
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design, synthesis and potent anti-pancreatic cancer activity of new pyrazole derivatives bearing chalcone, thiazole and thiadiazole moieties: gene expression, DNA fragmentation, cell cycle arrest and SAR†
Monica G. Kamela,
Farid M. Sroor *b,
Mahmoud KH. Hanafyc,
Karima F. Mahrousd and
Hamdi M. Hassaneen*a
*b,
Mahmoud KH. Hanafyc,
Karima F. Mahrousd and
Hamdi M. Hassaneen*a
aDepartment of Chemistry, Faculty of Science, Cairo University, Giza, Egypt. E-mail: hamdi_251@yahoo.com
bOrganometallic and Organometalloid Chemistry Department, National Research Centre, Cairo, 12622, Egypt. E-mail: faridsroor@gmx.de; fm.sroor@nrc.sci.eg
cBioassay-Cell Culture Laboratory, National Research Centre, Dokki, 12622, Egypt
dCell Biology Department, National Research Centre, Dokki, 12622, Egypt
First published on 27th August 2024
Abstract
Less than 5% of pancreatic cancer patients survive for more than five years after diagnosis. Therefore, there is an urgent need for novel therapeutic drugs to treat pancreatic cancer. Herein, we report the synthesis and full characterization of fifteen novel pyrazole derivatives bearing chalcone (4–10), thiazole (16–19) and thiadiazole (23–26) moieties. All the newly synthesized pyrazole derivatives were tested in vitro as anti-cancer agents against pancreatic cancer (PaCa-2), breast cancer (MCF-7), prostate cancer (PC3), and normal cell lines (BJ1). Three pyrazolyl-chalcone derivatives (4, 5, and 7) and a pyrazolyl-thiadiazole derivative (25) showed potent anti-cancer activity against the PaCa-2 cell line with IC50 values of 13.0, 31.5, 24.9, and 5.5 μg mL−1, respectively, compared with doxorubicin (IC50 = 28.3 μg mL−1). Compound 25 showed potent anti-cancer activity against the PC3 cell line with an IC50 value of 11.8 μg mL−1. In contrast, compounds 4, 5 and 7 are safer against the normal human-cell line (BJ1) with IC50 values of 74.2, 76.6 and 81.1 μg mL−1, respectively, compared with compound 25, which has an IC50 value of 23.7 μg mL−1. The mechanism of action of compounds 4, 5 and 7 against pancreatic cancer cells was studied by investigating gene expression, DNA fragmentation, comet assay and flow cytometry experiments using doxorubicin as a reference drug. Moreover, the structure–activity relationship between the structures of these compounds and their biological properties was discussed.
1. Introduction
Uncontrolled cell growth can generate cancer cells, which is a multifactorial disease and kills millions of people each year worldwide. Cancer is the cause of one in six deaths. In 2018, 18.1 million people were diagnosed with cancer globally, and it is expected that this number will increase to 29.4 million in 2040.1 Cancer treatments depend on a range of techniques, such as surgery, radiation, and chemotherapy, which can be used separately or together.1,2 Side effects and multidrug resistance (MDR) are the main obstacles in cancer therapy. Similarly, pancreatic cancer, also known as pancreatic adenocarcinoma (PDAC), is one of the most aggressive types of tumors in the world and is reported as the primary cause of cancer-related deaths.3,4 Moreover, less than 5% of pancreatic cancer patients have a survival rate exceeding five years.5–7 Therefore, there is an urgent need for novel therapeutic drugs to treat pancreatic cancer.Chalcone and its derivatives, which have an α,β-unsaturated system with three carbons, exhibit diverse biological properties and a preferred synthon to generate different kinds of heterocyclic rings.8–11 Chalcones are a member of the flavonoid family; moreover, the chemical structure of chalcone has been reported in many natural products, including vegetables, teas, fruits, and spices.12,13 Furthermore, they are used as anti-inflammatory,14 antiobesity,15 thioredoxin reductase inhibitory,16 antiprotozoal,17 antiplatelet,18 tubulin polymerization inhibitory,19 antidiabetic,20 antitubercular,21 antibacterial,8,22 anti-Alzheimer,23 and antimalarial agents24 (some biologically active chalcone derivatives are given in Fig. 1).
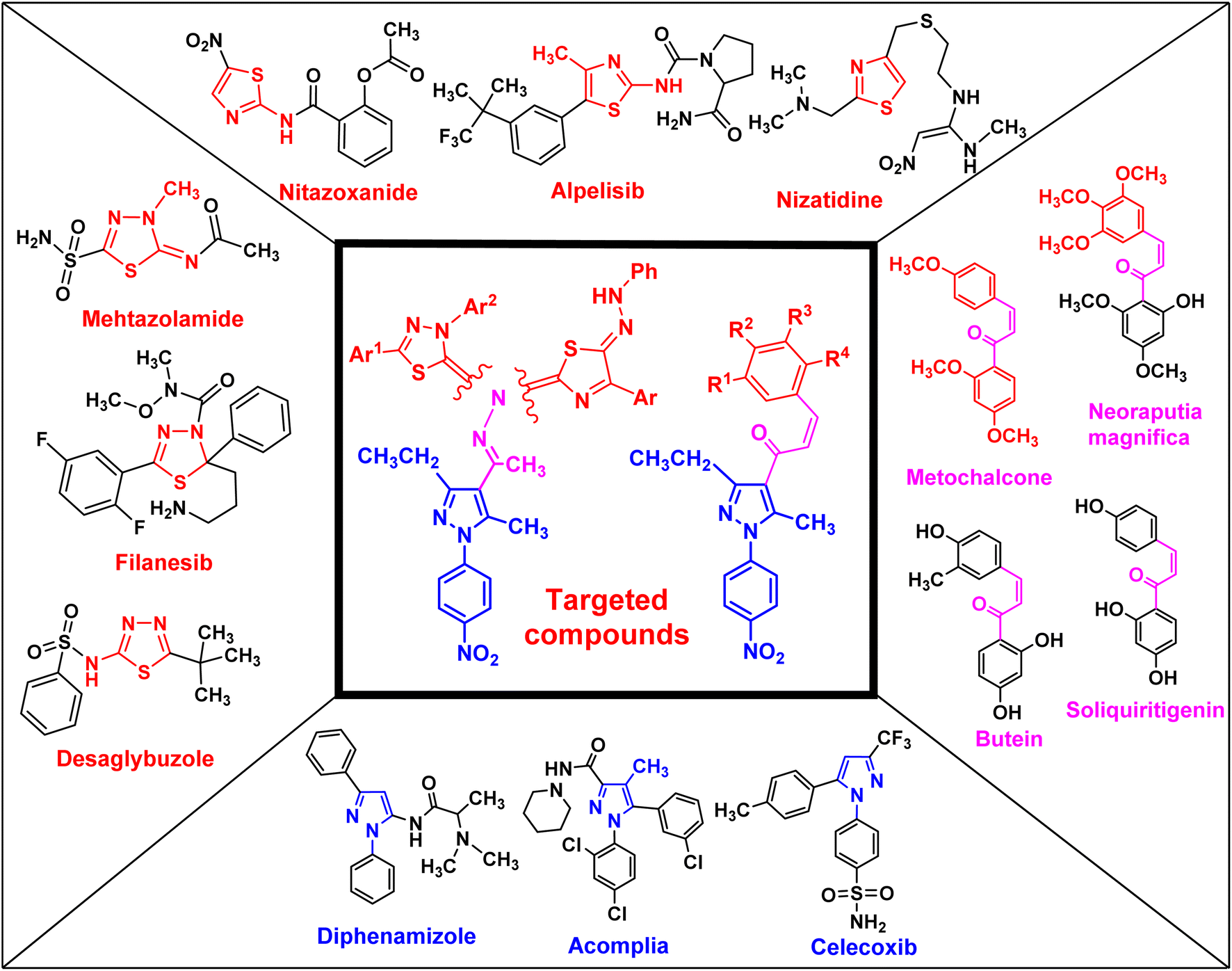 | ||
| Fig. 1 Rationale design of the targeted pyrazole derivatives using the molecular hybridization strategy. | ||
Heterocyclic compounds are an essential and unique class of compounds that cover a wide range of reactivity and stability and have a broad spectrum of chemical and biological characteristics.25–29 Heterocycle-based medications are adaptable enough to target a wide range of metabolic pathways and cellular processes involved in the development and progression of cancer.30–33 Therefore, numerous heterocyclic derivatives have been designed and synthesized for their possible uses as anti-cancer agents. Among these structures, the five-membered heterocyclic rings, such as pyrazole, thiazole, and thiadiazoles, are particularly important compounds.34–37 The pyrazole ring, as a five-membered heterocycle with two neighboring nitrogen atoms, is present in a wide range of compounds that possess different applications. Furthermore, it is commonly known that pyrazoles, both naturally occurring and synthesized, have a wide range of biological properties (some biologically active pyrazoles are given in Fig. 1).38 The thiazole moiety is a significant aromatic five-membered heterocycle. Its distinctive biological properties are established by the atoms of sulfur and nitrogen and the thiazole scaffold is found in over eighteen FDA-approved medications.39 It has been indicated that thiazole-containing compounds demonstrate a variety of biological properties, such as antifungal,40 antibacterial,41 anticancer,28 diuretic,42 anti-inflammatory,43 analgesic,44 neuroprotective,45 antimalarial,46 and antioxidant47 (some biologically active thiazoles are given in Fig. 1). The thiadiazole scaffold can be of four different types: 1,2,3-, 1,2,4-, 1,2,5- and 1,3,4-thiadiazole.48–50 The remarkable pharmacological characteristics of 1,3,4-thiadiazole derivatives are ascribed to their stability, high aromaticity, and absence of toxicity.51 Furthermore, due to the hydrogen binding domain of the 1,3,4-thiadiazole ring, it can be used as a potential agent in several FDA-approved medications, including methazolamide, sulfamethizole, desaglybuzole, litronesib, and filanesib (Fig. 1).34
The design and development of heterocyclic small molecules as anti-cancer agents has been an active area of research in recent years. Several studies have reported the synthesis and biological evaluation of pyrazole-containing compounds as potential therapeutics for various tumor types.52 For instance, Dabhade et al. described the preparation of novel pyrazole-chalcone hybrids and demonstrated their cytotoxic effects against a panel of breast cancer cell lines.53 Similarly, Yadav et al. developed pyrazole-based derivatives bearing thiazole moieties and found them to be promising anti-gastric agents.54 However, the incorporation of multiple heterocyclic substructures, such as chalcone, thiazole, and thiadiazole, within a single pyrazole scaffold as anti-pancreatic agents has been less explored. The present study builds upon these earlier efforts and presents a novel class of pyrazole derivatives that display enhanced anti-pancreatic cancer potency compared to previously reported pyrazole-based compounds.55 The synergistic effects arising from the combination of these heterocyclic fragments represent a unique structural design approach that sets this work apart from the state-of-the-art in this research area. The detailed structure–activity relationship analysis provided in this study further contributes to the growing body of knowledge on the development of heterocyclic small molecules as promising anti-pancreatic drug candidates.
In this study, we used the molecular hybridization strategy, a frequently utilized technique in the field of drug discovery, to synthesize effective pyrazole derivatives bearing chalcone, thiazole or thiadiazole moieties (Fig. 1). Fifteen novel hybrid pyrazole derivatives were designed and tested as anti-cancer agents against selected human cancerous cell lines of breast (MCF-7), pancreatic (PaCa-2), and prostate (PC3) relative to healthy noncancerous control skin fibroblast cells (BJ-1). Moreover, the mechanism of action of the most active compounds was investigated by studying the DNA fragmentation and gene expression as well as the structure–activity relationship (SAR).
2. Results and discussion
2.1. Chemistry
The targeted precursor of 2 was prepared in two steps as described in the literature.9,56 The bromination of propionaldehyde-4-nitrophenylhydrazone followed by the reaction of the produced N-(4-nitrophenyl)propionohydrazonoyl bromide with acetylacetone in the presence of sodium ethoxide afforded 1-(3-ethyl-5-methyl-1-(4-nitrophenyl)-1H-pyrazol-4-yl)ethan-1-one (2) in good yields. Claisen–Schmidt condensation of compound 2 with equimolar amounts of aryl-aldehyde derivatives (3) in ethanol and in the presence of sodium hydroxide solution afforded the targeted pyrazolyl-chalcone derivatives (4–10) (Scheme 1 and Fig. 2).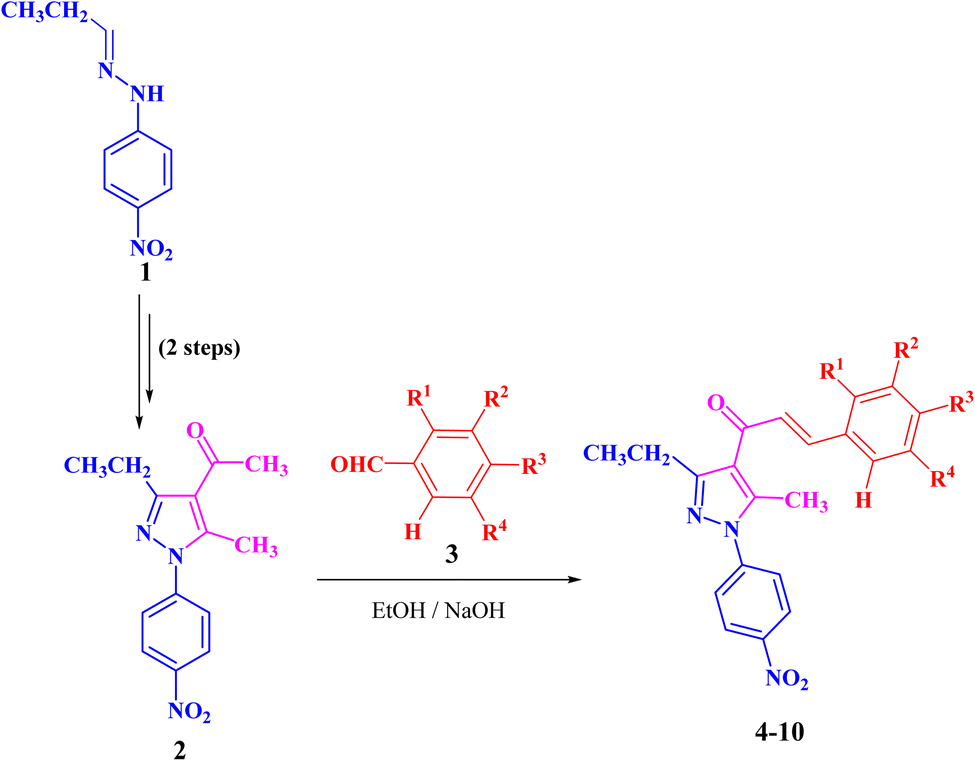 | ||
| Scheme 1 Synthesis of pyrazolyl-chalcone derivatives 4–10. | ||
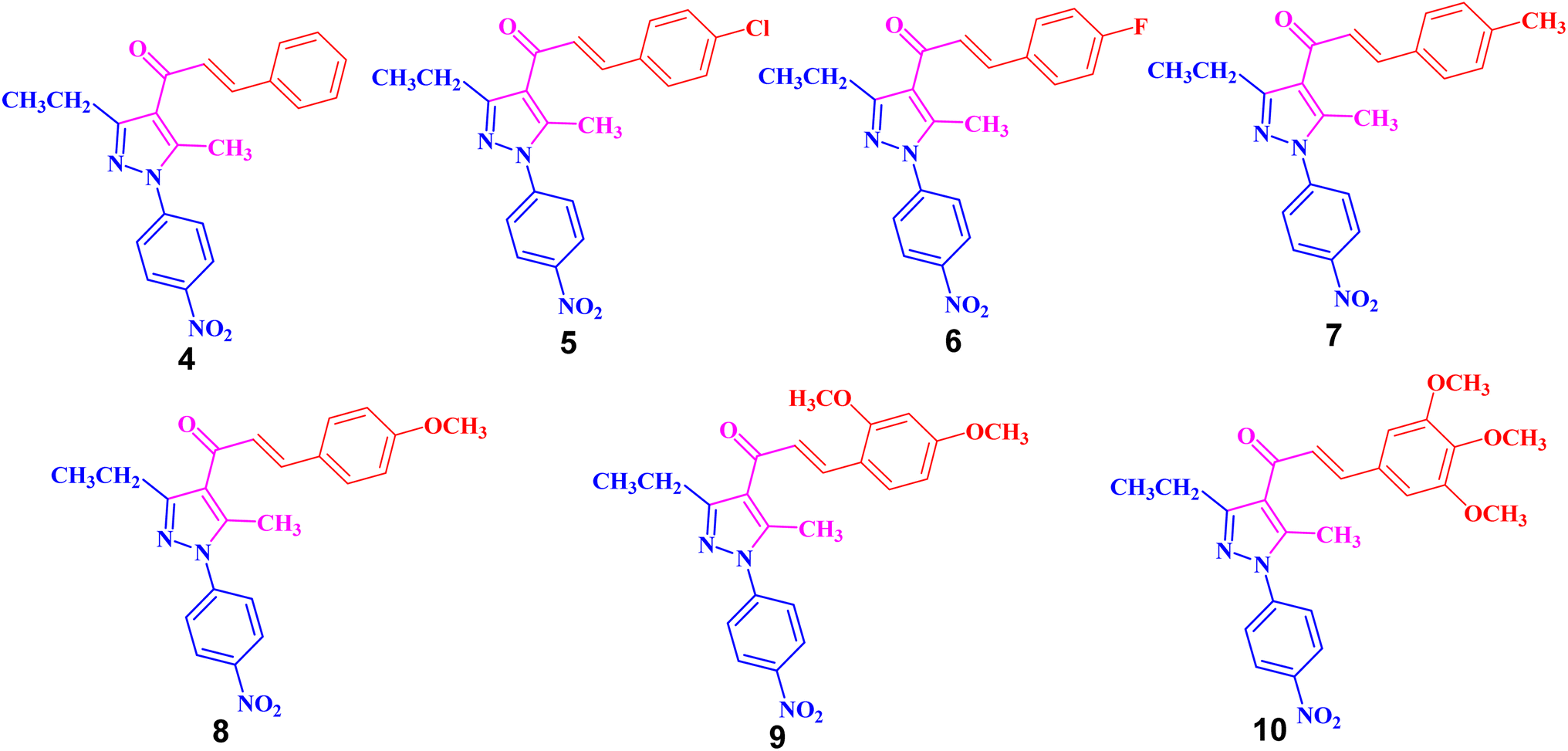 | ||
| Fig. 2 Chemical structures of pyrazolyl-chalcone derivatives 4–10. | ||
The structures of the formed pyrazolyl-chalcone derivatives 4–10 (Fig. 2) were confirmed from their spectral data, such as 1H NMR, 13C NMR, IR, MS, and elemental analysis. For instance, the 1H NMR spectrum of pyrazolyl-chalcone 4 as a representative example showed the following signals: a triplet at δ 1.35 (ppm) corresponding to the CH2CH3 group and a singlet signal at δ 2.62 (ppm) corresponding to the CH3 group. The methylene group (CH2CH3) was assigned as a quartet at δ 2.98 (ppm), while the aromatic protons were observed as a multiplet at δ 7.20–7.73 (ppm) and a doublet at δ 8.40 (ppm). The 13C NMR spectrum of 4 revealed 17 signals corresponding to asymmetric carbon atoms. Also, the mass spectrum (EI) of 4 showed a molecular ion peak at m/z = 361. In addition, the IR spectrum of 4 showed the band at 1654 (cm−1) corresponding to the conjugated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group.
O group.
Refluxing of 1-(3-ethyl-5-methyl-1-(4-nitrophenyl)-1H-pyrazol-4-yl)-ethan-1-one (2) with methyl hydrazinecarbodithioate (11)57,58 in absolute ethanol in the presence of a few drops of hydrochloric acid afforded methyl 2-(1-(3-ethyl-5-methyl-1-(4-nitrophenyl)-1H-pyrazol-4-yl)ethyl-idene)hydrazine-1-carbodithioate (13) (Scheme 2). The structure of compound 13 was elucidated from spectral data (1H NMR, 13C NMR, IR and MS) and elemental analysis. The 1H NMR spectrum of 13 revealed the following signals: a triplet at δ 1.34 (ppm) corresponding to the CH2CH3 group, four singlets at δ 2.35, 2.52, 2.61 and 10.03 (ppm), which could be assigned to three CH3 groups and NH group, respectively. A quartet signal of the methylene group (CH2CH3) was observed at δ 2.94 (ppm) and two doublets at δ 7.73 and 8.39 (ppm) corresponding to the protons of 4-NO2C6H4. The 13C NMR spectrum showed 14 signals corresponding to asymmetric carbon atoms. Also, the mass spectrum (EI) of 13 showed a molecular ion peak at m/z = 377. In addition, its IR spectrum showed a band at 3179 (cm−1), which could be attributed to the NH group.
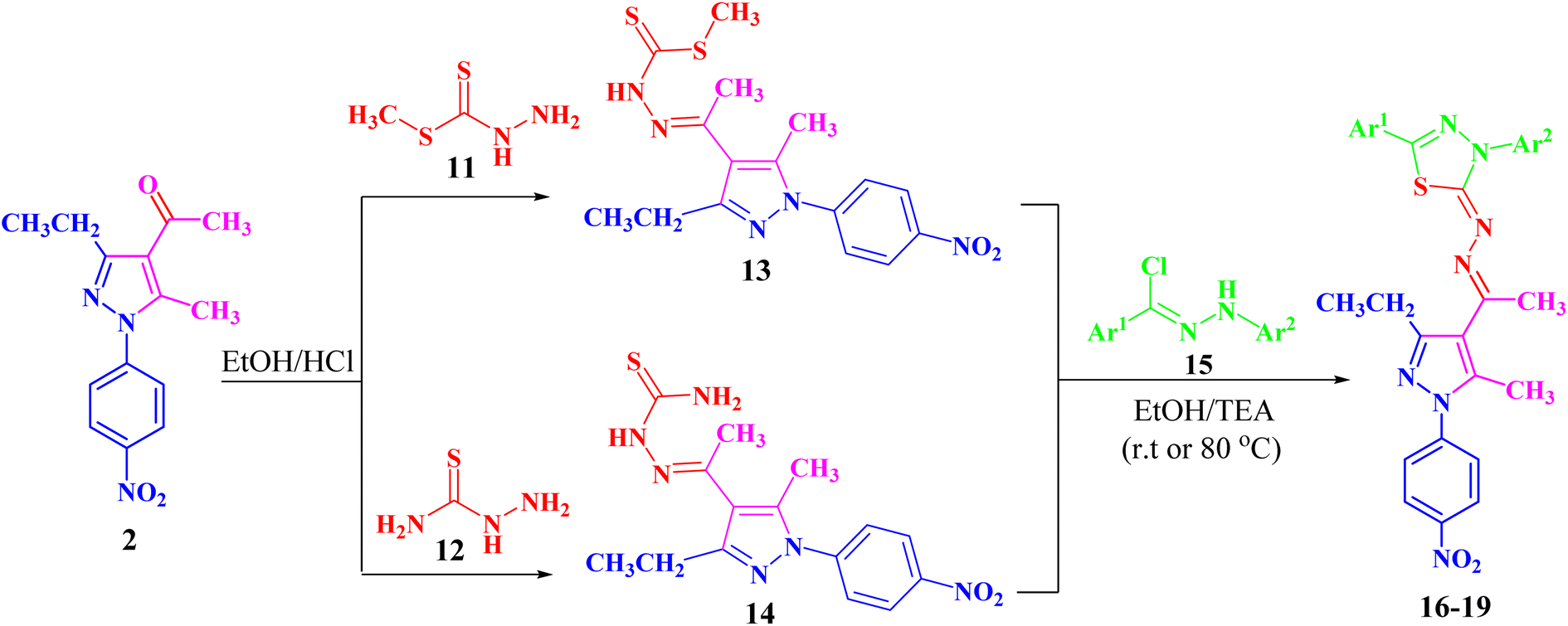 | ||
| Scheme 2 Synthesis of the targeted pyrazolyl-thiadiazole derivatives 16–19 in two different ways. | ||
Likewise, 2-(1-(3-ethyl-5-methyl-1-(4-nitrophenyl)-1H-pyrazol-4-yl)ethylidene)hydrazine-1-carbothioamide (14) was prepared via the treatment of acetylpyrazole 2 with hydrazinecarbothioamide 12 under the same conditions (Scheme 2). The structure of compound 14 was elucidated from spectral data (1H NMR, 13C NMR, IR and MS) and elemental analysis. The 1H NMR spectrum of compound 14 demonstrated the following signals: a triplet at δ 1.18 (ppm) corresponding to the methyl group (CH2CH3) and five singlets at δ 2.29, 2.44, 7.48, 8.23 and 10.16 (ppm) corresponding to two CH3, NH2 and NH groups, respectively. A quartet signal at δ 2.73 (ppm) refers to the methylene group (CH2CH3) and two doublets at δ 7.83 and 8.38 (ppm) correspond to the protons of 4-NO2C6H4. Its 13C NMR spectrum showed 13 signals corresponding to asymmetric carbon atoms. Also, the mass spectrum (EI) of 14 showed a molecular ion peak at m/z = 346. Its IR spectrum showed bands at 3464 cm−1 corresponding to the NH group and 3225 and 3298 (cm−1) referring to the NH2 group.
Both compounds 13 and 14 were used as suitable precursors to synthesize the targeted pyrazolyl-thiadiazole derivatives 16–19, as depicted in Scheme 2. Compound 13 was treated with appropriate hydrazonoyl chlorides (15) in absolute ethanol and in the presence of trimethylamine as a catalyst at ambient temperature to afford the corresponding pyrazolyl-thiadiazole derivatives 16–19 (Scheme 2 and Fig. 3). Compounds 16–19 could be obtained in different routes via the reaction of 14 with hydrazonoyl chlorides (15) at 80 °C in absolute ethanol and in the presence of trimethylamine (Scheme 2). Structures of pyrazolyl-thiadiazole derivatives 16–19 (Fig. 3) were elucidated from their spectral data (1H NMR, 13C NMR, IR and MS) and elemental analysis. For example, the 1H NMR spectrum of 16 (Fig. 3) showed the following signals: a triplet at δ 1.35 (ppm) corresponding to the methyl group (CH2CH3), two singlet at δ 2.51 and 2.60 (ppm) corresponding to the other methyl groups, and a quartet at δ 2.92 (ppm) corresponding to the methylene group (CH2CH3). The aromatic protons were assigned as a multiplet at the range δ 7.27–7.83 (ppm) corresponding to 10 protons and two doublets at δ 8.26 and 8.38 (ppm) belonging to 4 protons of NO2C6H4. Its 13C NMR spectrum showed 22 signals corresponding to asymmetric carbon atoms. In addition, the IR spectrum of 16 revealed the absence of both the NH and NH2 bands.
 | ||
| Fig. 3 Chemical structures of the targeted pyrazolyl-thiadiazole derivatives 16–19. | ||
In the same manner, the targeted pyrazolyl-thiazole derivatives 23–26 (Scheme 3 and Fig. 4) were prepared using methyl 2-(1-(3-ethyl-5-methyl-1-(4-nitrophenyl)-1H-pyrazol-4-yl)ethylidene)hydrazine-1-carbothioamide (14) as a suitable precursor by reaction with appropriate α-ketohydrazonoyl halides (20) in absolute ethanol and the presence of trimethylamine. The reaction involved condensation via elimination of water molecule to give the expected intermediate (21), which converted as soon as it was formed to the other intermediate (22), which gave the final products 23–26 via elimination of hydrogen halide (Scheme 3). Elemental analyses and spectral data (1H NMR, 13C NMR, IR and MS) verified the chemical structures of the synthesized pyrazolyl-thiazole derivatives 23–26 (Fig. 4).
 | ||
| Scheme 3 Synthesis of the targeted pyrazolyl-thiazole derivatives 23–26. | ||
 | ||
| Fig. 4 Chemical structures of the targeted pyrazolyl-thiazole derivatives 23–26. | ||
Compound 23 will be discussed in more detail as a representative example for a series of pyrazolyl-thiazole (23–26). In the 1H NMR spectrum of 23 (Fig. 4), a triplet signal at δ 1.34 (ppm) is due to the methyl group (CH2CH3), three singlet signals at δ 2.01, 2.61 and 2.62 (ppm) refer to the other methyl groups, and a quartet signal at δ 2.97 (ppm) for the methylene group (CH2CH3). The aromatic protons were assigned as a multiplet at range δ 7.03–7.37 (ppm) due to 5 protons and two doublets at δ 7.72 and 8.39 (ppm) belonging to 4-NO2C6H4, while the proton of the NH group was observed at δ 7.49 (ppm). The 13C NMR spectrum showed 20 signals corresponding to asymmetric carbon atoms. The IR spectrum revealed the presence of a band at 3427 (cm−1), referring to the NH group. Also, the mass spectrum (EI) of 23 depicted a molecular ion peak at m/z = 489.
2.2. Anti-cancer activity
| Comp. | PaCa2 | MCF7 | PC3 | BJ1 |
|---|---|---|---|---|
| 4 | 100 | 31.2 ± 0.77 | 5.3 ± 0.87 | 61.2 ± 0.44 |
| 5 | 88.3 ± 0.11 | 61.2 ± 0.94 | 5.7 ± 0.21 | 61.8 ± 0.27 |
| 6 | 51.2 ± 0.23 | 28.6 ± 0.16 | 8.4 ± 0.87 | |
| 7 | 91.2 ± 0.41 | 65.3 ± 0.19 | 31.5 ± 0.70 | 44.9 ± 0.62 |
| 8 | 90.5 ± 0.57 | 86.5 ± 0.32 | 30.7 ± 0.19 | 74.6 ± 0.28 |
| 9 | 53.8 ± 0.27 | 16.3 ± 0.52 | 27.6 ± 0.14 | |
| 10 | 63.4 ± 0.31 | 68.3 ± 0.63 | 48.6 ± 0.55 | |
| 16 | 74.6 ± 0.22 | 74.9 ± 0.87 | 1.5 ± 0.65 | |
| 17 | 45.6 ± 0.84 | 71.2 ± 0.14 | 51.2 ± 0.70 | |
| 18 | 46.3 ± 0.42 | 51.3 ± 0.18 | 40.8 ± 0.74 | |
| 19 | 5.7 ± 0.73 | 28.6 ± 0.79 | 4.2 ± 0.45 | |
| 23 | 94.2 ± 0.44 | 100 | 92.8 ± 0.21 | 83.5 ± 0.32 |
| 24 | 62.4 ± 0.25 | 73.9 ± 0.56 | 56.3 ± 0.29 | |
| 25 | 100 | 100 | 100 | 100 |
| DOX | 100 | 100 | 100 | 100 |
| Comp. | PaCa2 | MCF7 | PC3 | BJ1 |
|---|---|---|---|---|
| 4 | 13.0 ± 0.21 | — | — | 74.2 ± 0.11 |
| 5 | 31.5 ± 0.42 | 69.6 ± 0.72 | — | 76.6 ± 0.15 |
| 6 | — | — | — | — |
| 7 | 24.9 ± 0.15 | 78.3 ± 0.17 | — | 81.1 ± 0.13 |
| 8 | 33.9 ± 0.17 | 40.2 ± 0.10 | — | 44.9 ± 0.22 |
| 9 | — | — | — | — |
| 10 | — | — | — | — |
| 16 | 62.0 ± 0.35 | 54.8 ± 0.74 | — | — |
| 17 | — | 57.2 ± 0.54 | 83.7 ± 0.14 | — |
| 18 | — | — | — | — |
| 19 | — | — | — | — |
| 23 | 38.7 ± 0.78 | 29.9 ± 0.11 | 26.1 ± 0.65 | 42.9 ± 0.34 |
| 24 | — | 64.4 ± 0.18 | 82.1 ± 0.21 | — |
| 25 | 5.5 ± 0.22 | — | 11.8 ± 0.24 | 23.7 ± 0.41 |
| DOX | 28.3 ± 0.19 | 26.1 ± 0.1 | 23.8 ± 0.47 | 13.5 ± 0.52 |
| Treatment | DNA fragmentation (%) M ± SEM | Change | Inhibition (%) |
|---|---|---|---|
| a Means with different superscripts (a, b, and c) between locations in the same column are significantly different at P < 0.05. | |||
| PaCa-2 control (−ve) | 8.1 ± 0.32c | 0 | 0 |
| PaCa-2 + 4 | 22.5 ± 0.44ab | 14.4 | 23.21 |
| PaCa-2 + 5 | 20.2 ± 0.53b | 12.1 | 22.87 |
| PaCa-2 + 7 | 24.9 ± 0.69a | 16.8 | 28.24 |
| PaCa-2 + Dox (+ve) | 21.2 ± 0.64b | 13.1 | 24.01 |
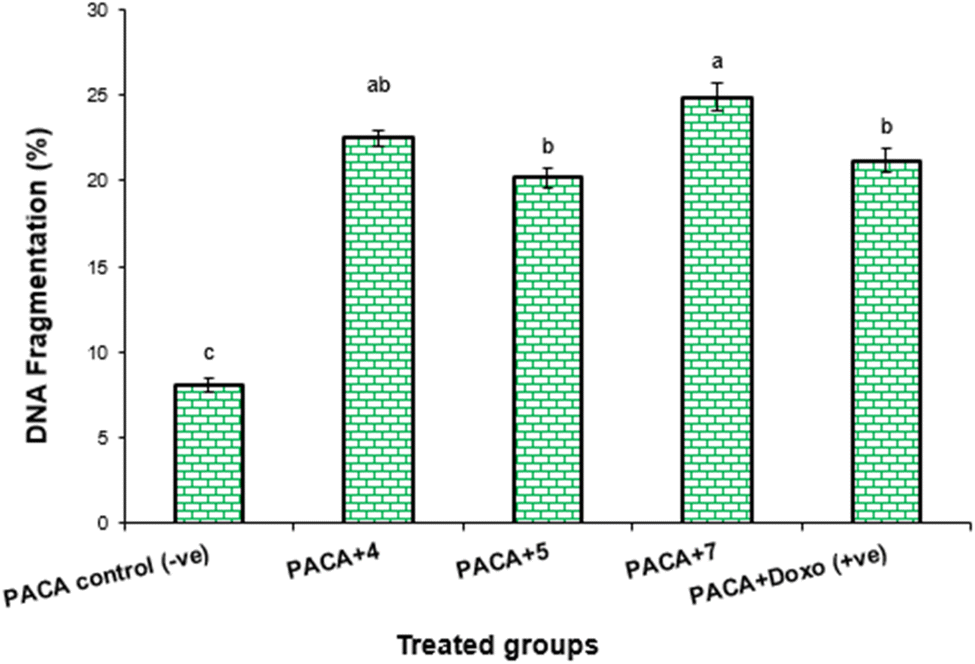 | ||
| Fig. 5 DNA fragmentation was detected in PaCa-2 cell lines treated with different compounds (4, 5 and 7) and Dox. Means with different superscripts (a,b,c) between locations in the same column are significantly different at P < 0.05. | ||
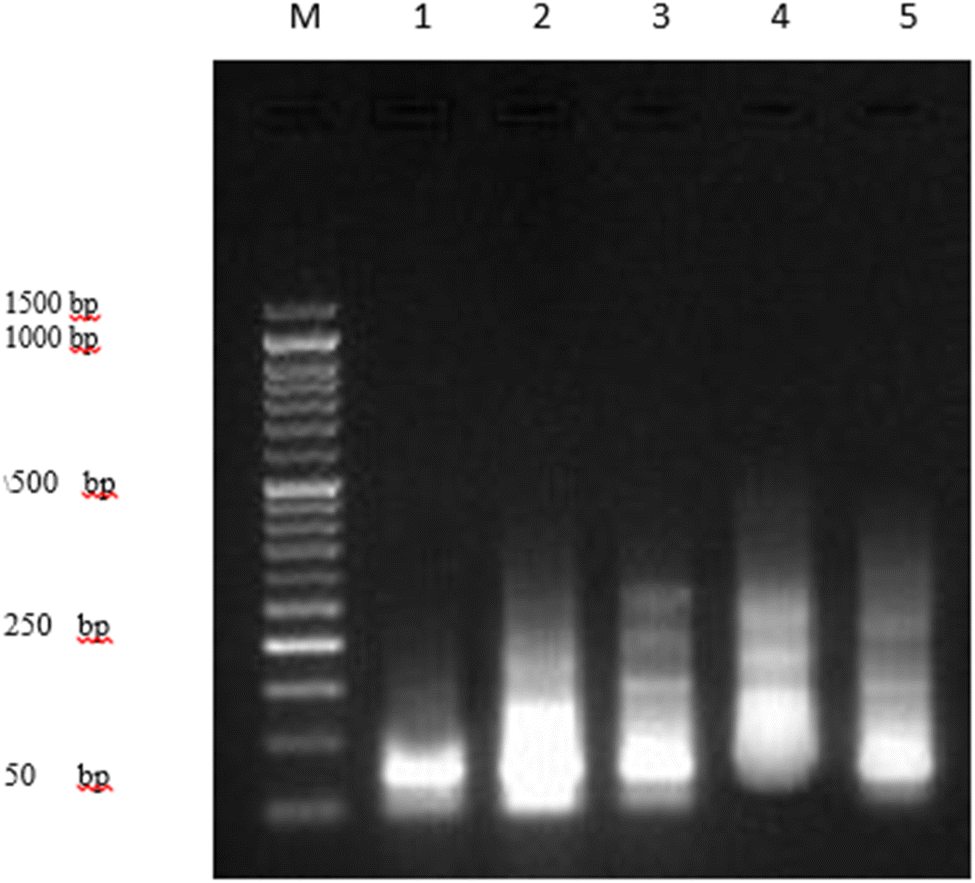 | ||
| Fig. 6 DNA fragmentation was detected with agarose gel in PaCa-2 cells treated with different compounds 4, 5 and 7 as well as Dox as the positive control. M: represent DNA marker, lane 1: represents PaCa-2 control (−ve), lane 2: represents PaCa + 4, lane 3: represents PaCa + 5, lane 4: represents PaCa + 7, lane 5: represents PaCa + Dox (+ve). | ||
Pyrazole and its derivatives have been synthesized for a wide range of biological activities, including antimicrobial, antifungal, anti-inflammatory, anti-cancer, neuroprotective, and anti-viral activity.59–61 Certain Food and Drug Administration (FDA)-approved drugs, such as celecoxib, deracoxib, etoricoxib, and atorivodine, possess pyrazole scaffolds as the effective functional moiety.62,63 Furthermore, this study showed that compounds 4, 5 and 7 induce apoptosis-related genes (Caspase-3 and Caspase-8) and anticancer-related genes (CDK6) as well as increased DNA damage response. So, our data suggest that compounds 4, 5 and 7 are novel pyrazolyl-chalcone derivatives that exert antitumor activity in pancreatic carcinoma.
Pyrazole moiety is a useful lead five-membered ring to synthesize many potent bioactive molecules for drug development with good safety profiles, particularly against different types of cancers.8,9,37,64,65 Anti-cancer activities of several pyrazole derivatives containing thiazoles and thiadiazoles compounds have been demonstrated in both in vitro and in vivo models, often resulting in promising lead compounds.64,66,67
Our data showed the novel compounds 4 and 7 had the most potent antitumor activity as they exhibited higher expression levels of Caspase-3, Caspase-8 and CDK6 genes in pancreatic cancer cell lines as well as higher DNA fragmentation rates, suggesting that they could inhibit the common apoptosis and oncogenic pathways. Moreover, it has been reported that the pyrazole compound (PCW-1001) increased apoptosis in several breast cancer cells, which is in agreement with our findings, and programmed cell death is an essential mechanism to eliminate cancer cells by anti-cancer drugs.68–70
It is well established that anti-cancer treatments, such as chemotherapy, induce DNA damage directly or indirectly in active proliferating cancer cells rather than non-proliferating normal cells.71,72 Our data showed that the newly synthesized compounds 4, 5 and 7 modulated apoptosis and anticancer-related genes in pancreatic cancer cell lines, suggesting that these compounds induce DNA damage response in pancreatic cancer cell lines due to the previous action of the gene modulation.
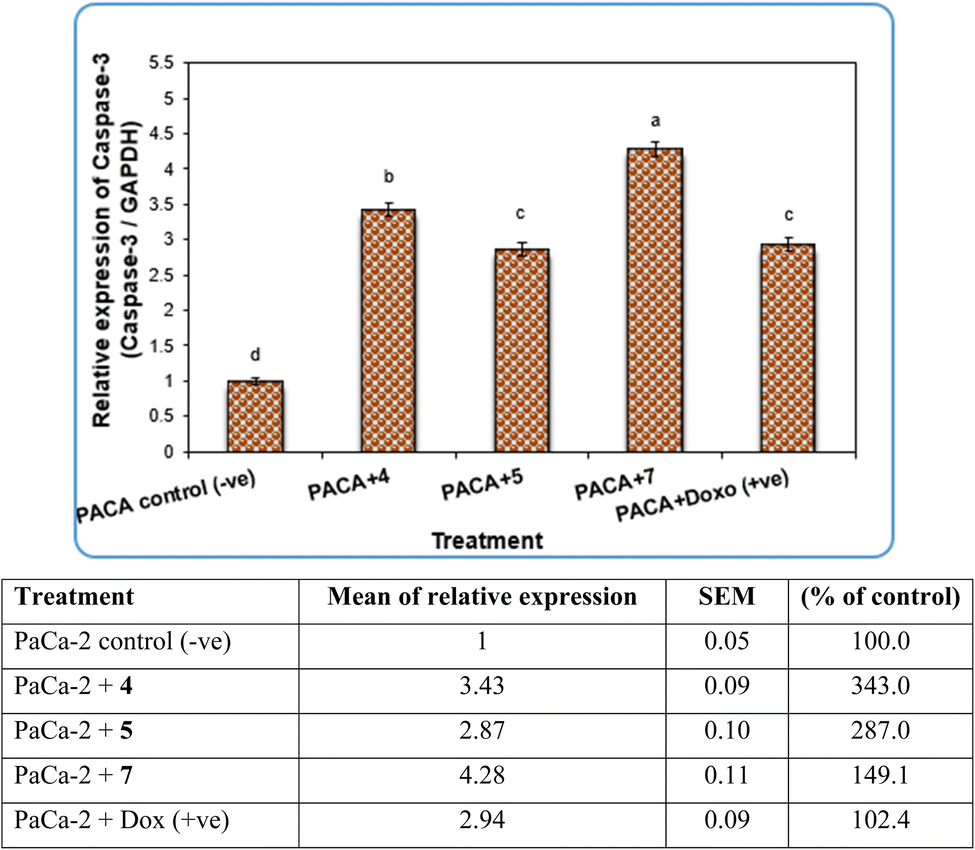 | ||
| Fig. 7 The alterations of the Caspase-3 gene in PaCa-2 cancer cell lines treated with compounds 4, 5 and 7 as well as Dox (+ve) control. Data are presented as mean ± SEM. a, b, c, d: mean values within tissue with unlike superscript letters were significantly different (P < 0.05). | ||
In contrast, the expression levels of the CDK6 gene were increased significantly (P < 0.01) in negative samples of PaCa-2 cancer cell lines compared with treated cell lines (Fig. 8). For the PaCa + 7 and PaCa + 4 groups, the expression levels of CDK6 decreased with high significant differences compared with the negative control. Moreover, for the PaCa + 4 and (+ve) control treated with doxorubicin, the expression levels of CDK6 were also decreased significantly compared with the negative control, but their expression levels were higher than those in the PaCa + 7 and PaCa + 4 groups (Fig. 8).
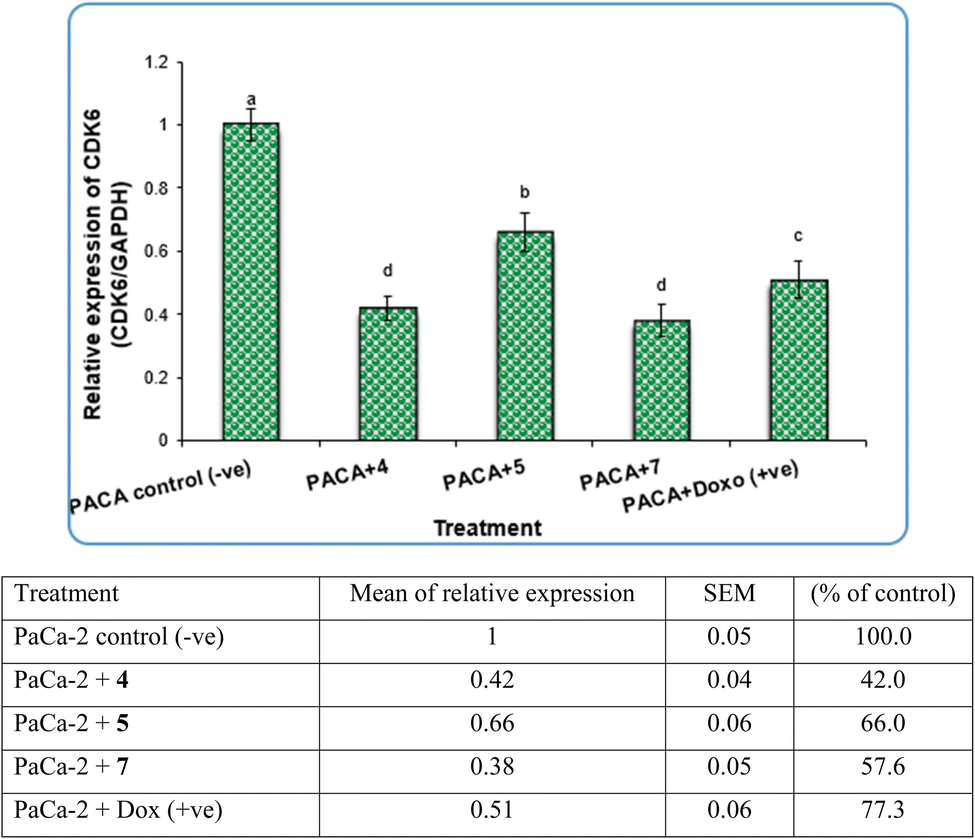 | ||
| Fig. 8 The alterations in the CDk6 gene in PaCa-2 cancer cell lines treated with compounds 4, 5 and 7 as well as Dox (+ve) control. Data are presented as mean ± SEM. a, b, c,d: mean values within tissue with unlike superscript letters were significantly different (P < 0.05). | ||
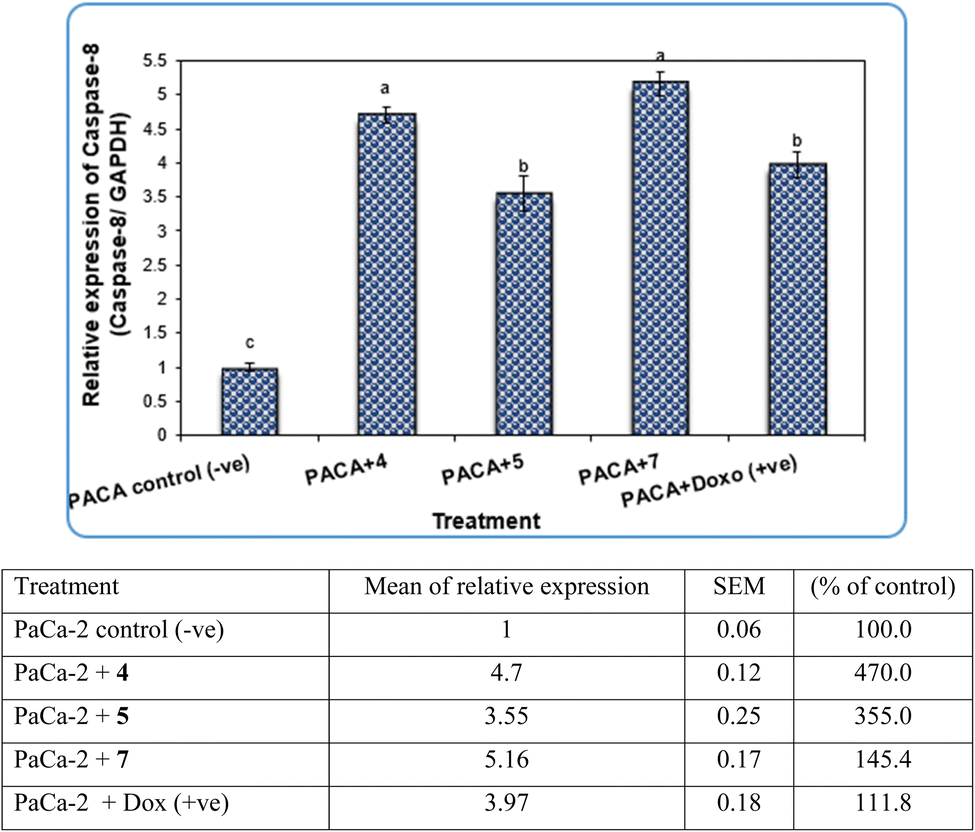 | ||
| Fig. 9 The alterations in the Caspase-3 gene in PaCa-2 cancer cell lines treated with compounds 4, 5 and 7 as well as Dox (+ve) control. Data are presented as mean ± SEM. a, b, c: mean values within tissue with unlike superscript letters were significantly different (P < 0.05). | ||
| Treatment | No of samples | No. of cells | Classb | DNA damaged cells% (mean ± SEM) | ||||
|---|---|---|---|---|---|---|---|---|
| Analyzeda | Comets | 0 | 1 | 2 | 3 | |||
| a Number of cells examined per group.b Class 0 = no tail; 1 = tail length < diameter of nucleus; 2 = tail length between 1× and 2× the diameter of nucleus; and 3 = tail length >2× the diameter of nucleus. Means with different superscripts (a, b and c) between groups in the same treatment are significantly different at P < 0.05. Data are presented as mean ± SEM. | ||||||||
| PaCa-2 control (−ve) | 4 | 420 | 47 | 373 | 31 | 10 | 6 | 11.19 ± 0.75c |
| PaCa-2 + 4 | 4 | 420 | 121 | 299 | 39 | 33 | 49 | 28.81 ± 1.11a |
| PaCa-2 + 5 | 4 | 420 | 89 | 331 | 42 | 26 | 21 | 21.19 ± 0.91b |
| PaCa-2 + 7 | 4 | 420 | 111 | 309 | 38 | 33 | 40 | 26.43 ± 0.66a |
| PaCa-2 + Dox (+ve) | 4 | 420 | 96 | 324 | 34 | 37 | 25 | 22.86 ± 1.75b |
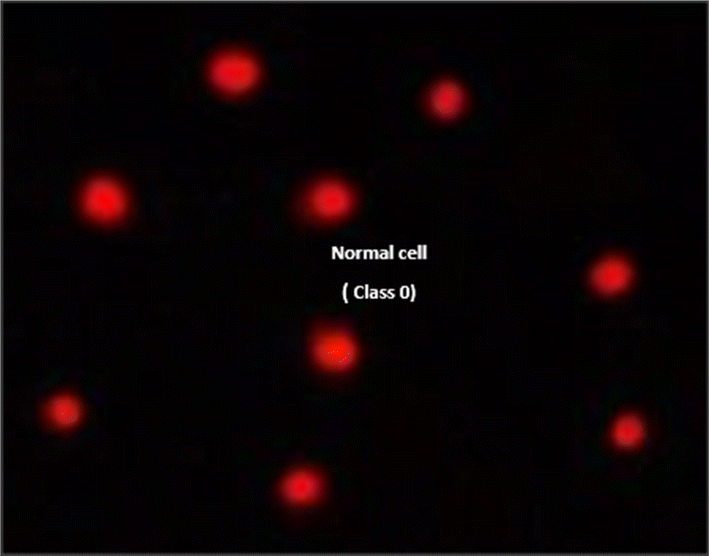 | ||
| Fig. 10 Visual score of normal DNA (class 0) using the comet assay in pancreatic cancer cell lines. | ||
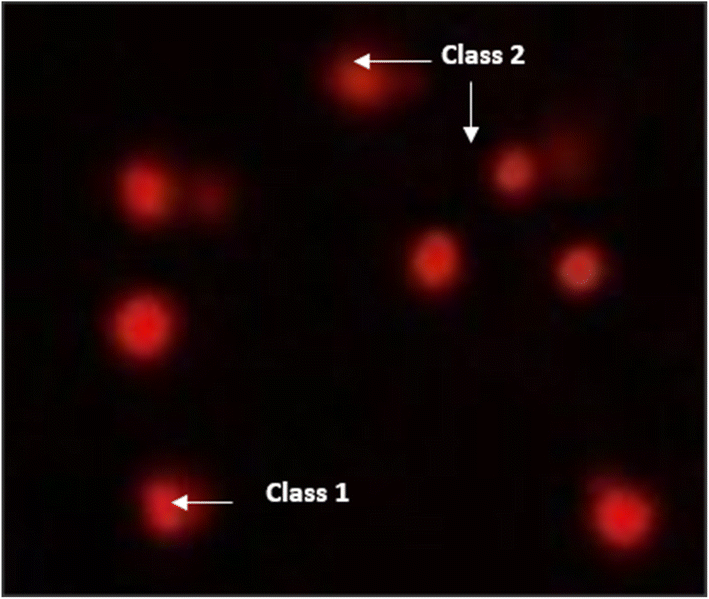 | ||
| Fig. 11 Visual score of DNA damage (classes 1 and 2) using the comet assay in pancreatic cancer cell lines exposed to 4, 5 and 7. | ||
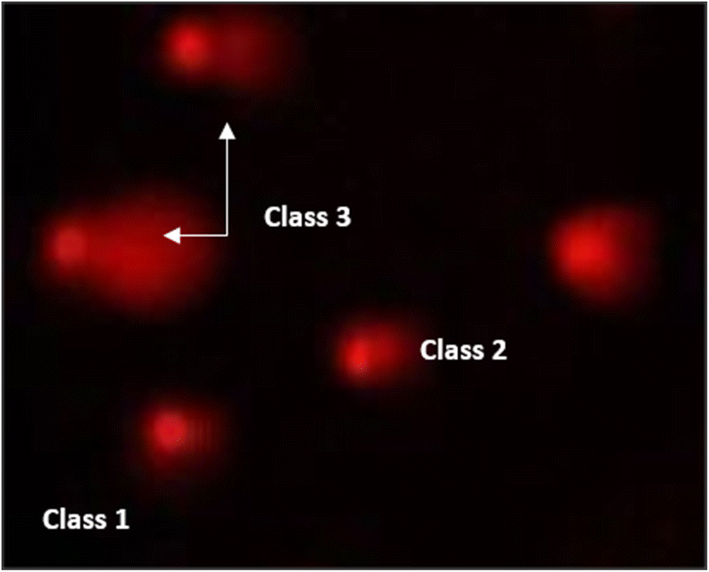 | ||
| Fig. 12 Visual score of DNA damage (classes 1, 2 and 3) using the comet assay in pancreatic cancer cell lines exposed to 4, 5 and 7. | ||
DNA damage and repair are among the challenges that living cells face at all times, and the long-term survival of a healthy cell depends on successfully overcoming this challenge. In a variety of cancer cell lines, DOX has been shown to induce different types of cell death/growth arrest, including apoptosis and mitotic catastrophe, through inhibition of apoptosis by overexpression of Bcl-2 and necrosis.73,74 DOX can inhibit the synthesis of both DNA and RNA. In addition, DOX exerts its effects by preventing the uncoiling of the DNA enzyme topoisomerase II.75 In agreement with our finding, DOX drug induced a significantly high rate of DNA damage in PACA2 + Dox as compared with negative PaCa-2.
Free radicals, such as ROS, are naturally produced in living cells but could be increased by external sources, such as chemotherapy drugs (e.g., DOX), X-rays, air pollutants or chemical compounds.76,77 So, the DNA damage induced by DOX in the present study could be induced by promoting ROS generation-mediated apoptosis. In the same trend, the used anti-cancer compounds 4, 5 and 7 exhibited more or less anti-cancer effects compared with DOX. Compound 4 revealed higher efficiency of anti-cancer effects as it induced DNA damage much more than DOX and other anti-cancer compounds. Thus, its anti-cancer effect could be attributed to its capacity to enhance ROS generation-mediated DNA damage and apoptosis.
 | ||
| Fig. 13 The cell distributions in the different phases of the cell cycle (G0/G1-S-G2/M) after 24 h analysed using flow cytometry: (A) 4-treated PaCa-2 cells, (B) PaCa-2 cells treated with doxorubicin as the positive (+ve) and (C) negative control (−ve). | ||
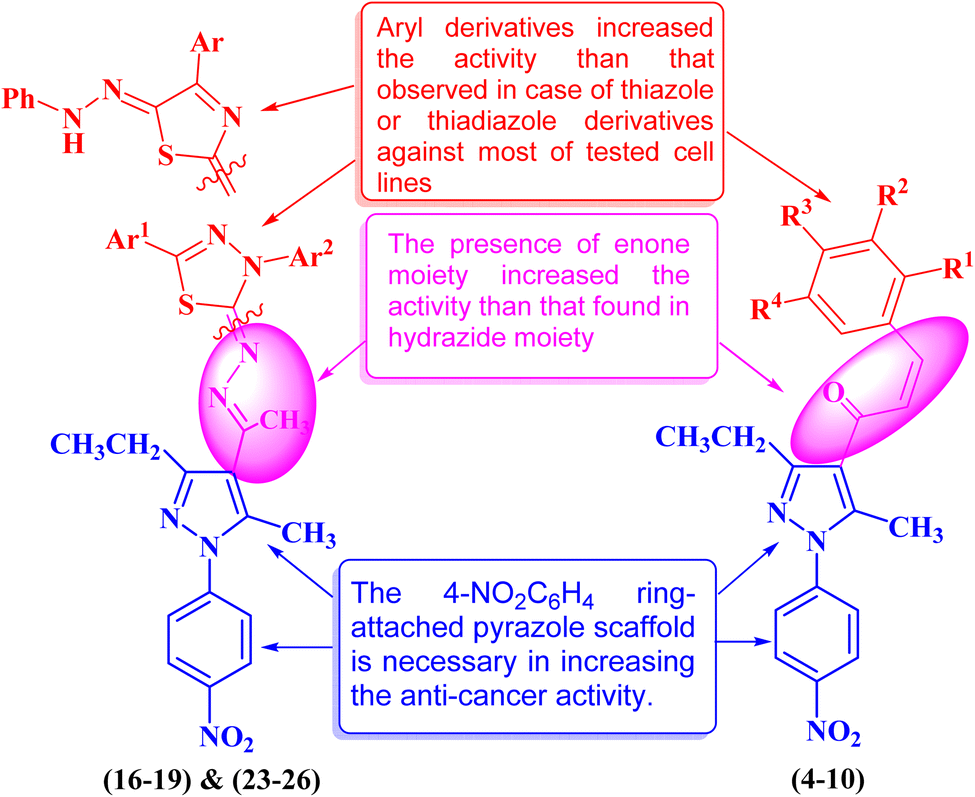 | ||
| Fig. 14 Structure–activity relationship study of the newly synthesized pyrazole derivatives. | ||
3. Materials and methods
3.1. Chemistry
Melting points were measured with an Electrothermal 9100 apparatus and were uncorrected. The IR spectra were recorded using FTIR Bruker-vector 22 spectrophotometer as KBr pellets. The 1H and 13C NMR spectra were recorded in CDCl3 or DMSO-d6 as a solvent on a Varian Gemini NMR spectrometer at 300 MHz and 75 MHz, respectively, using TMS as the internal standard. Chemical shifts were reported as δ values in ppm. Mass spectra were recorded with a Shimadzu GCMS-QP-1000 EX mass spectrometer in an EI (70 eV) model. The elemental analyses were performed at the Microanalytical Center, Cairo University. The progress of the reaction was tracked by exposing silica gel G-coated TLC plates. The reagents, 1-(3-ethyl-5-methyl-1-(4-nitrophenyl)-1H-pyrazol-4-yl)ethan-1-one 2,9,56 methyl hydrazinecarbodithioate 11,57,58 N-aryl-C-substituted methanohydrazonoyl chlorides 159,78,79 and α-ketohydrazonoyl halide derivatives 2080,81 were prepared according to the literature.O), 1597, 1535, 1519, 1381, 1334, 1049, 964, 856, 983, 686, 563. 1H NMR (300 MHz, CDCl3) δ 1.35 (t, 3H, CH3, J ≈ 8 Hz), 2.62 (s, 3H, CH3), 2.98 (q, 2H, CH2, J ≈ 8 Hz), 7.20–7.73 (m, 9H, Ar–H), 8.40 (d, 2H, Ar–H, J ≈ 9 Hz). 13C NMR (75 MHz, CDCl3) δ 13.3, 13.2, 21.7, 115.1, 121.5, 124.5, 124.6, 127.6, 128.2, 129.1, 135.0, 142.7, 143.5, 145.5, 146.1, 154.1, 188.8. MS (EI, 70 eV) m/z (%): 361 (M+, 100), 346 (M–CH3, 77.93), 284 (M–C6H5, 39.98), 270 (M–(CH3 + C6H5), 35.21), 212 (M–(C2H5 + 4-NO2–C6H4), 25.73), 103 (C6H5–CHCH2, 23.12), 77 (C6H5, 34.08). Anal. Calcd. for C21H19N3O3 (361.40): C, 69.79; H, 5.30; N, 11.63. Found: C, 69.87; H, 5.39; N, 11.55.O), 1597, 1512, 1334, 1049, 1010, 858, 825, 748, 686, 501. 1H NMR (300 MHz, CDCl3) δ 1.33 (t, 3H, CH3, J ≈ 8 Hz), 2.61 (s, 3H, CH3), 2.94 (q, 2H, CH2, J ≈ 8 Hz), 7.16–8.37 (m, 10H, Ar–H). 13C NMR (75 MHz, CDCl3) δ 13.2, 13.3, 21.8, 115.0, 121.1, 124.3, 124.5, 128.5, 129.1, 133.0, 133.3, 133.7, 142.5, 145.2, 145.2, 145.6, 154.0, 189.5. Anal. Calcd. for C21H18ClN3O3 (395.84): C, 63.72; H, 4.58; N, 10.62. Found: C, 63.80; H, 4.67; N, 10.71.O), 1589, 1527, 1334, 1049, 856, 694, 524, 501. 1H NMR (300 MHz, CDCl3) δ 1.34 (t, 3H, CH3, J ≈ 8 Hz), 2.40 (s, 3H, CH3), 2.61 (s, 3H, CH3), 2.95 (q, 2H, CH2, J ≈ 8 Hz), 7.14–7.72 (m, 8H, Ar–H), 8.39 (d, 2H, Ar–H, J ≈ 9 Hz). 13C NMR (75 MHz, CDCl3) δ 13.2, 13.3, 21.4, 21.9, 121.7, 124.6, 124.7, 125.2, 125.6, 128.2, 129.7, 131.8, 141.0, 142.9, 143.6, 146.5, 156.0, 187.5. MS (EI, 70 eV) m/z (%): 375 (M+, 100), 360 (M–CH3, 70.45), 284 (M–(C6H4–CH3), 22.41), 270 (M–(CH3 + C6H4–CH3), 17.85), 115 (CH3–C6H4–CHCH2, 20.77), 76 (C6H5, 14.51). Anal. Calcd. for C22H21N3O3 (375.43): C, 70.38; H, 5.64; N, 11.19. Found: C, 70.47; H, 5.72; N, 11.23.O), 1589, 1512, 1419, 1342, 1118, 1064, 987, 840, 758, 686, 609. 1H NMR (300 MHz, CDCl3) δ 1.34 (t, 3H, CH3, J ≈ 8 Hz), 2.60 (s, 3H, CH3), 2.93 (q, 2H, CH2, J ≈ 8 Hz), 3.91 (s, 3H, OCH3), 3.92 (s, 6H, 2OCH3), 6.83–7.72 (m, 6H, Ar–H), 8.39 (d, 2H, Ar–H, J ≈ 9 Hz). 13C NMR (75 MHz, CDCl3) δ 13.2, 13.4, 21.9, 56.2, 60.9, 105.5, 115.0, 121.7, 124.7, 125.2, 125.3, 130.1, 142.9, 143.8, 144.1, 146.6, 153.5, 156.0, 187.6. Anal. Calcd. for C24H25N3O6 (451.48): C, 63.85; H, 5.58; N, 9.31. Found: C, 63.97; H, 5.70; N, 9.42.C–CH3, 56.08), 76 (C6H4, or H2N–C(S)–NH, 76.87), 60 (H2N–CS, 100). Anal. Calcd. for C15H18N6O2S (346.41): C, 52.01; H, 5.24; N, 24.26. Found: C, 52.11; H, 5.33; N, 24.37.Method B: refluxing of 2-(1-(3-ethyl-5-methyl-1-(4-nitrophenyl)-1H-pyrazol-4-yl)ethylidene)hydrazine-1-carbothioamide 14 (0.3 g, 1 mmol) with the appropriate hydrazonoyl chlorides 15 (1 mmol) in absolute ethanol (20 mL) was performed in the presence of triethylamine for 4 h. Reaction progress was monitored by TLC till the disappearance of the starting material. The resulting solid product that precipitated was collected, washed with ethanol and crystallized from dimethylformamide to afford the same products 16–19. Moreover, the purity of the final products was tested using the TLC technique to have one spot for each compound. The compounds prepared are listed below.
3.2. Anti-cancer evaluation
Cell viability was assessed by the mitochondrial-dependent reduction of yellow MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) to purple formazan. Procedure: all the following procedures were done in a sterile area using a Laminar flow cabinet biosafety class II level (Baker, SG403INT, Sanford, ME, USA). Cells were suspended in DMEM-F12 medium (for PaCa-2, MCF7, and PC3) beside one normal cell line (BJ1), 1% antibiotic–antimycotic mixture (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U mL−1 potassium penicillin, 10000 μg mL−1 streptomycin sulfate and 25 μg mL−1 amphotericin B) and 1% L-glutamine at 37 °C under 5% CO2.82
000 U mL−1 potassium penicillin, 10000 μg mL−1 streptomycin sulfate and 25 μg mL−1 amphotericin B) and 1% L-glutamine at 37 °C under 5% CO2.82
Cells were cultured for 10 days, then seeded at a concentration of 10 × 103 cells per well in a fresh complete growth medium in 96-well microtiter plastic plates at 37 °C for 24 h under 5% CO2 using a water-jacketed carbon dioxide incubator (Sheldon, TC2323, Cornelius, OR, USA). Media was aspirated, fresh medium (without serum) was added, and cells were incubated either alone (negative control) or with different concentrations of the sample to give a final concentration of (100, 50, 25, 12.5, 6.25, 3.125, 0.78 and 1.56 μg mL−1). After 48 h of incubation, the medium was aspirated, 40 μL MTT salt (2.5 μg mL−1) was added to each well and incubated for a further four hours at 37 °C under 5% CO2. To stop the reaction and dissolve the formed crystals, 200 μL of 10% sodium dodecyl sulfate (SDS) in deionized water was added to each well and incubated overnight at 37 °C. A positive control composed of 100 μg mL−1 was used as a known cytotoxic natural agent that gives 100% lethality under the same conditions.82
The absorbance was then measured using a microplate multi-well reader (Bio-Rad Laboratories Inc., model 3350, Hercules, California, USA) at 595 nm and a reference wavelength of 620 nm. A statistical significance was tested between samples and the negative control using an independent t-test by the SPSS 11 program. DMSO is the vehicle used for the dissolution of plant extracts, and its final concentration in the cells was less than 0.2%. The percentage of change in viability was calculated according to the formula: ((reading of extract/reading of negative control) – 1) × 100. A probit analysis was carried out for IC50 determination using the SPSS 11 program. In the present study, the degree of selectivity of the synthetic compounds is expressed as SI = IC50 of a pure compound in a normal cell line/IC50 of the same pure compound in the cancer cell line, where IC50 is the concentration required to kill 50% of the cell population.
3.3. DNA fragmentation assay
The DNA fragmentation assay was performed in accordance with the protocol established by Yawata (1998)83 with some modifications. Briefly, after 24 h of exposure of PaCa-2 cell lines to the tested substances (H20) and doxorubicin in different Petri dishes (60 × 15 mm, Greiner), the cells were trypsinized, suspended, homogenized in 1 mL of medium and centrifuged (10 min at 800 rpm). Low-molecular-weight genomic DNA was extracted as described by Yawata (1998).83 Approximately 1 × 106 cells were plated and treated with the tested substances in various experiments. All the cells (including floating cells) were harvested by trypsinization and washed with Dulbecco's Phosphate Buffered Saline. Cells were lysed with the lysis buffer containing 10 mM Tris (pH 7.4), 150 mM NaCl, 5 mM ethylenediaminetetraacetic acid (EDTA), and 0.5% Triton X-100 for 30 min on ice. Lysates were vortexed and cleared by centrifugation at 10000g for 20 min. Fragmented DNA in the supernatant was extracted with an equal volume of neutral phenol:chloroform:isoamyl alcohol mixture (25:24:1) and analyzed electrophoretically on 2% agarose gels containing 0.1 μg mL−1 ethidium bromide.29,84
3.4. Gene expression analysis
Complete poly(A)+ RNA isolated from PaCa-2 cell line samples was reverse transcribed into cDNA in a total volume of 20 μL using RevertAid™ First Strand cDNA Synthesis Kit (Fermentas, Germany). An amount of total RNA (5 μg) was used with a master mix. The master mix consisted of 50 mM MgCl2, 10× RT buffer (50 mM KCl; 10 mM Tris–HCl; pH 8.3), 10 mM of each dNTP, 50 μM oligo-dT primer, 20 IU ribonuclease inhibitor (50 kDa recombinant enzyme to inhibit RNase activity) and 50 IU MuLV reverse transcriptase. The mixture of each sample was centrifuged for 30 s at 1000 g and transferred to the thermocycler. The RT reaction was carried out at 25 °C for 10 min, followed by 1 h at 42 °C, and finished with a denaturation step at 99 °C for 5 min. Afterward, the reaction tubes containing RT preparations were flash-cooled in an ice chamber until being used for cDNA amplification through quantitative real-time-polymerase chain reaction (qRT-PCR).
| Gene | Primer sequence | GenBank (accession no.) |
|---|---|---|
| a Caspase-3; CDK6: cyclin dependent kinase 6; aaspase-8; GAPDH: glyceraldehyde-3-phosphate dehydrogenase. | ||
| Caspase-3 | F: ACT GGA CTG TGG CAT TGA GA | AJ413269.1 |
| R: GCA CAA AGC GAC TGG ATG AA | ||
| CDK6 | F: TCT CCC GGC ACT TCT GAA AT | NM_001145306.2 |
| R: ACA CCA GGT AGA AGG ACT GC | ||
| Caspase-8 | F: TTG TGG TAC CCT GCC TAG TG | AH007578.2 |
| R: TGG TTT CTC TCT CAC GCC TT | ||
| GAPDH | F: CAC ATC GCT CAG ACA CCA TG | NM_001289746.2 |
| R: TGA CGG TGC CAT GGA ATT TG | ||
3.5. Determination of the DNA damage using comet assay
The DNA damage using comet assay was determined using pancreatic cancer cell lines (PaCa-2) according to Olive et al. (1990).88 After the trypsin treatment to produce a single cell suspension, approximately 1.5 × 104 cells were embedded in 0.75% low-gelling-temperature agarose and rapidly pipetted onto a pre-coated microscope slide. Cell samples were lysed for 4 h at 50 °C in 0.5% SDS and 30 mM EDTA at pH 8.0. After rinsing overnight at room temperature in tris/borate/EDTA buffer, pH 8.0, samples were electrophoresed for 25 min at 0.6 V cm−1, then stained with propidium iodide. Slides were viewed using a fluorescence microscope with a CCD camera, and 150 individual comet images were analyzed from each sample for the tail moment, DNA content, and percentage DNA in the tail. For each sample, about 100 cells were examined to determine the percentage of cells with DNA damage that appear like comets. The non-overlapping cells were randomly selected and were visually assigned a score on an arbitrary scale of 0–3 (i.e., class 0 = no detectable DNA damage and no tail; class 1 = tail with a length less than the diameter of the nucleus; class 2 = tail with a length between 1× and 2× the nuclear diameter; and class 3 = tail longer than 2× the diameter of the nucleus) based on perceived comet tail length migration and relative proportion of DNA in the nucleus (Collins et al., 1997).893.6. Cell cycle analysis assay of treated PaCa-2 cells
Tubes with cell suspensions were received from the client's own lab. Cells (1 × 106) were suspended in 0.5 mL 1× DPBS and aspirated several times with a Pasteur pipet and fixed with 70% ethanol on ice for 2 h. The ethanol-suspended cells were centrifuged for 5 min at 300g and decanted ethanol thoroughly. The cell pellets were re-suspended in 5 mL 1× DPBS for 30 s and were centrifuged at 300g for 5 min. The cell pellets were re-suspended in 1 mL of the PI staining solution and kept in the dark at room temperature for 30 min. Cells were then transferred to the CytoFLEX Flow Cytometer (Beckman Coulter Life Sciences, USA) to measure the cell fluorescence (BECKMAN COULTER Inc., Cairo, Egypt and Cat. No. 4238055-CB) for cell cycle analysis. The percentage of cells in the G0/G1, S, and G2/M phases of the cell cycle was calculated using CytExpert Software.3.7. Statistical analysis
All data were analyzed using the General Linear Models (GLM) procedure of Statistical Analysis System (1982),90 followed by the Scheffé test to assess significant differences between groups. The values are expressed as mean ± SEM. All statements of significance were based on the probability of P < 0.05.4. Conclusion
In summary, in this work, we investigated the synthesis and full characterization of a new set of fifteen pyrazole hybrids. All of these pyrazole derivatives bearing chalcone, thiazole and thiadiazole moieties were tested in vitro against PaCa-2, MCF-7 PC3 and BJ1. Three pyrazolyl-chalcone derivatives (4, 5, and 7) showed potent anti-cancer activity against PaCa-2 cells with IC50 values of 13.0, 31.5 and 24.9 μg mL−1, respectively, compared with doxorubicin (IC50, 28.3 μg mL−1). On the other hand, pyrazolyl-thiadiazole derivative 25 showed potent anti-cancer activity against PaCa-2 cells with IC50 = 5.5 μg mL−1 compared with doxorubicin (IC50, 28.3 μg mL−1), while compounds 23 and 25 showed potent anti-cancer activity against PC3 cells with IC50 values 26.1 and 11.8 μg mL−1, respectively, compared with doxorubicin (IC50, 23.8 μg mL−1). Moreover, the mechanism of action of the most active compounds against pancreatic cancer cells from the pyrazolyl-chalcone series was studied by investigating the gene expression, DNA fragmentation, and comet assay experiments using doxorubicin as a reference drug. These results indicated that positive control and compound 4 induced cell cycle arrest at the S and G2/M phases in the treated PaCa-2 cells. Finally, the structure–activity relationship between the structures of these compounds and their biological properties was discussed.Data availability
The data supporting this article have been included as part of the ESI.† Additional data are available upon request from the corresponding authors.Author contributions
Monica Kamel: chemical reactions, methodology, investigation. Farid Sroor: conceptualization, design, chemical reactions, methodology, writing – original draft, writing – review & editing, visualization, supervision, data curation, investigation. Mahmoud Hanafy: anti-cancer activity, flow cytometry. Karima Mahrous: gene expression, DNA damage, comet assay. Hamdi M. Hassaneen: supervision, investigation, and writing the original draft. All authors have seen and approved the submitted manuscript.Conflicts of interest
There are no financial or other relationships that might lead to a conflict of interest.References
- B. S. K. Aktar, Russ. J. Bioorg. Chem., 2023, 49, 1023–1033 CrossRef.
- S. P. Singh, A. Mishra, R. K. Shyanti, R. P. Singh and A. Acharya, Biol. Trace Elem. Res., 2020, 199, 1316–1331 CrossRef.
- C. L. Wolfgang, J. M. Herman, D. A. Laheru, A. P. Klein, M. A. Erdek, E. K. Fishman and R. H. Hruban, Ca-Cancer J. Clin., 2013, 63, 318–348 CrossRef.
- A. Speck-Planche, ACS Omega, 2019, 4, 3122–3132 CrossRef.
- C. Lepage, R. Capocaccia, M. Hackl, V. Lemmens, E. Molina, D. Pierannunzio, M. Sant, A. Trama, J. Faivre, M. Hackl, N. Zielonke, W. Oberaigner, E. Van Eycken, K. Henau, Z. Valerianova, N. Dimitrova, M. Sekerija, M. Zvolský, L. Dušek, H. Storm, G. Engholm, M. Mägi, T. Aareleid, N. Malila, K. Seppä, M. Velten, X. Troussard, V. Bouvier, G. Launoy, A. V. Guizard, J. Faivre, A. M. Bouvier, P. Arveux, M. Maynadié, A. S. Woronoff, M. Robaszkiewicz, I. Baldi, A. Monnereau, B. Tretarre, N. Bossard, A. Belot, M. Colonna, F. Molinié, S. Bara, C. Schvartz, B. Lapôtre-Ledoux, P. Grosclaude, M. Meyer, R. Stabenow, S. Luttmann, A. Eberle, H. Brenner, A. Nennecke, J. Engel, G. Schubert-Fritschle, J. Kieschke, J. Heidrich, B. Holleczek, A. Katalinic, J. G. Jónasson, L. Tryggvadóttir, H. Comber, G. Mazzoleni, A. Bulatko, C. Buzzoni, A. Giacomin, A. Sutera Sardo, P. Mancuso, S. Ferretti, E. Crocetti, A. Caldarella, G. Gatta, M. Sant, H. Amash, C. Amati, P. Baili, F. Berrino, S. Bonfarnuzzo, L. Botta, F. Di Salvo, R. Foschi, C. Margutti, E. Meneghini, P. Minicozzi, A. Trama, D. Serraino, L. Dal Maso, R. De Angelis, M. Caldora, R. Capocaccia, E. Carrani, S. Francisci, S. Mallone, D. Pierannunzio, P. Roazzi, S. Rossi, M. Santaquilani, A. Tavilla, F. Pannozzo, S. Busco, L. Bonelli, M. Vercelli, V. Gennaro, P. Ricci, M. Autelitano, G. Randi, M. Ponz De Leon, C. Marchesi, C. Cirilli, M. Fusco, M. F. Vitale, M. Usala, A. Traina, R. Staiti, F. Vitale, B. Ravazzolo, M. Michiara, R. Tumino, P. Giorgi Rossi, E. Di Felice, F. Falcini, A. Iannelli, O. Sechi, R. Cesaraccio, S. Piffer, A. Madeddu, F. Tisano, S. Maspero, A. C. Fanetti, R. Zanetti, S. Rosso, P. Candela, T. Scuderi, F. Stracci, F. Bianconi, G. Tagliabue, P. Contiero, A. P. Dei Tos, S. Guzzinati, S. Pildava, G. Smailyte, N. Calleja, D. Agius, T. B. Johannesen, J. Rachtan, S. Gózdz, R. Mezyk, J. Blaszczyk, M. Bebenek, M. Bielska-Lasota, G. Forjaz de Lacerda, M. J. Bento, C. Castro, A. Miranda, A. Mayer-da-Silva, F. Nicula, D. Coza, C. Safaei Diba, M. Primic-Zakelj, E. Almar, C. Ramírez, M. Errezola, J. Bidaurrazaga, A. Torrella-Ramos, J. M. Díaz García, R. Jimenez-Chillaron, R. Marcos-Gragera, A. Izquierdo Font, M. J. Sanchez, D. Y. L. Chang, C. Navarro, M. D. Chirlaque, C. Moreno-Iribas, E. Ardanaz, J. Galceran, M. Carulla, M. Lambe, S. Khan, M. Mousavi, C. Bouchardy, M. Usel, S. M. Ess, H. Frick, M. Lorez, S. M. Ess, C. Herrmann, A. Bordoni, A. Spitale, I. Konzelmann, O. Visser, L. van der Geest, R. Otter, M. Coleman, C. Allemani, B. Rachet, J. Verne, N. Easey, G. Lawrence, T. Moran, J. Rashbass, M. Roche, J. Wilkinson, A. Gavin, C. Donnelly, D. H. Brewster, D. W. Huws and C. White, Eur. J. Cancer, 2015, 51, 2169–2178 CrossRef.
- L. A. Torre, F. Bray, R. L. Siegel, J. Ferlay, J. Lortet-Tieulent and A. Jemal, Ca-Cancer J. Clin., 2015, 65, 87–108 CrossRef.
- W. Hartwig, J. Werner, D. Jäger, J. Debus and M. W. Büchler, Lancet Oncol., 2013, 14, e476–e485 CrossRef PubMed.
- M. G. Kamel, F. M. Sroor, A. M. Othman, K. F. Mahrous, F. M. Saleh, H. M. Hassaneen, T. A. Abdallah, I. A. Abdelhamid and M. A. M. Teleb, Chem. Mon., 2022, 153, 211–221 CrossRef CAS.
- M. G. Kamel, F. M. Sroor, A. M. Othman, H. M. Hassaneen, T. A. Abdallah, F. M. Saleh and M. A. M. Teleb, Chem. Mon., 2022, 153, 929–937 CrossRef CAS.
- K. Mezgebe, Y. Melaku and E. Mulugeta, ACS Omega, 2023, 8, 19194–19211 CrossRef CAS PubMed.
- N. Yasser, F. M. Sroor, H. M. El-Shorbagy, S. M. Eissa, H. M. Hassaneen and I. A. Abdelhamid, RSC Adv., 2024, 14, 21859–21873 RSC.
- C. Zhuang, W. Zhang, C. Sheng, W. Zhang, C. Xing and Z. Miao, Chem. Rev., 2017, 117, 7762–7810 CrossRef CAS PubMed.
- P. Singh, A. Anand and V. Kumar, Eur. J. Med. Chem., 2014, 85, 758–777 CrossRef CAS.
- D. Israf, T. Khaizurin, A. Syahida, N. Lajis and S. Khozirah, Mol. Immunol., 2007, 44, 673–679 CrossRef CAS PubMed.
- R. B. Birari, S. Gupta, C. G. Mohan and K. K. Bhutani, Phytomedicine, 2011, 18, 795–801 CrossRef CAS.
- F.-F. Gan, K. K. Kaminska, H. Yang, C.-Y. Liew, P.-C. Leow, C.-L. So, L. N. L. Tu, A. Roy, C.-W. Yap, T.-S. Kang, W.-K. Chui and E.-H. Chew, Antioxid. Redox Signaling, 2013, 19, 1149–1165 CrossRef CAS.
- F. Lunardi, M. Guzela, A. T. Rodrigues, R. r. Corrêa, I. Eger-Mangrich, M. r. Steindel, E. C. Grisard, J. Assreuy, J. o. B. Calixto and A. R. S. Santos, Antimicrob. Agents Chemother., 2003, 47, 1449–1451 CrossRef CAS PubMed.
- J. Yoon, E. A. Yoo, J.-Y. Kim, A. N. Pae, H. Rhim, W.-K. Park, J. Y. Kong and H.-Y. Park Choo, Bioorg. Med. Chem., 2008, 16, 5405–5412 CrossRef CAS.
- S. Ducki, Anti-Cancer Agents Med. Chem., 2009, 9, 336–347 CrossRef CAS.
- D. K. Mahapatra, V. Asati and S. K. Bharti, Eur. J. Med. Chem., 2015, 92, 839–865 CrossRef CAS.
- S. S. R. Alsayed, C. C. Beh, N. R. Foster, A. D. Payne, Y. Yu and H. Gunosewoyo, Curr. Mol. Pharmacol., 2019, 12, 27–49 CrossRef CAS.
- M. I. Abdullah, A. Mahmood, M. Madni, S. Masood and M. Kashif, Bioorg. Chem., 2014, 54, 31–37 CrossRef CAS PubMed.
- Y. Cao, W. Xu, Y. Huang and X. Zeng, Nat. Prod. Res., 2018, 34, 736–739 CrossRef.
- V. Tomar, G. Bhattacharjee, Kamaluddin, S. Rajakumar, K. Srivastava and S. K. Puri, Eur. J. Med. Chem., 2010, 45, 745–751 CrossRef CAS PubMed.
- F. M. Sroor, A. F. El-Sayed and M. Abdelraof, Arch. Pharm., 2024, 357(6), 2300738 CrossRef CAS.
- M. A. Elsayed, A. M. Elsayed and F. M. Sroor, Med. Chem. Res., 2024, 33, 476–491 CrossRef CAS.
- F. M. Sroor, W. M. Basyouni, H. F. Aly, S. A. Ali and A. F. Arafa, Med. Chem. Res., 2022, 31, 195–206 CrossRef CAS.
- F. M. Sroor, A. M. Othman, K. Mahmoud and K. F. Mahrous, J. Mol. Struct., 2023, 1294, 136516 CrossRef CAS.
- F. M. Sroor, K. F. Mahrous, H. A. M. A. El-Kader, A. M. Othman and N. S. Ibrahim, Sci. Rep., 2023, 13, 17560 CrossRef PubMed.
- F. M. Sroor, W. M. Tohamy, K. M. A. Zoheir, N. M. Abdelazeem, K. F. Mahrous and N. S. Ibrahim, BMC Chem., 2023, 17, 106 CrossRef PubMed.
- M. G. Kamel, H. M. Hassaneen, F. M. Sroor, T. A. Abdallah, M. A. M. Teleb and F. M. Saleh, J. Mol. Struct., 2023, 1287, 135722 CrossRef.
- M. T. Helmy, F. M. Sroor, H. M. Hassaneen, M. A. Mohamed Teleb and F. M. Saleh, Polycyclic Aromat. Compd., 2023, 1–14, DOI:10.1080/10406638.2023.2214284.
- F. M. Sroor, A. A. F. Soliman, E. M. Youssef, M. Abdelraof and A. F. El-Sayed, Sci. Rep., 2024, 14(1), 15441 CrossRef CAS PubMed.
- S. Haider, M. S. Alam and H. Hamid, Eur. J. Med. Chem., 2015, 92, 156–177 CrossRef CAS PubMed.
- A. Ansari, A. Ali, M. Asif and S. Shamsuzzaman, New J. Chem., 2017, 41, 16–41 RSC.
- F. M. Sroor, A. M. Othman, M. M. Aboelenin and K. F. Mahrous, Med. Chem. Res., 2022, 31, 400–415 CrossRef CAS.
- F. M. Sroor, M. F. Mohamed, G. K. Abdullah, K. F. Mahrous, K. M. A. Zoheir, S. A. Ibrahim, A. H. M. Elwahy and I. A. Abdelhamid, Polycyclic Aromat. Compd., 2022, 1–18, DOI:10.1080/10406638.2022.2046616.
- O. Ebenezer, M. Shapi and J. A. Tuszynski, Biomedicines, 2022, 10, 1124 CrossRef.
- A. Petrou, M. Fesatidou and A. Geronikaki, Molecules, 2021, 26, 3166 CrossRef PubMed.
- E. Pivovarova, A. Climova, M. Świątkowski, M. Staszewski, K. Walczyński, M. Dzięgielewski, M. Bauer, W. Kamysz, A. Krześlak, P. Jóźwiak and A. Czylkowska, Int. J. Mol. Sci., 2022, 23, 9844 CrossRef PubMed.
- K. Z. Laczkowski, A. Biernasiuk, A. Baranowska-Laczkowska, K. Misiura, A. Malm, T. Plech and A. Paneth, Med. Chem., 2016, 12, 553–562 CrossRef PubMed.
- A. Andreani, M. Rambaldi, G. Mascellani and P. Rugarli, Eur. J. Med. Chem., 1987, 22, 19–22 CrossRef CAS.
- Z. A. Muhammad, G. S. Masaret, M. M. Amin, M. A. Abdallah and T. A. Farghaly, Med. Chem., 2017, 13, 226–238 CrossRef CAS PubMed.
- M. Gopal, B. Padmashali, Y. N. Manohara and B. M. Gurupadayya, Ind. J. Pharm. Sci., 2008, 70, 572 CrossRef.
- M. Koufaki, C. Kiziridi, F. Nikoloudaki and M. N. Alexis, Bioorg. Med. Chem. Lett., 2007, 17, 4223–4227 CrossRef CAS.
- H. N. Karade, B. N. Acharya, M. Sathe and M. P. Kaushik, Med. Chem. Res., 2008, 17, 19–29 CrossRef CAS.
- A.-T. Pânzariu, M. Apotrosoaei, I. M. Vasincu, M. Drăgan, S. Constantin, F. Buron, S. Routier, L. Profire and C. Tuchilus, Chem. Cent. J., 2016, 10, 6 CrossRef.
- M. A. M. Teleb, F. M. Sroor, H. M. Hassaneen and F. M. Saleh, Egypt. J. Chem., 2022, 65(13B), 1045–1052 Search PubMed.
- M. T. Helmy, F. M. Sroor, A. M. Othman, H. M. Hassaneen, F. M. Saleh and M. A. M. Teleb, J. Heterocycl. Chem., 2022, 60(4), 585–595 CrossRef.
- M. GÜR, M. Zurnaci, E. AltinÖZ, N. ŞEner, Ç. ŞAhİN, M. ŞEnturan, İ. ŞEner, M. ÇAvuŞ and E. M. Altuner, Hittite J. Sci. Eng., 2023, 10, 99–116 CrossRef.
- A. K. Jain, S. Sharma, A. Vaidya, V. Ravichandran and R. K. Agrawal, Chem. Biol. Drug Des., 2013, 81, 557–576 CrossRef PubMed.
- Z. Xu, Y. Zhuang and Q. Chen, Eur. J. Med. Chem., 2023, 257, 115495 CrossRef CAS PubMed.
- P. S. Dabhade, M. P. Dabhade, L. S. Rathod, S. A. Dhawale, S. A. More, S. Y. Chaudhari and S. N. Mokale, Chem. Biodiversity, 2024, 21, e202400015 CrossRef CAS PubMed.
- S. Yadav, R. Lakhia, A. Bendi, H. Phougat, N. K. Verma, K. Singh, N. Raghav and R. Pundeer, J. Mol. Struct., 2024, 1316, 138847 CrossRef CAS.
- K. Karrouchi, S. Radi, Y. Ramli, J. Taoufik, Y. N. Mabkhot, F. A. Al-aizari and M. h. Ansar, Molecules, 2018, 23, 134 CrossRef.
- A. F. Hegarty, M. P. Cashman and F. L. Scott, J. Chem. Soc., Perkin Trans. 2, 1972, 1381, 10.1039/p29720001381.
- D. L. Klayman, J. F. Bartosevich, T. S. Griffin, C. J. Mason and J. P. Scovill, J. Med. Chem., 2002, 22, 855–862 CrossRef.
- L. F. Audrieth, E. S. Scott and P. S. Kippur, J. Org. Chem., 2002, 19, 733–741 CrossRef.
- İ. Koca, A. Özgür, K. A. Coşkun and Y. Tutar, Bioorg. Med. Chem., 2013, 21, 3859–3865 CrossRef PubMed.
- A. Tanitame, Y. Oyamada, K. Ofuji, M. Fujimoto, N. Iwai, Y. Hiyama, K. Suzuki, H. Ito, H. Terauchi, M. Kawasaki, K. Nagai, M. Wachi and J.-i. Yamagishi, J. Med. Chem., 2004, 47, 3693–3696 CrossRef PubMed.
- P. D. Sauzem, G. d. S. Sant'Anna, P. Machado, M. M. M. F. Duarte, J. Ferreira, C. F. Mello, P. Beck, H. G. Bonacorso, N. Zanatta, M. A. P. Martins and M. A. Rubin, Eur. J. Pharmacol., 2009, 616, 91–100 CrossRef.
- S. M. Sakya, H. Cheng, K. M. Lundy DeMello, A. Shavnya, M. L. Minich, B. Rast, J. Dutra, C. Li, R. J. Rafka, D. A. Koss, J. Li, B. H. Jaynes, C. B. Ziegler, D. W. Mann, C. F. Petras, S. B. Seibel, A. M. Silvia, D. M. George, A. Hickman, M. L. Haven and M. P. Lynch, Bioorg. Med. Chem. Lett., 2006, 16, 1202–1206 CrossRef.
- R. R. Ranatunge, D. S. Garvey, D. R. Janero, L. G. Letts, A. M. Martino, M. G. Murty, S. K. Richardson, D. V. Young and I. S. Zemetseva, Bioorg. Med. Chem., 2004, 12, 1357–1366 CrossRef CAS.
- M. Chauhan and R. Kumar, Bioorg. Med. Chem., 2013, 21, 5657–5668 CrossRef CAS PubMed.
- I. A. Abdelhamid, A. H. M. Elwahy, F. M. Sroor, M. F. Mohamed, S. E. Elsayed, K. F. Mahrous, L. Mageed, M. K. Hanafy and S. A. Ibrahim, Lett. Drug Des. Discovery, 2022, 19, 1007–1021 CrossRef.
- C. Harrison, J.-J. Kiladjian, H. K. Al-Ali, H. Gisslinger, R. Waltzman, V. Stalbovskaya, M. McQuitty, D. S. Hunter, R. Levy, L. Knoops, F. Cervantes, A. M. Vannucchi, T. Barbui and G. Barosi, N. Engl. J. Med., 2012, 366, 787–798 CrossRef CAS.
- J. E. Frampton, Drugs, 2013, 73, 2031–2051 CrossRef PubMed.
- A. Nicosia, G. Cavallaro, S. Costa, M. Utzeri, A. Cuttitta, G. Giammona and N. Mauro, Cancers, 2020, 12, 3114 CrossRef.
- X. Xu, L. Zhang, X. He, P. Zhang, C. Sun, X. Xu, Y. Lu and F. Li, Biochem. Biophys. Res. Commun., 2018, 502, 160–165 CrossRef.
- H. Moghtaderi, H. Sepehri and F. Attari, Biomed. Pharmacother., 2017, 88, 582–594 CrossRef.
- L. Carrassa and G. Damia, Cancer Treat. Rev., 2017, 60, 139–151 CrossRef CAS.
- M. Li, L. You, J. Xue and Y. Lu, Front. Pharmacol, 2018, 9, 522 CrossRef.
- S. A. Andrabi, H. C. Kang, J.-F. Haince, Y.-I. Lee, J. Zhang, Z. Chi, A. B. West, R. C. Koehler, G. G. Poirier, T. M. Dawson and V. L. Dawson, Nat. Med., 2011, 17, 692–699 CrossRef CAS.
- G. Kung, K. Konstantinidis and R. N. Kitsis, Circ. Res., 2011, 108, 1017–1036 CrossRef CAS.
- J. Cao, F. C. Wong, C. C. Woo, A. Hsu and B. K. H. Tan, PLoS One, 2013, 8, e78021 CrossRef.
- G. Pal, H. P. Mishra, T. K. Suvvari, A. Tanwar, T. Ghosh, P. Verma, A. Pal, K. Patial, C. Mahapatra, N. A. Amanullah, S. A. Shukoor, S. Kamal and V. Rohil, Cureus, 2023, 15(8), 43333 Search PubMed.
- V. Lobo, A. Patil, A. Phatak and N. Chandra, Pharmacogn. Rev., 2010, 4, 118 CrossRef PubMed.
- H. M. Hassaneen, H. A. H. Mousa, N. M. Abed and A. S. Shawali, Heterocycles, 1988, 27, 695 CrossRef.
- A. S. Shawali, H. M. Hassaneen and H. A. Ibrahim, Arch. Pharmacal Res., 1990, 13, 126–131 CrossRef CAS.
- F. Abdel-Galil, A. Sami Shawali, H. Mahmoud Hassaneen, A.-F. Shetta and A. Osman, Heterocycles, 1982, 19, 57 CrossRef.
- N. F. Eweiss and A. Osman, J. Heterocycl. Chem., 2009, 17, 1713–1717 CrossRef.
- M. I. Thabrew, R. D. Hughes and I. G. McFarlane, J. Pharm. Pharmacol., 1997, 49, 1132–1135 CrossRef CAS.
- A. Yawata, M. Adachi, H. Okuda, Y. Naishiro, T. Takamura, M. Hareyama, S. Takayama, J. C. Reed and K. Imai, Oncogene, 1998, 16, 2681–2686 CrossRef CAS.
- S. Fogli, P. Nieri, A. Chicca, B. Adinolfi, V. Mariotti, P. Iacopetti, M. C. Breschi and S. Pellegrini, FEBS Lett., 2006, 580, 1733–1739 CrossRef PubMed.
- T. Watanabe, T. Miura, Y. Degawa, Y. Fujita, M. Inoue, M. Kawaguchi and C. Furihata, Cancer Cell Int., 2010, 10, 2 CrossRef PubMed.
- Q. Yang, M. H. Feng, X. Ma, H. C. Li and W. Xie, Oncol. Lett., 2017, 14, 6071–6078 Search PubMed.
- F. M. Sroor, W. M. Basyouni, H. F. Aly, E. A. Younis, K. F. Mahrous and A. A. Haroun, Appl. Biol. Chem., 2022, 65, 68 CrossRef.
- P. L. Olive, J. P. Banáth and R. E. Durand, Radiat. Res., 2012, 178, AV35–AV42 CrossRef CAS PubMed.
- A. Collins, M. Dusinska, M. Franklin, M. Somorovska, H. Petrovska, S. Duthie, L. Fillion, M. Panayiotidis, K. Raslova and N. Vaughan, Environ. Mol. Mutagen., 1997, 30, 139–146 CrossRef CAS PubMed.
- SAS Institute, SAS User's Guide : Statistics, SAS Institute, Cary, NC, 1982 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03005b |
| This journal is © The Royal Society of Chemistry 2024 |