
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational screening of transition metal atom doped ZnS and ZnSe nanostructures as promising bifunctional oxygen electrocatalysts†
Feifei Xia *,
Li Shu,
Fengli Yang,
Yingpin Wen and
Chunzhi Zheng
*,
Li Shu,
Fengli Yang,
Yingpin Wen and
Chunzhi Zheng
School of Chemistry and Chemical Engineering, Jiangsu University of Technology, Changzhou 213001, Jiangsu, P. R. China. E-mail: ffxia@jsut.edu.cn
First published on 12th September 2024
Abstract
The design of bifunctional oxygen electrocatalysts showing high catalytic performance for the oxygen evolution reaction (OER) and oxygen reduction reaction (ORR) is of great significance for developing new renewable energy storage and conversion technologies. Herein, based on the first principles calculations, we systematically explored the electrocatalytic activity of a series of transition metal atom (Fe, Co, Ni, Cu, Pd and Pt)-doped ZnS and ZnSe nanostructures for OER and ORR. The calculated results revealed that Ni- and Pt-doped ZnS and ZnSe nanostructures exhibit promising electrocatalytic performance for both OER and ORR in comparison to the pristine ZnS and ZnSe nanostructures. Especially, the OER/ORR overpotentials of Ni-doped ZnS and ZnSe nanostructures are estimated to be 0.28/0.30 and 0.31/0.31 V, respectively, disclosing their great potential as bifunctional oxygen electrocatalysts. Moreover, it is found that Ni-doped ZnS and ZnSe nanostructures for OER and ORR are on the top of the volcano plots, evincing promising catalytic performance. Our results provide theoretical insights into a feasible strategy to synthesize highly efficient ZnS- and ZnSe-based bifunctional oxygen electrocatalysts in the future.
1. Introduction
Developing sustainable and green energy technologies is extremely urgent to solve the growing energy crisis and environmental pollution, which can ensure a safe and sustainable future.1–5 As two critical processes in the electrochemical energy conversion, oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) involve a four-electron process with complicated mechanisms and exhibit sluggish kinetics and high overpotentials,6–8 which requires highly active catalysts for the practical applications. Currently, RuO2 and IrO2 are popular as outstanding OER electrocatalysts,9–11 and noble metals such as Pt-based materials are the best ORR electrocatalysts.12,13 However, the high cost and low selectivity of these electrocatalysts considerably impede their large-scale commercial applications. Therefore, it is urgent to develop low-cost and high-selectivity OER/ORR electrocatalysts with promising catalytic performance.It is reported that transition metal chalcogenides (TMCs) containing sulfide and selenide have considerable electrical conductivity and tunable electronic configurations, which is beneficial to their wide application in electrocatalysis.14–22 Moreover, these TMCs can draw considerable attention from researchers due to their natural abundance and intrinsic activity.17 For instance, Garaj et al.18 demonstrated that the Gibbs free energy of hydrogen adsorption on the edges of MoS2 is close to zero, leading to the high catalytic activity for hydrogen evolution reaction (HER). Zheng et al.19 explored that nickel selenide with different stoichiometric ratios (NiSe, Ni0.5Se, Ni0.75Se and Ni0.85Se) exhibit HER and OER catalytic activity in an alkaline medium. Moreover, the synthesized NiS and FeSe materials20 exhibited excellent catalytic activity with overpotentials of 83 and 171 mV for HER and OER, respectively. All these reported results show that TMCs exhibit the potential catalytic performance and can serve as electrocatalysts for HER and OER.
TMCs, ZnS and ZnSe have become the electrode materials for HER, OER, and ORR because of their natural abundance, higher electrical conductivity and tunable electronic configurations.23–28 For example, Chen et al.23 presented that Ni and Sn anchored on the surface of ZnS materials can obviously improve their HER catalytic activity, while Sn and Pt supported on ZnS surface display higher OER catalytic activity. Yang et al.25 demonstrated that the ORR catalytic performance of ZnSe was ameliorated by the introduction of C and N atoms, which tunes the electronic properties of ZnSe and leads to the lower reaction energy barrier for the *O to *OOH conversion. These works suggest that metal or non-metal atoms can modulate the electronic properties of ZnS and ZnSe, resulting in the improvement of catalytic performance for HER, OER and ORR. Moreover, elemental doping is a facile and effective method to improve the intrinsic activity of catalysts during the reaction process.29 The introduction of metal or non-metal atoms can not only agitate the crystal lattice to expose more catalytic centers but also regulate their intrinsic electronic structure, which can activate the inherent catalytic activity.29,30 Motivated by these advantages, it is anticipated that transition metal (TM) single atoms (Fe, Co, Ni, Cu, Pd and Pt) doping can tune the electronic properties of ZnS and ZnSe nanostructures, which is beneficial to their catalytic activity.
Herein, we systemically explored the OER and ORR catalytic activity of pristine and TM (Fe, Co, Ni, Cu, Pd and Pt) doped ZnS and ZnSe nanostructures using the first principles calculations based on density functional theory (DFT).31,32 Firstly, the stabilities and catalytic active sites of TM (Fe, Co, Ni, Cu, Pd and Pt)-doped ZnS and ZnSe nanostructures were investigated. Subsequently, the OER/ORR catalytic activity of TM (Fe, Co, Ni, Cu, Pd and Pt)-doped ZnS and ZnSe nanostructures were examined. Then, the origin of OER/ORR catalytic activity was analyzed. Among all the Fe, Co, Ni, Cu, Pd and Pt-doped ZnS and ZnSe nanostructures, Ni and Pt doping obviously improves the OER/ORR catalytic activity of ZnS and ZnSe nanostructures. Interestingly, Ni-doped ZnS and ZnSe nanostructures possess the lowest OER/ORR overpotentials (0.28/0.30 and 0.31/0.31 V), suggesting their promising bifunctional catalytic performance. This work not only provides helpful theoretical guidance for developing inexpensive and effective OER/ORR electrocatalysts but also extends the possible application of ZnS- and ZnSe-based materials in water electrocatalysis fields.
2. Computational methods
All the geometry optimization and energy calculations were undertaken using the Vienna ab initio simulation package (VASP)33,34 based on the DFT. The electron exchange and correlation effects were handled with the Perdew–Burke–Ernzerhof (PBE) function of the generalized gradient approximation (GGA).35–37 Generally, the DFT + U method is more accurate than GGA density functionals to describe the partially filled d and f electronic states of transition metal elements.38 It has been reported that the effects of spin polarization on the free energies of each elementary step for OER/ORR can be ignored.39,40 However, our aim in this work is to screen the ideal OER/ORR catalysts from various ZnS and ZnSe candidates, and we are particularly interested in the catalytic tendency of these candidates for OER/ORR. Therefore, the spin polarization method was not adopted in this study. The van der Waals interaction was considered using Grimme's scheme (DFT-D3).41 The cutoff energy for the plane-wave basis was set as 500 eV. The energy and force convergence criteria were set to 10−6 eV and 0.01 eV Å−1, respectively. The vacuum space of 18 Å was selected to avoid the interaction between the periodical images. We performed our calculations using a 3 × 3 × 1 supercell of TM (Fe, Co, Ni, Cu, Pd and Pt)-doped ZnS and ZnSe nanostructures. Brillouin zone sampling is employed with Monkhorst–Pack special k-point meshes,42 and the 6 × 6 × 1 k-grid was chosen for the structural relaxation and frequency calculation.To evaluate the stability of the TM (Fe, Co, Ni, Cu, Pd and Pt)-doped ZnS and ZnSe nanostructures, the binding energy (Eb) was calculated using the following equation:
| Eb = ETM–ZnX − ETM − Evacancy-ZnX | (1) |
Free energies of OER/ORR intermediates in electrochemical reaction pathways were evaluated according to the computational hydrogen electrode (CHE) model developed by Nørskov et al.43,44 using the reversible hydrogen electrode (RHE) as a reference whose chemical potential (H+ + e−) is equivalent to that of 1/2H2 at 0 V and at all pH values. The Gibbs free energy change (ΔG) of each elementary step during the OER/ORR is defined as follows:
| ΔG = ΔE + ΔEZPE − TΔS + ΔGU + ΔGpH | (2) |
3. Results and discussion
3.1 Stabilities and catalytic active sites
The stability of ZnS and ZnSe surface structures was considered by the phonon spectra before their calculation of the electrocatalytic activity. To verify the dynamical stability of the ZnS and ZnSe surface structures as well as whether their structures are optimized, the phonon spectra were calculated based on the same DFT level and are shown in Fig. S1.† Note that there is no imaginary frequency in both the ZnS and ZnSe systems, which confirms their kinetic stability and well-optimized structures. To tune the catalytic performance of ZnS and ZnSe nanostructures, the substitutional doping method was employed. As shown in Fig. 1a, TM (TM = Fe, Co, Ni, Cu, Pd and Pt) atom substitutes the Zn atom in the first surface of the ZnS nanostructure and the similar substitution for ZnSe nanostructures (Fig. S2a†). The stable combination of TM atom as the active site on substrate materials is the premise for catalysts to maintain high catalytic activity for a long time.46 However, owing to the accumulation of a large number of positive charges, the TM atom is usually considered as the active site on substrate materials for OER/ORR,47 which is beneficial to form *OH, *O and *OOH intermediates. Therefore, Fe, Co, Ni, Cu, Pd and Pt sites were considered as the catalytic active site on the surface of the doped ZnS and ZnSe nanostructures during the OER/ORR process. Moreover, the binding energy (Eb) of the TM-doped ZnS and ZnSe nanostructures were calculated to investigate the stabilities of these materials (shown in Fig. 1b and S2b†). Results show that the Eb of all the TM atoms to the ZnS and ZnSe nanostructures is negative, thus demonstrating the better stability of these nanostructures, which can be considered as the substrate materials for catalysts. | ||
| Fig. 1 (a) Top and side views of the TM (TM = Fe, Co, Ni, Cu, Pd and Pt)-doped ZnS nanostructures, and the pink, gray and yellow atoms represent TM, Zn and S. (b) The binding energy of Fe, Co, Ni, Cu, Pd and Pt-doped ZnS nanostructures. | ||
3.2 OER/ORR catalytic activity
After confirming the stabilities and catalytic active sites of TM-doped ZnS and ZnSe nanostructures, the electrocatalytic activity of these materials for OER/ORR was systematically investigated. It is reported that the OER Gibbs free energy changes along the four-electron path for the previously screened systems.48,49 The computational results23–25,50 suggested that the four-electron step occurs on the surface of ZnS, ZnSe and CdS materials during the OER process, and TM atoms can improve their catalytic performance. Thus, the four elementary electrons for OER/ORR were adopted on the surface ZnS and ZnSe nanostructures in this work, as shown in Fig. 2, and the important *OH, *O, and *OOH intermediates are involved. Accordingly, the detailed reaction pathways, free energy equations and overpotentials for OER and ORR under an acidic condition are presented in Table 1. Moreover, free energy changes of each elementary step and overpotentials of ZnX (X = S and Se) nanostructures for OER and ORR at U = 0 V and pH = 0 are shown in Tables S1 and S2.† | ||
| Fig. 2 Schematic of the mechanism of OER/ORR on the surface of TM-doped ZnS and ZnSe nanostructures. The brown, gray, yellow, red and white atoms are TM, Zn, S, O and H. | ||
| Reaction | Reaction pathway | ΔG/eV | η/V |
|---|---|---|---|
| a Where ΔG*OH, ΔG*O, and ΔG*OOH are the adsorption energies of *OH, *O, and *OOH intermediates. These adsorption energies are defined as follows: ΔG*OH = G*OH + 0.5GH2 − G* − GH2O, ΔG*O = G*O + GH2 − G* − GH2O, ΔG*OOH = G*OOH + 1.5GH2 − G* − 2GH2O, where GH2O and GH2 are the free energies of H2O and H2 molecules, respectively. | |||
| OER | H2O(1) + * → *OH + H+ + e− | ΔG1 = ΔG*OH | 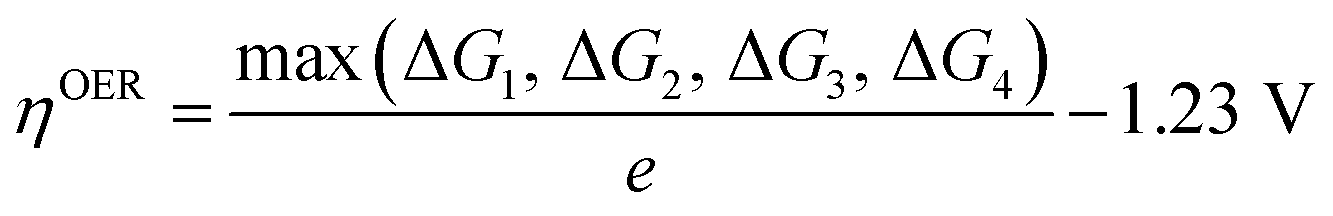 |
| *OH → *O + H+ + e− | ΔG2 = ΔG*O − ΔG*OH | ||
| H2O(1) + *O → *OOH + H+ + e− | ΔG3 = ΔG*OOH − ΔG*O | ||
| *OOH → * + O2 + H+ + e− | ΔG4 = 4.92 − ΔG*OOH | ||
| ORR | * + O2 + H+ + e− → *OOH | ΔG1 = ΔG*OOH − 4.92 |  |
| *OOH + H+ + e− → H2O(1) + *O | ΔG2 = ΔG*O − ΔG*OOH | ||
| *O + H+ + e− → *OH | ΔG3 = ΔG*OH − ΔG*O | ||
| *OH + H+ + e− → H2O(1) + * | ΔG4 = –ΔG*OH | ||
Furthermore, the OER/ORR catalytic activity can be directly determined by the adsorption Gibbs free energies of *OH, *O and *OOH intermediates (ΔG*OH, ΔG*O and ΔG*OOH), and too-strong or too-weak bonding between intermediates and reaction sites negatively impact the proceeding of reactions. Accordingly, ΔG*OH, ΔG*O and ΔG*OOH of pristine and TM-doped ZnS and ZnSe nanostructures for OER/ORR were calculated using the computational hydrogen electrode model proposed by Nørskov43 and presented in Table 2, which are indispensable for evaluating the ORR/OER activity. Moreover, the zero point energy (ZPE) corrections and entropy contributions (TS) of *OH, *O, and *OOH on the surface of ZnX (X = S and Se) nanostructures at 298.15 K by DFT calculations are presented in Tables S3 and S4.† As expected, *OH, *O, and *OOH intermediates are preferably adsorbed on the TM site of ZnS and ZnSe nanostructures owing to the moderate adsorption of Gibbs free energy (shown in Table 2), and the corresponding configurations are displayed in Fig. S3 and S4.† Moreover, the electrochemical interface between ZnX (X = S and Se) and water was investigated to determine how the inclusion of an explicit solvent (undissociated water molecule) influences the stability of adsorbed intermediates (*OH, *O and *OOH) in the OER and ORR. The results indicated that the inclusion of an explicit solvent can considerably stabilize the structure of adsorbed intermediates (*OH, *O and *OOH), which are capable of donating hydrogen bonds (as shown in Fig. S5†).
| Systems | ZnS | ZnSe | ||||
|---|---|---|---|---|---|---|
| ΔG*OH/eV | ΔG*O/eV | ΔG*OOH/eV | ΔG*OH/eV | ΔG*O/eV | ΔG*OOH/eV | |
| Pristine | 1.89 | 4.18 | 4.73 | 1.71 | 2.27 | 4.67 |
| Fe-doped | 0.01 | 1.43 | 3.29 | 0.02 | 1.43 | 3.36 |
| Co-doped | 0.50 | 1.76 | 3.69 | 0.46 | 1.61 | 3.67 |
| Ni-doped | 1.02 | 2.49 | 3.99 | 0.92 | 2.43 | 3.98 |
| Cu-doped | 1.72 | 3.74 | 4.73 | 1.82 | 2.47 | 4.75 |
| Pd-doped | 1.80 | 2.94 | 4.16 | 1.14 | 2.91 | 4.25 |
| Pt-doped | 1.24 | 2.56 | 4.06 | 0.85 | 2.39 | 4.12 |
To clarify OER/ORR catalytic activity and pathways, free-energy diagrams of pristine and TM-doped ZnS and ZnSe nanostructures for OER/ORR at U = 0 and 1.23 V are plotted and listed in Fig. 3–5. As mentioned above, the OER process involves four steps, including the formation of H2O → *OH → *O → *OOH → O2, while the ORR process is reversed. For pristine ZnS and ZnSe materials during the OER process, the potential-determining step (PDS) is the formation of *OH to *O and *O to *OOH, corresponding to the overpotential (ηOER) of 1.05 and 1.17 V, respectively. The steps of *OH to *O and *O to *OOH are uphill and endothermic at an equilibrium potential of 1.23 V, suggesting that additional applied electrode potential is required to ensure all the elementary steps are spontaneous. However, the ORR PDS is O2 → *OOH both for pristine ZnS and ZnSe nanostructures, and this step is downhill and endothermic, corresponding to the overpotentials (ηORR) of 1.04 and 0.98 V, respectively. The high ηOER (1.05 and 1.17 V) and ηORR (1.04 and 0.98 V) values suggest that the pristine ZnS and ZnSe nanostructures are not suitable for efficient oxygen electrocatalysts. Moreover, the associative and dissociative mechanism is involved in the ORR and thus the interaction between O2 molecule and ZnX (X = S and Se) nanostructures was considered. Our calculation suggests that, for both ZnS and ZnSe nanostructures, the *O2 dissociative pathway (*O2 → *O–*O) is endothermic, while the associative pathway (*O2 → *OOH) is exothermic, as shown in Fig. S6 in the ESI.† Therefore, the associative mechanism will be concerned with Fe, Co, Ni, Cu, Pd and Pt-doped ZnX (X = S and Se) nanostructures during the ORR process.
 | ||
| Fig. 3 Free energy diagram of (a) ZnS and (b) ZnSe nanostructures for OER and ORR at U = 0 and 1.23 V. The red and blue lines are the potential-determining step (PDS) for OER and ORR, respectively. | ||
 | ||
| Fig. 4 Free energy diagram of (a) Fe, (b) Co, (c) Ni, (d) Cu, (e) Pd and (f) Pt-doped ZnS nanostructures for OER and ORR at U = 0 and 1.23 V, respectively. The red and blue lines are the potential-determining steps (PDS) for OER and ORR. | ||
 | ||
| Fig. 5 Free energy diagram of (a) Fe, (b) Co, (c) Ni, (d) Cu, (e) Pd and (f) Pt-doped ZnSe nanostructures for OER and ORR at U = 0 and 1.23 V. The red and blue lines are the potential-determining steps (PDS) for OER and ORR, respectively. | ||
As shown in Fig. 4 and 5, the four steps of the OER on the surface of Fe, Co, Ni, Cu, Pd and Pt-doped ZnS and ZnSe nanostructures were uphill at U of 0 V, confirming that the whole process was endothermic. When the electrode potential is applied to the equilibrium potential of 1.23 V, there are energy barriers for OER (Fig. 4 and 5), disclosing that additional applied electrode potential is required to make all the elementary steps spontaneous. Interestingly, the formation of *O to *OOH is the PDS for all TM-doped ZnS and ZnSe nanostructures except for Cu/ZnS, Pd/ZnS and Pd/ZnSe during the OER process. For the ORR process, we found that the PDS of Fe and Co-doped ZnS occurred in the process of *OH → H2O, while that of Ni, Cu, Pd and Pt-doped ZnS is the formation of O2 to *OOH (Fig. 4). As can be seen in Fig. 5, during the process of ORR, the PDS of Fe-, Co- and Ni-doped ZnSe is the formation of *OH → H2O, while that of Cu- Pd- and Pt-doped ZnSe is the formation of O2 → *OOH. In general, the adsorption strength of the reaction intermediates plays a crucial role in the OER/ORR electrocatalytic activity. According to the adsorption Gibbs free energies of *OH, *O and *OOH calculated using the equations in Table 1, the computed ηOER and ηORR on Fe, Co, Ni, Cu, Pd and Pt-doped ZnS and ZnSe nanostructures are labeled in Fig. 4 and 5, respectively. Note that the OER/ORR overpotentials of Ni-doped ZnS and ZnSe are 0.30/0.28 and 0.31/0.31 V, respectively, which are lower than those of IrO2 (ηOER = 0.56 V) and Pt (ηORR = 0.45 V).51 This phenomenon shows that Ni/ZnS and Ni/ZnSe can be practically used as promising bifunctional oxygen electrocatalysts. Moreover, Pd and Pt substitutional doping obviously decreases the overpotential of ZnS and ZnSe nanostructures for OER and ORR, indicating their potential bifunctional catalytic performance.
3.3 Origin of OER/ORR catalytic activity
It has been well accepted that the scaling relationships among the adsorption-free energies of *OH, *O, and *OOH intermediates can provide insight into understanding the origin of catalytic activity.52 According to the above discussions, all the *OH, *O, and *OOH intermediates preferentially adsorb on the TM atoms site with TM–O binding (Fig. 1a and S1a†). Therefore, the scaling relationships of ΔG*OH with ΔG*O and ΔG*OOH were plotted and are displayed in Fig. 6 and 7. Remarkably, the significant correlation linear relationships among ΔG*OH, ΔG*O and ΔG*OOH can be obtained on ZnS nanostructures, namely, ΔG*O = 1.29ΔG*OH + 1.22 eV (R2 = 0.83) and ΔG*OOH = 0.68ΔG*OH + 3.30 eV (R2 = 0.83), as shown in Fig. 6a and b. However, the linear relationship between ΔG*OH and ΔG*O is poor with a weak determination coefficient (R2 = 0.39) for ZnSe nanostructures, while that between ΔG*OH and ΔG*OOH is good with a strong determination coefficient (R2 = 0.98). According to the universally recognized Sabatier principle, the adsorption strength of the *OH, *O, and *OOH intermediates plays a crucial role in the catalyst's OER/ORR electrochemical activity.53–56 Too-strong or too-weak adsorption of intermediates (*OH, *O and *OOH) on the surface of the catalyst will negatively impact the proceeding of OER and ORR. This indicates that an ideal catalyst should have a moderate ΔGi value to ensure suitable adsorption of intermediates during the reaction process. As shown in Table 2, the calculated adsorption Gibbs free energies of *OH, *O, and *OOH intermediates for Ni/ZnS (ΔG*OH = 1.02 eV, ΔG*O = 2.49 eV, ΔG*OOH = 3.99 eV) and Ni/ZnSe (ΔG*OH = 0.92 eV, ΔG*O = 2.43 eV, ΔG*OOH = 3.98 eV) are approximately equivalent to the optimal values for ideal OER/ORR catalysts. | ||
| Fig. 6 Scaling relationship between the adsorption-free energies of (a) ΔG*OH vs. ΔG*O and (b) ΔG*OH vs. ΔG*OOH on the surface of pristine and TM-doped ZnS nanostructures. The volcano plot of (c) ηOER vs. ΔG*O − ΔG*OH and (d) ηORR vs. ΔG*OH for OER and ORR on the surface of pristine and TM-doped ZnS nanostructures. | ||
 | ||
| Fig. 7 Scaling relationship between the adsorption-free energies of (a) ΔG*OH vs. ΔG*O and (b) ΔG*OH vs. ΔG*OOH on the surface of pristine and TM-doped ZnSe nanostructures. The volcano plot of (c) ηOER vs. ΔG*O − ΔG*OH and (d) ηORR vs. ΔG*OH for OER and ORR on the surface of pristine and TM-doped ZnSe nanostructures. | ||
Moreover, the volcano plots are considered a powerful guide to select and develop efficient electrocatalysts. Thus, the overpotentials volcano plots are displayed in Fig. 6c, d, 7c and d, which are established by ΔG*OH and ΔG*O − ΔG*OH as the descriptors of the OER/ORR catalytic activity of different ZnS and ZnSe nanostructures. Usually, the best-performance catalysts with the most moderate adsorption energy are located at the top of the volcano plot. The best OER catalyst has the ΔG*O − ΔG*OH value of ∼1.50 eV, and the ΔG*OH value of ∼1.00 eV is required for the best ORR catalyst. Obviously, the Ni-doped ZnS and ZnSe are at the top of the OER and ORR volcano plots, elucidating their superior catalytic performance for OER/ORR and implying the promising potential as bifunctional oxygen electrocatalysts. Moreover, Pt/ZnS (ηOER/ηORR = 0.27/0.37 V) and Pt/ZnSe (ηOER/ηORR = 0.43/0.50 V) are close to the peak of volcano plots with lower overpotentials, which is considerably better than other ZnS and ZnSe nanostructures for OER and ORR. As discussed above, the lower overpotential is favorable for OER and ORR, which can be used as an indicator of the catalytic performance of the electrocatalysts. Remarkably, Ni-doped ZnS and ZnSe nanostructures possess the lowest OER and ORR overpotential, suggesting that they are both selective for OER and ORR. Our results demonstrate that Ni/ZnS and Ni/ZnSe nanostructures can act as excellent bifunctional catalysts for OER and ORR, which can be beneficial for synthesizing efficient performance electrocatalysts in the future.
The insight into the origin of OER/ORR catalytic activity can provide a theoretical guide to improve the performance of catalysts. In general, the adsorption of *OH, *O, and *OOH intermediates on catalytic active sites is the key factor influencing the catalytic activity of OER/ORR. However, the d-band center of transition metal atoms (εd, the average position of the TM-d orbital) has been widely used to describe the bond strength between the adsorbed intermediates (*OH, *O and *OOH) and the surface of catalysts.57 Therefore, to understand the influence of different TM atoms as catalytic active sites on the catalyst performance, the εd of Fe, Co, Ni, Cu, Pd, and Pt in our studied systems were calculated and are shown in Fig. S7.† It can be found that different TM (Fe, Co, Ni, Cu, Pd and Pt)-doped ZnS and ZnSe can result in the different adsorption strengthen of intermediates. In particular, the Ni atom in the Ni/ZnS and Ni/ZnSe nanostructures with the moderate d-band center position displays appropriate adsorption strength, which leads to the lower overpotential (ηOER/ηORR = 0.28/0.30 V), suggesting the promising catalytic performance for OER/ORR.
4. Conclusions
In conclusion, the catalytic performance of TM (Fe, Co, Ni, Cu, Pd and Pt)-doped ZnS and ZnSe nanostructures for OER/ORR has been systematically evaluated by high-throughput theoretical screening. Our calculated results suggest that Fe, Co, Ni, Cu, Pd and Pt can be treated as the catalytic active sites on the surface of ZnS and ZnSe nanostructures during the OER/ORR process, which is available for the formation of *OH, *O and *OOH intermediates. Moreover, the free-energy diagrams show that the pristine ZnS and ZnSe nanostructures own high OER/ORR overpotentials, implying inefficient catalytic performance. In contrast, the OER/ORR overpotentials of Co, Ni, Pd and Pt-doped ZnS and ZnSe nanostructures obviously decrease. Particularly, the Ni/ZnS and Ni/ZnSe nanostructures own the lower OER/ORR overpotentials of 0.28/0.30 V and 0.31/0.31 V, respectively, demonstrating their promising bifunctional catalytic activity. Our findings not only provide a theoretical strategy to design the bifunctional oxygen electrocatalysts but also shed insight into the electrocatalytic application of ZnS and ZnSe-based materials in water electrolysis.Data availability
The authors confirm that the data supporting the findings of this study are available within its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21703087).Notes and references
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- N. L. Panwar, S. C. Kaushik and S. Kothari, Renewable Sustainable Energy Rev., 2011, 15, 1513–1524 CrossRef.
- G. S. Liu, S. J. You, Y. Tan and N. Q. Ren, Environ. Sci. Technol., 2017, 51, 2339–2346 CrossRef CAS PubMed.
- Z. Shu, H. F. Chen, X. Liu, H. X. Jia, H. J. Yan and Y. Q. Cai, Adv. Funct. Mater., 2023, 35, 2301493 CrossRef.
- Z. Shu, H. J. Yan, H. F. Chen and Y. Q. Cai, J. Mater. Chem. A, 2022, 10, 5470–5478 RSC.
- V. Rai, K. P. Lee, D. Safanama, S. Adams and D. J. Blackwood, ACS Appl. Energy Mater., 2020, 3, 9417–9427 CrossRef CAS.
- M. Tahir, L. Pan, F. Idrees, X. Zhang, L. Wang, J.-J. Zou and Z. L. Wang, Nano Energy, 2017, 37, 136–157 CrossRef CAS.
- J. Stacy, Y. N. Regmi, B. Leonard and M. Fan, Renewable Sustainable Energy Rev., 2017, 69, 401–414 CrossRef CAS.
- H. N. Sun and W. C. Jung, J. Mater. Chem. A, 2021, 9, 15506–15521 RSC.
- J. Ying, J. B. Chen, Y. X. Xiao, S. I. Cordoba de Torresi, K. I. Ozoemena and X. Y. Yang, J. Mater. Chem. A, 2023, 11, 1634–1650 RSC.
- Q. Dang, H. P. Lin, Z. L. Fan, L. Ma, Q. Shao, Y. J. Ji, F. F. Zheng, S. Z. Geng, S. Z. Yang, N. N. Kong, W. X. Zhu, Y. Y. Li, F. Liao, X. Q. Huang and M. W. Shao, Nat. Commun., 2021, 12, 6007 CrossRef CAS PubMed.
- C. L. Zhang, X. C. Shen, Y. B. Pan and Z. M. Peng, Front. Energy, 2017, 11, 268–285 CrossRef.
- J. W. Zhang, Y. L. Yuan, L. Gao, G. M. Zeng, M. F. Li and H. W. Huang, Adv. Mater., 2021, 33, 2006494 CrossRef CAS PubMed.
- K. C. Majhi and M. Yadav, ACS Eng. Au, 2023, 3, 278–284 CrossRef CAS.
- T. Ingsel and R. K. Gupta, Nanomaterials for Electrocatalysis (Micro and Nano Technologies). 2022, pp. 83–111 Search PubMed.
- Y. J. Liu, Y. J. Guo, Y. R. Liu, Z. L. Wei, K. Wang and Z. Q. Shi, Energy Fuels, 2023, 37, 2608–2630 CrossRef CAS.
- A. Azam, J. Yang, W. X. Li, J.-K. Huang and S. Li, Prog. Mater. Sci., 2022, 132, 101042 CrossRef.
- P. Zhang, H. Y. Xiang, L. Tao, H. J. Dong, Y. G. Zhou, T. S. Hu, X. L. Chen, S. Liu, S. Y. Wang and S. Garaj, Nano Energy, 2019, 57, 535–541 CrossRef CAS.
- X. Zheng, X. Han, H. Liu, J. Chen, D. Fu, J. Wang, C. Zhong, Y. Deng and W. Hu, ACS Appl. Mater. Interfaces, 2018, 10, 13675–13684 CrossRef CAS PubMed.
- S. Jo, K. B. Lee and J. I. Sohn, ACS Sustainable Chem. Eng., 2021, 9, 14911–14917 CrossRef CAS.
- Z. Y. Wang, S. L. Liu, W. Duan, Y. C. Xing, Y. L Hu and Y. J. Ma, Int. J. Hydrogen Energy, 2024, 60, 1414–1432 CrossRef CAS.
- X. Peng, Y. J. Yan, X. Jin, C. Huang, W. H. Jin, B. Gao and P. K. Chu, Nano Energy, 2020, 78, 105234 CrossRef CAS.
- M. Chen, Y. J. Yang, B. Xiong, Y. H. Huang, J. Liu and G. B. Wang, Int. J. Hydrogen Energy, 2024, 51B, 540–550 CrossRef.
- L. F. Cui, K. Xiang, X. M. Kang, K. K. Zhi, L. Wang, J. J. Zhang, X. Z. Fu and J. L. Luo, J. Colloid Interface Sci., 2022, 609, 868–877 CrossRef CAS PubMed.
- L. F. Yang, X. Y. Yao, C. L. Du, Z. L. Han, M. W. Jin, S. C. Peng, X. L. Ma, Y. Q. Zhu, M. S. Zou and C. B. Cao, Chem. Eng. J., 2024, 481, 148598 CrossRef CAS.
- F. F. Xia, L. Shu, Y. P. Wen, F. L. Yang and C. Z. Zheng, Mater. Adv., 2022, 3, 5772–5777 RSC.
- L. Hu, P. Zhong, X. Zhang, Y. Xiang, M.-S. Balogun, Y. X. Tong and H. Yang, Appl. Surf. Sci., 2023, 623, 157040 CrossRef CAS.
- S. Hussain, X. T. Yang, J. J. Yang and Q. Y. Li, Mater. Today Sustain., 2024, 25, 100686 Search PubMed.
- S. R. Ede and Z. P. Luo, J. Mater. Chem. A, 2021, 9, 20131–20163 RSC.
- X. Q. Du, H. Su and X. H. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 16917–16926 CrossRef CAS.
- P. Hohenberg and W. Kohn, Phys. Rev. B, 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed.
- B. Hammer, K. W. Jacobsen and J. K. Nørskov, Phys. Rev. Lett., 1993, 70, 3971–3974 CrossRef CAS PubMed.
- M. Cococcioni and S. de Gironcoli, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 035105 CrossRef.
- Z. Qin, Z. X. Wang and J. X. Zhao, Nanoscale, 2022, 14, 6902–6911 RSC.
- Y. W. Wang, W. Tian, J. Wan, G. Q. Xiong, H. J. Zhang and Y. Wang, Phys. Chem. Chem. Phys., 2022, 24, 10325–10333 RSC.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- V. Wang, N. Xu, J. C. Liu, G. Tang and W. T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- Y. Wang, J. Mao, X. G. Meng, L. Yu, D. Deng and X. H. Bao, Chem. Rev., 2019, 119, 1806–1854 CrossRef CAS PubMed.
- D. Y. Wang, K. X. Wang, Q. T. Liu, M. Y. Liu, G. Q. Zhang, K. Feng, K. Wang, X. W. Ding, H. M. Zhu, S. Yang, Y. H. Liu, T. H. Li, P. Gong, M. L. Wang, P. G. Wang, H. Z. Jin, W. Zhao and F. Yu, Adv. Sci., 2020, 7, 1901382 CrossRef PubMed.
- C. Feng, M. B. Faheem, J. Fu, Y. Q. Xiao, C. L. Li and Y. B. Li, ACS Catal., 2020, 10, 4019–4047 CrossRef CAS.
- C. Spöri, P. Briois, H. N. Nong, T. Reier, A. Billard, S. Kühl, D. Teschner and P. Strasser, ACS Catal., 2019, 9, 6653–6663 CrossRef.
- P. Garg, A. S. Nair, K. S. Rawat and B. Pathak, J. Phys. Chem. C, 2019, 123, 13419–13427 CrossRef CAS.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- Z. Qin and J. Zhao, J. Colloid Interface Sci., 2022, 605, 155–162 CrossRef CAS PubMed.
- J. Kari, J. P. Olsen, K. Jensen, S. F. Badino, K. B. R. M. Krogh, K. Borch and P. Westh, ACS Catal., 2018, 8, 11966–11972 CrossRef CAS.
- M. Bajdich, M. García-Mota, A. Vojvodic, J. K. Nørskov and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 13521–13530 CrossRef CAS PubMed.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- T. Zhang, B. Zhang, Q. Peng, J. Zhou and Z. Sun, J. Mater. Chem. A, 2021, 9, 433–441 RSC.
- H. Niu, X. H. Wan, X. T. Wang, C. Shao, J. Robertson, Z. F. Zhang and Y. Z. Guo, ACS Sustain. Chem. Eng., 2021, 9, 3590–3599 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04011b |
| This journal is © The Royal Society of Chemistry 2024 |