
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Repurposed organoselenium tethered amidic acids as apoptosis inducers in melanoma cancer via P53, BAX, caspases-3, 6, 8, 9, BCL-2, MMP2, and MMP9 modulations†
Saad Shaaban *ab,
Hanan A. Althikrallaha,
Amr Negma,
Ayman Abo Elmaatyc and
Ahmed A. Al-Karmalawy*de
*ab,
Hanan A. Althikrallaha,
Amr Negma,
Ayman Abo Elmaatyc and
Ahmed A. Al-Karmalawy*de
aDepartment of Chemistry, College of Science, King Faisal University, Al-Ahsa 31982, Saudi Arabia. E-mail: sibrahim@kfu.edu.sa
bDepartment of Chemistry, Faculty of Science, Mansoura University, 35516 Mansoura, Egypt
cMedicinal Chemistry Department, Faculty of Pharmacy, Port Said University, Port Said, 42511, Egypt
dDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Horus University–Egypt, New Damietta 34518, Egypt. E-mail: akarmalawy@horus.edu.eg
ePharmaceutical Chemistry Department, Faculty of Pharmacy, Ahram Canadian University, 6th of October City, Giza 12566, Egypt
First published on 10th June 2024
Abstract
Organoselenium (OSe) agents hold promise for preventing cancer due to their potential ability to fight cancer development and protect cells from oxidative damage. Herein, OSe-based maleanilic and succinanilic acids were tested to estimate their antitumor activities against fifteen cancer cell lines. Besides, their potential safety and selectivity were further investigated against two normal cell lines, namely, human skin fibroblasts (HSF) and olfactory ensheathing cell line (OEC) using the growth inhibition percentage (GI%) assay. Moreover, the apoptotic potential of the superior anticancer candidates (8, 9, 10, and 11) was evaluated against P53, BAX, Caspase-3, Caspase-6, Caspase-8, Caspase-9, BCL-2, MMP2, and MMP9 apoptotic markers. Additionally, to enhance our understanding and predict the inhibitory potential of the examined compounds as potential anticancer agents, a thorough structure–activity relationship (SAR) analysis was conducted. On the other hand, molecular docking and ADMET studies were performed for the examined candidates as well. Overall, our findings point to significant anticancer activities of the organoselenium tethered amidic acids, suggesting their promising cytotoxic potential as effective anticancer drugs.
1. Introduction
While advancements in cancer treatment have been significant, conquering this disease remains a major challenge for healthcare systems around the world.1 The burden of cancer is particularly felt in countries with developing or middle-income economies, highlighting the ongoing link between socioeconomic status and cancer's impact.2,3 Cancer ranks as the 2nd leading cause of death worldwide, following heart disease.4,5 A worrying trend suggests cancer cases could surge by over 50% in the years ahead.6,7 Furthermore, since anticancer drugs target rapidly dividing cells, they can unfortunately damage healthy tissues as well as cancerous ones. This can lead to a range of side effects, including a weakened immune system, hair loss, anemia, and nausea.8,9 In response to these challenges, researchers worldwide are dedicating significant effort to developing groundbreaking therapies for a wide range of cancers.Apoptosis, a form of controlled cell death, is overseen by two protein families: the BCL-2 family and the Caspase family. The release of cytochrome c triggers the caspase cascade, preparing it for the activation of cell death.10 Cells with built-in self-destruct mechanisms, known as programmed cell death, ensure our bodies eliminate old and malfunctioning cells, keeping everything in working order. Cancer cells, however, unlike normal cells, can disable this self-destruct program, allowing them to divide uncontrollably.11 Inside the cell's powerhouses, the mitochondria, a process called mitochondrial outer membrane permeabilization (MOMP) triggers cell suicide, also known as intrinsic apoptosis. MOMP leads to the formation of structures called apoptosomes, which activate Caspase-9. This, in turn, sets off a chain reaction involving other caspases, ultimately leading to cell death.10
Moreover, BAX is a key player in the BCL-2 family, a group of proteins that regulate cell death. When activated, BAX triggers MOMP, which leads to the release of pro-apoptotic proteins like cytochrome c. This release sets the stage for programmed cell death.10 On the other hand, P53 acts as a brake on cell growth. Located in the cell nucleus, it controls genes and promotes cell death when necessary. This makes P53 a classic tumor suppressor protein.12 When cells experience stress, like DNA damage, P53 accumulates in the nucleus. There, P53 acts like a tumor suppressor by triggering two important responses: cell cycle arrest (this gives the cell time to repair the damaged DNA) and apoptosis (P53 promotes cell death to prevent the spread of damaged cells, if the damage is too severe).12
Cyclic anhydrides are important heterocyclic scaffolds and versatile building blocks in organic synthesis, possessing a broad spectrum of uses in medicinal chemistry and materials science.13 They can readily react with various nucleophiles to afford diverse bifunctional intermediates used for the construction of complex molecular structures in the domains of materials science and pharmaceutical chemistry.14 For instance, maleic and succinic anhydrides are frequently utilized for the synthesis of dicarboxamides and cyclic imides via reaction with amines.15,16 The reaction typically involves a nucleophilic attack of the amine on the anhydride carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) carbon, followed by ring opening and the formation of the corresponding amidic acid intermediates i.e., N-maleanilic and N-succinanilic carboxylic acid derivatives.17,18 The latter is either subjected to further reaction with amines to furnish dicarboxamides or subsequent dehydration and intramolecular cyclization to give the corresponding cyclic imides (e.g., maleimides and succinimides).19
O) carbon, followed by ring opening and the formation of the corresponding amidic acid intermediates i.e., N-maleanilic and N-succinanilic carboxylic acid derivatives.17,18 The latter is either subjected to further reaction with amines to furnish dicarboxamides or subsequent dehydration and intramolecular cyclization to give the corresponding cyclic imides (e.g., maleimides and succinimides).19
Within this context, dicarboxamides are important synthetic intermediates and have shown prevalent applications in coordination, agrochemicals, and polymer chemistry.16,20 Furthermore, they exhibited anti-atherosclerotic, hemostatic, anti-inflammatory, and anticoagulant activities.21 Moreover, they were extensively used in coordination chemistry as efficient ligands for sensing and separation of toxic heavy metals.22 For example, 1,3-dicarboxamide I exhibited good antioxidant properties by increasing the expression of the cytochrome P-450 enzymes in the liver.16,23,24 Additionally, maleimides and succinimides are found in many natural products and pharmaceutical active drug molecules such as the farinomalein (II) natural pesticide and Zarontin (III), and oxaleimide A (IV) which are used as anticonvulsant drug candidates.17,25,26
Furthermore, scientists are increasingly excited about developing new compounds containing selenium (Se) (organoselenium (OSe) compounds). These compounds hold promise for preventing cancer due to their potential ability to fight cancer development (chemoprevention) and protect cells from damage (antioxidant activity).27–29 Hence, OSe hybrids are considered privileged scaffolds in drug discovery for cancer treatment.18,30–34 Recently, we reported the OSe-containing isomaleimide V with good antioxidant, cytoprotective, and antiapoptotic activity against oligodendrocytes.17,19,35 Additionally, the OSe-bearing N-succinimide VI, synthesized in our laboratory, exhibited good anticancer activity against HEPG2 cells.36 Additionally, we disclosed OSe-containing N-mealanilic acid (VII) and its zinc(II) chelate with promising antioxidant and antimicrobial activity against Staphylococcus aureus, Serratia marcescens, and Escherichia coli bacterial strains, and anticancer activities against different tumor cell lines (e.g., MCF-7, HCT116, HEPG2),37–39 Fig. 1.
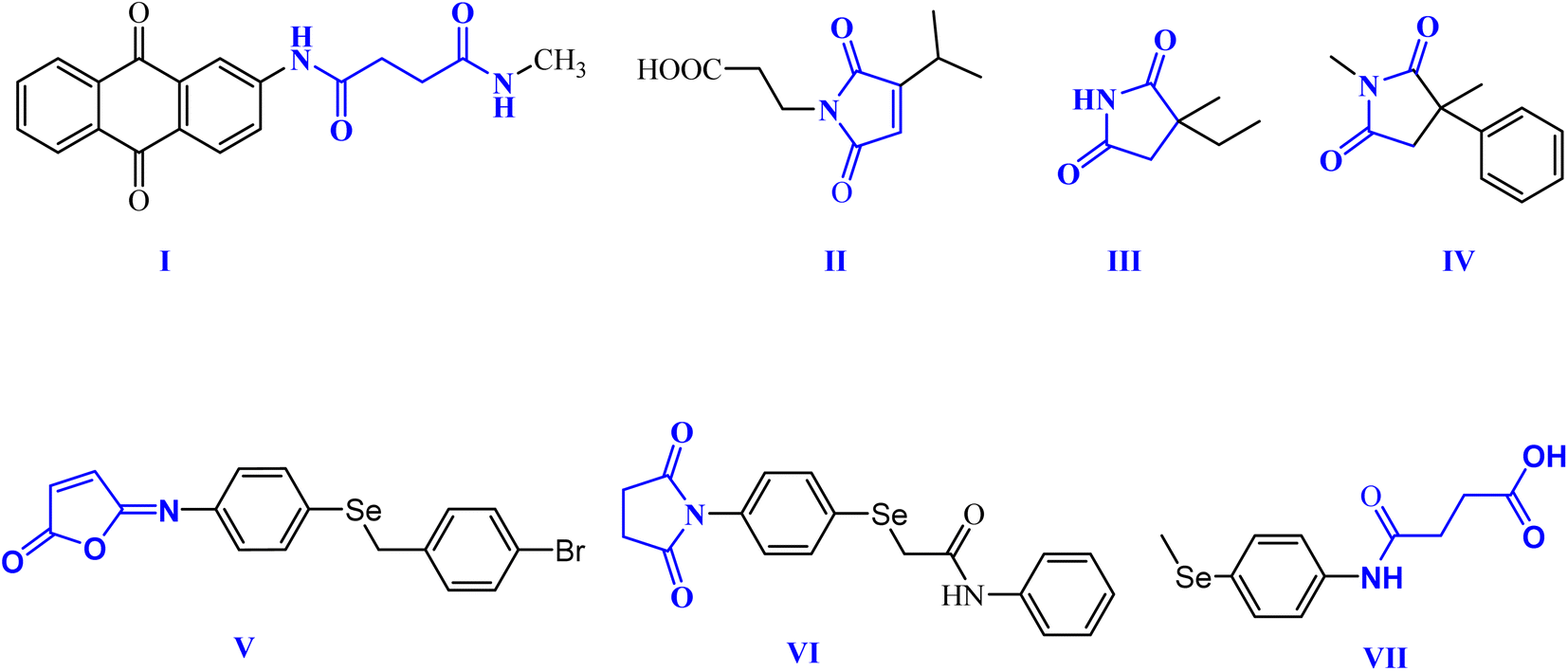 | ||
| Fig. 1 Biologically relevant amidic acid, dicarboxamide, and cyclic imides. | ||
Although OSe compounds manifested lower toxicity than inorganic-Se compounds; such toxicity is not enough to address the required specifications for drug development which is attributed to C–Se bond instability.30 Therefore, developing more stable OSe agents is of great interest. Furthermore, OSe agents induce cell death via multiple pathways including oxidative stress manipulation, apoptotic and antiapoptotic gene modulations, and death receptor activations, however, their specific target is still unclear.40 Accordingly, these challenges should be considered to better understand the underlying therapeutic potential of OSe compounds in cancer chemotherapy. The latter includes the estimation of their specific mode of action(s) and precise target(s) as well as exploring their possible use as chemosensitizers or in combination with other drugs in radiotherapy. Lately, we described OSe-bearing maleanilic and succinanilic acids as promising SARS-CoV-2 MPro inhibitors.41 Specifically, this preliminary study was restricted to computational calculations, and no pharmacological evaluations of the compounds were carried out to estimate their bioactivities.
Herein and as an extension of our earlier work, we aim to assess the antitumor activity of these OSe-based maleanilic and succinanilic acids against fifteen cancer cell lines. Also, their growth inhibition percentage (GI%) will be evaluated against two normal cell lines, namely, human skin fibroblasts (HSF) and olfactory ensheathing cell line (OEC), to estimate the potential safety and selectivity. Furthermore, the cytotoxic inhibitory concentration 50 (IC50) will be assessed against the cancer cell lines for compounds with the most outstanding GI% using the SRB assay. Moreover, the apoptotic potential of the superior anticancer candidates (8, 9, 10, and 11) will be evaluated against P53, BAX, Caspase-3, Caspase-6, Caspase-8, Caspase-9, BCL-2, MMP2, and MMP9 apoptotic markers. Furthermore, a molecular docking study will be performed to recommend the potential activity of the examined candidates to induce apoptosis as a recommended mechanism for their antitumor activity. Finally, ADMET studies will be applied to the examined candidates to investigate their physicochemical and pharmacokinetic properties.
1.1. Rational of design
The design rationale was based on combining different lead optimization approaches to improve the cytotoxic activity. Herein, it was displayed that the start compound; diphenyl diselenide (Ph2Se2) is very lipophilic with low oral bioavailability, toxicity issues due to off-target activities, and restricted physicochemical properties.42,43 Therefore, Ph2Se2 was simplified to 4-aminobenzeneselenol. Accordingly, a lead optimization tool was employed using substituent variation, chain elongation, and rigidification approaches. Thus, the hydrogen atom of the –SeH functionality was substituted with different alkyl groups to pursue the cytotoxic activity change (substituent variation approach). Besides, the 4-amino group was replaced with an amido-butanoic acid motif to improve the receptor binding affinity via affording extra hydrophobic and H-bond interactions (chain elongation approach). Furthermore, aiming to study the effect of compound flexibility change on cytotoxic activity, block bonds (olefenic bonds) were included between α and β carbons of butanoic acid elongated chain (rigidification approach), as shown in Fig. 2.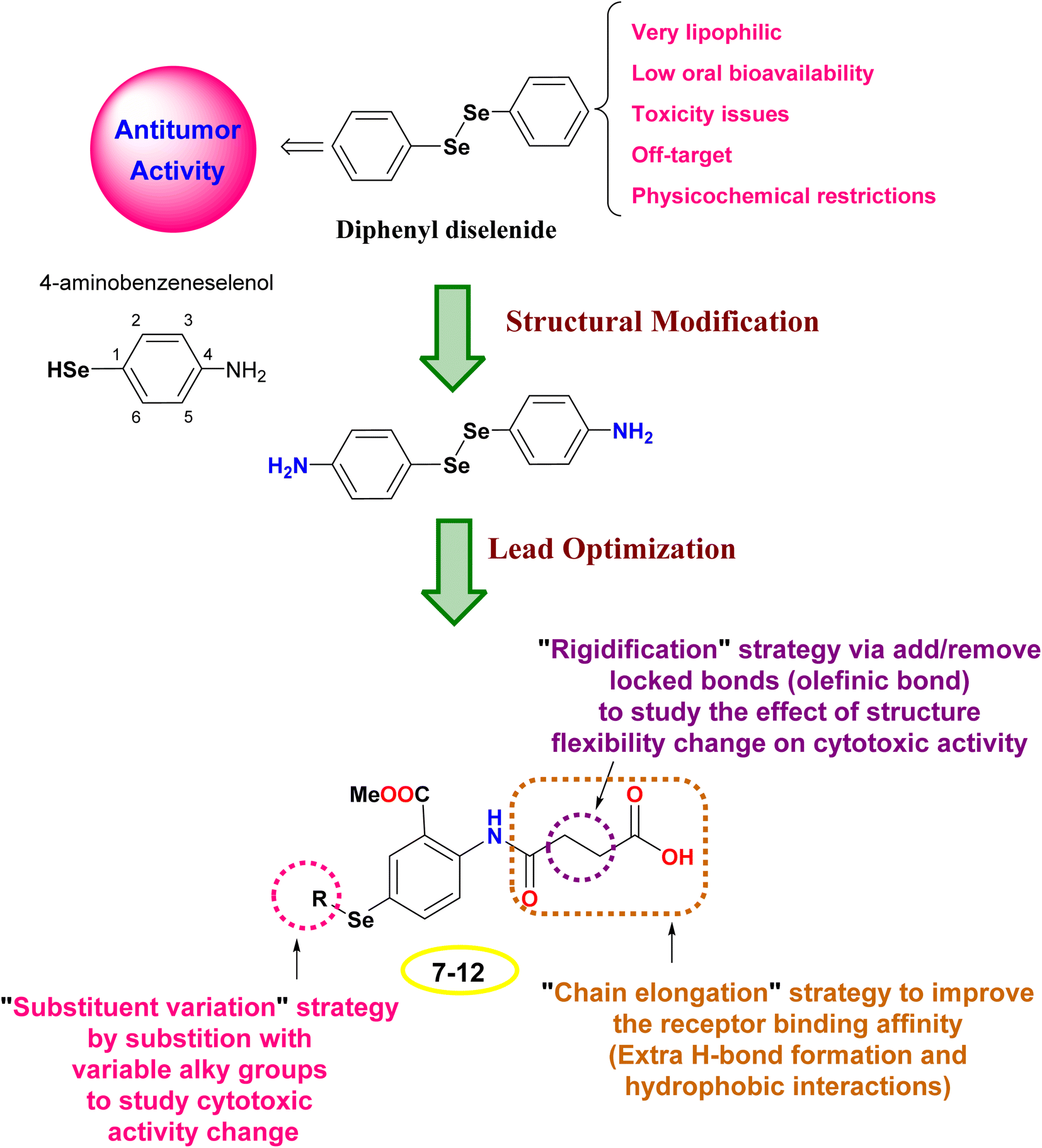 | ||
| Fig. 2 The design rationale of the investigated organoselenium compounds (7–12). | ||
2. Results and discussion
2.1. Chemistry
The interest in developing novel OSe candidates is growing stimulated by their privileged antitumor and antioxidant activities.44–46 Therefore, efficient, and innovative procedures are highly desired to prepare novel functionalized OSe agents and investigate their potential bioactivities. Although significant progress made in the synthesis of OSe compounds, their development has been frequently hindered by different synthetic complications.44,47 These challenges include the use of complicated reaction conditions (e.g. absence of O2/air or elevated temperature) as well as the hazardous, costly, sensitive, and limited functional group tolerance OSe reagents such as SeOCl2, KSeCN, SeCl4, and SeF6.48–50 Accordingly, the use of mild, and simple procedures employing stable OSe reagents compatible with a wide functional group is highly required. Diaryl diselenides are key precursors for diverse multifunctional OSe candidates such as aryl selenide halide (ArSeX) which in turn promotes selenocyclization of olefins and acetylenes to give structurally diverse selenaheterocycles.51 Their notable stability, safety, and ease of handling made them ideal for the optimization of new reactions.51 Indeed, diaryl diselenides can undergo a wide range of transformations and give access to various Se-containing reactive species such as RSe˙, RSe−, and RSe+ as well as Se-based functionalities (e.g. selenenic, seleninic, and selenonic acids).52,53 Likewise, amidic acid motifs play a fundamental role in the biological effectiveness of different biomolecules such as peptides, pseudopeptides, enzymes, and several pharmacologically active agents.36,54 Their exceptional activities stem from their unique electronic properties and their tendency to form hydrogen bonding. Therefore, it is hypothesized that the development of amidic acids-containing OSe scaffolds would result in enhanced antitumor activities compared to their respective amidic acids or OSe precursors alone. To this point, dimethyl 5,5′-diselanediylbis(2-aminobenzoate) (3) was proposed as the diselenide starting building block due to its diverse and reactive functional groups i.e. the amino and the ester groups. Furthermore, OSe 3 is prepared on a gram scale, and its high stability together with its good solubility in most organic solvents renders it a versatile synthon for various organic transformations.36,38,39 OSe 3 is obtained in excellent yield (92%) by the hydrolysis of selenocyanate derivative 2 using NaOH in C2H5OH at ambient temperature (Scheme 1). The reduction of the diselenide functionality in OSe 3 by NaBH4 led to the generation of the corresponding sodium arylselenolate. The latter is a strong and reactive nucleophile, however O2-sensitive, and therefore it was instantaneously trapped by the reaction with electrophiles such as iodomethane, α-chlorotoluene, and 2-chloroacetanilide to give the respective para-substituted primary aromatic OSe amines 4, 5, and 6 in very good yields (up to 96%) (Scheme 1). The nucleophilic attack of the amine functionality of the OSe compounds 4, 5, and 6 on the maleic and succinic anhydride carbonyl carbon resulted in ring opening and the subsequent formation of the N-amidic acids 7–12 in good yields (up to 95%) as shown in Scheme 1.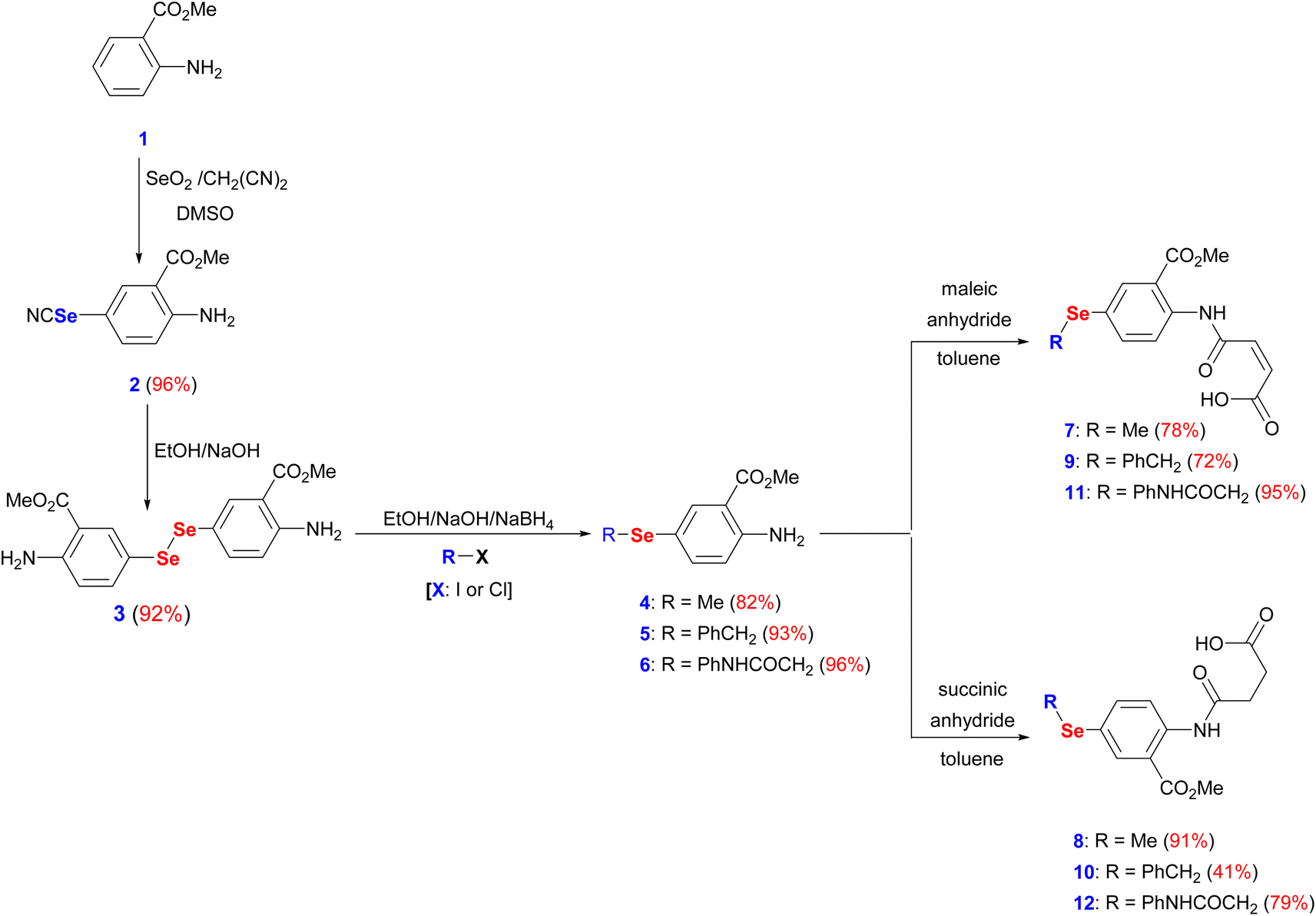 | ||
| Scheme 1 Synthesis of target amidic acids (7–12). | ||
2.2. Biological evaluation
| Comp no./cell line name | 7 | 8 | 9 | 10 | 11 | 12 | Dox |
|---|---|---|---|---|---|---|---|
| HNO97 | 55.60 | 53.62 | 53.37 | 55.83 | 49.98 | 56.83 | 70.50 |
| HN9 | 46.02 | 34.13 | 48.93 | 53.72 | 37.82 | 45.89 | 82.03 |
| MCF7 | 34.78 | 65.02 | 62.98 | 64.25 | 57.86 | 50.52 | 0.80 |
| CaCo2 | 48.44 | 45.22 | 41.18 | 45.20 | 41.69 | 33.70 | 2.81 |
| HEP2 | 60.04 | 67.71 | 66.25 | 68.87 | 62.91 | 60.78 | 62.10 |
| HEPG2 | 67.98 | 72.60 | 72.15 | 62.55 | 57.31 | 54.95 | 73.32 |
| A431 | 62.13 | 62.55 | 67.06 | 71.31 | 74.31 | 66.20 | 68.30 |
| A375 | 73.95 | 75.79 | 65.54 | 70.60 | 67.89 | 66.58 | 69.38 |
| H1299 | 57.88 | 58.32 | 63.14 | 60.39 | 64.79 | 48.88 | 94.96 |
| A549 | 42.72 | 60.07 | 68.23 | 73.05 | 73.49 | 62.56 | 86.43 |
| HCT116 | 79.20 | 77.64 | 79.73 | 68.18 | 53.95 | 72.04 | 71.01 |
| PC3 | 30.52 | 70.26 | 37.11 | 19.82 | 32.61 | 50.59 | 89.95 |
| FaDu | 58.11 | 79.39 | 71.29 | 52.65 | 64.81 | 60.51 | 92.94 |
| MDA-MB-468 | 92.05 | 91.94 | 90.95 | 89.39 | 86.74 | 73.61 | 96.02 |
| HeLa | 57.96 | 54.77 | 69.01 | 73.96 | 80.20 | 86.37 | 92.71 |
| OEC | 21.64 | 69.57 | 34.23 | 35.74 | 31.84 | 51.42 | 57.90 |
| HSF | 27.45 | 24.15 | 33.87 | 36.66 | 33.44 | 25.63 | 26.79 |
Intriguingly, among the investigated OSe compounds, 8 manifested the best mean GI% (64.60%) in comparison to Dox which displayed a GI% of 70.22%. Moreover, low GI% values were attained by the investigated OSe compounds against the utilized normal cell lines assuring their safety and selectivity to cancer cells.
2.2.1.1 Structure–activity relationship. To broaden our knowledge and predict the inhibitory potential of the examined compounds as potential anticancer agents, a thorough structure–activity relationship (SAR) analysis was conducted (Fig. 3). This analysis involved deliberate modifications to the 4-((4-hydroseleno-2-(methoxycarbonyl)phenyl)amino)-4-oxobutanoic acid scaffold. Various cancer cell lines were investigated to establish the correlation between structural modifications and anticancer activity based on:
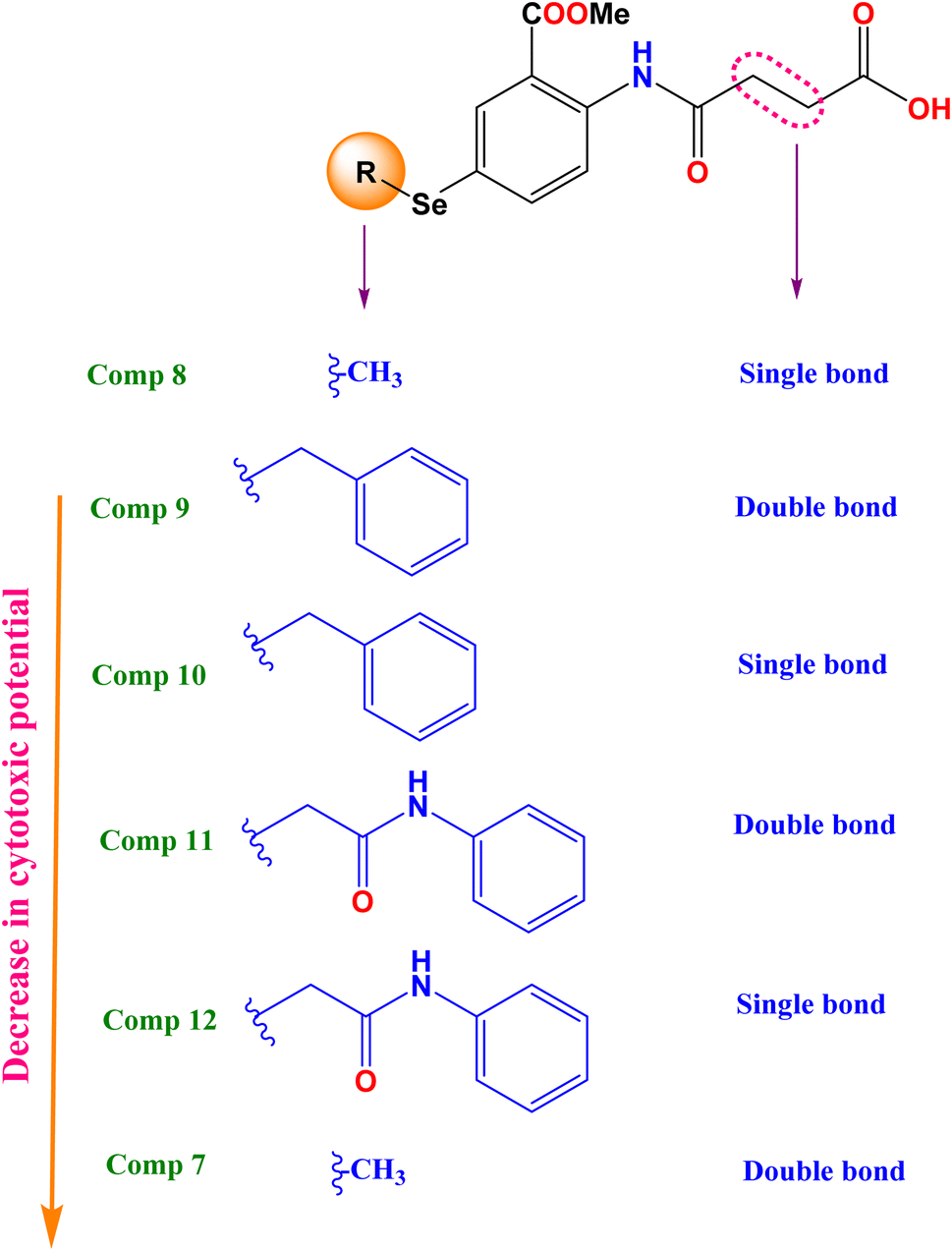 | ||
| Fig. 3 Structure cytotoxic relationship studies of the investigated compounds (7–12). | ||
(A) The mean growth inhibition (GI)%
(a) Interestingly, it was revealed that substituting the Se with a methyl group (compound 8), displayed the highest GI% and thus the highest cytotoxic potential, as shown in Fig. 3.
(b) However, a quite decrease in GI% was noticed by substituting the Se with the benzyl group (compound 10), by substituting the Se with the benzyl group along with α, β unsaturated acid derivative (compound 9), or by substituting the Se with phenyl acetamido group along with α, β unsaturated acid derivative (compound 11).
(c) Notably, the weakest GI% was displayed by substituting the Se with methyl along with α, β unsaturated acid derivative (compound 7), or substituting the Se with phenyl acetamido group (compound 12), as depicted in Fig. 3.
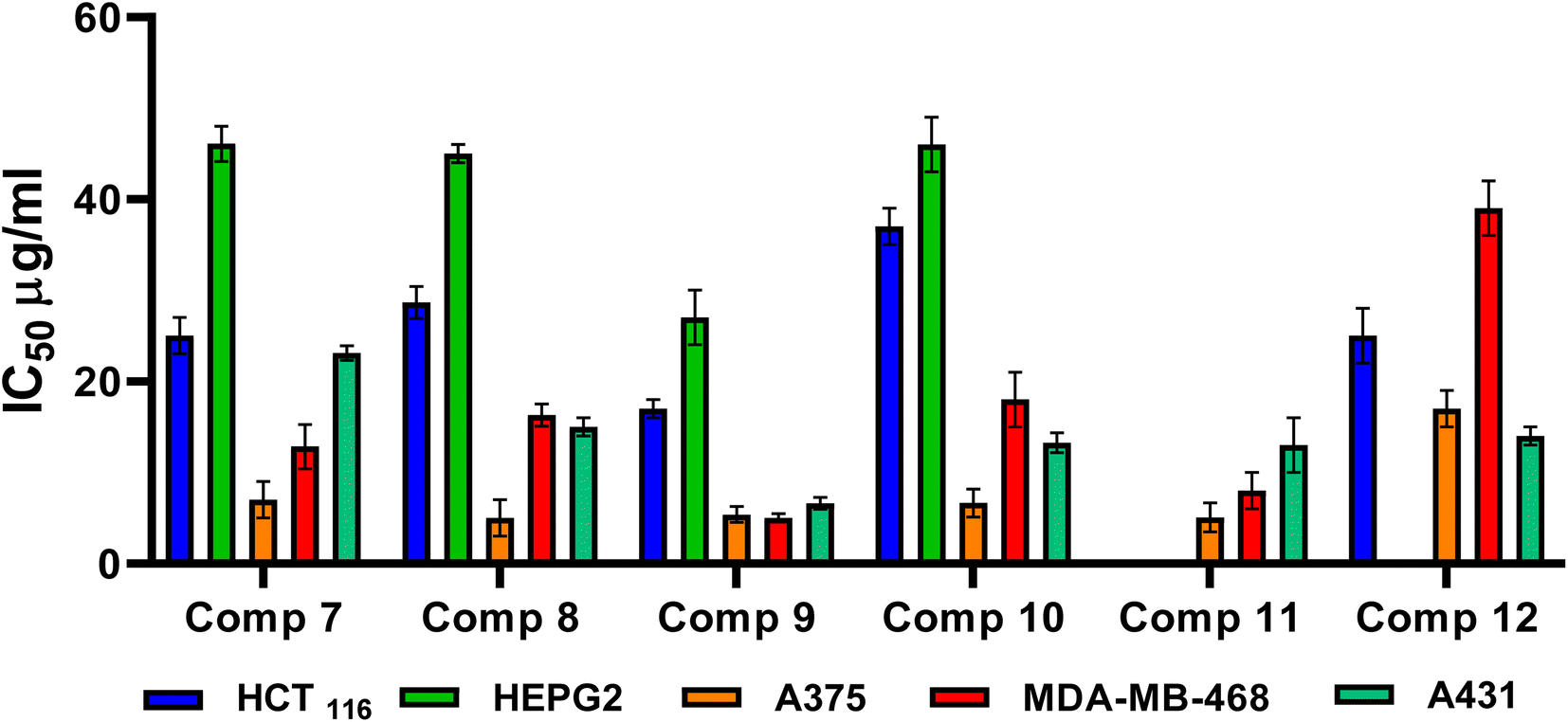 | ||
| Fig. 4 Cytotoxic inhibitory concentration 50 (IC50) evaluation of the assessed compounds (7–12) against HCT116, HEPG2, A375, MDA-MB-468, and A431 cancer cell lines. | ||
Compounds 8, 9, 10, and 11 treatments showed the upregulation of apoptotic proteins, as presented in Fig. 5. Thus, P53, BAX, Caspases-3, 6, 8, and 9 were upregulated by (2.19, 1.83, 3.35, 1.74, 3.07, and 1.79), (2.08, 2.06, 3.63, 1.78, 4.15, and 1.85), (2.32, 1.96, 3.17, 1.59, 6.76, and 1.69), and (2.01, 2.16, 3.79, 1.66, 7.64, and 1.69)-fold changes by compounds 8, 9, 10, and 11, respectively. However, compounds 8, 9, 10, and 11 expressed the downregulation of the investigated antiapoptotic proteins. Accordingly, BCL-2, MMP2, and MMP9 were downregulated by (0.62, 0.64, and 0.26), (0.42, 0.59, and 0.21), (0.46, 0.48, and 0.34), and (0.31, 0.67, and 0.25)-fold changes by compounds 8, 9, 10, and 11, respectively, Fig. 5.
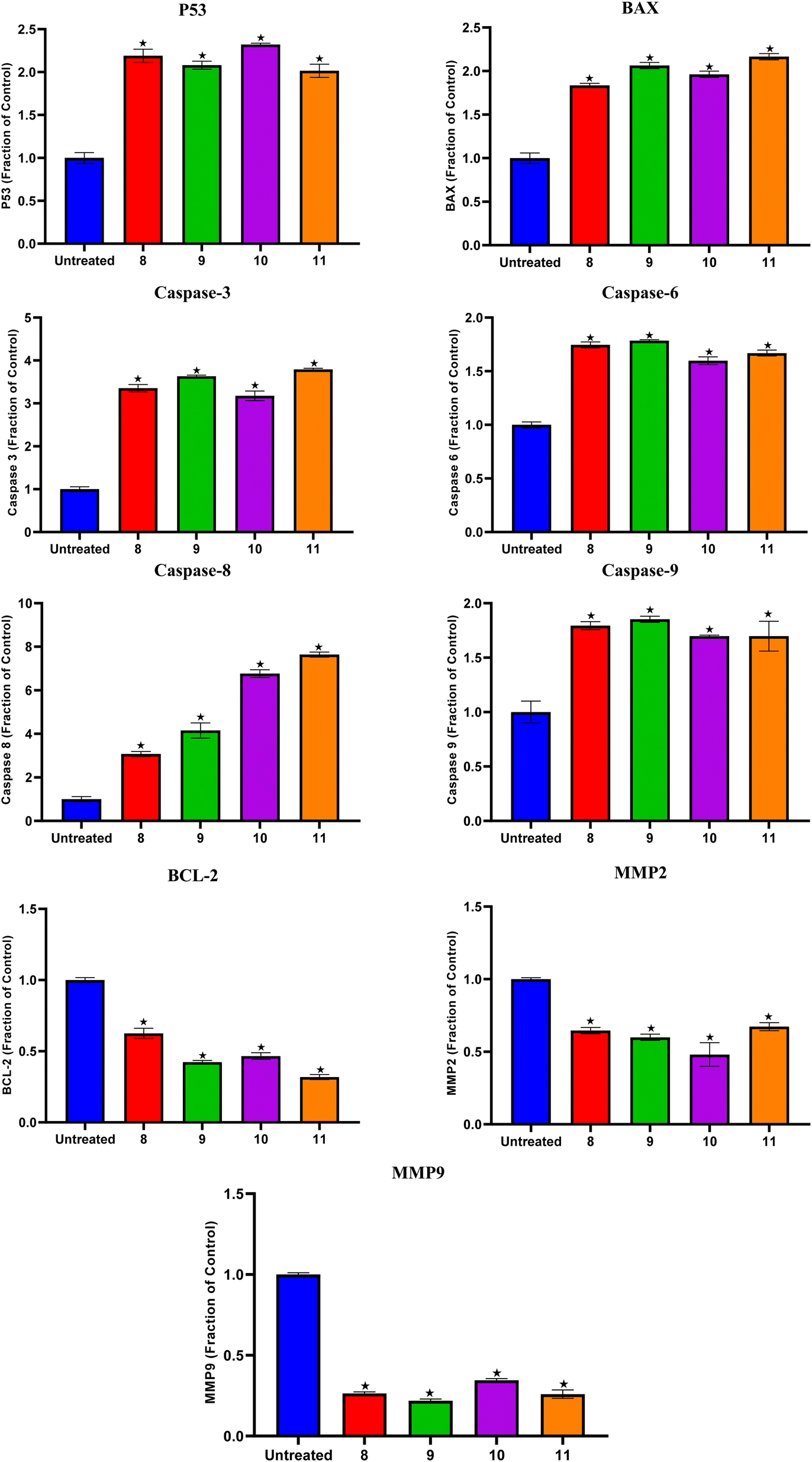 | ||
| Fig. 5 Compounds 8, 9, 10, and 11 protein expression levels for P53, BAX, Caspases-3, 6, 8, and 9, BCL-2, MMP2, and MMP9 in both the treated and untreated A375 cancer cell line. | ||
2.3. In silico studies
Compound 7 formed three hydrogen bonds with His219, Cys163, and Arg220, however, compound 8 formed seven hydrogen bonds with Cys163 (4), Arg220 (2), and Asp266. On the other side, compound 9 showed five hydrogen bonds with Cys163 (2), Arg220 (2), and His121, and one pi-hydrogen bond with His219, respectively. Besides, compound 10 described four hydrogen bonds with Cys163 (3) and Arg220. Moreover, compound 11 represented five hydrogen bonds with His219 (2), Cys163 (2), and Arg220, and compound 12 had four hydrogen bonds with Cys163, Arg220 (2), and Glu221. Notably, the docked co-crystallized inhibitor of Caspase-6 formed four hydrogen bonds with His219, Cys163 (2), and Arg220 (Fig. 6).
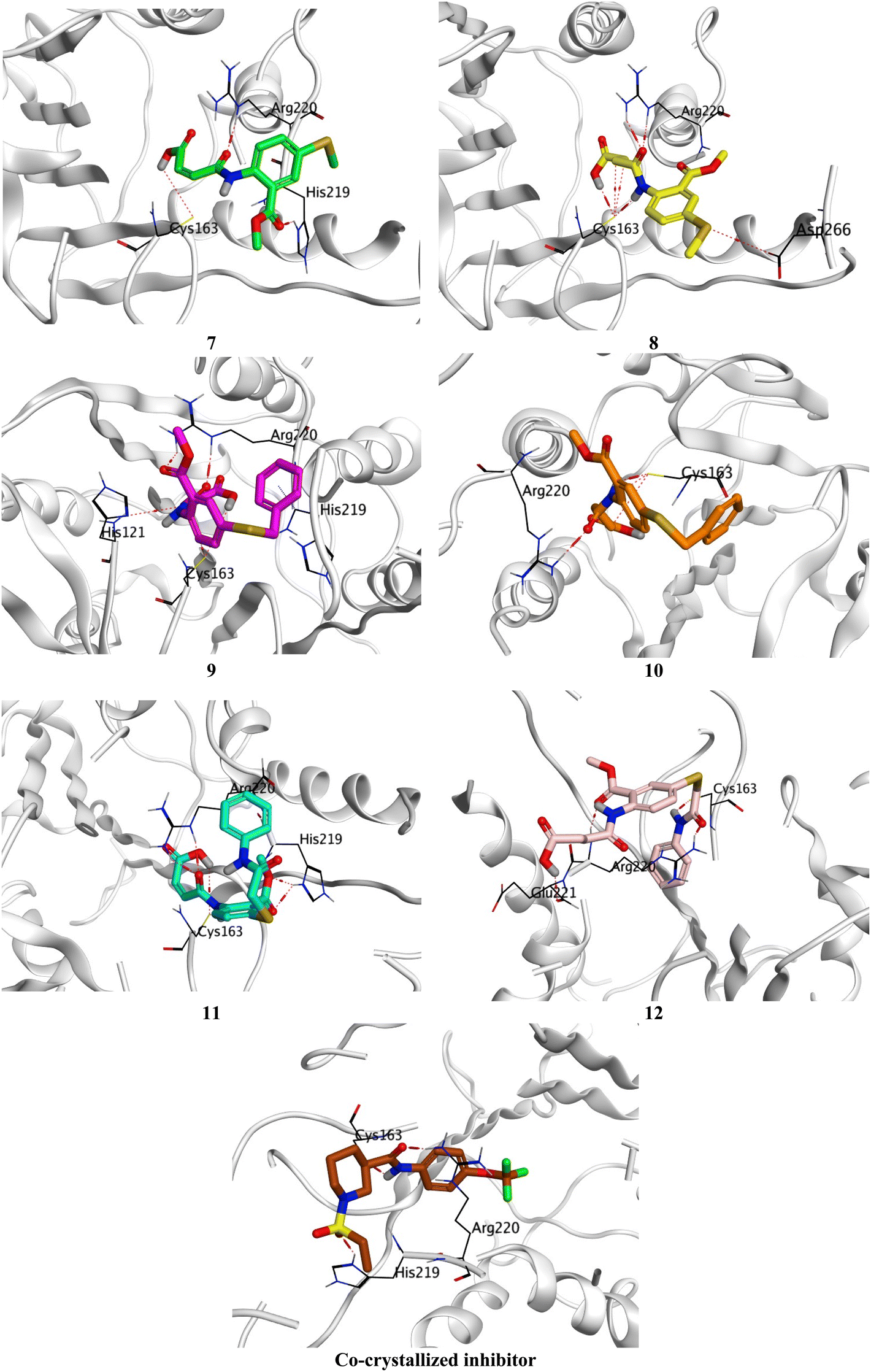 | ||
| Fig. 6 3D Binding interactions of the target candidates (7–12) within the binding site of human Caspase-6 (PDB ID: 8EG6) compared to the irreversible co-crystallized inhibitor. | ||
Based on the previously described binding scores and interactions of the studied candidates (7–12) compared to the co-crystallized inhibitor of Caspase-6; we could confirm the superior inhibitory potential and apoptotic induction towards the Caspase-6 target.
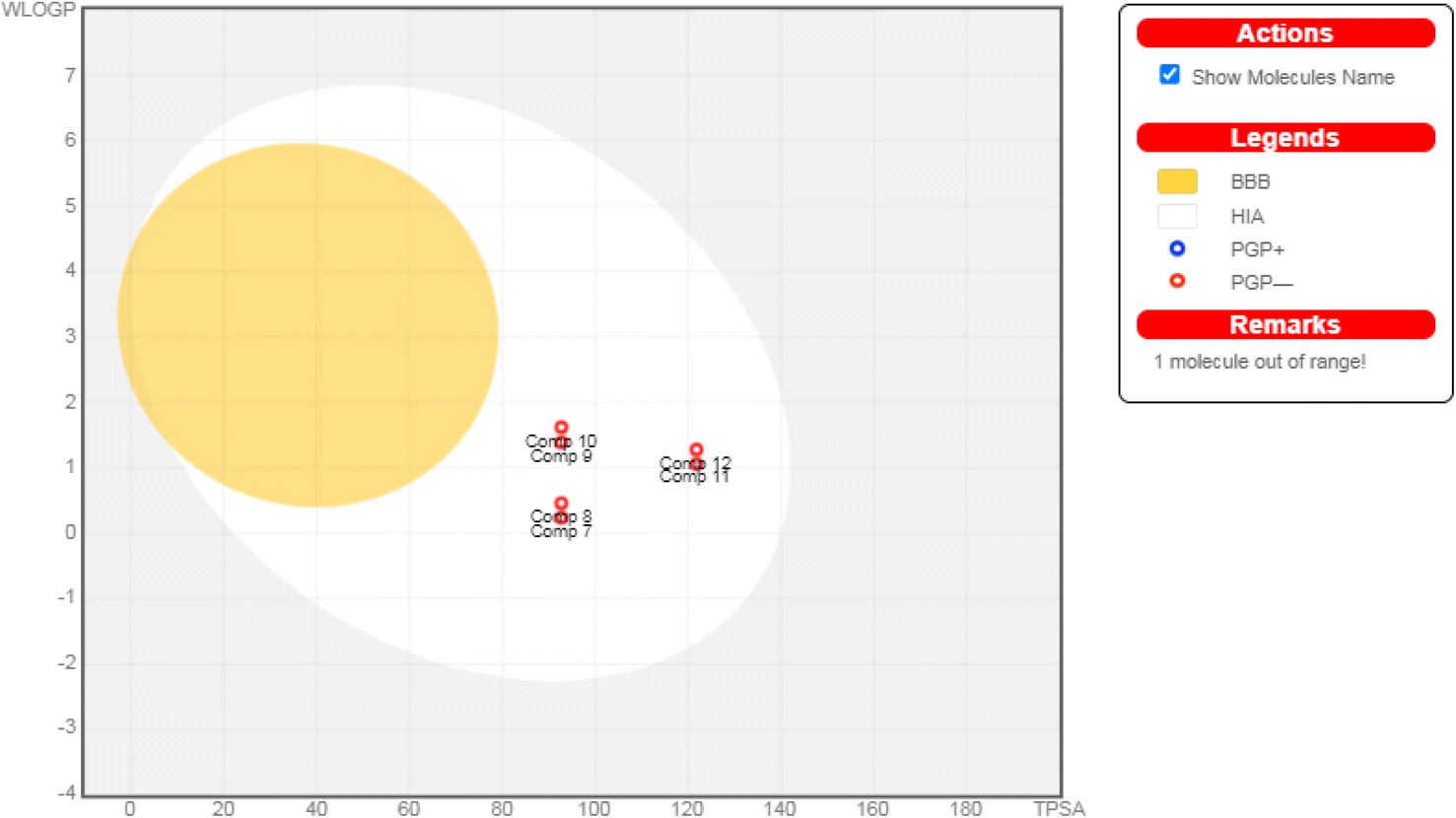 | ||
| Fig. 7 The boiled-egg diagram for all the afforded compounds (7–12) as well as doxorubicin as a reference control. | ||
Moreover, regarding their toxicity parameters, it was shown that all assessed compounds do not exhibit Ames toxicity, assuring their eligibility is not mutagenic.62 Additionally, all investigated compounds are fortunately non-inhibitors of hERG I and II, so they do not exhibit a cardiotoxic effect.63 Notably, investigated compounds are non-hepatotoxic assuring their safety, as depicted in Table 2.
| Comp 7 | Comp 8 | Comp 9 | Comp 10 | Comp 11 | Comp 12 | Dox | ||
|---|---|---|---|---|---|---|---|---|
| Molecular properties | Molar refractivity | 74.20 | 74.67 | 98.68 | 99.16 | 108.03 | 108.51 | 132.66 |
| TPSA (Az) | 92.70 | 92.70 | 92.70 | 92.70 | 121.80 | 121.80 | 206.07 | |
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Po/w (WLOGP) Po/w (WLOGP) |
0.24 | 0.46 | 1.39 | 1.62 | 1.06 | 1.28 | −0.32 | |
| Consensus logPo/w |
0.74 | 0.77 | 1.82 | 1.85 | 1.21 | 1.24 | 0.44 | |
| Water solubility | S | S | MS | S | S | S | S | |
| Pharmacokinetics parameters | GI absorption | High | High | High | High | High | High | Low |
| BBB permeant | No | No | No | No | No | No | No | |
| P-gp substrate | No | No | No | No | No | No | Yes | |
| CYP1A2 inhibitor | No | No | No | No | No | No | No | |
| CYP2C19 inhibitor | No | No | No | No | No | Yes | No | |
| CYP2C9 inhibitor | No | No | No | No | No | No | No | |
| CYP2D6 inhibitor | No | No | No | No | No | No | No | |
| CYP3A4 inhibitor | No | No | No | No | No | No | No | |
| Drug/Lead likeness | Drug likeness (lipinski) | Yes | Yes | Yes | Yes | Yes | Yes | No |
| Lead likeness | Yes | No | No | No | No | No | No | |
| Toxicity parameters | Ames toxicity | No | No | No | No | No | No | No |
| Max. Tolerated dose (log mg kg−1 per day) | 0.901 | 0.851 | 0.204 | 0.222 | −0.329 | −0.336 | 0.081 | |
| hERG I inhibitor | No | No | No | No | No | No | No | |
| hERG II inhibitor | No | No | No | No | No | No | Yes | |
| Oral rat acute toxicity (LD50) (mol kg−1) | 2.404 | 2.409 | 2.523 | 2.529 | 2.762 | 2.781 | 2.408 | |
| Oral rat chronic toxicity (LOAEL) (log mg per kg_bw per day) | 1.604 | 1.563 | 1.935 | 1.893 | 2.016 | 1.975 | 3.339 | |
| Hepatotoxicity | No | No | Yes | Yes | Yes | No | Yes | |
| Minnow toxicity (log mM) | 0.931 | 0.804 | 0.681 | 0.553 | −0.029 | −0.156 | 4.412 | |
3. Conclusion
N-Amidic acids 7–12 were prepared in good yields (up to 95%) in a two-step synthetic procedure starting from dimethyl 5,5′-diselanediylbis(2-aminobenzoate) (3). Intriguingly, compound 8 showed the best mean GI% (64.60%) in comparison to Dox which displayed a GI% of 70.22%. On the other side, compound 9 displayed the highest cytotoxic potential against the investigated cancer cell lines, in particular, MDA-MB-468 and A431 cell lines with IC50 values of 5.03 and 6.6 μg mL−1, respectively, assuring its anticancer potential. SAR analysis described that substituting the selenium with a methyl group (compound 8) displayed the highest GI% and thus the highest cytotoxic potential. However, a quite decrease in GI% was shown by substituting the selenium with the benzyl group (compound 10), by substituting the selenium with the benzyl group along with α, β unsaturated acid derivative (compound 9), or by substituting the selenium with phenyl acetamido group along with α, β unsaturated acid derivative (compound 11). Furthermore, it was shown that compounds 8, 9, 10, and 11 could induce the upregulation of the apoptotic proteins; P53, BAX, caspases-3, 6, 8, and 9. However, they can prompt the downregulation of the anti-apoptotic proteins; BCL-2, MMP2, and MMP9. In addition, the conducted ADMET studies assured eligible physicochemical and pharmacokinetic properties of the assessed compounds. Moreover, based on the molecular docking scores and interactions towards Caspase-6; we could confirm the superior inhibitory potential and apoptotic induction.4. Experimental
4.1. Synthesis of the amidic acid-containing OSe agents
The organic selenide-based maleanilic and succinanilic acids were prepared according to our reported literature method (see detailed experimental procedures in the ESI†).37,64 Details in ESI Data, SI 1.†4.2. Biological evaluation
4.3. In silico studies
Data availability
The raw/processed data generated in this work are available upon request from the corresponding author.Author contributions
Conceptualization and supervision: Saad Shaaban and Ahmed A. Al-Karmalawy; data curation and visualization: Saad Shaaban, Ayman Abo Elmaaty, and Ahmed A. Al-Karmalawy; methodology: Saad Shaaban, Hanan A. Althikrallah, Ayman Abo Elmaaty, Amr Negm, and Ahmed A. Al-Karmalawy; writing – review & editing: Saad Shaaban, Ayman Abo Elmaaty, and Ahmed A. Al-Karmalawy. Finally, all authors revised and approved the final submitted version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Funding
The research work was funded by the Deputyship for Research and Innovation, Ministry of Education in Saudi Arabia (Project number INST220).Acknowledgements
The authors extend their appreciation to the Deputyship for Research and Innovation, Ministry of Education in Saudi Arabia for funding this research work (Project number INST220).References
- The global challenge of cancer, Nat Cancer, 2020, 1, 1–2, DOI:10.1038/s43018-019-0023-9.
- Advancing Cancer Therapy, Nat Cancer, 2021, 2, 245–246, DOI:10.1038/s43018-021-00192-x.
- A. A. Al-Karmalawy, H. I. El-Subbagh, L. Logoyda, R. B. Lesyk and M. I. El-Gamal, Front. Chem., 2023, 11, 1328424 CrossRef PubMed.
- L. A. Torre, F. Bray, R. L. Siegel, J. Ferlay, J. Lortet-Tieulent and A. Jemal, Ca-Cancer J. Clin., 2015, 65, 87–108 CrossRef PubMed.
- A. A. Al-Karmalawy, M. H. A. Mousa, M. Sharaky, M. A. E. Mourad, A. M. El-Dessouki, A. O. Hamouda, R. Alnajjar, A. A. Ayed, M. A. Shaldam and H. O. Tawfik, J. Med. Chem., 2024, 67(1), 492–512 CrossRef CAS PubMed.
- F. Martínez-Jiménez, F. Muiños, I. Sentís, J. Deu-Pons, I. Reyes-Salazar, C. Arnedo-Pac, L. Mularoni, O. Pich, J. Bonet and H. Kranas, Nat. Rev. Cancer, 2020, 20, 555–572 CrossRef PubMed.
- T. Al-Warhi, A. A. Al-Karmalawy, A. A. Elmaaty, M. A. Alshubramy, M. Abdel-Motaal, T. A. Majrashi, M. Asem, A. Nabil, W. M. Eldehna and M. Sharaky, J. Enzyme Inhib. Med. Chem., 2023, 38, 176–191 CrossRef CAS PubMed.
- M. Demaria, M. N. O'Leary, J. Chang, L. Shao, S. Liu, F. Alimirah, K. Koenig, C. Le, N. Mitin and A. M. Deal, Cancer Discovery, 2017, 7, 165–176 CrossRef CAS PubMed.
- A. A. Al-Karmalawy, M. S. Nafie, M. A. Shaldam, A. A. Elmaaty, S. A. Antar, A. A. El-Hamaky, M. A. Saleh, A. Elkamhawy and H. O. Tawfik, J. Med. Chem., 2023, 66, 777–792 CrossRef CAS PubMed.
- M. Brentnall, L. Rodriguez-Menocal, R. L. De Guevara, E. Cepero and L. H. Boise, BMC Cell Biol., 2013, 14, 1–9 CrossRef PubMed.
- C. B. Bourguet, P.-L. Boulay, A. Claing and W. D. Lubell, Bioorg. Med. Chem. Lett., 2014, 24, 3361–3365 CrossRef CAS PubMed.
- T. Ozaki and A. Nakagawara, Cancers, 2011, 3, 994–1013 CrossRef CAS PubMed.
- M. Krasavin and D. Dar’in, Tetrahedron Lett., 2016, 57, 1635–1640 CrossRef CAS.
- M. Gonzalez-Lopez and J. T. Shaw, Chem. Rev., 2009, 109, 164–189 CrossRef CAS PubMed.
- N. I. Azmi, S.-Z. Zhan and M. R. Razali, J. Mol. Struct., 2022, 1264, 133240 CrossRef CAS.
- L. Kas’yan, A. Serbin, A. Kas’yan, D. Karpenko and E. Golodaeva, Russ. J. Org. Chem., 2008, 44, 340–347 CrossRef.
- S. Shaaban, H. Ferjani, H. M. Abd El-Lateef, M. M. Khalaf, M. Gouda, M. Alaasar and T. A. Yousef, Front. Chem., 2022, 10, 961787 CrossRef CAS PubMed.
- S. Shaaban, A. Negm, M. A. Sobh and L. A. Wessjohann, Eur. J. Med. Chem., 2015, 97, 190–201 CrossRef CAS PubMed.
- S. Shaaban, D. Vervandier-Fasseur, P. Andreoletti, A. Zarrouk, P. Richard, A. Negm, G. Manolikakes, C. Jacob and M. Cherkaoui-Malki, Bioorg. Chem., 2018, 80, 43–56 CrossRef CAS PubMed.
- J. Chi, B. Zhong, Y. Li, P. Shao, G. Liu, Q. Gao and B. Chen, Z. Anorg. Allg. Chem., 2021, 647, 1284–1293 CrossRef CAS.
- E. Carosati, P. Ioan, M. Micucci, F. Broccatelli, G. Cruciani, B. Zhorov, A. Chiarini and R. Budriesi, Curr. Med. Chem., 2012, 19, 4306–4323 CrossRef CAS PubMed.
- I. Lumb, B. S. Sran, H. Sood, D. S. Arora and G. Hundal, Polyhedron, 2017, 127, 153–166 CrossRef CAS.
- M. Mashkovskii, Novaya Volna, Moscow, 2002, vol. 39, p. 86 Search PubMed.
- M. Stasevych, V. Zvarych, O. Yaremkevych, M. Vovk, A. Vaskevych, T. Halenova and O. Savchuk, Acta Chim. Slov., 2022, 69, 584–595 CrossRef CAS PubMed.
- A. Redzicka, Ż. Czyżnikowska, B. Wiatrak, K. Gębczak and A. Kochel, Int. J. Mol. Sci., 2021, 22, 1410 CrossRef CAS PubMed.
- M. Sato, J. E. Dander, C. Sato, Y.-S. Hung, S.-S. Gao, M.-C. Tang, L. Hang, J. M. Winter, N. K. Garg and K. Watanabe, J. Am. Chem. Soc., 2017, 139, 5317–5320 CrossRef CAS PubMed.
- M. Sak, Y. S. Al-Faiyz, H. Elsawy and S. Shaaban, Antioxidants, 2022, 11, 1231 CrossRef CAS PubMed.
- S. Shaaban, S. M. Shabana, Y. S. Al-Faiyz, G. Manolikakes and F. F. El-Senduny, Bioorg. Chem., 2021, 109, 104713 CrossRef CAS PubMed.
- F. F. El-Senduny, S. M. Shabana, D. Rösel, J. Brabek, I. Althagafi, G. Angeloni, G. Manolikakes and S. Shaaban, Future Med. Chem., 2021, 13, 1655–1677 CrossRef CAS PubMed.
- Z. Chen, H. Lai, L. Hou and T. Chen, Chem. Commun., 2020, 56, 179–196 RSC.
- J. S. Alotaibi, Y. S. Al-Faiyz and S. Shaaban, Pharmaceuticals, 2023, 16, 367 CrossRef CAS PubMed.
- D. Plano, D. N. Karelia, M. K. Pandey, J. E. Spallholz, S. Amin and A. K. Sharma, J. Med. Chem., 2016, 59, 1946–1959 CrossRef CAS PubMed.
- X. He, M. Zhong, S. Li, X. Li, Y. Li, Z. Li, Y. Gao, F. Ding, D. Wen and Y. Lei, Eur. J. Med. Chem., 2020, 208, 112864 CrossRef CAS PubMed.
- Y. Pang, B. An, L. Lou, J. Zhang, J. Yan, L. Huang, X. Li and S. Yin, J. Med. Chem., 2017, 60, 7300–7314 CrossRef CAS PubMed.
- S. Shaaban, A. Zarrouk, D. Vervandier-Fasseur, Y. S. Al-Faiyz, H. El-Sawy, I. Althagafi, P. Andreoletti and M. Cherkaoui-Malki, Arabian J. Chem., 2021, 14(4), 103051 CrossRef CAS.
- M. Sak, Y. S. Al-Faiyz, H. Elsawy and S. Shaaban, Antioxidants, 2022, 11, 1231 CrossRef CAS PubMed.
- S. Shaaban, M. S. S. Adam and N. M. El-Metwaly, J. Mol. Liq., 2022, 363, 119907 CrossRef CAS.
- B. Al-Abdallah, Y. S. Al-Faiyz and S. Shaaban, Biomolecules, 2022, 12, 1765 CrossRef CAS PubMed.
- B. Al-Abdallah, Y. S. Al-Faiyz and S. Shaaban, Inorganics, 2022, 10, 246 CrossRef CAS.
- M. Soriano-Garcia, Curr. Med. Chem., 2004, 11, 1657–1669 CrossRef CAS PubMed.
- S. Shaaban, Y. S. Al-Faiyz, G. M. Alsulaim, M. Alaasar, N. Amri, H. Ba-Ghazal, A. A. Al-Karmalawy and A. Abdou, Inorganics, 2023, 11, 321 CrossRef CAS.
- O. d. R. A. Junior, E. Antônio, R. M. Mainardes and N. M. Khalil, J. Trace Elem. Med. Biol., 2017, 39, 176–185 CrossRef PubMed.
- C. W. Nogueira and J. B. Rocha, J. Braz. Chem. Soc., 2010, 21, 2055–2071 CrossRef CAS.
- S. Shaaban, H. Ba-Ghazal, Y. S. Al-Faiyz, A. A. Al-Karmalawy, N. Amri and I. Youssef, Tetrahedron, 2024, 133957 CrossRef CAS.
- S. Shaaban, H. M. A. El-Lateef, M. M. Khalaf, M. Gouda and I. Youssef, Polymers, 2022, 14, 2208 CrossRef CAS PubMed.
- S. Shaaban, M. A. Arafat, H. E. Gaffer and W. S. Hamama, Der Pharma Chem., 2014, 6, 186–193 Search PubMed.
- S. Shaaban, H. E. Gaffer, M. Alshahd and S. S. Elmorsy, Int. J. Res. Dev. Pharm. Life Sci., 2015, 4, 1654–1668 CAS.
- G. I. Giles, F. H. Fry, K. M. Tasker, A. L. Holme, C. Peers, K. N. Green, L. O. Klotz, H. Sies and C. Jacob, Org. Biomol. Chem., 2003, 1, 4317–4322 RSC.
- F. H. Fry and C. Jacob, Curr. Pharm. Des., 2006, 12, 4479–4499 CrossRef CAS PubMed.
- C. A. Collins, F. H. Fry, A. L. Holme, A. Yiakouvaki, A. Al-Qenaei, C. Pourzand and C. Jacob, Org. Biomol. Chem., 2005, 3, 1541–1546 RSC.
- A. D. Sonawane, R. A. Sonawane, M. Ninomiya and M. Koketsu, Dalton Trans., 2021, 50, 12764–12790 RSC.
- P. N. Makhal, A. Nandi and V. R. Kaki, ChemistrySelect, 2021, 6, 663–679 CrossRef CAS.
- J. M. Sonego, S. I. de Diego, S. H. Szajnman, C. Gallo-Rodriguez and J. B. Rodriguez, Chem.–Eur. J., 2023, 29, e202300030 CrossRef CAS PubMed.
- S. Shaaban, A. Negm, M. A. Sobh and L. A. Wessjohann, Adv. Anticancer Agents Med. Chem., 2016, 16, 621–632 CrossRef CAS PubMed.
- M. A. E. Mourad, A. Abo Elmaaty, I. Zaki, A. A. E. Mourad, A. Hofni, A. E. Khodir, E. M. Aboubakr, A. Elkamhawy, E. J. Roh and A. A. Al-Karmalawy, J. Enzyme Inhib. Med. Chem., 2023, 38, 2205043 CrossRef PubMed.
- S. El-Kalyoubi, M. M. Khalifa, M. T. Abo-Elfadl, A. A. El-Sayed, A. Elkamhawy, K. Lee and A. A. Al-Karmalawy, J. Enzyme Inhib. Med. Chem., 2023, 38, 2242714 CrossRef PubMed.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, JNCI, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS PubMed.
- K. T. Savjani, A. K. Gajjar and J. K. Savjani, Int. Scholarly Res. Not., 2012, 2012, 195727 Search PubMed.
- A. Scalbert, C. Morand, C. Manach and C. Rémésy, Biomed. Pharmacother., 2002, 56, 276–282 CrossRef CAS PubMed.
- M. Thanou, J. Verhoef and H. Junginger, Adv. Drug Delivery Rev., 2001, 52, 117–126 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- D. D. Levy, E. Zeiger, P. A. Escobar, A. Hakura, M. Bas-jan, M. Kato, M. M. Moore and K.-i. Sugiyama, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2019, 848, 403074 CrossRef CAS PubMed.
- S. Roy and M. Mathew, J. Biol. Chem., 2018, 293, 4289–4303 CrossRef CAS PubMed.
- S. Shaaban, A. Abdou, A. G. Alhamzani, M. M. Abou-Krisha, M. A. Al-Qudah, M. Alaasar, I. Youssef and T. A. Yousef, Life, 2023, 13, 912 CrossRef CAS PubMed.
- R. Huey, G. M. Morris and S. Forli, The Scripps Research Institute Molecular Graphics Laboratory, 2012, vol. 10550, p. 1000 Search PubMed.
- S. Yuan, H. S. Chan and Z. Hu, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 7, e1298 Search PubMed.
- M. F. El-Behairy, W. H. Abd-Allah, M. M. Khalifa, M. S. Nafie, M. A. Saleh, M. S. Abdel-Maksoud, T. Al-Warhi, W. M. Eldehna and A. A. Al-Karmalawy, J. Enzyme Inhib. Med. Chem., 2023, 38, 2157825 CrossRef PubMed.
- K. I. Eissa, M. M. Kamel, L. W. Mohamed, A. S. Doghish, R. Alnajjar, A. A. Al-Karmalawy and A. E. Kassab, Drug Dev. Res., 2023, 84(5), 937–961 CrossRef CAS PubMed.
- A. A. Al-Karmalawy, M. Rashed, M. Sharaky, H. S. Abulkhair, M. M. Hammouda, H. O. Tawfik and M. A. Shaldam, Eur. J. Med. Chem., 2023, 259, 115661 CrossRef CAS PubMed.
- A. A. Gaber, A. M. El-Morsy, F. F. Sherbiny, A. H. Bayoumi, K. M. El-Gamal, K. El-Adl, A. A. Al-Karmalawy, R. R. Ezz Eldin, M. A. Saleh and H. S. Abulkhair, Arch. Pharm., 2021, e2100258 CrossRef PubMed.
- M. H. El-Shershaby, A. Ghiaty, A. H. Bayoumi, A. A. Al-Karmalawy, E. M. Husseiny, M. S. El-Zoghbi and H. S. Abulkhair, Bioorg. Med. Chem., 2021, 42, 116266 CrossRef CAS PubMed.
- A. A. Gaber, A. M. El-Morsy, F. F. Sherbiny, A. H. Bayoumi, K. M. El-Gamal, K. El-Adl, A. A. Al-Karmalawy, R. R. Ezz Eldin, M. A. Saleh and H. S. Abulkhair, Archiv der Pharmazie, 2021, p. e2100258 Search PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed.
- D. E. Pires, T. L. Blundell and D. B. Ascher, J. Med. Chem., 2015, 58, 4066–4072 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: (SI1) Chemistry and experimental data (Fig. S1–S60); (SI2) GI% of the investigated organoselenium compounds (7–12) against a series of cancer and normal cell lines; (SI3) Cytotoxic inhibitory concentration 50 (IC50) evaluation against HCT116, HEPG2, A375, MDA-MB-468, and A431 cancer cell lines (Fig. S62–S67); (SI4) Protein expression of the apoptosis-related genes. See DOI: https://doi.org/10.1039/d4ra02944e. |
| This journal is © The Royal Society of Chemistry 2024 |