
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards biotechnological production of bio-based low molecular weight esters: a patent review
Mirko Zago ab,
Paola Branduardia and
Immacolata Serra*a
ab,
Paola Branduardia and
Immacolata Serra*a
aDepartment of Biotechnology and Biosciences, University of Milano-Bicocca, Piazza della Scienza 2, Milano, 20126, Italy. E-mail: immacolata.serra@unimib.it; m.zago@campus.unimib.it; zago@astrobiosolvent.com; paola.branduardi@unimib.it; Tel: +390264484140
bSoft Chemicals S.r.l., ASTROBIO™ Division, Via Sandro Pertini 14, Arsago Seprio, Varese, 21010, Italy
First published on 18th September 2024
Abstract
Low molecular weight (LMW) esters, like ethyl acetate, methyl formate or butyl acetate, are widespread bulk chemicals in many industries. Each of them is currently produced in huge amounts (millions of tons per year scale) starting from fossil-based feedstock and they are used mainly because of their low toxicity and complete biodegradability. Energy transition is just half of the story on the path of fighting climate change: 45% of the global greenhouse gas emissions are caused by the production and use of all the products, materials and food necessary for modern human life. If the world is to reach its climate goals, there is the need to leave underground a significant proportion of the fossil feedstock and minimize environmental impacts of chemical manufacturing. This is the reason why a lot of efforts have been made to find novel routes for LMW esters production starting from renewable raw materials (e.g. biomasses or off-gases) and exploiting low-impact manufacturing, such as microbial fermentation or enzymatic reactions. This review reports the most significant patents, in the field of white biotechnology, that will hopefully lead to the commercialization of bio-based LMW esters as well as novel strategies, current problems to be solved, newer technologies, and some patent applications aiming at possible future developments.
Introduction
Esters, or alkyl alkanoates, are useful and versatile organic compounds with various applications in the chemical and the food industry. Small carboxylate esters are ubiquitous in nature and are mainly responsible for the pleasant aroma of fruits and flowers.1,2 Alkyl alkanoates can be roughly classified, according to their chain length, in low molecular weight (LMW) esters and high molecular weight (HMW) esters. The first ones (LMW) are usually colourless and volatile liquids at ambient temperature and have chain length shorter than 10 carbon atoms, while the second ones (HMW) can be liquids or solids, like waxes, and are formed from long chain carboxylic acids and fatty alcohols.3 Ethyl acetate, ethyl lactate or butyl acetate are some of the most important examples of LMW esters for industrial applications. They are commonly used as fragrances and aroma compounds in the food industry,2 as carriers for pigments and resins in the coating industry but are also used as solvents in several other industrial applications due to their good solvency power for certain polymers and APIs, biodegradability and low toxicity for humans, animals and environment. Generally speaking, LMW esters are produced using the Fischer Speier esterification which involves the condensation between a carboxylic acid and an alcohol molecule with the aid of an acid catalyst, high temperatures and stoichiometric water release.4 Esterifications are reversible reactions dictated by an equilibrium that prevents complete conversion of reactants to products. Furthermore, produced water often impairs the catalyst, necessitating energy-intensive procedures to remove it from the reaction mixture in order to drive the reaction toward the desired product.2 Of course, other traditional routes for specific esters production are known and widely used in industrial practice, like the Tishchenko reaction or the dehydrogenation of ethanol for ethyl acetate synthesis. The use of corrosive catalysts, temperature, pressure and energy intensive product recovery procedures are common to all of them.2,5–7 To avoid the use of fossil resources, substrates could be obtained from renewable feedstocks, but even if certain organic acids (e.g. acetic acid or lactic acid) and alcohols (e.g. ethanol or butanol) are industrially available either from fermentation or green chemistry routes, it is still rare to find entirely bio-based LMW esters on the market. Most probably this is because the costs of the sustainable alternatives are still higher than fossil-based ones, but maybe other reasons are involved. Corbion N.V. and Shenzhen Esun Industrial Co. Ltd were among the first companies that were able to produce a completely bio-based ethyl lactate available to be used in EU countries (REACh registered).8,9 On the other hand, Godavari Biorefineries Ltd, SEKAB Biofuels & Chemicals AB and Rhodia Brasil S.A. (Solvay Group) seem to be the only three world producers of a completely bio-based ethyl acetate available on the European market.10–12 Industrial interest in bio-based bulk chemicals has increased enormously in the last decades and is mainly driven by the advantages that bioeconomy can offer in climate change mitigation, while maintaining growth and social wellbeing.13,14 As a matter of fact, energy transition is only part of the solution of fighting climate change: the production and use of all the products, materials, and food essential for modern human life account for about half of the global greenhouse gas (GHG) emissions.15,16 Using biomasses or off-gases as starting feedstock to produce chemicals results in a sustainable use of carbon. Very simplistically, this is mainly a question of timing: inorganic carbon is fixed by plants into organic molecules and then used to produce chemicals. Finally, this same carbon (so called “green” carbon) will then be released at the end of the chemical's life cycle without increasing the atmospheric carbon (i.e. CO2) concentration, as the overall process will last a few years or decades at most. On the other hand, fossil-based chemicals are produced using fossil (‘‘black’’) carbon, which was preserved underground over geological eras, and at the end of their life cycle they will release this additional CO2 to the atmosphere. It is reported in scientific literature that there is increasing evidence that a significant proportion of these fossil resources must stay underground if the world is to reach its climate goals.17–22 LMW esters production technologies are still in very early stages of development (proof of concepts in most cases) as clearly shown by this work. Despite extensive literature research has been performed for this review, life cycle assessments (LCAs) studies currently focus solely on traditional production processes of LMW esters. The key finding is that the use of renewable feedstocks versus fossil feedstocks offers clear advantages in terms of overall impacts, particularly when considering the end of life of the produced molecules.23,24LMW esters are also naturally produced by microorganisms, among which lactic acid bacteria and yeasts. These esters contribute to the taste and odour to fermented food and beverages and there are specific metabolic pathways or enzymes that can result in ester production starting from appropriate substrates.2 This potential has driven extensive research over the last 30 years to find new methods for producing bulk chemicals like LMW esters, starting from renewable feedstock, via microbial fermentation and/or using specific enzymes as biocatalysts. In this review, we will mainly focus on the critical analysis of patent literature claiming the production of LMW esters by the use of three different enzyme classes: esterases/lipases (EC 3.1.1.-), Baeyer Villiger monooxygenases (BVMOs, EC 1.14.13.-) and alcohol acyltransferases (AATs, EC 2.3.1.-) (Scheme 1).
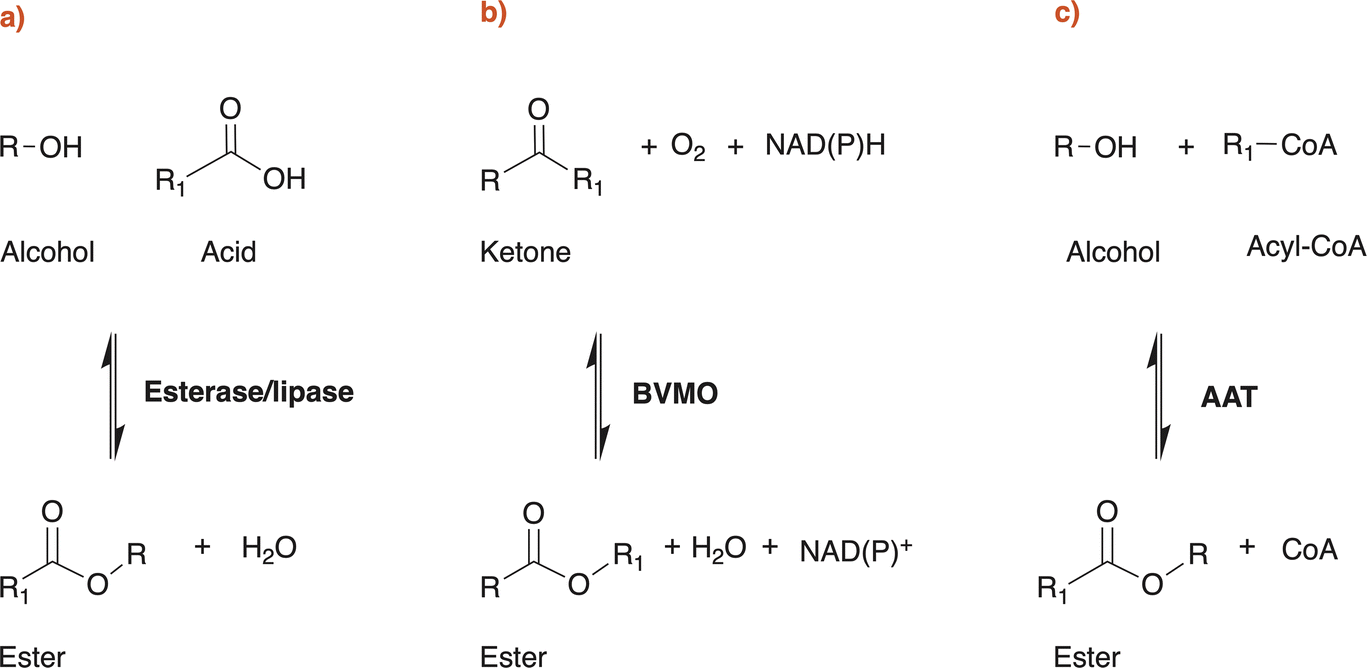 | ||
| Scheme 1 Enzymatic reactions described in patent literature to claim the biotechnological production of esters. Abbreviations: BVMO, Baeyer Villiger Monooxygenase; AAT, Alcohol Acyltransferase. | ||
The potential of these researches, patents and patent applications is significant. Excellent reviews reflecting scientific literature up to 2019 and 2020 were written by Lee et al. and Kruis et al. respectively.1,2 Plus, biosynthesis of esters can provide alternatives and more convenient routes for acids and alcohols production. These will be more than enough to replace a huge proportion of the market of the fossil based bulk chemicals with all the benefits related to climate change mitigation as well recognized by the international scientific community.17,19,25 Just to have an idea, the global market size of ethyl acetate only was about 3.32 billion dollars in 2019, with an average EXW (ex-works) price of 1$ per kg.26 Although great advances have been made in the field of acids and alcohols production from fermentation processes, and indeed some of them reached the market, the overall manufacturing of such substances is not yet ideal.27–29 Firstly, alcohols and acids can be very toxic to cells, which limits the concentration of such molecules achievable in fermentation broth. Secondly, small alcohols and acids tend to be very soluble in aqueous media and therefore can require energy intensive and expensive recovery procedures.2 These research efforts have led to the registration of several patents which may in the future allow biotechnological production of small (LMW) bio-based esters, but nowadays still a lot of technical problems remain unsolved. In this review we will discuss the most significant patents that in the future will hopefully lead to the commercialization of biotechnologically produced bio-based LMW esters as well as current problems to be solved, newer technologies, and some patents and patent applications aiming at possible future developments.
Enzymes and biochemical pathways
Microbial production of esters by yeasts and lactic acid bacteria is a well-established process that has historically been used in the production and conservation of foods and beverages (Fig. 1). Nonetheless, the existence of natural biochemical routes per sé is not sufficient to ensure their effective application in diverse sectors. LMW esters (i.e. volatile esters) production titres by wild-type microorganisms are typically low, although some exceptions exist. Yeasts like Kluyveromyces marxianus, Wickerhamomyces anomalus, Candida utilis and Cyberlindnera jadinii can produce significant amounts (i.e., in the range of g L−1) of ethyl acetate starting from sugars or ethanol.6 First report of this kind (ethyl acetate production by yeasts) dates back to the late 19th century and, since then, considerable knowledge has been gained.2 However, the potential of these microbial conversions has only recently been recognized for sectors other than the food industry as reviewed by Löser et al., 2014: i.e. production of bulk chemicals suitable as solvents or intermediates for industrial applications.6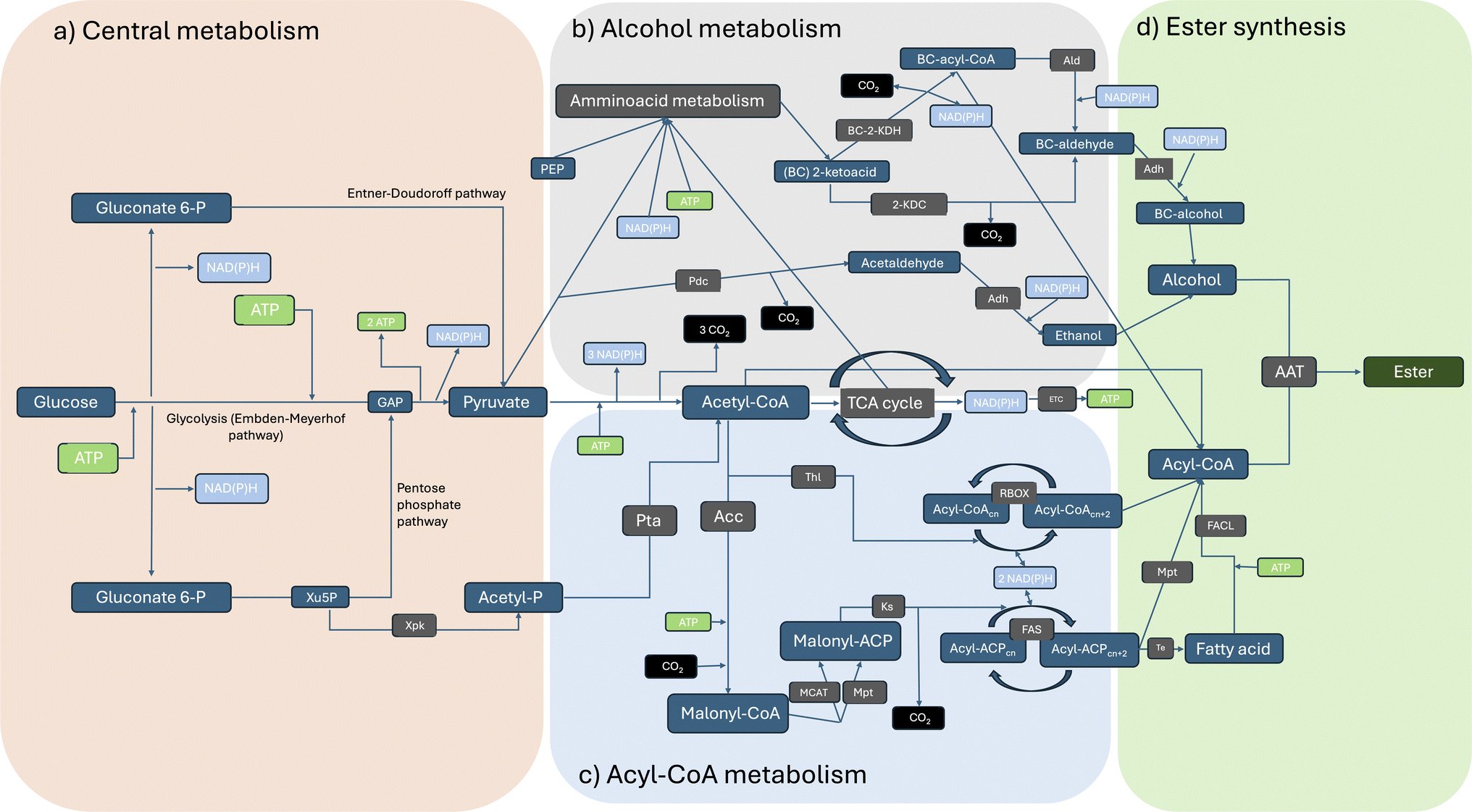 | ||
| Fig. 1 Overview of the main metabolic pathways that transform glucose into esters: (a) production of essential metabolites, redox equivalents and ATP from sugars; (b) generalised synthesis of longer-chain acyl-CoAs derived from acetyl-CoA; (c) general production of branched and higher alcohols from 2-ketoacids; (d) final ester production reaction facilitated by an AAT. Abbreviations: a-Ald – acetylating acetaldehyde dehydrogenase, AAT – alcohol acyl transferase, Acc – acetyl-CoA carboxylase, ACP – Acyl carrier protein, Adh – alcohol dehydrogenase, Ald – acetaldehyde dehydrogenase, BC – branched chain, BC-2-KDH – branched-chain 2-ketoacid dehydrogenase, ETC – electron transport chain, FACL – fatty acid CoA-ligase, FAS – fatty acid synthase, GAP – Glycerol 3-phosphate, 2-KDC – 2-ketoacid decarboxylase, Ks – ketoacyl synthase, MCAT – Malonyl-CoA acyl carrier protein transacylase, Mpt – malonyl/palmitoyl transferase, Pdc – pyruvate decarboxylase, PEP – phosphoenolpyruvate, Pta – phosphotransacetylase, RBOX – reverse β-oxidation, TCA – tricarboxylic acid cycle, Te – thioesterase, Thl – thiolase, Xpk – phosphoketolase, Xu5P – xylulose 5-phosphate. | ||
In patent literature, essentially three types of enzymes have been identified and successfully used, both in vivo and in vitro, for claiming the biotechnological production of esters (Scheme 1): esterases and lipases (EC 3.1.1.-), Baeyer Villiger monooxygenases (BVMOs, EC 1.14.13.-) and alcohol acyltransferases (AATs, EC 2.3.1.-). Esterases, lipases and AATs reactions are redox neutral, while BVMOs require NAD(P)H to perform their reaction. In fact, patents and patent applications mainly claim the use of these enzyme classes: Table 1 reports relevant information of the patents discussed in this work, together with additional literature data on the properties of the enzyme(s) exploited.
| Ester | Enzyme | Enzyme source | Substrate specificity | Relevant kinetic parameters (e.g. Km, kcat, etc.)b | Process type and technology readiness level (TRL)c | Maximum titre | Yield | Conversion | Patent/patent application |
|---|---|---|---|---|---|---|---|---|---|
| a n.d.: not disclosed; n.a.: not available.b Kinetic data reported are the ones available and related to the starting substrates of the esters listed in the table. Kinetic data are scarcely available in patent literature, so they were retrieved from scientific literature.c TRL is estimated from patent description.d Note: myristyl myristate, ethyl octanoate and geranyl acetate cannot be considered LMW esters, anyway technology disclosed in those patents can be considered relevant also for the synthesis of small esters. | |||||||||
| Ethyl acetate | n.d. | C. utilis, H. anomala | n.d. | n.d. | Fermentation; TRL3 | n.d. | n.d. | n.d. | US4720457A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 30 30 |
| Isoamyl acetate | AAT (Atf2) | S. cerevisiae | Primary alcohols; acetyl-CoA22 | Isoamyl alcohol:2,117 Km = 22.00 mM; kcat = 1.60 s−1; kcat/Km = 74.00 s−1 M−1 | Fermentation; TRL4 | 9.95 mM | n.d. | n.d. | US7569380B250 |
| Acetyl-CoA: n.a. | |||||||||
| Isobutyl acetate | AAT (Atf1) | S. cerevisiae | Primary alcohols; Acetyl-CoA22 | Isoamyl alcohol:2,118 Km = 32.00 mM; isoamyl alcohol:2,117 Km = 26.00 mM; kcat = 2.90 s−1; kcat/Km = 113.00 s−1 M−1 | Fermentation; TRL4 | 17.2 g L−1 | 0.334 g g−1 glucose | n.d. | WO2015031859A151 |
| Acetyl-CoA:2,119 Km = 0.06 mM kcat = 0.40 s−1 kcat/Km = 6656.00 s−1 M−1 | |||||||||
| n-Butyl methacrylate | AAT (MpAAT1) | Malus pumila | Short to medium chain alcohols (C3–C10); short to medium chain acyl-CoAs120 | n-Butanol:120 Km = 0.11 mM; Vmax = 20.00 nmol min−1 mgprotein−1; Vmax/Km = 181.80 10−6 L min−1 mgprotein−1 | Biocatalysis in vivo; TRL3 | ≈0.16 mM | n.d. | n.d. | WO2016185211A153 |
| Methacryloyl-CoA: n.a. | |||||||||
| n-Butyl methacrylate | AAT (engineered MpAAT1) | Malus pumila | Short to medium chain alcohols (C3–C10); short to medium chain acyl-CoAs120 | n-Butanol: n.a | Biocatalysis; TRL3 | n.d. | n.d | n.d. | EP3508585B163 |
| Methacryloyl-CoA: n.a. | |||||||||
| n-Butyl methacrylate | AAT (n.d. from crude leaf extract) | Durio zibethinus | Medium chain alcohols; short to medium chain acyl-CoAs121 | 1-Butanol:121 U mg−1 = 14.53 nmole CoASH g−1 FW | Biocatalysis; TRL3 | 14.0 μM | n.d. | n.d. | EP3115460A454 |
| Methacrylyl-CoA: n.a. | |||||||||
| n-Butylacrylate | AAT (various e.g. MpAAT1) | Plant origin (various e.g. Malus pumila) | Short to medium chain alcohols (C3–C10); short to medium chain acyl-CoAs120 | n-Butanol:120 Km = 0.11 mM; Vmax = 20.00 nmol min−1 mgprotein−1; Vmax/Km = 181.80 10−6 L min−1 mgprotein−1 | Fermentation; TRL3 | n.d. | n.d. | n.d. | WO2017167623A155 |
| Acryloyl-CoA: n.a. | |||||||||
| Isobutyl isobutyrate | AAT (LuxE) | Clarkia breweri | Primary and aromatic alcohols; medium chain and aromatic acyl-CoAs122 | Isobutanol: n.a. | Fermentation; TRL3 | 200 mg L−1 | n.d. | n.d. | US10006064B257 |
| Isobutyryl-CoA: n.a. | |||||||||
| Ethyl acetate | AAT (Eat1) | W. anomalus | Short to medium chain primary alcohols; acetyl-CoA2,59 | Ethanol:2,59 Km = 3.10 mM Acetyl-CoA2,59 Km = 2.40 mM | Fermentation; TRL4 | n.d. | 0.60 mole mole−1 glucose | n.d. | WO2018099719A15WO2018100097A158 |
| Myristyl myristate | Lipase (CalB) | C. antartica | Medium to long chain fatty acids; medium to long chain fatty alcohols67,123 | Myristyl alcohol:123 Km = 30.10 mM | Biocatalysis; TRL9 | n.d. | n.d. | 99.8% | EP2080807B167 |
| Myristic acid:123 Km = 25.60 mM | |||||||||
| Ethyl butyrate, butyl butyrate | Lipase (various e.g. CalB) | Yeasts (various e.g. C. antartica) | Medium to long chain fatty acids; medium to long chain fatty alcohols67,123 | Ethanol:124 Km = 540 mM kcat = 77.69 s−1 kcat/Km = 143.87 M−1 s−1 n-butanol: n.a. | Biocatalysis; TRL4 | Ethyl butyrate: 140 gL−1 | n.d. | Ethyl butyrate: 60% | US8357519B285 |
| Butyric acid: n.a | Butyl butyrate: 272 g L−1 | Butyl butyrate: 87% | |||||||
| Ethyl octanoate | Lipase (various e.g. CalB) | Yeasts (various e.g. C. antartica) | Medium to long chain fatty acids; medium to long chain fatty alcohols67,123 | Ethanol: n.a. | Biocatalysis; TRL3 | n.d. | n.d. | n.d. | US11214778B283 |
| Octanoic acid: n.a. | |||||||||
| Geranyl acetate | Lipase (various e.g. CalB) | Yeasts (various e.g. C. antartica) | Medium to long chain fatty acids; medium to long chain fatty alcohols67,123 | Geranyl alcohol: n.a. | Biocatalysis; TRL3 | n.d. | 98% | n.d. | US8506815B284 |
| Acetic acid: n.a. | |||||||||
| Methyl propionate | BVMO (AcCHMO) | Acinetobacter calcoaceticus (NCIMB 9871) | Small aliphatic ketones125 | 2-Butanone:126 Km = 2.4 mM | Biocatalysis; TRL3 | n.d. | 26% | n.d. | US9816115B289 |
Even though other ester forming enzymes are known, they have not been reported in patent literature for the production of such molecules. Very likely further research is needed to fully explore the effective potential of these niche ester-forming enzymes. Examples of such enzymes are the following: S-adenosyl methionine (SAM) dependent O-methyltransferases, hemiacetal dehydrogenases (HADH) and polyketide synthase associated proteins.2,3 SAM dependent O-methyltransferases are able to transfer the methyl group from SAM to free fatty acids to form FAME (fatty acids methyl esters).30–32 HADHs can oxidize hemiacetals to the correspondent esters through a NAD(P) dependent reaction.33,34 This type of enzymatic catalysis does not naturally occur as such, since HADH activity is a side reaction of alcohol dehydrogenases. Polyketide associated proteins finally, like PapA5, are better known for their ability to form complex esters with biological activity.35,36
Bioprocesses for LMW esters production involving AATs
As mentioned earlier, various microorganisms can produce esters from refined carbon sources like glucose and, potentially, from residual sugar-based biomasses such as whey sugars or cassava bagasse under specific conditions. According to scientific literature, iron limitation is a key factor that triggers bulk ethyl acetate production in certain yeasts, while in others oxygen limitation can also induce this process.2,37Since esters are a very heterogeneous group of molecules, it is not possible to assign a single physiological role to their natural microbial production. On one hand, high concentrations of volatile esters can repress growth of competitive organisms or can be used as metabolic intermediates during growth on alkanes or cyclic alcohols in certain bacterial species. On the other hand, some esters, like isoamyl acetate, may help yeasts to colonize other niches by attracting insects and exploiting their mobility. Finally, some more complex esters can serve as bacterial virulence factor and wax esters are usually produced as intracellular storage compounds (e.g. in Acinetobacter baylyi and Marinobacter hydrocarbonoclasticus).2,38 Since the titres of naturally produced esters are usually low, the direct conversion of sugars or alcohols by wild type organisms appears not a favourable way for industrial production of LMW esters. Nevertheless, these were the first approaches covered in patent literature. One of these patents discloses a process to produce ethyl acetate and acetaldehyde by microorganisms of the genus Candida or Hansenula starting from diluted ethanol wastes such as the ones coming from the brewing industry under conditions substantially free of dissolved iron.39 Inventors claimed that it is possible to switch from ethyl acetate to acetaldehyde production simply by altering the concentration of ethanol in the medium. Ethanol concentration below 35 g L−1 led to a preferential ethyl acetate production, while ethanol concentration above 35 g L−1 led to a preferential acetaldehyde production. Based on the result presented, it should be possible to obtain yields higher than 80% if ethanol is continuously fed to maintain a level of about 65 g L−1. Unfortunately, the invention does not provide information on the achievable ethyl acetate yield or the molecular mechanisms driving ester production (TRL3; i.e. proof of concept).39 Moreover, to our knowledge, it seems that no process has ever been commercialized using this technology.
Since then, other examples of direct microbial production of LMW esters starting either from glucose and/or from other feedstocks have been patented. Nearly all these processes utilise alcohol acyltransferases (AATs) to catalyse the final step of this microbial transformation.
AATs are ubiquitous enzymes that are capable to transfer an acyl moiety from an acyl-CoA molecule to an alcohol and thus catalyse the formation of the correspondent ester. The reaction is usually favoured in standard conditions (e.g. ΔrG′° = −23.6 kJ mol−1 for the formation of ethyl acetate starting from ethanol and acetyl-CoA) and does not require any cofactor.2 AATs belong to 2 unrelated protein families:2 the BAHD superfamily40–45 and the α/β hydrolase fold superfamily (Eht1, Eeb1 and Eat1 belong to this second family). This latter, characterized by the presence of a Ser-Asp-His catalytic triad, is the superfamily of hydrolytic enzymes such as proteases, esterases, lipases and peroxidases.46,47 BADH superfamily (and thus AATs like Atf1, Atf2 and AtfA) is characterised by the presence of HXXXD and DFGWG motifs, essential for protein function and structure, highly conserved in these enzymes from higher plants to yeasts. In the proposed catalytic mechanism, the HXXD motif is crucial as histidine takes part to the catalysis by deprotonating the hydroxyl group of the alcohol, while the aspartic acid is essential in keeping the solvent channel structure. Ester formation results from the transfer of the acyl group from the acyl-CoA substrate to the alcohol without the formation of a covalent acyl-protein intermediate (Scheme 2a).).2,48,49 α/β hydrolase fold-like AATs, on the other hand, have been proposed to form a covalent acyl-protein intermediate which is then transferred to the alcohol (Scheme 2b). Substrate specificities of AATs seem to be relatively broad regarding alcohols and narrower for acyl-CoAs2 and this is reflected also in patent literature (see Table 1 for some examples).
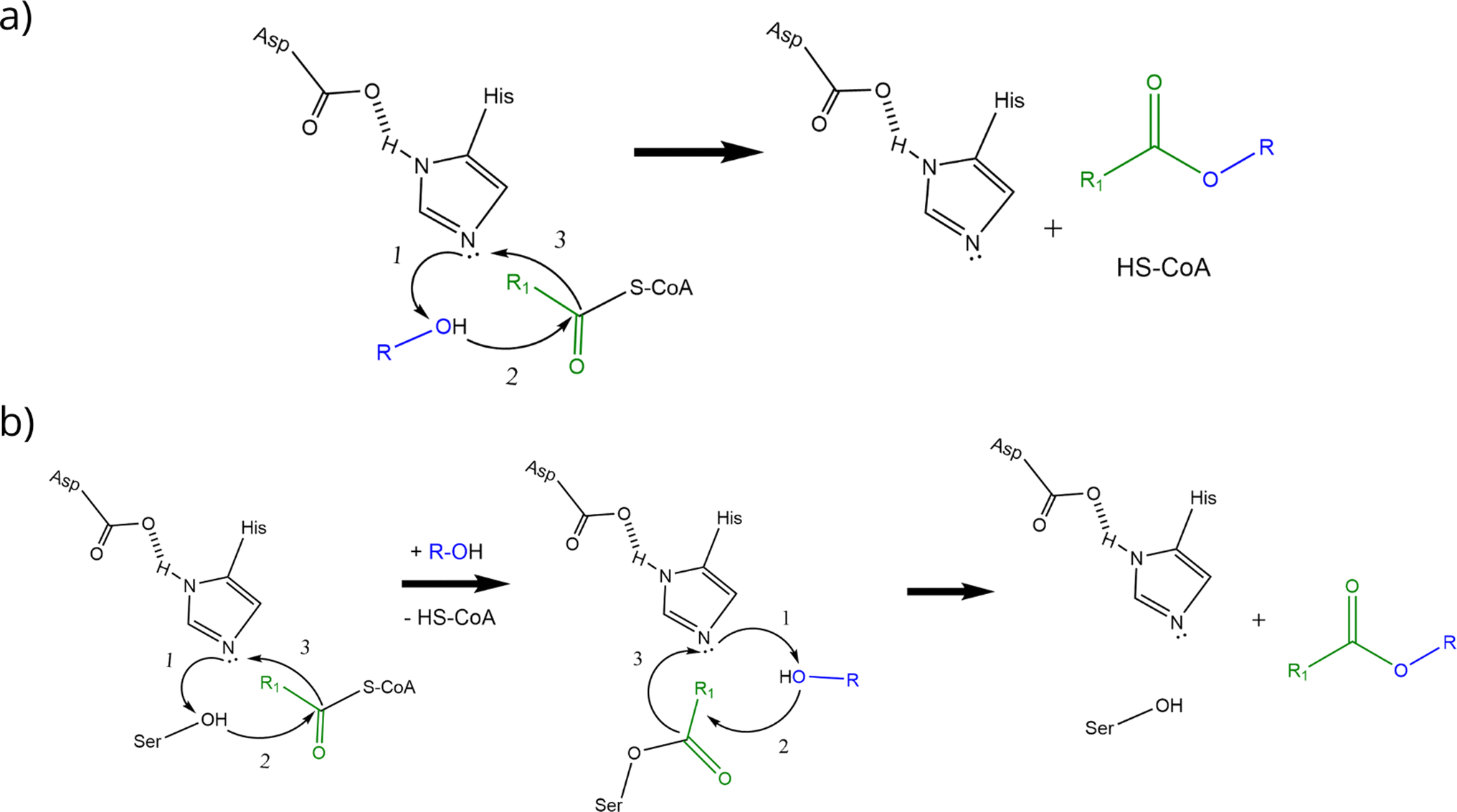 | ||
| Scheme 2 Proposed reaction mechanisms of BAHD-like AATs (a) and α/β hydrolase-like AATs (b) (re-drafted from (ref. 2)). Curved black arrows indicate the transfer of electrons during the reactions: transition states and water formation (b) are not shown to improve figure clarity (a and b). Alcohol activation is the first step in the catalytic mechanism of BAHD-like AATs; activated alcohol then reacts with the acyl-CoA leading to ester formation (a). In the α/β hydrolase like AATs, histidine activates the alcohol moiety on the serine residue which then can form a covalent bond with the acyl moiety of the acyl-CoA. The same happens to the alcohol substrate and finally reaction between the activated alcohol and the acyl-protein intermediate can take place thus forming the ester (b). | ||
One of the first attempts found in patent literature that makes use of these enzymes, reveals a process for the simultaneous production of isoamyl acetate and succinic acid.50 Inventors describe an in vivo method of producing esters from acetyl-CoA, such as isoamyl acetate, in E. coli. It has been observed, in literature, that the impairment of the acetate producing ackA-pta pathway leads to an increase in anaerobic isoamyl acetate production. However, an additional mutation in the lactate producing ldhA pathway reduced isoamyl acetate production to that of the wild-type strain. Additionally, the ldhA deletion activated the adhE pathway, presumably because the strain must still maintain the proper redox balance between NAD+ and NADH. In summary, inventors claimed to be able to obtain a recombinant strain with higher production levels of isoamyl acetate in respect to the wild-type strain by redirecting the NADH oxidation from ethanol to succinate production in order to balance the cofactors. According to authors this goal was reached by performing null mutations in ldhA, adhE and ackA-pta genes and performing an overexpression of pyruvate carboxylase (PYC) and alcohol acetyltransferase (ATF2) genes (Fig. 2).50
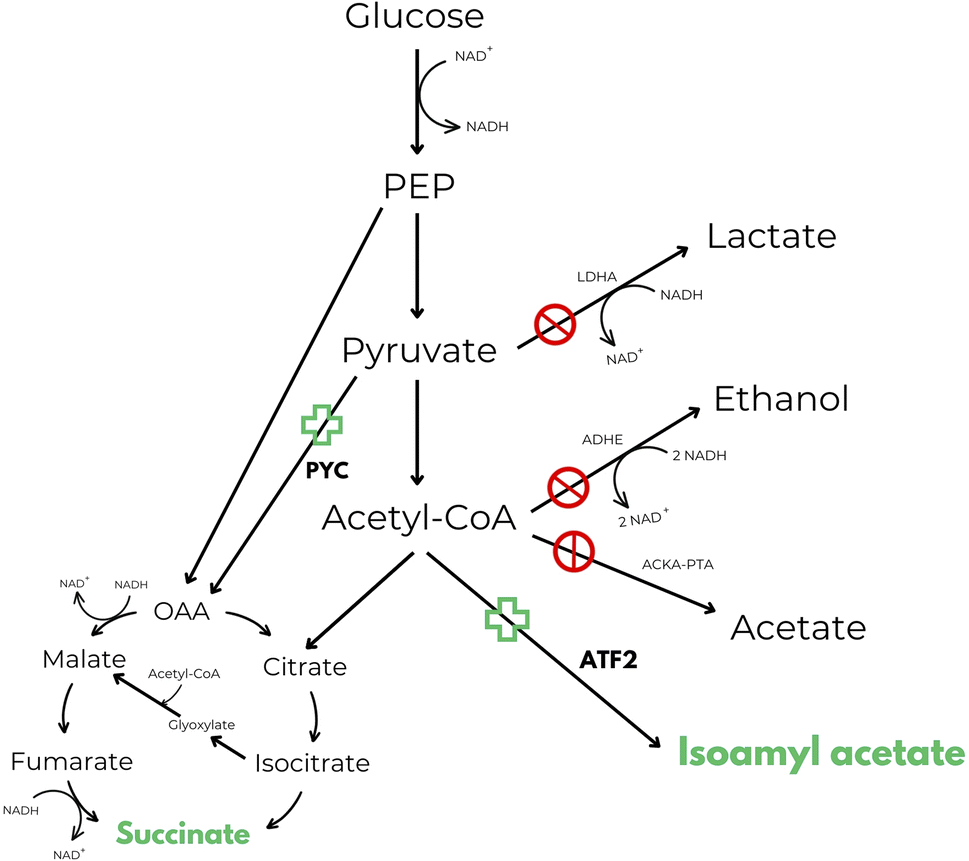 | ||
| Fig. 2 Central anaerobic metabolic pathway of E. coli including the novel isoamyl acetate and succinic acid production pathways disclosed in US 7569380 B2.50 Target products are highlighted in green. | ||
Even though the strain was able to reach almost the maximum theoretical ester yield, maximum titre of isoamyl acetate produced was very low (9.95 mM after 48 hours of incubation at 25 °C) and isoamyl alcohol was supplemented in the culture media at 10 mM concentration and not produced by the strain itself. Plus, when the amount of isoamyl alcohol supplemented was increased from 10 to 20 mM, the production of isoamyl acetate decreased in favour of succinic acid production. This is probably because isoamyl alcohol at appropriate concentration can increase the specific activity of succinate dehydrogenase, the enzyme responsible for the interconversion between fumarate and succinate.50 To the best of our knowledge, no process has ever been commercialized starting from this technology, and it is also impossible to know the cost effectiveness of the process without further tests (TRL3).
Pathway optimization and novel efficient AATs
As it may result from previous descriptions, still a lot of technical problems remain unsolved. First, much remains to be discovered about alcohol acyltransferases. Studies that focus on fundamental aspects of the enzymatic catalysis are few, controlling factors have been established only for a small number of AATs and finally it is unclear how efficiently or abundantly AATs are expressed in heterologous hosts. It is likely that some AATs may be catalytically inefficient towards ester synthesis because in many studies, or published patents, esters titres are very low. Finally, for an efficient engineering of ester production the balance between the supply of alcohols and acyl-CoA substrates is fundamental. Ideally, both substrates must be provided in 1:1 ratio to allow an efficient ester production in microbial cells.2
The first important step towards the development of an economically viable microbial production process of LMW esters is to find or engineer novel AATs with high activities towards the ester of interest. AATs can be found in the genetic pool of many different organisms (from yeasts and bacteria to fruits and vegetables). In patent literature we find several examples of different AATs used with this purpose and several examples of pathways engineering to make available AAT's substrates in the cellular pool of a microbial host (i.e. acyl-CoAs and alcohols).
The first patent that is worth mentioning is entitled “Bacteria engineered for ester production”:51 to our knowledge, it is the first example published in patent literature in which both the substrates (alcohols and acyl-CoAs) and the esters are produced in vivo by a microbial host. Herein, inventors disclose a method for the production of a recombinant bacterium with elevated alcohol acyl transferase activity (AAT) and either elevated 2-keto acid decarboxylase (KDC) or 2-ketoisovalerate dehydrogenase (KIVDH) activities that lead to an increased production of an acetate and/or an isobutyrate ester with respect to the wild-type strain. As an embodiment of the invention, the authors describe the optimization of isobutyl acetate production starting from glucose into a JCL88 E. coli strain. A complete isobutanol biosynthetic pathway (AlsS of B. subtilis, IlvCD of E. coli, Kdc of L. lactis, AdhA of L. lactis)52 was introduced onto a high copy plasmid (with a ColEl origin of replication, ∼40 copies per cell) while ATF1 from S. cerevisiae onto a medium copy plasmid (∼15 copies). To avoid toxicity of isobutyl acetate (E. coli is unable to grow in the presence of 3 g L−1 isobutyl acetate), a hexadecane layer was incorporated into the production culture to achieve in situ product removal. After 96 hours of incubation, a maximum titre of 17.2 g L−1 of isobutyl acetate was obtained. Final glucose consumption was measured, resulting in an isobutyl acetate yield of 0.334 g g−1 glucose (17.2 g L−1 isobutyl acetate produced/51.6 g L−1 glucose consumed), which is 80% of the theoretical maximum from glucose.51 This is the highest and practically the only titre that is worth noting reported in this patent for an ester production process at laboratory scale (TRL4). Nevertheless, from what is known, no process has ever been commercialized starting from this technology.
Both WO2016185211A153 from Mitsubishi Chemical UK Ltd and EP3115460A454 from Mitsubishi Chemical Corp disclose processes for the biological production of methacrylic acid esters, like methyl methacrylate, ethyl methacrylate and butyl methacrylate. Those LMW esters are important monomers widespread in the chemical industry to produce plastics for various applications. The most significant one is the casting, moulding or extrusion of polymethyl methacrylate (PMMA) to produce high optical clarity plastics. Inventors claimed to be able to produce methacrylic acid esters in E. coli, like n-butyl methacrylate, with the use of an AAT under EC group number 2.3.1.84 and preferably derived from plant origin selected from a group consisting of Lamiales, Vitales, Sapindales, Malvales, Magnoliales and Asterales.53,54 In WO2016185211A1, this kind of esters biologically derive from methacryloyl-CoA, which is formed from isobutyryl-CoA by the action of an appropriate oxidase (ACX4 from Arabidopsis thaliana), and an externally supplied n-butanol,53 while in EP3115460A4 the inventors describe an in vitro method for producing methacrylic acid esters by mixing a plant crude extract (which contains AATs) and appropriate substrates (methacryloyl-CoA and alcohols).53,54 According to both patents, it seems that plant AATs are usually more active toward methacryloyl-CoA (Km < 0.5 mM) than toward acetyl-CoA.53,54 In WO2016185211A1, inventors claimed to be able to produce almost 0.16 mM of butyl-methacrylate with MpAAT1 of Malus pumila after 25 hours reaction, while the highest titre reported in EP3115460A4 is of about 14 μM of butyl-methacrylate obtained with the crude plant extract of Durio zibethinus (probably containing different not characterized AATs) after 3 hours.53,54 Unfortunately, all the experiments described are merely at laboratory scale (TRL3).
The use of AATs for microbial esters production is fascinating from a technical and a commercial point of view. Theoretically speaking, one would be able to produce as many and diverse esters as needed (both symmetrical and asymmetrical esters) just by choosing the right AATs, with high activities toward the desired substrates and, of course, by engineering the appropriate number of microbial hosts for alcohols and acyl-CoAs production. Plus, as stated before, the doors for other bulk chemicals manufacturing will be opened, like acid and alcohols, simply by performing esters hydrolysis after the recovery.
An interesting example of what has been written so far can be found in patent literature, published in 2017 by BASF SE:55 the inventors are disclosing a method for the fermentative production of n-butyl acrylate, n-butyl propionate, n-butyl lactate and/or ethyl acetate combining appropriate AATs and strain engineering in S. cerevisiae. Strategy depicted in this patent is straightforward: by providing to a microbial strain a n-butanol production pathway, an acyl-CoA production pathway and a suitable AAT is it possible to produce the corresponding ester of interest. As an example of the invention, a method for engineering a yeast strain to produce n-butyl acrylate from glucose is described.55 They used a S. cerevisiae strain (TYC-185) engineered to produce butanol by reverse β-oxidation56 as a base to add an acryloyl-CoA pathway and an appropriate AAT. Acryloyl-CoA pathway essentially consists in a series of enzymatic reactions (methylmalonyl-CoA mutase, methylmalonyl-CoA decarboxylase and propionyl-CoA transferase) that are capable to increase the cellular propionyl-CoA pool, which is then oxidized to acryloyl-CoA by the aid of a plant acyl-CoA oxidase (ACO of Arabidopsis thaliana). Inventors do not disclose which is the AAT to use in this case to complete the n-butyl acrylate formation step, but from in vitro testing they said that it is possible to use a series of AATs coming mainly from plants and fruits (e.g. CmAAT1 and CmAAT2 of Cucumis melo, MpAAT1 and MpAAT2 of Malus pumila, VAAT of Fragaria vesca, BEBT and CbBEAT of Clarkia brewery).55 Again, maximum titres and achievable yields of esters are not provided, and all the experiments described are at laboratory scale (TRL3).
The same approach is also used in US10006064B2 in which the inventors disclose a method for engineering a microbial strain to produce any ester of interest.57 This is the first example in patent literature that does not limit the type of esters that can be obtained with this strategy. As in the previous description, one would need to introduce or increase the production of an acyl-CoA, introduce or enhance a pathway for the biosynthesis of an alcohol and, in this case, introduce or increase the expression of one or more appropriate AATs in a microbial strain to catalyse the formation of the ester of interest. As embodiments of the patent, examples describe the construction of three microbial strains capable of producing respectively higher levels of isobutyl isobutyrate, isobutyl acetate and isoamyl acetate from glucose, when compared to the wild type organisms. An interesting example that resulted in the production of 200 mg L−1 of isobutyl isobutyrate is the following: isobutyryl-CoA is produced by cloning branched chain ketoacid dehydrogenase complex BKDH from Pseudomonas putida in E. coli. To promote the biosynthesis of 2-ketoisovalerate, A1sS and l1vD genes were overexpressed and at the same time, to increase production of isobutanol from 2-ketoisovalerate, also Kivd and Yqhd genes were overexpressed. Finally, to catalyse the ester forming reaction, benzyl alcohol O-benzoyl transferase (BEBT or LuxE) from Clarkia breweri were also cloned in the same E. coli strain. Similar genetic engineering was used by the authors to produce isobutyl acetate, with exception of the isobutyryl-CoA forming pathway because acetyl-CoA is readily available in E. coli as a component of the TCA cycle. For the production of isoamyl acetate, on the other hand, the cell factory has been constructed to further express the LeuABCD pathway, which can elongate 2-ketoisovalerate by one carbon to form 2-keto-4-methylvalerate. In this case the combination of Kivd and Yqhd can catalyse the formation of isopentanol (i.e. isoamyl alcohol) from 2-keto-4-methylvalerate (see Fig. 3 for a scheme of all the 3 pathways described).
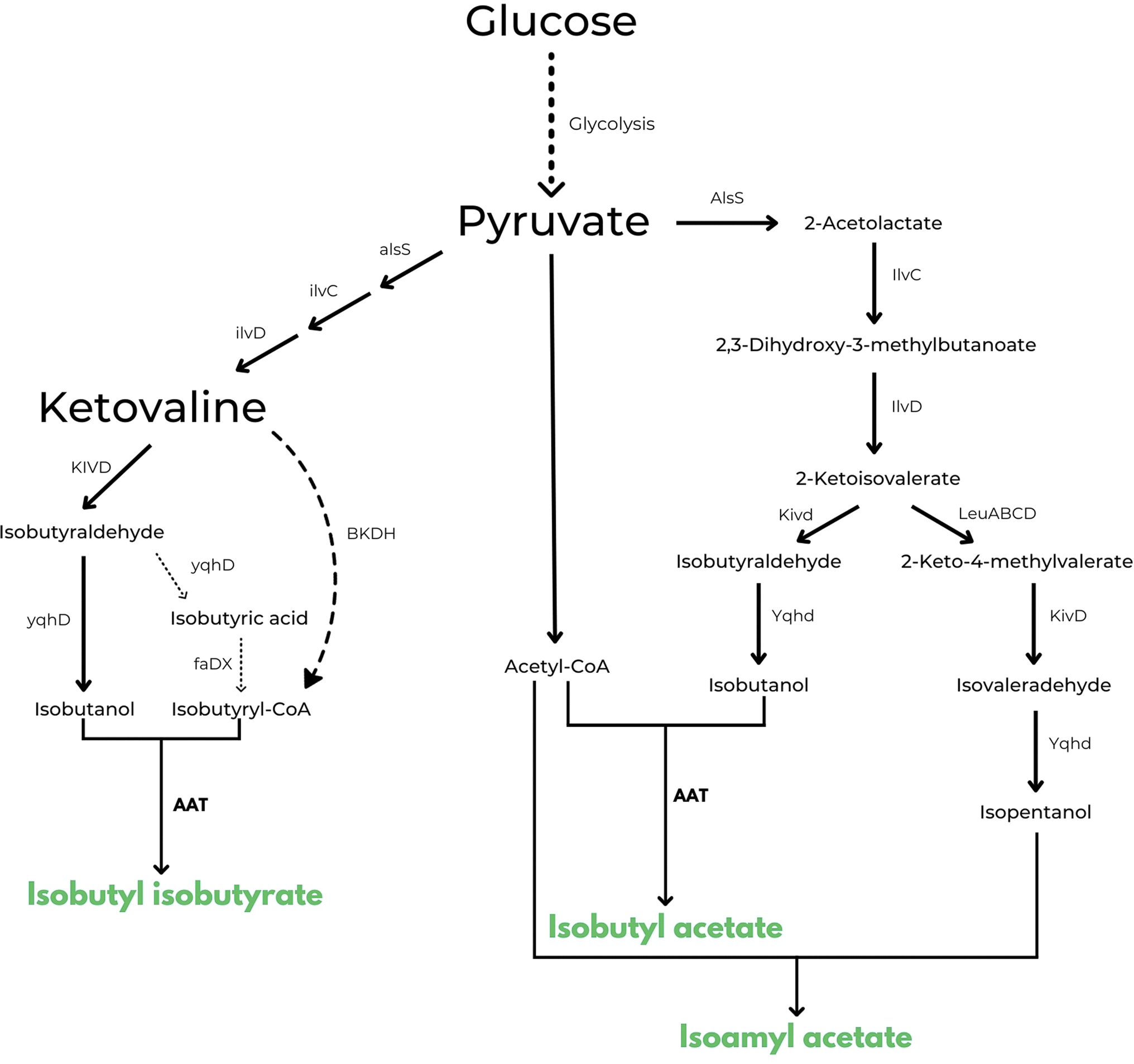 | ||
| Fig. 3 Enzymatic pathways disclosed in US 10006064 B2 for the microbial production of isobutyl isobutyrate, isobutyl acetate and isoamyl acetate respectively.57 Produced esters are highlighted in green. | ||
Highest titres of isobutyl acetate and isoamyl acetate (2.14 ± 0.17 g L−1 for isobutyl acetate) were obtained with the expression of ATF1 from S. cerevisiae.57 Even if interesting titres were reached in this patent, no other information was given by the authors about the scale and economic aspects of their experiments and from what we know thus far, no process has ever been commercialized using this technology (TRL3).
Discovery of new AATs is another tool that researchers can use to patent novel processes for LMW esters production. This is what happened for a couple of patent applications published in 2018 by the same inventors: WO2018099719A1 and WO2018100097A1.5,58 The first discloses a process for the general production of alkyl alkanoates (i.e. esters), while the second one discloses a process for ethyl acetate production only, using the same AAT enzyme that belongs to the novel Eat1 family of enzymes discovered by the authors.5,58 They started from the analysis of the sequenced and annotated genome of the ethyl acetate producing yeast Wickerhamomyces anomalus, strain DSM 6766. Its genome contains five putative Atf1 or Atf2 homologs and one Eht1 homolog. To see if they are involved in ethyl acetate production, the corresponding genes were expressed in a S. cerevisiae INVSc1 (MATa, his3D1, leu2, trp1-289, ura3-52, MAT, his3D1, leu2, trp1-289, ura3-52) strain. As transformants showed poor ethyl acetate production (0.005 g L−1 at most), maybe other enzymes are responsible for most ethyl acetate synthesis in W. anomalus. In the search of novel enzymes, inventors compared the transcriptome of W. anomalus DSM 6766 under ethyl acetate producing (oxygen limitations) and non-producing conditions in glucose limited continuous cultures. Known homologs of transcripts for Atf1, Atf2 and Eht1 did not show significant change in expression levels, while among the five most overexpressed genes there were two (wanomala_5543 and wanomala_7754) that encoded hypothetical proteins with an α/β hydrolase fold.
Both protein products resulted to be involved in ester metabolism in yeast and are 99% identical, meaning that the genes are most likely alleles in the diploid genome of W. anomalus. The enzyme encoded by Wanomala_5543 produced ethyl acetate in vitro in the presence of acetyl-CoA and ethanol, and therefore it was renamed Eat1 (Ethanol Acetyltransferase 1). A codon optimized version of Eat1 was expressed in E. coli BL21 (DE3) (pET26b:harmWanomala_5543-His) and this strain produced 4.87 ± 0.02 g L−1 of ethyl acetate by consuming 20 g L−1 glucose and 5.9 g L−1 ethanol, which corresponds to 32.93 ± 0.11% of the maximum yield.59 According to these inventions, 60% yields (moles of products over moles of consumed glucose) of ethyl acetate may be reached, but no maximum titres were reported in both patents. It is worth noting that culture conditions both in batch bioreactors (1.4 L) and continuous bioreactors (3 L) were given. Most probably, this means that at least tests in small pilot scale conditions were performed (TRL4).5,58
It is reported in non-patent literature that in the case of E. coli expressing AATs (particularly plant derived AATs), the large majority of enzymes are inactive insoluble proteins.60–62 Efficient expression of functional AATs in heterologous hosts is fundamental for the development of an in vivo ester production process. In EP3508585B1, published by Mitsubishi Chemical Corporation, inventors have tried to develop a solution to this problem by providing an engineered AAT with improved functionality and solubility in host cells. As an embodiment of the invention, it is described that is possible to enhance activity and solubility of an apple AAT (Malus pumila), by introducing the following mutations in its amino acid sequence:
• A substitution of cysteine at position 48 with alanine,
• A substitution of cysteine at position 150 with arginine,
• A substitution of cysteine at position 167 with alanine,
• A substitution of cysteine at position 270 with alanine,
• A substitution of cysteine at position 274 with alanine,
• A substitution of cysteine at position 447 with alanine,
• A substitution of alanine at position 64 by valine,
• A substitution of lysine at position 117 by glutamine,
• A substitution of valine at position 248 by alanine and,
• A substitution of glutamine at position 363 by lysine.
Activity of the enzyme was measured in vitro for the production of butyl methacrylate: 0.2 mL of the cell extract was added to 0.8 mL of a reaction solution containing 1 mM methacryl-CoA and 40 mM n-butanol. The mutant thus engineered exhibited approximately 30 times the activity of the wild-type enzyme (TRL3) and this was correlated also to the higher solubility assessed by measuring protein abundance in the soluble fraction versus the insoluble fraction of the cell extracts (SDS-page analysis).63
Processes for LMW esters production involving other enzymes
Enzymatic production of esters in patent literature is mainly focused on the use of lipases, but it is possible to find some examples also with the use of Baeyer Villiger monooxygenases (BVMO). Lipases are generally stable enzymes and can be purified in good yields from animals, plants and microorganisms. They are capable of catalysing ester formation with several mechanisms (esterification, interesterification and transesterification reactions) in non-aqueous conditions with good yields without the need of cofactors.65 Examples of such reactions in organic media, supercritical fluids, ionic liquids and deep eutectic solvents can be found in scientific literature, but also solvent free processes are available.65–68Lipases share together with some AATs (e.g. Eat1) and some other enzymes the so called α/β hydrolase fold with a Ser-Asp-His catalytic triad. Mechanism of action of lipase-catalysed esterification involves several key steps. The hydroxyl group of the serine acts as a nucleophile, attacking the carbonyl carbon of the fatty acid (or more in general, the carboxylic acid), which is activated by the enzyme. This forms a tetrahedral intermediate. The tetrahedral intermediate collapses, leading to the formation of a covalent acyl-enzyme intermediate, and the release of water. The alcohol can then displace the enzyme's active site residue (e.g., serine), leading to the regeneration of the free enzyme and the release of the ester product. Finally, the enzyme returns to its original state, ready to catalyse another reaction (see Scheme 3).69,70 Lipases are particularly known for their specificity towards certain fatty acid chains. Some are more effective on short-chain fatty acids, while others prefer to act on long-chain fatty acids. This is the same regarding the alcoholic substrate. CalB, for example, often mentioned in patent literature for ester production, has a large acyl binding cleft, but a narrow alcohol binding cleft, so it seems to be able to process longer fatty acids than other lipases. CalA, on the other hand, seems to be more specific about the fatty acids that can accommodate in its acyl binding pocket and less specific regarding alcohols.71,72
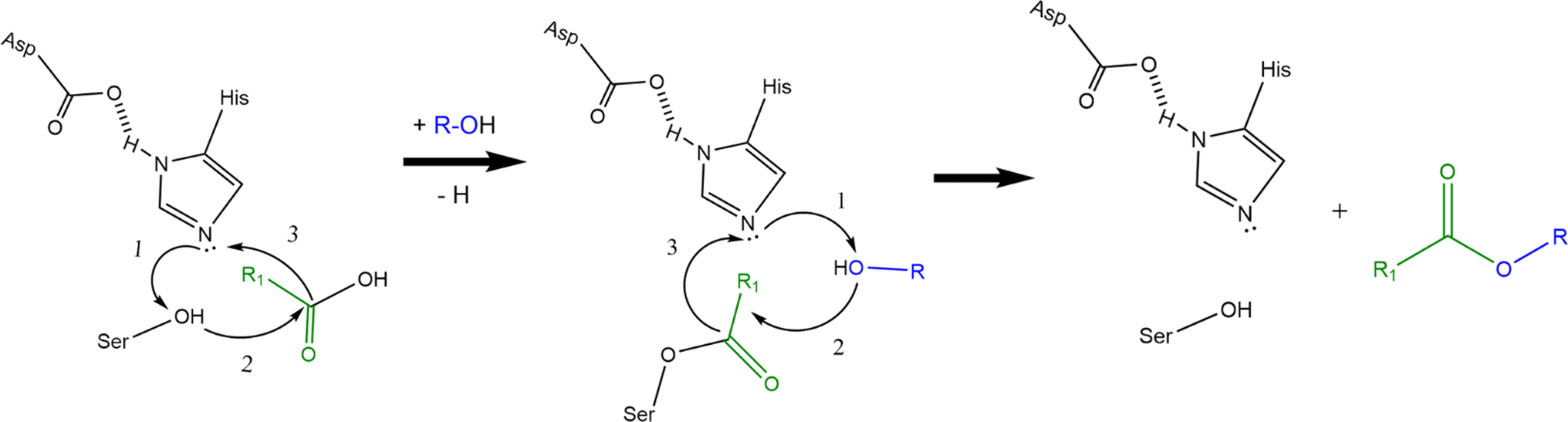 | ||
| Scheme 3 Ester formation reaction mechanism of lipases. Histidine activates the alcohol moiety on the serine residue which then can form a covalent bond with the (fatty) acid. The same happens to the alcohol substrate and finally reaction between the activated alcohol and the acyl-enzyme intermediate can take place thus forming the ester (b). Transition states and water formation are not shown to improve figure clarity. Curved black arrows indicate the transfer of electrons during the reactions. | ||
Baeyer Villiger monooxygenases (BVMOs), on the other hand, are flavin dependent enzymes capable of catalysing the insertion of an oxygen between a C–C bond in aldehydes and ketones to give the corresponding esters. Unfortunately, only very few potentially industrial applications of BVMOs are known and most of them are to produce high value compounds in the pharmaceutical industry, mainly because of their poor thermostability and their cofactor dependency.2,73 The 3D structure of BVMOs typically exhibits a characteristic α/β fold. Active site of BVMOs is located within a pocket formed by several conserved residues like the PXXXH motif in which the histidine is critical for the activation of the peroxide during the reaction.74 This site contains also the flavin adenine dinucleotide (FAD) cofactor, which is crucial for the enzyme function.75,76 The initial step of the catalysis involves the reduction of the flavin by NADPH, generating a reduced flavin (FADH2). The reduced flavin can then interact with molecular oxygen to form a peroxo-intermediate. The peroxo-flavin can then undergo a rearrangement in which the ketone substrate performs a nucleophilic attack. This is facilitated by amino acid residues, such as histidine, which can stabilize transition states and intermediates through hydrogen bonding and electrostatic interactions. Through the rearrangement and further interactions facilitated by the enzyme active site residues, an alkyl hydroperoxide intermediate is formed. The oxygens of the hydroperoxide intermediate can then rearrange, leading to the migration of the alkyl group and subsequent formation of the ester or lactone product while regenerating the oxidized flavin (see Scheme 4).77–79 BVMOs usually show preferences for certain ketone structures. For example, cyclohexanone monooxygenase (CHMO) preferentially oxidises cyclic ketones like cyclohexanone, even thought is clearly reported in patent and non-patent literature that CHMO (as other BVMOs) can oxidise also linear ketones like butanone.74,80
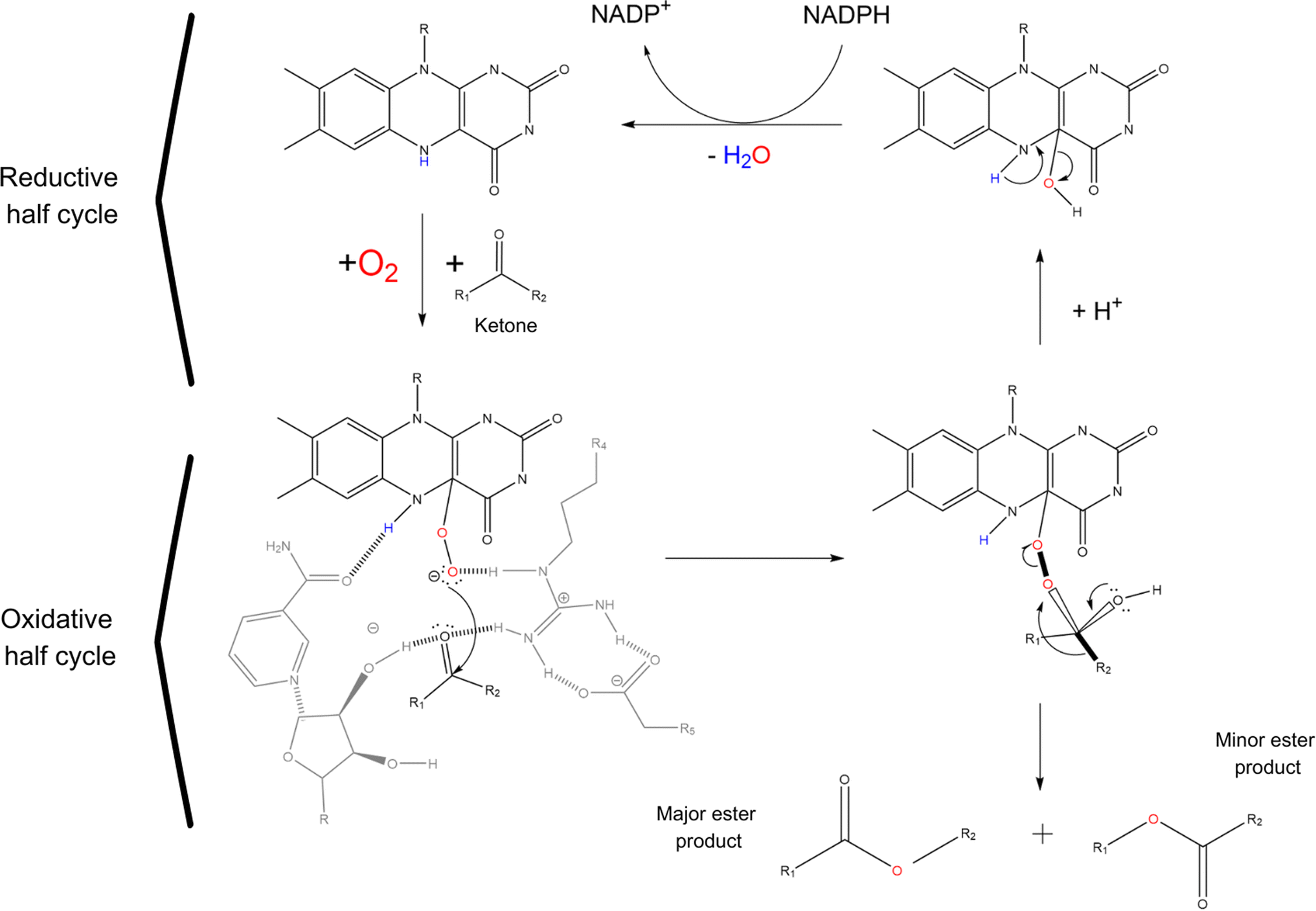 | ||
| Scheme 4 Ester formation reaction mechanism of BVMOs starting from a ketone. Catalytic cycle of BVMOs consists of 2 half reactions: an oxidative one in which the ketone is oxidised by a peroxy-flavin and a reductive one in which the oxy-flavin is regenerated to its reduced form with the aid of NADPH cofactor. Reaction involves an intermediate that determines regioselectivity. The migration of the bond relies on the anti-periplanar orientation (shown by thick bonds) of the migrating bond with the peroxy-bond and one of the lone pairs on the carbonyl oxygen. Curved black arrows represent electron movements during the reaction. | ||
LMW esters synthesis by lipase-catalysed reactions
Most of the patent literature published so far is devoted to the use of lipases for the production of biodiesel (i.e. FAME or FAEE of natural extracted oils like soy oil, coconut oil and sunflower oil just to name a few), polyesters or specialty chemicals. Only a few of these works are also applicable for the synthesis of LMW esters. We summarize the most significant ones in the context of this paper.Water activity, pH, substrates concentrations and temperature play a critical role in lipase catalysed ester formation. The lipase catalysed esterification is usually thermodynamically unfavourable under aqueous conditions (e.g. ΔrG′° = 16.1 kJ mol−1 for ethyl acetate starting from acetic acid and ethanol).2 For this reason the processes disclosed in patent literature are mostly performed in non-aqueous systems using organic solvents, high alcohol concentration (sometimes used also as the solvent for the reaction), relatively low acid concentration (to avoid lipase inactivation) and lipases immobilized on a solid support.65 EP2080807B1, published by Evonik, discloses a solvent-free process for the production of carboxylic acid esters by the use of immobilized lipases.67 Is it widely known that Evonik has been using this process since 2010 for the production of emollient esters for cosmetic applications, like oleyl erucate and myristyl myristate, in one of its German facilities, commercialized under the registered trademark TEGOSOFT®. The company has also expanded the production of this kind of esters with a new facility located in China in 2022 (TRL9).81,82 Even though emollient esters are obviously not LMW esters, according to the patent it is possible to produce also smaller esters like ethyl propanoate or others. Inventors were able to solve the main problems known in the art of lipase catalysed esterification (like impairment of the heterogeneous catalyst with standard mechanical agitation, water removal at low working temperature, slow and poor conversion rates) with a reactor design in which both the mixing of the reactants and the discharge of water occur by the introduction of a gas stream. In this way, mechanical and thermal stress caused by traditional stirrers are avoided, ensuring high reusability of the catalyst. The gas (e.g. air, lean air, nitrogen, argon, helium and/or carbon dioxide), which is introduced in the reactor by a sparger positioned at the bottom of the tank, must be inert against the other components of the reaction media and against the reactor material. The use of the gas sparger allows to reduce the reaction time, while maintaining a very high conversion of the reactants (higher than 99.6% in certain embodiments of the patent).67
One of the problems that may hinder the use of lipases for ester synthesis is reported by Wang and colleagues: several heterologously expressed lipases have lower ability to catalyse the synthesis of esters in comparison with wild type lipases.83 For this reason, they have patented a method for the preparation of lipases which have high ester synthesis activity by using surfactants. Inventors discovered that the interaction between surfactants and soluble lipases in aqueous environment, followed by lyophilization of the mixture afterwards, is able to change the microstructure of the lipases and enhance or restore the ester forming activity in non-aqueous media. According to the authors, almost any kind of zwitterionic, non-ionic and cationic surfactants could be used in appropriate concentrations (usually 10 mM) to perform the procedure, but each of them will give peculiar results. Lipase concentration should be around 0.2 mg mL−1 (between 0.1 and 0.4 mg mL−1) and pH should be kept between 6.5 and 8.0, with the use of an appropriate buffer if necessary. As an example, the authors were able to increase the ester synthesis activity of a commercial CALB powder by 700% (from 7.8 U mg−1 to 101.9 U mg−1), by adjusting the final protein concentration of CALB to 0.12 mg mL−1 and by using DDM (n-dodecyl-β-D-maltopyranoside) at 10 mM as a surfactant for the procedure. The target ester in this case was ethyl octanoate, which technically should not be defined as a LMW ester.83 It would be interesting to know if similar activity can be assessed for the synthesis of LMW esters, like ethyl acetate or butyl acetate. The authors do not disclose if this beneficial effect would be maintained if the processed enzymes would be immobilized on a solid support with a view to heterogeneous catalysis in an industrial process design (TRL3).
As already mentioned, water is produced during lipase-catalysed esterification, so it is important to control water concentration in the reaction media to reach appropriate conversion of the reactants. Most of the traditional approaches are energy intensive (e.g. fractional distillation) or operate cyclically and thus utilize oversized and redundant absorber units so that saturated ones can be taken offline to be regenerated (e.g. molecular sieves). Nemser and collaborators have developed an interesting process to remove water from other fluids using membrane technology that can be applicable, among the others, also to lipase catalysed esterification. In this kind of process one side of a selectively permeable membrane is in contact with the mixture to be separated. A driving force (e.g. pressure gradient or concentration gradient) causes the migration of one or more preferential components of the mixture from one side to the other side of the membrane. Inventors discovered that selectively permeable perfluoropolymer membranes are highly effective at separating water and/or methanol from heterogeneous mixtures with high performance and in a wide range of water concentrations (even below 0.5% wt). For example, in one embodiment they were able to increase the conversion of the lipase catalysed esterification reaction of geranyl alcohol and acetic acid for the production of geranyl acetate from 94% to 98% by the use of a hollow fiber module through which the reaction medium was recirculated for water separation (TRL4).84
Finally, in patent literature it is also possible to find integrated processes for the production of small esters that start from the production of the lipases' substrates (i.e. alcohols and acids) by fermentation and end with a lipase catalysed esterification in appropriate conditions.
US8357519B2 is one of these examples: the authors describe an integrated process for the production of small esters (e.g. ethyl butyrate, butyl butyrate, ethyl lactate or ethyl propionate) starting from biomass. It covers the production via fermentation of a target organic acid, which is then extracted and finally esterified with an alcohol by the aid of a suitable lipase. Inventors claimed that it is possible to increase productivity and concentration of the organic acid by coupling fermentation with extraction using membrane technology and a suitable organic solvent. This will prevent phase separation and, simultaneously, will keep constant the pH of the fermentation broth. With this method, butyric acid concentration, reached by extraction and enrichment, was higher than 300 g L−1. The organic acid is then stripped out with the use of base or with the use of a strong acid solution. Esterification is finally carried out with an immobilized lipase in a fibrous bed bioreactor and water content was controlled with the use of a molecular sieve. In one embodiment they were able to reach 272 g L−1 of butyl butyrate concentration after 24 hours reaction (TRL4).85
LMW esters synthesis by BVMO-catalysed reactions
Baeyer Villiger oxidative enzymes (BVMO) can be found in several organisms, from bacteria to plants, animals or fungi, and they are bioequivalent to chemical catalysts of Baeyer Villiger (BV) oxidations (i.e. peroxyacids or peroxides). The vast majority of the scientific literature is focused on the use of BVMOs for the oxidation of ring-based ketones (i.e. lactones), rather than on linear small ketones, even though appropriate BVMOs can clearly oxidize small ketones or aldehydes as well.86 This is reflected also in patent literature in which several examples of the use of BVMOs for the production of high value compounds or intermediates useful in the pharmaceutical, cosmetic and perfumery industries can be found. WO2022003017A1 for example, published by Givaudan, describes the use of an appropriate BVMO (from Pseudomonas veronii) for the preparation of homofarnesol, an important intermediate for the production of Ambrox, a terpenoid widely used in the formulation of perfumes.87 US20210355516A1, on the other hand, discloses a BVMO catalysed process for the synthesis of (1S,5R)-bicyclolactone: a key intermediate in the preparation of prostaglandin.88 As a matter of fact, we were able to find only one patent that discloses a process for the production of a LMW ester with the use of BVMOs: US9816115B2, by Mitsubishi Chemical UK Ltd.89 Inventors describe a methyl methacrylate production process which involves the oxidation of 2-butanone to methyl propionate with the use of a BVMO. Methyl propionate is then reacted with formaldehyde in anhydrous conditions to give methyl methacrylate as in the already known alpha process and commercially practiced by Mitsubishi itself. Baeyer Villiger oxidation of 2-butanone would usually result in ethyl acetate as a product but, according to the authors, certain BVMOs (e.g. cyclohexanone monooxygenase, 4-hydroxyacetophenone monooxygenase and cyclopentadecanone monooxygenase) are capable of oxidizing 2-butanone differently, to yield methyl propionate at industrially viable levels. The use of other solvents like methanol or an excess concentration of 2-butanone itself (i.e. 1000 times molar concentration relative to the BVMO) would lead to an increase in methyl propionate formation compared to ethyl acetate production (in some embodiments for example incubation with 5 mM 2-butanone yields an ethyl acetate:methyl propionate ratio of 5:1, while incubation with 1000 mM 2-butanone yields a ratio of 1.5:1). No reaction conditions are given in the patent as well as yields and maximum concentration of methyl propionate achievable with this technology89 and to our knowledge no process has ever been commercialized with the use of this reaction (TRL3). It would be interesting to evaluate the use of this kind of BVMOs also for ethyl acetate production or for other small esters preparation. Moreover, it seems feasible to switch between ethyl acetate and methyl propionate production, according to the reaction conditions and substrates concentrations.
Scale-up of fermentative and biocatalytic processes fostering AATs, lipases and BVMOs
Becoming economically competitive with their petrochemical counterparts is the final goal to attempt when dealing with the scale-up of biotechnologically produced esters. This is true for every synthetic strategy analysed in this review. High production titres, yields and volumetric productivity must be reached together with efficient downstream procedures to recover the products with desired purity. Those objectives are clearly not yet achieved in the patented processes regarding the use of AATs for microbial ester production or in the use of BVMOs for the same goal (mostly proof of concepts) and partially achieved in the use of lipase catalysed esterifications to manufacture LMW esters. Probably, this is the reason why no scale-up examples with AATs, BVMOs and lipases have been found in patent literature. Nevertheless, in scientific literature are present some interesting examples that is worth mentioning regarding AATs and lipases (unfortunately no scale-up example has been found with BVMOs for the production of LMW esters). Two of them, in a few liters bioreactors exploiting AATs,90,91 deal with E. coli strains optimization to increase ethyl acetate yields and volumetric productivity.64,90 Just one in a pilot scale bioreactor (70L) describing a wild-type microorganism (K. marxianus) cultivated for ethyl acetate production.64Ethyl acetate production from glucose in yeasts is an aerobic process that results in an NADH surplus.6,92–94 This excess is disposed by yeasts only through respiration, which leads to low ester production yields because glucose is mainly oxidised in the TCA cycle for growth.95 Plus, rate limiting step in large aerobic fermentations is often oxygen transfer rate due to low solubility of oxygen in culture media.96 To avoid these problems authors used E. coli because, like other bacteria, can correct redox imbalance anaerobically using pyruvate formate lyase (Pfl) and thus secreting formate.90,91 With this strategy, associated to the inactivation of competitive pathways (e.g. ackA and ldhA), they were able to reach 72% of the maximum pathway yield (or 3.8 g L−1) with Eat1 as AAT in 1.5L bioreactors.90,91
As already said, iron (Fe) and copper (Cu) limitation seems to be the main trigger for ethyl acetate synthesis in K. marxianus and other yeasts.64,97,98 This principle has been studied by Löser et al. in a 70L bioreactor using whey, waste of the dairy industry, as carbon and energy source. As expected, the highest ethyl acetate yield (0.265 g g−1 of lactose, corresponding to 51.4% of maximum pathway yield) was obtained at the lowest applied Fe concentration (53 μg L−1). No significant differences in yield or volumetric productivity were observed between the 70L pilot scale fermentation and laboratory scale experiments, and no particular issues arose during scale-up.64,97–100
There are indeed more reports focused on the scale-up of lipase-catalysed ester forming reactions; however, all these deal with the scale-up of processes for biodiesel or specialty esters for cosmetic applications.101–107 Nevertheless, some assumptions can also be applied to the synthesis of LMW esters. Lipase-catalysed esterification or transesterification may exhibit slow reaction rates (i.e. taking 10 to 30 h longer than the conventional alkaline catalysed biodiesel production) due to low mass transfer of the reactants to the enzyme active site. Intensification technologies like ultrasound irradiation,108 microwave irradiation,109,110 microreactors111,112 and supercritical CO2113,114 have been utilised to enhance product yield and shorten reaction times. Among these methods, ultrasonic irradiation and microreactor technology gave the most significant improvements with better scale-up potential107 and would likely also enhance LMW esters production processes with lipases. Another common issue in scale-up, frequently mentioned in literature, is the high cost of the enzymes,65,66,107,115 that could be overcome by recyclable immobilized lipases. Finally, it is interesting to notice that biodiesel is for all intent a bulk chemical, like LMW esters, and thus must have a competitive low price to gain market acceptance. Up to date, enzymatic biodiesel production has been successfully commercialised by Blue Sun Energy Ltd and Viese Fuel LLC.107 Most probably, the choice of the most suitable enzymes coupled to process engineering would improve catalytic efficiency and reduce costs, making lipase-mediated LMW ester production economically feasible on an industrial scale.
Concluding remarks and future developments
LMW esters are manufactured in a million of tons per year scale, starting from fossil based raw materials and with energy intensive chemical processes to enable massive production of products, food and materials.2,6 This consideration clearly exemplifies the fact that the energy transition will be not enough to tackle climate change and reach net zero GHG emissions by 2050. For these reasons there is urgent need to lower the environmental impact of such processes and this is the reason why extensive research has been performed to find more sustainable manufacturing routes for LMW ester production.13,14,17,18 In this review we reported the most significant patents that in the future will hopefully lead to the biotechnological production and commercialization of bio-based LMW esters (Table 1). Adopted strategies focused mainly on the use of three classes of enzymes that can enable LMW ester production: esterases/lipases (EC 3.1.1.-), Baeyer Villiger monooxygenases (BVMOs, EC 1.14.13.-) and alcohol acyltransferases (AATs, EC 2.3.1.-) (Fig. 1). Starting from AATs, patent literature is lacking yields, conversion data and final concentration of the targeted esters with very few exceptions. In addition, none of the patents found reported a strain engineering method capable to ensure the balance between the supply of alcohols and acyl-CoA substrates in the host cell. As already said, ideally both substrates must be produced in 1:1 ratio to allow an efficient ester production in microbial cells.2 Plus, all the promising processes found in patent literature, regarding AATs, started from refined glucose or from other intermediates that have or may have a high cost in view of an industrial production of bulk chemicals such as LMW esters. Even though it is theoretically possible to start from renewable carbon sources by providing an engineered host with the appropriate cellulolytic or ligninolytic enzymes, no practical examples of such processes have been found in patent literature.
Both scientific and patent literature is reach of documentation about lipase catalysed esterification, but most of them regards biodiesel, polyesters, or specialty chemicals production and very few are applicable to LMW esters preparation. The implementation of lipase technology to the biodiesel production industry is still in the early stages,116 while lipase technology for the production of LMW ester is still under evaluation by researchers all around the globe as clearly represented in this work. The only company that seems to be using this kind of technology for the production of specialty esters for cosmetic applications is Evonik, but not for the production of LMW esters.67 It can be argued that lipase mediated esterification to produce small esters has a great potential, but at the moment might not be an economically viable alternative, due to the elevated cost of several procedures like water removal and enzymes immobilization among others.
Patent literature about BVMOs catalysis for LMW esters production is truly at early stages. We were able to find just one example about methyl propionate synthesis.89 It would be interesting to know if BVMO technology for small ester production could be applied at an industrially viable level, not only in in vitro, but also in in vivo processes and possibly we will see developments in the coming years.
Despite all the efforts made so far, a microbial or an enzymatic process to produce LMW esters starting from renewable resources still seems a long way off. In some cases, further research is needed to understand molecular mechanisms of microbial esters production (i.e. AATs and BVMOs), while in others, enzyme engineering and optimization (i.e. lipase) is the main goal to tackle to realize a profitable industrial production of LMW esters. Proper production titres, high product yields and suitable volumetric productivity for industrial production still need to be achieved with practically all the strategies discussed in this work. Although an extensive patent literature search has been performed, just 16 patents have been found related to the biotechnological production of LMW esters: this is a clear sign of the young age of this research field.
Finally, production process of bulk chemicals is just one side of the coin in the story of a product's commercialization. The grade of purity required by the chemicals' users is also very important for its final price and therefore for its commercial viability. LMW esters are widely used compounds in many different industries. They are used as solvents for industrial cleaning applications or as ingredients in other formulations: in these cases, extreme purity is mainly not required. LMW esters can be used as intermediates or monomers (for further chemical synthesis) and take part in specific industrial applications for their peculiar physico-chemical properties, from fuel additives to extraction of APIs. The latter cases require the highest purity. The good news for LMW esters is that product removal and purification from reaction media or fermentation broth should not be as difficult as it is for acids and higher alcohols in aqueous environments. First, LMW esters usually have a poor or negligible solubility in water, so if produced at high titre they will separate autonomously from the aqueous reaction media, while if produced in low or medium concentrations extractability in an organic phase such hexane or decane (i.e. ISPR systems) is usually effective. Lastly, most of the times LMW esters are volatile molecules with low boiling points and high vapor pressure so standard energy intensive purification techniques, like distillation, or novel purification processes, like membrane separation systems, with low or moderate energy expenditure, could be efficiently applied to obtain any suitable purity.
In conclusion, it is very difficult to predict when and if the first LMW ester, produced entirely with a biotechnological route, will reach the market, as manufacture technologies are still in an early stage of development.
Abbreviations
| LMW | Low molecular weight |
| HMW | High molecular weight |
| EXW | Ex-works |
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
MZ, IS, PB: conceptualization. MZ: original draft preparation. MZ, IS and PB: writing, reviewing, editing.Conflicts of interest
The authors declare that there is no conflict of interest.Acknowledgements
This work was partially supported by the University of Milano-Bicocca with the FA (Fondo di Ateneo) to P. Branduardi and I. Serra. The Authors acknowledge the National Center 5 “National Biodiversity Future Center” (award number: Project code CN_00000033, Concession Decree No. 1034 of 17 June 2022 adopted by the Italian Ministry of University and Research, CUP H43C22000530001, Project title “National Biodiversity Future Center – NBFC. Funder: project funded under the National Recovery and Resilience Plan (NRRP), Mission 4 Component 2 Investment 1.4 – Call for tender No. 3138 of 16 December 2021, rectified by Decree n.3175 of 18 December 2021 of Italian Ministry of University and Research funded by the European Union – NextGenerationEU). This work was also supported by Soft Chemicals S.r.l. (Italy).References
- J. W. Lee and C. T. Trinh, Curr. Opin. Biotechnol., 2020, 61, 168 CrossRef CAS PubMed.
- A. J. Kruis, A. C. Bohnenkamp, C. Patinios, Y. M. van Nuland, M. Levisson and A. E. Mars, Biotechnol. Adv., 2019, 37, 107407 CrossRef CAS PubMed.
- S. Menendez-Bravo, S. Comba, H. Gramajo and A. Arabolaza, Appl. Microbiol. Biotechnol., 2017, 101, 3043 CrossRef CAS PubMed.
- E. Fischer, Ber. Dtsch. Chem. Ges., 1895, 28, 3252 CrossRef CAS.
- A. J. Kruis, M. Levisson, A. E. Mars, S. W. M. Kengen, J. Van Der Oost and J. P. M. Sanders, WO Pat., 2018099719A1, The Netherlands: WIPO, 2017 Search PubMed.
- C. Löser, T. Urit and T. Bley, Appl. Microbiol. Biotechnol., 2014, 98, 5397 CrossRef PubMed.
- A. Y. Adesina, I. B. Obot, A. Sorour Ahmad, S. Mtongana, S. B. Mamilla and A. A. Almathami, Eng. Failure Anal., 2021, 127, 105511 CrossRef CAS.
- Corbion Purasolv®, accessed on April 2024, https://www.corbion.com/Products/Biochemical-products/Purasolv.
- ESUN Ethyl lactate, accessed on April 2024, https://www.brightcn.net/en/product/item-87.html.
- Ethyl acetate - Godavari Biorefineries, accessed on April 2024, https://godavaribiorefineries.com/products/chemicals/bio-based-ethyl-acetate.
- Ethyl acetate – Sekab, accessed onApril 2024, https://www.sekab.com/en/products-services/product/ethyl-acetate/.
- Solsys® Bio Etac – Solvay, accessed on April 2024, https://www.solvay.com/en/brands/solsys-bio-etac.
- J. Philp, New Biotechnol., 2018, 40, 11 CrossRef CAS PubMed.
- P. Branduardi, Microb. Biotechnol., 2021, 14, 68 CrossRef PubMed.
- S. Gueye and N. Jeffries, Completing the picture: How the circular economy tackles climate change, accessed on April 2024, https://www.ellenmacarthurfoundation.org/completing-the-picture.
- B. Oberle, S. Bringezu, S. Hatfield-Dodds, S. Hellweg, H. Schandl and J. Clement, Global Resources Outlook 2019: Natural Resources for the Future We Want, accessed on April 2024, https://wedocs.unep.org/handle/20.500.11822/27517;jsessionid=EA5AE4DDE3E90FC53C7A0B41F6342546.
- M. Carus, Ind. Biotechnol., 2017, 13, 41 CrossRef.
- C. McGlade and P. Ekins, Nature, 2015, 517, 187 CrossRef CAS PubMed.
- E. A. R. Zuiderveen, K. J. J. Kuipers, C. Caldeira, S. V. Hanssen, M. K. van der Hulst and M. M. J. de Jonge, et al., Nat. Commun., 2023, 14, 8521 CrossRef CAS PubMed.
- B. G. Hermann, K. Blok and M. K. Patel, Environ. Sci. Technol., 2007, 41, 7915 CrossRef CAS PubMed.
- L. Yang, X. C. Wang, M. Dai, B. Chen, Y. Qiao and H. Deng, et al., Energy, 2021, 228, 120533 CrossRef.
- D. Peñaloza, M. Erlandsson and A. Falk, Constr. Build. Mater., 2016, 125, 219 CrossRef.
- F. K. Adom and J. B. Dunn, Biofuels, Bioprod. Biorefin., 2017, 11, 258 CrossRef CAS.
- N. T. Hong Thuy, Y. Kikuchi, H. Sugiyama, M. Noda and M. Hirao, Environ. Prog. Sustainable Energy, 2011, 30, 675 CrossRef.
- D. Nong, N. Escobar, W. Britz and J. Börner, J. Cleaner Prod., 2020, 272, 122738 CrossRef CAS.
- Ethyl acetate market size, share % COVID-19 impact analysis, by application, and regional forecast, 2020-2027, accessed on April 2024, https://www.fortunebusinessinsights.com/ethyl-acetate-market-104056.
- R. D. Di Lorenzo, I. Serra, D. Porro and P. Branduardi, Catalysts, 2022, 12, 234 CrossRef CAS.
- M. L. Lopes, S. C. Paulillo de L, A. Godoy, R. A. Cherubin, M. S. Lorenzi and F. H. C. Giometti, Braz. J. Microbiol., 2016, 47, 64 CrossRef CAS PubMed.
- C. E. de Farias Silva, E. Barbera and A. Bertucco, Bioethanol Production from Food Crops, Elsevier, 2019 Search PubMed.
- S. Sherkhanov, T. P. Korman, S. G. Clarke and J. U. Bowie, Sci. Rep., 2016, 6, 24239 CrossRef CAS PubMed.
- J. Li, C. Sun, W. Cai, J. Li, B. P. Rosen and J. Chen, Mutat. Res., Rev. Mutat. Res., 2021, 788, 108396 CrossRef CAS PubMed.
- C. Zhang, S. A. Sultan, T. R and X. Chen, Bioresources and Bioprocessing, 2021, 8, 72 CrossRef PubMed.
- M. Kusano, Y. Sakai, N. Kato, H. Yoshimoto, H. Sone and Y. Tamai, Biosci., Biotechnol., Biochem., 1998, 62, 1956 CrossRef CAS PubMed.
- M. Kusano, Y. Sakai, N. Kato, H. Yoshimoto and Y. Tamai, J. Biosci. Bioeng., 1999, 87, 690 CrossRef CAS PubMed.
- A. Miyanaga, F. Kudo and T. Eguchi, Nat. Prod. Rep., 2018, 35, 1185 RSC.
- A. M. Soohoo, D. P. Cogan, K. L. Brodsky and C. Khosla, Annu. Rev. Biochem., 2024, 93, 471 CrossRef PubMed.
- A. J. Kruis, A. E. Mars, S. W. M. Kengen, J. W. Borst, J. van der Oost and R. A. Weusthuis, Appl. Environ. Microbiol., 2018, 84, 1 CrossRef PubMed.
- Y. C. Park, C. E. H. Shaffer and G. N. Bennett, Appl. Microbiol. Biotechnol., 2009, 85, 13 CrossRef CAS PubMed.
- W. D. Armstrong, M. M. Stanley and Y. Hiroshi, US Pat., 4720457A, USPTO, 1986 Search PubMed.
- B. St-Pierre, P. Laflamme, A. Alarco, V. D and E. Luca, Plant J., 1998, 14, 703 CrossRef CAS PubMed.
- Q. Yang, K. Reinhard, E. Schiltz and U. Matern, Plant Mol. Biol., 1997, 35, 777 CrossRef CAS PubMed.
- H. Fujiwara, Y. Tanaka, Y. Fukui, M. Nakao, T. Ashikari and T. Kusumi, Eur. J. Biochem., 1997, 249, 45 CrossRef CAS PubMed.
- H. Fujiwara, Y. Tanaka, Y. Fukui, T. Ashikari, M. Yamaguchi and T. Kusumi, Plant Sci., 1998, 137, 87 CrossRef CAS.
- N. Dudareva, J. C. D'Auria, K. H. Nam, R. A. Raguso and E. Pichersky, Plant J., 1998, 14, 297 CrossRef CAS PubMed.
- G. Liu, L. Huang and J. Lia, Biotechnol. Biofuels Bioprod., 2023, 16, 93 CrossRef CAS PubMed.
- M. J. Knight, I. D. Bull and P. Curnow, Yeast, 2014, 31, 463 CrossRef CAS PubMed.
- S. M. G. Saerens, K. J. Verstrepen, S. D. M. Van Laere, A. R. D. Voet, P. Van Dijck and F. R. Delvaux, et al., J. Biol. Chem., 2006, 281, 4446 CrossRef CAS PubMed.
- I. Molina and D. Kosma, Plant Cell Rep., 2015, 34, 587 CrossRef CAS PubMed.
- A. Bayer, X. Ma and J. Stöckigt, Bioorg. Med. Chem., 2004, 12, 2787 CrossRef CAS PubMed.
- S. Ka-Yiu, A. Sanchez, N. G. Bennet and C. R. Dittrich, US Pat., 7569380B2, USPTO, 2005 Search PubMed.
- S. Atsumi, G. Rodriguez and Y. Tashiro, WO Pat., 2015031859A1, WIPO, 2014 Search PubMed.
- S. Atsumi, T. Y. Wu, E. M. Eckl, S. D. Hawkins, T. Buelter and J. C. Liao, Appl. Microbiol. Biotechnol., 2010, 85, 651 CrossRef CAS PubMed.
- R. G. Eastham, G. Stephens and A. Yiacoumetti, WO Pat., 2016185211A1, WIPO, 2016 Search PubMed.
- Y. Asano, E. Sato, F. Yu and W. Mizunashi, EU Pat., 3115460A4, EPO, 2015 Search PubMed.
- S. Saum, K. Woncheol, O. Zelder, J. Jaitzig and Z. Guo, WO Pat., 2017167623A1, WIPO, 2017 Search PubMed.
- V. Schadeweg and E. Boles, Biotechnol. Biofuels, 2016, 9, 257 CrossRef PubMed.
- K. Zhang, X. Mingyong and T. Yi-shu, US Pat., 10006064B2, USPTO, 2013 Search PubMed.
- J. A. Kruis, M. Levisson, E. A. Mars, S. W. M. Kengen, J. Van Der Oost and J. P. M. Sanders, WO Pat., 2018100097A1, WIPO, 2017 Search PubMed.
- A. J. Kruis, M. Levisson, A. E. Mars, M. van der Ploeg, F. Garcés Daza and V. Ellena, Metab. Eng., 2017, 41, 92 CrossRef CAS PubMed.
- Y. S. Tai, M. Xiong and K. Zhang, Metab. Eng., 2015, 27, 20 CrossRef CAS PubMed.
- E. J. F. Souleyre, D. R. Greenwood, E. N. Friel, S. Karunairetnam and R. D. Newcomb, FEBS J., 2005, 272, 3132 CrossRef CAS PubMed.
- D. Li, Y. Xu, G. Xu, L. Gu, D. Li and H. Shu, Phytochem., 2006, 67, 658 CrossRef CAS PubMed.
- F. Yu and W. Mizunashi, EU Pat., 3508585B1, EPO, 2017 Search PubMed.
- C. Löser, T. Urit, A. Stukert and T. Bley T., J. Biotechnol., 2013, 163, 17 CrossRef PubMed.
- P. Y. Stergiou, A. Foukis, M. Filippou, M. Koukouritaki, M. Parapouli and L. G. Theodorou, Biotechnol. Adv., 2013, 31, 1846 CrossRef CAS.
- A. A. Elgharbawy, F. A. Riyadi, M. D. Z. Alam and M. Moniruzzaman, J. Mol. Liq., 2018, 251, 150 CrossRef CAS.
- O. Thum, L. Hilterhaus and A. Liese, EU Pat., 2080807B1, EPO, 2008 Search PubMed.
- A. C. L. d. M. Carvalho, T. De Sousa Fonseca, M. C. de Mattos, M. D. C. Ferreira de Oliveira, T. L. Gomes de Lemos, F. Molinari, D. Romano and I. Serra, Int. J. Mol. Sci., 2015, 16, 29682 CrossRef PubMed.
- P. Y. Stergiou, A. Foukis, M. Filippou, M. Koukouritaki, M. Parapouli, L. G. Theodorou, E. Hatziloukas, A. Afendra, A. Pandey and E. M. Papamichael, Biotechnol. Adv., 2013, 31, 1846 CrossRef CAS PubMed.
- D. Romano, F. Bonomi, M. C. de Mattos, T. de Sousa Fonseca, M. D. C. Ferreira de Oliveira and F. Molinari, Biotechnol. Adv., 2015, 33, 547 CrossRef CAS PubMed.
- S. Naik, A. Basu, R. Saikia, B. Madan, P. Paul and R. Chaterjee, J. Mol. Catal. B: Enzym., 2010, 65, 18 CrossRef CAS.
- K. E. Jaeger, S. Ransac, B. W. Dijkstra, C. Colson, M. Heuvel and O. Misset, FEMS Microbiol. Rev., 1994, 15, 29 CrossRef CAS PubMed.
- T. Sakoleva, H. P. Austin, C. Tzima, M. Dörr and U. T. Bornscheuer, ChemBioChem, 2023, 24, 10 CrossRef PubMed.
- M. J. L. J. Fürst, A. Gran-Scheuch, F. S. Aalbers and M. W. Fraaije, ACS Catal., 2019, 9, 11207 CrossRef.
- M. L. Mascotti, W. J. Lapadula and M. Juri, PLoS One, 2015, 10, e0132689 CrossRef PubMed.
- J. Rebehmed, V. Alphand, V. de Berardinis and A. G. de Brevern, Biochimie, 2013, 95, 1394 CrossRef CAS PubMed.
- S. Schmidt S and U. T. Bornscheuer, Enzymes, 2020, 47, 231 Search PubMed.
- T. Sakoleva, H. P. Austin, C. Tzima, M. Dörr and U. T. Bornscheuer, ChemBioChem, 2023, 24, 10 CrossRef PubMed.
- F. Fiorentini, C. R. Nicoll and A. Mattevi, Biochemistry, 2021, 60, 3419 CrossRef CAS PubMed.
- S. Wang, M. M. Kayser, H. Iwaki and P. C. K. Lau, J. Mol. Catal. B: Enzym., 2003, 22, 211 CrossRef CAS.
- TEGOSOFT® OER, accessed on April 2024, https://corporate.evonik.com/en/media/press-releases/corporate/tegosoft-oer-living-up-to-our-responsibility-103212.html.
- Evonik expands production capacity of TEGOSOFT® MM MB to Shanghai, accessed on April 2024, https://personal-care.evonik.com/en/evonik-expands-production-capacity-of-tegosoft-mm-mb-to-shanghai-174450.html.
- D. Wang and Y. Xu, US Pat., 11214778B2, USPTO, 2017 Search PubMed.
- M. S. Nemser, S. Majumdar and K. J. Pennisi, US Pat., 8506815B2, USPTO, 2007 Search PubMed.
- S. T. Yang, US Pat., 8357519B2, USPTO, 2009 Search PubMed.
- M. Bučko, P. Gemeiner, A. Schenkmayerová, T. Krajčovič, F. Rudroff and M. D. Mihovilovič, Appl. Microbiol. Biotechnol., 2016, 100, 6585 CrossRef PubMed.
- E. Eichhorn and A. Goeke, WO Pat., 2022003017A1, WIPO, 2020 Search PubMed.
- F. Chen, K. Zhu, Z. Huang, D. Cheng, J. Wang and T. Yuan, US Pat., 20210355516A1, USPTO, 2020 Search PubMed.
- G. R. Eastham, D. W. Johnson, A. J. J. Straathof, M. W. Fraaije and R. T. Winter, US Pat., 9816115B2, USPTO, 2012 Search PubMed.
- A. C. Bohnenkamp, R. H. Wijffels, S. W. M. Kengen and R. A. Weusthuis, Biotechnol. Biofuels, 2021, 14, 192 CrossRef CAS PubMed.
- A. C. Bohnenkamp, A. J. Kruis, A. E. Mars, R. H. Wijffels, J. van der Oost and S. W. M. Kengen, et al., Biotechnol. Biofuels, 2020, 13, 65 CrossRef CAS PubMed.
- J. L. Peel, Biochem. J., 1951, 49, 62 CrossRef CAS PubMed.
- C. Löser, T. Urit, E. Gruner and T. Bley, Energy Sustain. Soc., 2015, 5, 2 CrossRef.
- R. Davies, E. A. Falkiner, J. F. Wilkinson and J. L. Peel, Biochem. J., 1951, 49, 58 CrossRef CAS PubMed.
- R. A. Weusthuis, I. Lamot I, J. van der Oost and J. P. M. Sanders, Trends Biotechnol., 2011, 29, 153 CrossRef CAS PubMed.
- F. Garcia-Ochoa and E. Gomez, Biotechnol. Adv., 2009, 27, 153 CrossRef CAS PubMed.
- H. Kallel-Mhiri, J. M. Engasser and A. Miclo, Appl. Microbiol. Biotechnol., 1993, 40, 2 CrossRef.
- A. Willetts, Antonie Van Leeuwenhoek, 1989, 56, 175 CrossRef CAS.
- T. Urit, C. Löser, M. Wunderlich and T. Bley, Bioprocess Biosyst. Eng., 2011, 34, 547 CrossRef CAS.
- C. Löser C, T. Urit, S. Förster, A. Stukert, T. Bley and T. Appl, Microbiol. Biotechnol., 2012, 96, 685 CrossRef.
- P. Keng, M. Basri, A. Ariff, M. Abdulrahman, R. Abdulrahman and A. Salleh, Bioresour. Technol., 2008, 99, 6097 CrossRef CAS.
- J. Price, M. Nordblad, H. H. Martel, B. Chrabas, H. Wang and P. M. Nielsen, et al., Biotechnol. Bioeng., 2016, 113, 1719 CrossRef CAS PubMed.
- E. de Sousa Cordeiro, R. O. Henriques, E. M. Deucher, D. Oliveira, L. A. Lerin and A. Furigo, Biotechnol. Appl. Biochem., 2021, 68, 1469 Search PubMed.
- Y. He, K. Li, J. Wang, L. Xu, J. Yan and M. Yang, et al., J. Cleaner Prod., 2022, 372, 133740 CrossRef CAS.
- J. López-Fernández, M. Dolors Benaiges and F. Valero, Bioresour. Technol., 2021, 334, 125233 CrossRef PubMed.
- P. Srimhan and T. Hongpattarakere, Catalysts, 2023, 13, 617 CrossRef CAS.
- M. Y. Liow, W. Gourich, M. Y. Chang, J. M. Loh, E. S. Chan and C. P. Song, J. Ind. Eng. Chem., 2022, 114, 1 CrossRef CAS.
- G. L. Maddikeri, A. B. Pandit and P. R. Gogate, Ind. Eng. Chem. Res., 2012, 51, 14610 CrossRef CAS.
- L. F. Chuah, J. J. Klemeš, S. Yusup, A. Bokhari and M. M. Akbar, J. Cleaner Prod., 2017, 146, 181 CrossRef CAS.
- V. G. Gude and E. Martinez-Guerra, Environ. Chem. Lett., 2018, 16, 327 CrossRef CAS.
- M. Gojun, M. Bačić, A. Ljubić, A. Šalić and B. Zelić, Micromachines, 2020, 11, 457 CrossRef PubMed.
- A. Hommes, T. de Wit, G. J. W. Euverink and J. Yue, Ind. Eng. Chem. Res., 2019, 58, 15432 CrossRef CAS.
- L. Quintana-Gómez, M. Ladero and L. Calvo, J. Supercrit. Fluids, 2021, 171, 105184 CrossRef.
- M. N. Varma, P. A. Deshpande and G. Madras, Fuel, 2010, 89, 1641 CrossRef CAS.
- A. L Paiva, D. van Rossum and F. X. Malcata, Biocatal. Biotransform., 2002, 20, 43 CrossRef.
- S. Hama, H. Noda and A. Kondo, Curr. Opin. Biotechnol., 2018, 50, 57 CrossRef CAS PubMed.
- Y. S. Tai, M. Xiong and K. Zhang, Metab. Eng., 2015, 27, 20 CrossRef CAS PubMed.
- J. Stribny, A. Querol and R. Pérez-Torrado, Front. Microbiol., 2016, 7, 7 Search PubMed.
- B. Nancolas, I. D. Bull, R. Stenner, V. Dufour and P. Curnow, Yeast, 2017, 34, 239 CrossRef CAS PubMed.
- E. J. F. Souleyre, D. R. Greenwood, E. N. Friel, S. Karunairetnam and R. D. Newcomb, FEBS J., 2005, 272, 3132 CrossRef CAS PubMed.
- W. Aschariyaphotha, S. Noichinda, K. Bodhipadma and C. Wongs-Aree, Plant Physiol. Biochem., 2024, 206, 108241 CrossRef CAS PubMed.
- J. C. D'Auria, F. Chen and E. Pichersky, Plant Physiol., 2002, 130, 466 CrossRef PubMed.
- T. Garcia, A. Coteron, M. Martinez and J. Aracil, Chem. Eng. Sci., 1996, 51, 2841 CrossRef CAS.
- A. Foukis, O. A. Gkini, P. Y. Stergiou and E. M. Papamichae, Mol. Catal., 2018, 455, 159 CrossRef CAS.
- E. Romero, J. R. G. Castellanos, A. Mattevi and M. W. Fraaije, Angew. Chem., Int. Ed., 2016, 55, 15852 CrossRef CAS PubMed.
- H. L. van Beek, E. Romero and M. W. Fraaije, ACS Chem. Biol., 2017, 12, 291 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |