
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
1,2,4-triazine derived binuclear lead(II) complexes: synthesis, spectroscopic and structural investigations and application in organic light-emitting diodes (OLEDs)†
Akbar Arkaka,
Moayad Hossaini Sadr a,
Mohammad Janghourib,
Farzin Marandi*c and
Daniel Fuhrmannd
a,
Mohammad Janghourib,
Farzin Marandi*c and
Daniel Fuhrmannd
aDepartment of Chemistry, Faculty of Science, Azarbaijan Shahid Madani University, Tabriz, Iran
bFaculty of Industrial Technologies, Urmia University of Technology, Band Road, Urmia, Iran
cInorganic Chemistry Department, Faculty of Chemistry, Urmia University, 57561-51818, Urmia, Iran. E-mail: f.marandi@gmail.com
dInstitut für Anorganische Chemie, Universität Leipzig, Johannisallee 29, 04103 Leipzig, Germany
First published on 12th July 2024
Abstract
Two novel binuclear complexes of Pb(II) were synthesized by reacting a 3-(2-pyridyl)-5-(4-methoxyphenyl)-1,2,4-triazine (PMPT) ligand with different anionic co-ligands (1: bromide, 2: acetate and isothiocyanate) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of PMPT ligands to lead(II) salts. The complexes, [Pb2(μ-PMPT)2Br4] (1) and [Pb2(μ-PMPT)2((μ-CH3COO)2(NCS)2] (2), were characterized using various physicochemical techniques such as CHN analysis, FT-IR spectroscopy, and 1H NMR spectroscopy. Additionally, their structures were determined using single-crystal X-ray diffraction. Based on the obtained structural parameters, complex 1 exhibited a PbN3Br2 environment, while complex 2 displayed a PbN4O3 environment, with holodirected and hemidirected coordination spheres, respectively. Within the crystal network of the complexes, there were interactions involving C–H⋯X (X: O, S, N) as well as π–π stacking. The Pb(II) complexes were further investigated for their potential use as the emitting layer in organic light-emitting devices (OLEDs). The current–voltage and luminescence-voltage characteristics, as well as the electroluminescence (EL) properties of the complexes, were studied.
:1 molar ratio of PMPT ligands to lead(II) salts. The complexes, [Pb2(μ-PMPT)2Br4] (1) and [Pb2(μ-PMPT)2((μ-CH3COO)2(NCS)2] (2), were characterized using various physicochemical techniques such as CHN analysis, FT-IR spectroscopy, and 1H NMR spectroscopy. Additionally, their structures were determined using single-crystal X-ray diffraction. Based on the obtained structural parameters, complex 1 exhibited a PbN3Br2 environment, while complex 2 displayed a PbN4O3 environment, with holodirected and hemidirected coordination spheres, respectively. Within the crystal network of the complexes, there were interactions involving C–H⋯X (X: O, S, N) as well as π–π stacking. The Pb(II) complexes were further investigated for their potential use as the emitting layer in organic light-emitting devices (OLEDs). The current–voltage and luminescence-voltage characteristics, as well as the electroluminescence (EL) properties of the complexes, were studied.
1. Introduction
π-Conjugated organic molecules possess highly versatile optoelectronic properties, such as light absorption and emission, that can be customized to a great extent. These molecules are already being used commercially in various areas, such as flexible electronics, optical displays, and logic circuits.1–3 Moreover, the application of organic compounds has witnessed remarkable interdisciplinary research in the fields of physics and materials science.1–6 Triazine derivatives are a well-known category of nitrogen-containing heterocyclic compounds. They serve as a fundamental component in a diverse array of applications, including optical bleaches, plastics, textiles, pharmaceuticals, rubber production, and surface-active agents.3–5 These derivatives have found extensive use as electron transport materials in organic light-emitting diodes (OLEDs), as thermally active delayed fluorescence emitters (TADF), and as donors in bulk heterojunction solar cells.3–8 An intriguing characteristic of triazine compound derivatives is their ability to form complexes with metal ions.9The derivatives of 3-pyridyl-1,2,4-triazine have garnered attention due to their diverse coordination chemistry. For example, 3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine forms colored complexes with certain transition metal ions, offering potential applications in colorimetric analysis.10 Other derivatives, such as 2,4,6-tris[bis(pyridin-2-yl)amino]-1,3,5-triazine,11 2,6-bis(tetramethylfuryl)-1,2,4-triazin-3-yl)pyridine,12 and 5,6-diphenyl-3-(2-pyridyl)-1,2,4-triazine,13 have been utilized in the construction of metal–organic frameworks (MOFs) with varying degrees of dimensionality.
Recently, complexes of lead(II) with various ligands such as 3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine (pdpt),14,15 3,5,6-tris(2-pyridyl)-1,2,4-triazine (tpt),16,17 3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine-p,p′-disulfonate (pdpts),18 3-(2-pyridyl)-5-phenyl-1,2,4-triazine (ppt),19 and 3-(2-pyridyl)-5,6-di(2-furyl)-1,2,4-triazine (pdft)20,21 were synthesized and characterized using X-ray crystallography. The influence of factors affecting the activity of the lead(II) 6s2 lone pair in the coordination sphere was discussed. Building on this recent work, we present the synthesis, spectroscopic analysis, and structural study using experimental methods of two new binuclear lead(II) complexes with the ligand 3-(2-pyridyl)-5-(4-methoxyphenyl)-1,2,4-triazine (PMPT): [Pb2(μ-PMPT)2I4] (1) and [Pb2(μ-PMPT)2(μ-CH3COO)2(NCS)2] (2). The optical and electrical properties of these complexes will be evaluated for potential application in organic light-emitting devices (OLEDs).
2. Experimental
2.1. Materials and measurements
All starting materials and solvents were purchased from commercial sources (Merck and Sigma-Aldrich) and used directly without any purification. The PXRD patterns were measured using a X-ray diffractometer (D500 S) utilizing Cu Kα (λ = 0.15418 nm) radiation source (30–40 kV and 40–50 mA) in the range of 2θ = 4–50°. The infrared spectra in the range 4000–400 cm−1 were recorded as KBr pellets with a FT-IR 8400-Shimadzu spectrometer. 1H NMR spectra were recorded using a Bruker DRX-250 Avance spectrometer at 250 MHz proton frequency; chemical shifts δ in ppm. 1H NMR spectra recorded in DMSO-d6 were referenced to the residual protonated signal of the solvent (2.47 ppm for DMSO-d6). Elemental analyses (C, H, N) were measured with a Thermo Finnigan Flash Elemental Analyzer 1112 EA. The melting points were determined with a Barnsted Electrothermal 9200 electrically heated apparatus. Thickness measurements were performed by DekTak 8000; Electrical and optical property of fabricated OLEDs were performed by USB2000 and HR4000 Ocean Optics. The current–voltage–luminance characteristics were checked by Keithley source meter 2400 model and optical meter Mastech-MS6612.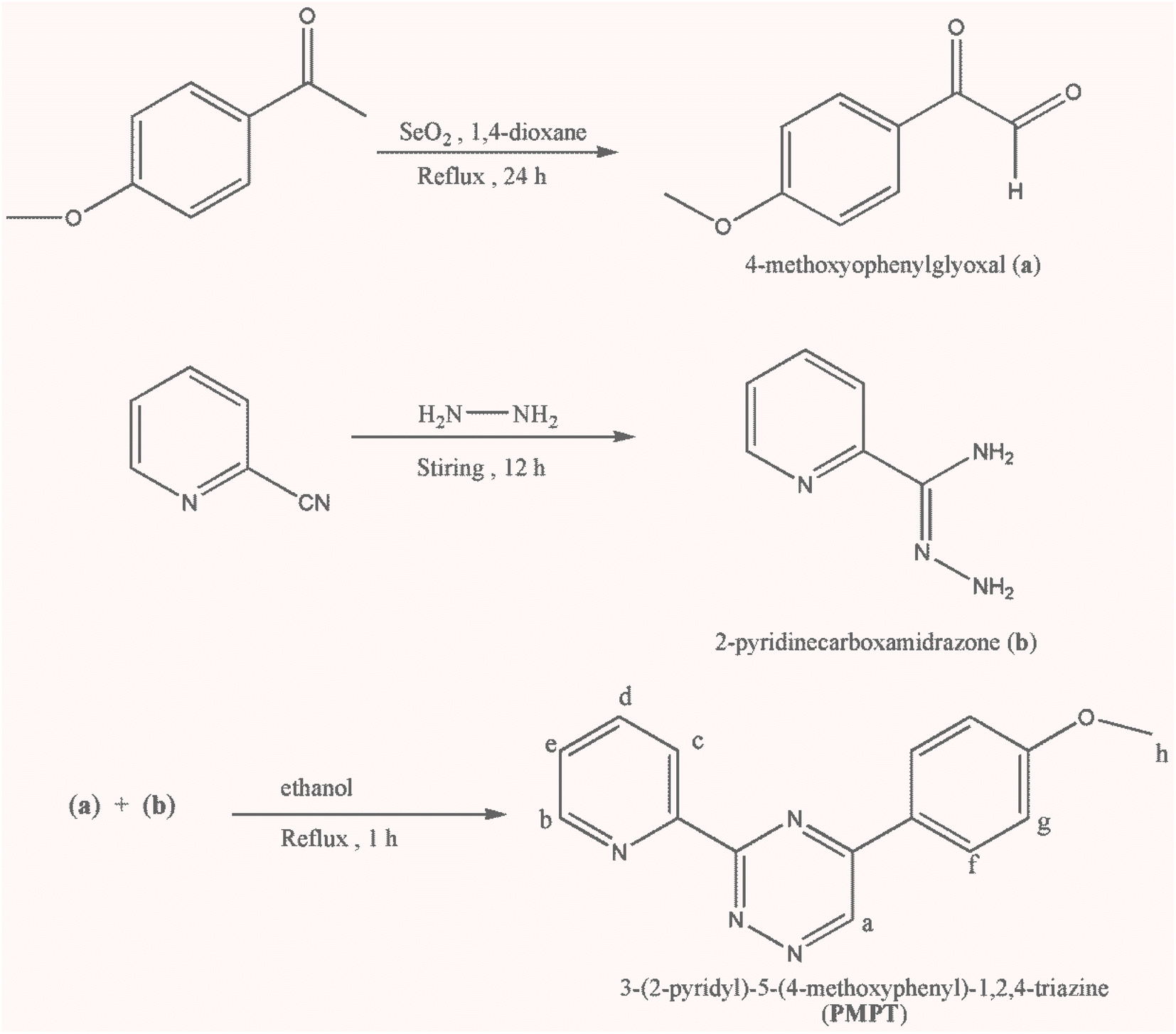 | ||
| Scheme 1 Synthesis steps of PMPT ligand. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1515 s (νCC)ar, 1266 s (νC–O), 849 m and 770 m (νpy). 1H NMR (250 MHz, DMSO-d6, ppm, Hz): δ = 10.02)s, 1Ha), 8.86)d, J = 2.5 Hz, 1Hb), 8.51)d, J = 7.8 Hz, 1Hc), 8.41) d, J = 8.5 Hz, 2Hf), 8.05)t, J = 7.5 Hz, 1Hd), 7.61 (t, J = 5.0 Hz, 1He), 7.15 (d, J = 8.5 2Hg), 3.85 (s, 3Hh).N), 1576 m (νasym(COO)), 1515 s (νCC), 1412 m (νsym(COO)), 1258 s (νC–O), 842 m and 774 m (νpy). 1H NMR (250 MHz, DMSO-d6, ppm, Hz): δ = 10.04)s, 1Ha), 8.83)d, J = 3.2 Hz, 1Hb), 8.52)d, J = 7.8 Hz, 1Hc), 8.42 (d, J = 8.8 Hz, 2Hf), 8.05)t, J = 7.8 Hz, 1Hd), 7.62 (t, J = 4.8 Hz, 1He), 7.16 (d, J = 8.8 Hz, 2Hg), 3.86 (s, 3Hh), 1.69 (s, 3Hacetate).
N), 1515 s (νCC)ar, 1266 s (νC–O), 849 m and 770 m (νpy). 1H NMR (250 MHz, DMSO-d6, ppm, Hz): δ = 10.02)s, 1Ha), 8.86)d, J = 2.5 Hz, 1Hb), 8.51)d, J = 7.8 Hz, 1Hc), 8.41) d, J = 8.5 Hz, 2Hf), 8.05)t, J = 7.5 Hz, 1Hd), 7.61 (t, J = 5.0 Hz, 1He), 7.15 (d, J = 8.5 2Hg), 3.85 (s, 3Hh).N), 1576 m (νasym(COO)), 1515 s (νCC), 1412 m (νsym(COO)), 1258 s (νC–O), 842 m and 774 m (νpy). 1H NMR (250 MHz, DMSO-d6, ppm, Hz): δ = 10.04)s, 1Ha), 8.83)d, J = 3.2 Hz, 1Hb), 8.52)d, J = 7.8 Hz, 1Hc), 8.42 (d, J = 8.8 Hz, 2Hf), 8.05)t, J = 7.8 Hz, 1Hd), 7.62 (t, J = 4.8 Hz, 1He), 7.16 (d, J = 8.8 Hz, 2Hg), 3.86 (s, 3Hh), 1.69 (s, 3Hacetate).2.2. Crystal structure determination
Crystallographic data are given in Table 1. Single-crystal X-ray diffraction measurements were performed on a STOE IPDS 2T image plate diffractometer system equipped with a sealed Mo X-ray tube and a graphite monochromator crystal (λ(Mo-Kα = 0.71073 Å). Data reduction and numerical absorption correction were done with STOE X-AREA software.25 All structures were solved by direct methods using SHELXS-2018 and refined with SHELXL-2018 (ref. 26) using WinGX27 as a graphical frontend. All non-hydrogen atoms were refined with anisotropic thermal. Hydrogen atoms were included on idealized positions applying the riding model. Olex2 software28 and Diamond 3.2k29 were used for structural analyses and visualization. CCDC 2350383 and 2350384 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/.| 1 | 2 | |
|---|---|---|
| Empirical formula | C30H24Br4N8O2Pb2 | C36H30N10O6Pb2S2 |
| Formula weight, g mol−1 | 1262.59 | 1177.2 |
| Crystal size, mm3 | 0.24 × 0.15 × 0.06 | 0.13 × 0.12 × 0.10 |
| Temperature, K | 200(2) | 200(2) |
| Crystal system | Triclinic | Triclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
|
||
| Unit cell dimensions (Å, °) | ||
| a | 7.7597(5) | 9.7327(6) |
| b | 9.1203(5) | 10.2844(6) |
| c | 12.5054(7) | 10.5326(7) |
| α | 102.924(4) | 81.463(5) |
| β | 97.329(4) | 68.068(5) |
| γ | 97.611(5) | 86.765(5) |
| Volume, Å3 | 843.60(9) | 967.1(2) |
| Z | 1 | 1 |
| Calculated density, g cm−3 | 2.485 | 2.021 |
| Absorption coefficient, mm−1 | 14.78 | 8.88 |
| F(000) | 580 | 560 |
| θ range for data collection (°) | 2.68–27.00 | 2.68–28.00 |
| h, k, l ranges | −9 ≤ h ≤ 9 | −12 ≤ h ≤ 12 |
| −11 ≤ k ≤ 11 | −13 ≤ k ≤ 13 | |
| −15 ≤ l ≤ 15 | −13 ≤ l ≤ 13 | |
| Reflections collected | 6695 | 9247 |
| Independent | 3657 | 4574 |
| Rint | 0.048 | 0.028 |
| Data/ref. parameters | 3657/209 | 4574/255 |
| Goodness-of-fit on F2 | 1.011 | 0.984 |
| Final R indexes [I >= 2σ (I)] | R1 = 0.0363 | R1 = 0.0254 |
| wR2 = 0.0988 | wR2 = 0.0556 | |
| Final R indexes [all data] | R1 = 0.0435 | R1 = 0.0348 |
| wR2 = 0.1016 | wR2 = 0.0572 | |
| Largest diff. peak/hole, e Å−3 | 1.88/−2.35 | 0.67/−1.57 |
2.3. Fabrication of OLEDs
ITO, were used as the conducting anodes. PEDOT: PSS as a hole injection layer was spin-coated on clean ITO substrate of 55 nm thickness and baked in an oven for 1 hour at 1200 °C. Afterward, PVK as a hole-transporting material and PBD as an electron-transporting material were doped with compound 1 and 2. PVK, PBD, and compound 1, 2 at ratio of 100:40:8 were blended in dimethylformamide (DMF) and then spin-coated and baked at 80 °C for 1 h. Finally, Al was evaporated by thermal evaporation method, respectively. Fig. 1 shows a schematic structure of the devices.
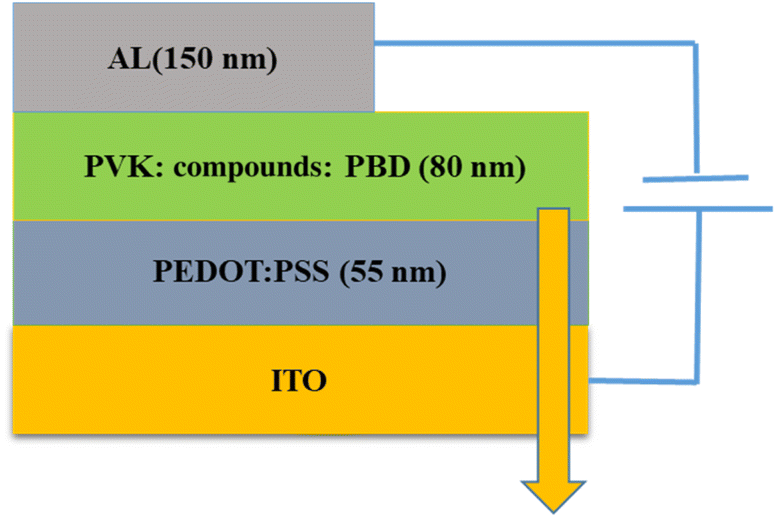 | ||
| Fig. 1 Schematic structures of the device. | ||
3. Results and discussion
3.1. Infrared spectra of complexes
The infrared (IR) spectra of compounds 1 and 2 were analyzed, and the selected frequencies are provided in Section 2. In the examined complexes, the stretching vibrations of the CC, CN, and NN groups were observed at significantly lower values compared to the vibrations of ν(CN) and ν(NN) in the 3-(2-pyridyl)-5-(4-methoxyphenyl)-1,2,4-triazine (PMPT) ligand. This observation supports the coordination of the triazine and pyridyl rings to the metal ion.30 The weak band observed at 2950–3000 cm−1 is attributed to the methoxy group of the PMPT ligand and the acetate ν(CH) mode in compound 2. In 2, the bands around 1576 and 1412 cm−1 correspond to the modes νasym(COO) and νsym(COO), respectively, indicating the presence of the acetate ligand in the molecule. Previous attempts have been made to correlate the positions of these modes or the frequency difference Δν (νasym − νsym) with the bonding type.31 The Δν value of acetate in the lead(II) complex is 164 cm−1, which aligns with the expected bidentate and bridging coordination of acetate. Additionally, the bands at a frequency of 2049 cm−1 in the IR spectrum of 2 provide evidence of N-coordination between the terminal isothiocyanate anions and the lead center, as supported by the crystal structure32 (Fig. S1 and S2†).
3.2. Crystal structure description
The crystalline phase purities for 1 and 2 were confirmed by the PXRD patterns. Fig. S5† shows that the powder patterns of the two complexes match quite well with those simulated from single-crystal X-ray data, indicating the bulk purities of the complexes.Scheme 2 illustrates the various possible coordination modes of derivatives of 3-(2-pyridyl)-1,2,4-triazine (PTZ) ligands. The PTZ ligands can form mononuclear lead complexes through a bidentate coordination site similar to 2,2′-bipyridine (referred as form A1). These mononuclear complexes are commonly described in the literature as derivatives involving PTZ ligands.14,15,18–21 By combining bidentate and monodentate coordination sites provided by the PTZ ligands, it is possible to obtain dinuclear complexes (referred to as forms C2 and D2). Only one binuclear complex of silver(I) in coordination form D2 has been reported.33 However, there are no examples in the literature where PTZ ligands coordinate to lead atoms in coordination forms B1, C2, and D2. In this study, we have synthesized two binuclear complexes where the PMPT ligand coordinates to the lead atom in coordination form D2.
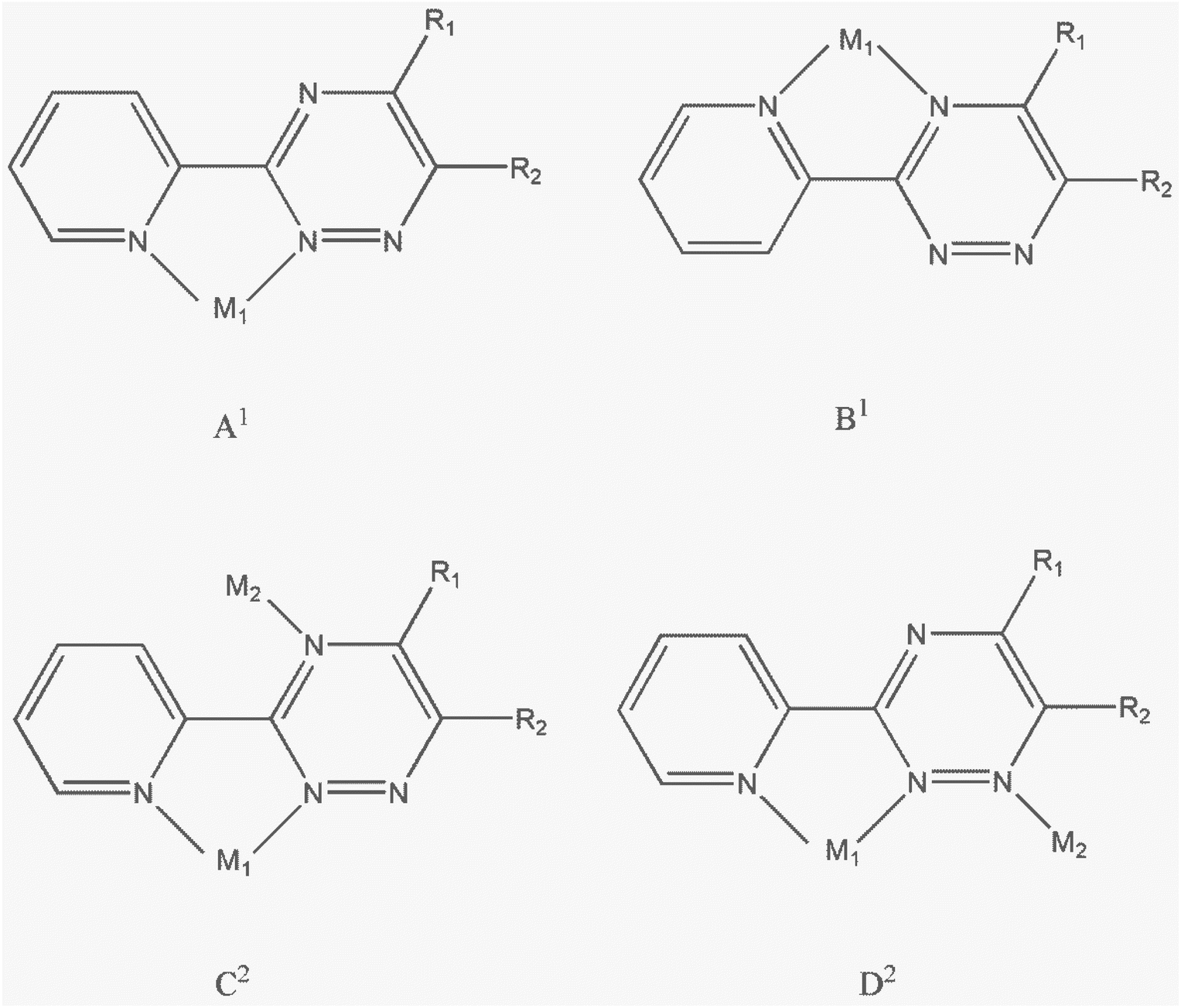 | ||
| Scheme 2 Possible coordination modes of derivatives of 3-(2-pyridyl)-1,2,4-triazine (PTZ) ligands to form mononuclear (A1, B1) or binuclear (C2, D2) metal complexes. | ||
Two novel lead(II) complexes with the newly developed triazine ligand, 3-(2-pyridyl)-5-(4-methoxyphenyl)-1,2,4-triazine (PMPT), and varied anions were synthesized and analyzed by their influences on the coordination chemistry of lead(II). X-ray single-crystal structure analyses indicate that [Pb2(μ-PMPT)2Br4] (1) crystallizes in the triclinic space group P (Table 1), with one formula unit per unit cell. A molecular view and selected bond parameters of 1 are presented in Fig. 2 and Table 2. Considering the PMPT ligand as a bidentate and monodentate N-donor ligand, along with two bromide anions, the compound [Pb2(μ-PMPT)2Br4] suggests the presence of a five-coordinate Pb atom. This coordination number is relatively low for Pb(II) in an N and Br-donor environment. The structure solution reveals the Pb atoms occurring in pairs that are approximately 3.900(2) Å apart. These pairs are formed through N-bridging in a centrosymmetric dimer unit consisting of PbN3Br2 entities. The structure of this “dimer” unit is quite remarkable, as depicted in Fig. 3, displaying a highly “hemidirected” coordination sphere. This observation suggests that this system might serve as an example of a stereochemically active lone pair. Within a “hemidirected” coordination sphere, the length of the Pb–X bonds varies and is generally greater than 0.4 Å. This variation is influenced by their proximity to a stereochemically active lone pair or their distance from it.34 However, in the case of compound 1, there is an approximate difference of 0.2 Å in the lengths of the five Pb–N and Pb–Br bonds. Notably, these longer bond lengths are indicative of a “holoidirected” coordination sphere.
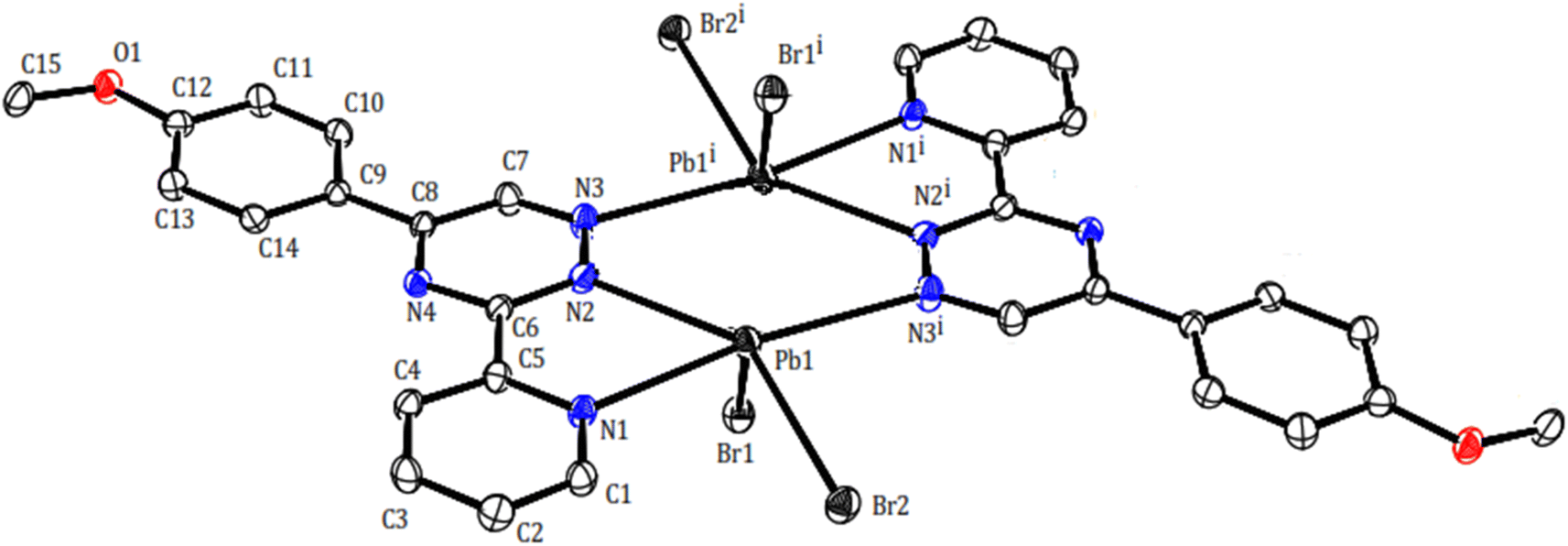 | ||
| Fig. 2 Molecular structure of compound 1 with atom numbering scheme. | ||
| 1 | |||||
| Pb1–N1 | 2.624(5) | Pb1–N2 | 2.743(5) | Pb1–N3i | 2.818(6) |
| Pb1–Br1 | 2.792(8) | Pb1–Br2 | 2.828(7) | Pb1⋯C14ii | 3.465(6) |
| Pb1⋯C13ii | 3.538(6) | N1–Pb1–N2 | 60.84(2) | N1–Pb1–Br1 | 97.69(1) |
| N2–Pb1–Br1 | 74.14(1) | N1–Pb1–Br2 | 88.72(1) | N2–Pb1–Br2 | 145.48(1) |
| Br1–Pb1–Br2 | 96.13(2) | N1–Pb1–N3i | 172.87(1) | Br2–Pb1–N3i | 84.33(1) |
| i 1 − x, 2 − y, 1 − z; ii 1 − x, 1 − y, 1 − z | |||||
|
|||||
| 2 | |||||
| Pb1–O1 | 2.402(4) | Pb1–O2 | 2.601(4) | Pb1–N1 | 2.624(5) |
| Pb1–N2 | 2.629(3) | Pb1–O2i | 2.746(3) | Pb1–N3 | 2.762(4) |
| Pb1–N4i | 2.951(3) | Pb1⋯S1ii | 3.441(2) | O1–Pb1–O2 | 51.38(1) |
| O1–Pb1–N1 | 77.54(1) | O2–Pb1–N1 | 115.90(1) | O1–Pb1–N2 | 76.37(1) |
| O2–Pb1–N2 | 113.00(1) | N1–Pb1–N2 | 83.65(1) | O1–Pb1–O2i | 131.13(1) |
| O2–Pb1–O2i | 82.27(1) | N1–Pb1–O2i | 146.72(1) | N1–Pb1–N4i | 89.70(1) |
| N2–Pb1–O2i | 116.12(1) | N2–Pb1–N3 | 61.26(1) | N3–Pb1–N4i | 123.59(1) |
| i 1 − x, 2 − y, 1 − z; ii 2 − x, 2 − y, 1 − z | |||||
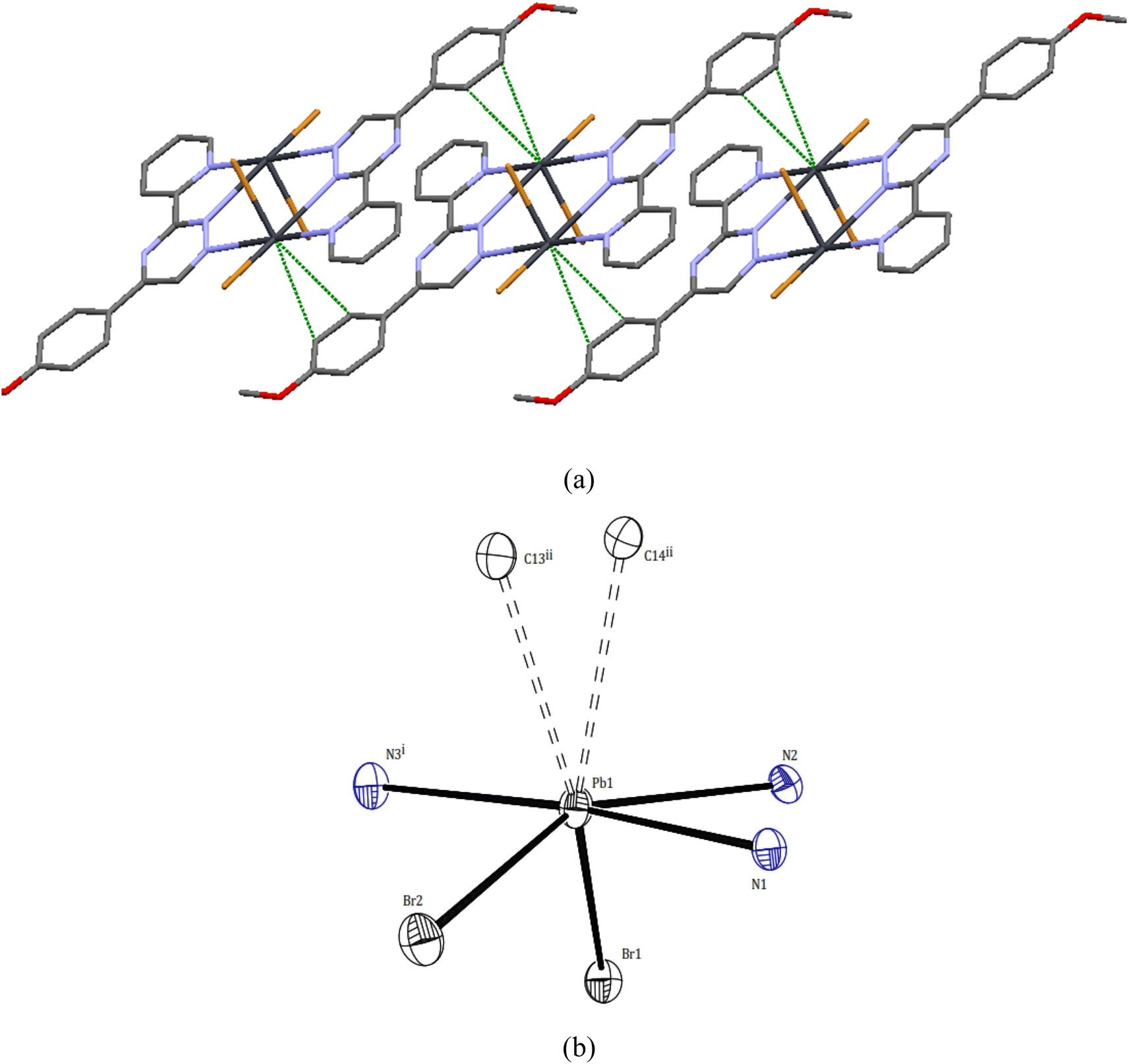 | ||
| Fig. 3 (a) Pb⋯C tetral bonding and π–π stacking (b) lead atoms geometries in 1. | ||
Upon further examination, it becomes clear that the interpretation of the crystal structure mentioned above is an oversimplification. The chains of Pb atom pairs, aligned parallel to the crystallographic c-axis, arise from close “intermolecular” contacts between such dimeric units. This arrangement is illustrated for a single adjacent pair in Fig. 3. While the parallel alignment of certain phenyl groups may initially suggest the presence of π-stacking interactions throughout the lattice, it is actually a consequence of two-hapto interactions occurring between the Pb atoms and phenyl groups originating from separate dimers.
A search was made generally for Pb⋯C approaches and it appears that Pb atoms in compound 1 may also be involved in η2 interaction with the phenyl groups of another dimer. Thus, the Pb atoms are linked to two carbon atoms of phenyl groups, with distances Pb1⋯C14ii and Pb1⋯C13ii of 3.465(6) and 3.538(6) Å, respectively. Hence, the PbII coordination sphere is completed and rather than a PbN3Br2 coordination sphere, the complex can be considered to contain a dihapto interactions (PbC2N3Br2) center with an irregular seven coordination number but ‘‘holodirected” coordination sphere (Fig. 3a and b). The reported Pb⋯C separations range is 3.083–4.05 Å in species [Pb(o-xylene)2(Cl2AlCl2)2], [Pb(η6-C6H6)(Cl2AlCl2)2]C6H6 (ref. 35) [Pb2{SeC6H2(CF3)3}4(toluene)2]36 and [Pb2(DBM)4].37 Thus, Pb⋯C interactions in compound 1 appear to be yet another factor which can make varying contributions to the stability of complexes of this metal ion.
Within the crystal structure of complex 1, adjacent complex molecules are connected through robust intermolecular hydrogen bond interactions. The primary intermolecular interactions observed in compound 1 include C–H⋯O and C–H⋯Br hydrogen bonds, as well as π⋯π stacking interactions between the molecules. These interactions play a crucial role in determining the overall arrangement of the crystal packing and contribute to the stabilization of the crystal structure in a two-dimensional supramolecular manner. For further details, refer to Fig. S6† and Table 3.
| D–H···A | d(D–H) | d(H⋯A) | <(DHA) | d(D⋯A) | Symmetry code on A atom |
|---|---|---|---|---|---|
| 1 | |||||
| C2–H2⋯O1 | 0.950 | 2.524 | 135.42 | 3.269(8) | −1 + x, y, −1 + z |
| C4–H4⋯Br1 | 0.950 | 2.848 | 148.42 | 3.691(7) | x, −1 + y, z |
| π–π (slipped face to face) between the phenyl and pyridyl rings | 3.370(2) | 2 − x, 1 − y, 1 − z | |||
|
|||||
| 2 | |||||
| C7–H7⋯O1 | 0.949 | 2.597 | 154.28 | 3.477(1) | 1 − x, 1 − y, 1 − z |
| C17–H17⋯O1 | 0.980 | 2.564 | 158.10 | 3.464(3) | 1 − x, 1 − y, 1 − z |
| π–π (slipped face to face) between the triazine and pyridyl rings | 3.359(2) | 1 − x, 1 − y, 1 − z | |||
In compound 2, the structure may be considered as a coordination polymer of lead(II) consisting of dimeric units with a building block of [Pb2(μ-PMPT)2(μ-CH3COO)2(NCS)2] (2). Similarly to compound 1, Two PMPT ligand doubly bridge two lead(II) ions via the N atoms (nitrogen of pyridine and triazine as chelating and another nitrogen of triazine as monodentate to another lead atom). The dimeric units are further linked across a center of inversion by two acetate anions, resulting are shown in Fig. 4. The Pb⋯Pb distances within the 2 moieties, those bridged by the acetate anions are 4.027(3) Å. Within the dimer unit, the carboxylate moiety of each acetate ligand acts as both bidentate, and bridging group (totally tridentate) in a μ-1,3 mode: both oxygen atoms of the carboxylate group coordinate to a lead(II) ions yielding the Pb2O2 core. Isothiocyanate anions as acts monodentate and as terminal N donor. Thus per lead atoms in 2 is seven-coordinated (PbN4O3) by two nitrogen of two PMPT ligands, nitrogen of isothiocyanate anion and three oxygen of two acetates with the Pb–O distances of 2.402(4), 2.601(4), 2.746(3) Å and Pb–N distances of 2.624(5), 2.629(3), 2.762(4) 2.762(4) and 2.951(3) Å (Table 2). This arrangement created a gap or hole in coordination geometry around the metal ions (presence of gap is clear), occupied possibly by a “stereoactive” lone pair of electrons on lead(II), and the coordination sphere is hemidirected. The bond length difference more than 0.4 Å in the coordination sphere and the observed shorting of Pb–O bonds on the side of Pb2+ ion opposite to the putative lone pair supports the presence of the lone pair electrons.34
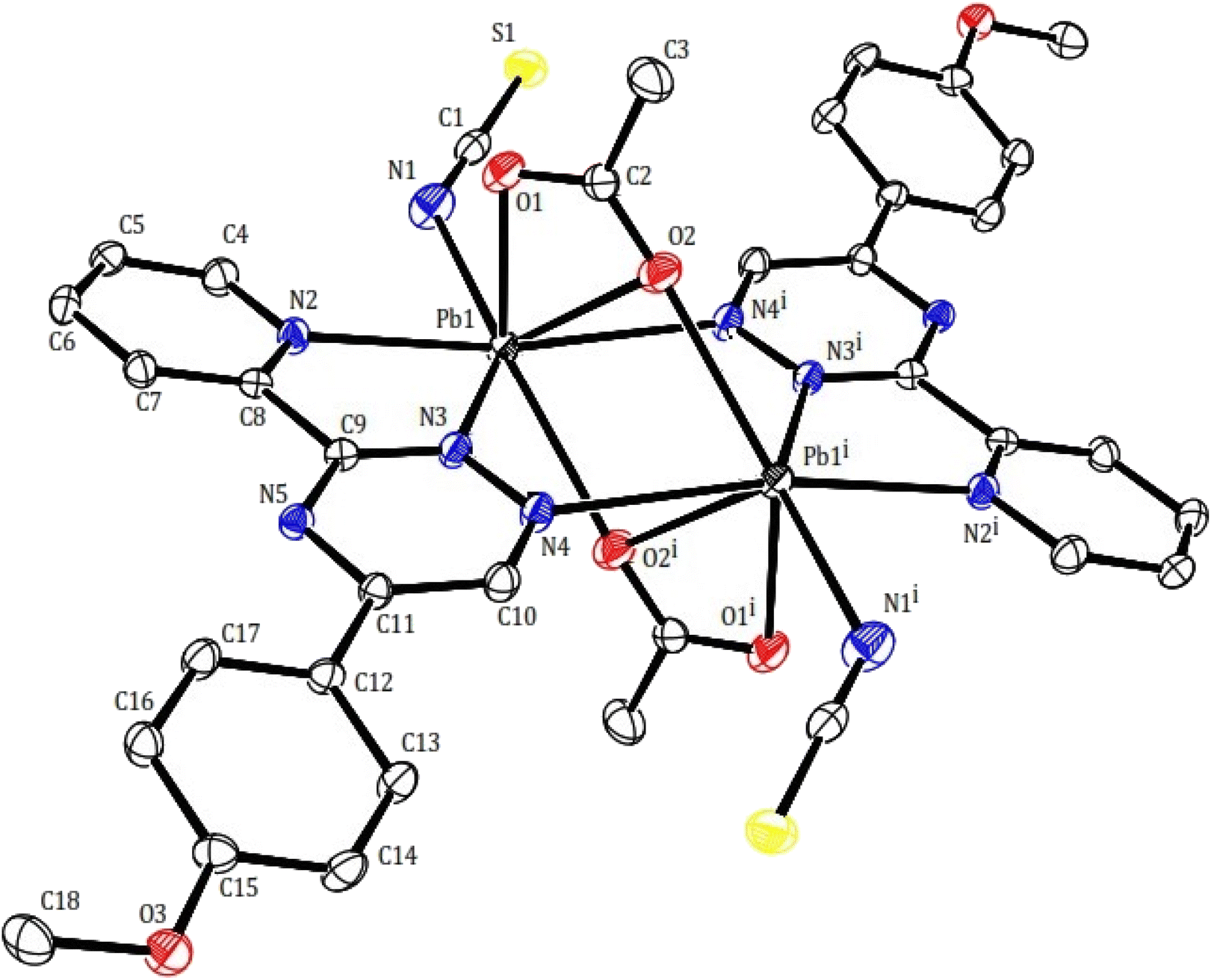 | ||
| Fig. 4 Molecular structure of compound 2 with atom numbering scheme. | ||
This particular environment provides a suitable space for forming bonds with other atoms. To explore potential donor centers, it is necessary to extend the bonding range. Within a limit of 3.5 Å (which is smaller than the van der Waals radius), there are Pb⋯S(thiocyanate) tetrel bonds observed in the crystallographic [010] direction. These bonds have distances of 3.441(2) Å, connecting the dimers and forming a polymeric chain. These distances fall within the sum of the van der Waals radii38 of the corresponding atoms. The Pb⋯S tetral distance in compound 2 is similar to that reported for lead(II) complexes with thiocyanate (Fig. 5).39 Additionally, the Pb⋯Pb distances within the [Pb2(μ-SCN)2]n moieties measure 6.716 Å.
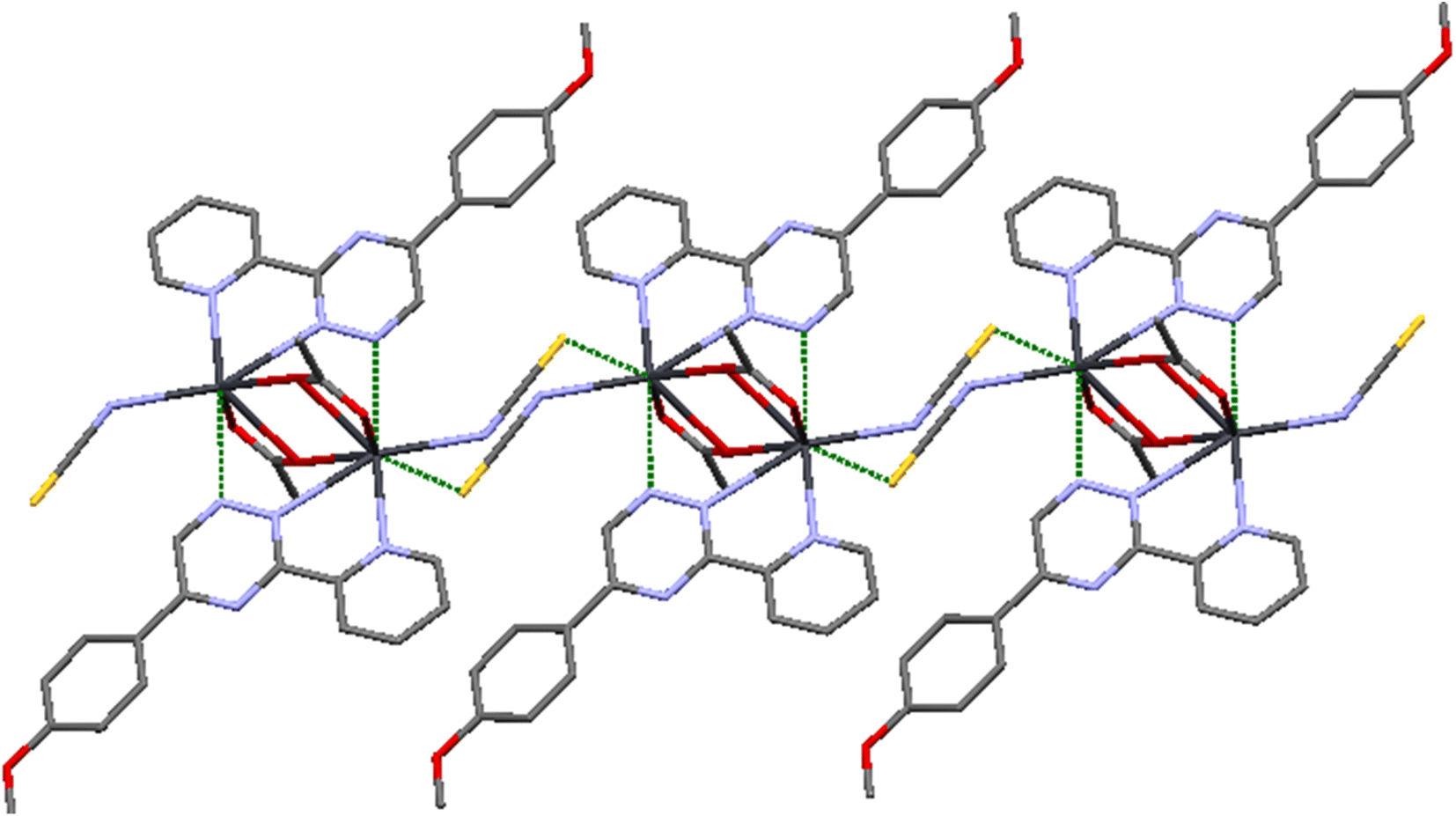 | ||
| Fig. 5 Pb1⋯S tetral bonding in 2. | ||
In compound 2, various types of interactions, including intermolecular, intramolecular, and π–π stacking interactions, contribute to the arrangement of the complex in the crystal lattice (refer to Table 3). To investigate the presence of weak directional intermolecular interactions in 2, Mercury programs were utilized. The analysis revealed the existence of C–H⋯O40 interactions and π–π stacking. The packing diagram of 2 demonstrates a two-dimensional self-assembled structure formed through slipped face-to-face π–π stacking. Notably, the distance between the triazine and pyridyl rings measures 3.359(2) Å, which is considerably shorter than the typical distance observed in normal π–π stacking.41 Furthermore, the two-dimensional supramolecular networks in 2 are constructed through C–H⋯O interactions. The distances for these interactions are 2.597 and 2.564 Å, significantly shorter than the van der Waals distance of 2.77 Å for H⋯O interactions.38 Refer to Fig. 7S† for visual representation.
3.3. Optical characteristics
The UV-Vis absorption spectra of complexes in ethanol solution are shown in Fig. 6. Compound 1 exhibit two absorption peaks at 380 nm and 360 nm. The UV-Vis spectrum of compound 2 is red shifted with respected to compound 1. All peaks can be attributed to the π–π* transitions of the aromatic ligand.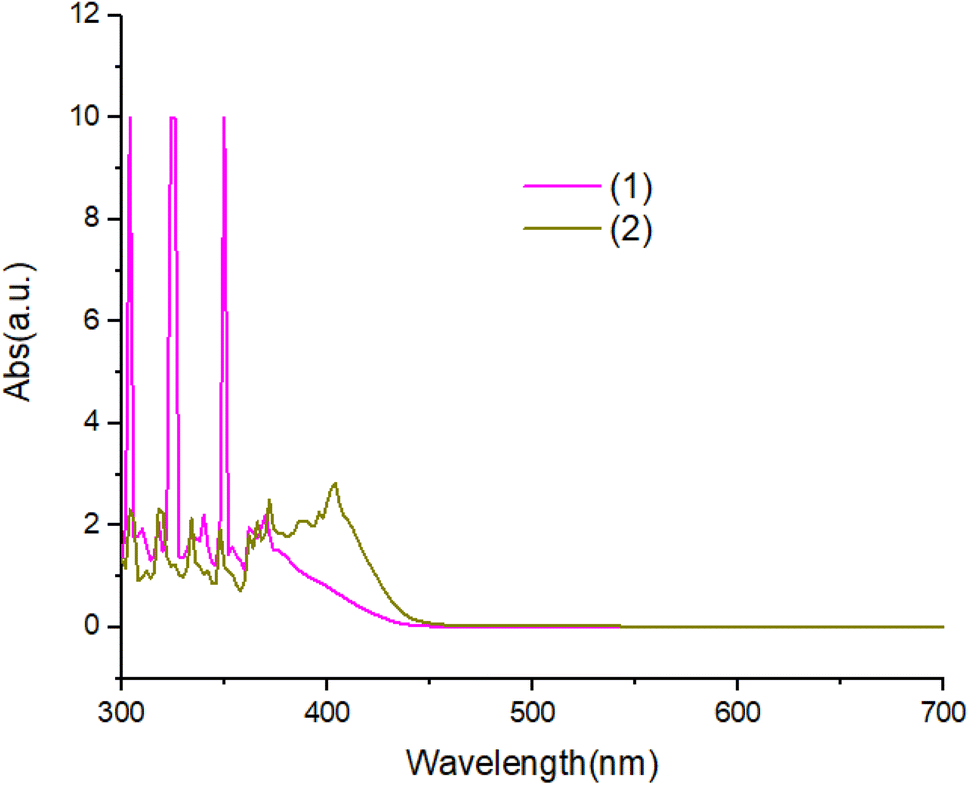 | ||
| Fig. 6 The UV-Vis absorption spectra of compounds. | ||
Fig. 7 and 8 shows the characteristics of the PL solution and solid state of compounds in the solution state by exciting with a wavelength of 405 nm, the PL spectra of compounds shows peak emission spectrum centered at 596 nm, but in the solid state, the PL spectra of compounds blue shifted about 65 nm with respect to solution.
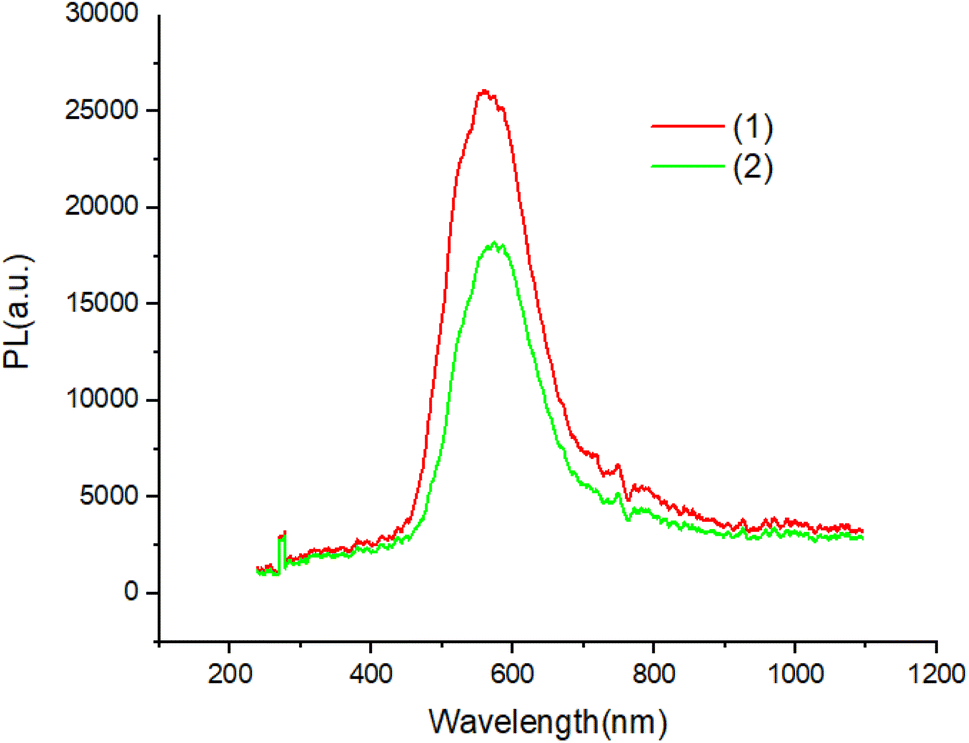 | ||
| Fig. 7 The PL of solution of compounds. | ||
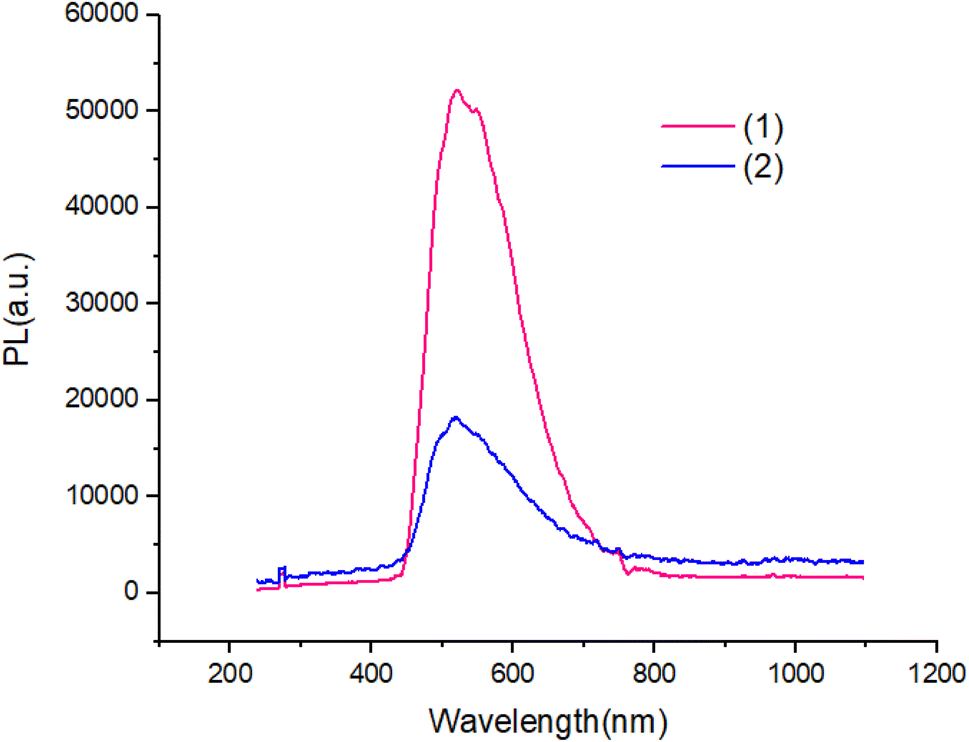 | ||
| Fig. 8 The PL of solid state of compounds. | ||
The PL quantum yields of the compounds were achievement using the equation established by Parker and Rees.42,43 The highest PL quantum yield for compounds 1 and 2 are measured as 0.67, and 0.38, respectively. The Förster radius energy transfer rate is commonly employed to showcase the power efficiency in OLED technology. The Förster radius is defined as:
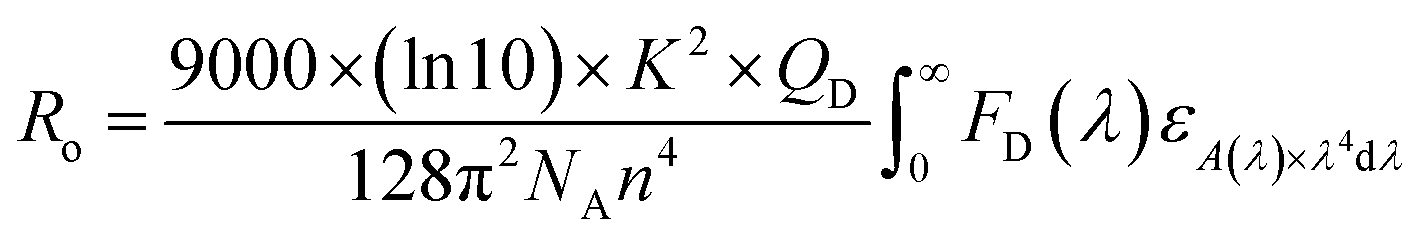
| Complex | k2 | NA | QD | n |
|
|---|---|---|---|---|---|
| 1 | 2/3 | 6.02 × 1023 | 0.67 | 1.73 | 2.5 × 10−32 |
| 2 | 2/3 | 6.02 × 1023 | 0.38 | 1.68 | 2.2 × 10−34 |
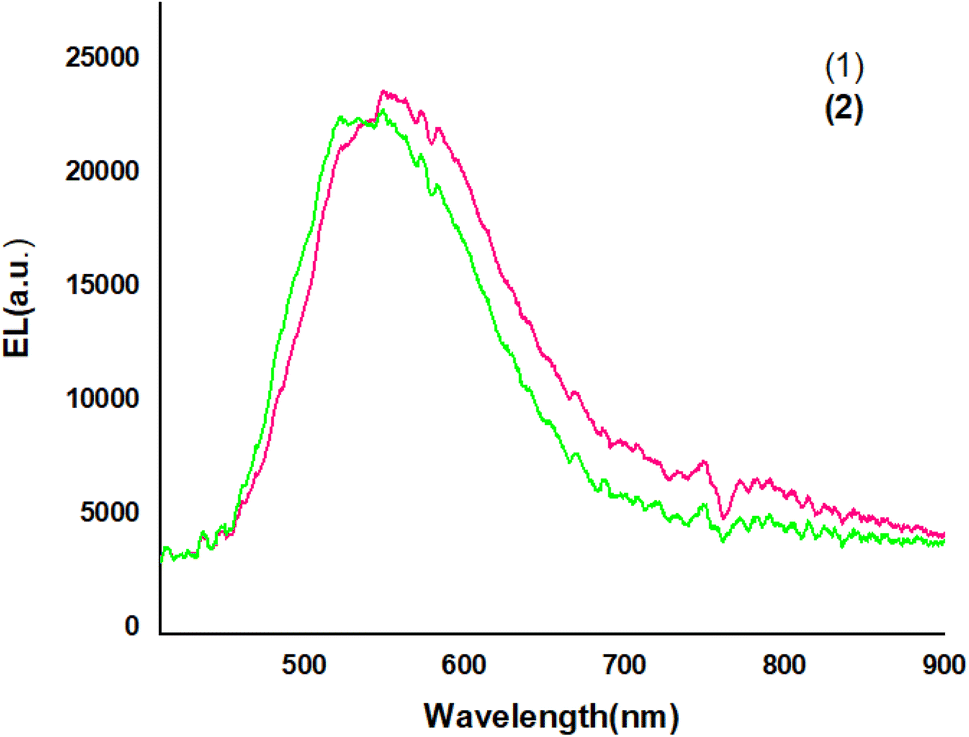
Fig. 9 shows The EL spectra of OLED devices. The EL of the compound 1 and 2-based devices showed a band in the green and yellow regions, respectively.
 | ||
| Fig. 9 The EL spectra of devices. | ||
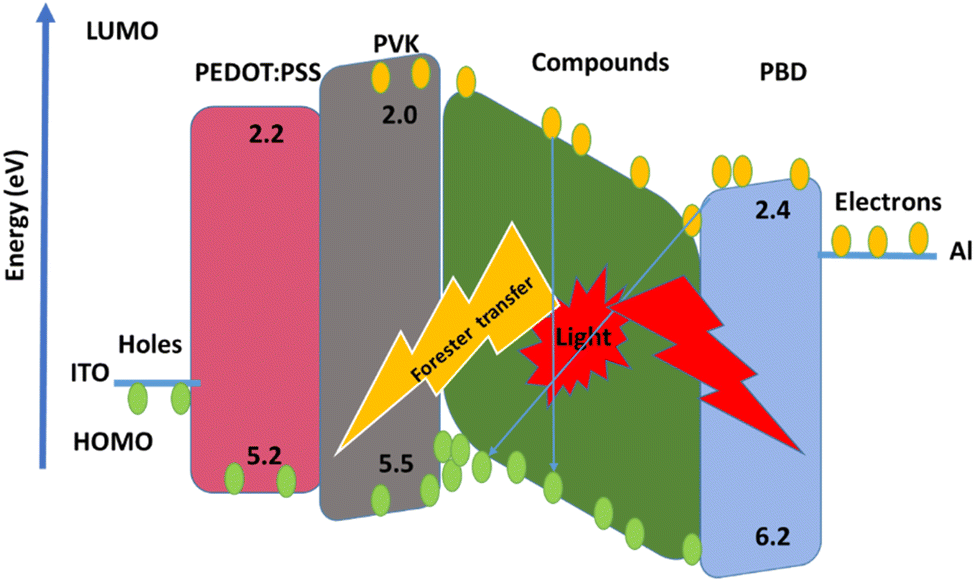
The EL spectra of the OLEDs depended on the PbN3Br2, 1 and PbN4O3, 2 of the environment of Pb complexes. The emission wavelength shifted from the green color to the yellow color when the functional group varied from PbN3Br2 to PbN4O3. (see the Fig. 10).
 | ||
| Fig. 10 Schematic energy levels. | ||
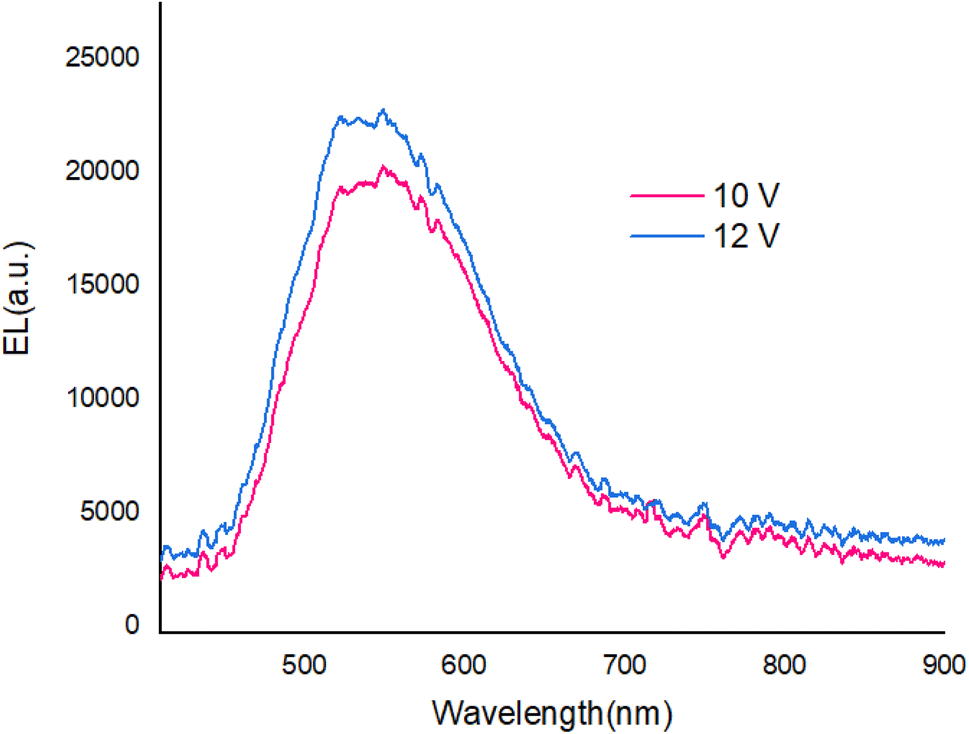
With applying of voltage, electrons (e) and holes (h) injected in the PEDOT:PSS and PBD layer, Finally e/h recombines at the compound molecules. Also, the EL intensity at 560 nm (Fig. 11) dependent on the applying voltage and with increasing voltage the EL intensity increases.
 | ||
| Fig. 11 The EL spectrum at various voltages. | ||
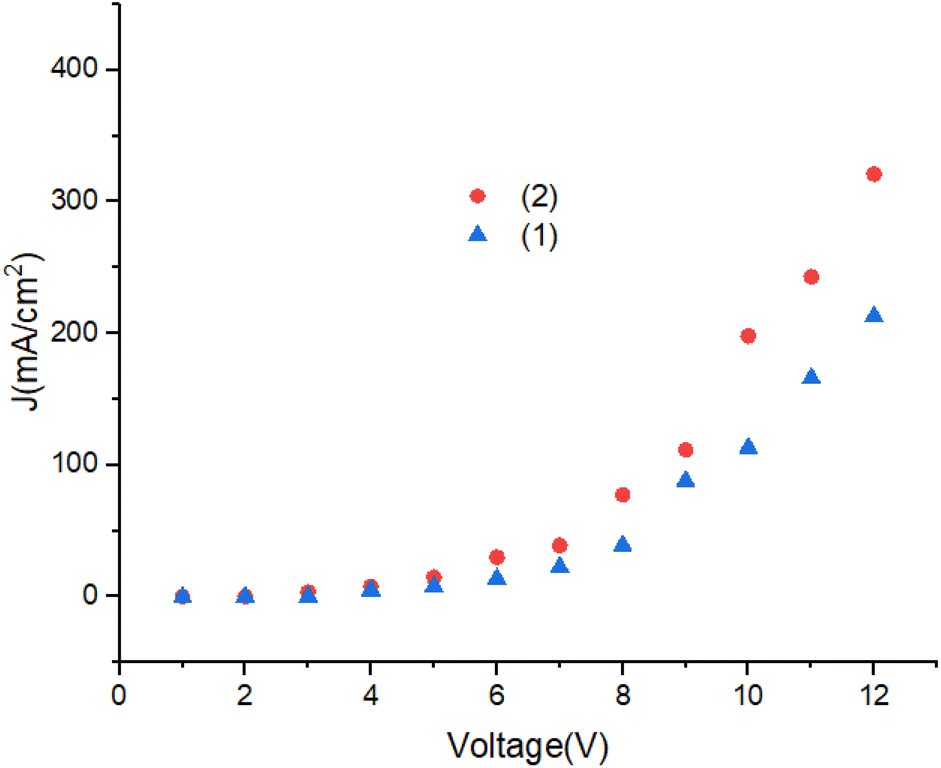
Fig. 12 shows the current density of the devices. Turn on voltage of the OLEDs is lower than 4.5 V. With increasing of voltage the current density increases. The J–V characteristics change from ohmic region to the space charge limited current (SCLC) region, respectively. A significant improvement in the electrical conductivity in OLEDs can be achieved by modification of the molecular structure.44–47
 | ||
| Fig. 12 The current density–voltage characteristic of device. | ||
Fig. 13 shows luminescence efficiency – current density of devices. The differences between the luminescence are ascribed to the role of the PbN3Br2 to PbN4O3 in compounds. Also, with the increase in applied current density the luminescence remains reliably stable.
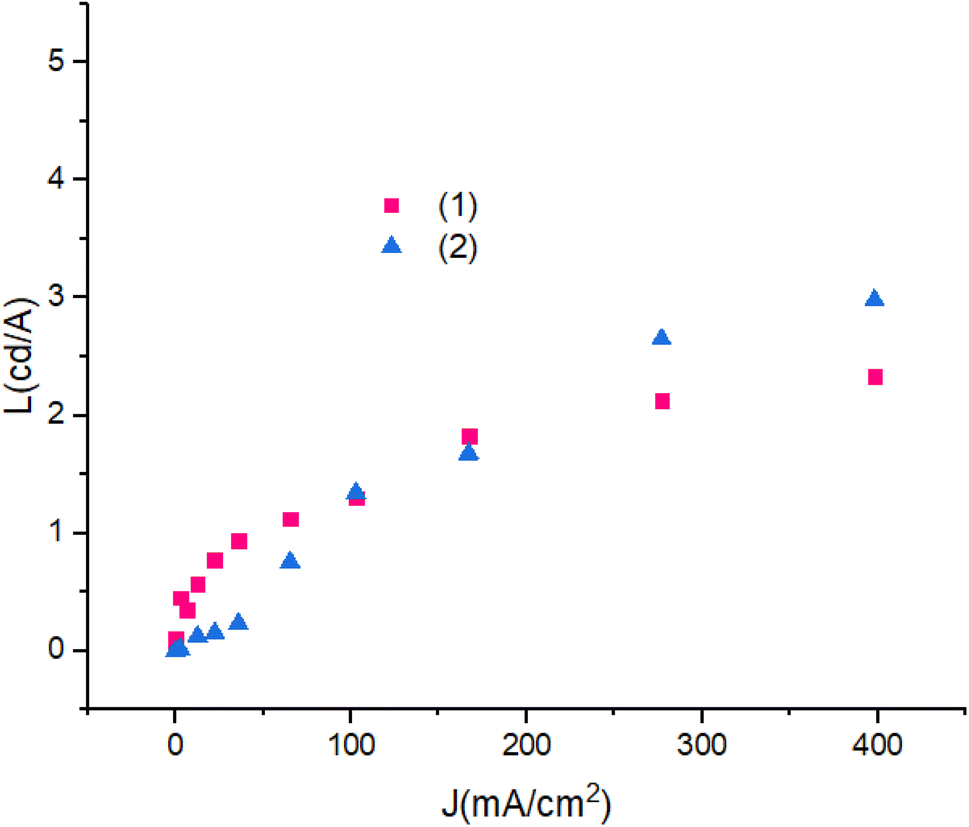 | ||
| Fig. 13 Luminescence efficiency–current density characteristic of devices. | ||
4. Conclusion
In conclusion, we report the synthesis and physicochemical structural characterization of two new binuclear Pb(II) complexes with PMPT ligand and auxiliary anionic co-ligands: bromide in 1, acetate and isothiocyanate in 2. In all complexes, per PMPT ligand coordinate two lead atoms by three nitrogen atoms (new coordination mode in lead complexes). Based on the obtained structural parameters, complex 1 exhibited a PbC2N3Br2 environment, while complex 2 displayed a PbN4O3S environment, with holodirected and hemidirected coordination spheres, respectively. It seems that anions play an important role in the activity of lead(II) coordination sphere by creating various intermolecular interactions. Upon closer examination, it becomes evident that tetrel bonding, although not adequately recognized until now, plays a significant role in the study of lead's solid-state chemistry. This newfound understanding has the potential to greatly aid in the manipulation and design of supramolecular architectures and organometallic frameworks that rely on lead coordination complexes. The prepared complexes show high luminescence efficiency at room temperature and have good stability, which make it suitable for the fabrication of OLEDs. It should be noted that auxiliary ligands are a factor for changing the optical properties of compounds. The results of this work show that these compounds can be used as a precursor in the manufacture of optical devices.Data availability
CCDC 2350383 and 2350384 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/.Conflicts of interest
There are no conflicts to declare.References
- S. Antohe, S. Iftimie, L. Hrostea, V. A. Antohe and M. Girtan, Org. Electron., 2017, 642, 219–231 CAS.
- O. Marinov, M. J. Deen, J. A. Jiménez-Tejada and C. H. Chen, Phys. Rep., 2020, 844, 1–105 CrossRef CAS.
- H. Chen, S. Yang, Z. Tsai, W. Hung, T. Wang and K. Wong, J. Mater. Chem., 2009, 19, 8112–8118 RSC.
- K. M. Omer, S. Ku, Y. Chen, K. Wong and A. J. Bard, J. Am. Chem. Soc., 2010, 132, 10944–10952 CrossRef CAS.
- S. Ren, D. Zeng, H. Zhong, Y. Wang, S. Qian and Q. Fang, J. Phys. Chem. B, 2010, 114, 10374–10383 CrossRef CAS PubMed.
- H. Zhong, H. Lai and Q. Fang, J. Phys. Chem. C, 2011, 115, 2423–2427 CrossRef CAS.
- Z. Zhang, R. Liu, X. Zhu, Y. Li, J. Chang, H. Zhu, L. Ma, W. Lv and J. Guo, J. Lumin., 2014, 156, 130–136 CrossRef CAS.
- G. M. Ziarani, R. Moradi, N. Lashgari and H. G. Kruger, Metal Free Synthetic Organic Dyes, Elsevier, Amsterdam, 2018, pp. 197–217 Search PubMed.
- A. O. Ibrahim, Y. Zhou, F. Jiang, L. Chen, X. Li, W. Xu, O. O. E. Onawumi, O. A. Odunola and M. Hong, Eur. J. Inorg. Chem., 2011, 2011, 5000–5005 CrossRef CAS.
- P. L. Croot and K. A. Hunter, Anal. Chim. Acta, 2000, 406, 289–302 CrossRef CAS.
- P. Gamez, P. de Hoog, O. Roubeau, M. Lutz, W. L. Driessen, A. L. Spek and J. Reedijk, Chem. Commun., 2002, 1488–1489 RSC.
- A. M. Fedosseev, M. S. Grigoriev, I. A. Chartishnikova, N. A. Budantseva, Z. A. Starikova and P. Moisy, Polyhedron, 2008, 27, 2007–7014 CrossRef CAS.
- B. Machura, R. Kruszynski, J. Kusz, J. Klak and J. Mrozinski, Polyhedron, 2007, 26, 4427–4435 CrossRef CAS.
- S. Taheri, F. Marandi, H.-K. Fun and R. Kia, Z. Anorg. Allg. Chem., 2009, 635, 1352–1354 CrossRef CAS.
- A. A. Soudi, F. Marandi, A. Morsali and G. P. A. Yap, J. Coord. Chem., 2006, 59, 1139–1148 CrossRef.
- F. Marandi, Chin. J. Struct. Chem., 2014, 33, 1184–1190 CAS.
- F. Marandi and H.-K. Fun, J. Coord. Chem., 2009, 62, 1972–1979 CrossRef CAS.
- F. Marandi, H.-K. Fun and C. K. Quah, J. Coord. Chem., 2010, 63, 2113–2121 CrossRef CAS.
- F. Marandi, K. Moeini, B. Mostafazadeh and H. Krautscheid, Polyhedron, 2017, 133, 146–154 CrossRef CAS.
- F. Marandi, K. Moeini, Z. Mardani and H. Krautscheid, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2019, 75, 1023–1030 CrossRef CAS PubMed.
- F. Marandi, K. Moeini, Z. Mardani and H. Krautscheid, J. Coord. Chem., 2019, 72, 1876–1889 CrossRef CAS.
- V. K. Sharma and S. B. Chandalia, J. Chem. Technol. Biotechnol., 1986, 36, 456–460 CrossRef CAS.
- G. M. Sheldrick, Acta Cryst., 2008, 64, 112–122 CrossRef CAS PubMed.
- R. Hage, J. Haasnoot and J. Reedijk, Inorg. Chim. Acta, 1990, 172, 19–23 CrossRef CAS.
- F. Marandi, F. Amoopour, I. Pantenburg and G. Meyer, J. Mol. Struct., 2010, 973, 124–129 CrossRef CAS.
- Stoe & Cie GmbH, X-area (Version 1.90), Darmstadt, Germany, 2014 Search PubMed.
- L. J. Farrugia, J. Appl. Cryst., 2012, 45, 849–854 CrossRef CAS.
- O. Dolomanov, L. Bourhis, R. Gildea, J. Howard and H. Puschmann, J. Appl. Cryst., 2009, 42, 339–341 CrossRef CAS.
- K. Brandenburg and H. Putz, Diamond Version 3.2k, CRYSTAL IMPACT, Bonn, Germany, 2014 Search PubMed.
- B. Machura, A. Świtlicka, R. Kruszynski, J. Mroziński, J. Kłak and J. Kusz, Polyhedron, 2008, 27, 2959–2967 CrossRef CAS.
- G. B. Deacon and R. J. Philips, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
- S. Wöhlert, L. Fink, M. Schmidt and C. Näther, CrystEngComm, 2013, 15, 945–957 RSC.
- F. Marandi, N. Hosseini, H. Krautscheid, D. Lassig, J. Lincke, M. Rafiee and Y. A. Asl, J. Mol. Struct., 2011, 1006, 324–329 CrossRef CAS.
- L. Shimoni-Livny, J. P. Glusker and C. W. Bock, Inorg. Chem., 1998, 37, 1853–1867 CrossRef CAS.
- A. G. Gash, P. F. Rodesiler and E. L. Amma, Inorg. Chem., 1974, 13, 2429–2433 CrossRef CAS.
- D. Labahn, F. M. Bohnen, R. Herbst-Irmer, E. Pohl, D. Stalke and H. W. Roesky, Z. Anorg. Allg. Chem., 1994, 620, 41–47 CrossRef CAS.
- J. M. Harrowfield, S. Maghaminia and A. A. Soudi, Inorg. Chem., 2004, 43, 1810–1812 CrossRef CAS PubMed.
- S. S. Batsanov, Inorg. Mater., 2001, 37, 871–885 CrossRef CAS.
- M. S. Gargari, V. Stilinović, A. Bauz, A. Frontera, P. McArdle, D. V. Derveer, S. W. Ng and G. Mahmoudi, Chem. Eur. J., 2015, 21, 17951–17958 CrossRef.
- F. Marandi and H. Krautscheid, Z. Naturforsch., B: J. Chem. Sci., 2020, 75, 1043–1048 CrossRef CAS.
- F. Marandi, M. Mazrehali, S. Pourbeyram, C. K. Quah, H.-K. Fun and J. Inorg, Organomet. Polymer., 2014, 24, 940–947 CrossRef CAS.
- C. A. Parker and W. Rees, Analyst, 1960, 85, 587–600 RSC.
- L. J. Zhu, J. Wang, T. G. Reng, C. Y. Li, D. C. Guo and C. C. Guo, J. Phys. Org. Chem., 2010, 23, 190–194 CrossRef CAS.
- S. Singha, R. Jana, R. Mondal, P. P. Ray, P. P. Bag, K. Gupta, N. Pakhira, C. Rizzoli, A. Mallick, S. Kumar and R. Saha, CrystEngComm, 2021, 23, 3510–3523 RSC.
- S. Singha, B. Khanra, S. Goswami, R. Mondal, R. Jana, A. Dey, S. K. Dey, P. P. Ray, C. Rizzoli, R. Saha and S. Kumar, New J. Chem., 2021, 45, 20596–20611 RSC.
- S. Singha, S. Goswami, S. K. Dey, R. Jana, P. Ray, I. Saha, C. Rizzoli, P. Bag, S. Kumar and R. Saha, CrystEngComm, 2020, 22, 8197–8207 RSC.
- S. K. Dey, R. Saha, S. Biswas, A. Layek, S. Middya, I. M. Steele, M. Fleck, P. P. Ray and S. Kumar, Cryst. Growth Des., 2014, 14(1), 207–221 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2350383 and 2350384. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra03383c |
| This journal is © The Royal Society of Chemistry 2024 |