
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of Fc region affinity of protein-based antibody-recruiting molecules on antibody-dependent cellular cytotoxicity†
Hiroshi Tagawa‡
a,
Riku Saeki‡a,
Chihaya Yamamoto‡a,
Kenta Tanitoa,
Chihiro Tanakab,
Shoki Munekawaa,
Teruki Niiac,
Akihiro Kishimuraacde,
Hiroshi Murakami bf,
Takeshi Mori*acd and
Yoshiki Katayama*acd
bf,
Takeshi Mori*acd and
Yoshiki Katayama*acd
aGraduate School of Systems Life Sciences, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: mori.takeshi.880@m.kyushu-u.ac.jp; katayama.yoshiki.958@m.kyushu-u.ac.jp
bDepartment of Biomolecular Engineering, Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan
cDepartment of Applied Chemistry, Faculty of Engineering, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
dCenter for Future Chemistry, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
eInternational Research Center for Molecular Systems, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
fInstitute of Nano-Life-Systems, Institutes of Innovation for Future Society, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan
First published on 22nd July 2024
Abstract
Previously, we reported anticancer molecules, Fc-binding antibody-recruiting molecules (Fc-ARMs), which crosslink proteins on cancer cells with endogenous immunoglobulin Gs (IgGs) via their Fc region. The mobilized IgGs on cancer cells can accommodate natural killer cells to induce antibody-dependent cellular cytotoxicity (ADCC). Because previous Fc-ARMs utilized Fc-binding peptides, their affinity to IgGs is weak, which resulted in the limited induction capability of ADCC. Previous Fc-ARMs also unitized small molecular ligands to cancer cells, which limited their universal applicability to any cancer cells. A recent study reported that protein-based Fc-ARMs might overcome the issues associated with non-proteinous Fc-ARMs. Here, we examined the universality of a protein-based Fc-ARM by replacing its tumor-binding domain with a human epidermal growth factor receptor 2 (HER2)-specific affibody (ZHER2:342). We also examined the requirement of its Fc-binding domain affinity. We found that the Fc-ARMs accepted an affibody as a tumor-binding domain to induce ADCC. Furthermore, the required residence time of the complex between Fc-ARM and IgG was ∼102 min, which was comparable to that when monoclonal antibodies bind to their specific antigens. However, we found that the extent of ADCC induced by Fc-ARM was lower than that of conventional IgG-mediated ADCC, indicating that further enhancement of the affinity of the antibody-binding terminus and tumor-binding terminus of Fc-ARM may be needed to achieve ADCC equivalent to that of conventional IgG-mediated ADCC.
1. Introduction
Monoclonal antibody (mAb) drugs are immunoglobulin Gs (IgGs) that specifically bind to antigens on cancer cells. The Fc regions of the bound IgGs are recognized by natural killer (NK) cells via FcγRIIIa (CD16a), leading to the killing of cancer cells, termed antibody-dependent cellular cytotoxicity (ADCC).1,2 Because mAb drugs need to be expressed from mammalian cell lines and require quality control related to the carbohydrate chains on mAb that govern ADCC capability, mAb drugs are expensive to produce.3 Spiegel et al. proposed the use of antibody-recruiting molecules (ARMs) to overcome the difficulty of using mAb.4–7 ARMs are non-proteinous bispecific molecules that mobilize endogenous IgGs on cancer cells via cancer antigens to induce ADCC.8 ARMs can bind to endogenous IgGs in the blood, which results in their extended blood circulation time allowing them to reach target cancer cells efficiently.8 ARMs contain a tumor-binding terminus (TBT) and antibody-binding terminus (ABT) (Fig. 1A). Few endogenous IgGs recognize ABT, and therefore, a limited fraction of endogenous IgGs is utilized for ADCC.9–12 Recently, we developed a novel ARM, termed Fc-ARM, whose ABT targets the Fc region of IgGs.13,14 Because the sequence of the Fc region is constant in each IgG subtype, Fc-ARM can utilize a large fraction of IgGs (Fig. 1B and S3†).15 | ||
| Fig. 1 (A) Structure of Fc-ARM containing a target-binding terminus (TBT) and antibody-binding terminus (ABT). (B) Schematic illustration showing the activation of an NK cell by Fc-ARM via the crosslinking of FcγRIIIa and antigen. A complex model of HER2/Fc-ARM/IgG/FcγRIIIa is shown in Fig. S3.† (C) 3D structure predictions of two types of Fc-ARMs. ZHER2:342 and Z domain were used as the TBT (red) and ABT (orange), respectively. | ||
The disadvantage of ARM and Fc-ARM is the instability of ternary complexes of antigen/Fc-ARM/IgG formed on the cancer cell surface (left side of Fig. 1B) because of the usage of small molecules in the TBT and ABT.14 The instability of the ternary complex might explain the weak induction ability of ADCC. Stable binding of a mAb to a cancer cell over a long period is required to induce cell signaling via FcγRIIIa, an immunoreceptor tyrosine-based activation motif receptor (right side of Fig. 1B).16 Thus, the overall stability of the ternary complex should satisfy the threshold to induce cell signaling via FcγRIIIa. In this sense, protein-based Fc-ARM should satisfy the threshold because of the more stable binding of proteins to targets than that of small molecules. Recently, Hong et al. reported the use of two tandemly connected Z domains as the ABT and an epidermal growth factor receptor (EGFR)-targeting nanobody as the TBT.17 The Z domain was a molecule that originated from Protein A of Staphylococcus aureus, with high affinity for the Fc region.18–20 Nanobody is an antibody-mimicking small protein that uses the Fv region of a camel antibody as a scaffold. A high affinity nanobody to a target antigen can be obtained from a library of mutants. Fc-ARMs designed by Hong et al.17 were small and could be expressed by Escherichia coli (E. coli). These Fc-ARMs successfully induced ADCC in vitro and suppressed tumor growth in vivo. Protein-based Fc-ARMs are expected to be universally designed for each target antigen because a TBT with a high affinity can be obtained by screening a library of antibody-mimicking small proteins.
This study was performed to extend our knowledge of protein-based Fc-ARMs by examining their universality by changing the TBT to an affibody, another platform of antibody-mimicking small proteins using protein Z as a scaffold. In this study, we used ZHER2:342, which has a high affinity to human epidermal growth factor receptor 2 (HER2).21 We also investigated the importance of having two Z domains in Fc-ARMs to induce ADCC. We designed two Fc-ARMs, A-Z and A-ZZ, which have single and double Z domains, respectively (Fig. 1C). We found that the affibody was accepted as a TBT of Fc-ARM to induce ADCC and double Z domains in Fc-ARM are required for the induction of ADCC.
2. Material and methods
2.1 Preparation of Fc-ARMs
Genes encoding the fusion proteins of each protein-based Fc-ARM were designed (Fig. S1†) and 15 bp of homologous sequence for in-fusion were inserted at the N- and C-termini of the genes, which were purchased from Integrated DNA Technologies. The purchased genes were inserted into a pET22b (+) vector. The purchased genes and vector were amplified by PCR using Takara PCR Thermal Cycler Dice (Takara). First, 0.5 μL of DpnI (New England Biolabs) was added to the PCR product of the vector and incubated at 37 °C for 1 h. Then, 5 μL of 6× DNA loading buffer dye (Thermo Scientific) was added to the vector and insert sequences. Each reaction solution was loaded onto a 1% agarose gel containing ethidium bromide (EtBr) prepared using Agarose Kanto S. Agarose electrophoresis was performed at 100 V for 30 min. The band at the target position was cut out using a gel band cutter (Nippon Genetics), and gene extraction was performed according to the Qiagen protocol. Then, the extracted DNA fragments were synthesized using In-Fusion® Snap Assembly Master Mix (Clontech) according to the manufacturer's manual. Next, 5 μL of the prepared plasmid solution was mixed with 50 μL of E. coli JM 109 competent cell (Takara) and transformed by heat shock. The transformants were then cultured overnight on LB agar medium (Kanto Chemical) containing ampicillin sodium (100 μg mL−1). The resulting colonies were selected and incubated (37 °C, 120 rpm, 14 h) in LB medium containing sodium ampicillin (100 μg mL−1). The culture medium was centrifuged at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 1 min, and the bacteria were collected and used for plasmid extraction using plasmid buffer (Qiagen) according to the manufacturer's protocol. Sequence analysis of the obtained plasmid DNA was performed by Eurofins Genomics (Tokyo, Japan), and the plasmid was confirmed to have the sequence of interest. Each Fc-ARM was prepared using an E. coli expression system. Prepared plasmids were added to BL21(DE3) competent E. coli (New England Biolabs), and transformation was performed by heat shock (42 °C, 30 s). The E. coli were then cultured on LB agar medium containing sodium ampicillin (100 μg mL−1) at 37 °C overnight. One of the resulting colonies was selected and incubated (37 °C, 120 rpm, 6 h) in 5 mL of LB medium containing sodium ampicillin (100 μg mL−1). The whole solution was added to 1 L of LB medium containing 100 μg mL−1 sodium ampicillin and incubated (37 °C, 120 rpm) until the OD value reached 0.6. Isopropyl β-D-thiogalactopyranoside was then added at a final concentration of 0.5 mM and the cultures were incubated (17 °C, 120 rpm, overnight). Bacteria expressing the protein of interest were then collected by centrifugation (4 °C, 2000×g, 20 min), suspended in 25 mL of PBS, and ultrasonicated. Centrifugation (4 °C, 8000×g, 20 min) was then performed and the supernatant containing the protein of interest was collected, filtered through a 0.22 μm filter, and purified by a chromatography system for proteins (BioLogic Duo Flow 10F QD system; Bio-Rad) using a His-Tag column (His-Trap FF crude, 5 mL; Cytiva). The purification was performed according to the protocol in the user manual of the column. After chromatography, fractions with absorbance at 280 nm were collected, and the molecular weight of the proteins was confirmed by SDS-PAGE. The fractions containing the protein of interest were then concentrated by ultrafiltration, the solvent replaced with 1 × PBS using PD-10 desalting columns packed with Sephadex G-25 resin (Cytiva), and the concentration determined by a UV-vis spectrophotometer (Denovix Inc., USA).
000 rpm for 1 min, and the bacteria were collected and used for plasmid extraction using plasmid buffer (Qiagen) according to the manufacturer's protocol. Sequence analysis of the obtained plasmid DNA was performed by Eurofins Genomics (Tokyo, Japan), and the plasmid was confirmed to have the sequence of interest. Each Fc-ARM was prepared using an E. coli expression system. Prepared plasmids were added to BL21(DE3) competent E. coli (New England Biolabs), and transformation was performed by heat shock (42 °C, 30 s). The E. coli were then cultured on LB agar medium containing sodium ampicillin (100 μg mL−1) at 37 °C overnight. One of the resulting colonies was selected and incubated (37 °C, 120 rpm, 6 h) in 5 mL of LB medium containing sodium ampicillin (100 μg mL−1). The whole solution was added to 1 L of LB medium containing 100 μg mL−1 sodium ampicillin and incubated (37 °C, 120 rpm) until the OD value reached 0.6. Isopropyl β-D-thiogalactopyranoside was then added at a final concentration of 0.5 mM and the cultures were incubated (17 °C, 120 rpm, overnight). Bacteria expressing the protein of interest were then collected by centrifugation (4 °C, 2000×g, 20 min), suspended in 25 mL of PBS, and ultrasonicated. Centrifugation (4 °C, 8000×g, 20 min) was then performed and the supernatant containing the protein of interest was collected, filtered through a 0.22 μm filter, and purified by a chromatography system for proteins (BioLogic Duo Flow 10F QD system; Bio-Rad) using a His-Tag column (His-Trap FF crude, 5 mL; Cytiva). The purification was performed according to the protocol in the user manual of the column. After chromatography, fractions with absorbance at 280 nm were collected, and the molecular weight of the proteins was confirmed by SDS-PAGE. The fractions containing the protein of interest were then concentrated by ultrafiltration, the solvent replaced with 1 × PBS using PD-10 desalting columns packed with Sephadex G-25 resin (Cytiva), and the concentration determined by a UV-vis spectrophotometer (Denovix Inc., USA).
2.2 Cell culture
SK-BR-3 human breast cancer cells and MDA-MB-231 human breast cancer cells were purchased from the American Type Culture Collection (ATCC). KHYG-1/CD16a-158V cells were kindly provided by Dr Y. Mishima (Japanese Foundation for Cancer Research). SK-BR-3 cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 (Nacalai Tesque). MDA-MB-231 cells were cultured in Dulbecco's Modified Eagle Medium (DMEM)-high (Nacalai Tesque). KHYG-1/CD16a-158V cells were cultured in RPMI-1640 containing 10 ng mL−1 recombinant human IL-2 (PeproTech). All the media were supplemented with 10% heat-inactivated fetal bovine serum (Nichirei Bioscience), and 1% Antibiotic-Antimycotic Mixed Stock Solution (Nacalai Tesque). Cells were cultured in a humidified atmosphere containing 5% CO2 and 95% air at 37 °C. The cell lines were checked for mycoplasma contamination using the MycoAlert mycoplasma detection kit (Lonza).2.3 Antibodies
The following antibodies were used: anti-CD20 (Ofatumumab, Novartis), anti-HER2 (Trastuzumab, Chugai), and fluorescein-labeled IgG1 from human serum (Sigma-Aldrich).2.4 Biolayer interferometry
Affinity measurements were performed against biotinylated human IgG1 Fc protein (IG1-H8213, ACROBiosystems), biotinylated human HER2/ErbB2 protein (HE2-H82 × 102, ACROBiosystems), and biotinylated human EGFR protein (EGR-H82 × 103, ACROBiosystems) immobilized on Octet® SA Biosensor (18-5019, Sartorius), using an Octet RED96 System (ForteBio), following the manufacturer's instructions. The buffer of each sample for biolayer interferometry was changed to buffer D (50 mM HEPES-KOH pH 7.5, 300 mM NaCl, 0.05% (v/v) Tween 20, and 0.1% (w/v) PEG6000) using Bio-Gel P-6 Gel (1504134, Bio-Rad) before the affinity measurement. The binding assay was performed at 30 °C in buffer D. The binding assay steps were as follows: equilibration for 60 s, association for 600 s, and dissociation for 600 s. Analysis was performed using 1:1 global fitting in ForteBio Data Analysis software v. 10.0.
2.5 Fluorescent microscopy
SK-BR-3 or MDA-MB-231 cells were seeded at 5 × 104 cells per well in folate-free RPMI-1640 medium onto a 96-well glass bottom microplate (Greiner Bio-One) and incubated for 24 h. The cells were washed twice with 100 μL of PBS (−), and then incubated with Fc-ARM (100 nM) and fluorescein-labeled hIgG (IgG-Fluor) (1000 nM) in RPMI-1640 medium containing 1% Super Low IgG FBS for 30 min at 4 °C. After washing, cells were stained with Hoechst 33342 (Life Technologies) and analyzed by a BZ-8000 fluorescent microscope (Keyence, Osaka, Japan).
2.6 ADCC assay
SK-BR-3 cells were washed with measurement medium (RPMI-1640 medium containing 1% Super Low IgG FBS (Cytiva)). After cell counting, SK-BR-3 cells were re-suspended in RPMI-1640 medium containing 1% Super Low IgG FBS and diluted to 1 × 105 cells per mL. This suspension was seeded into 96-well U-bottom plates (Greiner Bio-One) at 5 × 103 cells per well (50 μL per well). Then, 25 μL per well of Fc-ARM was added and incubated for 10 min. Next, 25 μL per well of measurement medium containing anti-HER2 antibody or anti-CD20 antibody was added, and incubated for 10 min. Subsequently, KHYG-1/CD16a-158V was added (100 μL per well at the indicated effector/target ratio), and the plates were centrifuged (200×g, 5 min). After incubation at 37 °C under 5% CO2 for 16 h, the plates were centrifuged and 100 μL of the supernatant was transferred to a new 96-well plate (Thermo Fisher Scientific). ADCC was evaluated using Cytotoxicity LDH Assay Kit-WST (Dojindo) according to the manufacturer's instructions. The absorbance at 490 nm was measured by Infinite® 200 PRO M Plex (TECAN). The final concentration of each sample and control is shown below.2.7 Sample
Fc-ARMs (A-Z, A-ZZ): 10 nM, anti-CD20 mAb: 100 nM, anti-HER2 mAb: 10 nM.Cytotoxicity (%) was calculated using the following formula:
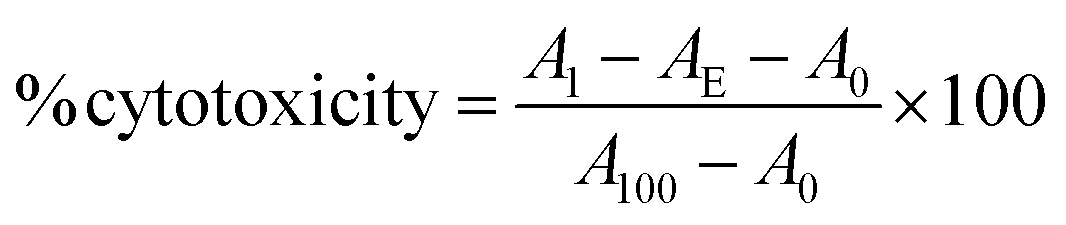
3. Results and discussion
We designed two Fc-ARMs with single and double Z domains (Z or ZZ) as the ABT and HER2-targeting affibody21 as the TBT (Fig. S1† for the sequences). After expression by E. coli, each Fc-ARM was purified using a His-tag. The molecular weights of the Fc-ARMs were confirmed by SDS-PAGE (Fig. S2†). We used biolayer interferometry (BLI) to measure the affinities of Fc-ARMs against the HER2 and Fc region of human IgG1 (hIgG1). We functionalized BLI sensor surface with each target and Fc-ARMs were allowed to flow over the surface. The results of the dissociation constant (Kd) and resident time (τ) of binding of the TBT and ABT of Fc-ARMs are summarized in Table 1. The activation of NK cells is controlled by the residence time of antibody binding to antigens. Usually, the residence time of mAbs to antigens is in the range of 101–102 min.22,23 Although the Kd of Z (A-Z) was equivalent to that of the Fc-binding cyclic peptide (peptide Fc-ARM in Table 1),12 the residence time of Z was longer than that of the cyclic peptide, indicating the slow dissociation rate of Z to IgG. The dissociation rate of protein–protein interactions is usually slower than that of protein–peptide interactions.13,24 Notably, the residence time of ZZ was longer by approximately two orders of magnitude than that of the cyclic peptide, which is comparable to the residence time of the binding of mAb to its target. Thus, the resident time of ZZ was sufficient to induce ADCC. Furthermore, the affinity of TBT (affibody) to HER2 was comparable to a previously reported value,21 and the residence time of TBT (101 min) was sufficient to induce ADCC.Next, we verified whether the ternary complex of the target antigen, Fc-ARM, and IgG could be formed on the cell surface. As shown in Fig. 2, fluorescence-labeled IgG was bound to SK-BR-3 cells (HER2-positive) in the presence of each Fc-ARM. In contrast, each Fc-ARM did not recruit IgG to MDA-MB-231 cells (HER2-negative). These results showed that the ternary complex formation is driven by the bispecific affinities of Fc-ARMs to the target antigen and IgG.
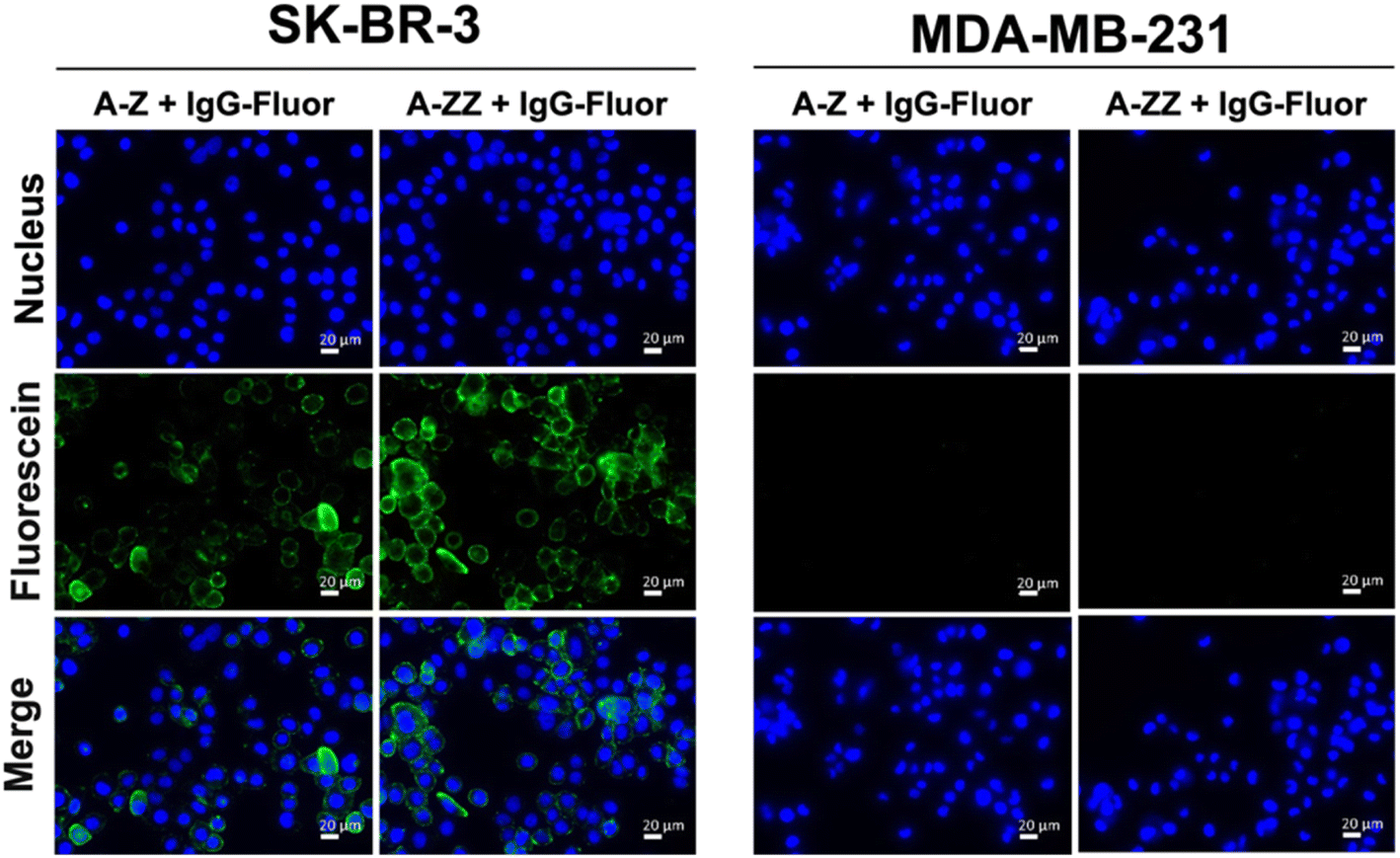 | ||
| Fig. 2 Recruitment of fluorescence-labeled IgG (IgG-Fluor) via A-Z or A-ZZ to SK-BR-3 and MDA-MB-231 cells, which are HER2 positive and negative, respectively. Cells were seeded and incubated overnight. Fluorescence-labeled IgG (1.0 μM) and each Fc-ARM (100 nM) were added. Nuclei were stained with Hoechst 33342 (blue). Scale bar = 20 μm. | ||
We conducted a quantitative evaluation of the ternary complex formation on SK-BR-3 cells. As shown in Fig. 3A and B, 10 nM of A-Z and A-ZZ achieved the maximum fluorescent intensity in the presence of 1 μM fluorescence-labeled IgG. When the Fc-ARM concentration was fixed at 10 nM, the recruited amount of IgG was saturated at 100 nM (Fig. 3C and D). Based on these results, ADCC was induced by 10 nM of Fc-ARMs and 100 nM of IgG.
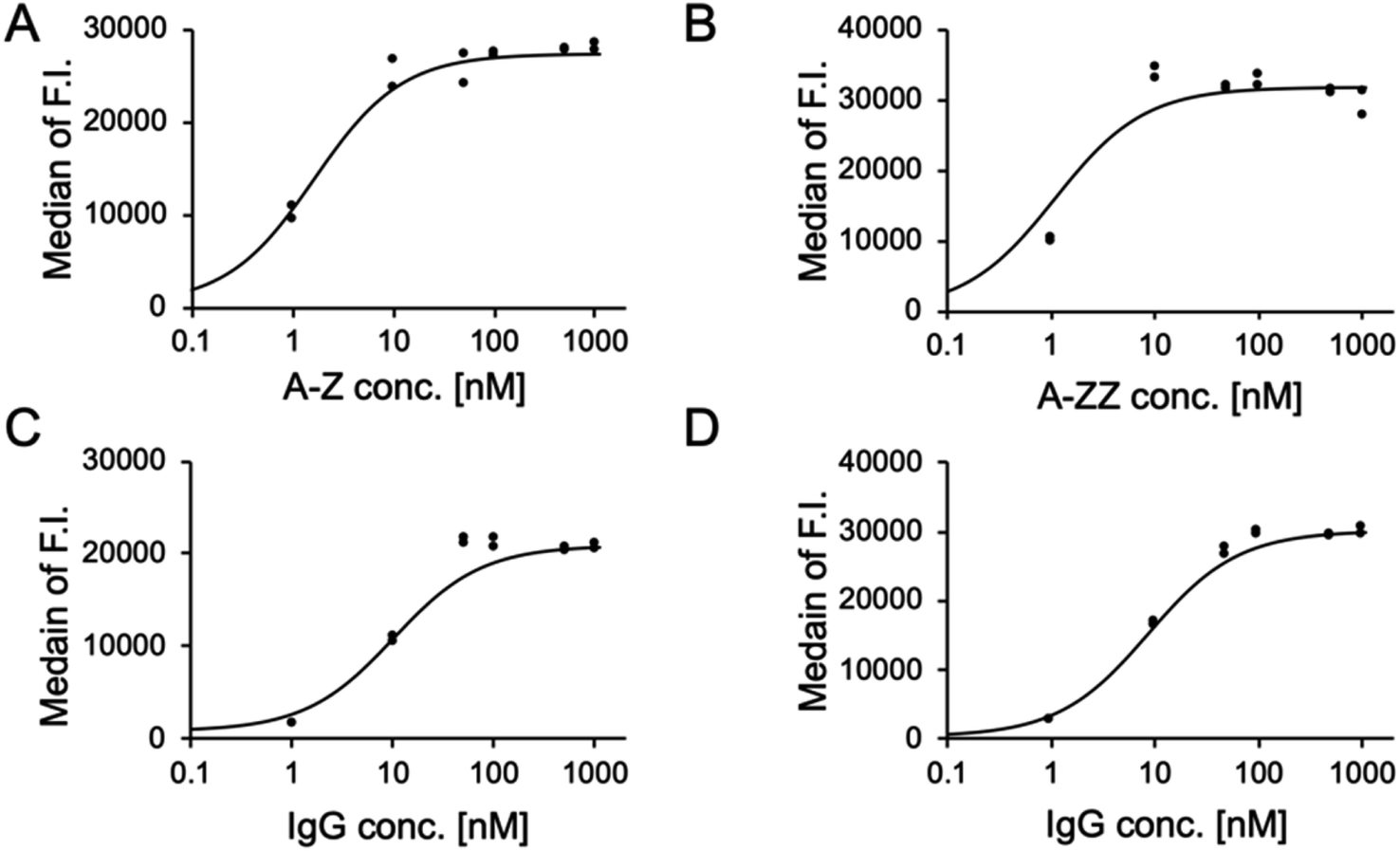 | ||
| Fig. 3 The effect of the concentration of Fc-ARMs and fluorescence-labeled IgG on ternary complex formation on SK-BR-3. (A) and (B) SK-BR-3 cells were treated with increasing concentrations of Fc-ARM with an excess amount of fluorescence-labeled IgG (1.0 μM). (C) and (D) SK-BR-3 cells were treated with increasing concentrations of IgG with Fc-ARM at a concentration of 10 nM. The y-axis indicates the median fluorescence intensity and the x-axis indicates the concentration of each sample. Two experimental repeats were performed. | ||
ADCC activity was evaluated using Fc-ARM and an approved mAb drug (ofatumumab, anti-CD20 human IgG1). Ofatumumab exhibits ADCC-inducing ability25 and does not bind to SK-BR-3 because it is a CD20-negative cell line. An anti-HER2 mAb (Herceptin), used as a positive control, was previously shown to induce ADCC in SK-BR-3 cells.26 SK-BR-3 cells were co-cultured with human NK cells (KHYG-1/CD16a-158V) to induce ADCC.27 Importantly, the binding sites of the Z domain and Fcγ receptor against the Fc region of the antibody do not overlap.28,29 A complex model of HER2/Fc-ARM/IgG/FcγRIIIa showed that this complex can be formed on the interface between cancer and NK cells without steric hindrance (Fig. S3†).
As shown in Fig. 4, anti-HER2 induced ADCC, and its extent increased with NK cell/cancer cell ratio (E/T ratio). However, anti-CD20 did not induce ADCC because of the absence of CD20 on SK-BR-3 cells. In contrast, in the mixture of Fc-ARM and anti-CD20, ADCC was observed for A-ZZ and not for A-Z. This result reflected the difference in the affinity of both Fc-ARMs to the antibody.
 | ||
| Fig. 4 ADCC induced by the combination of Fc-ARM and non-target IgG. SK-BR-3 cells were treated with Fc-ARM (A) A-Z and (B) A-ZZ, and IgG. Fc-ARM (10 nM) was added to the cells and incubated for 30 min. After unbound molecules were washed away, IgG (100 nM) was added. The y-axis indicates the cellular cytotoxicity ratio and the x-axis indicates the ratio of the number of NK cells to cancer cells. | ||
ADCC induced by the combination of A-ZZ and anti-CD20 was weaker than that induced by Herceptin. One of the reasons for this might be that the overall stability of the ternary complex was lower than that of the binary complex of Herceptin/HER2 (residence time of Herceptin/HER2 was 250 min).30 Of note, the Fc region is a dimer, but each Z domain of ZZ cannot bind simultaneously to the Fc region because of the short spacer length between each Z domain (Fig. S4†). Thus, elongating the spacer length of ZZ to allow simultaneous binding may extend its residence time to induce more effective ADCC.
Although the protein-based Fc-ARMs examined here induced inferior ADCC than the conventional IgG-mediated ADCC, the Fc-ARMs have advantages over mAb drugs including their ease of preparation (expression by E. coli and no saccharide chain) and smaller weight dose. Fc-ARMs were reported to have extended blood retention probably via an FcRn-mediated mechanism.13,17 Protein-based biologics generally suffer from the induction of anti-drug antibodies that neutralize their effects. We have not yet examined the induction of anti-drug antibodies for Fc-ARMs but it was previously reported that the antigenicity of antibody-mimicking small proteins such as nanobodies and affibodies was low.31,32
4. Conclusion
Here, we designed protein-based Fc-ARMs composed of a HER2-targeting affibody and Fc-binding Z or ZZ domain. We observed that ZZ had a much longer residence time (∼100 min) to Fc than that of Z, which was a critical difference in the induction of ADCC. Because we also showed that TBT was exchangeable, the protein-based Fc-ARM is a universal molecular design, in which a small protein platform specifically designed for targets can be installed. ADCC induced by Fc-ARM was inferior to Herceptin-mediated ADCC, indicating that further enhancement of the affinity of the ABT and TBT of Fc-ARM will be needed to induce ADCC equivalent to that of conventional IgG-mediated ADCC.Data availability
Raw data were generated at Kyushu University. Derived data supporting the findings of this study are available from the corresponding author T. Mori on request.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by JSPS KAKENHI (grant number: 21H02061) and a research grant from the Canon Foundation.References
- H. Mitoma, T. Horiuchi, H. Tsukamoto, Y. Tamimoto, Y. Kimoto, A. Uchino, K. To, S. Harashima, N. Hatta and M. Harada, Mechanisms for cytotoxic effects of anti-tumor necrosis factor agents on transmembrane tumor necrosis factor alpha-expressing cells: comparison among infliximab, etanercept, and adalimumab, Arthritis Rheum., 2008, 58, 1248–1257 CrossRef CAS PubMed.
- R. Vazquez-Lombardi, C. Loetsch, D. Zinkl, J. Jackson, P. Schofield, E. K. Deenick, C. King, T. G. Phan, K. E. Webster, J. Sprent and D. Christ, Potent antitumour activity of interleukin-2-Fc fusion proteins requires Fc-mediated depletion of regulatory T-cells, Nat. Commun., 2017, 8, 15373 CrossRef CAS PubMed.
- C. A. Challener, Tackling the challenge of HOS determination, BioPharm Int., 2014, 27, 20–24 Search PubMed.
- P. J. McEnaney, C. G. Parker, A. X. Zhang and D. A. Spiegel, Antibody-recruiting molecules: an emerging paradigm for engaging immune function in treating human disease, ACS Chem. Biol., 2012, 7, 1139–1151 CrossRef CAS.
- R. P. Murelli, A. X. Zhang, J. Michel, W. L. Jorgensen and D. A. Spiegel, Chemical Control over Immune Recognition: A Class of Antibody-Recruiting Small Molecules That Target Prostate Cancer, J. Am. Chem. Soc., 2019, 131, 17090–17092 CrossRef.
- A. Dubrovska, C. Kim, J. Elliott, W. Shen, T. H. Kuo, D. I. Koo, C. Li, T. Tuntland, J. Chang, T. Groessl, X. Wu, V. Gorney, T. Ramirez-Montagut, D. A. Spiegel, C. Y. Cho and P. G. Schultz, A Chemically Induced Vaccine Strategy for Prostate Cancer, ACS Chem. Biol., 2011, 6, 1223–1231 CrossRef CAS PubMed.
- C. G. Parker, R. A. Domaoal, K. S. Anderson and D. A. Spiegel, An antibody-recruiting small molecule that targets HIV gp120, J. Am. Chem. Soc., 2009, 131, 16392–16394 CrossRef CAS.
- J. T. Sockolosky, S. Kivimäe and F. C. Szoka, Fusion of a short peptide that binds immunoglobulin G to a recombinant protein substantially increases its plasma half-life in mice, PLoS One, 2014, 9, e102566 CrossRef.
- U. Galili, E. A. Rachmilewitz, A. Peleg and I. Flechner, A unique natural human IgG antibody with anti-alpha-galactosyl specificity, J. Exp. Res., 1984, 160, 1519–1531 CrossRef CAS.
- W. Parker, D. Bruno, Z. E. Holzknecht and J. L. Platt, Characterization and affinity isolation of xenoreactive human natural antibodies, J. Immunol., 1994, 153, 3791–3803 CrossRef CAS.
- U. Galili, The α-gal epitope ant the anti-Gal antibody in xenotransplantation and in cancer immunotherapy, Immunol. Cell Biol., 2005, 83, 674–686 CrossRef CAS.
- E. Ortega, M. Kostovetzky and C. Larralde, Natural DNP-binding immunoglobulins and antibody multispecificity, Mol. Immunol., 1984, 21, 883–888 CrossRef CAS PubMed.
- K. Sasaki, M. Harada, Y. Miyashita, H. Tagawa, A. Kishimura, T. Mori and Y. Katayama, Fc-binding antibody-recruiting molecules exploit endogenous antibodies for anti-tumor immune responses, Chem. Sci., 2020, 11, 3208–3214 RSC.
- K. Sasaki, M. Harada, T. Yoshikawa, H. Tagawa, Y. Harada, Y. Yonemitsu, T. Ryujin, A. Kishimura, T. Mori and Y. Katayama, Fc-Binding Antibody-Recruiting Molecules Targeting Prostate-Specific Membrane Antigen: Defucosylation of Antibody for Efficacy Improvement, ChemBioChem, 2021, 22, 496–500 CrossRef CAS.
- Y. Jung, H. J. Kang, J. M. Lee, S. O. Jung, W. S. Yun, S. J. Chung and B. H. Chung, Controlled antibody immobilization onto immunoanalytical platforms by synthetic peptide, Anal. Biochem., 2008, 374, 99–105 CrossRef CAS PubMed.
- L. B. Ivashkiv, How ITAMs inhibit signaling, Sci. Signal., 2011, 4, pe20 Search PubMed.
- H. Hong, C. Li, L. Gong, J. Wang, D. Li, J. Shi, Z. Zhou, Z. Huang and Z. Wu, Universal endogenous antibody recruiting nanobodies capable of triggering immune effectors for targeted cancer immunotherapy, Chem. Sci., 2021, 12, 4623–4630 RSC.
- B. Nilsson, T. Moks, B. Jansson, L. Abrahmsén, A. Elmblad, E. Holmgren, C. Henrichson, T. A. Jones and M. Uhlén, A synthetic IgG-binding domain based on staphylococcal protein A, Protein Eng., 1987, 1, 107–113 CrossRef CAS.
- K. Nord, E. Gunneriusson, J. Ringdahl, S. Ståhl, M. Uhlén and P. A. Nygren, Binding proteins selected from combinatorial libraries of an alpha-helical bacterial receptor domain, Nat. Biotechnol., 1997, 15, 772–777 CrossRef CAS PubMed.
- P. Arora, T. G. Oas and J. K. Myers, Fast and faster: a designed variant of the B-domain of protein A folds in 3 μsec, Protein Sci., 2004, 13, 847–853 CrossRef CAS.
- A. Orlova, M. Magnusson, T. L. Eriksson, M. Nilsson, B. Larsson, I. Höidén-Guthenberg, C. Widström, J. Carlsson, V. Tolmachev, S. Ståhl and F. Y. Nilsson, Tumor Imaging Using a Picomolar Affinity HER2 Binding Affibody Molecule, Cancer Res., 2006, 66, 4339–4348 CrossRef CAS.
- X. Zhuang, Z. Wang, J. Fan, X. Bai, Y. Xu, J. J. Chou, T. Hou, S. Chen and L. Pan, Structure-guided and phage-assisted evolution of a therapeutic anti-EGFR antibody to reverse acquired resistance, Nat. Commun., 2022, 13, 4431 CrossRef CAS PubMed.
- J. Bostrom, L. Haber, P. Koenig, R. F. Kelley and G. Fuh, High affinity antigen recognition of the dual specific variants of Herceptin is entropy-driven in spite of structural plasticity, PLoS One, 2011, 6, e17887 CrossRef CAS.
- A. Norman, C. Franck, M. Christie, P. M. E. Hawkins, K. Patel, A. S. Ashhurst, A. Aggarwal, J. K. K. Low, R. Siddiquee, C. L. Ashley, M. Steain, J. A. Triccas, S. Turville, J. P. Mackay, T. Passioura and R. J. Payne, Discovery of Cyclic Peptide Ligands to the SARS-CoV-2 Spike Protein Using mRNA Display, ACS Cent. Sci., 2021, 7, 1001–1008 CrossRef CAS.
- M. J. Barth, C. Mavis, M. S. Czuczman and F. J. Hernandez-Ilizaliturri, Ofatumumab exhibits enhanced in vitro and in vivo activity compared to rituximab in preclinical models of mantle cell lymphoma, Clin. Cancer Res., 2015, 21, 4391–4397 CrossRef CAS.
- S. Boero, A. Morabito, B. Banelli, B. Cardinali, B. Dozin, G. Lunardi, P. Piccioli, S. Lastraioli, R. Carosio, S. Salvi, A. Levaggi, F. Poggio, A. D'Alonzo, M. Romani, L. Del Mastro, A. Poggi and M. P. Pistillo, Analysis of in vitro ADCC and clinical response to trastuzumab: possible relevance of FcγRIIIA/FcγRIIA gene polymorphisms and HER-2 expression levels on breast cancer cell lines, J. Transl. Med., 2015, 13, 1–14 CrossRef.
- Y. Mishima, Y. Terui, Y. Mishima, R. Kuniyoshi, S. Matsusaka, M. Mikuniya, K. Kojima and K. Hatake, High reproducible ADCC analysis revealed a competitive relation between ADCC and CDC and differences between FcγRllla polymorphism, Int. Immunol., 2012, 24, 477–483 CrossRef CAS PubMed.
- M. Ultsch, A. Braisted, H. R. Maun and C. Eigenbrot, 3-2-1: Structural insights from stepwise shrinkage of a three-helix Fc-binding domain to a single helix, Protein Eng. Des. Sel., 2017, 30, 619–625 CrossRef CAS.
- F. Mimoto, S. Kadono, H. Katada, T. Igawa, T. Kamikawa and K. Hattori, Crystal structure of a novel asymmetrically engineered Fc variant with improved affinity for FcγRs, Mol. Immunol., 2014, 58, 132–138 CrossRef CAS.
- F. Troise, V. Cafaro, C. Giancola, G. D'Alessio and C. De Lorenzo, Differential Binding of Human Immunoagents and Herceptin to the ErbB2 Receptor, FEBS J., 2008, 275, 4967–4979 CrossRef CAS.
- J. Zhu, S. Kamara, D. Cen, W. Tang, M. Gu, X. Ci, J. Chen, L. Wang, S. Zhu, P. Jiang, S. Chen, X. Xue and L. Zhang, Generation of novel affibody molecules targeting the EBV LMP2A N-terminal domain with inhibiting effects on the proliferation of nasopharyngeal carcinoma cells, Cell Death Dis., 2020, 11, 213 CrossRef CAS.
- J. P. Salvador, L. Vilaplana and M. P. Marco, Nanobody: outstanding features for diagnostic and therapeutic applications, Anal. Bioanal. Chem., 2019, 411, 1703–1713 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03391d |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2024 |