
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdsorption behavior of different cresols on bismuthene: a DFT study†
Ukkasha Iqrara,
Usman Masooda,
Saleh S. Alarfajib,
Tahir Iqbalc,
Abdul Majidc and
Muhammad Isa Khan *a
*a
aDepartment of Physics, The Islamia University of Bahawalpur, Rahim Yar Khan Campus, Bahawalpur, Pakistan. E-mail: ui939msc@gmail.com; usmanmasoodahmed@gmail.com; Muhammad.isa@iub.edu.pk
bDepartment of Chemistry, Faculty of Science, King Khalid University, P. O. Box 9004, Abha 61413, Saudi Arabia. E-mail: ssalarvagi@kku.edu.sa
cDepartment of Physics, University of Gujrat, Gujrat 50700, Pakistan. E-mail: tahir.awan@uog.edu.pk; abdulmajid40@uog.edu.pk
First published on 11th June 2024
Abstract
Phenolic compounds present in wastewater were utilized for first-principle calculations based on DFT to observe adsorption effects. Results indicate that bismuthene exhibits different adsorption characteristics for different compounds. Following the adsorption process, the aromatic ring remains in the same plane, while CH3 and OH groups move upward, causing slight changes in the molecules' overall position. The calculated results show that bisphenol A has the least atomic distance (4.00 Å) from the bismuthene surface and the highest adsorption energy value (12.8509 eV), indicating the stability and smoothness of the adsorption process. The electronic properties results reveal that phenolic compounds exhibit overlapping peaks at a distance from the Fermi level, describing the stability of the adsorption system. Additionally, the charge transfer results mirror the adsorption energy calculation results, showing that the bisphenol A adsorption system accepts a greater amount of (−0.116e) charge from the bismuthene surface, demonstrating a strong adsorption effect.
1. Introduction
With the swift progress of industrialization, a diverse array of carbon-based and non-organic materials, serving as agents, networks, and reagents, find extensive application within crucial industrial sectors such as petrochemicals, cloth dyeing and coloring, polymer manufacturing, biopharmaceuticals, and process engineering.1–3 The previously discussed manufacturing processes often generate polluted water alongside intricate components. Nevertheless, contaminated water with sophisticated constituents frequently comprises a multitude of organic contaminants that prove challenging for disintegrate and are extremely poisonous.4,5Among these, phenolic compounds are prone towards being overlooked. Allowing their discharge can result in severe repercussions for aquatic life, soil, and the ecological surroundings, presenting a potential hazard to fitness. Various soluble inorganic and organic substances serve as primary contributors to water contamination. Among these, phenol derivatives find widespread use within manufacturing processes, such as the creating pigments, antiseptics, pesticides, detergents, and antioxidants. The discharge of such toxic unprocessed particles into surroundings6 through contaminated water release can have enduring detrimental effects on water ecosystems, potentially leading to irreversible consequences.
Currently, various methods, encompassing procedural, organic, and chemical techniques such as organic carbon adherence,7 sophisticated oxidation processes,8 doping alteration,9,10 composite heterojunction design,11,12 membrane-based process,13 and the reverse osmotic transfer,14 are employed for the elimination of carbon-based contaminants from polluted water. Nevertheless, stability of structure of phenolic pollutants poses a challenge, rendering the application of these decomposition approaches not only lengthy, expensive as well as prone towards byproduct formation.15 In recent decades, adsorption treatment has emerged as a prominent method among previous approaches due to its simplicity of operation and cost-effectiveness.16–18
Monolayer materials, characterized by numerous operative sites and greater region, are considered excellent adsorptive material in favor of water medication. Ghafry et al.19 Examined the photochemical breakdown of phenol using nanomaterials of ZnO doped with Ag, revealing that even a minor Ag doping enhances the photocatalytic efficiency of ZnO nanomaterials. Albedah et al.20 investigated the adherence of phenol from industrial wastewater on two-dimensional substances (ZrS3, CdS2, SnS2, MoS2) using particle simulation. The study revealed that CdS2 exhibits most effective adherence of phenol and is a promising new material for phenol removal from contaminated water. Furthermore, metallic or non-metallic contaminating has been shown to enhance the adsorption properties of various adsorbents.
Aside from experimental approaches, computational simulation techniques are employed to examine and elucidate the entire reaction process at the nano scale. First-principles calculations and particle dynamics simulations aid in gaining deeper insights into the adherence and decomposition mechanisms of phenol containing compounds.21–23 Ghahghaey et al.24 investigated theoretical phenol extraction capability of various graphene types and discovered that graphene materials modified with functional groups exhibited superior adherence durability and competence for phenol. While theoretical investigations into phenol adsorption have been abundant, there is a scarcity of first-principles analyses concerning the adherence of various methyl phenol molecule types upon both natural and doped surfaces.
Studies of the interaction between phenol and intrinsic graphene, phenol and aluminum-doped graphene, and phenol with hexagonal boron nitride (BN) were conducted using first-principles total energy calculations within periodic density functional theory. Results indicate that the direct interaction between oxygen and aluminum yields the ground state geometry with the phenol molecule adsorbed on the graphene layer. Binding energies and DOS structures also demonstrate that the ground state configuration is characterized by an O–Al interaction with a separation distance of 1.97 Å. Furthermore, results of the binding energies indicate that phenol is chemisorbed when interacting with the BN sheet.25,26
Yuan et al.27 uses molecular dynamics simulation to model and optimize the parameters of the phenol and p-cresol separation process, resulting in more precise separation parameters. Abadee et al.28 explored the adhesion behavior of phenol compounds on graphene layers. The findings reveal that the adherence potency of phenol upon graphene, nano-buds located on the apex of the bucky ball, in proximity to the cervix, and upon the graphene, layer was greater compared to those in water. Zhang et al. explored the adsorption of cresols found in wastewater on the Au (111) plane. Analysis of the adherence energy graph illustrates the varying adsorption behavior of different cresols on the surface.
Bismuthene derived from monolayers of group V elements represents a relatively new addition to the family of two dimensional materials. Its conductive, physical, and photonic properties have undergone extensive scrutiny among both empirical and hypothetical investigations in recent times.29 Research suggests that bismuthene monolayers maintain stability in a buckled hexagonal phase and are anticipated to possess a direct, narrow band gap.
The widespread utilization of various phenolic compounds alongside the challenges in their removal, the progress of reasonable, competent, and environmentally amiable materials in favor of organic pollutant removal has emerged as a significant challenge in industrial production. In this study, we systematically investigate the interactions between different phenolic compounds, and bismuthene atoms. Our primary aim is to examine the variations in adherence energy, adherence distance, and electron arrangement resulting from various numbers and forms of phenolic compounds adhere upon the bismuthene layer, using first-principles calculations. This evaluation allows us to assess distinct adsorption behaviors. The findings of this research offer a potential method for removing phenolic pollutants from wastewater.
2. Computational detail
The Amsterdam Density Functional package (ADF-BAND)30 was utilized to determine energy coefficients, configurations, gas absorption, and electronic emulsion models using the Density Functional Theory (DFT) approach. ADF's Slater-type orbitals were applied to accurately represent the response of orbitals surrounding the nucleus in studied composition.31 The study utilized the triple zeta polarisation (TZP) basis set, standard numeric characteristics, and the unfrozen core implement to determine fundamental and electron state functions.32 For evaluating the influence of charge density upon ion substitution interaction energies, the Perdew–Burke–Ernzerhof (PBE) form of the Generalized Gradient Approximation (GGA) was employed.33 Effectiveness of approximating distant interaction among the bismuthene surface plus phenolic molecules significantly improves when considering van der Waals (vdW) interactions with the D3-Grimme correction (DFT-D3).34Crystal parameters and atomic arrangements are completely relaxed. Computations utilized five (05) k-points, chosen to determine numerical accuracy in ADF, as the k-grid, basis vectors, and density fitting coefficients are foundational. These k-points, determined through Becke's fuzzy cell technique,35 sample Brillouin zone. Convergence standards were defined as energy (10−5 eV), gradient (0.02 eV Å−1), and step-convergence (10−3 Å) for overall structural decompression processes.
To identify most stable adsorption arrangement, the adsorption energy (Eads) was computed. The equation to determining the adherence energy of various organic compounds adhere on bismuthene layer is described like this:
| Eads = Eadsorbates/slab − Eslab − Eadsorbates | (1) |
Disparity in electron density may be characterized as:
| Δρ = ρslab/adsorbates − ρslab − ρadsorbates | (2) |
Charge transfer formula can be defined as:
| Qt = Qa − Qb | (3) |
The harmonic vibrational frequencies were calculated at the same level to confirm that the obtained structures are minima on the potential energy surface.
3. Results and discussion
3.1 Structure analysis of pure bismuthene
Prior to initiating a discussion on bismuthene, it is essential to delve into and assess its characteristics. Bismuthene pristine structure manifests as a buckled hexagon, wherein the atoms appear arranged in a hexagonal pattern from a top-down perspective but are not situated within the same plane; this disparity in atomic positioning, termed “buckling,” entails a distance between bonded atoms along the z-direction.We have already explored bismuthene for different applications in our previous work. The bismuthene has a buckled hexagonal structure with calculated lattice constants of a = b = 4.34 Å and a Bi–Bi interatomic bond length of 3.07 Å. The buckling height, defined as the vertical distance between the lower and higher bismuth atomic planes, is Δ = 1.73 Å. These calculated structural parameters are consistent with relevant literature36–40 and given in Table 1.
Fig. 1(b) shows the density of states of freestanding bismuthene, indicating its semiconducting nature with a direct bandgap of 0.54 eV, consistent with previous research. The structure is stabilized, and the electronic properties of the Bi atom in bismuthene are similar to those in arsenene and antimonene. Fundamentally, π-type bonding states cannot form due to the presence of a lone pair of electrons and three σ-bonding orbitals. Consequently, the Bi monolayer exhibits a buckled structure rather than a planar one like graphene. As illustrated in Fig. 1(b), the p orbital predominantly influences the material's electronic properties, with minimal contributions from the s and d orbitals.43
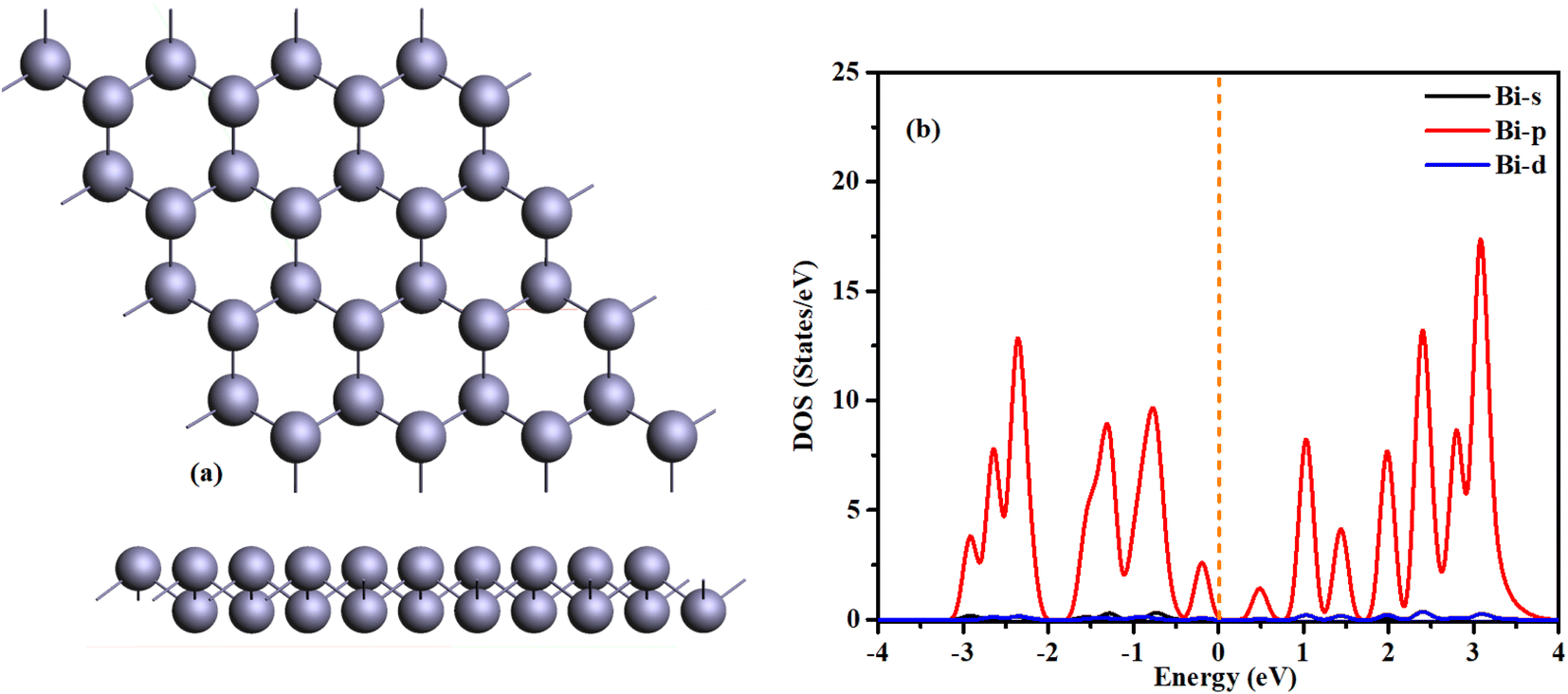 | ||
| Fig. 1 (a) Optimized structure of bismuthene z and y-view (b) density of states of bismuthene. | ||
3.2 Configuration of organic compounds and adsorption models
To explore adherence characteristics of carbon-based compounds on the bismuthene layer, molecular structure models were constructed for each compound. The figures presented in Fig. 2 illustrate the structural models resulting from the geometric optimization of these organic compounds. In these models, hydrogen atoms (H) are depicted purple, carbon particles navy blue, N atoms royal blue, O atoms red, and Cl particle light green colors. The organic compound o-nitrophenol (C6H5NO3) comprises six carbon, five hydrogen, three oxygen, and one nitrogen atom. Catechin (C6H6O2) consists of six carbon, six hydrogen, and two oxygen atoms. Phenol (C6H6O) contains six carbon, six hydrogen, and one oxygen atom. Bisphenol A (C15H16O2) includes fifteen carbon, sixteen hydrogen, and two oxygen atoms. Xylenol (C8H10O) contains eight carbon, ten hydrogen, and one oxygen atom. Dichlorophenol (C6H4Cl2) comprises six carbon, four hydrogen, two chlorine, and one oxygen atom.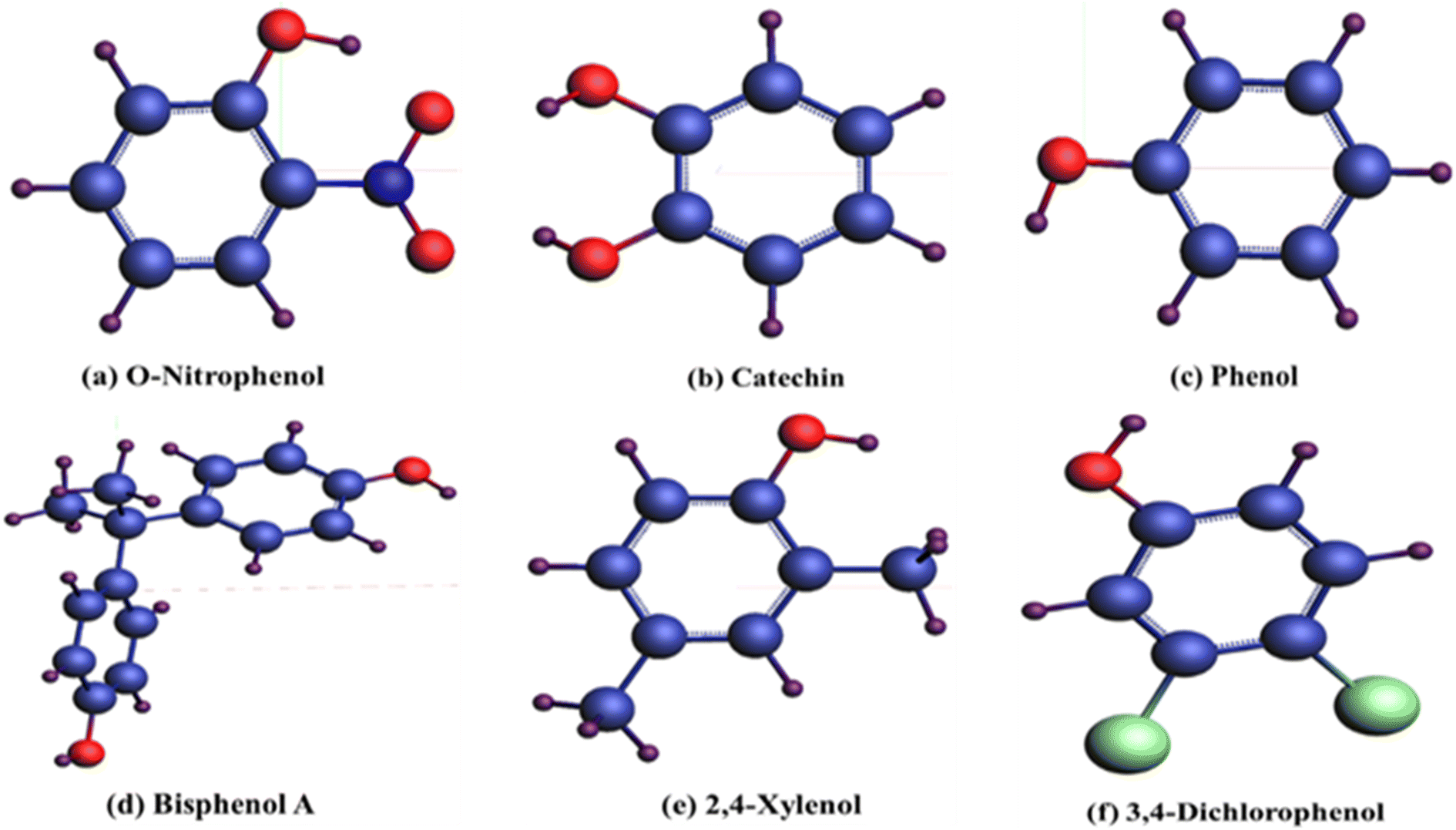 | ||
| Fig. 2 Structures of phenolic compounds after geometric optimization. | ||
After performing geometric optimization, determine the adsorption energy of different organic compounds using eqn (1), showcasing the interconnection of compound with upper layer atom. Computed adherence energy values, obtained post-geometry optimization, are predominantly positive, indicating varying degrees of interaction strength and weakness across different organic compounds. The adsorption distance of these organic compounds from the bismuthene upper layer atom falls within the range of (3–5) Å, with the adsorbent being positioned parallel to the upper layer of the slab atom.
In order to exemplify the modeling concept introduced in this paper, an exhibition of the adsorption process was carried out. Fig. 3 exhibits the optimized arrangement of different organic compounds adhered to the bismuthene surface. The visual representation offers both a y and z perspective of the structure, showcasing the adsorbents attached to the top surface of the bismuthene substrate. These adsorbents are oriented parallel to the atoms on the upper surface, aimed at improving the efficiency of the adsorption process.
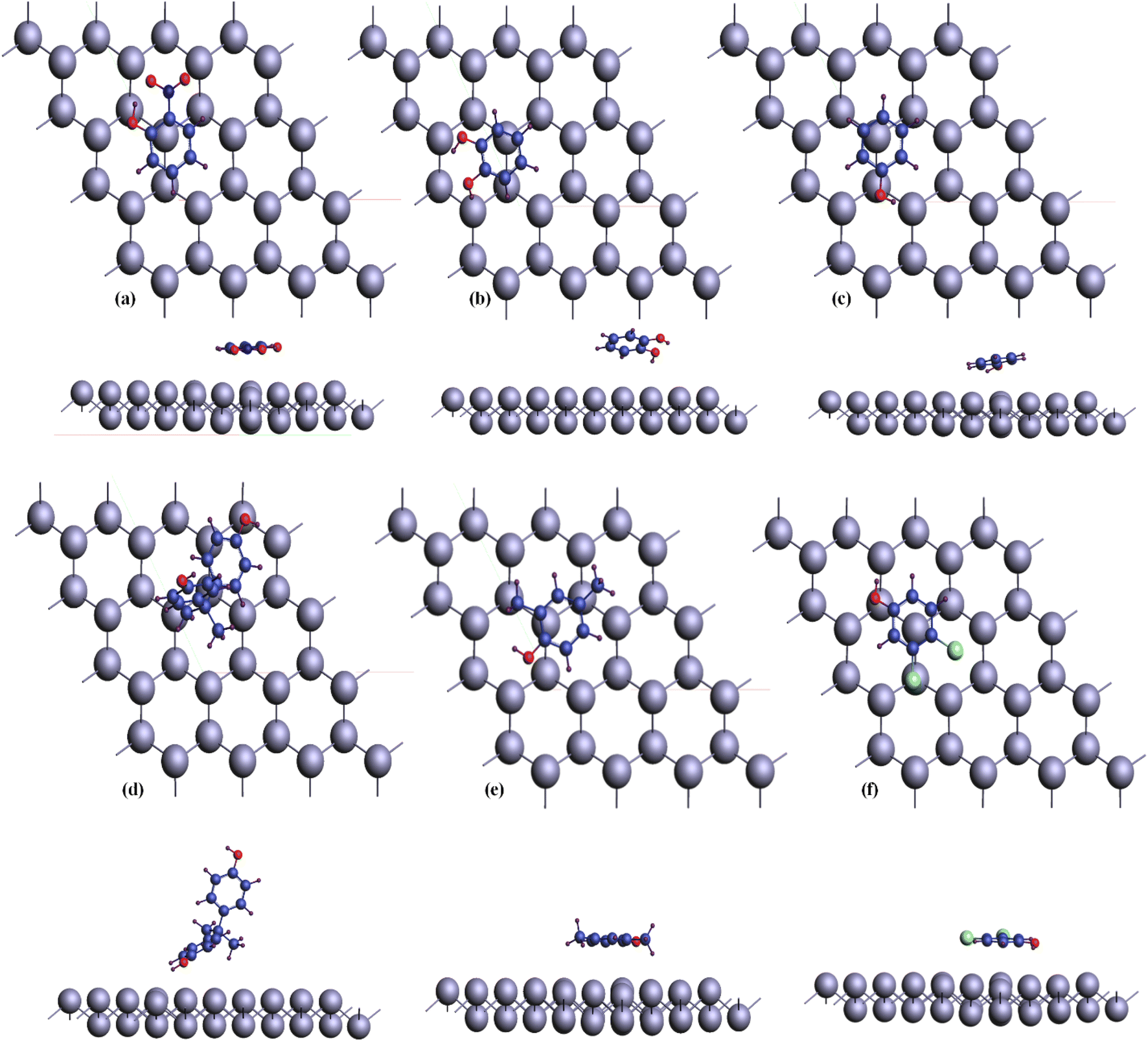 | ||
| Fig. 3 Configuration of phenolic compounds adsorbed on bismuthene surface. (a) o-nitrophenol (b) catechin (c) phenol (d) bisphenol A (e) 2,4-xylenol (f) dichlorophenol. | ||
3.3 Parameters of organic compounds adsorbed on bismuthene surface
To analyze the interaction of the specified organic compounds on the bismuthene surface, we calculated the adsorption energy in both vacuum and water. Fig. 4(a) illustrates the adsorption energy values for the interaction of organic compounds on the bismuthene surface in both vacuum and water. Fig. 4(b) shows the minimum adsorption distance from the surface of the material to the organic compounds.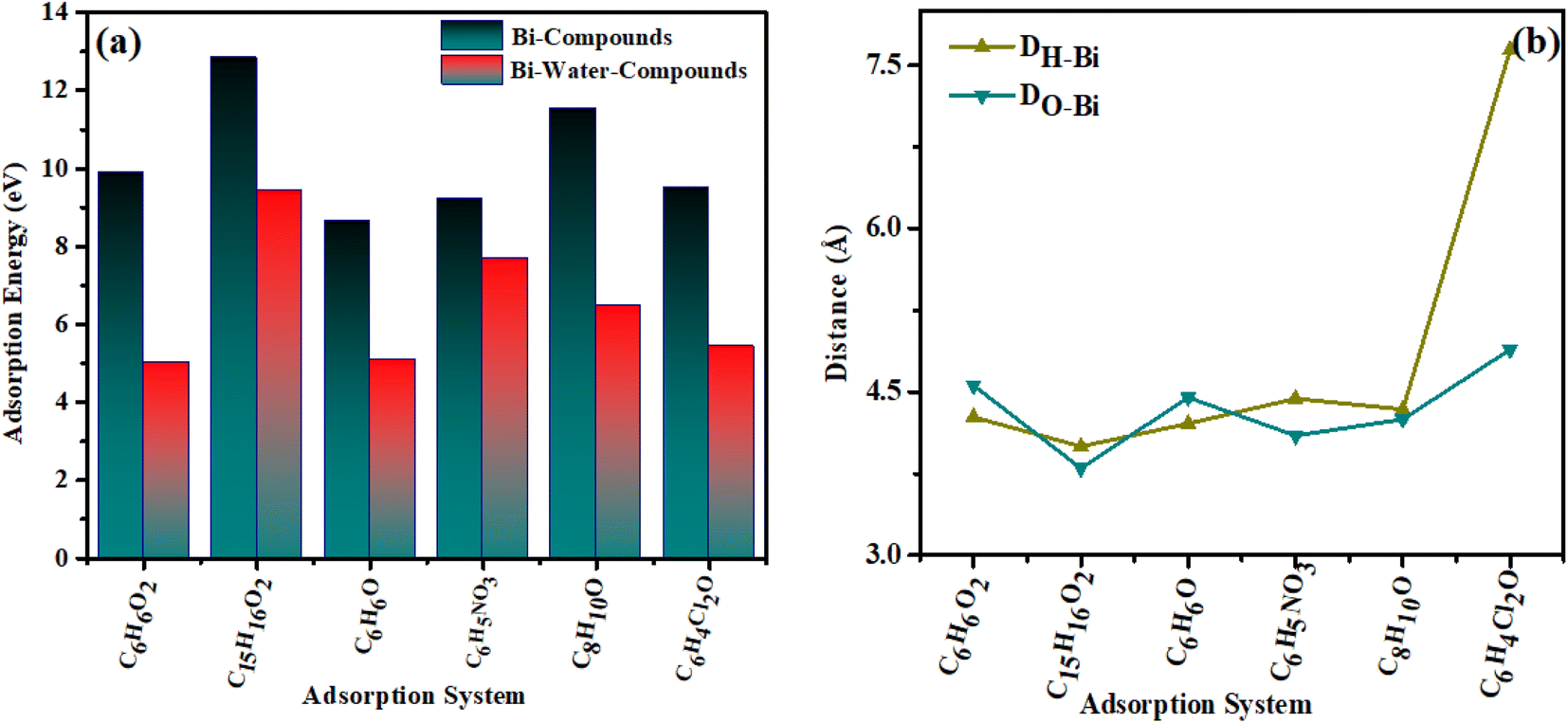 | ||
| Fig. 4 (a) adsorption energy data (b) distance curves of organic compounds adsorbed on bismuthene. | ||
Following the adsorption of organic compounds, there is an increase in the adsorption distance of the absorbent atoms, causing the adsorbent to shift towards the hydroxyl group. Additionally, the hydroxyl group exhibits an upward tilt while the methyl group tilts downward towards the slab atoms. The order of adsorption energy in vacuum for the organic compounds is as follows: bisphenol A > o-nitrophenol > dichlorophenol > catechin > xylenol > phenol. While the adsorption energy values in aqueous medium water are of the order bisphenol A > o-nitrophenol > xylenol > dichlorophenol > phenol > catechin. It is found that positive values of adsorption energy are found according to literature.45,46 The adsorption energy values are given in the Table 2.
Dalton et al. investigated the adsorption of nitrophenols on 2-propanol, calculating an adsorption energy of 2.72 eV. Their study aimed to analyze the photochemical degradation and electronic structure of nitrophenol.47 Sharma et al. studied activated carbon to analyze the adsorption behavior of catechin, known as catechol. According to their calculated results, the adsorption energy for catechol is −44.869 kJ mol−1 (−0.468 eV). Their study reveals that the carboxyl group has the greatest effect on the adsorption process. Erwin et al. investigated the adsorption response of phenol on pristine and metal-doped phosphorene. Their results indicate that metal-doped phosphorene exhibits excellent phenol adsorption, with an adsorption energy of −0.90 eV in the gas phase and −1.20 eV in the aqueous phase.48 Ahangar et al. studied the effect of doping on the adsorption process of phenol by comparing pristine and Cu-doped phosphorene. Their results show that the adsorption energy of phenol on pristine phosphorene is −0.602 eV, while Cu doping increases the adsorption energy to −1.14 eV.49 In our studies, the adsorption energy increases with decreasing atomic distance. Additionally, when an aqueous medium is introduced, the adsorption energy values shift towards more negative, indicating significant changes in adsorption energy. The adsorption energy values indicate that all adsorbed organic compounds exhibit values greater than 1, indicating chemical adsorption. Analyzing adsorption parameters enables us to swiftly and efficiently identifying highly reliable adsorption process. In gas absorption process, higher adsorption energy corresponds to shorter equilibrium adsorption distances.
3.4 Electronic properties of organic compounds adsorbed on bismuthene surface
The investigation delved into the electronic characteristics of phenolic compounds upon adsorption onto the bismuthene surface, offering a comprehensive analysis. We have applied spin polarized calculation to the molecules adsorbed systems and found their behavior as non-magnetic and DOS results are added as ESI I.†Fig. 5(a)–(f) presents the computed results of the total density of states (TDOS). Observing these figures reveals minimal alterations in TDOS among various systems post-adsorption, with their structures closely resembling that of the Bi surface. Notably, the primary contribution to TDOS stems from the adsorbents.
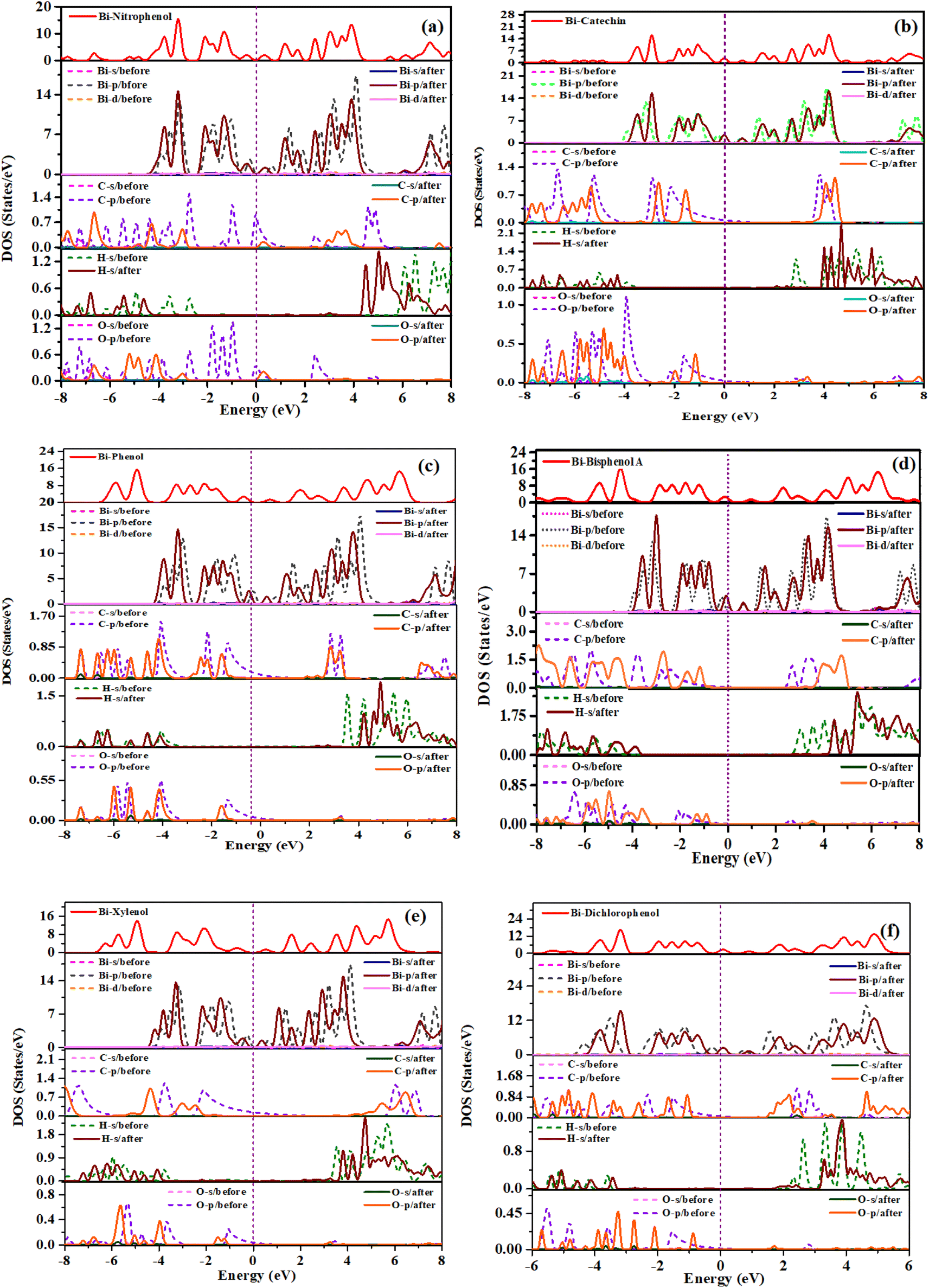 | ||
| Fig. 5 Density of states of phenolic compounds adsorbed on bismuthene (a) o-nitrophenol (b) catechin (c) phenol (d) bisphenol A (e) xylenol (f) dichlorophenol. | ||
As shown in Fig. 5(a)–(f), for nitrophenol, p orbital of C, O and s orbital of H exhibit overlapping peaks within the value of (−7 to −4.8) eV. For catechin, p orbital of C, O and s orbital of H displays overlapping peaks within the values (−7.9 to −4.4) eV. For phenol, p orbital of C, O and s orbital of H shows overlapping peaks upon (−6.5 to −4 and −1.9 to −1) eV. As can be seen from figure the overlapping peaks have much more contribution in total density of states. For bisphenol A, p orbital of C, O and s orbital of H exhibits overlapping peaks upon (−6 to −4) eV. For xylenol p orbital of C, O and s orbital of H presents overlapping peaks upon (−6.7 to −4 and −1.75 to −1) eV. Also for dichlorophenol, p orbital of C, O and s orbital of H demonstrates overlapping peaks upon (−5.5 to −4 and −3 to −1) eV. These compounds display dominant peaks in valence energy band, with overlapping peaks making valuable input to aggregate electronic states density, along with hybrid peaks.45
The direction of charge transfer during adsorption is indicated by the positive and negative values. The study demonstrates that in the adsorption process, charge transfers from the bismuthene surface to the adsorbents. Nitrophenol take charge of amount (−0.098e), catechin take charge of value (−0.092e), phenol take charge of value (−0.079e), xylenol accepts charge from bismuthene surface of amount (−0.097e), while in case of dichlorophenol, this compound take charge of amount (−0.04e). The magnitude of charge transfer for each system is relatively low, which aligns with the earlier analysis of adsorption energy. Specifically, for bisphenol A, the higher amounts of (−0.116e) charge displacement among the bismuthene surface and adsorptive material indicate a stronger adherence effect between them.46
3.5 Electron localization function
Electron density function, is associated with the density of kinetic energy.50 It serves like measure also the confinement and scattering of charge.51 Electron density function values range starts from 0.5 to 1.0, representing localized electron regions involved in bonding and nonbonding interactions. For instance, electron localization often occurs within nucleus shells, electron-sharing bonds, or unshared pairs. Weak van der Waals forces are characterized by electron density function values from 0.0 to 0.5, whereas subatomic particle dispersion spans across electron shells.52 The ELF map offers insight into how electron density on surfaces and adsorbents changes as complexes form, highlighting regions of higher electron density as shown in Fig. 6.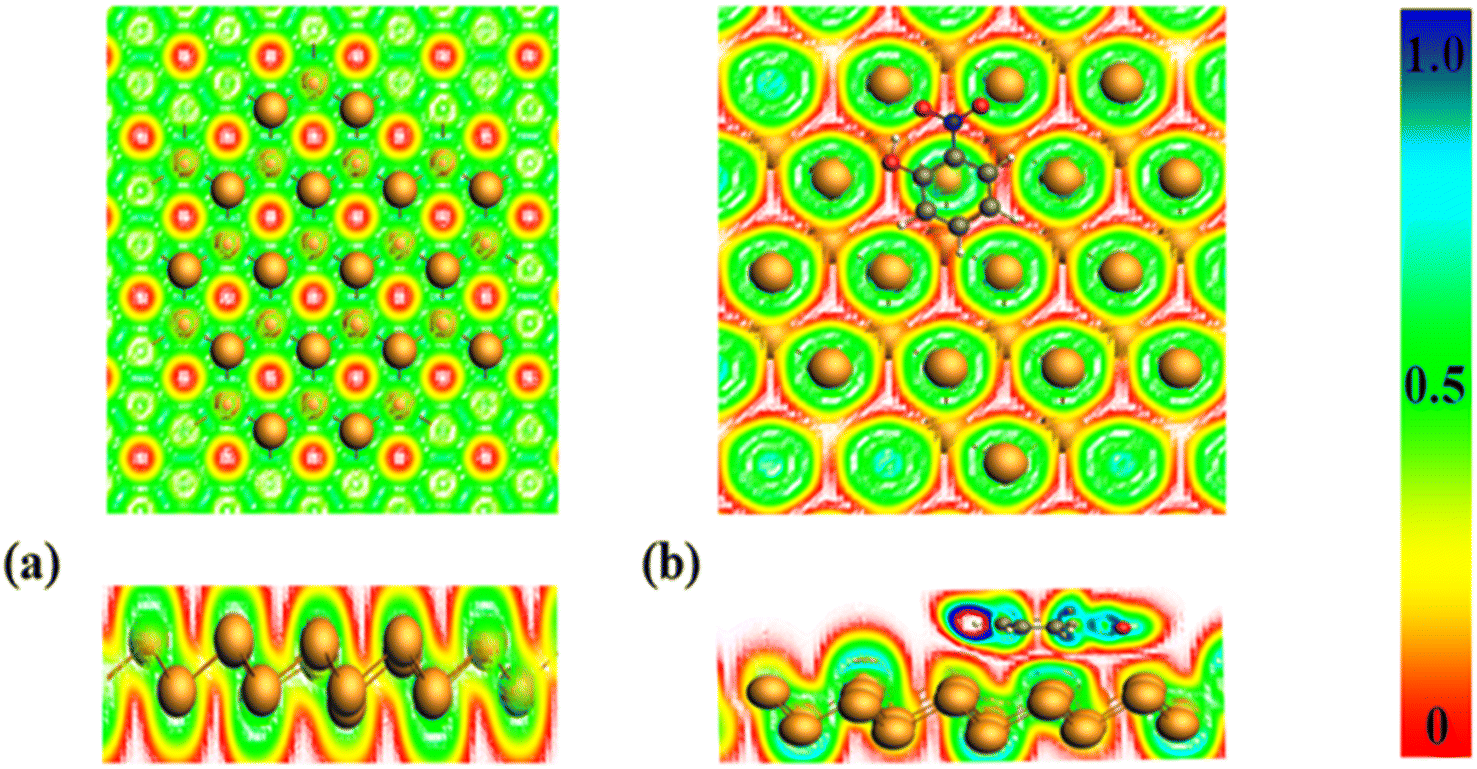 | ||
| Fig. 6 (a) ELF of pure bismuthene surface (b) ELF of nitrophenol adsorbed bismuthene. | ||
As depicted in the preceding Fig. 6, the pure bismuthene surface exhibits a predominance of red coloration, indicating a higher charge density on its surface, while the green color signifies a range of <0.5, confirming the existence of covalent bonds.53 Following the adsorption of nitrophenol, there is a noticeable change in color, with the appearance of a light blue shade, indicating charge displacement arising from bismuthene layer toward phenolic compounds. Additionally, a faint red color indicates reduction of electron density on the surface. Likewise, other phenolic compounds demonstrate analogous electron localization function behavior as described previously.
3.6 Vibrational frequency analysis
The analysis of the potential energy surface, derived from harmonic vibrational frequencies, for both the phenolic compounds and the molecule-absorbing system, verifies the energy minimum. The degree of molecule interaction becomes evident through comparing the vibrational frequencies before and after gas molecule adsorption onto bismuthene layer.In the bismuthene with bisphenol A molecules, the calculated stretching frequency registers at 2237 cm s−1. Stretching frequencies for catechin molecules, before and after adsorption onto bismuthene layer, are 213 cm s−1 and 2503 cm s−1, respectively. This outcome underscores the strong interaction tendency of catechin, as previously inferred from charge transfer and binding energy calculations. Regarding nitrophenol, the calculated frequency ranges between 1343 cm s−1 and 957 cm s−1, indicating the significant impact of adsorption. In contrast, phenol exhibits a frequency shift from 1276 cm s−1 in its free state to 2159 cm s−1 upon binding with bismuthene. With xylenol, a similar trend is observed, transitioning from 1230 cm s−1 to 2269 cm s−1 when adsorbed and interacting with other molecules. Likewise, for dichlorophenol, the stretching frequency is calculated to change from 1457 cm s−1 to 1870 cm s−1. This shift in vibrational properties could serve as a criterion for molecules detection.
Hence, the preceding discourse confirms that every molecule, barring catechin and xylenol, engages in a chemical bonding interaction with bismuthene, leading to notable modifications in their vibrational attributes.38
3.7 Stability of bismuthene before and after adsorption
To assess the efficiency of bismuthene monolayer in adsorbing phenolic compounds according to environmental standards, we created a simulation environment comprising phenolic molecule on bismuthene monolayer. The simulation involved optimizing all atoms in the system without constraints. The energy (E) of the system was then graphed against simulation time (fs) and are show in Fig. 7.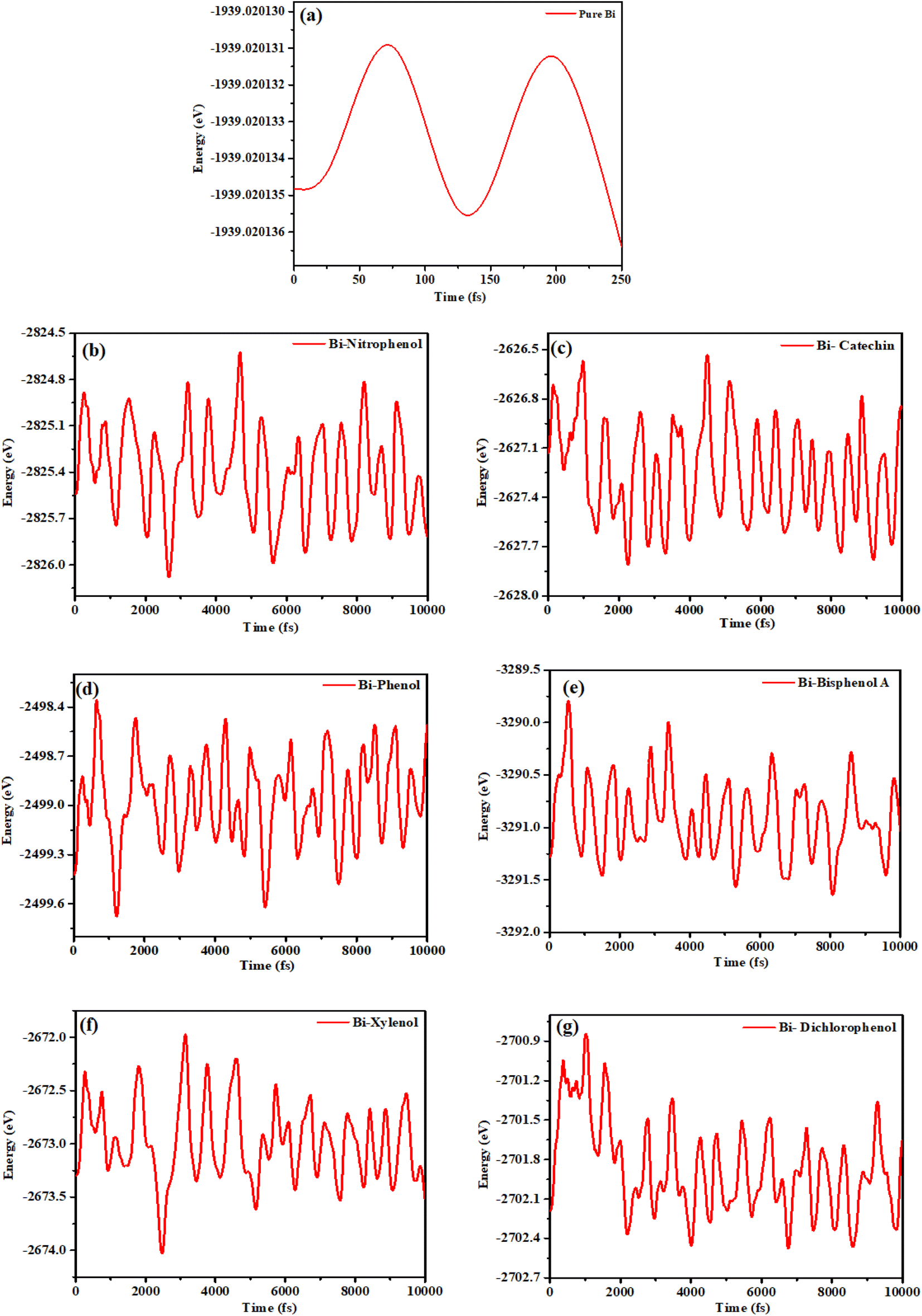 | ||
| Fig. 7 Stability graphs (a) bismuthene (b) o-nitrophenol (c) catechin (d) phenol (e) bisphenol A (f) xylenol (g) dichlorophenol. | ||
As shown in Fig. 7(a) the maximum energy peak of pure bismuthene is analyzed at the value of −1939 eV and there is continuous smooth peak for energy graph. After the adsorption of nitrophenol Fig. 7(b) it can be seen that the energy graph gets its maximum peak at the energy value of −2824.6 eV, and also it can be concluded that the energy value fluctuates within (−2824.8 to −2826.0) eV. After the adsorption of catechin Fig. 7(c) on bismuthene layer, it can be concluded from the results that the energy approaches to maximum value −2626.5 eV. The overall graph peaks fluctuates within the range of (−2626.6 to 2627.8) eV. After adsorbing phenol Fig. 7(d) the results shows that the system get maximum energy of −2498.4 eV, and the overall energy of the system varies within the range (−2498.5 to 2499.6) eV. In case of bisphenol A adsorption Fig. 7(e) the results conclude that the maximum energy for this system is −3289.6 eV, after analysis of energy graph it can be seen that energy value fluctuates within (−3290 to 3291.6) eV. After adsorption of xylenol Fig. 7(f) the results concluded that the maximum energy of the system is −2672 eV, and from the overall results of the energy graph it is concluded that energy value varies within (−2672.25 to −2674.0) eV. In case of dichlorophenol adsorption the maximum energy value for the adsorption system is −2700.8 eV while the overall energy value varies within the range of (−2701.0 to −2702.4) eV. The variation in energy around a constant value indicates that the simulated system has achieved an energetically equilibrium state, affirming the reliability of the resulting data obtained.54
4. Conclusion
Theoretical calculations have been conducted to analyze adherence among carbon-based compound from polluted water onto the bismuthene surface. Calculation confirms that various phenolic molecules can be adsorbed simultaneously on the bismuthene surface. While adsorbing phenolic molecules, the aromatic ring structure predominantly maintains level positioning, whereas the CH3 and OH groups exhibit an elevated tilt. Observation made for nearest atomic distances in various adsorption setup indicates the occurrence of chemisorption. Of these, the bisphenol A (C15H16O2) system exhibits the shortest atomic distance of 4.00 Å, suggesting that configuration among adsorption setup is reliable. Furthermore, bisphenol A (C15H16O2) system demonstrates a maximum adsorption energy of 12.8509 eV, indicating the adsorption mechanism of that arrangement looks most favorable to happen. By analyzing electronic states density, it is evident the fact overlapping crests of C-p, H-s, and O-p orbitals are located away from the Fermi energy level in valence band, indicating the system's stability and contributing to the aggregate electronic states density. The conclusion drawn from the charge transfer indicates that bisphenol A exhibits more stable adsorption on the bismuthene surface.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors express their appreciation to the Deanship of Scientific Research at King Khalid University, Saudi Arabia, for funding this work through research group program under grant number RGP 2/538/45 to Saleh S. Alarfaji.References
- Y. Cao, Y. Wang, F. Zhou, J. Huang and M. Xu, Acylamino-functionalized hyper-cross-linked polymers for efficient adsorption removal of phenol in aqueous solution, Sep. Purif. Technol., 2022, 303, 122229 CrossRef CAS.
- Y. Yuan, W.-g. Pan, R.-t. Guo, L.-f. Hong, Z.-d. Lin and X.-y. Ji, Flower spherical-like Bi7O9I3/AgI S-scheme heterojunction for phenol photodegradation: the synergetic effect of dual surface plasmon resonance and photothermal property, Sep. Purif. Technol., 2022, 297, 121538 CrossRef CAS.
- H. Ma, W. Zhang and D. Chen, Catalytic hydrodeoxygenation of phenolic compounds over Ru-MoFeP/Al2O3 catalyst, Catal. Today, 2023, 408, 50–57 CrossRef CAS.
- J.-z. Shu, Y.-j. Zhang, Y.-l. Yang, Z.-f. Zhang, Q.-l. Zhang, W.-w. Gao, L.-j. Feng and X.-y. Wei, A new eco-friendly method for efficient recovery and reuse of phenols in a semi-coke wastewater, Environ. Technol. Innovation, 2022, 28, 102951 CrossRef CAS.
- S. Liu, J. Wang, W. Huang, X. Tan, H. Dong, B. A. Goodman, H. Du, F. Lei and K. Diao, Adsorption of phenolic compounds from water by a novel ethylenediamine rosin-based resin: Interaction models and adsorption mechanisms, Chemosphere, 2019, 214, 821–829 CrossRef CAS PubMed.
- S. Krishnan, P. Homroskla, K. Saritpongteeraka, O. Suttinun, M. Nasrullah, Y. Tirawanichakul and S. Chaiprapat, Specific degradation of phenolic compounds from palm oil mill effluent using ozonation and its impact on methane fermentation, Chem. Eng. J., 2023, 451, 138487 CrossRef CAS.
- M. C. da Silva, C. Schnorr, S. F. Lütke, S. Knani, V. X. Nascimento, É. C. Lima, P. S. Thue, J. Vieillard, L. F. Silva and G. L. Dotto, KOH activated carbons from Brazil nut shell: preparation, characterization, and their application in phenol adsorption, Chem. Eng. Res. Des., 2022, 187, 387–396 CrossRef CAS.
- Y. Wang, H. Duan, H. Li, J. Zhen and W. Lv, Efficient activation of peroxymonosulfate by MoS2 intercalated MgCuFe layered double hydroxide for phenol pollutant control, J. Environ. Chem. Eng., 2023, 11(2), 109502 CrossRef CAS.
- X. Tian, H. Zhang, C. Hu and Y. Yan, Preparation of microfiber composite nitrogen doped carbon nanotube membranes and their degradation properties of phenol in the structured fixed bed, J. Environ. Chem. Eng., 2023, 11(1), 109255 CrossRef CAS.
- X. Song, Y. Fu, Y. Pang and L. Gao, Preparation of La-Zn/HZSM-5 zeolite and its application in photocatalytic degradation of phenol, Chem. Phys. Lett., 2022, 805, 139947 CrossRef CAS.
- C. Xu, Z. Jin, J. Cui, F. Guo, R. Hu, W. Meng, J. Hu and X. Shen, One step grinding method to prepare BiFeO3/α-Fe2O3 type-II heterojunction for enhancing phenolic wastewater degradation, Mater. Sci. Semicond. Process., 2023, 155, 107242 CrossRef CAS.
- Y. Wang, X. Fan, W. Dong, Q. Zhang, J. Liu, R. Li, Y. Wang, X. Zhang and C. Fan, CeO2 nanoparticles decorated Bi4O7 nanosheets for enhanced photodegradation performance of phenol, Mater. Lett., 2022, 322, 132465 CrossRef CAS.
- P. Praveen and K.-C. Loh, Trioctylphosphine oxide-impregnated hollow fiber membranes for removal of phenol from wastewater, J. Membr. Sci., 2013, 437, 1–6 CrossRef CAS.
- S. Khan, M. A. Al-Obaidi, C. Kara-Zaïtri and I. M. Mujtaba, Optimisation of design and operating parameters of reverse osmosis process for the removal of phenol from wastewater, S. Afr. J. Chem. Eng., 2023, 43(1), 79–90 Search PubMed.
- D. Bharali, S. Saikia, R. Devi, B. M. Choudary, N. K. Gour and R. C. Deka, Photocatalytic degradation of phenol and its derivatives over ZnFe layered double hydroxide, J. Photochem. Photobiol., A, 2023, 438, 114509 CrossRef CAS.
- J. Guo, Q. Peng, H. Fu, G. Zou and Q. Zhang, Heavy-metal adsorption behavior of two-dimensional alkalization-intercalated MXene by first-principles calculations, J. Phys. Chem. C, 2015, 119(36), 20923–20930 CrossRef CAS.
- M. Zeng, M. Chen, D. Huang, S. Lei, X. Zhang, L. Wang and Z. Cheng, Engineered two-dimensional nanomaterials: an emerging paradigm for water purification and monitoring, Mater. Horiz., 2021, 8(3), 758–802 RSC.
- Y. Cheng, M. Li and Y. Song, Theoretical study of M2CO2 MXenes stability and adsorption properties for heavy metals ions removal from water, Comput. Mater. Sci., 2023, 220, 112042 CrossRef CAS.
- S. S. Al Ghafry, H. Al Shidhani, B. Al Farsi, R. S. Sofin, A. S. Al-Hosni, Z. Alsharji, J. Al-Sabahi and M. Z. Al-Abri, The photocatalytic degradation of phenol under solar irradiation using microwave-assisted Ag-doped ZnO nanostructures, Opt. Mater., 2023, 135, 113272 CrossRef CAS.
- M. Albedah, M. R. Hamoudi, S. H. Sadon, E. Oussama and Q. H. Le, Study of phenol removal from wastewater petroleum industry using molecular dynamics method for two-dimensional adsorbents from the aqueous environment, Eng. Anal. Bound. Elem., 2023, 147, 69–75 CrossRef.
- F. Anaya, L. Zhang, Q. Tan and D. E. Resasco, Tuning the acid–metal balance in Pd/and Pt/zeolite catalysts for the hydroalkylation of m-cresol, J. Catal., 2015, 328, 173–185 CrossRef CAS.
- A. Miri-Jahromi, D. M. Maklavany, Z. Rouzitalab, S. G. Khiavi, E. Ghasemy, M. Khedri, S. Rezvantalab, S. Sharafinia, A. Rashidi and R. Maleki, Engineering of two-dimensional monolayers to phenolic compounds removal from wastewater: an experimental and computational insight, J. Mol. Liq., 2022, 362, 119784 CrossRef CAS.
- A. A. Melaibari, A. S. Elamoudi, M. E. Mostafa and N. H. Abu-Hamdeh, Utilization of various waste sources in Saudi Arabia as a new clean and renewable energy source: adsorption of phenol pollutants and removal from petroleum industrial wastes via molecular dynamics simulation, Eng. Anal. Bound. Elem., 2023, 147, 164–170 CrossRef.
- Z. Ghahghaey, M. Hekmati and M. D. Ganji, Theoretical investigation of phenol adsorption on functionalized graphene using DFT calculations for effective removal of organic contaminants from wastewater, J. Mol. Liq., 2021, 324, 114777 CrossRef CAS.
- J. M. Galicia Hernández, G. H. Cocoletzi and E. C. Anota, DFT studies of the phenol adsorption on boron nitride sheets, J. Mol. Model., 2012, 18, 137–144 CrossRef PubMed.
- J. M. G. Hernández, E. C. Anota, M. T. R. de la Cruz, M. G. Melchor and G. H. Cocoletzi, First principles studies of the graphene-phenol interactions, J. Mol. Model., 2012, 18, 3857–3866 CrossRef PubMed.
- Z. Yuan, Y. Yamamoto, T. Yajima and Y. Kawajiri, Estimation and statistical analysis of model parameters using sequential Monte Carlo for phenol and p-cresol separation, J. Chromatogr. A, 2023, 1688, 463703 CrossRef CAS PubMed.
- Z. G. N. Abadee, M. Hekmati and M. D. Ganji, Removing phenol contaminants from wastewater using graphene nanobuds: DFT and reactive MD simulation investigations, J. Mol. Liq., 2019, 286, 110872 CrossRef.
- E. Aktürk, O. Ü. Aktürk and S. Ciraci, Single and bilayer bismuthene: stability at high temperature and mechanical and electronic properties, Phys. Rev. B, 2016, 94(1), 014115 CrossRef.
- G. t. Te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. van Gisbergen, J. G. Snijders and T. Ziegler, Chemistry with ADF, J. Comput. Chem., 2001, 22(9), 931–967 CrossRef CAS.
- M. Güell, J. M. Luis, M. Sola and M. Swart, Importance of the basis set for the spin-state energetics of iron complexes, J. Phys. Chem. A, 2008, 112(28), 6384–6391 CrossRef PubMed.
- E. Van Lenthe and E. J. Baerends, Optimized Slater-type basis sets for the elements 1–118, J. Comput. Chem., 2003, 24(9), 1142–1156 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865 CrossRef CAS PubMed.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27(15), 1787–1799 CrossRef CAS PubMed.
- M. Franchini, P. H. T. Philipsen and L. Visscher, The Becke fuzzy cells integration scheme in the Amsterdam Density Functional program suite, J. Comput. Chem., 2013, 34(21), 1819–1827 CrossRef CAS PubMed.
- M. I. Khan, G. Nadeem, A. Majid and M. Shakil, A DFT study of bismuthene as anode material for alkali-metal (Li/Na/K)-ion batteries, Mater. Sci. Eng., B, 2021, 266, 115061 CrossRef CAS.
- M. I. Khan, S. M. Zaigam, A. Majid and M. B. Tahir, Computational insights of alkali metal (Li/Na/K) atom decorated buckled bismuthene for hydrogen storage, Int. J. Hydrogen Energy, 2021, 46(56), 28700–28708 CrossRef CAS.
- M. Isa, I. Ashfaq, A. Majid, M. Shakil and T. Iqbal, A DFT study of silver decorated bismuthene for gas sensing properties and effect of humidity, Mater. Sci. Semicond. Process., 2022, 145, 106635 CrossRef CAS.
- I. khan Muhammad, K. Swera and M. Abdul, Computational study of 4d transition metals doped bismuthene for spintronics, Phys. E, 2021, 126, 114464 CrossRef.
- M. A. Tamerd, A. Marjaoui, A. El Kasmi, M. Assebban, M. Diani and M. Zanouni, First-principles investigations of structural, electronic and thermoelectric properties of Sb/Bi2Se3 van der Waals heterostructure, Mater. Sci. Semicond. Process., 2022, 142, 106472 CrossRef.
- N. Mounet, M. Gibertini, P. Schwaller, D. Campi, A. Merkys, A. Marrazzo, T. Sohier, I. E. Castelli, A. Cepellotti and G. Pizzi, Two-dimensional materials from high-throughput computational exfoliation of experimentally known compounds, Nat. Nanotechnol., 2018, 13(3), 246–252 CrossRef CAS PubMed.
- M.-Y. Liu, Y. Huang, Q.-Y. Chen, Z.-Y. Li, C. Cao and Y. He, Strain and electric field tunable electronic structure of buckled bismuthene, RSC Adv., 2017, 7(63), 39546–39555 RSC.
- S. M. Mozvashi, S. I. Vishkayi and M. B. Tagani, Antimonene/bismuthene vertical van-der Waals heterostructure: a computational study, Phys. E, 2020, 118, 113914 CrossRef CAS.
- A. Marjaoui, M. Zanouni, A. El Kasmi, M. Jbilou and M. Diani, Na adsorption on bismuthene monolayer for battery applications: a first-principles study, FlatChem, 2021, 27, 100251 CrossRef CAS.
- Z. Zhang, L. Wang and X. Zhou, Study on adsorption behaviors of different cresols: first-principles calculation, J. Mol. Liq., 2023, 390, 123120 CrossRef CAS.
- Z. Zhang, W. Teng, J. Cao and X. Zhou, First-principles calculation of cresol contaminants removal from wastewater environment, J. Phys. Chem. Solids, 2024, 184, 111696 CrossRef CAS.
- A. B. Dalton, S. M. Le, N. V. Karimova, R. B. Gerber and S. A. Nizkorodov, Influence of solvent on the electronic structure and the photochemistry of nitrophenols, Environ. Sci.: Atmos., 2023, 3(2), 257–267 CAS.
- E. García-Hernández, Pristine and X-doped (X = B, N) phosphorene as platform materials to the removal of phenol: a theoretical insight, J. Mol. Liq., 2023, 374, 121280 CrossRef.
- R. M. Ahangar and D. Farmanzadeh, Theoretical study for exploring the adsorption behavior of aniline and phenol on pristine and Cu-doped phosphorene surface, Appl. Surf. Sci., 2023, 614, 156194 CrossRef.
- B. Silvi and A. Savin, Classification of chemical bonds based on topological analysis of electron localization functions, Nature, 1994, 371(6499), 683–686 CrossRef CAS.
- C. S. Abraham, S. Muthu, J. C. Prasana, S. Armaković, S. J. Armaković and B. Geoffrey, Computational evaluation of the reactivity and pharmaceutical potential of an organic amine: a DFT, molecular dynamics simulations and molecular docking approach, Spectrochim. Acta, Part A, 2019, 222, 117188 CrossRef CAS PubMed.
- M. Vatanparast and A.-R. Nekoei, RAHB concept and σ-skeleton in some oximes of 3-hydroxy fulvene; DFT, AIM, ELF and NBO studies, Struct. Chem., 2015, 26, 1039–1048 CrossRef CAS.
- M. I. Khan, M. I. Akber, M. Gul, T. Iqbal, S. S. Alarfaji and A. Mahmood, Exploring the sensing potential of Fe-decorated h-BN toward harmful gases: a DFT study, RSC Adv., 2024, 14(10), 7040–7051 RSC.
- H. Liu, W. He, H. Xu, X. Wang and Q. An, Assessment of g-Mg3N2 membrane performance for efficient removal of dioxane contaminant from wastewater: DFT-MD simulation treatments, Curr. Appl. Phys., 2023, 54, 75–83 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02933j |
| This journal is © The Royal Society of Chemistry 2024 |