
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoswitchable hydrazones with pyridine-based rotors and halogen substituents†
Lucie Kotásková *a,
Pawel Jewulaa,
Radovan Herchelb,
Ivan Nemec*ab and
Petr Neugebauera
*a,
Pawel Jewulaa,
Radovan Herchelb,
Ivan Nemec*ab and
Petr Neugebauera
aCentral European Institute of Technology, Brno University of Technology, Purkyňova 656/123, 61200 Brno, Czech Republic. E-mail: ivan.nemec@upol.cz; lucie.kotaskova1@ceitec.vutbr.cz
bDepartment of Inorganic Chemistry, Palacký University, 17 listopadu 1192/12, 77900 Olomouc, Czech Republic
First published on 1st July 2024
Abstract
The Z,E-photoisomerization of pyridine-based hydrazone switches is typically suppressed due to the presence of pyridine-based rotors. The crystal structures of studied compounds were investigated using theoretical methods combining DFT and QT-AIM calculations to unveil the nature and properties of the intramolecular hydrogen bonding. In this study, we introduced a new series of pyridine-based hydrazones anchored with o-halogen substituents (2-X) and investigated their photoswitching abilities using 1H NMR and UV-Vis spectroscopy. The efficiency of the photoisomerization from initial 2-X-Z to the 2-X-E isomer varied, with the highest yield observed for 2-Cl-E (55%). Our findings, supported by DFT calculations, revealed the formation of a new diastereomer, 2-X-E*, upon back-photoisomerization. We demonstrated that hydrazones from the 2-X series can be reversibly photoswitched using irradiation from the UV-Vis range, and additionally, we explored the effect of the halogen atom on their switching capabilities and also on their thermodynamics and kinetics of photoswitching, determining their molecular solar thermal energy storage potential.
Introduction
Versatility, potential for multiple applications, straightforward synthesis, and resistance to hydrolysis have propelled hydrazones into the spotlight across various research fields.1,2 Notably, hydrazones feature the distinctive C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–NH group, endowing the carbon atom with nucleophilic and electrophilic characteristics, and the nitrogen atom with nucleophilic properties.1 This dual nature enables hydrazones to be utilized in both cation3,4 and anion5,6 sensing applications. Hydrazones serve as valuable biologically active compounds in drug development, particularly as antimicrobial agents.7,8 Moreover, they play a crucial role in organic synthesis by facilitating the activation of organocatalytic reactions.9
N–NH group, endowing the carbon atom with nucleophilic and electrophilic characteristics, and the nitrogen atom with nucleophilic properties.1 This dual nature enables hydrazones to be utilized in both cation3,4 and anion5,6 sensing applications. Hydrazones serve as valuable biologically active compounds in drug development, particularly as antimicrobial agents.7,8 Moreover, they play a crucial role in organic synthesis by facilitating the activation of organocatalytic reactions.9
The ability of hydrazones to undergo Z,E-isomerization allows them to function as molecular switches. The distinctiveness of the hydrazone functional group is accentuated by its ability to control rotary motion through Z,E-isomerization triggered by both light and chemical stimuli. This precise modulation of motion at the molecular level positions hydrazones as promising candidates for the development of molecular machines.1 Hydrazones also play a crucial role in the advancement of optoelectronic devices10 and serve as fundamental building blocks for the synthesis of covalent organic frameworks.11 Moreover, this characteristic makes them valuable in supramolecular chemistry as systems exhibiting multiple dynamics properties.12
The structural modularity of hydrazones enables a wide range of applications, including their incorporation into adaptive materials like emissive hydrogels,13 drug delivery systems,14 pH probes,15 and molecular actuators.16 Recent work by Aprahamian et al. has demonstrated that the common drawback of a small difference in Gibbs energies between the Z- and E-isomers of hydrazone photoswitches can be overcome, allowing their use in energy storage applications.17
Isomerization in the Z,E-configuration can occur through three mechanisms: inversion (lateral shift), rotation, and hydrazone-azo tautomerization followed by rotation around the C–N bond.18 It has been demonstrated that this type of molecular switch exhibits reversible switching with high fatigue resistance.19 The key parameter governing Z,E-isomerization is N–H⋯N intramolecular hydrogen bonding (IMHB). The impact of electron-donating or electron-withdrawing para-substituents on the strength of IMHB in hydrazones was investigated.20 This structure–property analysis provided insights into the varying strength of IMHB, as indicated by the chemical shift values of N–H in E- and Z-isomers. The strength of IMHB increased from electron-withdrawing groups (EWGs) to electron-donating groups (EDGs) in the E-isomer, following the nomenclature in our work. Conversely, in the Z-isomer, both EWGs and EDGs increased the strength of IMHB. This difference is attributed to the absence of resonance-assisted hydrogen bonding in the Z-isomer due to the presence of a pyridinyl ring, where its aromaticity is diminished. The structure of the hydrazone appears to play a significant role in determining reversible photochemical isomerization.
While Courtot's work demonstrated that acylhydrazones can be photoswitched,21 Aprahamian and co-workers confirmed that the N–H⋯N intramolecular hydrogen bond suppresses photoswitching. Irradiation of the ethyl-(2-phenylhydrazinylidene)(pyridin-2-yl)acetate at 412 nm for 65 minutes or at 340 nm for 36 minutes did not induce isomerization. However, replacing the pyridinyl ring with a phenyl in the rotor part provided photoswitchable hydrazone with excellent thermal stability.19
In this study, we present a theoretical and experimental examination of a new family of hydrazone switches bearing o-halogen substituents (see Scheme 1). Our research pursues two main objectives: firstly, we aim to investigate whether these pyridine rotor-based hydrazones might undergo photoisomerization upon UV-Vis irradiation, in contrast to previous contradictory findings,19 and to elucidate the mechanism of photoswitching, both theoretically and experimentally. Secondly, we explore the influence of intramolecular hydrogen bonding, particularly in the context of alterations in halogens at the ortho position, on photoswitching behavior and its impact on thermodynamic parameters. Understanding these factors is crucial for characterizing the tunability and potential of these hydrazones as energy storage media, especially considering the possible significant effects of adjacent group modifications. Halogen atoms were deliberately chosen as hydrogen bond acceptors due to their anticipated weak strength and ease of tunability through variation.
 | ||
| Scheme 1 Reaction scheme for the preparation of compounds 1, 2-F, 2-Cl, 2-Br, and 2-I; (i) lithium diisopropylamide (2.05 eq.), diethyl carbonate (3 eq.), THF, Ar, −84 °C; (ii) (1) 2-halogen aniline, NaNO2 (1.1 eq.), HCl, H2O, 0 °C, (2) 1, NaOAc (9 eq.), EtOH, H2O, 0 °C. | ||
Experimental
Materials
The abbreviation used are the following: lithium diisopropylamide (LDA), column chromatography (CC), tetrahydrofuran (THF), thin layer chromatography (TLC), dichloromethane (DCM). The preparation of compound 1 was carried out under an argon atmosphere, using Schlenk technique. Analytical-grade chemicals were obtained from commercial suppliers and used without further purification. The solvents were dried as follows: THF by reflux over sodium/benzophenone ketyl followed by distillation under argon, then stored under argon on 3 Å molecular sieves. Manual chromatographic separations were performed by column chromatography using 60–200 μm silica gel 60. The reaction progress was monitored by a TLC on pre-coated aluminium sheets and visualized using the UV lamp (254 and 365 nm).Instrumentation
Melting points were measured using a digital Buchi B-540 Melting Point Apparatus. The melting points were determined based on the three measurements. 1H, 13C{1H} and 19F{1H} NMR spectra were recorded on Bruker spectrometer operating at 500 MHz (Bruker Avance III) using CDCl3 as solvent. Chemical shifts on the δ scale (ppm) relative to tetramethylsilane were referenced internally with respect to either the protio resonance of residual CHCl3 (δH = 7.26 ppm and δC = 77.16 ppm) or CF3COOH (δF = −78.50 ppm). UV-Vis spectra were recorded using UV/Vis/NIR Spectrophotometer Jasco V-770 with a single monochromator, deuterium, and wolfram-halogen lamp, operating in the range of 190 to 2700 nm. The samples were inserted into quartz cuvettes (10 mm). The mass spectra were collected on an LCQ Fleet mass spectrometer (Thermo Scientific, Waltham, MA, USA) equipped with an electrospray ion source and three-dimensional (3D) ion-trap detector. Spectra were acquired in the positive mode in the m/z range 50–2000 using following settings: ionization spray voltage = 5 kV [−4.83 (−), 5.22 (+)]; capillary temperature = 275 °C; capillary voltage = 50 V; tube lens = 120 V (+), −100 V (−). Photoswitching experiments were conducted within an irradiation chamber assembled with high-power LED diodes operating at wavelengths of 365, 420 or 460 nm. These experiments were performed in a refrigerated environment to prevent overheating and exposure to the ambient light. The photoswitching experiments were conducted in either an NMR tube or a cuvette for UV-Vis measurements, with the sample exposed to irradiation at 365 nm or 420 nm to initiate Z,E-isomerization. Spectra were recorded after transferring the sample from the irradiation chamber to the spectrometer, ensuring that the sample was protected from light during the transfer process. The spectra recorded after irradiation at 365 nm and 420 nm were comparable, and the most representative data are presented in this study. The reverse E,Z-isomerization induced by 460 nm irradiation was carried out in the same manner. The ratios of Z- and E-isomers (Table 2) were determined by integrating the intensities of the respective NN–H signals in the 1H NMR spectra. Irradiation times were chosen to be 30 minutes for long irradiation cycles and shorter durations (20 seconds, 1 minute, 3 minutes) for measuring photofatigue resistance.
Crystallography
The collection of the X-ray diffraction data (see ESI, Table S1†) for the selected single crystals was performed using an XtaLAB Synergy-I diffractometer (Rigaku Corporation, Tokyo, Japan) with a HyPix3000 hybrid pixel array detector and a microfocused PhotonJet-I X-ray source (Cu Kα) at 100.0(2) K. The data integration, scaling, and absorption for correction were applied using the program CrysAlisPro 1.171.40.82a.22 The crystal structures were solved using the SHELXT23 and all non-hydrogen atoms were refined anisotropically on F2 using and refined by the full matrix least-squares procedure with Olex2.refine24 in OLEX2 (version 1.5).25 All hydrogen atoms were found in differential Fourier maps and their parameters were refined using a riding model with Uiso(H) = 1.2(–CH2) or 1.5(–CH3)Ueq. To obtain structures suitable for the calculations we finalized their refinement using the non-spherical structure refinement method called Hirshfeld atom refinement26,27 incorporated into the module NoSpherA2 (ref. 28) (level of theory: B3LYP and def2-TZVPD for 2-Br, B3LYP and x2c-TZVPP for 2-I). All the structural files, either classically or NoSpherA2 refined, were submitted to Cambridge Structural Database (CSD) with the following deposition numbers: 2335394 (2-Br), 2335395 (2-Brnosph), 2335397 (2-I), 2335396 (2-Inosph). The molecular structures and packing diagram were drawn with MERCURY.29Compound 1
Diisopropylamine (13 ml, 0.092 mol, 2.1 eq.) was placed into Schlenk flask (250 ml) and dissolved in 40 ml of THF that was added via syringe under argon atmosphere. Then, to the stirred solution was added dropwise n-butyllithium (36 ml, 0.091 mol, 2.05 eq.) via syringe under argon atmosphere at −84 °C. The resulting mixture was warmed to 0 °C and stirred at this temperature for 1 hour. 2-Picoline (4.3 ml, 0.044 mol, 1 eq.) and diethyl carbonate (16 ml, 0.132 mol, 3 eq.) were dissolved in 40 ml of THF in a separate Schlenk flask (250 ml). The solution of lithium diisopropylamide was cannulated into the mixture of 2-picoline and diethyl carbonate at −84 °C. The resulting solution was allowed to stir for 1 hour at −84 °C. Then, the solution was warmed to room temperature and stirred at this temperature overnight. The reaction mixture was diluted with a saturated aqueous solution of NH4Cl (120 ml) and demineralized water (120 ml), followed by the extraction with ethyl acetate (3 × 100 ml). The combined organic layers were dried over MgSO4 and concentrated in vacuo. Purification by column chromatography (petroleum ether/ethyl acetate, 7/3, v/v) resulted in bright yellow oil (3.91 g, 54%). 1H NMR (500 MHz, CDCl3, 298 K): δ 1.22 (t, 3J = 7.2 Hz, 3H, H9), 3.84 (s, 2H, H6), 4.15 (q, 3JH–H = 7.2 Hz, 2H, H8), 7.14–7.17 (m, 3JH–H = 7.2 Hz, 3JH–H = 5.0 Hz, 4JH–H = 1.0 Hz, 1H, H4), 7.27 (d, 3JH–H = 7.2 Hz, 1H, H2), 7.62 (td, 3JH–H = 7.2 Hz, 4JH–H = 1.0 Hz, 1H, H3), 8.52 (dd, 3JH–H = 5 Hz, 4JH–H = 1.0 Hz, 1H, H5) ppm. 13C NMR (125 MHz, CDCl3, 298 K): δ 14.2 (s, C9), 44.0 (s, C6), 61.1 (s, C8), 122.1 (s, C4), 123.9 (s, C2), 136.7 (s, C3), 149.5 (s, C5) 154.6 (s, C1) ppm.General procedure for synthesis of hydrazones (2)
o-Halogen substituted aniline (3 mmol, 1 eq.) was dissolved in 3.6 ml of demineralized water and 1.8 ml of concentrated HCl and the solution was stirred at 0 °C for ca. 15 minutes. The cold solution of NaNO2 (3.3 mmol, 1.1 eq.) in 0.9 ml of demineralized water was added dropwise over a period of 30 minutes. The resulting diazonium salt was added dropwise at 0 °C to the mixture of 1 (3 mmol, 1 eq.) and sodium acetate (27 mmol, 9 eq.) in 13.5 ml of ethanol/demineralized water (v/v, 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). After addition of the whole portion of the diazonium salt, the reaction mixture was let to stir overnight at room temperature. The filtration and washing of the precipitate with cold demineralized water and diethyl ether led to isolation of hydrazones 2-Cl and 2-Br that were crystallized from methanol solution and isolated in sufficient purity. Compounds 2-F and 2-I were purified by CC as described below for individual products. Atom labelling of 2-X compounds is displayed in the inset of Scheme 1.
:1, v/v) furnished yellow-orange liquid (379 mg, 48%). 1H NMR (500 MHz, CDCl3, 298 K): δ 1.45 (t, 3J = 7.2 Hz, 3H, H9), 4.41 (q, 3JH–H = 7.2 Hz, 2H, H8), 6.92–6.97 (m, 1H, H12), 7.05–7.10 (m, 1H, H14), 7.16 (t, 3JH–H = 7.7 Hz, 1H, H13), 7.28 (ddd, 3JH–H = 7.5 Hz, 3JH–H = 4.9 Hz, 4JH–H = 1.0 Hz, 1H, H4), 7.78–7.84 (m, 2H, H11 and H3), 8.23 (d, 3JH–H = 8.3 Hz, 1H, H2), 8.66–8.68 (dq, 1H, H5), 15.00 (s, 1H, NH) ppm. 19F NMR (470 MHz, CDCl3, 298 K): δ −137.0 ppm. 13C NMR (125 MHz, CDCl3, 298 K): δ 14.5 (s, C9), 61.3 (s, C8), 115.3 (d, 2JC–F = 17.7 Hz, C14), 115.7 (d, 3JC–F = 2.2 Hz, C11), 122.6 (d, 4JC–F = 6.5 Hz, C12), 122.7 (s, C4), 124.5 (s, C2) 125.1 (d, 3JC–F = 3.3 Hz, C13), 127.5 (s, C6), 132.1 (d, 2JC–F = 9.8 Hz, C10), 137.0 (s, C3), 146.7 (s, C5), 151.4 (d, 1JC–F = 243.2 Hz, C15), 152.5 (s, C1), 165.7 (s, C7) ppm. MS m/z (+): calcd for C15H14N3O2F 310.10 [M + Na]+; found 310.02. λmax(CHCl3)/nm 370 (ε/dm3 mol−1 cm−1 28622).293).955).:5, v/v) furnished yellow solid (90 mg, 10%), mp 98–99 °C, 1H NMR (500 MHz, CDCl3, 298 K): δ 1.45 (t, 3JH–H = 7.1 Hz, 3H, H9), 4.41 (q, 3JH–H = 7.1 Hz, 2H, H8), 6.76 (t, 3J = 7.6 Hz, 1H, H12), 7.28–7.31 (m, 1H, H4), 7.37 (t, 3JH–H = 7.7 Hz, 1H, H13), 7.74 (d, 3JH–H = 8.0 Hz, 2H, H11 and H14), 7.82–7.86 (m, 1H, H3), 8.26 (dd, 3JH–H = 8.3 Hz, 1H, H2), 8.72–8.75 (m, 1H, H5), 15.34 (s, 1H, NH) ppm. 13C NMR (125 MHz, CDCl3, 298 K): δ 14.5 (s, C9), 61.3 (s, C8), 83.6 (s, C15), 115.9 (s, C11), 122.7 (s, C4), 124.2 (s, C12), 124.5 (s, C2), 127.4 (s, C6), 129.5 (s, C13) 137.1 (s, C3), 139.2 (s, C14), 144.6 (s, C10), 147.1 (s, C5), 152.3 (s, C1), 165.7 (s, C7) ppm. MS m/z (+): calcd for C15H14N3O2I 418.00 [M + Na]+; found 417.94. λmax(CHCl3)/nm 370 (ε/dm3 mol−1 cm−1 23106).
:1). After addition of the whole portion of the diazonium salt, the reaction mixture was let to stir overnight at room temperature. The filtration and washing of the precipitate with cold demineralized water and diethyl ether led to isolation of hydrazones 2-Cl and 2-Br that were crystallized from methanol solution and isolated in sufficient purity. Compounds 2-F and 2-I were purified by CC as described below for individual products. Atom labelling of 2-X compounds is displayed in the inset of Scheme 1.
:1, v/v) furnished yellow-orange liquid (379 mg, 48%). 1H NMR (500 MHz, CDCl3, 298 K): δ 1.45 (t, 3J = 7.2 Hz, 3H, H9), 4.41 (q, 3JH–H = 7.2 Hz, 2H, H8), 6.92–6.97 (m, 1H, H12), 7.05–7.10 (m, 1H, H14), 7.16 (t, 3JH–H = 7.7 Hz, 1H, H13), 7.28 (ddd, 3JH–H = 7.5 Hz, 3JH–H = 4.9 Hz, 4JH–H = 1.0 Hz, 1H, H4), 7.78–7.84 (m, 2H, H11 and H3), 8.23 (d, 3JH–H = 8.3 Hz, 1H, H2), 8.66–8.68 (dq, 1H, H5), 15.00 (s, 1H, NH) ppm. 19F NMR (470 MHz, CDCl3, 298 K): δ −137.0 ppm. 13C NMR (125 MHz, CDCl3, 298 K): δ 14.5 (s, C9), 61.3 (s, C8), 115.3 (d, 2JC–F = 17.7 Hz, C14), 115.7 (d, 3JC–F = 2.2 Hz, C11), 122.6 (d, 4JC–F = 6.5 Hz, C12), 122.7 (s, C4), 124.5 (s, C2) 125.1 (d, 3JC–F = 3.3 Hz, C13), 127.5 (s, C6), 132.1 (d, 2JC–F = 9.8 Hz, C10), 137.0 (s, C3), 146.7 (s, C5), 151.4 (d, 1JC–F = 243.2 Hz, C15), 152.5 (s, C1), 165.7 (s, C7) ppm. MS m/z (+): calcd for C15H14N3O2F 310.10 [M + Na]+; found 310.02. λmax(CHCl3)/nm 370 (ε/dm3 mol−1 cm−1 28622).293).955).:5, v/v) furnished yellow solid (90 mg, 10%), mp 98–99 °C, 1H NMR (500 MHz, CDCl3, 298 K): δ 1.45 (t, 3JH–H = 7.1 Hz, 3H, H9), 4.41 (q, 3JH–H = 7.1 Hz, 2H, H8), 6.76 (t, 3J = 7.6 Hz, 1H, H12), 7.28–7.31 (m, 1H, H4), 7.37 (t, 3JH–H = 7.7 Hz, 1H, H13), 7.74 (d, 3JH–H = 8.0 Hz, 2H, H11 and H14), 7.82–7.86 (m, 1H, H3), 8.26 (dd, 3JH–H = 8.3 Hz, 1H, H2), 8.72–8.75 (m, 1H, H5), 15.34 (s, 1H, NH) ppm. 13C NMR (125 MHz, CDCl3, 298 K): δ 14.5 (s, C9), 61.3 (s, C8), 83.6 (s, C15), 115.9 (s, C11), 122.7 (s, C4), 124.2 (s, C12), 124.5 (s, C2), 127.4 (s, C6), 129.5 (s, C13) 137.1 (s, C3), 139.2 (s, C14), 144.6 (s, C10), 147.1 (s, C5), 152.3 (s, C1), 165.7 (s, C7) ppm. MS m/z (+): calcd for C15H14N3O2I 418.00 [M + Na]+; found 417.94. λmax(CHCl3)/nm 370 (ε/dm3 mol−1 cm−1 23106).Results and discussion
Synthesis and characterization
The Japp–Klingemann reaction produced a series of compounds 2-X. Compounds 2-Cl and 2-Br were obtained through filtration and water washing from the reaction mixture, demonstrating sufficient purity without requiring further purification, with yields of 64% and 86%, respectively. The synthesis of 2-F yielded a dark red liquid as the crude product, subsequently purified via CC on silica gel. The resulting yellow-orange liquid was isolated with a 48% yield. 2-I was isolated as a light brown solid. Crystallization of this crude product from a petroleum ether/acetone solvent mixture (95:5, v/v) led to the isolation of yellow ribbon-like and orange square-shaped crystals, both suitable for X-ray diffraction analysis. They were later identified as 2-I and a side product 2-{(Z)-[(E)-(2-iodophenyl)diazenyl][2-(2-iodophenyl)hydrazinylidene]methyl}pyridine (2-I_azo),30 respectively. The similar polarity of these components made purification using CC to be a rather complicated process allowing us to isolate only limited amount of 2-I (10%) as a pure fraction. Unfortunately, 2-I_azo could not be isolated as a pure product and cannot be considered a successful synthesis outcome.
Mass analysis, 1H and 13C and NMR spectra of hydrazones 2-F, 2-Cl, 2-Br, and 2-I confirmed isolation of the desired products. Additionally, 2-F was investigated by 19F NMR (Fig. S1–S27†). All 2-X series examined by ESI mass spectrometry showed one set of peaks. The most intense mass peak corresponded to the [M + Na]+ ion. In all cases, the experimental monoisotopic distribution patterns perfectly matched the calculated ones. Mass spectrometry analysis indicated the isolation of a mixture of compounds 2-I and 2-I_azo (Fig. S28†). 1H NMR spectroscopy analysis confirmed the formation of the CN hydrazone bond in all 2-X compounds, evidenced by the disappearance of the corresponding singlet at 3.84 ppm for compound 1. Another piece of evidence for hydrazone formation was the downfield shift of signals observed in the 1H NMR spectra of compounds 2-X, with the most significant shifts occurring for H2 atoms (Δδ ≈ 1 ppm) in comparison to compound 1. As expected, the 1H NMR analysis also confirmed that compounds 2-X were not isolated as 100% isomerically pure compounds.
Singlets in the range between 15.35 and 12.23 ppm revealed the isolation of hydrazones in both isomers: 91%, 92%, 93%, 91% of Z-isomer and 9%, 8%, 7%, 9% of E-isomer for 2-F, 2-Cl, 2-Br, and 2-I, respectively. 19F NMR of 2-F also confirmed the presence of two isomers E- and Z-, −137.6 and −137.0 ppm, respectively.
Carbon signals for the pyridine ring in 13C NMR spectra are shifted downfield in 2-X series compared with 1. Signals for C-X in hydrazones (referring to C15) exhibited the largest chemical shift difference depending on the selected halogen substituent. The more electronegative substituent, the more downfield shift was observed: 151.4 ppm for 2-F, 120.1 ppm for 2-Cl, 109.7 ppm for 2-Br and 83.6 ppm for 2-I. The opposite trend can be found for signals C10 and C14, which are the neighbouring signals to C15–X. Additionally, the signals for all carbons of the stator part (C10–C15) in 2-F were observed as doublets due to coupling with the fluorine atom.
Crystal structures
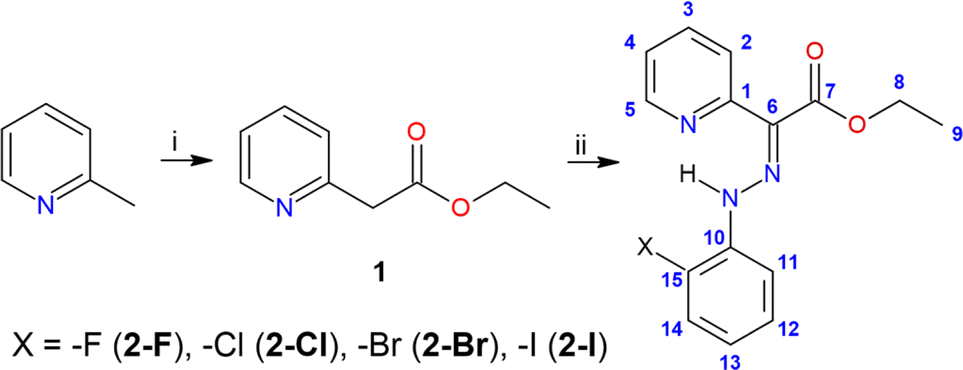
The compounds 2-Cl, 2-Br, and 2-I crystallize in the form of soft ribbon-like crystals that tend to stack and cluster. This, coupled with their noted softness, poses a significant obstacle to obtaining high-quality diffraction data. Therefore, good quality X-ray diffraction data were collected only for 2-Br and 2-I, while the crystal structure of 2-Cl was submitted to Cambridge Structural Database (CSD) as CSD Communication.31 Nevertheless, also in the case of 2-Cl, the structure and connectivity were undoubtedly confirmed.The crystal structures of 2-Cl, 2-Br, and 2-I are very similar, composed solely of the organic molecules adopting the Z-isomer configuration (Fig. 1A–C). The pyridine rotor part of the molecule is involved in intramolecular hydrogen bonding (IMHB) formed between the hydrazone amine group and pyridine nitrogen atom. The donor⋯acceptor distances for this contact are similar (in Å): d(N⋯N) = 2.614(5) and 2.628(5) in 2-Cl, 2.563(3) in 2-Br, and 2.559(6) in 2-I. The distances between the hydrazone nitrogen and the halogen atom are more influenced by the type of the halogen atom (in Å): d(N⋯X) = 2.941(4) and 2.946(4) in 2-Cl, 3.038(2) in 2-Br, 3.197(2) in 2-I. These interactions were investigated using Quantum Theory of Atoms in Molecules (QT-AIM).32 The wavefunctions were calculated using fragments from the experimental X-ray diffraction studies. In cases where the diffraction data exhibited sufficient quality, the structures were ultimately refined using Hirshfeld refinement implemented as NoSpherA2 module in Olex2. In the case of 2-Cl, the positions of hydrogen atoms were optimized using Density Functional Theory (DFT) at B3LYP level of theory (def2-SVP basis set). For the wavefunction calculations, DFT was used at the B3LYP level of theory and with the ZORA relativistic approximation33 and ZORA-def2-TZVP basis27 for all the atoms other than hydrogen and carbon (ZORA-def2-SVP). The SARC/J Coulomb fitting basis set was utilized as an auxiliary basis set.34 The calculations were performed using the Orca 4.2.1. package,35 and the Multiwfn software36,37 was used for QT-AIM calculations. For visualisation of the calculated properties the AIMAll software was used.38
 | ||
| Fig. 1 A perspective view of the molecular structures of hydrazone switches in crystal structures 2-Cl (A), 2-Br (B), and 2-I (C). Used color code: bromine (brown), carbon (light brown), chlorine (green), hydrogen (white), iodine (violet), nitrogen (blue), oxygen (red). Contour plots for 2-Cl (D), 2-Br (E) and 2-I (F) represent the Laplacian of electron density ∇2ρ(r) with shown (3, −1) bond critical points (brown dots) and bond paths (black solid and dashed lines). The perspective view is the same as that used for displaying the molecular structures. | ||
The investigation of the calculated electron density (ρ(r)) topology revealed (3, −1) bond critical points together with respective bond paths among all the involved atoms (Fig. 1D–F). Both types of contacts, either N–H⋯N or N–H⋯X, exhibit density properties corresponding to their non-covalent nature (∇2ρ(r) > 0). However, for the N–H⋯N contacts, small and negative values of potential electron density he(r), and a ratio of potential and kinetic energy densities |V(r)|/G(r) slightly larger than 1.0, were calculated. This indicates small covalent contribution and a large strength of these hydrogen bonds (Table 1). The interaction energies were calculated from V(r) using approximation (Eint = V(r)/2)39 confirming that the N–H⋯N hydrogen bonds are the strongest, with their strength increasing from 2-Cl (11.3 kcal mol−1) to 2-I (13.2 kcal mol−1, Table 1). The N–H⋯X contact is significantly weaker, with energies ranging between 3.5–4.1 kcal mol−1 (Table 1).
| ∇2ρ(r) | he(r) | G(r) | V(r) | |V(r)|/G(r) | ε | Eint/kcal mol−1 | ||
|---|---|---|---|---|---|---|---|---|
| 2-Cl mol1 | N–H⋯N | 0.130 | −0.0018 | 0.034 | −0.036 | 1.06 | 0.007 | 11.3 |
| N–H⋯Cl | 0.066 | +0.0020 | 0.014 | −0.012 | 0.86 | 1.142 | 3.8 | |
| 2-Cl mol2 | N–H⋯N | 0.0126 | −0.0012 | 0.033 | −0.034 | 1.03 | 0.008 | 10.7 |
| N–H⋯Cl | 0.066 | +0.0020 | 0.014 | −0.012 | 0.86 | 1.123 | 3.8 | |
| 2-Br | N–H⋯N | 0.142 | −0.0012 | 0.037 | −0.038 | 1.03 | 0.007 | 11.9 |
| N–H⋯Br | 0.059 | +0.0011 | 0.014 | −0.013 | 0.93 | 0.563 | 4.1 | |
| 2-I | N–H⋯N | 0.144 | −0.0029 | 0.039 | −0.042 | 1.08 | 0.061 | 13.2 |
| N–H⋯I | 0.051 | 0.0009 | 0.012 | −0.011 | 0.92 | 0.515 | 3.5 | |
Photoisomerization study
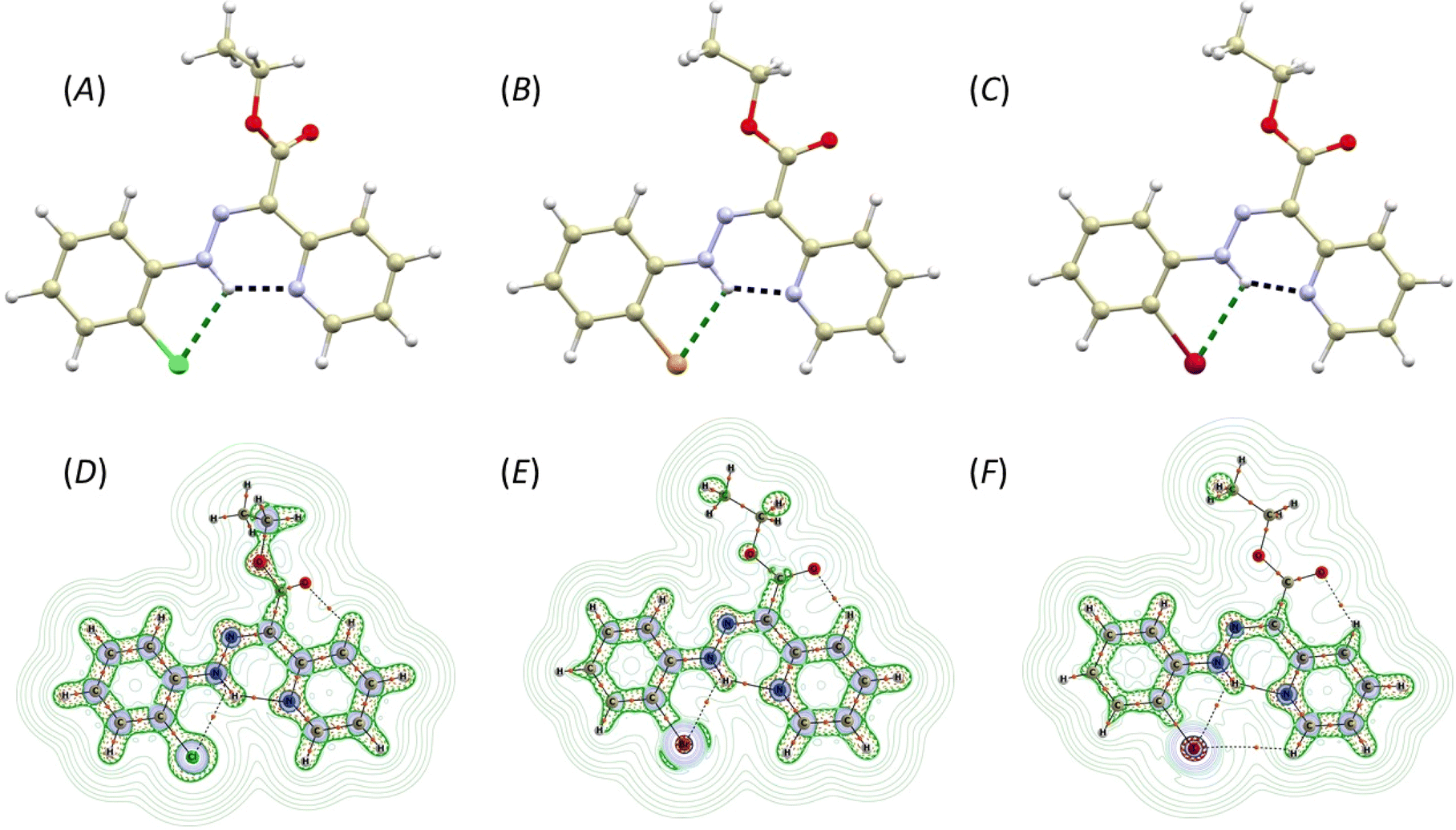
The Z,E-isomerization of compounds 2-X, induced by irradiation, was monitored using UV-Vis and 1H and 19F NMR spectroscopy. The spectra can be found in ESI – Fig. S29–S45.†The study of photoswitching phenomena (Fig. 2A) of 2-F, 2-Cl, 2-Br, and 2-I using UV-Vis spectroscopy was performed by dissolving samples in chloroform (2.73 × 10−5 M for 2-F, 2-Cl, 2-Br, and 1.36 × 10−5 M for 2-I).
 | ||
| Fig. 2 (A) Scheme for Z,E-isomerization induced by irradiation from UV-Vis range for 2-F (X = –F), 2-Cl (X = –Cl), 2-Br (X = –Br) and 2-I (X = –I). (B) Structure of diastereomer 2-X-E* isolated after photoswitching at 460 nm. (C) UV-Vis spectrum of 2-Cl (2.73 × 10−5 M) in chloroform. (D) UV-Vis spectrum of 2-Br (2.73 × 10−5 M) in chloroform. | ||
The spectra of all studied hydrazones exhibited a similar trend with λmax = 370 nm (2-F: ε370 = 28622 mol−1 dm3 cm−1, 2-Cl: ε370 = 26293 mol−1 dm3 cm−1, 2-Br: ε370 = 21955 mol−1 dm3 cm−1, 2-I: ε370 = 23106 mol−1 dm3 cm−1). These values shifted hypsochromically upon Z → E isomerization (2-F: ε363 = 26880 mol−1 dm3 cm−1, 2-Cl: ε363 = 24582 mol−1 dm3 cm−1, 2-Br: ε364 = 20650 mol−1 dm3 cm−1, 2-I: ε365 = 22182 mol−1 dm3 cm−1), upon irradiation for 30 minutes at 420 nm. Conversely, the absorption bands shifted bathochromically upon E → Z back isomerization after 30 minutes of irradiation at 460 nm (2-F: ε373 = 30543 mol−1 dm3 cm−1, 2-Cl: ε372 = 26292 mol−1 dm3 cm−1, 2-Br: ε375 = 23289 mol−1 dm3 cm−1, 2-I: ε376 = 24229 mol−1 dm3 cm−1). For 2-Cl, the E → Z isomerization process was found to be completely reversible (Fig. 3C).
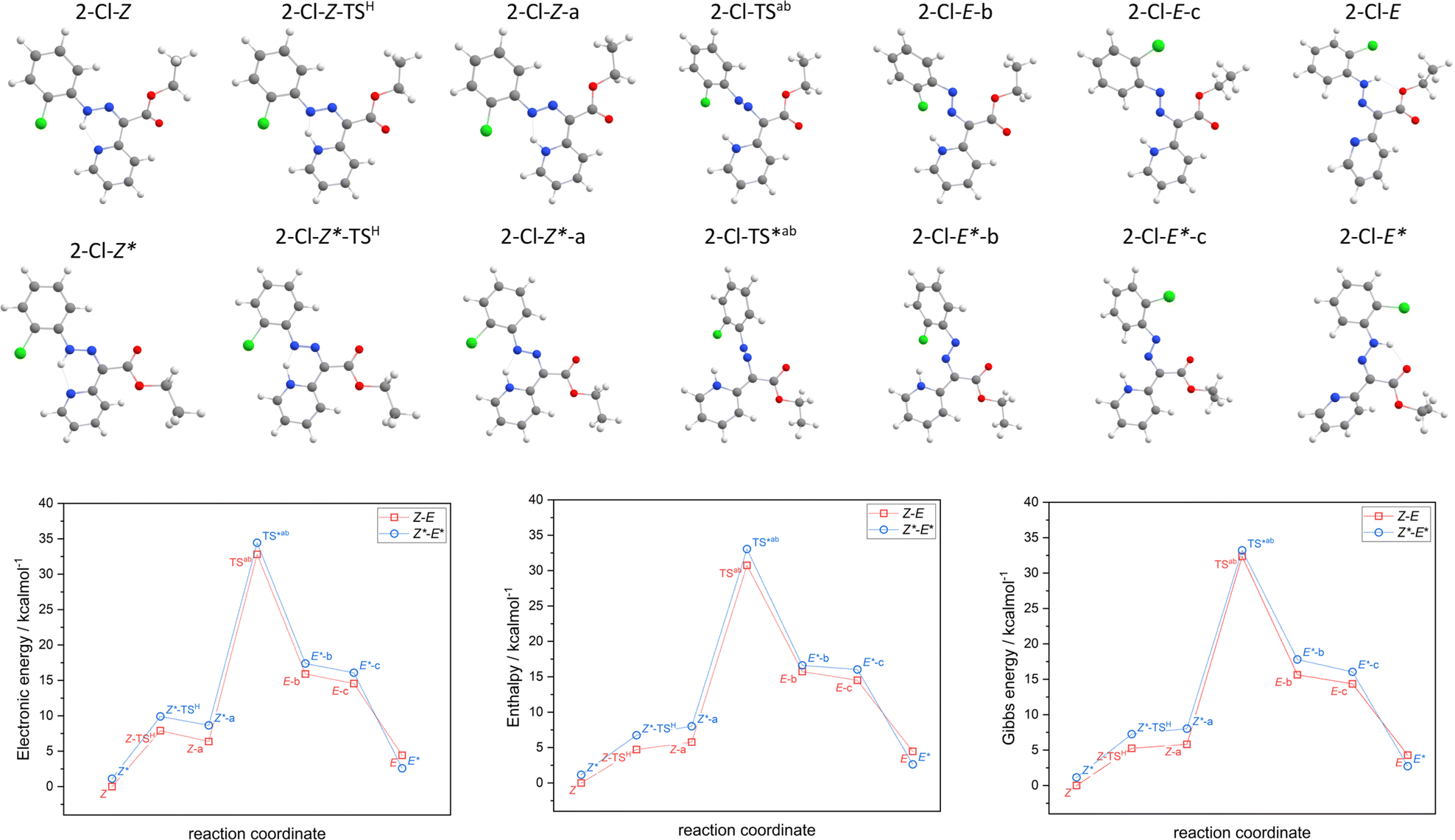 | ||
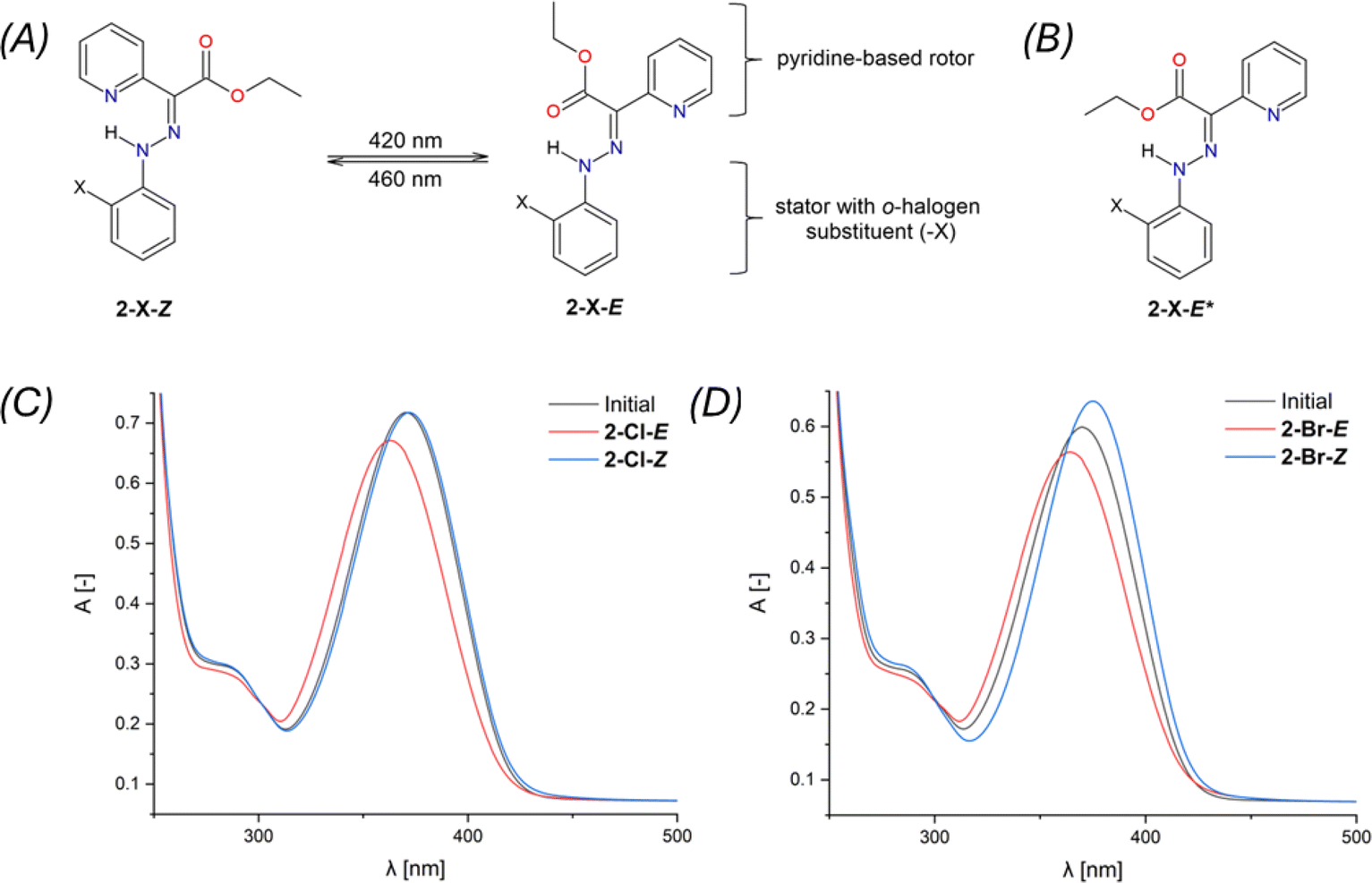
| Fig. 3 The minimal energy pathway for Z,E-isomerization for 2-Cl calculated with CAM-B3LYP+D4/def2-TZVP/C-PCM(CHCl3). Top – the respective molecular geometries. Bottom – relative energies, enthalpies, and Gibbs energies. | ||
The photoswitching efficiency of 2-X hydrazones was tested and demonstrated (see ESI, Fig. S36–S45†). Unfortunately, only partial reversibility was observed, particularly for compounds 2-Cl and 2-Br (Fig. S39 and S43†). Optimizing irradiation conditions or selecting different solvents did not achieve full reversibility for 2-Cl and 2-Br (Fig. S40–S42 and S44†). It is noteworthy that this behavior has been previously reported for hydrazone photoswitches.40 We did not investigate this trend in more detail, as it is not the primary focus of this study.
Isomerization induced by irradiation at wavelengths of 420 and 460 nm, as investigated via 1H NMR, is summarized in Table 2. The N–H signals were chosen to elucidate the isomerization process. Initial equilibrium for Z- and E-isomers was determined in the range of 15.00–15.35 ppm and 12.23–12.48 ppm, respectively. The δN–H(E) values displayed negligible changes after irradiation at 420 nm, except for 2-I-E with Δδ = 0.14 ppm. The signal intensity values for N–H(E) increased for all 2-X-E compounds with the highest ratio observed for 2-Cl-E (55%). Intriguingly, irradiation at 460 nm induced back-isomerization into the primary 2-X-Z and a minor 2-X-E* isomer (Fig. 2B). The back-isomerization of 2-X compounds proved to be efficient, notably yielding 2-I-Z isomer in 100% yield. NMR assignment of diastereomers 2-X-E and 2-X-E* was corroborated by DFT calculations (vide infra). The diastereomer 2-X-Z* was not detected by 1H NMR in this study. Isomerization of 2-F was also monitored by 19F NMR spectroscopy. Isomers 2-F-Z and 2-F-E appeared as singlets at −137.0 and −137.6 ppm, respectively, and remained unchanged after irradiation at 420 nm (Fig. S30†). An expected increase in signal intensity was observed for 2-F-E. Upon isomerization to the initial state, a multiplet was observed at −137.0 ppm for 2-F-Z. This technique does not distinguish between diastereomers 2-F-E and 2-F-E*. Comparingly to structurally similar systems,19 the photoisomerization of pyridine rotor-based hydrazone, such as ethyl-(2-phenylhydrazinylidene)(pyridin-2-yl)acetate, was observed to be considerably inefficient, yielding only an additional 2% of the minor isomer, with the back isomerization even being suppressed. However, when a quinoline stator was incorporated into the hydrazone structure, the photoisomerization became notably more efficient, shifting the E/Z ratio (following the nomenclature used in this work19) from 90/10% to 5/95%. Importantly, the back isomerization was not induced, likely due to the absence of a suitable irradiation wavelength.19 In summary, our investigation confirmed the photoswitchibility of o-halogen hydrazones with pyridine-based rotors, as well as the reversibility of this process.
| 2-F | 2-Cl | 2-Br | 2-I | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Initial | 420 nm | 460 nm | Initial | 420 nm | 460 nm | Initial | 420 nm | 460 nm | Initial | 420 nm | 460 nm | |
| a Formation of the 2-X-E*.b Measured but not detected. n.d. not detected (the measurement was not performed). | ||||||||||||
| δN–H(Z) | 15.00 | 15.00 | 15.00 | 15.31 | 15.31 | 15.31 | 15.35 | 15.35 | 15.34 | 15.34 | 15.34 | 15.36 |
| δN–H(E) | 12.23 | 12.25 | 12.96a | 12.48 | 12.45 | 13.31a | 12.45 | 12.43 | 13.38a | 12.29 | 12.43 | —b |
| δC15 | 151.4 | n.d. | n.d. | 120.1 | n.d. | n.d. | 109.7 | n.d. | n.d. | 83.6 | n.d. | n.d. |
| % Z/E | 91/9 | 53/47 | 93/7 | 92/8 | 44/55 | 91/9 | 93/7 | 51/49 | 90/10 | 91/9 | 62/38 | 100/0 |
| δF(Z) | −137.0 | −137.0 | −137.0 | — | — | — | — | — | — | — | — | — |
| δF(E) | −137.6 | −137.5 | —b | — | — | — | — | — | — | — | — | — |
| λmax | 370 | 363 | 373 | 370 | 363 | 372 | 370 | 364 | 375 | 370 | 365 | 376 |
| ε | 28622 |
26880 |
30543 |
26293 |
24582 |
26292 |
21955 |
20650 |
23289 |
23106 |
22182 |
24229 |
Photostability study
The presence of diastereomer 2-X-E*, determined from photoisomerization studies of 2-X, was supported by DFT calculations. The obtained findings, based on 1H NMR spectra, suggested possible photodegradation upon prolonged exposure to 460 nm irradiation. Consequently, we chose to investigate the photostability of 2-Br as a model system due to its satisfactory yield.Initially, 2-Br (0.02 M) was subjected to 460 nm irradiation for 30 minutes, confirming the observed trend with back-isomerization to 2-Br-Z and the formation of diastereomer 2-Br-E* (Fig. S32†). This was corroborated by the downfield shift of the initial N–H(E) signal from 12.45 to a new signal at 13.38 ppm. However, upon extending the exposure of the sample to 460 nm irradiation for 3 hours, a significant alteration in the proton spectrum was observed, indicating probable hydrazone decomposition (Fig. S46†). Notably, the N–H signal characteristic for the hydrazone was undetectable, while the remaining signals remained unaffected.
MS analysis supported the proton NMR findings, revealing signals for fragments of the rotor part and confirming decomposition of the CN bond (Fig. S47 and S48†). The sample concentration (0.02 M) was deemed inadequate for additional supporting measurements (e.g., 15N, 1H–15N HSQC, and HMBC) aimed at further investigating this process. Consequently, the sample concentration was increased to 0.1 M. However, measurements of this sample following exposure to 460 nm irradiation for 3 hours did not confirm sample degradation (Fig. S49–S51†). This limitation hindered the verification of hydrazone group degradation.
An identical experiment was performed for 2-F (0.02 M) to verify and compare the effect of o-halogen substituents on photostability. Elevated signals corresponding to the initial reactant 1 were observed, suggesting the decomposition of the hydrazone. However, the N–H signals for both isomers remained intact, indicating the robustness of the hydrazone when exposed to 460 nm irradiation for 3 hours. This contrasts with the findings obtained for 2-Br. Prolonged exposure of 2-F to 460 nm irradiation did not reveal any changes in this regard.
Computational studies
The hydrazone switching was also investigated theoretically at DFT and TD-DFT level of theory using ORCA 5.0 software.41 Herein, CAM-B3LYP range-separated hybrid functional was used42 together with the atom-pairwise dispersion correction (D4).43 The def2-TZVP basis set was used for all atoms.44 The calculations were speed-up using def2/J Coulomb fitting basis set34 and RIJCOSX approximation.45 The largest integration grid (DefGrid3) and tightSCF convergence criteria were used in all calculations. Also, the implicit solvation model C-PCM was used during geometry optimization.46,47 The calculated data were visualized with VESTA 3 program.48In order to elucidate the impact of different halogen atoms on the isomerization process, the geometry optimization and energetics calculations were done for the reaction mechanism involving: (i) the proton transfer from azo-group to pyridine, hence tautomerization (2-X-Z → 2-X-Z-a), where also respective transition state was identified (2-X-Z-TSH), (ii) rotation of azo-group (2-X-Z-a → 2-X-E-b) and locating the transition state (2-X-TSab), (iii) followed by the rotation of X-phenyl group (2-X-E-b → 2-X-E-c), (iv) and finished again by the tautomerization (2-X-E-c → 2-X-E). In case of first tautomerization, the intramolecular transfer of proton was considered which enabled to locate its 2-X-Z-TSH, whereas the intermolecular transfer of proton is required for the second tautomerization, thus such transition state was not pursued. Moreover, analogous reaction path was studied for respective diastereomers, 2-X-Z* ↔ 2-X-E*. The results of the calculations are depicted in Fig. 3 for X = –Cl, and in Fig. S52–S54† for the rest of the compounds.
Having thermodynamic data available, the potential of these compounds for Molecular solar thermal (MOST) energy storage materials was evaluated – Fig. 4, Tables S2 and S3.† Therefore, the enthalpy of the back-isomerization process (E → Z) 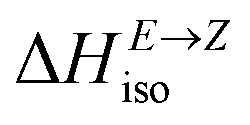 defining energy storage capacity was calculated as
defining energy storage capacity was calculated as 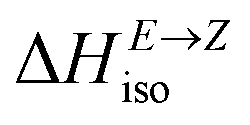 = H(2-X-Z) − H(2-X-E). The values of
= H(2-X-Z) − H(2-X-E). The values of 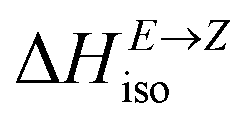 were found in narrow range, 18.27–18.90 kJ mol−1 (4.37–4.52 kcal mol−1) and these values are similar to other systems based on the hydrazones. Next, the activation Gibbs energies of the back-isomerization process ΔG‡ E→Z defining the rate of releasing the stored energy were calculated as ΔG‡ E→Z = G(2-X-TSab) − G(2-X-E). In contrast to
were found in narrow range, 18.27–18.90 kJ mol−1 (4.37–4.52 kcal mol−1) and these values are similar to other systems based on the hydrazones. Next, the activation Gibbs energies of the back-isomerization process ΔG‡ E→Z defining the rate of releasing the stored energy were calculated as ΔG‡ E→Z = G(2-X-TSab) − G(2-X-E). In contrast to 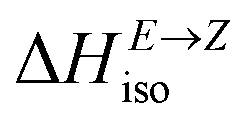 , significant variations in ΔG‡ E→Z values are observed for various halogen substituents –X, ΔG‡ E→Z = 88–138 kJ mol−1 (21–33 kcal mol−1) indicating the potential to tune the activation energy height of photoisomerization by selecting appropriate ortho substituent on the stator. Analogous calculations were performed for back-isomerization of diastereomers (E* → Z*) with similar trends.
, significant variations in ΔG‡ E→Z values are observed for various halogen substituents –X, ΔG‡ E→Z = 88–138 kJ mol−1 (21–33 kcal mol−1) indicating the potential to tune the activation energy height of photoisomerization by selecting appropriate ortho substituent on the stator. Analogous calculations were performed for back-isomerization of diastereomers (E* → Z*) with similar trends.
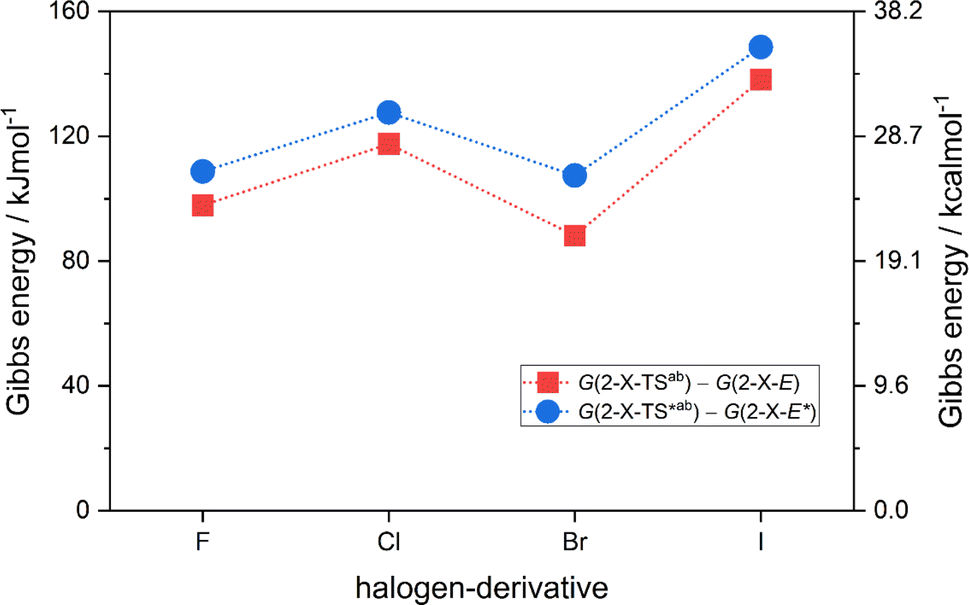 | ||
| Fig. 4 The variation of activation Gibbs energies of the back-isomerization process ΔG‡ E→Z and ΔG‡ E*→Z* for 2-X compounds based on CAM-B3LYP/pcSseg-2/C-PCM(CHCl3) calculations. | ||
Moreover, the 1H NMR shifts were calculated for all reactants, products and intermediates using the already optimized molecular geometries, but the basis set more suitable for such calculations, namely pcSseg-2.49 In case of iodine atoms, such basis was not available, therefore def2-TZVP together with respective ECP was used.50 The outcome of these calculations are presented in Tables S4–S7.† The variation of 1H NMR shifts along Z–E (Z*–E*) pathway for the hydrogen atom bonded either to azo-group or to pyridine moiety is highlighted in Fig. 5. The most acidic signal is found for 2-X-Z-a (2-X-Z*-a) compounds, whereas the lowest values are found for 2-X-E compounds. Interestingly, the 1H NMR shifts are significantly different for diastereomers 2-X-E vs. 2-X-E*, in contrast to the difference found for 2-X-Z vs. 2-X-Z* diastereomers.
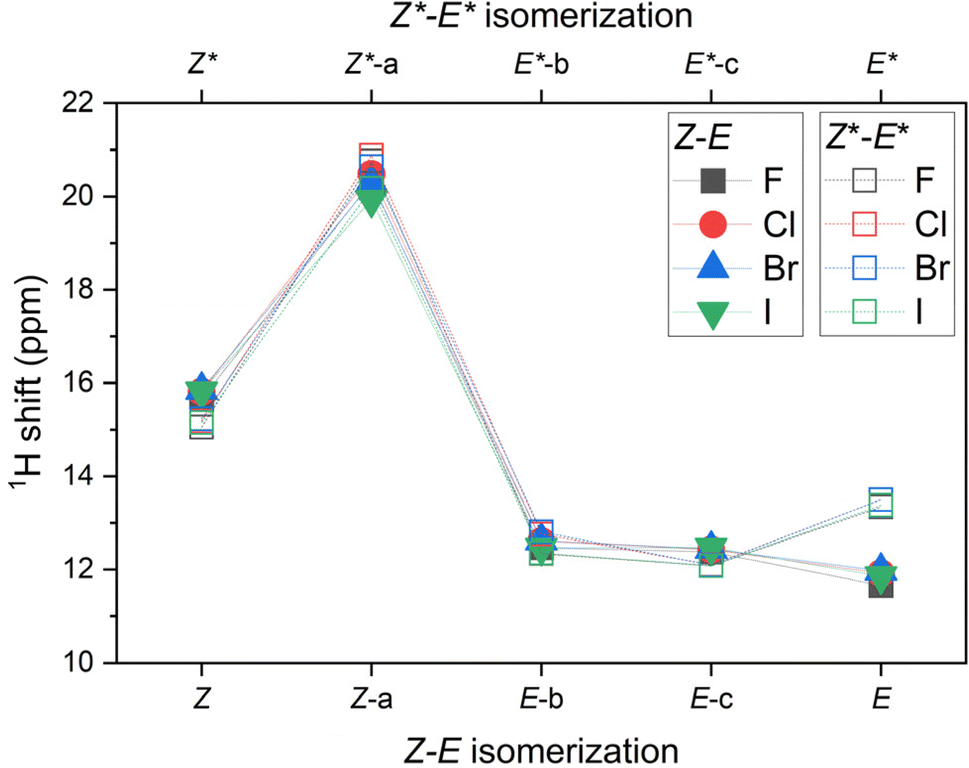 | ||
| Fig. 5 The variation of 1H NMR shifts along Z–E (Z*–E*) pathway for the hydrogen atom bonded either to azo-group or to pyridine moiety of 2-X compounds based on CAM-B3LYP/pcSseg-2/C-PCM(CHCl3) calculations. | ||
Furthermore, TD-DFT calculations were performed to compute the excited states and predict the absorption spectra for the most stable species according to Gibbs energy, which are 2-X-Z and 2-X-E*. The intensities calculated from the TD-DFT oscillator strengths were transformed into the molar absorption coefficients as implemented in software Multiwfn.36 The resulted plots are showed in Fig. 6. Generally, the main transition located at ca. 31000 cm−1 for 2-X-Z and at ca. 32000 cm−1 for 2-X-E* show little dependence on halogen substituent –X. Anyway, these calculations show that these dominant transitions are shifted to higher energies for 2-X-E* which agrees with the experiments.
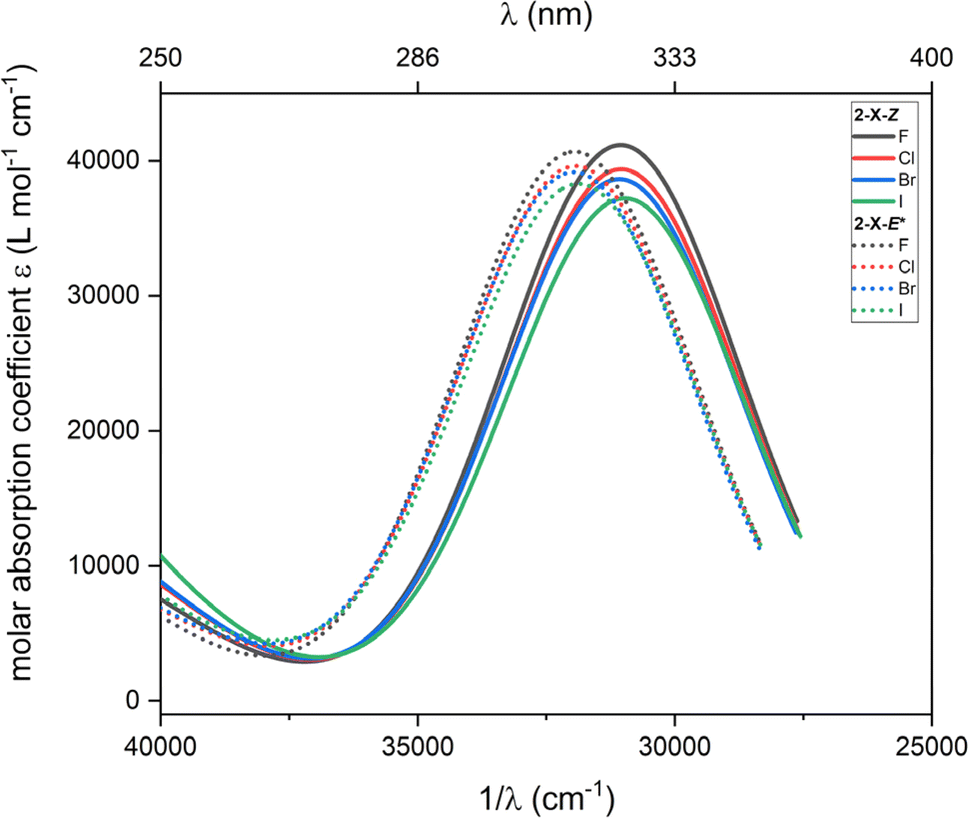 | ||
| Fig. 6 The TD-DFT UV-Vis spectra of 2-X-Z and 2-X-E* species calculated with CAM-B3LYP/def2-TZVP/C-PCM(CHCl3). | ||
The origin of these electron transitions was inspected for 2-Cl-Z and 2-Cl-E* with the method called interfragment charge transfer during electron excitation (IFCT).51 Herein, the molecules were divided into four regions: X-phenyl group (Xph), pyridine group (py), N–N–C atoms (azo) and the acetyl-ester group (ac) as depicted in Fig. S55.† Now, the UV-Vis spectra can be decomposed into the individual contribution of the excitation within the defined groups – intra-group states (IaG), and into the charge transfer between the groups – inter-groups states (IeG) as showed in Fig. S55.† Thus, the most significant absorption band at ∼31000 cm−1 for 2-Cl-Z is dominated by IeG(Xph → azo), IeG(Xph → py), IaG(azo) and IeG(azo → py) whereas the situation for 2-Cl-E* is slightly different, the main contributions are IeG(Xph → azo), IaG(azo), IeG(Xph → ac), IeG(azo → ac) – Fig. S55.†
The optimized structures in the Z- and E-configurations (vide supra) were subjected QT-AIM calculations to investigate the nature and strength of intramolecular interactions of hydrazone switches dissolved in CHCl3. Additionally, since the crystal structure of 2-F is unknown, understanding the calculated molecular structure and its properties offers valuable insights.
The optimized structures of the hydrazone switches in the Z-conformation closely resemble the structures of 2-Cl, 2-Br, and 2-I as determined by X-ray diffraction. However, a notable difference lies in the lower planarity of the optimized molecular structures. This was quantified by measuring the angle between the least-square planes of the phenyl and aromatic ring. In the experimental structures, this angle ranged from 3.9° in 2-Cl to 6.3° in 2-I, whereas in the optimized structures, the angle exhibited more uniform values, ranging from 13.6° in 2-F to 14.6° in 2-Cl. We examined the topology of the calculated electron density in the optimized structures to analyse intramolecular hydrogen bonding. Our investigation revealed the presence of bond paths and (3, −1) critical points between the hydrazone amino group and the pyridine nitrogen and halogen atoms in the molecules of 2-Cl, 2-Br, and 2-I (Fig. S56†). The interaction energies, Eint, for N–H⋯N contacts were found to be lower (Eint = 10.2–10.3 kcal mol−1) and with smaller covalent contributions (|V(r)|/G(r) = 1.03, Table S8†) compared to those calculated for the experimental crystal structures (Table 1). The N–H⋯X interactions exhibit similar energies to those observed in the experimental crystal structures, with the lowest Eint observed for 2-I (3.1 kcal mol−1, Table S8†) In the case of 2-F, the calculated electron density of the optimized structure lacks bond path and critical point for the N–H⋯F contact (Fig. S56†). However, Eint for the N–H⋯N hydrogen bond is very similar to other members of the series (10.1 kcal mol−1, Table S8†).
The photo switched molecules are in the E-configuration and based on the donor⋯acceptor distances, it could be expected that they exhibit two intramolecular hydrogen bonds: one between the hydrazone amine group and carboxylic oxygen atom and the second between the hydrazone amine group and halogen atom. Topological analysis of the calculated electron density revealed that, similarly to molecules in the Z-configuration, the bond paths and (3, −1) critical points between N–H group and halogen atom are observed only in the case of the 2-Cl, 2-Br, and 2-I molecules (Fig. S57†). Their Eint values are similar to those observed for Z-configuration, ranging from the smallest in 2-I (3.2 kcal mol−1) to 3.9 kcal mol−1 for 2-Cl (Table S9†). The N–H⋯O are weaker than N–H⋯N hydrogen bonds, with EInt adopting uniform values ranging between 9.2 and 9.4 kcal mol−1 (Table S9†).
To determine the nature of the interaction between the fluorine atoms and the hydrazone group, despite the absence of a (3, −1) critical point, we can evaluate its characteristics using Non-Covalent Interactions (NCI) index. This method utilizes the analysis of ρ(r) using the reduced density gradient function (s) to distinguish between attractive and repulsive non-covalent interactions.52 For the 2-Cl, 2-Br, and 2-I compounds, the sign(λ2)ρ values indicates transition from a weak van der Waals type of repulsive interaction observed for smaller halogen atoms (sign(λ2)ρ > 0) to weakly attractive for bigger halogen atoms (sign(λ2)ρ < 0, Table S10†). In the absence of a critical point in 2-F, we are left to visually compare the calculated gradient isosurfaces (s = 0.42 au, Fig. S58 and S59†). The interaction between the fluorine atom and the hydrazone group in both conformations is primarily repulsive, indicating that this interaction does not contribute to the stability of either conformation. Furthermore, when we consider the nature of the N–H⋯X interactions in the Z-configuration for entire series (2-F, 2-Cl, 2-Br dominantly repulsive, vs. attractive for 2-I), we can speculate that these differences among the members of the series may contribute to a non-monotonous dependence of Gibbs energies of tautomerization, with the largest value predicted for 2-I.
Conclusions
A series of novel photoswitchable o-halogen-substituted hydrazone switches (2-F, 2-Cl, 2-Br, and 2-I) were investigated. We confirmed the photoswitching capability of all studied compounds upon irradiation in the UV-Vis range using 1H NMR and UV-Vis spectroscopy. We observed that 2-X-E isomers were obtained in yields of 47%, 55%, 49%, and 38% after irradiation at 420 nm. The photoswitching experiments provided compelling evidence of their reversible isomerization to 2-X-Z when irradiated at 460 nm, with 100% back isomerisation of 2-I to 2-I-Z. Furthermore, we discovered that back-isomerization produced a new diastereomer, 2-X-E* (for X = –F, –Cl, and –Br), which was supported by DFT calculations, and we also demonstrated the photofatigue resistance of 2-X. Our findings indicate that o-halogen substituents in hydrazones slightly alter photoswitchability. For 2-F, 2-Cl, 2-Br, the Z → E conversion rate is very similar, ranging from 47 to 55%; however, for 2-I, the conversion was suppressed to below 38%. Conversely, the E → Z conversion was complete only for 2-I. We investigated the structure of the studied hydrazone switches in both the solid state and in CHCl3 solution using theoretical methods that combine DFT and QT-AIM calculations. We revealed that the N–H⋯N hydrogen bonding in Z-conformations is stronger than the N–H⋯O hydrogen bonding in E-conformations. The N–H⋯X hydrogen bonding is significantly weaker and adopts similar values (3.1–4.1 kcal mol−1) for both conformations and phases. A closer examination of the nature of the N–H⋯X interactions using the NCI method revealed that the weakly attractive interactions for both conformations in solution are found only in 2-I. These results could be correlated with non-monotonous dependence of Gibbs energies of tautomerization as calculated by DFT, with the largest value predicted for 2-I. The DFT calculations also suggest that these derivatives are promising materials for molecular solar thermal energy storage. They demonstrate a significant impact of the X-substituents on the kinetics of the isomerization process, particularly on the height of the photoisomerization activation energy barrier, which exhibits values either comparable to or larger than those observed for structurally strained macrocyclic hydrazones.53Data availability
The data supporting this article have been included in the ESI.† Crystallographic data for 2-Br and 2-I has been deposited at the CCDC with the following deposition numbers: 2335394, 2335397.Author contributions
All authors discussed the results and contributed to the final manuscript. Investigation, visualization, conceptualization – L. K., R. H., I. N. Methodology – R. H., I. N. Formal analysis – L. K., P. J., R. H., I. N. Writing – original draft, writing – review and editing – L. K., R. H., I. N. Supervision – I. N. Funding acquisition – L. K., P. N.Conflicts of interest
There are no conflicts to declare.Note added after first publication
This article replaces the version published on 01 Jul 2024, which contained errors in the footnotes of Table 2.Acknowledgements
L. K. acknowledges financial support from an internal grant from Brno University of Technology CEITEC VUT-J-23-8383. L. K., P. J. and P. N. acknowledge financial support from the Grant Agency of the Czech Republic Grant No. 21-20716X. We acknowledge Josef Dadok National NMR Centre of CIISB, Instruct-CZ Centre, supported by MEYS CR (LMs2023042) and European Regional Development Fund-Project “UP CIISB” (No. CZ.02.1.01/0.0/0.0/18_046/0015974). We acknowledge CzechNanoLab Research Infrastructure supported by MEYS CR (LM2023051). I. N. acknowledges financial support from the Grant Agency of the Czech Republic Grant No. 22-23760S. R. H. and I. N. acknowledge the financial support from the institutional sources of the Department of Inorganic Chemistry, Palacký University Olomouc, Czech Republic. We would also like to express our gratitude to Doc. Ing. Ivan Šalitroš, DrSc., for generously providing us with access to the irradiation chamber.References
- X. Su and I. Aprahamian, Chem. Soc. Rev., 2014, 43, 1963 RSC.
- B. Shao and I. Aprahamian, Chem, 2020, 6, 2162–2173 CAS.
- X. Chen, Z. Li, Y. Xiang and A. Tong, Tetrahedron Lett., 2008, 49, 4697–4700 CrossRef CAS.
- H. Li, J. Fan, F. Song, H. Zhu, J. Du, S. Sun and X. Peng, Chem.–Eur. J., 2010, 16, 12349–12356 CrossRef CAS.
- Y. Sun, Y. Liu and W. Guo, Sens. Actuators, B, 2009, 143, 171–176 CrossRef.
- Q. Li, Y. Guo, J. Xu and S. Shao, J. Photochem. Photobiol., B, 2011, 103, 140–144 CrossRef CAS.
- S. Rollas and S. Küçükgüzel, Molecules, 2007, 12, 1910–1939 CrossRef CAS.
- A. S. Elgubbi, E. A. E. El-Helw, A. Y. A. Alzahrani and S. K. Ramadan, RSC Adv., 2024, 14, 5926–5940 RSC.
- M. de Gracia Retamosa, E. Matador, D. Monge, J. M. Lassaletta and R. Fernández, Chem.–Eur. J., 2016, 22, 13430–13445 CrossRef.
- R. Lygaitis, V. Getautis and J. V. Grazulevicius, Chem. Soc. Rev., 2008, 37, 770 RSC.
- D. N. Bunck and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 14952–14955 CrossRef CAS PubMed.
- J.-M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 2836–2850 CrossRef CAS PubMed.
- H. Qian and I. Aprahamian, Chem. Commun., 2015, 51, 11158–11161 RSC.
- X. Guo, B. Shao, S. Zhou, I. Aprahamian and Z. Chen, Chem. Sci., 2020, 11, 3016–3021 RSC.
- D. Janasik, K. Jasiński, W. P. Węglarz, I. Nemec, P. Jewula and T. Krawczyk, Anal. Chem., 2022, 94, 3427–3431 CrossRef CAS.
- A. Ryabchun, Q. Li, F. Lancia, I. Aprahamian and N. Katsonis, J. Am. Chem. Soc., 2019, 141, 1196–1200 CrossRef CAS.
- Q. Qiu, S. Yang, M. A. Gerkman, H. Fu, I. Aprahamian and G. G. D. Han, J. Am. Chem. Soc., 2022, 144, 12627–12631 CrossRef CAS.
- S. M. Landge, E. Tkatchouk, D. Benítez, D. A. Lanfranchi, M. Elhabiri, W. A. Goddard and I. Aprahamian, J. Am. Chem. Soc., 2011, 133, 9812–9823 CrossRef CAS.
- H. Qian, S. Pramanik and I. Aprahamian, J. Am. Chem. Soc., 2017, 139, 9140–9143 CrossRef CAS.
- X. Su, M. Lõkov, A. Kütt, I. Leito and I. Aprahamian, Chem. Commun., 2012, 48, 10490 RSC.
- R. Pichon, J. Le Saint and P. Courtot, Tetrahedron, 1981, 37, 1517–1524 CrossRef CAS.
- Rigaku Oxford Diffraction, CrysAlisPro 1.171.40.82a, 2020 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 59–75 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- D. Jayatilaka and B. Dittrich, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 383–393 CrossRef CAS.
- S. C. Capelli, H.-B. Bürgi, B. Dittrich, S. Grabowsky and D. Jayatilaka, IUCrJ, 2014, 1, 361–379 CrossRef CAS PubMed.
- F. Kleemiss, O. V. Dolomanov, M. Bodensteiner, N. Peyerimhoff, L. Midgley, L. J. Bourhis, A. Genoni, L. A. Malaspina, D. Jayatilaka, J. L. Spencer, F. White, B. Grundkötter-Stock, S. Steinhauer, D. Lentz, H. Puschmann and S. Grabowsky, Chem. Sci., 2021, 12, 1675–1692 RSC.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS.
- L. Kotásková and I. Nemec, CCDC 2335567: Experimental Crystal Structure Determination, 2024, DOI:10.5517/ccdc.csd.cc2jdbw5.
- L. Kotásková and I. Nemec, CCDC 2334065: Experimental Crystal Structure Determination, 2024, DOI:10.5517/ccdc.csd.cc2jbsf3.
- R. F. W. Bader, Atoms in Molecules, Oxford University Press, 1994 Search PubMed.
- C. van Wüllen, J. Chem. Phys., 1998, 109, 392–399 CrossRef.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Mol. Graphics Modell., 2012, 38, 314–323 CrossRef CAS PubMed.
- T. A. Keith, AIMAll (Version 19.10.12), TK Gristmill Software, Overland Park KS, USA, 2019, https://www.aim.tkgristmill.com Search PubMed.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
- B. Shao and I. Aprahamian, ChemPhotoChem, 2019, 3, 361–364 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, 1–15 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2019, 150, 154122 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- M. Garcia-Ratés and F. Neese, J. Comput. Chem., 2020, 41, 922–939 CrossRef.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- F. Jensen, J. Chem. Theory Comput., 2015, 11, 132–138 CrossRef CAS PubMed.
- K. A. Peterson, D. Figgen, E. Goll, H. Stoll and M. Dolg, J. Chem. Phys., 2003, 119, 11113–11123 CrossRef CAS.
- Z. Liu, X. Wang, T. Lu, A. Yuan and X. Yan, Carbon, 2022, 187, 78–85 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS.
- Q. Li, H. Qian, B. Shao, R. P. Hughes and I. Aprahamian, J. Am. Chem. Soc., 2018, 140, 11829–11835 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 19F, 13C NMR, UV-Vis, and MS spectra, results of DFT calculations. CCDC 2335394–2335397. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra02909g |
| This journal is © The Royal Society of Chemistry 2024 |