
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEndowing rubber with intrinsic self-healing properties using thiourea-based polymer†
Afreen Shagufta abcd,
Lei Wangabcd,
Senbiao Fangacd,
Qingshan Kong*acd and
Haibo Zhang*abcd
abcd,
Lei Wangabcd,
Senbiao Fangacd,
Qingshan Kong*acd and
Haibo Zhang*abcd
aQingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, China
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cQingdao New Energy Shandong Laboratory, Qingdao 266101, China
dShandong Energy Institute, Qingdao 266101, China
First published on 19th August 2024
Abstract
Self-healing polymers are extensively researched for the sustainability of materials. The introduction of dynamic networks instead of traditional cross-linkers for an autonomous healing mechanism in elastomers is a promising strategy for improving rubber properties. However, exchangeable covalent bonds in a dynamic network generally rely on external stimulants and fillers, which can compromise the material's performance. Herein, we introduce a mechanically strong yet resilient and independent self-healing polymeric network by dual cross-linking of bonds based on covalent and non-covalent dual interaction. The thiourea-based polymer polyether thiourea ethylene glycol (PTUEG) was blended with natural rubber (NR) and epoxidized natural rubber (ENR) to strengthen the mechanical characteristics of the material NR-ENR-PTUEG3. In the material, the thermoplastic polymer PTUEG3 applied the thiourea linkage as a hydrogen bonding and dynamic covalent motif together to enhance mechanical adaptability in a self-healing polymer network exhibiting stiffness, toughness, and resilience, thereby extending its longevity. The resulting mechanical characteristics of the NR-ENR-PTUEG3 with 25 phr PTUEG3 exhibited tensile stress 4.8 ± 0.3 MPa and high elongation at break 833 ± 0.1%, demonstrating far better performance than that of pristine NR, and 85% recovery of its original strength at ambient temperature. The healing behaviour is strongly influenced by thiourea-based polymer contents, enabling autonomous self-healing at ambient temperature, exhibiting in situ load-bearing efficiency in the repaired material, and maintaining their mechanical characteristics.
1. Introduction
Natural rubber (NR) stands out as a significant and extensively used bio-based elastomer due to its mechanical strength and resilience properties. In its uncured condition, it is a biodegradable resource, making it renewable.1–3 However, to achieve desired mechanical, thermal, and chemical resistance properties like other rubbers, it typically undergoes vulcanization, additive cross-linking, or chemical processing. This treatment makes it suitable for extensive industrial applications such as tires, tubing, sealing joints, shock-absorbing layers, and insulation.4–8 The traditional vulcanization process typically involves the use of fillers such as carbon black, silica, sulphur or peroxide, and similar materials as curing agents to establish a permanent, irreversible cross-linking network, resulting in a mechanically strong material.9 However, they pose challenges such as filler dispersion, energy consumption, and environmental pollution due to the formation of an irreversible network.10,11 Additionally, these products are susceptible to damage from various causes, including mechanical stress, chemical exposure, heat, light, radiation, or a combination of these factors.12,13 Such damage can lead to the formation of micro-cracks or deep fractures within the material, making detection and external intervention by conventional methods challenging or impossible. The internal damage not only reduces the material's performance but also serves as a catalyst for further catastrophes like macro-cracks, moisture swelling, and de-bonding.14 Traditional repair methods such as welding, gluing, or patching can extend the material's lifespan but often compromise its mechanical strength in the repaired region.15Addressing this need, Cordier has initiated the development of self-healing rubber using a dynamic supramolecular self-assembly approach involving multiple hydrogen bonds in the network.16 These relatively weak bonds collectively form a network at room temperature, allowing the material to autonomously repair damage without requiring external stimulation. This innovative approach opens up new possibilities for the development of widespread applications of self-healing materials. However, the healing process can only operate once the capsules are broken, and the mechanism is no longer present to participate in the same region. Subsequently, various methods have been developed to achieve self-healing properties in rubber for instance by introducing dynamic covalent bonds, such as TEMPO-oxidized cellulose nanocrystals and dicarboxylic acid, into the rubber network. Dynamic covalent cross-linking involves establishing exchangeable covalent bonds in a dynamic network that may break and reconstruct under specific circumstances. This enables rubber to repair damage under certain stimulations, resulting in excellent self-healing efficiency.17 These modifications allow rubber to efficiently repair damage under stimulation, thereby significantly enhancing its durability and demonstrating its potential for high-temperature environments. However, while these approaches demonstrated good efficacy with external triggers such as heat,17 ultraviolet radiation,18 light,19,20 current,21 and catalysts22 to initiate the self-healing process, these stimuli often compromised mechanical strength and restricted the mobility of molecular chains. This limitation hinders mechanical performance and limits potential applications.15,23 Thermoplastics materials are often weak in toughness and resilience, while having strong mechanical stiffness and strength. Although elastomers are very resilient, their stiffness and strength are constrained therefore utilizing the dynamic covalent bond with a non-covalent network in materials aids in addressing the challenge of growing demand for sustainable rubber-based materials that exhibit resilient like elastomers and mechanically strong like thermoplastics. It also eliminates the dependency on external stimulants and additives or fillers for a wide range of desirable rubber-based materials.
In a recent study, Yanagisawa reported a series of poly (ether thiourea)s incorporating ethylene glycol (TUEG) as a spacer.24 This novel material exhibits remarkable self-healing properties and robustness without relying on external stimulation. Notably, it possesses low glass transition temperatures (Tg) and sufficient chain segment mobility, enabling macroscopic flow that aids the self-healing process. Consequently, damaged regions rapidly reconnect through the reshuffling of chemical interactions, leading to partial or complete material recovery. Presently, TUEG has been found to improve the performance of numerous material properties, including ion-conducting, shape memory, antibacterial, Li batteries, and solar cells, endowing them with both mechanically robust and self-healing properties.25–32 However, polymeric binders lacking three-dimensional cross-linking have the potential to dissolve due to their low cross-linking density. Introducing PTUEG3 into the rubber network may form a dual network containing hydrogen bonds and dynamic covalent networks, and improve significant properties of the rubber. However, to our knowledge, there have been no reports of high-performance self-healing rubber materials obtained by blending PTUEG3 with rubber.
A material blended with NR, epoxidized natural rubber (ENR), and PTUEG3 was prepared. The thiourea moieties of PTUEG3 in the material may form dense non-crystalline hydrogen bonding interactions and undergo dynamic covalent bond exchange with the epoxy moieties of ENR.33 This interaction will facilitate the bond exchange between thermosets and thermoplastics that improve the mechanical performance and allow the recovery of the material. The autonomous recovery under ambient conditions eliminates the need for external stimuli and conventional vulcanizing agents or additives. Additionally, it helps to improve mechanical properties, such as high elasticity, resilience, and damping characteristics, making it more convenient for various application scenarios.
2. Experiment
2.1. Materials
NR and ENR (50 mol% epoxidation degree) were purchased from Qingdao Kekaida Rubber & Plastic Co., Ltd. 2-Bis(2-aminoethoxy)ethane and 1,1-thiocarbonyldiimidazole were purchased from Aladdin Scientific Co., Ltd. Additional reagents, including chloroform, diethyl ether absolute, dimethyl formamide, and absolute methanol, were purchased from Sinopharm Chemical Reagent Co., Ltd. All the procured reagents were of analytical grade and used as received.2.2. Synthesis of thermoplastic polymer
PTUEG3 was synthesized following the route described in the literature.24 In brief, 1,2-bis(2-aminoethoxy)ethane (160 mg) and 1,1′-thiocarbonyl diimidazole (175 mg) were dissolved in a 50 mL solution of DMF. The resulting mixture was stirred at 30 °C for 2 h, followed by additional stirring at 25 °C for 24 h. The resulting precipitates were dissolved in 50 mL of chloroform, and the mixture was then re-precipitated in diethyl ether (1 L) to obtain the purified polymer. The obtained precipitate was further dried in a vacuum oven at 140 °C for 12 h (Fig. S1†).2.3. In situ reaction and torque rheometer blending to prepare NR-ENR-PTUEG3
To establish dynamic networks in NR-based materials, NR, ENR, and PTUEG3 were initially masticated separately in an internal mixer and then combined at various formulations (Table 1) using a two-roll mill machine. The mastication procedure commenced with NR being subjected to an internal mixer set to 60 °C and a rotor speed of 50 rpm for 2 min. Subsequently, after masticating NR and ENR, was introduced into the mixing chamber, followed by PTUEG3. Mastication was continued for an additional 2 min to achieve the desired formulation. After the blending and reaction process, material was allowed to blend and react until the torque stabilized, then removed from the internal mixer and cooled to room temperature.| Sample | NR (phr) | ENR (phr) | PTUEG3 (phr) |
|---|---|---|---|
| NR | 100 | 0 | 0 |
| NR-ENR | 50 | 50 | 0 |
| NR-ENR-PTUEG3 [5] | 50 | 45 | 5 |
| NR-ENR-PTUEG3 [15] | 50 | 35 | 15 |
| NR-ENR-PTUEG3 [25] | 50 | 25 | 25 |
2.4. Curing of material by compression molding
Preliminary curing of material tested from 120 °C to 180 °C varying temperatures and times, according to the nature of the material 160 °C for 10 minutes (including 7 min under compression) provided the best balance of mechanical properties, including tensile strength, elasticity, and hardness, while preventing over-curing or thermal degradation.At a curing temperature of 160 °C, all the blended rubber were made using a hydraulic press equipped with an electric heating system. Rubber compression molding (hot press) was performed using a mold spacer of 3 mm thickness with dimensions of 100 mm × 100 mm, applying a pressure of 150 kg cm−2, and curing at 160 °C for 10 min (including 7 min under compression).34 The samples were then extracted after the mold assembly was left compressed, and the cured material was cooled using a cold press at 30 °C for 10 min. After processing, the material was cut into a dog-bone shape for further characterization.
2.5. Characterization
Initially, PTUEG3 was characterized using 1H and 13C nuclear magnetic resonance (NMR) spectroscopy (Fig. S2†). The spectra were recorded using a Bruker Avance III 600 NMR spectrometer at 500 MHz and 125 MHz, respectively, and the chemical shifts (δ in ppm) were determined using a non-deuterated solvent.The chemical structural changes of the material were analysed by Fourier Transform Infrared Spectroscopy (FT-IR) on a Nicolet 6700 FT-IR spectrometer (Thermo Fisher, USA) using the attenuated total reflectance (ATR) model. All spectra were recorded in the spectral range of 600 cm−1 to 4000 cm−1 with a resolution of 4 cm−1 and an average of 64 scans.
Furthermore, the thermal properties of natural rubber and the NR-ENR and NR-ENR-PTUEG3 were investigated using thermogravimetric analysis (TGA) (STA449F5 Jupiter, Netzsch, Germany). Samples weighing 2–3 mg were precisely measured, placed in an aluminium tray, and sealed. The aluminium tray was scanned from 30–800 °C at a rate of 20 °C min−1 in an inert atmosphere under a nitrogen flow rate of 20 mL min−1.
An index of the crosslinking density of the NR-ENR-TUEG3 material was determined through equilibrium swelling tests. The swelling experiment was carried out by first cutting the samples to the nearest 0.5 g and immersing them in 50 mL of toluene at room temperature for 48 h to achieve an equilibrium swelling condition. Subsequently, each sample was removed from the solvent, gently blotted to remove the excess solvent from the surface, and weighed. The sample weight was then recorded and dried to a constant weight in an oven at 60 °C for 24 h, after which the weight was recorded again. The swelling ratio (Q) was calculated using eqn (1):
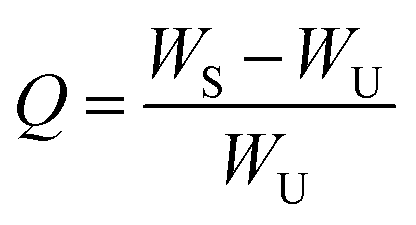 | (1) |
The tensile strength was determined using a microcomputer-controlled Instron universal tester 5966 (Instron, Norwood, MA, USA). The tensile strength of each blended rubber was tested at a speed of 400 mm min−1 at room temperature without humidity control until the specimen broke. Each specimen was cut into a dog-bone-shaped gauge measuring 75 mm in effective length, 20 mm in width, and 3 mm in thickness. Young's modulus was calculated from the initial slope of the stress–strain curve. To assure data accuracy, each test was repeated at least three times.
For the self-healing behaviour test, dog-bone-shaped specimens were cut in the middle using a clean knife, and the damaged samples were immediately re-joined. Manual pressure was applied for 30 seconds, and then the specimens were left at room temperature for 12 h to self-heal. The healing process was monitored at intervals of every 3 h using the DMS-756TR optical microscope (Shanghai Yanfeng Precision Instrument Co., Ltd, China) to visualize the self-healing lines on the surface of the material. Subsequently, stress–strain testing was conducted. The healing efficiency of each specimen was calculated as the ratio of the maximum stress of the original (σ pristine) and healed (σ healed) specimens obtained from the tensile test, as expressed in eqn (2):
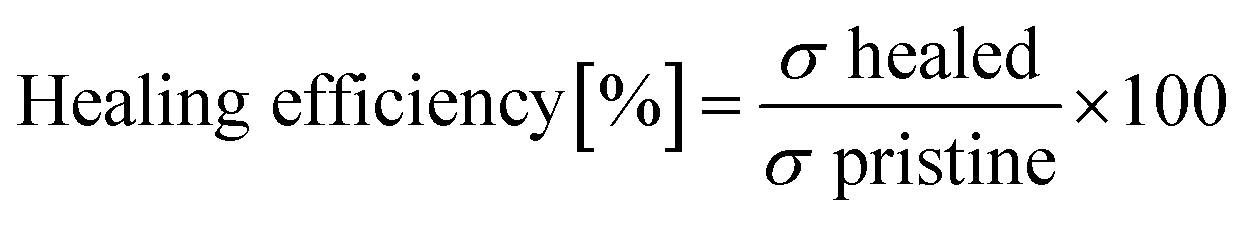 | (2) |
Furthermore, in situ load testing was carried out with three different weights: 250 g, 500 g, and 750 g on NR, NR-ENR and NR-ENR-PTUEG3.
3. Result and discussion
3.1. Preparation of the rubber NR-ENR-PTUEG3
PTUEG3 can be blended with various polymers to establish a dual crosslinked polymer network based on hydrogen bonding and dynamic covalent crosslinking, enabling materials to achieve self-healing properties. Blending PTUEG3with NR and ENR may also have a similar promoting effect. Initially curing of material tested from 120 °C to 180 °C varying temperatures and times, and at 160 °C for 10 minutes (including 7 min under compression) provided the best balance of the material performance, including tensile strength, elasticity, and hardness, while preventing over-curing or thermal degradation. Further the effect of PTUEG3 concentration was assessed through analysis of swelling, mechanical properties, and self-healing efficiency. The light brown NR-ENR-PTUEG3 obtained by blending NR, ENR, and PTUEG3 has a rubber based texture (Fig. 1a and b). The thiourea moieties in NR-ENR-PTUEG3 help activating the epoxy ring, leading to nucleophilic attack and playing an important role in self-healing (Fig. 1c). In addition, the hydrogen on the nitrogen in the PTUEG3 structure can form hydrogen bonds with sulfur, oxygen, etc., providing additional attraction and further stabilizing the physical and chemical properties of the material. These interconnected networks will promote significant self-healing and mechanical strength within the rubber material structure. | ||
| Fig. 1 Mechanism of self-healing rubber NR-ENR-PTUEG3. (a) Chemical structure of the component used in the synthesis of NR-ENR-PTUEG3. (b) Self-healing mechanism of prepared NR-ENR-PTUEG3. (c) Chemical reaction between epoxy and thiourea motif. | ||
3.2. Characterisation of the rubber NR-ENR-PTUEG3
To confirm the potential structure in the NR-ENR-PTUEG3, the precursor blending of pristine PTUEG3, NR and ENR were analysed by FT-IR using the ATR model (Fig. 2a). In the NR-ENR-PTUEG3 spectra, the epoxy ring undergoes a ring-opening reaction with the amine, forming a three-dimensional polymer network. Unsaturated hydrocarbons, such as alkenes have C–H bonds that absorb at higher frequencies, usually above 3000 cm−1. The absence of C–H stretching above 3000 cm−1 indicates the absence of unsaturated carbon, while the presence of three peaks suggests the presence of methyl and methylene groups. Specifically, these peaks are assigned as follows: the CH3 asymmetric stretch at 2963 cm−1, the CH2 asymmetric stretch at 2917 cm−1, and the CH3 symmetric stretch at 2853 cm−1. Additionally, characteristic absorption peaks of ENR are observed at 1663, 1447, 1377, and 840 cm−1, corresponding to the cis-1,4-polyisoprene structure.35 The peak at 1663 cm−1 represents C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching, while the peak at 1377 cm−1 corresponds to the methyl group umbrella mode. The intense peak at 840 cm−1 signifies the C–H wagging vibration of an alkene group, which diminished further in the blended material due to crosslinking between hydrogen and the oxygenous group of ENR and the H-bond group of thiourea.36 A small peak is observed at 1017 cm−1, which shifts towards 1020 cm−1 in the NR-ENR-PTUEG3 structure (Fig. 2a). This shift is attributed to the involvement of the 1133 cm−1 peak in the ENR stretching C–O–C from the epoxy ring, indicating the formation of the β-hydroxyl ring-opening reaction. The opening of the epoxy ring is important indication that reveals the cross-linking mechanism between PTUEG3 and NR-ENR. Furthermore, the ring-opening reaction promotes the possibility of hydrogen bonding between ENR and PTUEG3 molecules, allowing more active sites for further dynamic cross-link network, which is favourable for the self-healing of the blended material.
C stretching, while the peak at 1377 cm−1 corresponds to the methyl group umbrella mode. The intense peak at 840 cm−1 signifies the C–H wagging vibration of an alkene group, which diminished further in the blended material due to crosslinking between hydrogen and the oxygenous group of ENR and the H-bond group of thiourea.36 A small peak is observed at 1017 cm−1, which shifts towards 1020 cm−1 in the NR-ENR-PTUEG3 structure (Fig. 2a). This shift is attributed to the involvement of the 1133 cm−1 peak in the ENR stretching C–O–C from the epoxy ring, indicating the formation of the β-hydroxyl ring-opening reaction. The opening of the epoxy ring is important indication that reveals the cross-linking mechanism between PTUEG3 and NR-ENR. Furthermore, the ring-opening reaction promotes the possibility of hydrogen bonding between ENR and PTUEG3 molecules, allowing more active sites for further dynamic cross-link network, which is favourable for the self-healing of the blended material.
 | ||
| Fig. 2 Characterization of NR-ENR-PTUEG3. (a) FTIR spectra of NR, ENR, PTUEG3 and NR-ENR-PTUEG3. (b) Toluene swelling for crosslinking density. (c) TGA curves, and (d) DTG curves. | ||
In PTUEG3, the broad vibrational bands appearing around 3289 and 2920 cm−1 are attributed to –NH bending. The symmetric and asymmetric CS stretching vibrations at 940 cm−1, characteristic of thiourea, are observed to shift to a lower frequency of 933 cm−1 in the NR-ENR-PTUEG3. The former is diagnostic of NH deformation vibration of nonlinearly H-bonded thiourea units, while the latter is characteristic of NH stretching vibration.37 This shift suggests that densely located thiourea units tightly cross-link the polymer main chains via H-bonding interactions without inducing crystallization in the blended material, as the resultant H-bonded arrays are nonlinear and less ordered.24 Additionally, peaks observed around 1552 cm−1, 1093 cm−1, and 822 cm−1 are attributed to –CN stretching and –CS groups present in PTUEG3. Consequently, PTUEG3 with multiple reactive groups on its surface served as a multifunctional linkage, facilitating the formation of covalent bonding between the C and S group present in thiourea and epoxy groups, thus forming a hybrid rubber network. The presence of multiple reactive groups in PTUEG3 enhances its interfacial compatibility with the NR-ENR matrix, as the β-hydroxyl ester linkages regulate bond exchange reactions and activate copolymerization. These findings provide evidence supporting the confirmation of PTUEG3 grafting with ENR and NR. Furthermore, the crosslinking equilibrium swelling experiment provided confirmation of the crosslinking status of the NR-ENR-PTUEG3 containing NR, ENR, and PTUEG3.
The total crosslinking density of the vulcanized rubber sample was determined by swelling experiment using a toluene solution. As anticipated, the control NR (pristine) completely dissolved within 12 h after immersed in toluene for 48 h due to a lack of structural linkage holding the rubber chain together.34 On the other hand, the materials prepared with ENR and PTUEG3 exhibited swelling, indicating the formation of cross-links to varying degrees. The degree of crosslinking depends on the concentration of the epoxy and thiourea group formulations present in the NR network. Therefore, various formulations of NR-ENR-PTUEG3 with different compositions were investigated (Table 1). The NR-ENR-PTUEG3 absorbed less toluene compared to the NR-ENR and exhibited reduced swelling. The crosslinking density analysis results (Fig. 2b) demonstrates that the extent of crosslinking density increases with the presence of ENR and PTUEG3 in the formulations. The dynamic covalent linkage and epoxy group inhibited chain mobility upon contact with toluene, resulting in decreased toluene absorption, reduced swelling volume, and crosslinking density significantly increased. Compared to pristine NR, NR-ENR and NR-ENR-PTUEG3 form a more cross-linked network.
The TGA curves of the blended rubber material NR-ENR-PTUEG3 indicate a single-stage degradation process characterized by well-defined initial and final degradation temperatures. Within the temperature range of 100–270 °C, there is no discernible difference in the percentage mass loss. The initial degradation temperatures for NR and NR-ENR are noted at 300 and 310 °C, respectively (Fig. 2c). Subsequently, the maximum initial degradation temperature of NR-ENR-PTUEG3 is notably observed at 370 °C. Although there is no disparity in the percentage mass loss within the temperature range of 370–420 °C in all the material, the incorporation of PTUEG3 into the rubber impacted its thermal property. In contrast to the NR-ENR-PTUEG3, the NR and NR-ENR blend initiate degradation earlier at high temperatures, primarily due to the presence of CC bonds in the backbone. Despite PTUEG3 being thermally less stable than NR-ENR, blending it with the NR-ENR slightly improved the thermal stability of PTUEG3 due to changes in its chain mobility. It is reasonable to assume that the presence of a soft molecular segment can also contribute to improving the thermal stability of the material. Thus, thiourea emerged as our choice for executing our concept, given its recognition as a soft molecular chain suitable for NR vulcanization due to the presence of sulphur atoms and low glass transition temperature that assist sufficient chain segment mobility of thiourea that enable macroscopic flow to assist the inherent self-healing process in rubber. The DTG curves of the NR-ENR network improve the thermal stability comparatively to pristine NR shows a maximum weight loss temperature at 389 °C, while the incorporation of PTUEG3 into rubber makes maximum weight loss towards higher temperature at 391 °C (Fig. 2d). The improved thermal stability in ENR-NR network may due to the formation of hydrogen bonding strengthen the interaction between polymeric chain in the network. These findings indicate that while the incorporation of PTUEG3 into rubber moderately affects their thermal stability, it also helps to improve the overall thermal stability of the rubber.
In the rubber network, the chemical interaction between NR-ENR and PTUEG3 involves thiourea SC NH2 carbonyl groups in thermoplastic polymers linking with the thermosets of ENR, thereby opening the epoxy ring along with the rubber chains. This prepared thermoplastic polymer serves as a synthetic intermediate for polymers, and forming a cross-linked network.38–40 Consequently, PTUEG3, with its reactive carbonyl sites, acts as a multifunctional linkage and contributes to the formation of a dual network mechanism. The combined action of covalent bonding and hydrogen bonding results in a blended structure that reconfigures the network topology. This configuration endows the material with the ability to rapidly fill rubber cracks at room temperature without requiring thermally activation. Additionally, it enhances its rheological, mechanical, morphological, and maturing characteristics.
3.3. Mechanical analysis of NR-ENR-PTUEG3
Mechanical characteristics obtained through tensile testing provide significant insights into the effects of the prepared materials at different formulations. Fig. 3a–d illustrate the mechanical performance of NR-ENR and various NR-ENR-PTUEG3 formulations compared to NR, which typically exhibits low tensile strength and high elastic modulus, as is typical for elastomeric materials. In the comparative study, pristine NR exhibited a stress of 0.19 MPa at 336% elongation at break, notably poor performance compared to the NR-ENR blend, which exhibited a stress of 2.27 MPa at 470% elongation at break. This improvement maybe due to the chain entanglement resulting from the non-covalent network, as demonstrated in the swelling analysis. On the other hand, the blended rubber network shows potential as a multi-functional cross-linking network, facilitated by the presence of multiple reactive sites on PTUEG3. These sites contribute to the formation of a tough and resilient network. Consequently, an increase in PTUEG3 concentration in the network led to improvements in maximum stress, toughness, and elongation at break.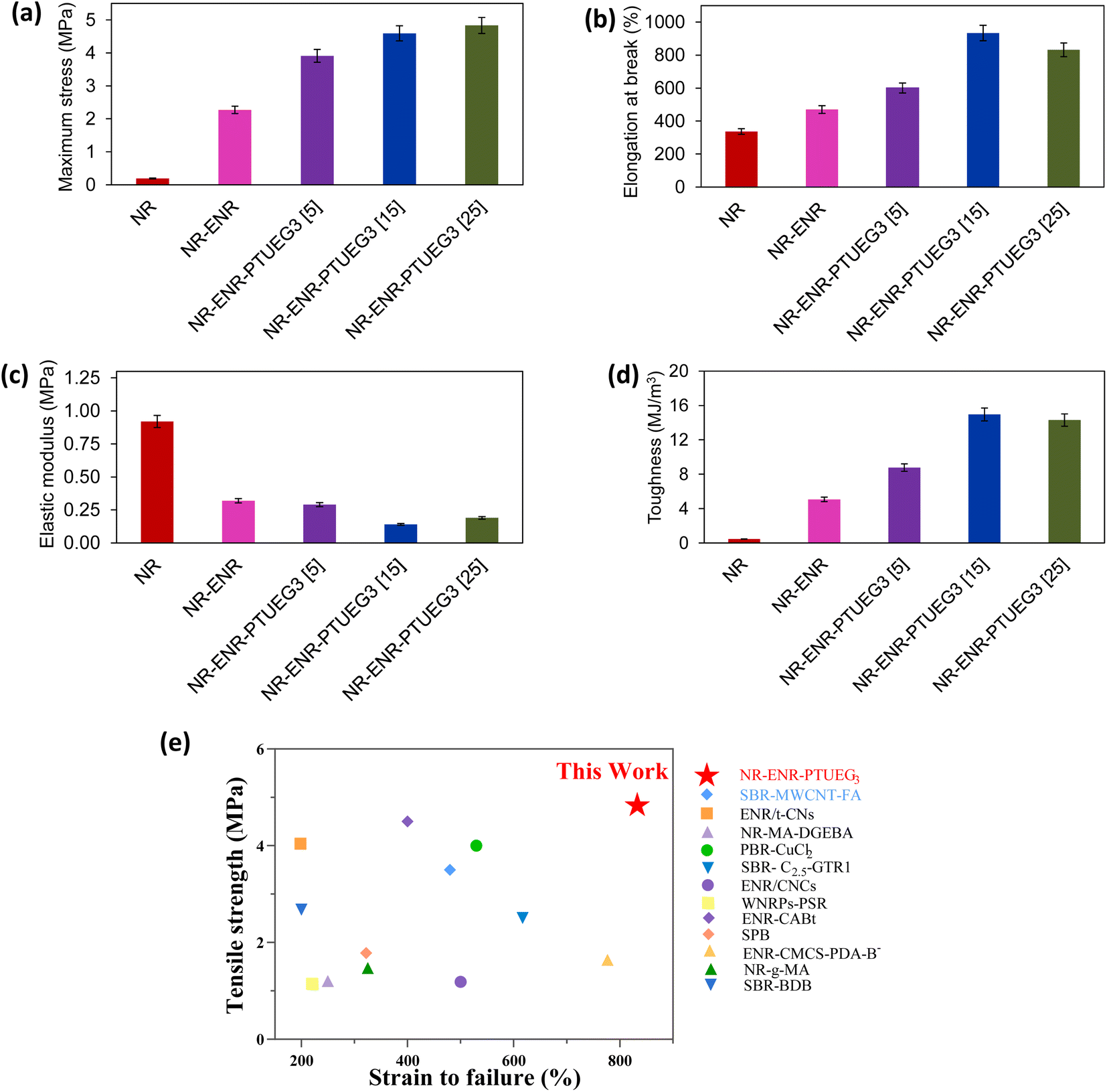 | ||
| Fig. 3 Typical stress–strain curves of NR, NR-ENR and NR-ENR-PTUEG3 with different concentrations of PTUEG3 (a) maximum stress, (b) elongation at break, (c) elastic modulus, (d) toughness, and (e) comparative study between this work and reported self-healing rubber materials. Detailed information is summarized in Table S1.† | ||
Significant results observed for the NR-ENR-PTUEG3 at various formulations of 5 phr, 15 phr, and 25 phr of PTUEG3 demonstrated that tensile strength increases with higher concentrations, reaching 3.91, 4.59, and 4.83 MPa, respectively. Corresponding, elongations at break were 600%, 934%, and 833%, showing significant improvements compared to NR-ENR, attributed to the enhanced crosslinking with the thiourea group to form dynamic networks. The combination of NR-ENR reinforced with PTUEG3 is noted for simultaneously enhancing strength and elongation. The presence of short ENR chains linking PTUEG3 together provides additional stress resistance during stretching, while longer NR chains contribute to the elasticity of the material. The network formed by hydrogen bonding in NR is considerably weaker compared to the robust sulfur covalent crosslink network in PTUEG3. When the material is subjected to external stretching, the weaker hydrogen bonding network is the first to be disrupted. This preferential breakage of hydrogen bonds allows the material to absorb and dissipate energy efficiently, preventing immediate failure. As a result, this mechanism helps to enhance the overall mechanical properties of NR-ENR- PTUEG3, such as its toughness and resilience. The energy dissipation through the breaking of hydrogen bonds allows the sulfur covalent crosslink network to maintain its integrity, contributing to the durability and elasticity of the material under stress.41 This hybrid rubber network, coupled with the covalent network, significantly improves the strength of the NR-ENR-PTUEG3. The maximum improvements in tensile strength and toughness were observed at higher concentration of PTUEG3, indicating increased crosslinking within the thiourea network.
In contrast, the decrease in elastic modulus observed in each blended material with increasing concentration of PTUEG3 and ENR compared to pristine NR network exhibiting their elastomeric characteristics. This dual reaction mechanism, involving both covalent and non-covalent networks, significantly contributes to the mechanical properties of NR-ENR-PTUEG3, which exhibit superior mechanical performance compared to the pristine NR material. Moreover, mechanical strength plays a crucial role in the self-healing properties of the blended material. The optimum rubber demonstrates enhanced tensile strength and high strain percentage compared to previously reported works (Fig. 3e). Comparative analysis with other reported studies also highlights typical tensile strength, elongation at break, and self-healing efficiency over time (Table S1†).
3.4. Self-healing property of NR-ENR-PTUEG3
To demonstrate the self-healing ability of the blended rubber, pristine NR, NR-ENR, and various formulations of NR-ENR-PTUEG3 were initially cut in the middle using a clean knife. The damaged samples were immediately re-joined, and manual pressure was applied for 30 seconds. The specimens were then left at room temperature for 12 h to self-heal. Tensile testing was conducted to evaluate the mechanical characteristics of the repaired material (Fig. 4a). The results from the tensile test showed that the healed samples preserved the mechanical characteristics of the original samples, mostly recovering compared to the pristine NR and NR-ENR material. Formulations with 5 phr, 15 phr, and 25 phr of PTUEG3 exhibited significant strengths of 1.07, 3.92, and 3.36 MPa, respectively, after 12 h. The corresponding elongations at break were 325%, 724%, and 599%, respectively, due to the further bond exchange reaction with the thiourea group forming dynamic networks. In comparison, NR and NR-ENR exhibited strength of 0.07 and 0.26 MPa, with corresponding elongation at break of 818% and 325%, respectively, indicating poor recovery compared to the blended rubber material. Although some healing effects were observed in NR and NR-ENR blend, indicating the broken hydrogen bonds cannot completely recover in a short time, thus fewer weak bonds participate and exhibited the contribution of polar epoxy groups that limited in the chain diffusion of NR-ENR, resulting in poor self-healing. The interaction of epoxy groups with thiourea in the healing process of the NR-ENR-PTUEG3 was highly effective. For a more intuitive comparison, the stress–strain curve of the healed NR-ENR-PTUEG3 with 15 phr was recorded from 0 h to 12 h at 3 h intervals to observe the significant time for maximum self-healing (Fig. 4b). The healing efficiency initially exhibited 5%, 20%, 36%, and 85% MPa at 0 h, 3 h, 6 h, and 12 h, respectively, compared to its original strength. This indicates that the healing efficiency and crosslinking network improved with prolonged healing time, aligning with findings in self-healing materials with different healing times.42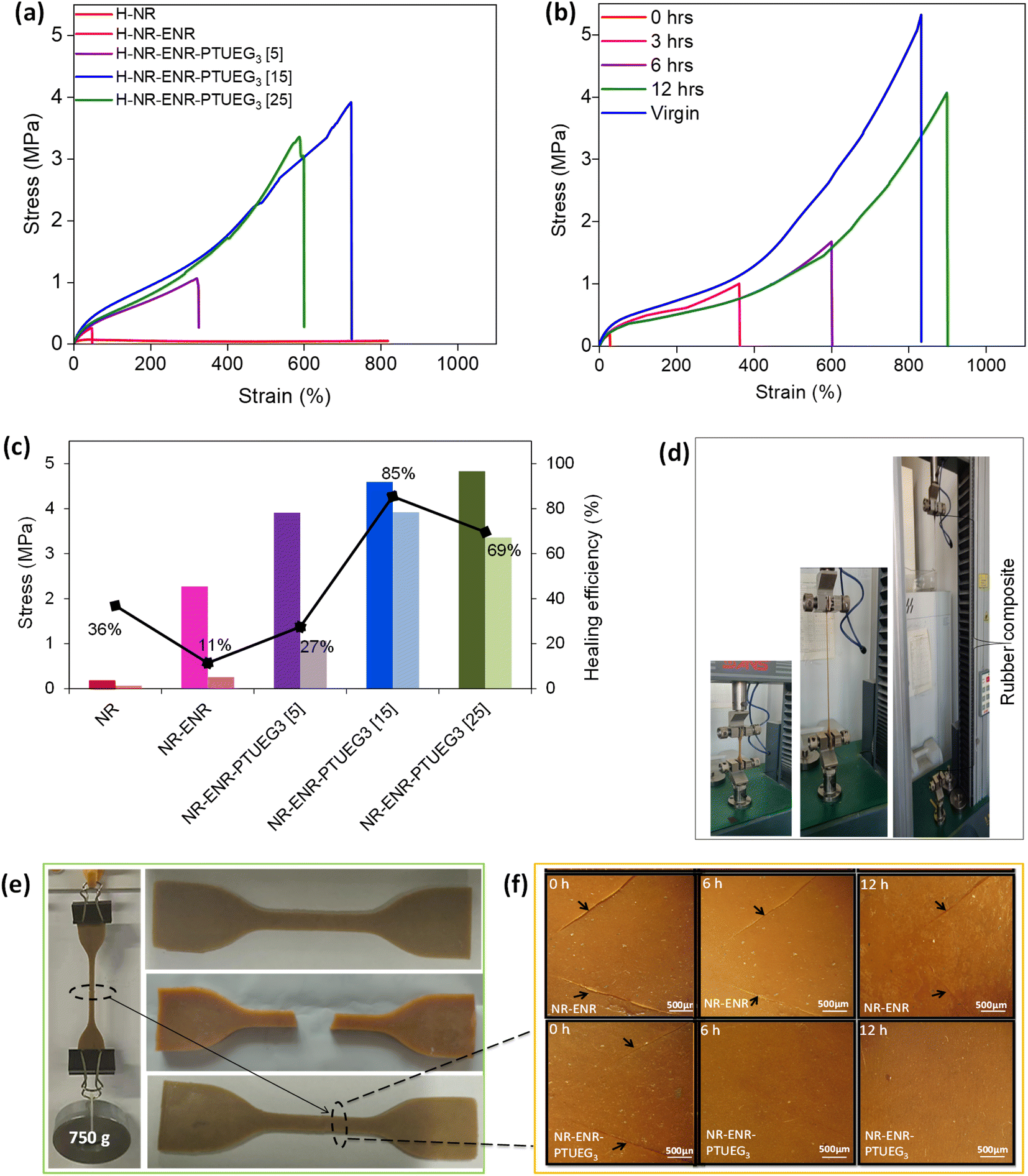 | ||
| Fig. 4 Self-healing property of NR-ENR-PTUEG3. (a) Typical stress–strain test of the self-healing rubber. (b) Self-healing time study of NR-ENR-PTUEG3. (c) Strength recovery of the repaired material. (d) The tensile test performance of the self-healing NR-ENR-PTUEG3. (e) Image of the NR-ENR-PTUEG3 material performing in situ load-lifting test, and the different colors of the samples are due to varying lighting conditions at the time of photography. (f) Optical microscopy image showing the healing of the repaired material. | ||
The initial 5% MPa at 0 h indicates relatively inferior self-healing due to weak crosslinking with the thiourea and hydrogen networks. However, the substantial improvement to 85% MPa at 12 h demonstrated extensively improved self-healing at room temperature. This indicates that the healing efficiency and crosslinking network improved with prolonged healing time, aligning with findings in self-healing materials with different healing times.42 And the reformation of the hydrogen-bond network plays a primary role in the recovery process, as these bonds can quickly re-establish themselves, contributing to the initial stages of healing. In contrast, the dynamic covalent bonds take a longer time to reform due to their more complex nature, which involves the gradual reorganization of molecular structures. This disparity in the recovery times of different bond types results in a slower overall elasticity recovery of the rubbers at room temperature. The slower reformation of covalent bonds suggests that while initial strength and integrity are regained relatively quickly, full mechanical restoration and elasticity take longer to achieve.43 The self-healing efficiency was calculated using eqn (2), comparing the stress and strain curves before and after 12 h of healing at room temperature (Fig. 4c). As a result, the self-healing NR-ENR-PTUEG3 exhibited excellent recovery (27%, 85%, and 69%) at PTUEG3 concentration of 5 phr, 15 phr, and 25 phr in the rubber. However, a tensile test indicated that 15 phr PTUEG3 loading resulted in improved elongation at break and healing efficiency, while an increase in PTUEG3 loading to 25 phr led to a decline in strain and healing efficiency. Nonetheless, more detailed studies are needed to understand the mechanism of the self-healing process.
To further examine the mechanical strength of the repaired material, in situ load-bearing tests were conducted at three different loads (250 g, 500 g, and 750 g) in pristine NR. These tests exhibited an inability to support an initial weight of 250 g, breaking from the repaired region. Similarly, NR-ENR was unable to support a load of 500 g from the attach region; however, it withstood the load test at 250 g for a few seconds due to the chain entanglement of the rubber. In contrast, the repaired NR-ENR-PTUEG3 after 12 h significantly sustained a 750 g load without breaking from the damaged region for several hours (Fig. 4e), demonstrating the presence of a dual healing network based on dynamic covalent and hydrogen bonds, which enhances the self-healing ability of NR-ENR-PTUEG3. In this network, the PTUEG3 chains maintain high mobility at room temperature, resulting in network rearrangement. Overall, the NR-ENR-PTUEG3 exhibited excellent self-healing capabilities, with mechanical strength attributed to the dynamic covalent and non-covalent networks compared to pristine natural rubber.
The healing process was monitored at intervals of every 3 h using optical microscopy to visualize the self-healing lines of the blended material on the surface (Fig. 4f). In both pristine NR and NR-ENR, a sharp crack line between the cuts is visibly apparent from the top surface within the cut and re-joined regions. Interestingly, at room temperature, the healed NR-ENR-PTUEG3 gradually fades the damaged region over a span of 12 h, affirming the ability to self-heal without any external stimulation and showcasing the excellent stiffness of the material after being healed. Consequently, this clearly illustrated the significant role of the thiourea network in the rubber network.
4. Conclusions
In this study, a thiourea-based rubber material, NR-ENR-PTUEG3, was successfully synthesized through a dynamic covalent network and hydrogen bond interaction, enabling self-healing without external stimulants, additives, or fillers. Pristine NR has poor self-healing and mechanical properties and required additives to vulcanize the rubber and enhance its strength. However, this compromises its healing efficiency due to irreversible crosslinking after vulcanization. Therefore, thiourea was formulated to cross-link the NR-ENR network, curing the elastomer and improving its mechanical strength and inherent self-healing efficiency. The blending exhibited autonomous self-healing in rubber with good mechanical strength due to the presence of a dual network mechanism. Spectra findings revealed that the incorporation of PTUEG3 served as a multifunctional linkage, facilitating the formation of covalent bonds between the C and S groups in the epoxy and thiourea, as well as non-covalent bonds between the H and O groups in the thiourea and oxygenous epoxy group, respectively. This formed a reversible network that increased toughness and durability by establishing a hybrid rubber network. While the TGA results indicate that the blended material exhibits good thermal stability, it is important to note that the specific contribution of molecular motion to this stability is not fully understood. Given the lack of detailed research on molecular motion, these conclusions should be considered preliminary. Further studies are needed to elucidate the molecular dynamics that contribute to the observed thermal behavior. The NR-ENR-PTUEG3 at 15 phr exhibited good tensile strength (4.8 ± 0.3 MPa) before healing and tensile strength (3.92 ± 0.6 MPa) after healing without any external stimulation or additives at room temperature. However, it was observed that the network also exhibited limited efficiency at 25 phr PTUEG3 loading, slightly reducing elongation at break and healing efficiency. On the other hand, toughness and mechanical stress increases with the increase in PTUEG3 loading up to 25 phr. The blended rubber material demonstrated the ability to heal when broken surfaces were in contact at room temperature, regaining mechanical strength and exhibiting healing efficiency of up to 85% without any triggering agent. This approach accelerated the self-healing process, eliminated the dependency of traditional additives and external stimulation such as heat, light, radiation, etc. and its inherent self-healing efficiency shows promising outcomes in advanced material applications where safety, performance, and longer fatigue life are major concerns.Data availability
Data will be available on request.Conflicts of interest
The authors declare no known competing financial or personal relationship interests that could influence the work reported in this paper.Acknowledgements
This research work was supported by the CAS-TWAS Presidents Fellowship and sponsored by the National Key Research and Development Program of China (Project No. 2021YFC2101900).Notes and references
- R. Blanchard, E. O. Ogunsona, S. Hojabr, R. Berry and T. H. Mekonnen, ACS Appl. Polym. Mater., 2020, 2, 887–898 CrossRef CAS.
- A. Adibi, D. Valdesueiro, L. Simon, C. P. Lenges and T. H. Mekonnen, ACS Sustain. Chem. Eng., 2022, 10, 10718–10732 CrossRef CAS.
- H. Chen, C. Liu, M. Li, H. Zhang, M. Xian and H. Liu, RSC Adv., 2019, 9, 8934 RSC.
- E. Ducrot, Y. Chen, M. Bulters, R. P. Sijbesma and C. Creton, Science, 2014, 344, 186–189 CrossRef CAS PubMed.
- P. Jagadeesh, M. Puttegowda, S. Mavinkere Rangappa and S. Siengchin, Polym. Compos., 2021, 42, 5691–5711 CrossRef CAS.
- R. Li, Z. Wang and Y. Wang, The Innovation, 2021, 2, 100095 CrossRef CAS PubMed.
- Y. Hao, The Innovation, 2024, 5, 100613 CrossRef CAS.
- W. Pang, C. Yuan, Y. Zheng, T. Zhong and P. Lai, Innov. Life, 2024, 2, 100058 CrossRef.
- R. Fujio, M. Kitayama, N. Kataoka and S. Anzai, Rubber Chem. Technol., 1979, 52, 74–83 CrossRef CAS.
- L. Chen, T. Bi, E. Lizundia, A. Liu, L. Qi, Y. Ma, J. Huang, Z. Lu, L. Yu, H. Deng and C. Chen, The Innovation, 2024, 5, 100655 CrossRef CAS.
- J. Guo, S. Ali and M. Xu, Green Carbon, 2023, 1, 150–153 CrossRef.
- S. Vishnu, B. Prabu and M. Pugazhvadivu, Hybrid Adv., 2023, 3, 100062 CrossRef.
- Z. Yuan, Y. Liu and H. Zhou, Innov. Mater, 2023, 1, 100025 CrossRef.
- Y. Zhu, K. Cao, M. Chen and L. Wu, ACS Appl. Mater. Interfaces, 2019, 11, 33314–33322 CrossRef CAS.
- B. Willocq, J. Odent, P. Dubois and J.-M. Raquez, RSC Adv., 2020, 10, 13766–13782 RSC.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS.
- L. Cao, J. Fan, J. Huang and Y. Chen, J. Mater. Chem. A, 2019, 7, 4922–4933 RSC.
- L. Yang, X. Lu, Z. Wang and H. Xia, Polym. Chem., 2018, 9, 2166–2172 RSC.
- Y. Amamoto, J. Kamada, H. Otsuka, A. Takahara and K. Matyjaszewski, Angew. Chem., Int. Ed., 2011, 50, 1660–1663 CrossRef CAS.
- Z. Zheng, L. Lian and M. Xie, Innov. Life, 2024, 2, 100053 CrossRef.
- J. Zhang, L. Cao and Y. Chen, Eur. Polym. J., 2022, 168, 111103 CrossRef CAS.
- D. M. Beaupre and R. G. Weiss, Molecules, 2021, 26, 3332 CrossRef CAS.
- Y. Peng, S. Gu, Q. Wu, Z. Xie and J. Wu, Acc. Mater. Res., 2023, 4, 323–333 CrossRef CAS.
- Y. Yanagisawa, Y. Nan, K. Okuro and T. Aida, Science, 2018, 359, 72–76 CrossRef CAS PubMed.
- B. P. Finkenauer, Y. Gao, X. Wang, Y. Tian, Z. Wei, C. Zhu, D. J. Rokke, L. Jin, L. Meng and Y. Yang, Cell Rep. Phys. Sci, 2021, 2, 100320 CrossRef CAS.
- H. Chen, Z. Wu, Z. Su, S. Chen, C. Yan, M. Al-Mamun, Y. Tang and S. Zhang, Nano Energy, 2021, 81, 105654 CrossRef CAS.
- B. Kim, Y. Cho, D.-G. Kim and J.-H. Seo, Mater. Today Chem., 2023, 30, 101583 CrossRef CAS.
- J. H. Hwang, E. Kim, E. Y. Lim, W. Lee, J. O. Kim, I. Choi, Y. S. Kim, D. G. Kim, J. H. Lee and J. C. Lee, Adv. Sci., 2023, 10, 2302144 CrossRef CAS.
- E. J. Cha, D. S. Lee, H. Kim, Y. H. Kim, B. G. Kim, Y. Yoo, Y. S. Kim and D.-G. Kim, RSC Adv., 2019, 9, 15780–15784 RSC.
- J. Du, Y. Li, J. Wang, C. Wang, D. Liu, G. Wang and S. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 26966–26972 CrossRef CAS PubMed.
- Y. Fujisawa, A. Asano, Y. Itoh and T. Aida, J. Am. Chem. Soc., 2021, 143, 15279–15285 CrossRef CAS PubMed.
- D. S. Lee, Y.-S. Choi, J. H. Hwang, J.-H. Lee, W. Lee, S.-k. Ahn, S. Park, J.-H. Lee, Y. S. Kim and D.-G. Kim, ACS Appl. Polym. Mater., 2021, 3, 3714–3720 CrossRef CAS.
- S. S. Chimni, N. Bala, V. A. Dixit and P. V. Bharatam, Tetrahedron, 2010, 66, 3042–3049 CrossRef CAS.
- B. Trinh, P. Owen, A. vanderHeide, A. Gupta and T. H. Mekonnen, ACS Appl. Polym. Mater., 2023, 5, 8890–8906 CrossRef CAS.
- S. Chuayjuljit, C. Yaowsang, N. Na-Ranong and P. Potiyaraj, J. Appl. Polym. Sci., 2006, 100, 3948–3955 CrossRef CAS.
- M. Bijarimi, S. Ahmad and R. Rasid, J. Elastomers Plast., 2014, 46, 338–354 CrossRef CAS.
- R. Custelcean, M. G. Gorbunova and P. V. Bonnesen, Chem.–Eur. J., 2005, 11, 1459–1466 CrossRef CAS PubMed.
- R. Mogaki, K. Okuro and T. Aida, Chem. Sci., 2015, 6, 2802–2805 RSC.
- P. Hashim, K. Okuro, S. Sasaki, Y. Hoashi and T. Aida, J. Am. Chem. Soc., 2015, 137, 15608–15611 CrossRef CAS PubMed.
- R. Mogaki, P. Hashim, K. Okuro and T. Aida, Chem. Soc. Rev., 2017, 46, 6480–6491 RSC.
- S.-Q. Huang, J.-Q. Zhang, Y. Zhu, L.-M. Kong, L.-S. Liao, F.-Q. Zhang, Z.-T. Xie and J.-R. Wu, Chin. J. Polym. Sci., 2024, 42, 457–467 CrossRef CAS.
- D. Wang, J. Guo, H. Zhang, B. Cheng, H. Shen, N. Zhao and J. Xu, J. Mater. Chem. A, 2015, 3, 12864–12872 RSC.
- J. Zhang, C. Huang, Y. Zhu, G. Huang and J. Wu, Polymer, 2021, 231, 124114 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03808h |
| This journal is © The Royal Society of Chemistry 2024 |