
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInhalable drug-loaded silk fibroin carriers for pulmonary drug delivery
Ilenia D'Onofrio ab,
Giuseppe De Giorgio*a,
Roman Sajapin†
a,
Davide Vurroa,
Aris Liboàab,
Elena Dembecha,
Giovanna Trevisia,
Maddalena Bottiac,
Vardan Galstyanad,
Giuseppe Tarabella*a and
Pasquale D'Angeloa
ab,
Giuseppe De Giorgio*a,
Roman Sajapin†
a,
Davide Vurroa,
Aris Liboàab,
Elena Dembecha,
Giovanna Trevisia,
Maddalena Bottiac,
Vardan Galstyanad,
Giuseppe Tarabella*a and
Pasquale D'Angeloa
aInstitute of Materials for Electronics and Magnetism, IMEM-CNR, P.co Area delle Scienze 37/A, 43124, Parma, Italy. E-mail: giuseppedegiorgio@cnr.it; giuseppe.tarabella@cnr.it
bGraduate School in Science and Technologies of Materials, Department of Chemistry, Life Sciences and Environmental Sustainability, University of Parma, Parco Area delle Scienze, 11/A, 43121, Parma, Italy
cDepartment of Veterinary Medical Sciences, University of Parma, Via del Taglio, 10, 43121, Parma, Italy
dDepartment of Engineering “Enzo Ferrari”, University of Modena and Reggio Emilia, Via Vivarelli 10, 41125 Modena, Italy
First published on 30th August 2024
Abstract
The design and development of engineered micro and nano-carriers offering superior therapeutic performance compared to traditional delivery forms, are crucial in pharmaceutical research. Aerosolization and inhalation of carriers with improved solubility/stability of insoluble drugs, has huge potential for targeted drug delivery (DD) in various pulmonary diseases. Indeed, dedicated carriers must meet specific dimensional rules for proper lung delivery. Particles between 2–10 μm in size are normally deposited in the tracheobronchial region, while particles of 0.5–2 μm may be properly deposited in the alveoli. In this work, we report the development of inhalable nanostructured carries made of a ‘green’ bio-inspired polymer from aqueous solutions, i.e. silk fibroin (SF), efficiently loaded with a hydrophobic drug, i.e. the thyroid hormone levothyroxine (L-T4), a drug for the treatment of idiopathic pulmonary fibrosis. The aim is to optimize a standard method for the synthesis of SF-based nanocarriers with controlled size and shape, where a fine control of their geometrical properties is aimed at efficiently controlling the pulmonary DD. L-T4 loaded SF particles were synthesized through a one-pot co-precipitation method. Optimized systems were determined by varying the chemo–physical parameters during the synthesis. Ethylenediaminetetraacetic acid (EDTA) was used to remove CaCO3 cores. The proposed synthesis routes have led to two SF structures, whose structural heterogeneity and nanostructured morphology have been demonstrated using fluorescence microscopy, DLS, SEM and EDX. Two systems with varying shape and size have been obtained: (i) a flat disk-like SF structure with an irregular surface and an in-plane length of about 1–2 μm; (ii) solid SF nanospheres, obtained using ethylene glycol as additive, showing two size populations (main diameters of 0.5 μm and 1.7 μm). Solid nanospherical systems, in particular, show a tendency to arrange into agglomerates that, when loosely bound into smaller particles, can facilitate the delivery at the alveoli. Both formulations exhibited similar drug loading efficiencies, evaluated by HPLC analysis. However, SF nanospheres showed greater in vitro drug release after 24 hours. The demonstrated control over the characteristics imparted to the proposed DD systems may be critical to select the most suitable size/shape to achieve high rates of delivery to the appropriate lung compartment.
1 Introduction
Engineered carriers such as microparticles (MPs) and nanoparticles (NPs) have attracted enormous attention in pharmaceutical technology fields for use in drug delivery (DD) as they provide higher therapeutic and diagnostic performance compared to conventional DD forms.1 MPs and NPs can be used to protect drugs from degradation while allowing their targeted delivery through several strategies, obtaining enhanced bioavailability, efficient drug uptake, and sustained release in proximity of the target site. Other advantages of these new drug formulations include reduced toxicity, increased therapeutic efficacy and tolerability, and improved drug solubility.2,3 MPs and NPs can be prepared by using synthetic biodegradable polymers, such as poly-(lactic acid) (PLA), poly-(ε-caprolactone) (PCL) and poly-(glycolic acid) (PGA), or natural polymers, such as polysaccharides, including cellulose, chitosan, hyaluronic acid, alginate, dextran and starch, or proteins such as collagen, gelatine, elastin, albumin, and silk fibroin.4 Silk fibroin (SF) represents an interesting natural polymer commonly produced by some lepidoptera larvae like silkworms, spiders, scorpions, mites, and flies.5 The most characterized SF derives from Bombyx mori cocoons.6 Bombyx mori SF is obtained from raw silk by a thermochemical process, called degumming, through the separation of sericin.7 The primary structure of Bombyx mori SF is mainly composed of glycine (Gly) (43%), alanine (Ala) (30%) and serine (Ser) (12%). SF is a heterodimeric protein composed of a heavy (H) chain (∼325 kDa) and a light (L) chain (∼25 kDa) interconnected by a single disulfide bond and by P25 protein, a 25 kD glycoprotein associated with the H–L complex by noncovalent interactions. The SF H-chain primary sequence contains alternating hydrophobic and hydrophilic aminoacidic patterns. The peculiar disposition of hydrophobic patterns in β-crystallites provides crystalline features in the silk thread, responsible for its mechanical properties. While the L-chain is hydrophilic and relatively elastic, the P25 protein could play a significant role in maintaining the integrity of the complex.4,7,8 SF shows unique properties suitable for DD applications such as natural accessibility, physical–chemical properties, biocompatibility, and versatility.9 In a pharmacological context, drug loading in SF engineered carriers, and the associated release, can be controlled by tuning the crystalline composition and secondary structure of the SF, so that the properties of the SF are suitable for use in different types of formulations.6,7,10Herein, we evaluated the chemical properties and morphological structure of two inhalable SF-based formulations designed for lung delivery of drugs by inhalation. Inhalation micro-based DD systems offer a new way to treat several pulmonary diseases. When inhaled, the encapsulated drug can enter the lungs, bypassing the bloodstream, with a high local deposition rate.11
Despite the above advantages, the deposition and distribution of inhaled particle systems in the lungs is still a challenge as it is affected by many factors, such as the respiratory rate, the lung volume and the individuals’ health. In addition, particulate systems can deposit in different regions of the respiratory system depending on the particle size and airflow. A good distribution throughout the lung requires particles with an average size distribution ranging between 1 and 5 μm. Smaller particles can reach the alveolar region where Brownian diffusion controls their motion, while particles exceeding 5 μm in size are typically subject to inertial impact in the oropharyngeal region or sedimentation in the bronchial region.11,12 It is worth mentioning that nano-sized particles (average size <100 nm) would be suitable for access to the alveolar tissue, allowing to get high deposition rates in lungs; the uptake rate of these nano-sized nanoparticles by alveolar type II cells is greater than that of alveolar macrophages. On the other hand, it is important to overcome the exhalation tendency, which can significantly reduce the number nanoparticles delivered.13,14
This work aims to develop new inhalable delivery systems with tailored size and shape for the administration of drugs effective in the treatment of pulmonary diseases. The proposed DD systems are fabricated using porous calcium carbonate microparticles (CaCO3 MPs) as templates. The CaCO3 was employed in shaping the particles, and ethylenediaminetetraacetic acid (EDTA) was used to remove the CaCO3 template core through its dissolution. Levothyroxine (3,5,3′,5′-tetraiodothyronine), an L-isomer of thyroxine T4 (L-T4), identical to the endogenous hormone produced by the thyroid gland, served as the model drug for encapsulation.15–17 L-T4 drug was selected18 due to its hydrophobicity that is expected to assist the drug load within amphiphilic SF.19 Previous in vivo studies on murine models have shown that inhalation of L-T4 can mitigate lung damage in idiopathic pulmonary fibrosis.20–22 Encapsulating L-T4 within engineered MCs with potential enhancements in bioavailability and therapeutic efficacy is proposed. The size distribution of the two formulations was characterized using dynamic light scattering (DLS), and the particle morphology, composition, and L-T4 distribution were examined using optical and fluorescence microscopy, scanning electron microscopy (SEM), and energy-dispersive X-ray spectroscopy (EDX).
2 Materials and methods
2.1. Materials
Calcium chloride anhydrous (CaCl2), methanol (MeOH) and sodium fluorescein were purchased from Carlo Erba (Val de Reuil, France). Sodium chloride (NaCl) was purchased from VWR (Carnaxide, Portugal). Rhodamine B (RhoB), 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS) and levothyroxine (L-T4) were purchased from Sigma-Aldrich (St. Louis MO, USA). Ethylenediaminetetraacetic acid (EDTA) was purchased from Acros Organics (Geel, Belgium). Acid 2-(N-morpholino)-ethanesulfonic acid (MES) and dimethyl sulfoxide (DMSO) were purchased from Merck (Darmstad, Germany). Phosphate-buffered saline (PBS, pH 7.4) was purchased from Thermo Fisher (Waltham, USA). Purified water (Milli-Q) was used to perform the experiments.2.2. Purification and fluorescent labelling of SF
Aqueous SF stock solutions were prepared following the steps described in the Rockwood experimental protocol.23 Accordingly, cocoons of Bombyx mori were first cut into small pieces and afterwards boiled for 20 min in an aqueous solution of 0.02 M sodium carbonate to remove the glue-like cladding made of sericin. The obtained degummed fibroin was rinsed thoroughly with Milli-Q water and finally left to dry in a fume hood overnight. After drying, the extracted silk fibroin was dissolved in 9.3 M LiBr solution at 60 °C. To remove the salt, the obtained solution was dialyzed against Milli-Q water for 3 days, using Spectra/Por 6 dialysis membranes with Molecular Weight Cutoff (MWCO) of 10 kDa and flat width of 45 mm (VWR, USA). The solution, appearing optically clear after dialysis, was centrifuged twice at 5000 rpm for 20 min to remove the unsolved portion of fibroin. The final concentration of silk fibroin was determined by weighing the residual solid of a known volume of solution after drying.For fluorescent labelling, 9.8 μl of EDC was added in 4.5 ml of 0.1 M Rhodamine B and 2-(N-morpholino)-ethanesulfonic acid (MES) solution (pH 5.6) with stirring, and the reaction was continued for 15 min. Subsequently, 13.1 mg of NHS was added to the solution and reacted for 1 hour. 500 μl of 1% (v/v) acetic acid was added to the solution to quench the unreacted EDC and allowed to react for another hour. After the reaction, 10 ml of 1.6% (v/v) fibroin solution was added. The reaction continued for 2 h under slow stirring at room temperature. Finally, the solution was dialyzed against Milli-Q water for 1 day using Spectra/Por 6 dialysis membranes with Molecular Weight Cutoff (MWCO) of 10 kDa and a flat width of 45 mm (VWR, USA). The solution (Rho-SF) was optically pink after dialysis.
2.3. Drug fluorescent labelling
L-T4 was labelled following the same standard procedure employed for SF. After the reaction, 10 ml of a solution prepared by dissolving 50 mg of L-T4 in 10 ml of DMSO was added. The reaction continued for 2 hours under slow stirring at room temperature. Finally, the solution was added to 200 ml of Milli-Q water under magnetic stirring to obtain a precipitate. The suspension was centrifuged at 3000 rpm for 3 min to collect T4, which was then washed three times with Milli-Q water, followed by drying at 60 °C for 24 hours. The dry powder (Fluo-L-T4) was optically yellow after drying.2.4. One-pot preparation of L-T4 loaded SF-CaCO3 microcapsules
Silk fibroin-CaCO3 microcapsules (SF-CaCO3 MCs) were synthesized via a co-precipitation reaction between CaCl2 and Na2CO3 precursors in the presence of SF. Briefly, 1.5 ml of SF aqueous solution at a concentration of 0.5% (w/v) was added to 5 ml of 0.1 M aqueous solution of CaCl2. Next, 5 ml of 0.1 M Na2CO3 solution was added to the CaCl2 reaction mixture using a magnetic stirrer, and CaCO3 microparticles (CaCO3 MPs) were formed by the interaction of calcium ions and carbonate ions. The synthesis was carried out using a magnetic stirring speed of 800 rpm for 1 min. The suspension was centrifuged for 3 min at 3000 rpm at room temperature to collect pellets which were then reacted for 30 min with 1.5 ml of 50% (v/v) water/methanol solution to induce the formation of SF β-sheet structure (Silk II) through dehydration.24,25 After 30 min, the solution was centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 min and the supernatant was removed. The as-collected microcapsules (MCs) were resuspended in water and then subjected to sonication (ultrasonic frequencies 37 kHz and power 60%) to prevent aggregation. To synthetise SF-CaCO3 MCs with L-T4 (L-T4@SF-CaCO3 MCs), 500 μl of 0.02 M L-T4/DMSO solution was added in a Becher with CaCl2 and Na2CO3 water solutions and SF, again following the just described one-pot co-precipitation method.
000 rpm for 5 min and the supernatant was removed. The as-collected microcapsules (MCs) were resuspended in water and then subjected to sonication (ultrasonic frequencies 37 kHz and power 60%) to prevent aggregation. To synthetise SF-CaCO3 MCs with L-T4 (L-T4@SF-CaCO3 MCs), 500 μl of 0.02 M L-T4/DMSO solution was added in a Becher with CaCl2 and Na2CO3 water solutions and SF, again following the just described one-pot co-precipitation method.
2.5. Preparation of L-T4-loaded SF-CaCO3 nanospheres
:5, v/v). The Na2CO3 solution was added to a Becher and reacted with the CaCl2 solution using a magnetic stirrer at 1000 rpm, for 30 min. The synthesized particles were collected by sequential washing with ethanol and acetone at 10000 rpm for 5 min to remove unreacted ions and cosolvent molecules.000 rpm for 5 min, washed with ethanol and acetone and then resuspended in Milli-Q water.000 rpm and the obtained pellet was first washed once with 1 ml Milli-Q water to remove excess fibroin, and then reacted for 30 min with 1.5 ml of 50% (v/v) water/methanol solution. Next, the solution was centrifuged at 12000 rpm for 5 min and the supernatant was removed. Finally, the particles were resuspended in water and sonicated (ultrasonic frequencies 37 kHz and power 60%) to prevent aggregation.2.6. EDTA treatment
To dissolve the calcium carbonate core, 50 ml of 0.1 M EDTA solution in 0.4 M KOH in water was initially prepared. Subsequently, L-T4@SF-CaCO3 MCs and L-T4@SF-EG-CaCO3 NSs were centrifuged for 5 min at 12000 rpm to collect pellets. Then, the pellets were then reacted for 30 min with EDTA. After 30 min, the solution was centrifuged at 12000 rpm for 5 min, the supernatant was removed, and the particles were resuspended in Milli-Q water.
2.7. Characterization
The shape, size, and aggregation of particles were analyzed by optical microscopy on dried samples with a Nikon Eclipse Ni-E optical microscope. Image acquisition was performed via NIS-Element software. To identify the fibroin coating and the loaded drug, the particles labelled with fluorophores were examined by a Nikon Eclipse Ni-E microscope with fluorescence imaging optical filters FITC and TRITC. The imaging filters were chosen based on the Fluorescein and Rhodamine B-labelled substrates, respectively. The size and the polydispersity index (PDI) of the samples were determined by dynamic light scattering (DLS, Brookhaven Zeta Plus) with a 665 nm laser source and 15 mW power. Prior to the analysis, the suspensions were first sonicated for 5 min and then diluted to different volumetric ratios with 2 ml of Milli-Q water in dedicated cuvettes. The experiments were replicated three times. The surface and internal morphology of the particles was examined by a crossbeam Zeiss Auriga compact equipped with a Field Emission Scanning Electron Microscope and a Focused Ion Beam (FESEM-FIB). The Electron High Tension (EHT) and magnitude (Mag) used to acquire SEM images are indicated in the captions. FIB sections of selected microparticles were obtained by using a 30 keV accelerated Ga ion beam with a current of 2 pA. After preparation of the particles-based solutions, a drop of each formulation was dispensed on a Si/SiO2 (native oxide) support, left to dry at room temperature and placed on a SEM holder. The analysis of the particles chemical composition was carried out using the Energy Dispersive X-ray Spectroscopy (EDX). The EDX analysis was performed using the above SEM microscope, which is equipped with an Oxford Xplore30 detector, using EHT = 10 keV. Samples were prepared following the same experimental procedure described for SEM.2.8. Determination of drug entrapment efficiency
The particles were redispersed in 4 ml of 0.01 M phosphate-buffered saline (PBS, pH 7.4) containing 10% methanol and sonicated at 82% amplitude for 1 min with a UP200St Hielscher sonicator. The solutions were filtered with disposable syringe filters (Macherey-Nagel, Germany) with pore size of 0.45 μm and the L-T4 concentration in the filtrate was quantified by high-performance liquid chromatography-mass spectrometry (HPLC-MS) system (Agilent LC/MSD XT).2.9. In Vitro drug release studies
The release of L-T4 from the particles was assessed utilizing the dialysis membrane method. Both L-T4@SF-CaCO3 MCs and L-T4@SF-EG-CaCO3 NSs were redispersed in 4 ml of Milli-Q water and then put in a Spectra/Por 6 dialysis membranes with Molecular Weight Cutoff (MWCO) of 10 kDa and flat width of 45 mm (VWR, USA). The membranes were suspended in a Becher containing 250 ml of 0.01 M phosphate-buffered saline (PBS, pH 7.4) containing 10% methanol at 37 °C using magnetic stirrer at 100 rpm. After 0, 6, 12 and 24 hours, 1 ml of release medium was collected for analysis and replaced with an equal amount of fresh release medium to maintain sink condition. The L-T4 content in the release medium was quantified using the previously mentioned HPLC-MS system.3 Results and discussion
3.1. Synthesis and characterization of L-T4 loaded CaCO3 and SF microcapsules
The first method employed in this study was specifically developed for the synthesis of L-T4 loaded SF microcapsules using CaCO3 as sacrificial template. Thanks to their high porosity, non-toxicity, and biocompatibility, porous CaCO3 MPs can be exploited as templates for synthesizing polymer shells designed to encapsulate various drugs.27 However, the use of calcium carbonate for pulmonary DD in inhalation therapy requires the ability to control the size of crystals synthesized from calcium chloride and sodium carbonate precursors. It's crucial to emphasize that a complete exploitation of CaCO3 platforms as a support for the synthesis of inhalable polymeric systems requires that the particles size must not exceed 5 μm. The particles' size, in fact, is one of the main parameters that influence the biodistribution of the drug to the lung.12 The tuning of particles' size can be obtained by varying synthesis parameters such as the mixing speed, precursors' concentration, pH and temperature, as well as by the addition of additives such us divalent cations, organic solvents and synthetic or natural macromolecules.28–30 Co-precipitation is one of the simplest methods to prepare CaCO3 particles via mixing CaCl2 and Na2CO3 in water solution, often in the presence of additives. The resulting particles are polydisperse with a diameter of about 2–3 μm.28–30In the first part of this work, the CaCO3 MPs were synthesized by a one-pot co-precipitation reaction between CaCl2 and Na2CO3 in the presence of SF. The particles were characterized using SEM to determine their size and morphology. The introduction of SF during the one-pot co-precipitation step led to a regular and homogeneous SF-CaCO3 MCs shape with a size ranging between 2 and 3 μm. The particles showed a spherical morphology with a highly porous structure, typical of vaterite particles, and a high aggregation tendency. However, some particles exhibited a rhombohedral morphology structure with laminated surface, characteristic of calcite particles, one of the most stable CaCO3 polymorphs (Fig. 1A and B).27 SEM analysis suggests that size and shape of SF-CaCO3 MCs are influenced by the presence of SF during the one-pot reaction, probably due to interactions between Ca2+ cations with acidic SF residues.31 SF, which initially had a mostly random coil structure (Silk I), after the methanol treatment switches in its β-sheet conformation state (Silk II), replicating the shape and dimensionality of the inorganic core and forming a coating layer around it. The driving force behind the formation of β-sheet crystallites is attributed to the formation of intermolecular hydrogen bond. The addition of methanol facilitates the removal of water surrounding silk fibroin chains, accelerating the formation of β-sheet nuclei.24,25
 | ||
| Fig. 1 SEM images of SF-CaCO3 MCs without L-T4 at low magnification ((A), Mag 7.63k×, EHT 1.00 kV) and high magnification ((B), Mag 20.00k×, EHT 1.00 kV). | ||
The as-fabricated SF-CaCO3 MCs were employed as a template for drug encapsulation. L-T4 was previously dissolved in DMSO due to the drug's limited solubility in water or organic solvents such as methanol or ethanol.18 Upon encapsulation of L-T4, the geometry, morphology, size and state of aggregation of the L-T4-loaded SF-CaCO3 MCs remained unaltered (Fig. 2A), suggesting that the drug may be absorbed inside the particles during their formation under a one-pot co-precipitation reaction. The relatively high entrapment efficiency of L-T4 within SF-CaCO3 MCs might be ascribed to the hydrophobic nature of L-T4, which could initially favour its attachment to the bulk hydrophobic domains of SF via hydrophobic interactions.32 These hydrophobic interactions can be consequently enhanced and stabilized after the methanol treatment, which promotes the strengthening of hydrophobic sites upon removal of water and assists the formation of hydrophobic SF β-sheet crystallites, respectively. In this manner, the drug coordinates with the hydrophobic region of fibroin that forms a coating layer around the CaCO3 core.
 | ||
| Fig. 2 SEM images of SF-CaCO3 MCs with L-T4 before (A) and after (B) 0.1 M EDTA treatment for 30 minutes. Optical microscopy images in transmitted light of MCs before treatment (C1) and after treatment (D4). Fluorescence microscopy images: green colour indicates the presence of fluorescein-labelled L-T4 on the surface of the MCs before (C2) and after treatment (D5), red colour indicates the presence of rhodamine B-labelled fibroin layer before (C3) and after treatment (D6). EDX spectrum before EDTA treatment indicates the presence of iodine and calcium (E), while after the EDTA treatment it indicates the presence of iodine and nitrogen, and the absence of calcium (F). | ||
As reported in the materials and methods section, the SF and the drug were tagged with Rhodamine B and Fluorescein by EDC-NHS coupling. Fluorescent microcapsules were examined using fluorescence microscopy to confirm the absorption of the drug and to visualize its spatial arrangement within them. Fig. 2C reports the optical and fluorescence images of the microcapsules with Rho-SF and Fluo-L-T4, showing aggregates of spherical and rhombohedral particles. Fluorescence from the particles' surface confirms the presence of the polymer and demonstrates successful drug adsorption. From the acquired micrographs, it is also visible the conformational switching of some CaCO3 MPs from vaterite to a rhombohedral morphology typical of calcite particles.33,34 It is worth noting that although some CaCO3 particles have a rhombohedral morphology, they are covered with SF to the same extent as amorphous particles. Moreover, L-T4 and SF are homogeneously adsorbed onto the template surface, suggesting that L-T4 is solubilized in the hydrophobic regions of crystallized SF in a stable manner. The lower fluorescence seen in the central part of the microcapsules and the higher fluorescence at the edges, suggest that the SF-L-T4 coating interacts exclusively with the surface regions of the CaCO3 templates. Thus, it is likely that CaCO3 MPs are quickly generated during the one-pot synthesis process and that their coating by the SF–L-T4 complex, which is favoured by the interaction between SF and CaCO3, takes place after the formation of templates.
EDX spectroscopy indicates the formation of peaks corresponding to the presence of calcium, oxygen, and carbon, which are markers for the CaCO3 core, and peaks corresponding to the presence of iodine, which are a sign of the drug. Nitrogen, which could be exploited as a marker for proteins, was not detected through the EDX analysis, being hidden by the higher calcium and carbon peaks because of its low atomic mass resulting in a weak X-ray emission. For this reason, it is very difficult to clearly detect nitrogen in the EDX spectrum and to discriminate it from the background noise (Fig. 2E).
To obtain hollow L-T4 loaded SF MCs (L-T4@SF MCs), the cores were removed by EDTA solution. EDTA was chosen due to its chelating properties; it acts as an agent capable of fixing and sequestering various metal ions, such as Ca2+ and Fe3+, forming stable and water-soluble complexes.35,36 After core removal, SEM images showed the folded nature of the hollow fibroin and the complete dissolution of the CaCO3 core. The hollow L-T4@SF MCs exhibited a flat disk-like structure with an irregular and rough surface, and an in-plane size of about 1–2 μm, being the out-of-plane size far lower. Moreover, the core removal significantly affected the particle size, approximately reducing it by 50% (Fig. 2B). Some studies have highlighted that the particles shape37 and morphology38 can influence the aerosolization and deposition of inhaled powders. On one hand, the particles' shape plays an effective role in governing the aerodynamics of inhalable carriers. On the other hand, the increase in surface roughness of particles may be attributed to a smaller aerodynamic diameter and, contextually, a reduced aerodynamic diameter implies that particles are more likely to reach the deep lung.39
Fluorescence analysis and EDX spectrometry were carried out to identify the presence of SF, the drug adsorption and subsequent removal of the CaCO3 core after the EDTA treatment. Micrographs reported in Fig. 2D show aggregates of flat spherical particles, as previously highlighted by the SEM analysis. Red and green fluorescence on the surface of microcapsules demonstrated that SF and L-T4 were not dissolved after the removal of CaCO3 templates by EDTA treatment (Fig. 2D). The EDX graph displayed a peak corresponding to the presence of the iodine. A peak corresponding to nitrogen was detected thanks to the almost complete elimination of calcium ions, resulting in a lower background not affecting the appearance of the nitrogen peak (Fig. 2F). Finally, the comparison between EDX analysis of filled (Fig. 2E) and hollow (Fig. 2F) MCs confirms that EDTA treatment results in a significant decrease in calcium atomic percentage, confirming the dissolution of the CaCO3 core.
3.2. Synthesis and characterization of L-T4-loaded CaCO3 and SF nanospheres
The second part of this work was targeted at the tuning of the particle's dimensions and structure. This is because the features of an efficient delivery system must respect some restrictions in particles' dimension, and micro and nano carriers must respond to specific structural characteristics aimed at avoiding interaction with macrophages while, maintaining a strong target interaction and an easily controlled drug release. To obtain these features, we have developed an optimized synthesis method for implementing potentially more effective delivery systems. Specifically, some synthesis parameters such as reaction time and mixing speed were adapted, while the synthesis of CaCO3 templates was modified by adding EG and completed by inducing the reaction with SF in a two-step synthesis process. The use of EG prevents the conversion of vaterite into calcite upon suppressing the related crystals growth. In this way, CaCO3 maintained a spherical morphology, and a reduced polydispersity of CaCO3 templates was previously observed.29,30 The resulting particles were analysed using DLS to obtain the average diameter and the PDI, and successively they were imaged by SEM. We obtained uniform and monodispersed EG-CaCO3 spheres, with a diameter of 1177.3 ± 2 nm and a PDI of 0.005 (Table 1). SEM analysis showed dispersed EG-CaCO3 MPs with a diameter of about 800–900 nm and a highly porous surface (Fig. 3A and B). As expected, SEM images on dry samples showed a smaller size of the EG-CaCO3 particles compared with results from DLS, as the latter technique provides an estimation of the hydrodynamic diameter of the particles.40 Images acquired upon Focused Ion Beam (FIB) milling of the EG-CaCO3 MPs also showed the presence of internal porosity in the analysed sample (Fig. 3C). The average pore size of 50 nm is comparable to the size of the grains observed on the particles surface (Fig. 3C). Thus, the direct mixing of soluble salts of Ca2+ and CO32− resulted in an amorphous precipitate that induces the formation of aggregated CaCO3 particles, also characterized by an extremely porous morphology.| Sample | Particles size (nm) | Polydispersity index |
|---|---|---|
| EG-CaCO3 | 1177.3 ± 2 | 0.005 |
| L-T4@EG-CaCO3 | 156.8 ± 11; 1977.6 ± 163 | 0.38 |
| L-T4@SF-EG-CaCO3 | 602 ± 78; 3502.38 ± 461 | 0.4 |
| L-T4@SF NSs | 493.1 ± 21.4; 1736.8 ± 81.3 | 0.25 |
 | ||
| Fig. 3 SEM images at different magnification of porous EG-CaCO3 MPs: top view ((A), Mag 20.00k×, EHT 2.00 kV, (B) Mag 50.00k×, EHT 2.00 kV), cross-section ((C1), Mag 80.00k×, EHT 2.00 kV, (C2), Mag 250.00k×, EHT 2.00 kV), porous surface ((C3), 80.00k×, EHT 2.00 kV). | ||
To evaluate the drug adsorption capability of the EG-CaCO3 MPs, we investigated the encapsulation of the L-T4 model molecule. The drug was first conjugated with fluorescein through EDC-NHS coupling and then dissolved in DMSO. Next, CaCl2 and Na2CO3 were mixed with an L-T4/DMSO solution, facilitating the deposition of the drug onto the CaCO3 core. In the case of L-T4@EG-CaCO3, we obtained two particles' populations with an average diameter of 156.8 ± 11 nm and 1977.6 ± 163 nm, with a PDI of 0.38 (Table 1). The presence of two different populations and the increase in the PDI from 0.005 to 0.38 index after drug deposition could be ascribed to the aggregation effects of the particles. The aggregation tendency of the particles was confirmed by SEM. We obtained spherical, aggregate, and porous loaded particles, with a slight solubilization. The drug was absorbed inside the porous particle during co-precipitation reaction, being unreacted excess of L-T4 dispersed on the spherical EG-CaCO3 MPs due to their flat and thin sheet-like configuration (Fig. 4A). This can be caused by the EG-CaCO3 high insolubility in water.
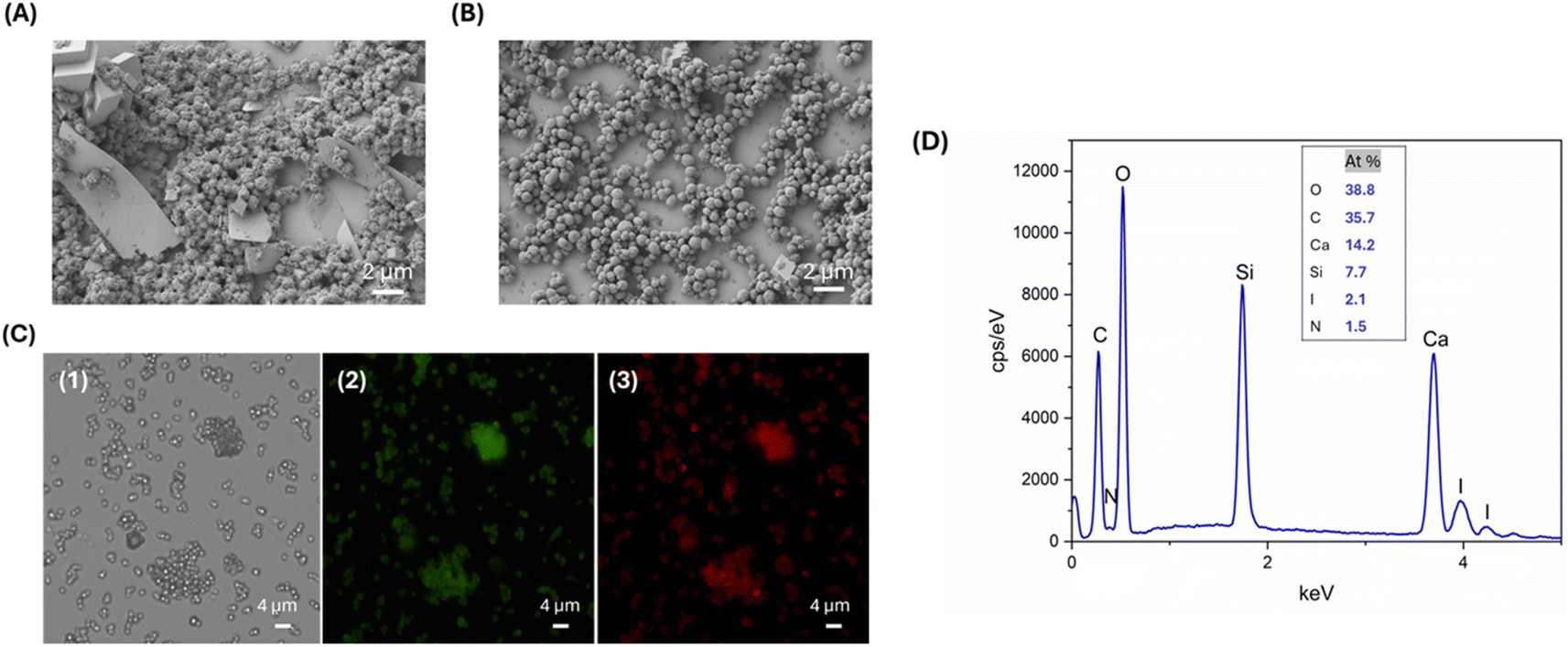 | ||
| Fig. 4 SEM images of L-T4@EG-CaCO3 particles without (A) and with SF (B). Optical microscopy image in transmitted light of L-T4@EG-CaCO3 particles with SF before EDTA treatment (C1). Fluorescence microscopy images of L-T4@EG-CaCO3 with SF before EDTA treatment: green colour indicates the presence of fluorescein-labelled L-T4 (C2), red colour indicates the presence of rhodamine B-labelled SF (C3). Representative measurement of the EDX spectrum before EDTA treatment which indicates the presence of iodine and nitrogen (D). | ||
The porosity of the L-T4@EG-CaCO3 particles could be exploited to adsorb the coating biopolymer and, consequently, to obtain solid fibroin spheres loaded by the drug.
The deposition of SF to L-T4@EG-CaCO3 particles was achieved by adding 0.5% rhodamine-labelled SF into an aqueous suspension of L-T4@EG-CaCO3, followed by a methanol treatment. This method allows the adsorption of the polymer on the surface of nanometre-sized pores inside the CaCO3 template, which is driven by molecular interactions.33 The effect of SF on the morphology and size of the particles was assessed by DLS and SEM analyses. The SEM images of the sample in Fig. 4B showed uniform and homogeneously sized, highly porous nanospheres (NSs) that tend to aggregate each other. Comparing the results from particles with and without SF coverage, we observed that SF in L-T4@EG-CaCO3 hampered the template solubilization, protecting in this way the particle structure of loaded T4. In this respect, the L-T4@SF-EG-CaCO3 spheres were 600 nm in size and showed a more defined structure (Fig. 4B), suggesting that SF switching to a β-sheet conformation promotes an interaction within the porous structure of CaCO3 core that confers higher stability to the whole system. The DLS analysis confirmed the particle size and their state of aggregation, as found by SEM, with two particle distributions (PDI of 0.4) having diameters of 602 ± 78 nm and 3502.38 ± 461 nm (Table 1).
Drug loading efficiency and deposition of SF onto the CaCO3 core were analyzed by fluorescence microscopy and EDX. Fig. 4C illustrates the optical and the fluorescence images of the particles with Fluo-L-T4 and Rho-SF. The presence of green and red fluorescence on the L-T4@EG-CaCO3 particles with SF confirms the spatial distribution and large adsorption of the drug and polymer into the CaCO3 particles. The EDX plot displayed a peak corresponding to the presence of the iodine, a fingerprint for L-T4, and a peak related to the presence of the nitrogen, characteristic of SF (Fig. 4D). The comparison of the EDX results from the two DD systems (Fig. 2D and 4D) reveals that a nitrogen peak missing in the first case is instead observed in the second one. These findings suggest that in the second case the concentration of calcium may be lower, as expected because of the porous nature of the CaCO3 template, also indicating that a higher SF content due to its adsorption in the porous template may take place at the same time.
Finally, the residual CaCO3 template was dissolved using EDTA solution to obtain L-T4 loaded SF NSs (L-T4@SF NSs). SEM images of L-T4@SF NSs after EDTA treatment are reported in Fig. 5, panels A and B. The template removal had an important impact on size reduction. The acquired images revealed the formation of solid L-T4-filled SF NSs with a diameter varying from 100 to 400 nm, arranged in clusters of microstructures of 1.5 μm in size. It is worth noting that the solid nature of as-prepared carriers confirms that the intercalation of SF into CaCO3 pores occurred. Single detached nanoparticles with size of about 100 nm are also observed, suggesting that a weak interaction between the NSs composing the observed clusters takes place. Consequently, the aggregation by weak interactions may have a potential enhancing effect in a DD scenario. Indeed, when the aggregate is close to the target tissue, a detachment of small NSs composing it, may exponentially improve the interaction within the tissue. At the same time, small NSs reduce the occurrence of phagocytosis events that are physiologically expected in case of large objects circulating through the body. DLS analysis confirmed the presence of two different particles populations with diameters of 493.1 ± 21.4 nm and 1736.8 ± 81.3 nm, respectively, and a polydispersity of 0.25 (Table 1).
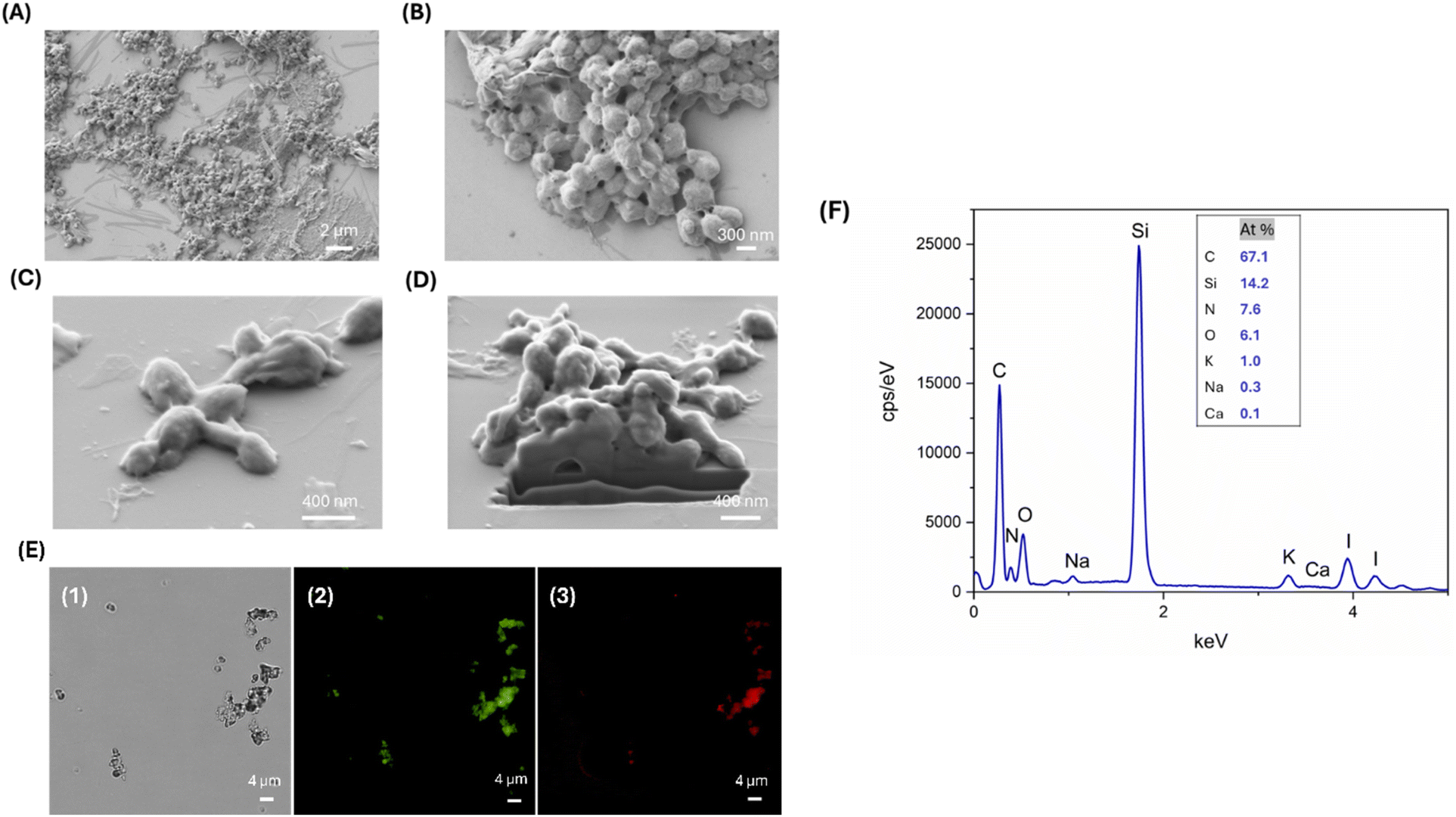 | ||
| Fig. 5 Analysis of L-T4@SF NSs aggregates, after treatment with 0.1 M EDTA for 30 minutes. SEM images at different magnification: top view ((A), Mag 10.00k×, EHT 1.00 kV, (B), Mag 50.00k×, EHT 1.00 kV), surface ((C), Mag 112.50k×, EHT 2.00 kV) cross-section ((D), Mag 80.00k×, EHT 2.00 kV). Optical microscopy image in transmitted light (E1). Fluorescence microscopy images: green colour indicates the presence of fluorescein-labelled L-T4 (E2), red colour indicates the presence of rhodamine B-labelled SF (E3). Representative measurement of the EDX spectrum after EDTA treatment which indicate the presence of iodine and nitrogen (F). | ||
The solid nature of the particles demonstrated that there was a filling-in of the rough and porous CaCO3 surface due to diffusion and subsequent adsorption of both the model drug and SF. However, when the template was removed, the shells still retained the structure of the core. Fig. 5C and D shows a SEM image acquired after performing a FIB micro sectioning. This shows a lack of internal porosity in the EDTA-treated particles, suggesting that the sample consists only of SF and L-T4 and not calcium carbonate.
Images from the fluorescence microscopy show that it is clearly visible the maintenance of L-T4 loading after the template removal by EDTA treatment (Fig. 5E). This evidence confirms the direct interplay between SF and L-T4, consisting of a direct interaction of L-T4 molecules through hydrophobic interaction within the β-crystallites of SF particles. The final composition of L-T4@SF NSs after EDTA treatment is shown in Fig. 5F. The EDX plot displayed a nitrogen peak ascribable to the presence of SF and an iodine peak indicating the presence of L-T4. Furthermore, the absence of a calcium peak confirms the successful dissolution of calcium carbonate cores upon the EDTA treatment.
3.3. Drug loading capacity
Levothyroxine is a drug characterized by poor water solubility, high lipophilicity, and low stability, which makes it suitable for encapsulating SF-particles. SF-based particulate carriers have been reported as particularly promising for the delivery of various drugs, especially hydrophobic ones.41,42 In order to compare the two different formulations, we decided to quantify the amount of drug entrapped within the particles using an ultrasonic sonicator, with the aim of disrupting the particulate suspensions to allow the drug release into the medium. Based on the HPLC-MS analyses, it was found that at pH 7.4, the concentration of levothyroxine in the medium was 16.16 μg ml−1 for L-T4@SF MCs and 12.51 μg ml−1 for L-T4@SF NSs (Table 2).| Sample | Drug loading (μg ml−1) | Drug released after 24 h (ng ml−1) | Drug released after 24 h (%) |
|---|---|---|---|
| L-T4@SF MCs | 16.16 ± 0.10 | 485 ± 13.84 | 47 ± 2.9 |
| L-T4@SF NSs | 12.51 ± 0.67 | 565 ± 20.68 | 73 ± 3.7 |
3.4. In vitro drug release characterization
In vitro drug release studies on L-T4@SF MCs and L-T4-@SF NSs were conducted at 37 °C in PBS buffer with 10% of MeOH at pH 7.4 to mimic the physiological pH environment. After 24 hours, L-T4@SF MCs and L-T4@SF NSs released 47% and 73% of the drug, respectively. These results indicate that L-T4@SF NSs exhibits a higher drug release percentage compared that L-T4@SF MCs. The release of a higher percentage of the drug by the optimized system is reasonably due to the greater surface-to-volume ratio and the spatial distribution of the drug, in addition the smaller primary particle size provides a more efficient release.Literature shows various SF-based systems employed as carriers for drug delivery. Within our experimental approach and the proposed methods, we focused on synthesizing two distinct particulate systems based on SF. The aim was to primarily achieve spherical bio-polymeric carriers of suitable dimensions for drug encapsulation and release in the lungs. Comparing our optimized nanoparticles with those from other experimental study, we highlight significant differences in size43,44 and even in morphology.25 Our method led to the assembling of solid particles of spherical shape and reduced dimensions, potentially enhancing drug release efficacy and bioactivity in the desired therapeutic context. Preliminary drug release analyses for the herein proposed systems have shown that both of them can release an adequate quantity of drug upon in vitro tests (Table 2).
The as-found contents of entrapped and released T4 are in line with a previous in vivo experimental study in which the aerosolized thyroid hormone was administered by inhalation to mice at a dose of 40 μg kg−1, resulting in a drug concentration in the mouse plasma of 5 ng ml−1.20 These findings indicate that in this work, the amounts of drug encapsulated and released after 24 hours by both systems meet the requirements for targeted therapy in the lungs. In particular, it is possible to benefit from all the advantages of a nanostructured delivery systems, in so far as they stabilize the drug and deliver it much more effectively to the target tissue.
4 Conclusions
DD carriers are currently implemented using phospholipids and bio-inspired materials, among which SF has found fertile ground as a promising material for developing delivery systems. The application of SF-based systems effective in DD in the lungs has been achieved in this study. In particular, two systems consisting of SF carriers loaded with the hydrophobic L-T4 molecule (a drug for the treatment of IPF) and having different size and shape, have been obtained using a one-pot co-precipitation method involving CaCO3 templates, SF and L-T4 and a two-step coating method. These methods aim to promote the controlled assembly of SF micro/nano carriers efficiently loaded with T4 after dissolution of the solid template by EDTA treatment. We first showed a flat disk-like structure with an irregular and rough surface, having an average in-plane length of about 1–2 μm, which is compatible with delivery to the pulmonary region. Importantly, the demonstrated system is aerodynamically suited to implement an efficient delivery upon aerosolization. Furthermore, we have proven that the use of additives, in this specific case EG, allows obtaining porous CaCO3 templates as a first stage of the particle formation by the same one-pot precipitation method. Upon dissolution of the templates, we found that SF inclusion in their porous structure promoted the assembly of solid carriers loaded into their bulk by L-T4, thanks to hydrophobic interactions between the drug and the β-sheet crystallites in the SF. Such carriers are indeed characterized by two different size populations of about 500 and 1750 nm (i.e. again compatible with delivery to the lungs) and are presented predominantly in the form of clusters in which the particles' aggregation is governed by weak interactions. The latter aspect may be important in terms of the promoting efficient delivery systems that, once at the treatment site, can undergo a process of disaggregation, favouring the tissue–drug interaction and reducing the adverse events of phagocytosis. The in vitro release also indicated a significantly higher percentage release by the optimized solid carriers compared to the non-optimized system. This finding strengthens the potential application of SF-based nanospheres as effective release systems in a plethora of pathological scenarios.Data availability
Data for this article are available at zenodo, https://zenodo.org/api/records/12772524/draft/files/DASDataAvailabilityStatement.zip/content.Author contributions
I. D. O. writing – original draft, investigation, methodology, formal analysis, visualization; G. D. G. writing – original draft, investigation, methodology, formal analysis; R. S. conceptualization, methodology; D. V. writing – review & editing, validation, formal analysis; A. L. writing – review & editing, validation, formal analysis; E. D. writing – review & editing, validation, formal analysis; G. Trevisi investigation, visualization; M. B. writing – review & editing, funding acquisition; V. G. writing – review & editing, validation; G. Tarabella supervision, writing – review & editing, funding acquisition; P. D. A. conceptualization, supervision, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was carried out under the PRIN project SMARTPROSYS: Progetti di ricerca di Rilevante Interesse Nazionale – Bando 2022, No. 2022T5MN35, CUP: D53D23012000006. G. Tarabella and P. D. A. gratefully acknowledge the support from the European Union's Horizon Europe research and innovation program under the Pathfinder Challenge grant agreement No. 101115545—IV-Lab. We wish to thank Prof. Ruggero Bettini, University of Parma, for providing L-T4, and Dr Salvatore Iannotta for fruitful discussions.Notes and references
- R. Y. P. da Silva, D. L. B. de Menezes, V. da S. Oliveira, A. Converti and Á. A. N. de Lima, Int. J. Mol. Sci., 2023, 24, 5441 CrossRef CAS PubMed.
- H. Rahmani, A. Fattahi, K. Sadrjavadi, S. Khaledian and Y. Shokoohinia, Adv. Pharm. Bull., 2019, 9, 601–608 CrossRef CAS PubMed.
- R. Singh and J. W. Lillard, Exp. Mol. Pathol., 2009, 86, 215–223 CrossRef CAS PubMed.
- Z. Zhao, Y. Li and M. Bin Xie, Int. J. Mol. Sci., 2015, 16, 4880–4903 CrossRef CAS PubMed.
- G. H. Altman, F. Diaz, C. Jakuba, T. Calabro, R. L. Horan, J. Chen, H. Lu, J. Richmond and D. L. Kaplan, Biomaterials, 2003, 24, 401–416 CrossRef CAS PubMed.
- E. Wenk, H. P. Merkle and L. Meinel, J. Controlled Release, 2011, 150, 128–141 CrossRef CAS PubMed.
- A. Reizabal, C. M. Costa, L. Pérez-Álvarez, J. L. Vilas-Vilela and S. Lanceros-Méndez, Adv. Funct. Mater., 2023, 33, 2210764 CrossRef CAS.
- E. S. Sashina, A. M. Bochek, N. P. Novoselov and D. A. Kirichenko, Russ. J. Appl. Chem., 2006, 79, 869–876 CrossRef CAS.
- G. De Giorgio, B. Matera, D. Vurro, E. Manfredi, V. Galstyan, G. Tarabella, B. Ghezzi and P. D’Angelo, Bioengineering, 2024, 11, 167 CrossRef CAS PubMed.
- L. Lin, Y. Zhou, G. Quan, X. Pan and C. Wu, Int. J. Pharm., 2021, 607, 120974 CrossRef CAS PubMed.
- Q. Wan, X. Zhang, D. Zhou, R. Xie, Y. Cai, K. Zhang and X. Sun, J. Nanobiotechnol., 2023, 21, 215 CrossRef CAS PubMed.
- G. Li, D. Liu and Y. Y. Zuo, Nanomedicine, 2022, 469–499 Search PubMed.
- M. Beck-Broichsitter, O. M. Merkel and T. Kissel, J. Controlled Release, 2012, 161, 214–224 CrossRef CAS PubMed.
- M. Griese and D. Reinhardt, J. Drug Target., 1998, 5, 471–479 CrossRef CAS PubMed.
- A. J. HULBERT, Biol. Rev. Cambridge Philos. Soc., 2000, 75, 519–631 CrossRef CAS PubMed.
- S. Mondal and G. Mugesh, Angew. Chem., 2015, 127, 10983–10987 CrossRef.
- A. Aleskndrany and I. Sahin, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183245 CrossRef CAS PubMed.
- A. Barreira, A. F. M. Santos, M. Dionísio, A. R. Jesus, A. R. C. Duarte, Ž. Petrovski, A. B. Paninho, M. G. Ventura and L. C. Branco, Int. J. Mol. Sci., 2023, 24, 8822 CrossRef CAS PubMed.
- B. Subia and S. C. Kundu, Nanotechnology, 2013, 24, 035103 CrossRef CAS PubMed.
- G. Yu, A. Tzouvelekis, R. Wang, J. D. Herazo-Maya, G. H. Ibarra, A. Srivastava, J. P. W. De Castro, G. Deiuliis, F. Ahangari, T. Woolard, N. Aurelien, R. A. E. Drigo, Y. Gan, M. Graham, X. Liu, R. J. Homer, T. S. Scanlan, P. Mannam, P. J. Lee, E. L. Herzog, A. C. Bianco and N. Kaminski, Nat. Med., 2018, 24, 39–49 CrossRef CAS PubMed.
- O. Barca-Mayo, X. H. Liao, C. DiCosmo, A. Dumitrescu, L. Moreno-Vinasco, M. S. Wade, S. Sammani, T. Mirzapoiazova, J. G. N. Garcia, S. Refetoff and R. E. Weissa, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1321–1329 CrossRef PubMed.
- L. Li, X. Nie, M. Yi, W. Qin, F. Li, B. Wu and X. Yuan, Front. Oncol., 2020, 10, 528686 CrossRef PubMed.
- D. N. Rockwood, R. C. Preda, T. Yücel, X. Wang, M. L. Lovett and D. L. Kaplan, Nat. Protoc., 2011, 6, 1612–1631 CrossRef CAS PubMed.
- X. Chen, Z. Shao, D. P. Knight and F. Vollrath, Proteins: Struct., Funct., Genet., 2007, 68, 223–231 CrossRef CAS PubMed.
- B. Park, S. K. Ramesh, S. W. Rhee and J. Kim, Bull. Korean Chem. Soc., 2023, 44, 274–279 CrossRef CAS.
- F. Persano, C. Nobile, C. Piccirillo, G. Gigli and S. Leporatti, Nanomaterials, 2022, 12, 1494 CrossRef CAS PubMed.
- Y. Q. Niu, J. H. Liu, C. Aymonier, S. Fermani, D. Kralj, G. Falini and C. H. Zhou, Chem. Soc. Rev., 2022, 51, 7883–7943 RSC.
- P. Fadia, S. Tyagi, S. Bhagat, A. Nair, P. Panchal, H. Dave, S. Dang and S. Singh, 3 Biotech, 2021, 11, 457 CrossRef PubMed.
- F. Persano, C. Nobile, C. Piccirillo, G. Gigli and S. Leporatti, Nanomaterials, 2022, 12, 1494 CrossRef CAS PubMed.
- D. B. Trushina, T. V. Bukreeva and M. N. Antipina, Cryst. Growth Des., 2016, 16, 1311–1319 CrossRef CAS.
- C. Cheng, Z. Shao and F. Vollrath, Adv. Funct. Mater., 2008, 18, 2172–2179 CrossRef CAS.
- A. Moin, S. U. D. Wani, R. A. Osmani, A. S. Abu Lila, E. S. Khafagy, H. H. Arab, H. V. Gangadharappa and A. N. Allam, Drug Delivery, 2021, 28, 1626–1636 CrossRef CAS PubMed.
- A. M. Ferreira, A. S. Vikulina and D. Volodkin, J. Controlled Release, 2020, 328, 470–489 CrossRef CAS PubMed.
- Y. Mori, T. Enomae and A. Isogai, Mater. Sci. Eng., C, 2009, 29, 1409–1414 CrossRef CAS.
- C. Oviedo and J. Rodríguez, Quim. Nova, 2003, 26, 901–905 CrossRef CAS.
- D. V. Volodkin, A. I. Petrov, M. Prevot and G. B. Sukhorukov, Langmuir, 2004, 20, 3398–3406 CrossRef CAS PubMed.
- S. K. Shukla, A. Sarode, D. D. Kanabar, A. Muth, N. K. Kunda, S. Mitragotri and V. Gupta, Mater Sci Eng C Mater Biol Appl., 2021, 128, 112324 CrossRef CAS PubMed.
- A. Negi, S. Nimbkar and J. A. Moses, Pharmaceutics, 2023, 15, 2706 CrossRef CAS PubMed.
- M. S. Hassan and R. W. M. Lau, AAPS PharmSciTech, 2009, 10, 1252–1262 CrossRef CAS PubMed.
- W. E. Soliman, S. Khan, S. M. D. Rizvi, A. Moin, H. S. Elsewedy, A. S. Abulila and T. M. Shehata, Nanomaterials, 2020, 10, 1–13 Search PubMed.
- A. B. Mathur and V. Gupta, Nanomedicine, 2010, 5, 807–820 CrossRef CAS PubMed.
- J. G. Hardy, L. M. Römer and T. R. Scheibel, Polymer, 2008, 49, 4309–4327 CrossRef CAS.
- Z. Cao, X. Chen, J. Yao, L. Huang and Z. Shao, Soft Matter, 2007, 3, 910–915 RSC.
- P. Shi and J. C. H. Goh, Int. J. Pharm., 2011, 420, 282–289 CrossRef CAS PubMed.
Footnote |
| † Present address: Henesis srl, Strada Budellungo, 2, 43123, Parma, Italy. |
| This journal is © The Royal Society of Chemistry 2024 |