
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInhibition of apoptosis and biofilm formation in Candida auris by click-synthesized triazole-bridged quinoline derivatives†
Mohmmad Younus Wani *a,
Vartika Srivastavab,
Waleed Ahmed El-Saida,
Abdullah Saad Al-Bogamia and
Aijaz Ahmadbc
*a,
Vartika Srivastavab,
Waleed Ahmed El-Saida,
Abdullah Saad Al-Bogamia and
Aijaz Ahmadbc
aDepartment of Chemistry, College of Science, University of Jeddah, 21589 Jeddah, Saudi Arabia. E-mail: mwani@uj.edu.sa
bDepartment of Clinical Microbiology and Infectious Diseases, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, South Africa
cDivision of Pulmonary, Allergy, Critical Care, and Sleep Medicine, Department of Medicine, University of Pittsburgh Medical Center, Pittsburgh, PA 15213, USA
First published on 4th July 2024
Abstract
Candida auris, a recent addition to the Candida species, poses a significant threat with its association to numerous hospital outbreaks globally, particularly affecting immunocompromised individuals. Given its resistance to existing antifungal therapies, there is a pressing need for innovative treatments. In this study, novel triazole bridged quinoline derivatives were synthesized and evaluated for their antifungal activity against C. auris. The most promising compound, QT7, demonstrated exceptional efficacy with a minimum inhibitory concentration (MIC) and minimum fungicidal concentration (MFC) of 0.12 μg mL−1 and 0.24 μg mL−1, respectively. Additionally, QT7 effectively disrupted mature biofilms, inhibiting them by 81.98% ± 8.51 and 89.57 ± 5.47 at MFC and 2× MFC values, respectively. Furthermore, QT7 induced cellular apoptosis in a dose-dependent manner, supported by various apoptotic markers such as phosphatidylserine externalization, mitochondrial depolarization, and reduced cytochrome c and oxidase activity. Importantly, QT7 exhibited low hemolytic activity, highlighting its potential for further investigation. Additionally, the physicochemical properties of this compound suggest its potential as a lead drug candidate, warranting further exploration in drug discovery efforts against Candida auris infections.
1. Introduction
Candida auris, an incipient yeast responsible for causing nosocomial bloodstream infections, is a matter of serious concern, globally. This species of Candida was first isolated in the year 2009 and from then its presence is known in thirty-five countries excluding Antarctica.1,2 The invasive infections caused by this pathogen are coupled with high rates of mortality. Moreover, C. auris is well known for its multi-drug resistant properties against commonly used antifungal agents.3 In the past few years, the increasing incidence of non-albicans Candida species linked infection and colonization is assumed to be a result of the unwarranted use of preventive antifungals such as fluconazole.4 Also, yeast identification techniques utilized in various laboratories mainly misidentify C. auris thus making the recognition and control of this microbe challenging.C. auris generally spreads from person to person and according to experts the transmission capability of this pathogen varies from other species of Candida.5 Also, unlike several other species of Candida, C. auris is not a member of healthy microflora and is not naturally present within the gastrointestinal tract of humans.6 This species of Candida is tagged with various pathogenic attributes that are very similar to C. albicans, factors such as hydrolytic enzyme secretion, nutrient acquisition, siderophore-based iron acquisition, tissue penetration, two-component histidine kinase system, and pathways involved in cell wall modelling are commonly reported.7,8 C. auris also displayed the capability to form biofilms allowing for the colonization on biotic and abiotic surfaces.9 Additionally, evidence of C. auris escaping the host immune response has been published in the past.10,11 Thus, alternative strategies for prevention and better management of the global fungal burden are in high demand.12
The Centers for Disease Control and Prevention (CDC) breakpoint evaluation of C. auris isolates in the United States of America revealed remarkably elevated minimal inhibitory concentration (MIC) for commonly used antifungal drugs.13 Thus, the foremost challenge in managing invasive C. auris infections is the high level of drug resistance along with its capacity to swiftly acquire resistance to the three main classes of anti-Candida drugs. This suggests that developing an effective management strategy for this multi-drug resistant species of Candida would be extremely challenging. Consequently, there is an unequivocal need for new antifungal molecules with distinct and alternative mechanisms of antifungal action. Creating new antifungal treatments is a formidable and demanding activity due to the numerous resemblances between yeast and human cellular machinery. A pragmatic approach to combat this multi-drug resistant threat has been the structural modification of established antifungal drug classes, significantly aiding in circumventing the mechanisms of antifungal drug resistance.
Triazoles have long been used as pivotal components in the arsenal against fungal infections, owing to their broad-spectrum activity and favorable pharmacokinetic properties.14,15 However, the rise of drug-resistant fungal strains necessitates continuous innovation in antifungal therapeutics.16 One promising avenue lies in the development of novel triazole derivatives incorporating quinoline moieties17 as depicted in Fig. 1. Quinolines possess diverse pharmacological activities, including antimicrobial effects, and their integration with triazoles offers the potential to enhance antifungal efficacy.18,19 Furthermore, click chemistry techniques, such as the CuAAC reaction, facilitate the synthesis of these hybrid molecules with improved efficiency and precision.20 By harnessing the synergistic properties of triazoles and quinolines, and in continuation of our search for better antifungal leads,21–24 in this study, we synthesized new triazole bridged quinoline derivatives and evaluated them for their antifungal activity profile. Furthermore, we investigated their mechanisms of action and predicted their physicochemical properties to pinpoint lead antifungal molecules with drug-like properties.
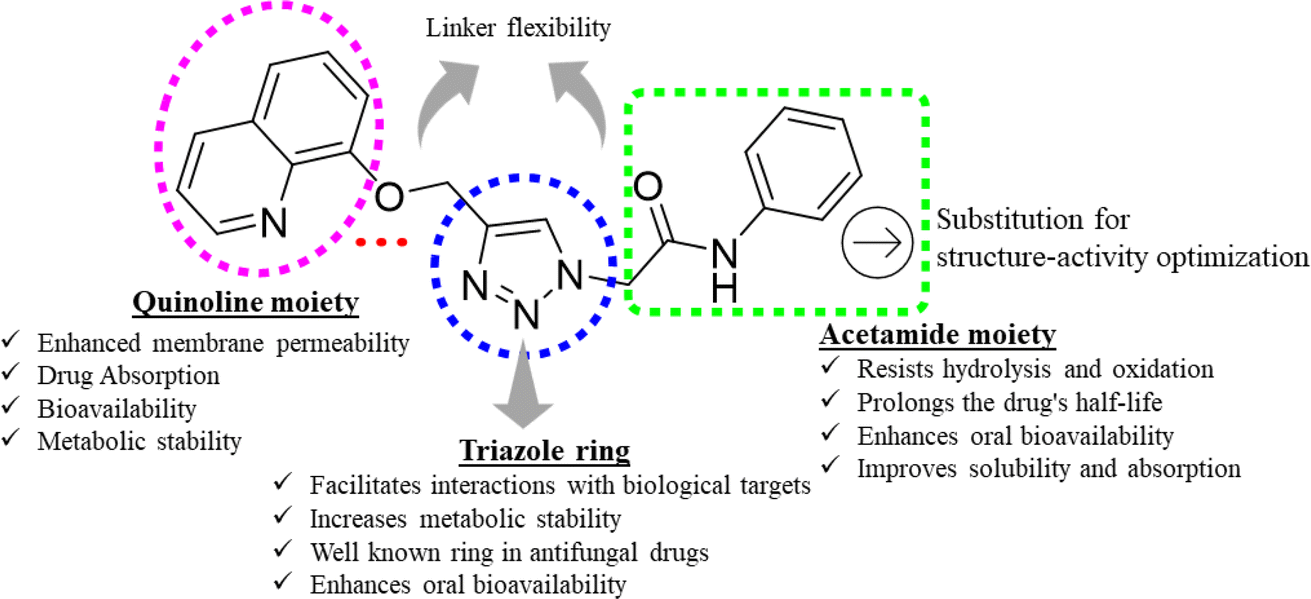 | ||
| Fig. 1 Design and structural features of the triazole-bridged quinoline derivatives. | ||
2. Result and discussion
2.1 Chemistry
The outline synthesis of the target derivatives (QT1–QT10) is given in Scheme 1. Compound 2 (2-(4-((quinolin-8-yloxy)methyl)-1H-1,2,3-triazol-1-yl)acetyl chloride) was obtained through copper-xatalyzed azide–alkyne cycloaddition (CuAAC) click chemistry approach from (8-(prop-2-yn-1-yloxy)quinoline) (2) and with 2-azidoacetyl chloride. The target derivatives QT1–QT10 were obtained in 80–85 percent yield through amide coupling reaction between different substituted phenylamines and the acyl group of the triazole derivative (2). All the reactions were performed in anhydrous solvents considering the moisture sensitivity of the acyl group. Click reactions are one of the most important green routes to the synthesis of 1,2,3-triazoles with higher reaction yields and high purity.25,26 The structures of all the derivatives were established by different physical and spectroscopic techniques. The structure of the triazole (2) was confirmed by FTIR, 1HNMR, 13CNMR and ESI MS. Presence of stretching bands at 3185 (C–H triazole), 2850 (C–H), 1725 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and absence of any stretching bands for azide (NNN at or around 2100–2160 cm−1) or alkyne (around 2100–2140 cm−1) indicated the cyclization of 8-(prop-2-yn-1-yloxy)quinoline with 2-azidoacetyl chloride. The formation of the triazole ring was also confirmed by the characteristic absorption band in the region 3165–3135 cm−1 in the FTIR spectra due to the C–H stretching of the triazole ring in the target derivatives. The presence of a singlet peak at 8.20 (1H, s, triazole ring) in 1HNMR spectrum and the presence of peaks at 147.6 and 123.4 in 13CNMR spectrum corresponding to the carbon atoms in the triazole ring established the structure of the compound 2. Positive ion mass peak at m/z: [M+ + H] 303.75 further confirmed the structure. The structures of the target compounds were also established in a similar way. The formation of the amide bonds in the target compounds was established by the characteristic stretching frequencies for CO and N–H, represented as amide I and amide II, characteristic of amides. The 1HNMR, 13CNMR and ESI MS spectra were also in agreement with the established structures of the new molecules as given in the experimental section and ESI.†
O) and absence of any stretching bands for azide (NNN at or around 2100–2160 cm−1) or alkyne (around 2100–2140 cm−1) indicated the cyclization of 8-(prop-2-yn-1-yloxy)quinoline with 2-azidoacetyl chloride. The formation of the triazole ring was also confirmed by the characteristic absorption band in the region 3165–3135 cm−1 in the FTIR spectra due to the C–H stretching of the triazole ring in the target derivatives. The presence of a singlet peak at 8.20 (1H, s, triazole ring) in 1HNMR spectrum and the presence of peaks at 147.6 and 123.4 in 13CNMR spectrum corresponding to the carbon atoms in the triazole ring established the structure of the compound 2. Positive ion mass peak at m/z: [M+ + H] 303.75 further confirmed the structure. The structures of the target compounds were also established in a similar way. The formation of the amide bonds in the target compounds was established by the characteristic stretching frequencies for CO and N–H, represented as amide I and amide II, characteristic of amides. The 1HNMR, 13CNMR and ESI MS spectra were also in agreement with the established structures of the new molecules as given in the experimental section and ESI.†
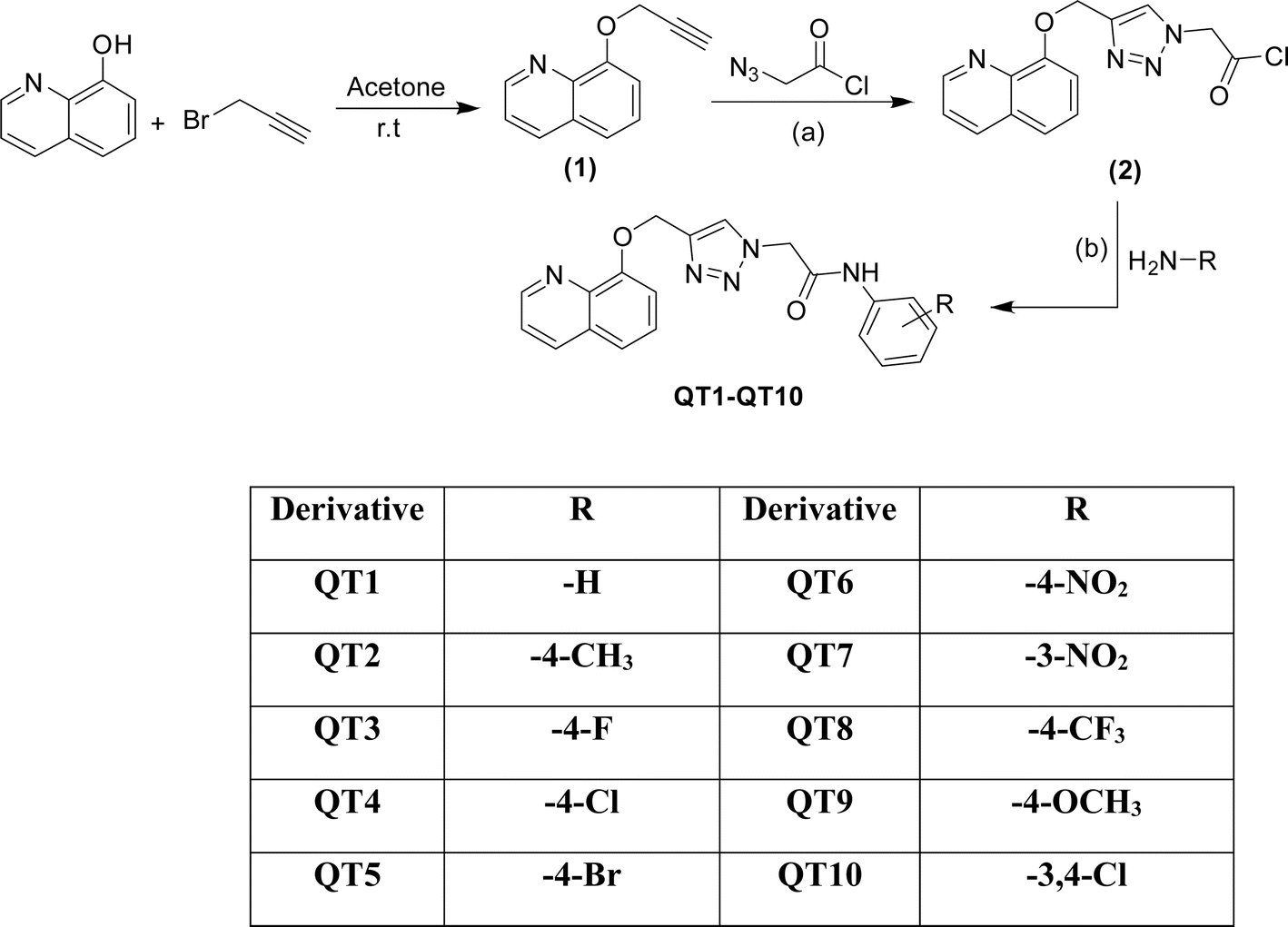 | ||
| Scheme 1 Synthesis of triazole-bridged quinoline derivatives (QT1–QT10); reagents and conditions: (a) sodium ascorbate, CuSO4, DMF (b) HATU/DIPEA, DMF, 60 °C. | ||
2.2 Antifungal activity
| Derivative | MIC (μg mL−1) | MFC (μg mL−1) | Derivative | MIC (μg mL−1) | MFC (μg mL−1) |
|---|---|---|---|---|---|
| QT1 | 15.62 | 62.5 | QT6 | 0.97 | 1.95 |
| QT2 | 15.62 | 62.5 | QT7 | 0.12 | 0.23 |
| QT3 | 3.90 | 7.81 | QT8 | 7.81 | 15.62 |
| QT4 | 7.813 | 15.62 | QT9 | 15.62 | 62.5 |
| QT5 | 7.813 | 15.62 | QT10 | 15.62 | 62.5 |
| DMSO | — | — | FLZ | 125 | NA |
 | ||
| Fig. 2 Biofilm inhibition rates of QT7 against C. auris strain. Five different dilutions (1/4× MIC, 1/2× MIC, MIC, MFC and 2× MFC) were used to examine the anti-biofilm activities of the test compound. Untreated control showed no inhibition. *p < 0.05. | ||
Furthermore, micrographs obtained from confocal microscopy supported the crystal violet assay and confirmed the anti-biofilm activity of QT7 (Fig. 3). A large number of accumulated and metabolically active C. auris cells were observed in the untreated control sample. The micrograph of the untreated control sample showed a well-defined and dense biofilm matrix with bright green fluorescence (produced by ConA-Alexa Fluor 488 conjugate dye) and the cells embedded into the matrix were fluoresced red (generated by FUN-1 dye). The images of the C. auris biofilm treated with various doses of compound (MIC to 2× MFC) revealed the biofilm disruptive ability of QT7. At higher concentrations, MFC and 2× MFC, the compound destroyed the mature biofilm which was evident from faded green and red fluorescence as compared to the untreated control. Therefore, these results expose the fact that QT7 can penetrate the biofilm matrix thus impacting the sustainability of yeast cells embedded in the biofilm matrix and therefore resulting in successful anti-biofilm activity.
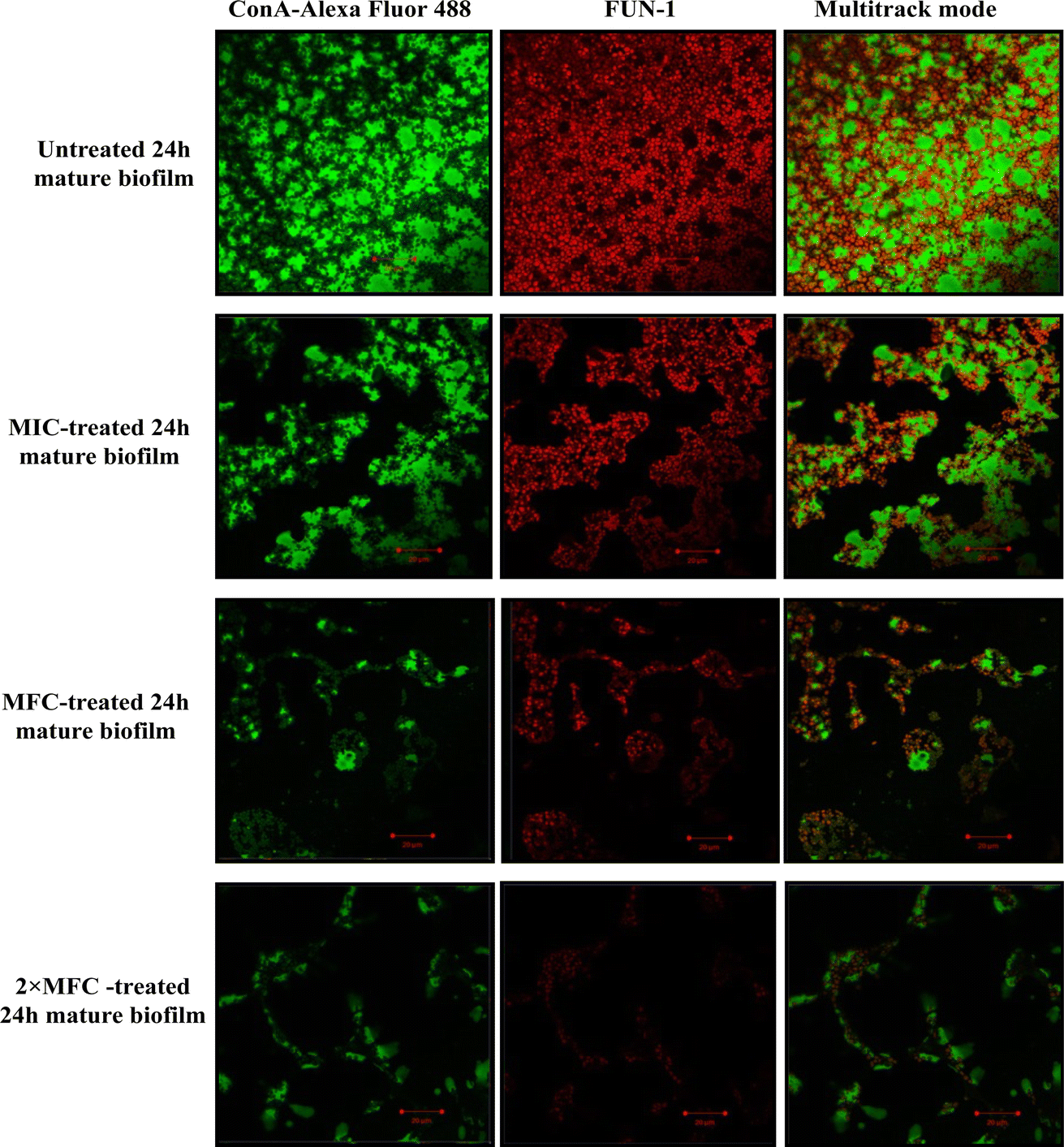 | ||
| Fig. 3 Confocal microscopy of untreated and compound-treated 24 h mature C. auris biofilm. The green fluorescence (produced by ConA-Alexa Fluor 488) in the micrographs represents the C. auris biofilm matrix whereas the red fluorescence (produced by FUN-1) represents viable/metabolically active cells. | ||
The MMP is directly proportional to the mitochondrial energetic status and is used to gauge the activity of energy-dependent proton pumps, electron transport systems and mitochondrial permeability. The thrashing of MMP is considered important for cellular sustainability and is an indispensable step for the onset of the apoptotic cascade. Hence, to examine the apoptotic potential of QT7 its impact on the MMP was evaluated. The constant MMP in viable cells permits the JC-10 dye to aggregate (red fluorescence) whereas, in the case of apoptotic cells (lower MMP) the JC-10 dye stays in its monomeric form (green fluorescence). In the present experiment the MMP was quantified in terms of the ratio between JC-10 aggregates and JC-10 monomers and a decrease in the output compared to the untreated control indicated deprotonation of MMP (Fig. 4). The average RF to GF ratio in the untreated control cells was high (RF/GF = 2.43, 40.27) representing intact mitochondrial membrane whereas, the value for the compound-treated cells and the positive control was lower representing the compromised MMP. The average RF/GF values for 1/2× MIC, MIC and positive control were 2.12 C 0.14, 1.8, 10.07 and 1.95 ± 0.06 respectively. These findings advocate the ability of QT7 to reduce the mitochondrial membrane potential in C. auris by disintegrating the mitochondrial membrane. The depolarization of the mitochondrial membrane triggers the leakage of cytochrome c along with other crucial factors into the cytoplasm and finally results in apoptosis.32
 | ||
| Fig. 4 Bar graph representing fluorescence ratio between the average values of RF (Ex/Em = 540/590 nm, red fluorescence (RF)) to GF (Ex/Em = 490/530 nm, green fluorescence (GF)). The average value of RF represents the JC-10 aggregates whereas, the average value of GF represents the JC-10 monomers. Exposure to MIC and sub-MIC values resulted in mitochondrial membrane depolarization in C. auris cells as compared to the untreated control. UC: untreated control, PC: positive control. | ||
Evaluation of cytochrome c oxidase activity helps in understanding if the cells are undergoing apoptosis. In the present work, we quantified the amount of cytochrome c leaching out from mitochondria into the cytosol in the untreated control, compound-treated (1/2× MIC, MIC) and positive control cells (Fig. 5). According to the data obtained, cells treated with QT7 showed a significant surge in cytosolic cytochrome c levels as compared to the untreated control cells. The cytosolic and mitochondrial cytochrome c values for the untreated control cells were considered 1.0. The treatment with QT7 at different concentrations resulted in a significant oozing out of cytochrome c from the mitochondria and the average relative values for mitochondrial cytochrome c were 0.92 we0.07 and 0.84 ± 0.03 at 1/2× MIC and MIC respectively whereas, the values for cytosolic cytochrome c were 1.16 we0.09 and 1.21 d 0.06 at 1/2× MIC and MIC respectively. In the positive control, the level of cytochrome c in mitochondria was found to be 0.88 in0.04 and in the cytosol, the value was 1.19 d 0.02. In yeast cells, cytochrome c controls both cellular metabolism as well as apoptotic pathways and is also responsible for electron transfer from complex III to IV in mitochondria therefore the discharge of cytochrome c disturbs the electron transport chain and triggers apoptosis.33
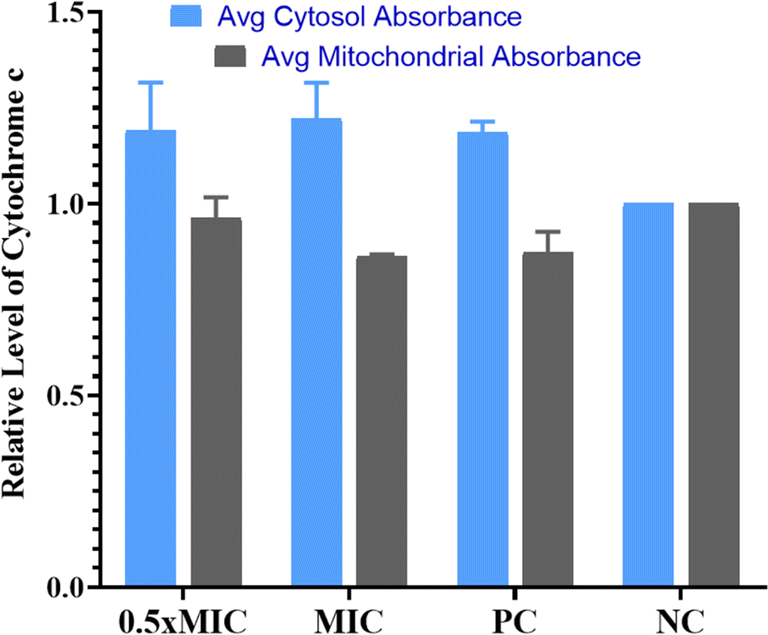 | ||
| Fig. 5 Exposure of QT7 results in the activation of apoptotic factors in C. auris. The mitochondrial and cytosolic cytochrome c were quantified at 550 nm and the data showed a decrease in the cytochrome c level of mitochondria whereas an increase in the cytochrome c level in the cytosol. NC: negative control; PC: positive control. | ||
The onset of apoptosis exposes membrane protein (phosphatidylserine) to the outer side of the plasma membrane.34 Thus, we included this experiment to validate the apoptotic property of QT7. The exposure of phosphatidylserine was determined with the help of the Annexin V + PI dual staining assay. During the process, Annexin V stains the exposed phosphatidylserine while PI validates plasma membrane integrity. Hence, this technique can discriminate between apoptotic, late apoptotic and necrotic cells (Fig. 6). In the untreated control, the majority of cells (95.3%) were confined to Q4 representing the presence of live cells in the sample. Whereas treatment with QT7 at 1/2× MIC resulted in the migration of cells from Q4 (71.7%) to Q2 (15.6%), Q3 (3.05%) and Q1 (9.62%), representing the onset of cellular apoptosis and at a higher concentration of compound (MIC) the maximum cellular population was found sitting in Q2 (65.9%) followed by Q1 (18.4%), Q4 (12.8%) and Q3 (2.83%). The data suggested that QT7 has exerted dose-dependent cellular apoptosis in C. auris. Similarly, the cellular population in the positive control was dispersed in all the quadrants (37.9%, Q1; 27.4%, Q2; 1.49%, Q3 and 33.2%, Q4) suggesting exposure to H2O2 triggered necrosis in C. auris.
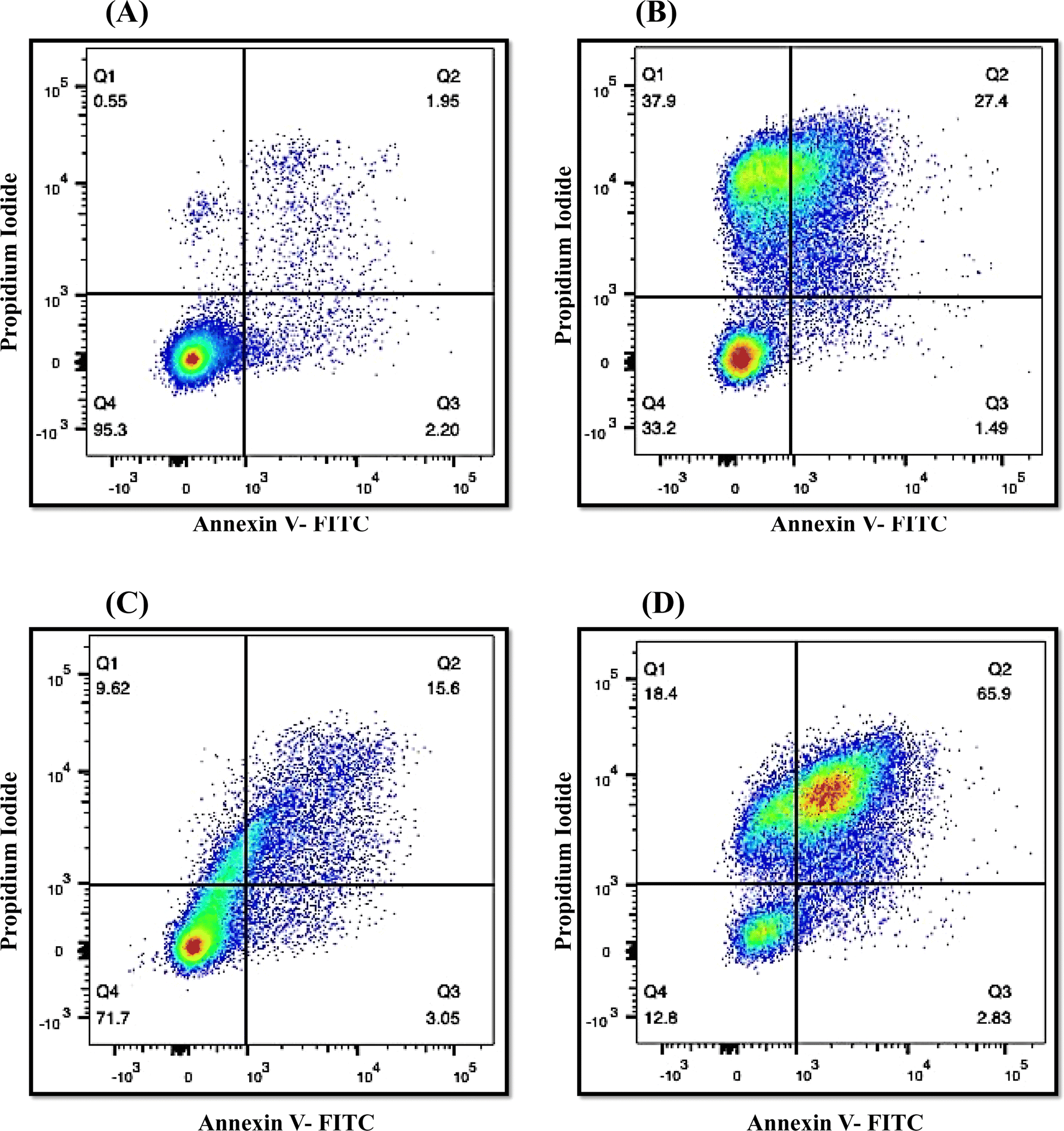 | ||
| Fig. 6 Determination of the apoptotic activity of QT7 by Annexin V-FITC + PI double staining technique. (A) Untreated control cells, (B) H2O2 (10 mM) treated positive control and C. auris treated with 1/2× MIC (C) and MIC (D) values of QT7. Q1 represents necrosis (Annexin V−/PI+), Q2 represents late apoptosis (Annexin V+/PI+), Q3 represents early apoptosis (Annexin V+/PI−) and Q4 represents viable cells (Annexin V−/PI−). | ||
 | ||
| Fig. 7 Hemolytic activity of QT7. The hemolytic potential of QT7 at various concentrations was evaluated against h-RBCs in the presence of 10% Triton X-100 (PC, positive control) and PBS (NC, negative control). | ||
3. Pharmacokinetic studies/ADMET profile
Pharmacokinetic and ADMET properties were assessed using pkCSM, an online resource provided by Cambridge University and validated by MedChemDesigner™ software version 3.0 (ref. 35) and SwissADME36 was employed to validate the predictions with high precision. The results obtained for all the molecules indicate promising solubility potential of the molecules within a desirable range, both in water (−4.16 to −3.73 log mol L−1) and in terms of permeability of Caco-2 (1.22 to 0.14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logPapp (cm s−1) × 10) (Table S1 in the ESI†). Furthermore, the intrinsic water solubility (logS0) was determined to be within the range of −4.83 to −3.78. It is a very important property that describes the solubility of a compound in an uncharged state and is pH independent. The molecules also exhibit favorable characteristics such as high intestinal absorption (approximately 93% absorbed in humans), skin permeability ranging around −2.74 (logKp), blood–brain barrier (BBB), permeability from −0.87 to 0.67 (logBB), and central nervous system (CNS) permeability around −2.74 (logPS). Notably, the clearance values, which represent the rate of drug removal relative to its plasma concentration, were found to be quite favorable for all derivatives (0.06 to 0.29 mL min−1 kg−1). This suggests that the accumulation of these molecules will not occur in the body, indicating their non-hepatotoxic and non-toxic behavior. The oral rat acute toxicity (LD50) ranged from 2.47 to 3.06 mol kg−1, while the oral rat chronic toxicity (LOAEL) was observed between 0.83 to 1.38 (mg kg−1 day−1), with a maximum tolerated dose (human) ranging from 0.06 to 0.48 mg kg−1 day−1. All these findings demonstrate that these molecules possess favorable ADMET profile, which is essential for a compound to exhibit drug-like characteristics.
logPapp (cm s−1) × 10) (Table S1 in the ESI†). Furthermore, the intrinsic water solubility (logS0) was determined to be within the range of −4.83 to −3.78. It is a very important property that describes the solubility of a compound in an uncharged state and is pH independent. The molecules also exhibit favorable characteristics such as high intestinal absorption (approximately 93% absorbed in humans), skin permeability ranging around −2.74 (logKp), blood–brain barrier (BBB), permeability from −0.87 to 0.67 (logBB), and central nervous system (CNS) permeability around −2.74 (logPS). Notably, the clearance values, which represent the rate of drug removal relative to its plasma concentration, were found to be quite favorable for all derivatives (0.06 to 0.29 mL min−1 kg−1). This suggests that the accumulation of these molecules will not occur in the body, indicating their non-hepatotoxic and non-toxic behavior. The oral rat acute toxicity (LD50) ranged from 2.47 to 3.06 mol kg−1, while the oral rat chronic toxicity (LOAEL) was observed between 0.83 to 1.38 (mg kg−1 day−1), with a maximum tolerated dose (human) ranging from 0.06 to 0.48 mg kg−1 day−1. All these findings demonstrate that these molecules possess favorable ADMET profile, which is essential for a compound to exhibit drug-like characteristics.
The parameters of pharmacokinetic characteristics and adherence to Lipinski's filters, which encompass essential physicochemical properties, are very significant for in vitro and in silico assessment of drug like characteristics.37–39 Lipinski's rule of five serves as a valuable tool in the drug discovery process, aiding in predicting the oral bioavailability of potential drug candidates. By considering factors like the number of H-bond acceptors and donors, lipophilicity and molecular weight, this rule assists in the identification of compounds with a higher likelihood of successful oral delivery.
While not an absolute determinant of a drug's fate, adhering to these guidelines helps streamline the initial stages of drug development and prioritize compounds with a greater potential for effective oral administration. A compound that does not exhibit favorable pharmacokinetic parameters or comply with Lipinski's filters is likely to face obstacles in the last stages of the development of drug. Based on the projected pharmacokinetic parameters and after subjecting the molecules to Lipinski's filters (Table 2), it is apparent that all the compounds possess favorable drug-like characteristics and hold promise as potential candidates for further investigation and study.
| Compound | MWt | ClogPa |
logDb |
HBA | HBD | RBs | tPSA | Ro5 (Y/N) |
|---|---|---|---|---|---|---|---|---|
| a At pH 7.4.b Determined with ChemAxon logD predictor.c Standard drug; HBA: hydrogen bond acceptor, HBD: hydrogen bond donor, obtained by Marvin Sketch 23.16; RB: rotatable bonds; Ro5 (Y/N): rule of five followed or not; Y: yes; N: no; veber filter: rotatable bonds ≤ 10, tPSA ≤ 140 in Å2. |
||||||||
| QT1 | 359.14 | 2.519 | 2.74 | 5 | 1 | 7 | 81.93 | Y |
| QT2 | 373.42 | 3.018 | 3.25 | 5 | 1 | 7 | 81.93 | Y |
| QT3 | 377.38 | 2.919 | 2.88 | 6 | 1 | 7 | 81.93 | Y |
| QT4 | 393.83 | 3.489 | 3.34 | 6 | 1 | 7 | 81.93 | Y |
| QT5 | 438.29 | 3.639 | 3.51 | 6 | 1 | 7 | 81.93 | Y |
| QT6 | 404.39 | 2.814 | 2.68 | 7 | 1 | 8 | 127.75 | Y |
| QT7 | 404.39 | 2.814 | 2.68 | 7 | 1 | 8 | 125.07 | Y |
| QT8 | 427.39 | 3.853 | 3.62 | 8 | 1 | 8 | 78.65 | Y |
| QT9 | 389.42 | 2.594 | 2.58 | 6 | 1 | 8 | 87.88 | Y |
| QT10 | 428.27 | 4.173 | 3.95 | 7 | 1 | 7 | 81.93 | Y |
| Fluconazolec | 306.27 | 0.50 | 0.56 | 7 | 1 | 5 | 81.93 | Y |
The assessment of molecules using the bioavailability radar tool from SwissADME revealed that certain molecules demonstrated promising physicochemical properties related to oral bioavailability. When examining the most active molecule (QT7), its bioavailability radar plot displayed favorable characteristics within the pink-colored area (as depicted in Fig. 8).40,41 This pink area demonstrates the desired range for six important physicochemical parameters, crucial for oral bioavailability: flexibility, lipophilicity, solubility polarity size and saturation.
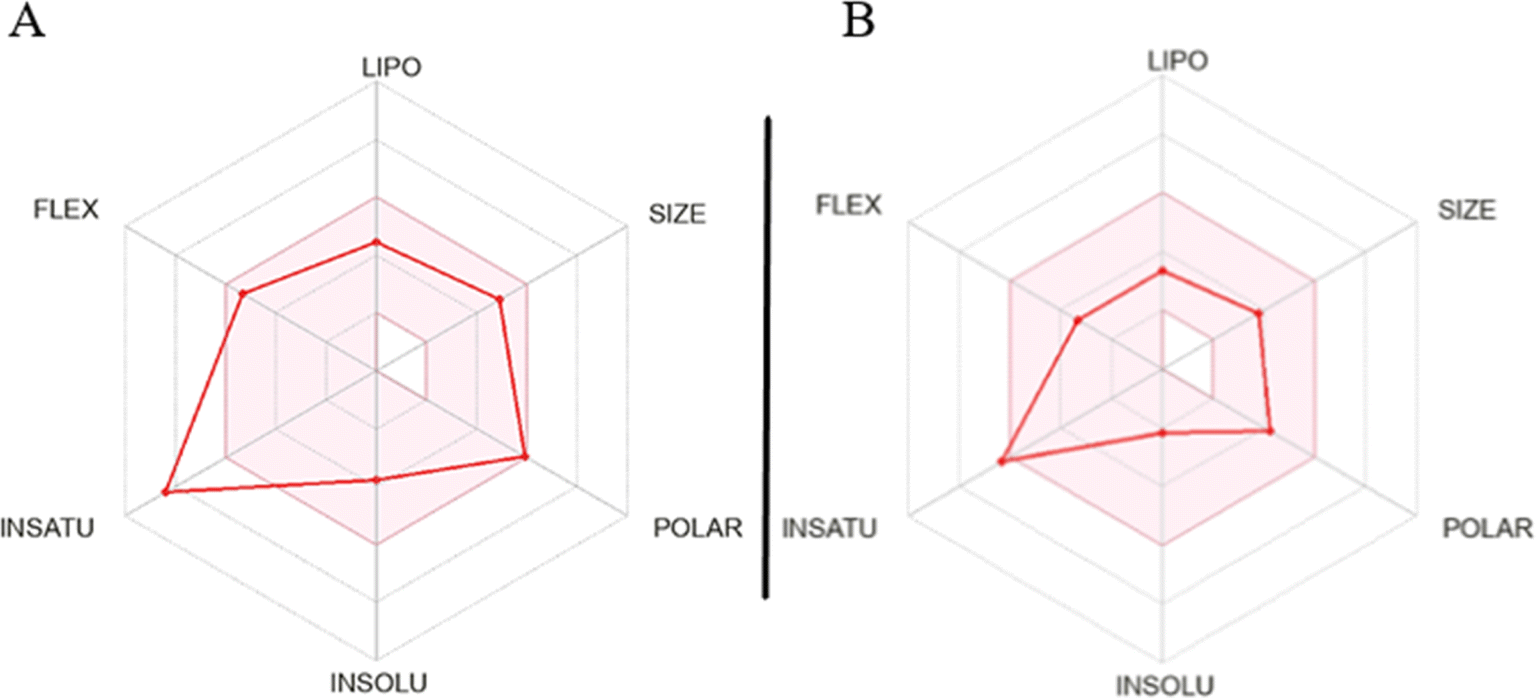 | ||
| Fig. 8 (A) Bioavailability radar plot of QT7 (most active compound) and (B) fluconazole (standard drug). LIPO (lipophilicity), POLAR (polarity), FLEX (flexibility), INSATU (saturation) and INSOLU (solubility). | ||
For a compound to be considered as drug like, its radar plot should fall completely within the pink area, indicating optimal values for every property. These properties include polarity (TPSA) between 20 and 130 Å2, solubility (logS) not exceeding 6, size (MW) between 150 and 500 g mol−1, number of rotatable bonds (flexibility) no more than 9, fraction of carbons in sp3 hybridization (saturation) not less than 0.25, and lipophilicity (XLOGP3) between −0.7 and +5.0. The molecules exhibiting a bioavailability radar plot within the pink area demonstrate desirable physicochemical properties associated with enhanced oral bioavailability. This suggests that they have the potential to be effectively absorbed and distributed in the body, increasing their chances of success as drug candidates. It is important to note that while the molecules meet the criteria within the pink area of the bioavailability radar, further comprehensive evaluations are necessary to assess other aspects of drug development, including pharmacological activity, efficacy, safety, and potential drug interactions.
The findings of the study reveal that the triazole-bridged quinoline derivative QT7, featuring a nitro group at position-3 of the phenyl ring of the acetamide moiety, exhibits exceptional efficacy. QT7 demonstrated minimum inhibitory concentration (MIC) and minimum fungicidal concentration (MFC) values of 0.12 μg mL−1 and 0.24 μg mL−1, respectively, indicating its high potency in inhibiting and killing C. auris cells. This potency is notably superior compared to other derivatives that are structurally similar but vary in the substituents on the phenyl ring. Specifically, the presence of a nitro group at position-4 of the phenyl ring in QT6 increased the MIC and MFC to 0.97 μg mL−1 and 1.95 μg mL−1, respectively, suggesting that the position of the nitro group significantly influences efficacy. The derivative QT1, which has an unsubstituted phenyl ring, exhibited much higher MIC and MFC values of 15.62 μg mL−1 and 62.5 μg mL−1, respectively. Substitutions with a methyl (QT2) or methoxy group (QT9) at position-4 did not markedly improve efficacy, with MIC values of 7.813 μg mL−1 and 15.62 μg mL−1, respectively. However, substitutions with chloro (QT4), bromo (QT5), and trifluoromethyl groups (QT8) led to MIC and MFC values of 7.813 μg mL−1 and 15.62 μg mL−1 respectively, indicating varied impacts on antifungal activity. Interestingly, the substitution with a fluoro group further decreased the MIC and MFC to 3.90 μg mL−1 and 7.81 μg mL−1, respectively. The efficacy of QT10, which features a 2,4-dichlorophenyl ring, was found to be similar to that of the unsubstituted phenyl ring.
These observations suggest that the nitro group at position-3 of the phenyl ring is the most favorable for achieving optimal binding interactions with the target. The electronic effects of the substituents likely play a critical role in determining the efficacy of the molecules, indicating that both the nature and position of the substituents are crucial for antifungal activity. Furthermore, the ability QT7 to disrupt mature biofilms by 81.98% at MFC and 89.57% at 2× MFC highlights its potential in addressing biofilm-associated infections, which are notoriously resistant to treatment. The mode of action of QT7 is particularly noteworthy, as it induces cellular apoptosis in C. auris through various apoptotic pathways. Key markers such as phosphatidylserine externalization, mitochondrial depolarization, and reduced cytochrome c and oxidase activity collectively contribute to the apoptotic process. This multifaceted mechanism not only enhances our understanding of the antifungal effect of QT7 but also opens avenues for targeting similar pathways in other resistant fungal pathogens.
The promising physicochemical properties of QT7 further support its potential as a lead drug candidate. These properties, including solubility, stability, and bioavailability, are critical in the drug development process. Additionally, QT7 exhibited low hemolytic activity, indicating minimal toxicity towards human red blood cells (h-RBCs). This finding is crucial, suggesting that QT7 can be administered at therapeutic concentrations without causing significant harm to human cells, thereby enhancing its candidacy for further development. Future studies should focus on in vivo applications of QT7 to assess the efficacy and safety in animal models, providing crucial data for potential clinical trials. Moreover, exploring the efficacy of QT7 against a broader spectrum of fungal pathogens could establish its role as a versatile antifungal agent.
4. Conclusion
The synthesis of triazole bridged quinoline derivatives via CuAAC reaction, followed by amide coupling, yielded excellent results. Among the derivatives tested for antifungal activity against a C. auris strain, QT7 demonstrated significant efficacy, inducing cellular apoptosis by targeting crucial yeast apoptotic markers, such as mitochondrial membrane potential, movement of cytochrome c from mitochondria to cytosol, and flipping PS from inner side to the outer leaflet of the cell membrane. Furthermore, QT7 disrupted the mature biofilm after 24 hours and most importantly, it exhibited low toxicity towards h-RBCs. These findings, coupled with promising physicochemical properties, suggest the potential of these derivatives as lead drug candidates. Particularly noteworthy is QT7, which warrants further in-depth investigation for potential in vivo applications. These encouraging results advocate for expanded research to explore the efficacy of QT7 against various fungal pathogens, underscoring its potential as a lead molecule for antifungal therapy.5. Materials and method
5.1 Chemistry
The chemical reagents and solvents were procured from Sigma Aldrich and Merck Germany. TLC plates used were precoated aluminium sheets (silica gel 60 F254, Merck Germany) and visualization was done by UV light in a UV cabinet. Heraeus Vario EL III analyser was used for elemental analysis. Bruker ALPHA FT-IR spectrometer (Eco-ATR) was used for FTIR analysis. Bruker AVANCE 400 spectrometer (400 MHz) was used for 1H and 13C NMR spectra using DMSO-d6/CDCl3 as solvent with TMS (tetramethylsilane) as standard. ESI-MS positive ion mode was recorded on Micromass Quattro II triple quadrapole mass spectrometer.Yield: 95%; anal. calc. for C14H11ClN4O2: C 55.52, H 3.65, N 18.50%; found: C 55.40, H 3.68, N 18.65%; FTIR νmax cm−1: 3185 (C–H triazole ring), 2850 (C–H), 1725 (CO), 1630 (CN); 1H NMR (DMSO-d6) δ (ppm): 8.69 (dd, 1H, quinoline ring), 8.20 (1H, s, triazole ring), 8.11 (d, 1H, quinoline ring), 7.51–7.05 (m, 4H, quinoline ring), 5.86 (2H, s, CH2), 5.29 (2H, s, CH2); 13CNMR (DMSO-d6) δ (ppm): 167.4 (CO), 152.7 (C–O), 147.6 (CN), 143.2, 139.6, 129.3, 127.5, 125.8, 125.0, 123.4, 122.6, 121.5, 111.7, 58.3, 54.3; ESI-MS m/z: [M+ + H] 303.75.
5.1.2.1 2-(4-((Quinolin-8-yloxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(4-phenyl)acetamide (QT1). Yield: 85%; anal. calc. for C20H17N5O2: C 66.84, H 4.77, N 19.49%; found: C 66.54, H 4.55, N 19.70%; FTIR νmax cm−1: 3284 (N–H), 3155 (C–H triazole ring), 2862 (C–H), 1765 (C
O), 1630 (CN); 1H NMR (DMSO-d6) δ (ppm): 8.96 (s, 1H, NH), 8.69 (dd, 1H, quinoline ring), 8.20 (1H, s, triazole ring), 8.11 (d, 1H, quinoline ring), 7.53–7.42 (m, 4H, quinoline ring), 7.40–7.24 (m, 5H, Ph), 5.46 (2H, s, CH2), 5.29 (2H, s, CH2); 13CNMR (DMSO-d6) δ (ppm): 168.4 (CO), 152.7 (C–O), 147.6 (CN), 143.2, 139.6, 137.5, 129.8, 129.0, 127.5, 125.7, 125.0, 123.9, 122.7, 121.8, 120.1, 111.7, 59.3, 57.3; ESI-MS m/z: [M++H] 360.14.
5.1.2.2 2-(4-((Quinolin-8-yloxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(4-methylphenyl)acetamide (QT2). Yield: 87%; anal. calc. for C21H19N5O2: C 67.55, H 5.13, N 18.76%; found: C 67.31, H 5.26, N 18.55%; FTIR νmax cm−1: 3385 (N–H), 3160 (C–H triazole ring), 2862 (C–H), 1750 (C
O), 1632 (CN); 1H NMR (DMSO-d6) δ (ppm): 9.70 (s, 1H, NH), 8.68 (dd, 1H, quinoline ring), 8.21 (1H, s, triazole ring), 8.09 (d, 1H, quinoline ring), 7.50–7.42 (m, 4H, quinoline ring), 7.40–7.23 (m, 4H, Ph), 5.46 (2H, s, CH2), 5.29 (2H, s, CH2) 2.35 (3H, s, CH3); 13CNMR (DMSO-d6) δ (ppm): 168.4 (CO), 152.7 (C–O), 147.6 (CN), 143.2, 139.6, 137.5, 129.8, 129.0, 127.5, 125.7, 125.0, 123.9, 122.7, 121.8, 120.1, 111.7, 59.3, 52.9, 20.73; ESI-MS m/z: [M+ + H] 374.18.
5.1.2.3 2-(4-((Quinolin-8-yloxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(4-fluorophenyl)acetamide (QT3). Yield: 75%; anal. calc. for C20H16FN5O2: C 63.65, H 5.03, N 18.56%; found: C 63.75, H 5.26, N 18.66%; FTIR νmax cm−1: 3385 (N–H), 3160 (C–H triazole ring), 2858 (C–H), 1760 (C
O), 1628 (CN); 1H NMR (DMSO-d6) δ (ppm): 9.72 (s, 1H, NH), 8.69 (dd, 1H, quinoline ring), 8.20 (1H, s, triazole ring), 8.10 (d, 1H, quinoline ring), 7.52–7.44 (m, 4H, quinoline ring), 7.38–7.23 (m, 4H, Ph), 5.46 (2H, s, CH2), 5.29 (2H, s, CH2); 13CNMR (DMSO-d6) δ (ppm): 169.5 (CO), 152.5 (C–O), 147.5 (CN), 143.6, 139.2, 137.0, 129.3, 129.1, 127.6, 125.8, 125.0, 123.2, 122.4, 121.4, 120.6, 111.5, 58.6, 52.5; ESI-MS m/z: [M+ + H] 377.13.
5.1.2.4 2-(4-((Quinolin-8-yloxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(4-chlorophenyl)acetamide (QT4). Yield: 82%; anal. calc. for C20H16ClN5O2: C 61.00, H 4.10, N 17.78%; found: C 61.14, H 4.15, N 17.70%; FTIR νmax cm−1: 3382 (N–H), 3158 (C–H triazole ring), 2862 (C–H), 1765 (C
O), 1630 (CN); 1H NMR (DMSO-d6) δ (ppm): 9.55 (s, 1H, NH), 8.67 (dd, 1H, quinoline ring), 8.21 (1H, s, triazole ring), 8.10 (d, 1H, quinoline ring), 7.58–7.46 (m, 4H, quinoline ring), 7.40–7.23 (m, 4H, Ph), 5.46 (2H, s, CH2), 5.29 (2H, s, CH2); 13CNMR (DMSO-d6) δ (ppm): 168.5 (CO), 152.7 (C–O), 147.6 (CN), 143.1, 139.6, 137.1, 129.3, 128.1, 127.5, 125.7, 125.1, 122.8, 121.8, 111.7, 59.3, 52.9; ESI-MS m/z: [M+ + H] 394.10.
5.1.2.5 2-(4-((Quinolin-8-yloxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(4-bromophenyl)acetamide (QT5). Yield: 75%; anal. calc. for C20H16BrN5O2: C 54.81, H 3.68, N 15.98%; found: C 54.64, H 3.55, N 16.05%; FTIR νmax cm−1: 3380 (N–H), 3152 (C–H triazole ring), 2860 (C–H), 1765 (C
O), 1632 (CN); 1H NMR (DMSO-d6) δ (ppm): 9.58 (s, 1H, NH), 8.66 (dd, 1H, quinoline ring), 8.21 (1H, s, triazole ring), 8.11 (d, 1H, quinoline ring), 7.56–7.44 (m, 4H, quinoline ring), 7.38–7.23 (m, 4H, Ph), 5.45 (2H, s, CH2), 5.29 (2H, s, CH2); 13CNMR (DMSO-d6) δ (ppm): 168.1 (CO), 152.7 (C–O), 147.6 (CN), 143.2, 139.6, 137.2, 129.4, 128.5, 127.5, 125.5, 125.0, 122.4, 121.8, 111.7, 59.6, 52.6; ESI-MS m/z: [M+ + H] 438.05.
5.1.2.6 2-(4-((Quinolin-8-yloxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(4-nitrophenyl)acetamide (QT6). Yield: 80%; anal. calc. for C20H16N6O4: C 59.40, H 3.99, N 20.78%; found: C 59.24, H 4.15, N 20.10%; FTIR νmax cm−1: 3380 (N–H), 3155 (C–H triazole ring), 2860 (C–H), 1760 (C
O), 1632 (CN); 1H NMR (DMSO-d6) δ (ppm): 9.58 (s, 1H, NH), 8.69 (dd, 1H, quinoline ring), 8.21 (1H, s, triazole ring), 8.10 (d, 1H, quinoline ring), 8.01–7.79 (m, 4H, Ph), 7.38–7.23 (m, 4H, quinoline ring), 5.48 (2H, s, CH2), 5.23 (2H, s, CH2); 13CNMR (DMSO-d6) δ (ppm): 168.1 (CO), 152.7 (C–O), 147.6 (CN), 143.2, 142.6, 139.2, 129.4, 127.5, 125.5, 125.0, 122.4, 121.8, 118.2, 111.7, 59.3, 53.0; ESI-MS m/z: [M+ + H] 405.39.
5.1.2.7 2-(4-((Quinolin-8-yloxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(3-nitrophenyl)acetamide (QT7). Yield: 80%; anal. calc. for C20H16N6O4: C 59.40, H 3.99, N 20.78%; found: C 59.55, H 4.05, N 20.06%; FTIR νmax cm−1: 3285 (N–H), 3152 (C–H triazole ring), 2865 (C–H), 1762 (C
O), 1630 (CN); 1H NMR (DMSO-d6) δ (ppm): 9.56 (s, 1H, NH), 8.68 (dd, 1H, quinoline ring), 8.25 (1H, s, triazole ring), 8.12 (d, 1H, quinoline ring), 8.02–7.76 (m, 4H, Ph), 7.42–7.24 (m, 4H, quinoline ring), 5.50 (2H, s, CH2), 5.22 (2H, s, CH2); 13CNMR (DMSO-d6) δ (ppm): 168.0 (CO), 153.3 (C–O), 148.6 (CN), 147.5, 143.1, 139.2, 138.8, 130.4, 129.5, 127.5, 126.1, 125.7, 125.0, 122.7, 121.6, 117.8, 114.5, 111.7, 59.3, 53.0; ESI-MS m/z: [M+ + H] 405.39.
5.1.2.8 2-(4-((Quinolin-8-yloxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(4-trifluoromethylphenyl) acetamide (QT8). Yield: 78%; anal. calc. for C21H16F3N5O2: C 59.02, H 3.77, N 16.39%; found: C 59.14, H 3.85, N 16.50%; FTIR νmax cm−1: 3280 (N–H), 3155 (C–H triazole ring), 2865 (C–H), 1765 (C
O), 1632 (CN); 1H NMR (DMSO-d6) δ (ppm): 9.56 (s, 1H, NH), 8.66 (dd, 1H, quinoline ring), 8.21 (1H, s, triazole ring), 8.10 (d, 1H, quinoline ring), 7.76–7.47 (m, 4H, Ph), 7.38–7.14 (m, 4H, quinoline ring), 5.49 (2H, s, CH2), 5.23 (2H, s, CH2); 13CNMR (DMSO-d6) δ (ppm): 168.1 (CO), 152.7 (C–O), 147.6 (CN), 143.1, 139.6, 138.6, 129.3, 127.5, 126.5, 125.7, 125.0, 122.7, 121.8, 120.3, 111.7, 59.3, 52.9; ESI-MS m/z: [M+ + H] 428.13.
5.1.2.9 2-(4-((Quinolin-8-yloxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(4-methoxyphenyl)acetamide (QT9). Yield: 80%; anal. calc. for C21H19N5O3: C 64.77, H 4.92, N 17.98%; found: C 64.55, H 4.75, N 17.75%; FTIR νmax cm−1: 3285 (N–H), 3152 (C–H triazole ring), 2860 (C–H), 1762 (C
O), 1628 (CN); 1H NMR (DMSO-d6) δ (ppm): 9.51 (s, 1H, NH), 8.69 (dd, 1H, quinoline ring), 8.21 (1H, s, triazole ring), 8.11 (d, 1H, quinoline ring), 7.51–7.42 (m, 4H, quinoline ring), 7.36–6.96 (m, 4H, Ph), 5.46 (2H, s, CH2), 5.29 (2H, s, CH2) 3.80 (3H, s, OCH3); 13CNMR (DMSO-d6) δ (ppm): 168.2 (CO), 156.4 (C–O), 152.5 (C–O), 147.6 (CN), 143.1, 139.6, 133.5, 129.3, 127.5, 125.7, 125.1, 123.2, 122.7, 121.8, 120.1, 112.2, 59.3, 52.9, 50.2; ESI-MS m/z: [M+ + H] 390.15.
5.1.2.10 2-(4-((Quinolin-8-yloxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(3,4-dichlorophenyl)acetamide (QT10). Yield: 85%; anal. calc. for C20H15Cl2N5O2: C 56.09, H 3.53, N 16.35%; found: C 56.24, H 3.59, N 16.50%; FTIR νmax cm−1: 3284 (N–H), 3155 (C–H triazole ring), 2860 (C–H), 1760 (C
O), 1630 (CN); 1H NMR (DMSO-d6) δ (ppm): 9.50 (s, 1H, NH), 8.68 (dd, 1H, quinoline ring), 8.21 (1H, s, triazole ring), 8.10 (d, 1H, quinoline ring), 7.50–7.42 (m, 4H, quinoline ring), 7.38–7.16 (m, 3H, Ph), 5.46 (2H, s, CH2), 5.29 (2H, s, CH2); 13CNMR (DMSO-d6) δ (ppm): 168.0 (CO), 152.7 (C–O), 147.6 (CN), 143.1, 139.6, 136.4, 132.5, 129.7, 127.5, 125.7, 125.3, 122.7, 122.2, 121.8, 120.0, 111.7, 59.3, 53.0; ESI-MS m/z: [M++H] 429.07.
5.2 Anti-Candida activity evaluation
| Biofilm inhibition rate = (OD control − OD sample/OD control) × 100 |
The crystal violet assay was further confirmed using confocal laser scanning microscopy.9 In a parallel experiment, the compound-treated (MIC, MFC and 2× MFC) and untreated 24 h mature biofilm was stained with fluorescent dyes FUN-1 and ConA-Alexa Fluor 488 conjugate (45 minutes in dark). The slides were visualised using a confocal microscope CLSM-780 and Airyscan. The images were captured in a multitrack mode (FUN-1, Ex/Em = 543/560 nm; ConA, Ex/Em = 488/505 nm). The ConA dye has the ability to conjugate with the biofilm matrix and fluoresces green whereas FUN-1 stains the live cells and gives red fluorescence.
The MMP was measured with the help of the JC-10 kit (Abcam, UK) and the method was adopted from the user's manual. As suggested, ninety microliters of both treated and untreated C. auris cells were mixed with fifty microliters of JC-10 dye in black-walled and transparent-bottom microtiter plates (Thermo Fisher Scientific, Germany) and kept at room temperature in the dark for one hour. Afterwards, all the wells of the microtiter plate were supplemented with fifty microliters of buffer B and the plate was spun at 800 rpm for 2 minutes. The result was captured using a microplate reader (Ex/Em = 490/530 nm, green fluorescence (GF); 540/590 nm, red fluorescence (RF)). The MMP was calculated as the ratio of the mean value of RF to GF and a decreased ratio compared to the untreated control signified the depolarization of MMP.22
For examining the leakage of cytochrome c oxidase from mitochondria to the cytoplasm, all the samples were washed twice with PBS and homogenized in buffer X (EDTA, 1 mM; PMSF, 1 mM; tris base 50 mM; pH = 7.5). Then the cells were spun (4000 rpm and 10 minutes), and the supernatant was secured and again subjected to centrifugation for 45 minutes at 15000 rpm. The clear supernatant was aliquoted in clean 1.5 mL microcentrifuge tubes and was used to evaluate the concentration of cytochrome c in the cytoplasm. Parallelly, the pellet was resuspended in buffer Y (EDTA, 2 mM; tris base 50 mM; pH = 5.0) and was used to quantify the concentration of cytochrome c in the mitochondria. Before analysis, ascorbic acid (500 mg mL−1) was added to the samples to reduce the amount of cytochrome c present, and the relative amount of the reduced cytochrome c was analyzed at 550 nm.44
The transfer of phosphatidylserine to the outer portion of the cell membrane denotes the onset of apoptosis and this phenomenon can be traced by using Annexin V-FITC Apoptosis Detection Kit I (BD, USA) [9]. Briefly, the treated cells (0.5–2.5 × 103 cells per mL) were mixed in 1× binding buffer and mixed with 5 μL each of propidium iodide (PI) and Annexin V-FITC. Thereafter, samples were kept in a dark room for 15 min. Binding buffer (400 μL) was again added to each sample followed by analysis through a Flow cytometer (BD, USA) and FlowJo_V10 software was used for analyzing the results. The obtained cell population was segregated into 4 quadrants (Q), Q 1: necrosis (Annexin V−/PI+); Q2: late apoptosis (Annexin V+/PI+); Q3: early apoptosis (Annexin V+/PI−), Q4: viable cells (Annexin V−/PI−).
| %Hemolysis = [(ODTest − ODnegative) ÷ (ODpositive − ODnegative)] × 100 |
5.3 Data management
All the above-mentioned experiments contained at least two technical replicates and were repeated at least thrice for statistical power. GraphPad Prism was used to generate graphs and for statistical analysis.5.4 Pharmacokinetic studies/ADMET profile
Pharmacokinetic and ADMET properties were assessed using pkCSM, an online resource provided by Cambridge University to evaluate the drug-like potential of the freshly prepared compounds.35 Additionally, the MedChemDesigner™ software version 3.0 (ref. 35) and SwissADME,36 an online tool from Swiss Institute of Bioinformatics was used to validate the results.Data availability
The data supporting the findings of this study will be made available upon request. Requests for data should be directed to the corresponding author.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was funded by the University of Jeddah, Jeddah, Saudi Arabia, under grant No. (UJ-23-DR-170). Therefore, the authors thank the University of Jeddah for its technical and financial support.References
- K. Satoh, K. Makimura, Y. Hasumi, Y. Nishiyama, K. Uchida and H. Yamaguchi, Candida auris sp. nov., a novel ascomycetous yeast isolated from the external ear canal of an inpatient in a Japanese hospital, Microbiol. Immunol., 2009, 53(1), 41–44, DOI:10.1111/j.1348-0421.2008.00083.x.
- R. Sabino, C. Veríssimo, Á. A. Pereira and F. Antunes, Candida auris, an agent of hospital-associated outbreaks: which challenging issues do we need to have in mind?, Microorganisms, 2020, 8(2), 181, DOI:10.3390/microorganisms8020181.
- V. Srivastava, R. K. Singla and A. K. Dubey, Emerging virulence, drug resistance and future anti-fungal drugs for candida pathogens, Curr. Top. Med. Chem., 2018, 18(9), 759–778, DOI:10.2174/1568026618666180528121707.
- S. C. Deorukhkar, S. Saini and S. Mathew, Non-albicans Candida infection: an emerging threat, Interdiscip. Perspect. Infect. Dis., 2014, 2014, 615958, DOI:10.1155/2014/615958.
- E. S. Spivak and K. E. Hanson, Candida auris: an emerging fungal pathogen, J. Clin. Microbiol., 2018, 56(2), e015888, DOI:10.1128/JCM.01588-17.
- A. M. Day, M. M. McNiff, A. da Silva Dantas, N. A. R. Gow and J. Quinn, Hog1 regulates stress tolerance and virulence in the emerging fungal pathogen Candida auris, mSphere, 2018, 3(5), e00506–e00518, DOI:10.1128/mSphere.00506-18.
- S. Chatterjee, S. V. Alampalli, R. K. Nageshan, S. T. Chettiar, S. Joshi and U. S. Tatu, Draft genome of a commonly misdiagnosed multidrug resistant pathogen Candida auris, BMC Genomics, 2015, 16(1), 686, DOI:10.1186/s12864-015-1863-z.
- C. Sharma, N. Kumar, R. Pandey, J. F. Meis and A. Chowdhary, Whole genome sequencing of emerging multidrug resistant Candida auris isolates in India demonstrates low genetic variation, New Microbes New Infect., 2016, 13, 77–82, DOI:10.1016/j.nmni.2016.07.003.
- V. Srivastava and A. Ahmad, Abrogation of pathogenic attributes in drug resistant Candida auris strains by farnesol, PLoS One, 2020, 15(5), e0233102, DOI:10.1371/journal.pone.0233102.
- C. J. Johnson, J. M. Davis, A. Huttenlocher, J. F. Kernien and J. E. Nett, Emerging fungal pathogen Candida auris evades neutrophil attack, mBio, 2018, 9(4), e01403–e01418, DOI:10.1128/mBio.01403-18.
- M. J. Navarro-Arias, M. J. Hernández-Chávez, L. C. García-Carnero, D. G. Amezcua-Hernández, N. E. Lozoya-Pérez, E. Estrada-Mata, I. Martínez-Duncker, B. Franco and H. M. Mora-Montes, Differential recognition of Candida tropicalis, Candida guilliermondii, Candida krusei, and Candida auris by human innate immune cells, Infect. Drug Resist., 2019, 12, 783–794, DOI:10.2147/IDR.S197531.
- R. Kumar and V. Srivastava, Application of anti-fungal vaccines as a tool against emerging anti-fungal resistance, Front. Fungal Biol., 2023, 4, 1241539, DOI:10.3389/ffunb.2023.1241539.
- F. Chaabane, A. Graf, L. Jequier and A. T. Coste, Review on antifungal resistance mechanisms in the emerging pathogen Candida auris, Front. Microbiol., 2019, 10, 2788, DOI:10.3389/fmicb.2019.02788.
- L. R. Peyton, S. Gallagher and M. Hashemzadeh, Triazole antifungals: a review, Drugs Today, 2015, 51(12), 705–718, DOI:10.1358/dot.2015.51.12.2421058 . PMID: 26798851.
- D. Allen, D. Wilson, R. Drew and J. Perfect, Azole antifungals: 35 years of invasive fungal infection management, Expert Rev. Anti Infect. Ther., 2015, 13(6), 787–798, DOI:10.1586/14787210.2015.1032939.
- A. Vitiello, F. Ferrara, M. Boccellino, A. Ponzo, C. Cimmino, E. Comberiati, A. Zovi, S. Clemente and M. Sabbatucci, Antifungal drug resistance: an emergent health threat, Biomedicines, 2023, 11(4), 1063, DOI:10.3390/biomedicines11041063.
- T. Vanzolini and M. Magnani, Old and new strategies in therapy and diagnosis against fungal infections, Appl. Microbiol. Biotechnol., 2024, 108(1), 147, DOI:10.1007/s00253-023-12884-8.
- A. Marella, O. P. Tanwar, R. Saha, M. R. Ali, S. Srivastava, M. Akhter, M. Shaquiquzzaman and M. M. Alam, Quinoline: a versatile heterocyclic, Saudi Pharm. J., 2013, 21(1), 1–12, DOI:10.1016/j.jsps.2012.03.002.
- B. Suliphuldevara Matada, R. Pattanashettar and N. G. Yernale, A comprehensive review on the biological interest of quinoline and its derivatives, Bioorg. Med. Chem., 2021, 32, 115973, DOI:10.1016/j.bmc.2020.115973.
- G. A. Sandip, R. M. Suleman and S. P. Vandana, Click chemistry: 1,2,3-triazoles as pharmacophores, Chem.–Asian J., 2011, 6, 2696–2718, DOI:10.1002/asia.201100432.
- Y. W. Mohmmad, S. S. A. Majed, S. Vartika, A. Aijaz, M. A. Faisal and S. Al-B. Abdullah, Click synthesis of pyrrolidine-based 1,2,3-triazole derivatives as antifungal agents causing cell cycle arrest and apoptosis in Candida auris, Bioorg. Chem., 2023, 136, 106562, DOI:10.1016/j.bioorg.2023.106562.
- H. Alam, V. Srivastava, W. Sekgele, M. Y. Wani, A. S. Al-Bogami and J. Molepo, et al., Cellular apoptosis and cell cycle arrest as potential therapeutic targets for eugenol derivatives in Candida auris, PLoS ONE, 2023, 18(6), e0285473, DOI:10.1371/journal.pone.0285473.
- Y. W. Mohmmad, A. Aijaz, M. A. Faisal and S. Al-B. Abdullah, Modulation of key antioxidant enzymes and cell cycle arrest as a possible antifungal mode of action of cinnamaldehyde based azole derivative, Bioorg. Med. Chem. Lett., 2022, 73, 128922, DOI:10.1016/j.bmcl.2022.128922.
- S. Vartika, Y. W. Mohmmad, S. Al-B. Abdullah and A. Aijaz, Piperidine based 1,2,3-triazolylacetamide derivatives induce cell cycle arrest and apoptotic cell death in Candida auris, J. Adv. Res., 2021, 29, 121–135, DOI:10.1016/j.jare.2020.11.002.
- J. E. Moses and A. D. Moorhouse, The growing applications of click chemistry, Chem. Soc. Rev., 2007, 36(8), 1249–1262, 10.1039/B613014N.
- M. K. Shameer, B. Shubham, Y. Ce, L. Dongning, F. Zach and W. Binghe, Click chemistry and drug delivery: a bird's-eye view, Acta Pharm. Sin. B, 2023, 13, 1990–2016, DOI:10.1016/j.apsb.2022.10.015.
- T. Ni, Z. Ding, F. Xie, Y. Hao, J. Bao, J. Zhang and D. Zhang, Design, synthesis, and in vitro and in vivo antifungal activity of novel triazoles containing phenylethynyl pyrazole side chains, Molecules, 2022, 27(11), 3370 CrossRef CAS PubMed.
- F. Xie, Y. Hao, J. Bao, J. Liu, Y. Liu, R. Wang, X. Chi, X. Chai, T. Wang, S. Yu, Y. Jin, L. Yan, D. Zhang and T. Ni, Design, synthesis, and in vitro evaluation of novel antifungal triazoles containing substituted 1,2,3-triazole-methoxyl side chains, Bioorg. Chem., 2022, 129, 106216, DOI:10.1016/j.bioorg.2022.106216.
- J. Bao, Y. Hao, T. Ni, R. Wang, J. Liu, X. Chi, T. Wang, S. Yu, Y. Jin, L. Yan, X. Li, D. Zhang and F. Xie, Design, synthesis and in vitro biological studies of novel triazoles with potent and broad-spectrum antifungal activity, J. Enzyme Inhib. Med. Chem., 2023, 38(1), 2244696, DOI:10.1080/14756366.2023.2244696.
- D. Osmaniye, N. Baltacı Bozkurt, S. Levent, G. Benli Yardımcı, B. N. Sağlık, Y. Ozkay and Z. A. Kaplancıklı, Synthesis, antifungal activities, molecular docking and molecular dynamic studies of novel quinoxaline-triazole compounds, ACS Omega, 2023, 8(27), 24573–24585, DOI:10.1021/acsomega.3c02797.
- D. A. Brown, N. Yang, and S. D. Ray, Apoptosis, in Encyclopedia of Toxicology, Elsevier, 2014, pp. 287–94, doi: DOI:10.1016/B978-0-12-386454-3.00242-6.
- H. U. Simon, A. Haj-Yehia and F. Levi-Schaffer, Role of reactive oxygen species (ROS) in apoptosis induction, Apoptosis, 2000, 5, 415–418, DOI:10.1023/A:1009616228304.
- M. Huttemann, P. Pecina, M. Rainbolt, T. H. Sanderson, V. E. Kagan and L. Samavati, et al., The multiple functions of cytochrome c and their regulation in life and death decisions of the mammalian cell: from respiration to apoptosis, Mitochondrion, 2011, 11, 369–381, DOI:10.1016/j.mito.2011.01.010.
- C. Jia, J. Zhang, L. Yu, C. Wang, Y. Yang and X. Rong, et al., Antifungal activity of coumarin against Candida albicans is related to apoptosis, Front. Cell. Infect. Microbiol., 2019, 8, 445, DOI:10.3389/fcimb.2018.00445.
- D. E. V. Pires, T. L. Blundell and D. B. Ascher, pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures, J. Med. Chem., 2015, 58, 4066–4072, DOI:10.1021/acs.jmedchem.5b00104.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- N. A. Meanwell, Improving drug candidates by design: a focus on physicochemical properties as a means of improving compound disposition and safety, Chem. Res. Toxicol., 2011, 24, 1420–1456, DOI:10.1021/tx200211v.
- E. H. Kearns and L. Di, Drug-like Properties: Concepts, Structure Design and Methods, Elsevier, USA, 2008, ISBN: 978-0-12-369520-8 Search PubMed.
- M. P. Gleeson, Generation of a set of simple, interpretable ADMET rules of thumb, J. Med. Chem., 2008, 51, 817–834, DOI:10.1021/jm701122q.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717, DOI:10.1038/srep42717.
- H. T. Abdel-Mohsen, A. Abood, K. J. Flanagon, A. Meindl, M. O. Senge and H. I. El Diwani, Synthesis, crystal structure, and ADME prediction studies of novel imidazopyrimidines as antibacterial and cytotoxic agents, Arch. Pharm., 2020, 353, 1900271, DOI:10.1002/ardp.201900271.
- M. Aydin, A. Ozturk, T. Duran, U. O. Ozmen, E. Sumlu, E. B. Ayan and E. N. Korucu, In vitro antifungal and antibiofilm activities of novel sulfonyl hydrazone derivatives against Candida spp, J. Med. Mycol., 2023, 33, 101327, DOI:10.1016/j.mycmed.2022.101327.
- A. Ozturk, O. Abdulmajed and M. Aydin, Investigation of antifungal, antibiofilm and anti-filamentation activities of biocides against Candida isolates, Ann. Med. Res., 2020, 27, 2041–2046, DOI:10.5455/annalsmedres.2020.05.478.
- D. G. Yun and D. G. Lee, Silibinin triggers yeast apoptosis related to mitochondrial Ca2+ influx in Candida albicans, Int. J. Biochem. Cell Biol., 2016, 80, 1–9, DOI:10.1016/j.biocel.2016.09.008.
- I. P. Sæbø, M. Bjørås, H. Franzyk, E. Helgesen and J. A. Booth, Optimization of the hemolysis assay for the assessment of cytotoxicity, Int. J. Mol. Sci., 2023, 24(3), 2914, DOI:10.3390/ijms24032914.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03728f |
| This journal is © The Royal Society of Chemistry 2024 |