sPS, and then led to the formation of much more sPS crystallites. In the process of non-isothermal cooling crystallization, the presence of D-MXene will facilitate the generation of α-form of sPS. An investigation of the dynamic mechanical properties indicated enhanced nanofiller–matrix interactions. The improvement of dispersion after MXene modification was beneficial for improving thermal stability and flame retardancy. And as the load content of D-MXene increased, its thermal stability and flame retardancy became better.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Tongtong Zhang: conceptualization, methodology, investigation, formal analysis, writing – original draft, writing – review & editing. Shuo Wang: investigation. Runkai Qiu: methodology. Xiaolong Yang: software. Fangming Zhu: supervision, project administration, funding acquisition, resources, data curation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51773228) and Shenzhen Sunway Communication Co., Ltd. (Contract No. HT-99982021-0269).References
- E. P. Giannelis, Adv. Mater., 1996, 8, 29–35 CrossRef CAS.
- A. Okada and A. Usuki, Mater. Sci. Eng. C, 1995, 3, 109–115 CrossRef.
- E. P. Giannelis, Appl. Organomet. Chem., 1998, 12, 675–680 CrossRef CAS.
- G. Keledi, J. Hári and B. Pukánszky, Nanoscale, 2012, 4, 1919–1938 RSC.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- C. Hu, Z. Wei, L. Li and G. Shen, Adv. Funct. Mater., 2023, 33, 2302188 CrossRef CAS.
- L. Li, W. Liu, K. Jiang, D. Chen, F. Qu and G. Shen, Nano-Micro Lett., 2021, 13, 100 CrossRef CAS PubMed.
- T. Bashir, S. A. Ismail, J. Wang, W. Zhu, J. Zhao and L. Gao, J. Energy Chem., 2023, 76, 90–104 CrossRef CAS.
- Z. Duan, M. Yuan, Z. Liu, W. Pei, K. Jiang, L. Li and G. Shen, Small, 2024, 20, 2470185 CrossRef.
- C. Hu, Z. Du, Z. Wei, L. Li and G. Shen, Appl. Phys. Rev., 2023, 10, 021402 CAS.
- G. Milano and G. Guerra, Prog. Mater. Sci., 2009, 54, 68–88 CrossRef CAS.
- Y. Chatani, Y. Shimane, T. Ijitsu and T. Yukinari, Polymer, 1993, 34, 1625–1629 CrossRef CAS.
- C. De Rosa, G. Guerra, V. Petraccone and P. Corradini, Polym. J., 1991, 23, 1435–1442 CrossRef CAS.
- C. De Rosa, M. Rapacciuolo, G. Guerra, V. Petraccone and P. Corradini, Polymer, 1992, 33, 1423–1428 CrossRef CAS.
- C. R. Tseng, H. Y. Lee and F. C. Chang, J. Polym. Sci. B Polym. Lett., 2001, 39, 2097–2107 CrossRef CAS.
- C. R. Tseng, S. C. Wu, J. J. Wu and F. C. Chang, J. Appl. Polym. Sci., 2002, 86, 2492–2501 CrossRef CAS.
- S. Kumar, T. Rath, R. N. Mahaling and C. K. Das, Compos. Appl. Sci. Manuf., 2007, 38, 1304–1317 CrossRef.
- C. L. Huang and C. Wang, Eur. Polym. J., 2011, 47, 2087–2096 CrossRef CAS.
- Y.-C. Chiu, C.-L. Huang and C. Wang, Compos. Sci. Technol., 2016, 134, 153–160 CrossRef CAS.
- C. Harito, D. V. Bavykin, B. Yuliarto, H. K. Dipojono and F. C. Walsh, Nanoscale, 2019, 11, 4653–4682 RSC.
- W. Ma, W. Cai, W. Chen, P. Liu, J. Wang and Z. Liu, Chem. Eng. J., 2021, 425, 131699 CrossRef CAS.
- X. Wang and S. C. Jana, ACS Appl. Mater. Interfaces, 2013, 5, 6423–6429 CrossRef CAS PubMed.
- A. K. Kota, B. H. Cipriano, M. K. Duesterberg, A. L. Gershon, D. Powell, S. R. Raghavan and H. A. Bruck, Macromolecules, 2007, 40, 7400–7406 CrossRef CAS.
- H.-B. Zhang, W.-G. Zheng, Q. Yan, Z.-G. Jiang and Z.-Z. Yu, Carbon, 2012, 50, 5117–5125 CrossRef CAS.
- M. J. McAllister, J.-L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud'homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396–4404 CrossRef CAS.
- C.-L. Huang and C. Wang, Carbon, 2011, 49, 2334–2344 CrossRef CAS.
- M. Malanga, Adv. Mater., 2000, 12, 1869–1872 CrossRef CAS.
- G. Sun, G. Chen, Z. Liu and M. Chen, Carbon, 2010, 48, 1434–1440 CrossRef CAS.
- H. Wang, C. Wu, D. Cui and Y. Men, Macromolecules, 2018, 51, 497–503 CrossRef CAS.
- R. H. Lin and E. M. Woo, Polymer, 2000, 41, 121–131 CrossRef CAS.
- X. Liao, J. He and J. Yu, Polymer, 2005, 46, 5789–5796 CrossRef CAS.
- G. Guerra, V. M. Vitagliano, C. D. Rosa, V. Petraccone and P. J. M. Corradini, Macromolecules, 1990, 23, 1539–1544 CrossRef CAS.
- Y. S. Sun and E. M. Woo, Macromolecules, 1999, 32, 7836–7844 CrossRef CAS.
- S. Stack, O. O'Donoghue and C. Birkinshaw, Polym. Degrad. Stab., 2003, 79, 29–36 CrossRef CAS.
- J. H. Woo, N. H. Kim, S. I. Kim, O.-K. Park and J. H. Lee, Composites, Part B, 2020, 199, 108205 CrossRef CAS.
- R. E. Lyon and R. N. Walters, J. Anal. Appl. Pyrolysis, 2004, 71, 27–46 CrossRef CAS.
- L. Qiu, Y. Gao, P. Lu, D. O'Hare and Q. Wang, J. Appl. Polym. Sci., 2018, 135, 46204 CrossRef.
- D. Xu, G. Huang, L. Guo, Y. Chen, C. Ding and C. Liu, Adv. Compos. Hybrid Mater., 2022, 5, 113–129 CrossRef CAS.
- H. Lu, C. A. Wilkie, M. Ding and L. Song, Polym. Degrad. Stab., 2011, 96, 1219–1224 CrossRef CAS.
- R. E. Lyon, R. N. Walters and S. I. Stoliarov, Polym. Eng. Sci., 2007, 47, 1501–1510 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04190a |
| This journal is © The Royal Society of Chemistry 2024 |