
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium nanoparticles for the synthesis of phenanthridinones and benzo[c]chromenes via C–H activation reaction†
Eva D. Díaz-Vázquezab,
Micaela A. Cuellarab,
Micaela D. Heredia ab,
Silvia M. Baroloab,
Aday González-Bakkerc,
José M. Padrónc,
María E. Budén*ab,
Sandra E. Martín*ab and
Paula M. Uberman*ab
ab,
Silvia M. Baroloab,
Aday González-Bakkerc,
José M. Padrónc,
María E. Budén*ab,
Sandra E. Martín*ab and
Paula M. Uberman*ab
aDepartamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Haya de La Torre y Medina Allende, Ciudad Universitaria, X5000HUA, Córdoba, Argentina. E-mail: paula.uberman@unc.edu.ar; eugebuden@unc.edu.ar; sandra.martin@unc.edu.ar
bInstituto de Investigaciones en Fisicoquímica de Córdoba-INFIQC-CONICET-Universidad Nacional de Córdoba, Haya de La Torre y Medina Allende, Ciudad Universitaria, X5000HUA, Córdoba, Argentina
cBioLab, Instituto Universitario de Bio-Orgánica “Antonio González” (IUBO-AG), Universidad de La Laguna, C/Astrofísico Francisco Sánchez 2, E-38206 La Laguna, Spain
First published on 11th June 2024
Abstract
In the present work, derivatives of phenanthridine-6(5H)-ones and benzo[c]chromenes were efficiently prepared through an intramolecular C–H bond functionalization reaction catalyzed by photochemically synthesized Pd-PVP nanoparticles. The heterocycles were obtained via intramolecular arylation of the corresponding N-methyl-N-aryl-2-halobenzamide or aryl-(2-halo)benzyl ethers using K2CO3 as base in a mixture of H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMA as solvent without additives or ligands. High yields of the heterocyclic compounds were achieved (up to 95%) using a moderately low catalyst loading (1–5 mol%) under an air atmosphere at 100 °C. The reaction exhibited very good tolerance to diverse functional groups (OMe, Me, tBu, Ph, OCF3, CF3, F, Cl, –CN, Naph), and both bromine and iodine substrates showed great reactivity. Finally, the in vitro antiproliferative activity of phenanthridine-6(5H)-ones and benzo[c]chromenes was evaluated against six human solid tumor cell lines. The more active compounds exhibit activity in the low micromolar range. 1-Isopropyl-4-methyl-6H-benzo[c]chromene was identified as the best compound with promising values of activity (GI50 range 3.9–8.6 μM). Thus, the benzochromene core was highlighted as a novel organic building block to prepare potential antitumor agents.
:DMA as solvent without additives or ligands. High yields of the heterocyclic compounds were achieved (up to 95%) using a moderately low catalyst loading (1–5 mol%) under an air atmosphere at 100 °C. The reaction exhibited very good tolerance to diverse functional groups (OMe, Me, tBu, Ph, OCF3, CF3, F, Cl, –CN, Naph), and both bromine and iodine substrates showed great reactivity. Finally, the in vitro antiproliferative activity of phenanthridine-6(5H)-ones and benzo[c]chromenes was evaluated against six human solid tumor cell lines. The more active compounds exhibit activity in the low micromolar range. 1-Isopropyl-4-methyl-6H-benzo[c]chromene was identified as the best compound with promising values of activity (GI50 range 3.9–8.6 μM). Thus, the benzochromene core was highlighted as a novel organic building block to prepare potential antitumor agents.
Introduction
In the last decades, Pd-catalyzed C–H bond activation reactions have emerged as a sustainable tool to achieve the functionalization of complex molecules in organic synthesis.1–5 These Pd-catalyzed C–H functionalization methodologies efficiently enabled the development of total synthesis of natural compounds and biologically interesting molecules with elevated atomic and step economy.6–11 Particularly, Pd-catalyzed C–H bond activation reactions were intensively studied in the synthesis and functionalization of heterocyclic compounds.In the extensive world of heterocycles, phenanthridine-6(5H)-ones and benzo[c]chromenes derivatives are important structural scaffolds found in many natural alkaloids and therapeutically active compounds. On one hand, phenanthridine-6(5H)-ones derivatives have a wide range of pharmacological properties including anticancer,12,13 antileukemic,14 antiviral and anti-HIV,15 antibacterial and antifungal,16 antiplasmodial,17 anti-inflammatory,18 among others. For example, oxyavacine alkaloid (I, Fig. 1A) exhibited analgesic and anti-inflammatory activity and it is used for the treatment of ophthalmic disorders.19,20 On the other hand, benzo[c]chromenes are also present in numerous natural products being important structures in modern pharmaceutical chemistry.21 In this way, cannabinol (II, Fig. 1B) has antimicrobial activity;22 pulchrol and pulchral have antiparasitic activity;23 and others benzochromenes show a variety a biological activity.24,25
 | ||
| Fig. 1 Biologically active examples of phenanthridinone (A) and benzochromene (B) and common synthetic strategies of Pd-catalyzed C–H bond activation reactions (C and D). | ||
Phenanthridine-6(5H)-ones can be successfully synthesized by Pd-catalyzed C–H bond activation reactions in a direct arylation process.26,27 In these reactions, the catalysts are usually homogeneous Pd-complexes with ligands such as phosphines,17,28,29 N-heterocyclic carbene,30 pincer-type ligands,31 or carboxylic acid derivatives,32,33 among others.34 The most commonly used synthetic strategy to obtain phenanthridine-6(5H)-ones scaffolds by Pd-catalyzed C–H bond activation includes the cyclization reaction of prefabricated N-substituted o-halobenzanilides (path i, Fig. 1C).17,28–31,33,35–38 Other strategies involve consecutive C–C and C–N bond formations, as in the carbonylation reaction of 2-aminobiphenyls (path ii, Fig. 1C);39 in the Pd-catalyzed multicomponent process with N-alkyl-2-iodoaniline, benzyne precursor III and CO insertion reaction (path iii, Fig. 1C);40 or in the combination of N-methoxybenzamides and aryl derivatives (path iv, Fig. 1C),41,42 along with others.19,26,27
Benzo[c]chromenes were also obtained by Pd-catalyzed C–H bond activation reactions with Pd organometallic complexes.28–30,33,34,43–45 Synthetic strategies generally involve the C–C bond formation using 2-halobenzylaryl ethers as substrates (path v, Fig. 1D). Catellani reaction, employing norbornene as the transient mediator, was also used in the synthesis of benzochromenes.11 Another strategy consists in the carboxyl-directed C–H activation/C–O cyclization to construct biaryl lactones, followed by a reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond with NaBH4 (path vi, Fig. 1D).46
O bond with NaBH4 (path vi, Fig. 1D).46
Although the above-mentioned strategies allowed to successfully synthesize phenanthridine-6(5H)-ones and benzo[c]chromenes, it is essential to develop more economical, simpler and selective synthetic methods to obtain compounds with applications in medicinal chemistry.7 As Pd-catalyzed C–H bond activation reactions proved to be versatile, reliable and efficient methods to obtain heterocyclic compounds, it is necessary to continue developing new catalytic methods avoiding the use of expensive or sensitive ligands or additives, reducing reaction temperatures and minimizing the cost associated to the use of large amounts of Pd in these reactions (>5 mol%). In that sense, the use of heterogeneous47 and colloidal catalysts such as metal nanoparticles (NPs)48,49 are useful tools to achieve eco-friendly reaction conditions or improve catalysts recovery. Due to unique properties of Pd NPs such as high reactivity, selectivity, stability and recyclability, the use of Pd nanocatalyst has been extensively investigated in organic synthesis.50 For instance, Pd NPs were recently explored in C–H bond activation reactions for heterocyclic compounds functionalization,51–61 or heterocycles synthesis via cyclization reactions.62–65
The synthesis of phenanthridine-6(5H)-ones and benzo[c]chromenes by direct arylation reaction catalyzed by Pd NPs was recently reported. Saka et al. described the synthesis of phenanthridine-6(5H)-ones employing binaphthyl-supported Pd NPs (Pd-BNP) in a multiple C–H activation reaction (by consecutive C–C and C–N bond formations) with N-methoxybenzamides and aryl iodides (path iv; Fig. 1C).66 The reactions conditions involved the use of 1.5 mol% of Pd-BNP and Ag2O as oxidant in acetic acid at 110 °C for 24–36 h, obtaining phenanthridine-6(5H)-ones in 40–80% of yield. Regarding benzo[c]chromenes, Cano et al.67 used 2-bromobenzyl phenyl ether as substrate (path v, Fig. 1D) and Pd impregnated on magnetite as catalyst (10 mol%) to achieve benzo[c]chromene in 85% of yield.67 DMA was employed as solvent and KOAc as base at 140 °C for 48 h.
Considering all above mentioned, it is evident that the design of Pd-nanocatalyzed systems efficient and selective is still necessary. These nanocatalytic systems should allow lower temperatures without the use of additives or ligands in the synthesis of heterocyclic compounds.
Recently, we developed a simple and straightforward photochemical method to obtain Pd-PVP NPs with outstanding catalytic activity in Suzuki–Miyaura cross-coupling reaction under mild reaction conditions.68 The synthesis of Pd-PVP NPs was carried out under visible light irradiation (3 W blue LED), in aqueous solution with sodium citrate and PVP for one hour at room temperature, obtaining Pd-PVP NPs with a mean diameter of (2.8 ± 0.8) nm by TEM analysis. The colloidal dispersion of Pd-PVP NPs has been demonstrate to be stable and can be stored under air for several months. This photochemical method has proven to being a rapid, and cost-effective methodology for generating small Pd-PVP NPs in aqueous media.
Our ongoing research on the development of Pd-nanocatalysts for organic transformations,69–73 prompted us to explore the synthesis of phenanthridine-6(5H)-ones and benzo[c]chromenes using Pd-PVP NPs. Thus, herein, the reactivity of these NPs in the Pd-catalyzed C–H bond activation for the intramolecular arylation reaction of benzamides and aryl benzyl ethers is reported. The reaction proceeds under mild reaction conditions and does not require the use of expensive or sensitive additives, ligands or oxidants. Furthermore, aqueous reaction mixtures are used and desired heterocycles obtained with good to excellent yields and high selectivity.
Results and discussion
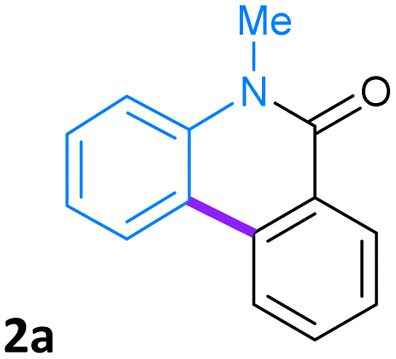
First, the Pd-PVP NPs catalyzed cyclization reaction of N-methyl-N-phenyl-2-iodobenzamide (1a) to obtain the phenanthridin-6(5H)-one ring 2a was evaluated (eqn (1), Table 1).
|
||||
|---|---|---|---|---|
| Entry | Substrate | Catalyst loading (mol%) | Solvent (1:1) |
Yield 2ab (%) |
| a Reactions were carried out under air atmosphere using 1a-1b (1 equiv., 0.2 mmol), K2CO3 (3 equiv.), and solvent (Vf = 4 mL), heating in an oil bath at 100 °C for 24 h.b Yields were quantified by 1H NMR using 4-nitroacetophenone as internal standard.c The substrate was recuperated in ca. 100% yield.d Reaction time = 12 h.e AcOK as base.f The reaction was carried out at 65 °C.g Reduced substrate 4 was observed in 18% yield. | ||||
| 1 | 1a (X = I) | 2 | H2O:DMA |
90 |
| 2c | — | H2O:DMA |
— | |
| 3 | 0.5 | H2O:DMA |
58 | |
| 4 | 1 | H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :DMA :DMA |
95 | |
| 5d | 1 | H2O:DMA |
61 | |
| 6e | 1 | H2O:DMA |
<5 | |
| 7f | 2 | H2O:EtOH |
52g | |
| 8f | 2 | H2O:THF |
<5 | |
| 9 | 1b (X = Br) | 1 | H2O:DMA |
30 |
| 10 | 5 | H2O:DMA |
60 | |
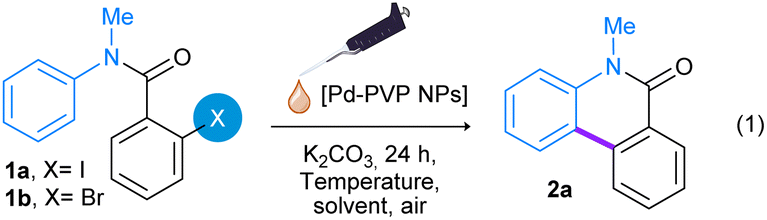
The initial reaction conditions involved the use of 3 equivalents of K2CO3 as base, 2 mL of aqueous dispersion of Pd-PVP NPs (2 mol%) and 2 mL dimethylacetamide (DMA), achieving a mixture of H2O:DMA (1:1) as solvent. The reaction was carried out under air atmosphere at 100 °C for 24 h (Table 1, entry 1). To our delight, under these reaction conditions product 2a was obtained in 90% yield. Furthermore, when the reaction was carried out without Pd nanocatalyst, substrate 1a was recovered in almost 100% yield (entry 2). As Pd NPs efficiently produce phenanthridinone 2a in the absence of any additional additive or ligand, the reaction conditions were optimized starting with the catalyst loading. Reactions were carried out with 0.5 mol% and 1 mol% of Pd-PVP NPs, giving 58% and 95% of product 2a respectively (entries 3 and 4).
Secondly, employing 1 mol% of Pd-PVP NPs, the reaction time was studied. After 12 h of reaction, Pd-PVP NPs produced 61% yield of 2a (entry 5). The base effect was also evaluated. When AcOK was used in replacement of K2CO3 as base, 2a was afforded in very low yield, less than 5% (entries 6 vs. 4). Next, the nature of solvent was evaluated (entries 7–8). The use of co-solvents such as EtOH or THF produce lower yields of product 2a (entries 7 and 8 vs. entry 4).
Thus, the optimal reaction conditions involve 3 equivalents of K2CO3 as base, 1 mol% of Pd-PVP NPs in a mixture of H2O:DMA (1:1), at 100 °C for 24 h. It is worth noting that this procedure yields a clean reaction mixture that is very simple to extract and purify, where only product 2a was observed with a small amount of substrate 1a. Furthermore, the reduction of the substrate, commonly observed in Pd-catalyzed C–H bond activation reactions, was not detected.36 Additionally, the Pd-catalyzed direct arylation was performed in an air atmosphere, representing an extra practical advantage.
Under optimal reaction conditions (Table 1, entry 4), substrate N-methyl-N-phenyl-2-bromobenzamide (1b) was also evaluated. However, product 2a was only obtained in 30% yield (entry 9) and when the catalyst loading was increased to 5 mol%, product 2a yielded 60% (entry 10). These lower yields could be explained considering an oxidative addition step of Pd on aryl halide substrates, and taking into account the lower reactivity of bromine derivatives compared to iodinated ones.
Next, the effect of N-protection on the amide group in the Pd-catalyzed C–H functionalization reaction was studied employing N-phenyl-2-iodobenzamide (1c) and N-(2-iodophenyl)-benzamide (1d) as substrates (eqn (2) and (3)). Under the optimized reaction conditions, substrate N-phenyl-2-iodobenzamide (1c) produced 8% of dehalogenated N-phenylbenzamide 4 (detected by GC-MS) and almost 85% of unreacted substrate was recovered (eqn (2)). In this case, the presence of unprotected N–H bond probably allows the N–Pd coordination, which inhibits the activation reaction of the C–H bond.32,34,36
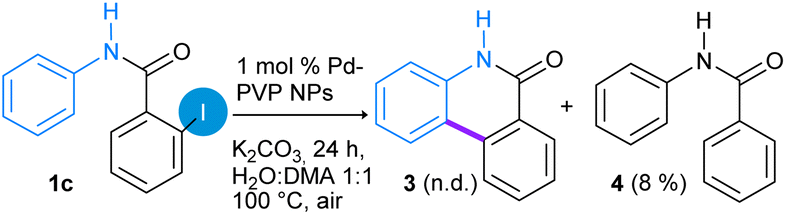 | (2) |
 | (3) |
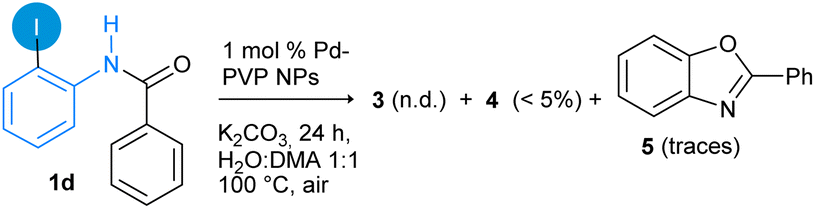
Likewise, substrate N-(2-iodophenyl)-benzamide (1d) produces small amounts of N-phenylbenzamide 4 and 2-phenylbenzo[d]oxazole (5), identified by GC-MS analysis,36 and substrate 1d was recovered in 87% of yield (eqn (3)). Benzoxazole 5 is possibly derived from Pd-catalyzed intramolecular C–O bond formation, by the reaction of amide oxygen and ortho-iodine fragment, as frequently was observed in Cu-catalyzed reactions.74,75 These results indicate that the N–H protection in the benzamide group is crucial to achieve the intramolecular C–H bond activation catalyzed by Pd-PVP NPs, as it was previously reported for other related catalytic systems.38
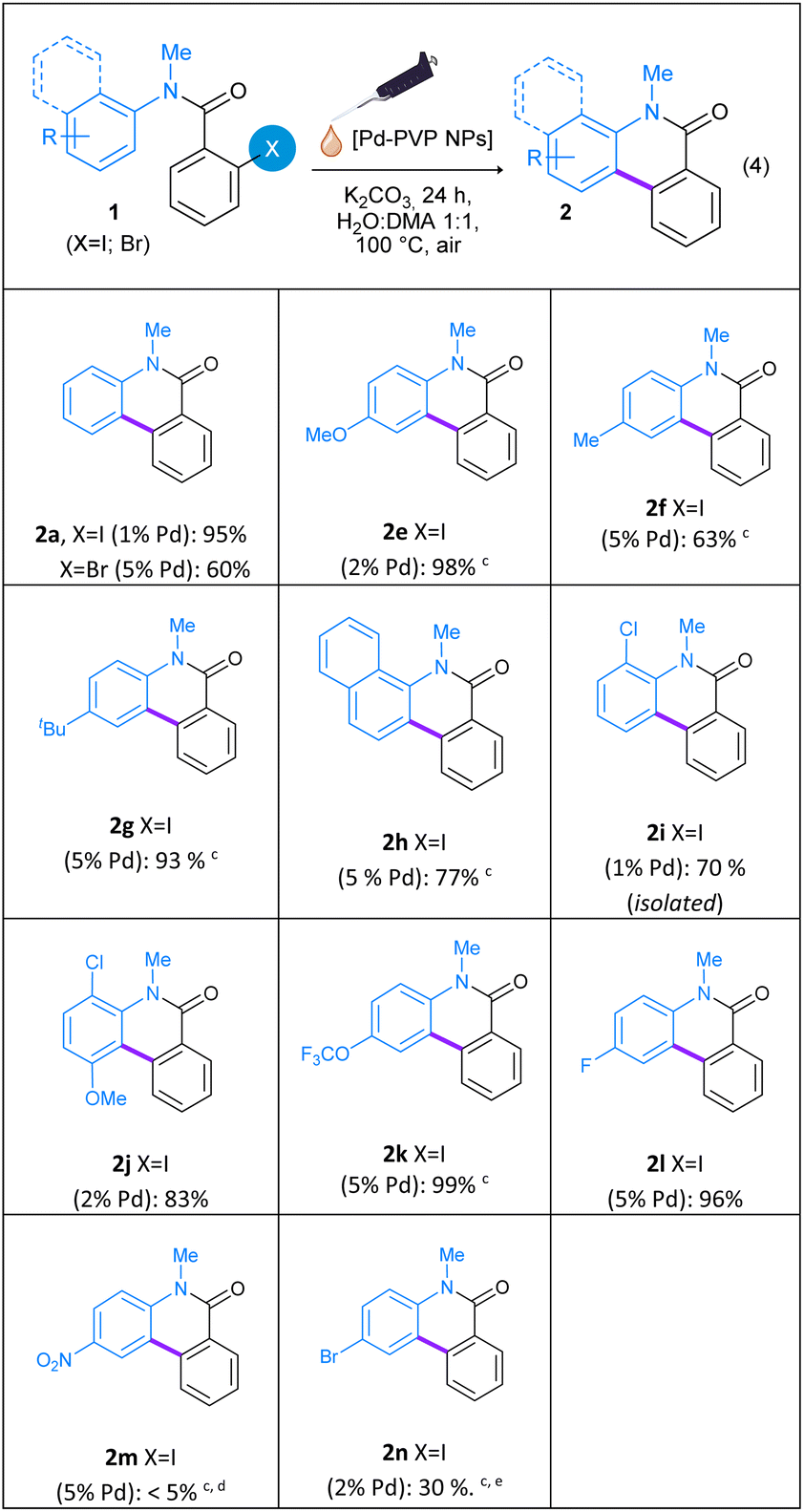
Following, the reaction scope was evaluated employing a variety of substituted N-methyl-N-aryl-2-iodobenzamides (1e–n) as starting materials. As summarized on Table 2, both electron-withdrawing and electron-donating groups were well tolerated and cyclization into the corresponding phenanthridine-6(5H)-ones 2e–n was efficiently achieved in good to excellent yields. The catalyst loading and reaction time were optimized for each benzamide (1e–n), in order to obtain better yields of phenanthridine-6(5H)-ones 2e–n. As shown on Table 2, the reaction proceeds with excellent yields in the presence of various substituents such as –OMe (1e), –Me (1f), –tBu (1g), –OCF3 (1k), and –F (1l) on para position in the N-aryl ring. The electronic properties of the substituents on the aniline aromatic rings affected the products yields. Great reactivity at low Pd loading was observed for amides with strong donating groups, such as –OMe (1e), or with weak withdrawing groups on ortho position (1i and 1j). Electron-withdrawing groups on para position (1k and 1l), weak electron-donating groups (1f and 1g) and bulky groups (1h) require a higher Pd loading (up to 5 mol% Pd). However, the presence of a strong electron-withdrawing group such as –NO2 group (1m) on the N-aryl ring results in a very low reactivity, even with the use of 5 mol% of Pd nanocatalyst and a reaction time of 48 h. In this case, N-methyl-4-nitroaniline was obtained as the main product by decomposition of 1m. The sterically hindered substrates N-methyl-N-(naphthyl)-2-iodobenzamide (1h), N-methyl-N-(2-chlorophenyl)-2-iodobenzamide (1i) and N-methyl-N-(2-chloro-4-methoxyphenyl)-2-iodobenzamide (1j) afforded phenanthridinones 2h–j in very good yields (77%, 70% and 83%, respectively). With regard to the reaction with the halogenated substrate N-methyl-N-(4-bromophenyl)-2-iodobenzamide (1n), cyclization product 2n was achieved in only 30% together with 11% of phenanthridinone 2a. When the amount of Pd was increased to 5 mol%, only 35% of dehalogenated cyclization product 2a was found.
| a Reactions carried out under air atmosphere using 1e–n (1 equiv., 0.2 mmol), K2CO3 (3 equiv.); and solvent (Vf = 4 mL), heating in an oil bath for 24 h. Pd loading in brackets.b Yields were quantified by 1H NMR using 4-nitroacetophenone as internal standard.c Reaction time: 48 h.d Substrate decompose under this reaction conditions.e Dehalogenated cyclization product 2a was observed in 11% yield. |
|---|
 |
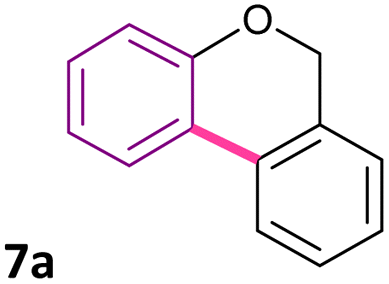
Considering the good catalytic activity of Pd-PVP NPs in the intramolecular arylation of 2-halobenzamides, and in an attempt to extend the synthetic applications of this nanocatalytic system, the cyclization of 2-halobenzyl aryl ethers to obtain benzo[c]chromenes by the Pd-catalyzed C–H bond activation was studied (eqn (5)). In the first place, the reactivity of aryl bromides against aryl iodides, using the corresponding 2-halobenzyl phenyl ethers 6a (X = Br) and 6b (X = I) as substrates was evaluated (Table 3). Initially, the previously optimized reaction conditions for the synthesis of phenanthridinones were applied, using 5 mol% of Pd-PVP NPs and for 48 h of reaction. In both cases, benzo[c]chromene 7a was obtained together with the reduced product (8a). Remarkably, aryl bromide 6a gave a similar yield for benzo[c]chromene 7a as aryl iodide 6b (62% vs. 58% yield, respectively). Nevertheless, the by-product benzyl phenyl ether 8a was generated in a 7% yield for bromide 6a and 30% for iodide 6b, indicating a higher selectivity of bromide analog 6a over iodide substrate 6b.
| a Reactions were carried out under air atmosphere using 6a–k (1 equiv., 0.2 mmol), K2CO3 (3 equiv.); and solvent (Vf = 4 mL), heating in an oil bath for 48 h.b Yields were quantified by 1H NMR using 4-nitroacetophenone as internal standard.c The benzyl phenyl ether (8a) was observed in 7% yield.d The benzyl phenyl ether (8a) was detected in 30% yield.e Yields were quantified by GC using 4-nitroacetophenone as internal standard.f The substrate was recuperated in ca. 100% yield.g n.d.: not detected. |
|---|
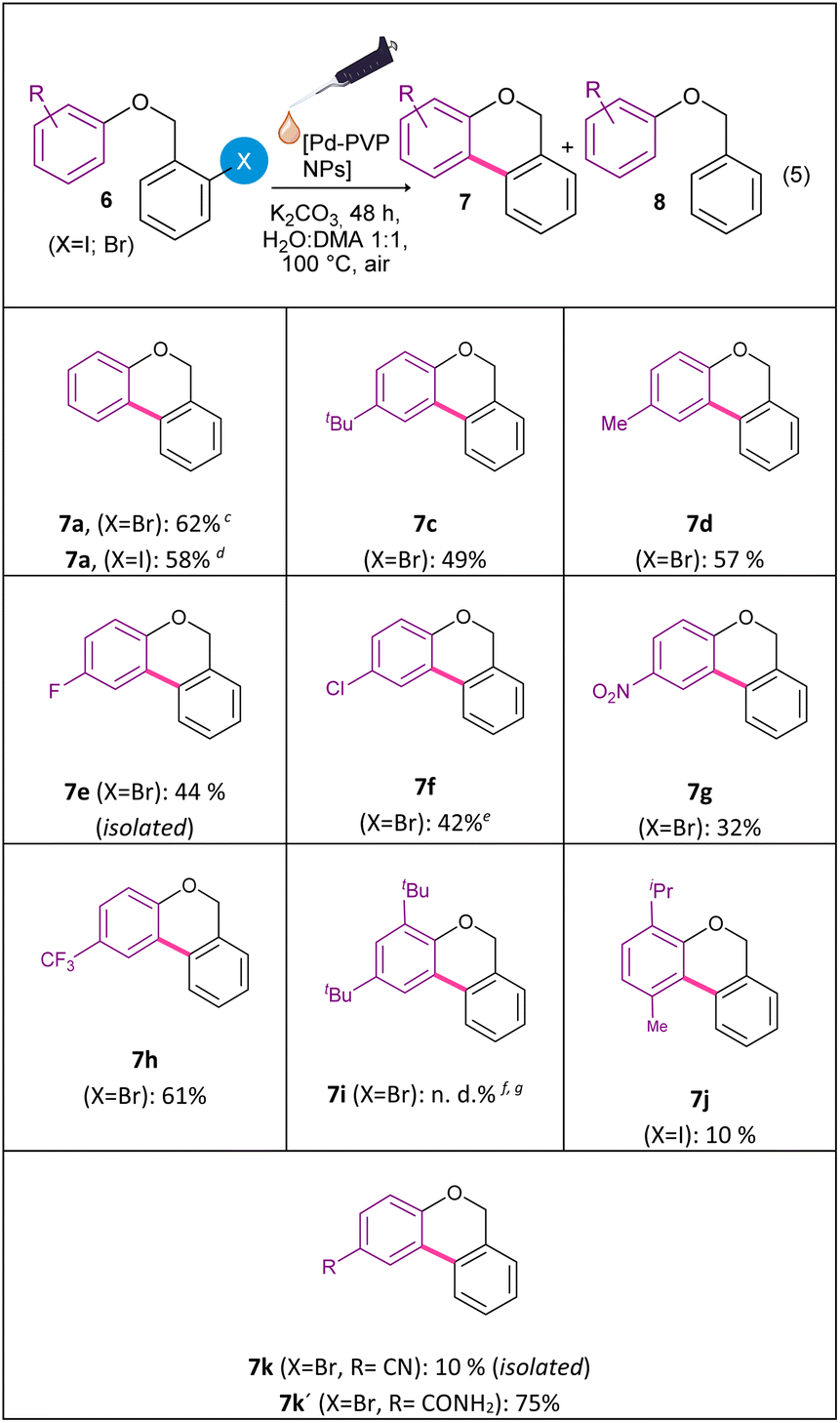 |
Fagnou et al., explored the reactivity of 2-halobenzyl phenyl ethers 6a and 6b using a homogeneous Pd-complex as a catalyst. They observed that aryl bromide 6a gave higher yields of 7a compared to the iodine derivative 6b.28 Although the oxidative addition of Pd in a C–X bond is typically favored in aryl iodides, in this particular case, the poor reactivity of the iodine substrate was attributed to the accumulation of iodide anions in the reaction mixture. The authors proposed that I− anions could poison the active catalytic species.
In our catalytic system, the reaction with N-methyl-N-phenyl-2-iodobenzamide (1a) efficiently produced the cyclization product 2a (eqn (1)), indicating that the presence of iodide anion did not inhibit the catalytic activity of the Pd-PVP NPs. Furthermore, the reactivity of aryl halides in the synthesis of phenanthridinones followed the expected trend, with iodide 1a being more reactive than bromide 1b. In addition, when the reaction of Pd-PVP NPs with iodide phenyl ether 6b was carried out, a higher conversion was observed compared to the reaction with bromide phenyl ether 6a. Therefore, the oxidative addition step occurs efficiently with the iodide substrate 6b, however, there is significant competition between the reduction process and the C–C bond formation, observing a ratio of 7a:8a of 2:1. In contrast, aryl bromide 6a showed a better selectivity ratio 7a:8a, which was of 9:1.
It is important to emphasize that the increased selectivity of brominated compounds provides the advantage of being able to use more accessible and economical starting materials. Additionally, the higher selectivity facilitate the purification process. Recently, we reported the synthesis of benzochromenes from 2-iodobenzyl aryl ethers in the presence of KOtBu and visible light; however, this methodology did not enable the efficient formation of the cyclized product when starting from brominated derivatives.76
The reaction conditions were attempted to be optimized by exploring different solvent mixtures, bases, and additives in the cyclization reaction of 6a (Table S3†). However, in all cases, lower yields and selectivity of 7a were obtained compared to the aforementioned conditions. In addition, when the reaction was carried out in the absence of Pd nanocatalyst substrate 6a was recovered in almost 100% yield (Table S3,† entry 1).
To extend the scope of the intramolecular Pd-catalyzed C–H bond activation reaction different aryl bromides ethers derivatives 6c–6k were synthetized (Table 3). The Pd-catalyzed cyclization of 2-halobenzyl aryl ethers gave benzochromenes 7c–7k in moderate to good yields, with high selectivity. Remarkably, the electronic nature of the functional group on the aryl ring did not significantly affects product yields. Both electron-withdrawing (F, Cl, CF3 and NO2) and electron-donating groups (t-Bu, Me) gave benzochromenes 7c–7h in similar yields. On the other hand, steric hindrance had a noticeable effect on this cyclization reaction. The hindered substrates 6i and 6j, in particular, did not react or react in lower yield under the same reaction conditions.
In the presence of –CN group as substituent (6k), the reaction afforded the amide product 7k′ (RCONH2) in a 75% of yield, along with the expected product 7k (RCN) in a 10% of isolated yield. Probably, product 7k′ was obtained from the hydrolysis of –CN group in the cyclized product 7k during the reaction.
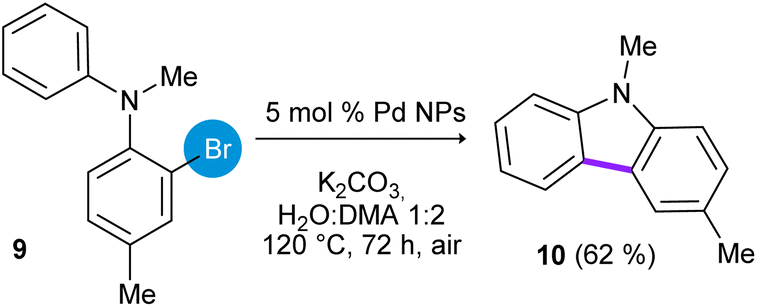
In addition, the synthesis of 3,9-dimethyl-9H-carbazole (10) was studied employing N-methyl-N-(2-bromotolyl)-aniline (9) as substrate (eqn (6)). In this case, the cyclization product 10 was obtained in 62% of yield after heating at 120 °C for 72 h.
 | (6) |
Based on the obtained results and several reports of Pd-catalyzed C–H bond activation reactions, the general mechanism shown in Fig. 2 can be proposed for phenanthridinones synthesis.77,78
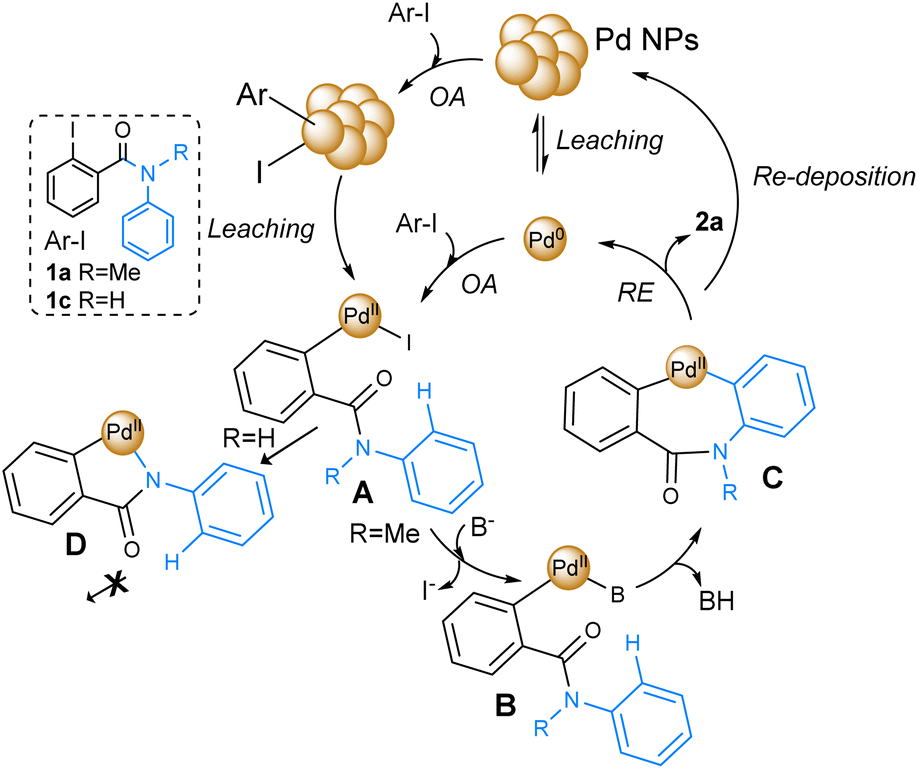 | ||
| Fig. 2 Plausible mechanism for Pd-catalyzed C–H bond activation with Pd NPs and benzamide 1a and 1c. | ||
Initially, the catalytic cycle involves the oxidative addition onto the C–I bond of 1a by the Pd(0) active catalytic species. The role of Pd NPs as active catalysts in these reactions is still under discussion.48 Some studies suggest that NPs might actually act as a reservoir of Pd, from which active species are leached as mononuclear/oligomeric Pd species or clusters, which, after the oxidative addition of 1a, give rise to the formation of complex A.79 Another possibility is that oxidative addition occurs on the surface of Pd NPs, followed by the leaching of generated PdII species, resulting in the formation of the same complex A.80,81 Additionally, it has been proposed that Pd NPs are the active site for catalysis.82 Thus, a “cocktail”-type mechanism can occurs, in which several species can contribute to the formation of product 2a.83 Following the oxidative addition step, a base-assisted anion exchange takes place on complex A, leading to the liberation of iodine and the formation of complex B. Subsequently, activation of the Csp2–H bond occurs, yielding intermediate C. This step, involving C–H bond cleavage, is also facilitated by the base, resulting in the formation of BH. Finally, the Pd (0) is regenerated through a reductive elimination step, and the desired product 2a is released (Fig. 2). On the other hand, the reaction of substrate 1c was completely inhibited, probably due to the formation of Pd–N intermediate D after oxidative addition onto 1c.
It is important to note that the Pd NPs employed as nanocatalyst are stabilized with PVP polymer. Generally, NPs are prepared in the presence of capping and/or stabilizing agents, like PVP, to stabilize and control the growth of metal NPs.84 These capping agents can stabilize the NPs by enclosing their surfaces and providing individual space through repulsive forces, such as electrostatic or steric repulsion. However, the presence of these agents can block the active sites and inhibit mass transfer of reactants, hindering access to the catalyst surface. Therefore, from a catalytic perspective, the presence of capping agents is considered to have a negative impact on the performance of NPs. However, several studies have revealed an unexpected potential of capping agents, which might act as promoters or selectivity modifiers.84 For instance, Han et al. investigated the effect of PVP in the direct synthesis of H2O2 catalyzed by a supported Pd-nanocatalyst.85 They found that the presence of an optimal amount PVP does not inhibit the catalytic activity of Pd NPs, but rather, even a moderate presence of PVP resulted in improvements in catalytic activity compared to the complete elimination of the polymer. In the Suzuki–Miyaura cross-coupling reaction, Narayanan and El-Sayed observed that the presence of PVP prevents catalyst aggregation and deactivation.86,87 In our previous reports, we found that Pd NPs with high catalytic activity were prepared in the presence of PVP as a stabilizer.68,70,71 Thus, this capping agent represents an appropriate choice to obtain stable and active nanocatalyst.
In order to highlight the advantage of our catalytic system, a comparison of the catalytic activity of the Pd-PVP NPs with other catalytic systems, including nanocatalysts, Pd-complexes, or heterogeneous systems, for the C–H bond activation reactions by a direct arylation process, is presented on Table 4. Regarding the synthesis of phenanthridinones, our nanocatalytic system exhibited excellent catalytic activity and selectivity to obtain these heterocyclic derivatives. The use of homogeneous catalysts typically requires expensive, sensitive, or non-commercial ligands, elevated temperatures (>110 °C), inert atmospheres, or anhydrous conditions. In general, Pd-catalyzed C–H bond activation reactions involve the use of several equivalents of a base, which are needed to eliminate the hydrogen atom after the C–H insertion step in the reaction mechanism.
| Heterocyclic compound | Catalyst | Reaction conditions | Examples | Halogen in the starting material | Yield | Ref |
|---|---|---|---|---|---|---|
 |
Pd(OAc)2 (2 mol%) | DMA (10 mL), 135 °C | 1 | X = Br | 83% | 29 |
| Ligand (Ph2PC6H4-C6H4NMe2) (4 mol%) | K2CO3 (2 equiv.), overnight, N2 | |||||
| PdCl2 (5 mol%) | DMI, 25 °C | 2 | X = Br | 89% | 33 | |
| Ligand (R-COOH) (30 mol%) | K2CO3 (1.5 equiv.), 24 h, Ar | |||||
| Pd(OAc)2 (10 mol%) | DMA, 170 °C | 2 | X = I | 50% | 34 | |
| Na2CO3 (1.2 equiv.), 2.5 h, N2 | ||||||
| Pd(OH)2/C (10 mol%) | DMA, 140 °C | 2 | X = Br | 80% | 45 | |
| KOAc (2 equiv.), 24 h, N2 | ||||||
| Pd/C (10 mol%) | DMA, 125 °C | 12 | X = I | 98% | 88 | |
| KOAc (2 equiv.). 24 h, N2 | ||||||
| Pd-PVP NPs (1 mol%) | H2O:DMA 1:1 (4 mL), 100 °C |
11 | X = I | 95% | This work | |
| K2CO3 (3 equiv.), 24 h, air | ||||||
 |
Pd(OAc)2 (2 mol%) | DMA (10 mL), 125 °C | 7 | X = Br | 96% | 29 |
| Ligand (Ph2PC6H4-C6H4NMe2) (4 mol%) | K2CO3 (2 equiv.), 2 h, N2 | |||||
| PdCl2 (5 mol%) | DMI (0.5 mL), 25 °C | 10 | X = Br | 95% | 33 | |
| Ligand (R-COOH) (30 mol%) | Rb2CO3 (1.5 equiv.), 40 h, Ar | |||||
| PdCl2(MeCN)2 (5 mol%) | DMA (5 mL), 120 °C | 17 | X = Br | 96% | 44 | |
| P(p-FC6H4)3 (5 mol%) | K2CO3 (3 equiv.), PivOH (30 mol%), 3 h, Ar | |||||
| Pd(OH)2/C (10 mol%) | DMA (5 mL), 130 °C | 5 | X = I | 92% | 45 | |
| KOAc (2 equiv.), 12 h, Ar | ||||||
| PdO-Fe3O4 (10 mol%) | DMA (2 mL), 140 °C | 9 | X = Br | 85% | 67 | |
| KOAc (2 equiv.), 48 h | ||||||
| Pd-PVP NPs (5 mol%) | H2O:DMA 1:1 (4 mL), 100 °C |
10 | X = Br | 62% | This work | |
| K2CO3 (3 equiv.), 48 h, air |
The Pd-PVP NPs catalytic system demonstrated notable enhancements in the overall reaction conditions for synthesizing phenanthridinones. These encompass reduced catalyst loading, elimination of costly co-catalysts or ligands, the use of aqueous medium (H2O:DMA), operation under atmospheric air, and the use of a lower reaction temperature when compared to other recently published strategies. Additionally, this nanocatalyst is straightforward to prepare and easy to handle. It does not require manipulation under a controlled atmosphere and can be stored on the workbench for months without any special care, without losing its catalytic activity.
For the synthesis of benzochromenes by Pd-catalyzed C–H bond activation by direct arylation reactions, typically high catalyst loading (>5 mol%) and elevated temperatures (>110 °C) are necessary. Furthermore, the use of expensive or non-commercial ligands are usually employed. Herein, Pd-PVP NPs exhibited moderate to good yields to obtain 7a–k derivatives under milder reaction conditions, involving the use of aqueous medium, ligand-free conditions and under air.
A very important aspect is that phenanthridinones and benzochromenes revealed as promising scaffolds in the search of small molecules with biological activity as mentioned before.89,90 Therefore, compounds of the series 2 and 7 were evaluated for their in vitro antiproliferative activity against the human cancer cell lines A549 (lung), HBL-100 (breast), HeLa (cervical), SW1573 (lung), T-47D (breast), and WiDr (colon). The cisplatin (CDDP) and 5-fluorouracil (5-FU) were used as reference compounds. The most relevant results, expressed as 50% growth inhibition (GI50) of selected phenanthridinones (2) and benzo[c]chromenes (7) are summarized in Table 5.
| Comp. | A549 | HBL-100 | HeLa | SW1573 | T-47D | WiDr |
|---|---|---|---|---|---|---|
| a For all compounds, GI50 values are given in μM and are means of two to four experiments (mean ± SD).b Obtained by KOtBu (3 equiv.), blue-LEDs in DMSO (ref. 76). | ||||||
| 2a | 47 ± 6.8 | 58 ± 12 | 49 ± 7.9 | 54 ± 10 | 53 ± 9.8 | 48 ± 13 |
| 2e | 27 ± 4.8 | 34 ± 8.5 | 26 ± 3.3 | 28 ± 9.7 | 31 ± 8.4 | 31 ± 7.9 |
| 2f | 37 ± 7.5 | 44 ± 9.8 | 39 ± 8.0 | 47 ± 18 | 35 ± 16 | 32 ± 12 |
| 2i | >100 | >100 | >100 | >100 | >100 | >100 |
| 2k | >100 | >100 | 48 ± 18 | >100 | >100 | >100 |
| 2l | 73 ± 34 | 77 ± 34 | 76 ± 36 | >100 | 83 ± 35 | >100 |
| 7a | >100 | >100 | 75 ± 16 | >100 | >100 | >100 |
| 7c | >100 | >100 | 56 ± 18 | >100 | 56 ± 21 | 48 ± 11 |
| 7d | >100 | >100 | >100 | >100 | >100 | >100 |
| 7e | >100 | >100 | >100 | >100 | >100 | >100 |
| 7f | >100 | >100 | 43 ± 6.9 | >100 | 79 ± 29 | 62 ± 0.4 |
| 7h | >100 | >100 | >100 | >100 | >100 | >100 |
| 7ib | 24 ± 10 | 24 ± 3.3 | 13 ± 4.2 | 15 ± 3.8 | 17 ± 6.1 | 21 ± 9.1 |
| 7jb | 3.9 ± 0.3 | 8.6 ± 3.4 | 3.6 ± 0.2 | 4.4 ± 0.04 | 6.2 ± 0.4 | 6.8 ± 1.5 |
| 7k (CN) | 82 ± 15 | >100 | 43 ± 5.4 | 88 ± 14 | 67 ± 23 | 75 ± 32 |
| CDDP | 4.9 ± 0.6 | 1.9 ± 0.2 | 1.8 ± 0.5 | 2.7 ± 0.4 | 17 ± 3.3 | 23 ± 4.3 |
| 5-FU | 2.2 ± 0.3 | 4.4 ± 0.7 | 19 ± 1.2 | 3.3 ± 1.2 | 8.2 ± 2.0 | 49 ± 6.7 |
As observed, phenanthridinones 2i and 2k, and benzochromenes 7a, 7d, 7e, and 7h are entirely inactive, with GI50 values exceeding 100 μM in all cell lines. On the other hand, phenanthridinones 2a, 2e, 2f, 2l, and benzo[c]chromenes 7c, 7f, and 7k exhibit moderate or low activity, showing GI50 values in the range 26–88 μM in some cell lines. Finally, the disubstituted benzochromenes 7i and 7j demonstrate antiproliferative activity comparable to that of cisplatin (CDDP) and 5-fluorouracil (5-FU) across the six studied cell lines. In the case of 2,4-di-tert-butyl-6H-benzo[c]chromene 7i, activities in the range of 13–24 μM were identified. Meanwhile, the derivative 4-isopropyl-1-methyl-6H-benzo[c]chromene 7j exhibits the best antiproliferative activity tested, reaching values between 3.6 and 8.6 μM, establishing itself as the most active compound in the entire series of synthesized benzochromenes. Noteworthy, the presence of two substituents on the phenyl ring contributed to improve GI50 values. However, this cannot be considered a trend, since compound 7i exhibited larger GI50 values. The study of further compounds with diverse substitution patterns could give a more precise structure–activity relationship.
Experimental
General methods
Purification of desired compounds was carried out by column chromatography on silica gel or by High Performance Liquid Chromatograpy (HPLC) preparative. Gas chromatographic (GC) analysis were performed with a flame-ionization detector, on 30 m capillary column of a 0.32 mm × 0.25 μm film thickness, with a 5% phenylpolysiloxane phase. Gas chromatography-mass spectroscopy (GC-MS) analysis were performed employing an electronic impact (EI) ionization method and a 30 m × 0.32 mm × 0.25 μm column with a 5% phenylpolysiloxane phase. 1H NMR and 13C NMR {1H} spectra were recorded on 400 MHz in spectrometer with CDCl3, acetone-d6 or DMSO-d6 as solvents with TMS as internal standard. Coupling constants are given in Hz and chemical shifts are reported in δ values in ppm. Data are reported as followed: chemical shift, multiplicity (s = singlet, s br = broad singlet, d = doublet, t = triplet, dd = double doublet, dt = double triplet, ddd = double double doublet, m = multiplet), coupling constants (Hz), and integration. All unknown products were further characterized by high resolution mass spectrometry (HRMS). HRMS analyses were carried out using a time-of-flight mass spectrometry (TOF-MS) instrument with an electrospray ionization (ESI) source.Materials
Aniline, N-methylaniline, 4-methoxyaniline, 4-methylaniline, 4-tert-butylaniline, 1-naphtylamine, 2-chloroaniline, 2-chloro-5-methoxyaniline, 4-(trifluoromethoxy)aniline, 4-fluoroaniline, 4-nitroaniline, 4-bromoaniline, 2-iodoaniline, formaldehyde, 2-iodobenzoic acid, 2-bromobenzoic acid, benzoic acid, triethylamine, methyl iodide, NaBH4, KOtBu, K2CO3, H2PdCl4, sodium citrate, polyvinylpyrrolidone (PVP), 4-nitroacetophenone, phenol, 4-(tert-butyl)phenol, 2-bromobenzyl bromide, 2-iodobenzyliodide, p-cresol, 4-fluorophenol, 4-chlorophenol, 4-nitrophenol, 4-(trifluoromethoxy)phenol, 2,4-di-tert-butylphenol, 2-isopropyl-5-methylphenol, 4-hydroxybenzonitrile, 18-crown-6, 2-bromo-4-methylaniline, iodobenzene, NaOtBu, bis[(2-diphenylphosphino)phenyl] ether (DPEphos), Pd(OAc)2, potassium acetate and anhydrous Na2SO4 were purchased from commercial suppliers and used without further purification. Acetone, DCM, EtOAc and SOCl2, were previously distilled. DMSO and toluene were distilled and dried under molecular sieves (3 Å). DMA, DMF, THF, EtOH, MeOH and MeCN HPLC were previously filtered. All solvents were analytical grade. The silica used in column chromatography corresponds to silica gel 60 (0.063–0.200 mm).Typical procedures for the synthesis of compounds 1, 6 and 9
Synthesis of N-methyl-N-aryl-2-halobenzamidesa (1a–1n)
Upon completion of the reaction, the mixture was cooled and used directly for a nucleophilic acyl substitution reaction, without further purification.
Previously synthesized N-methylaniline (1 equiv., 2 mmol, see Section 1.3.1) and triethylamine (3 equivs, 6 mmol) in 10 mL of DCM were added to a round-bottomed flask and cooled to 0 °C in an ice bath. After 5 minutes of stirring, the toluene solution of acid chloride (1.0 equiv.) was added dropwise to the reaction mixture and allowed to warm to room temperature overnight.
After that, water was added and the layers were separated. The organic layer was washed with water (3 × 20 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure to yield the crude reaction product.
Upon completion of the reaction, the mixture was cooled and used directly for a nucleophilic acyl substitution reaction, without further purification.
Aniline (1 equiv., 2 mmol), triethylamine (3 equivs, 6 mmol), and 10 mL of DCM were added to a round-bottomed flask and cooled to 0 °C in an ice bath. After 5 min of stirring, the toluene solution of acid chloride (1.0 equiv.) was added dropwise to the reaction mixture and allowed to warm to room temperature.
After that, water was added and the layers were separated. The organic layer was washed with water (3 × 20 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure to yield the crude reaction product. The amide derivative was purified by column chromatography on silica gel eluting with hexane/EtOAc and used without characterization in the methylation reaction to obtain the corresponding N-methyl-N-phenylbenzamides.
:20 → 60:40). A mixture of rotamers (3.5:1) as a light-yellow oil was obtained in 73% yield (353 mg, 0.97 mmol). 1H NMR (400 MHz, CDCl3) major isomer: δ 7.66 (d, J = 7.9 Hz, 1H), 7.18 (d, J = 8.3 Hz, 2H), 7.12–7.07 (m, 3H), 7.02 (dd, J = 7.7, 1.8 Hz, 1H), 6.84 (td, J = 7.6, 1.7 Hz, 1H), 3.49 (s, 3H), 1.21 (s, 9H); minor isomer: δ 7.86 (d, J = 8.0, 1H), 7.48–7.36 (m, 7H), 3.18 (s, 3H), 1.34 (s, 9H). 13C NMR {1H} (101 MHz, CDCl3) major isomer: δ 170.3, 150.1, 142.4, 140.5, 139.1, 129.6, 128.5, 127.2, 126.4, 125.8, 93.7, 37.4, 34.4, 31.1. GC/MS (EI) m/z 394 (M+ +1, 13), 393 (M+, 60), 378 (18), 250 (10), 244 (37), 231 (100), 210 (50), 203 (24), 146 (16), 105 (10), 91 (17), 77 (28), 76 (54), 57 (26), 50 (12). HRMS (ESI-TOF+) m/z: [M + H]+ calcd for C18H21INO 394.0662, found 394.0668.:10 → 70:30). A mixture of rotamers (5.5:1) as a brown solid was obtained with an overall yield of 89% (45.8 mg, 0.12 mmol). 1H NMR (400 MHz, CDCl3) major isomer: δ 8.04 (d, J = 8.4 Hz, 1H), 7.82 (d, J = 8.2 Hz, 1H), 7.69–7.62 (m, 3H), 7.56–7.51 (m, 2H), 7.29–7.25 (m, 1H), 6.84 (dd, J = 7.6, 1.8 Hz, 1H), 6.77 (td, J = 7.5, 1.3 Hz, 1H), 6.71 (td, J = 7.6, 1.8 Hz, 1H), 3.58 (s, 3H); minor isomer: δ 7.93 (d, J = 8.0 Hz, 2H), 7.89 (d, J = 8.5 Hz, 1H), 7.69–7.62 (m, 1H), 7.59–7.51 (m, 5H), 7.29–7.21 (m, 1H); 7.20–7.14 (m, 1H), 3.27 (s, 3H). 13C NMR {1H} (101 MHz, CDCl3) major isomer: δ 171.0, 142.1, 139.7, 139.2, 134.4, 129.7, 128.7, 128.6, 127.3, 126.9, 126.5, 126.5, 125.8, 125.5, 125.1, 122.8, 93.9, 37.5. CG/MS (EI) m/z 387 (M+, 42), 245 (12), 244 (78), 231 (100), 203 (26), 154 (18), 128 (23), 127 (26), 77 (24), 76 (70), 75 (12), 50 (20). HRMS (ESI-TOF+) m/z: [M + H]+ calcd for C18H15INO 388.0193, found 388.0198.:10 → 70:30). A mixture of rotamers (4.1:1) as a yellow oil was obtained in 74% yield (268.5 mg. 0.72 mmol). 1H NMR (400 MHz, CDCl3) δ 7.69 (dd, J = 7.9, 1.2 Hz, 1H), 7.45–7.42 (m, 1H), 7.35–7.32 (m, 1H), 7.18–7.02 (m, 4H), 6.84 (ddd, J = 7.9, 7.4, 1.7 Hz, 1H), 3.42 (s, 3H); minor isomer: δ 7.88 (dd, J = 8.1, 1.1 Hz, 1H), 7.52 (dd, J = 7.8, 1.6 Hz, 2H), 7.48–7.30 (m, 4H), 7.17–7.05 (m, 1H), 3.12 (s, 3H). 13C NMR {1H} (101 MHz, CDCl3) major isomer: δ 170.1, 141.9, 140.9, 139.2, 132.3, 130.3, 130.1, 130.0, 129.4, 127.8, 127.4, 126.6, 93.8, 36.0. CG/MS (EI) m/z 371 (M+, 1), 337 (16), 336 (99), 230 (61), 203 (19), 111 (7), 104 (9), 78 (6), 77 (49), 76 (100), 75 (25), 74 (9), 63 (10), 51 (20), 50 (38). HRMS (ESI-TOF+) m/z: [M + H]+ calcd for C14H12ClINO 371.9647, found 371.9652.:10 → 60:40). A mixture of rotamers (5:1) as a brown oil was obtained in 78% yield (312 mg. 0.78 mmol). 1H NMR (400 MHz, CDCl3) major isomer δ 7.70 (dd, J = 7.9, 0.8 Hz, 1H), 7.19 (d, J = 9.0 Hz, 1H), 7.18–7.15 (m, 1H), 7.09 (td, J = 7.5, 1.0 Hz, 1H), 7.00 (d, J = 3.0 Hz, 1H), 6.87 (td, J = 7.6, 1.8 Hz, 1H), 6.66 (dd, J = 8.9, 3.0 Hz, 1H), 3.69 (s, 3H), 3.41 (s, 3H); minor isomer: δ 7.87 (d, J = 7.8 Hz, 1H), 7.45 (td, J = 7.4, 0.9 Hz, 1H), 7.42–7.39 (m, 2H), 7.14–7.10 (m, 1H), 7.04–7.03 (m, 1H), 6.87–6.86 (m, 1H), 3.96 (s, 3H), 3.83 (s, 3H). 13C NMR {1H} (101 MHz, CDCl3) major isomer: δ 170.1, 158.7, 142.1, 141.3, 139.1, 130.6, 130.4, 130.2, 127.6, 126.7, 116.4, 114.4, 93.8, 55.8, 35.9. GC/MS (EI) m/z 401 (M+, 1), 367 (18), 366 (100), 238 (6), 231 (27), 203 (12), 77 (18), 76 (55), 75 (8), 63 (10), 51 (9), 50 (18). HRMS (ESI-TOF+) m/z: [M + H]+ calcd for C15H14ClINO2 401.9752, found 401.9757.:10 → 60:40). A brown oil was obtained in 44% yield (185 mg. 0.44 mmol). 1H NMR (400 MHz, CDCl3) major isomer δ = 7.67 (d, J = 8.0 Hz, 1H), 7.23–7.12 (m, 3H), 7.03 (d, J = 8.0 Hz, 3H), 6.92–6.86 (m, 1H), 3.51 (s, 3H); minor isomer: δ 7.88 (d, J = 8.0 Hz, 1H), 7.57–7.43 (m, 3H), 7.41–7.28 (m, 3H), 7.23–7.10 (m, 1H), 3.19 (s, 3H). 13C NMR {1H} (101 MHz, CDCl3) major isomer δ 170.1, 147.6, 142.1, 141.8, 139.3, 130.0, 128.5, 127.5, 121.3, 120.3 (q, J = 259.8 Hz), 93.5, 37.4. GC/MS (EI) m/z 421 (M+, 45), 231 (100), 203 (20), 95 (13), 77 (26), 76 (85), 75 (15), 50 (27). HRMS (ESI-TOF+) m/z: [M + H]+ calcd for C15H12F3INO2 421.9859, found 421.9865.Synthesis of aryl-2-halobenzyl ethers (6a–k)
To a solution of the corresponding phenol (1 equiv., 2 mmol) with K2CO3 (1 equiv., 2 mmol, 276 mg) in DMF (7.2 mL), 18-crown-6 (1 equiv., 2 mmol, 426 μL) and the required 2-halobenzyl bromide (1 equiv., 2 mmol) were added. The mixture was stirred followed by heating to 120 °C for 17 h. The reaction mixture was then cooled to room temperature and water was added and the layers separated. The organic layer was washed with water (3 × 20 mL), dried over Na2SO4 and concentrated under reduced pressure to afford the reaction crude which was analyzed by TLC, GC and isolated with column chromatography over silica gel.Synthesis of the Pd nanoparticle suspension (Pd-PVP NPs)
The Pd-PVP NPs synthesis was performed following the procedure previously described.68 Into a 10 mL scintillation vial equipped with a magnetic stirrer, 44.0 mg PVP (2% w/v), 11.0 mg of sodium citrate (molar ratio Pd2+:citrate = 1:10) and 2 mL of a feedstock aqueous solution of H2PdCl4 (2 mM) were placed. Then, the vial was sealed and high purity nitrogen was bubbled for 5 min to saturate the solution. The reaction mixture was irradiated under vigorous magnetic stirring for 1 hour in a photochemical reactor equipped with a 3 W blue LED (462 nm). The color of the mixture changed to the characteristic dark brown of Pd NPs. Finally, the vial was opened to the air, and the Pd-PVP NPs dispersion was stored in a Falcon tube to be used as a catalyst without further purification. The Pd NPs were characterized by transmission electron microscopy (TEM) using a JEM-JEOL 1120 microscope operating at 80 kV, available at the Research Institute IPAVE-INTA-CIAP in Córdoba, Argentina.
Intramolecular arylation reactions catalyzed by Pd-PVP NPs
:0 → 0:100) and recrystallized in acetone. White solid was isolated in 55% yield (24.8 mg, 0.11 mmol). 1H NMR (400 MHz, CDCl3) δ 8.29 (d, J = 2.2 Hz, 1H), 7.79 (d, J = 7.7 Hz, 1H), 7.63 (dd, J = 8.4, 2.2 Hz, 1H), 7.46–7.37 (m, 1H), 7.33 (td, J = 7.5, 1.2 Hz, 1H), 7.17 (d, J = 7.4 Hz, 1H), 7.02 (d, J = 8.4 Hz, 1H), 5.67 (s br, 2H), 5.19 (s, 2H). 13C NMR {1H} (101 MHz, CDCl3) δ 168.9, 158.0, 131.0, 129.2, 128.9, 128.5, 128.4, 127.2, 124.9, 123.6, 123.1, 122.4, 117.6, 68.8. GC/MS EI m/z 226 (M+ +1, 15), 225 (M+, 100), 224 (85), 209 (26), 181 (11), 153 (32), 152 (41), 151 (18), 104 (45), 90 (23), 77 (10), 76 (50), 75 (12), 63 (18). HRMS (ESI-TOF+) m/z: [M + H]+ calcd for C14H12NO2 226.0863, found 226.0868.Antiproliferative tests
The human solid tumor cell lines used in this study were the non-small cell lung cancer A549 and SW1573, the cervix cancer HeLa, the breast cancer cell lines HBL-100 and T-47D, and the colon cancer WiDr. Cell lines were obtained from Prof. Godefridus J. Peters (VUmc, Amsterdam, NL). The maintenance of cell cultures was in 60 mm Petri dishes in a humidified air incubator (37 °C, 5% CO2, 95% humidity). The cell culture medium used was RPMI 1640 supplemented with 5% heat inactivated FCS, 2 mM l-glutamine, 100 U mL−1 penicillin and 0.1 mg mL−1 streptomycin. Cell cultures were passaged biweekly using 0.05% trypsin and maintained at low passage. Single cell suspensions were counted using Moxi Z automated cell counter.On day 0, cells were inoculated in a volume of 100 μL per well at densities of 2500 (A549, HBL-100, HeLa and SW1573) or 5000 (T-47D and WiDr) cells per well, based on their doubling times. Stock solutions of compounds 2 and 7 were prepared in DMSO at 40 mM. Each compound was tested in triplicate at different dilutions in the range 1–100 μM. Cisplatin and 5-fluoruracil served as positive controls. Control cells received an equivalent concentration of DMSO (0.25% v/v, negative control). The drug treatment was started on day 1 after plating and incubation times was 48 h. Then, the SRB colorimetric method of the NCI was performed.92 The optical density (OD) of each well was measured in dual mode (530 & 620 nm), using BioTek's PowerWave XS Absorbance Microplate Reader. Values were corrected for background OD from wells only containing medium. Antiproliferative activity of the compounds expressed as GI50 was calculated according to NCI formulas.93
Conclusions
In conclusion, this study demonstrates the effective use of photochemically-synthesized Pd nanoparticles stabilized with poly(vinyl)pyrrolidone (Pd-PVP NPs) as catalysts for the intramolecular C–H bond activation reaction of aromatic amides, ethers and amines, leading to the formation of phenanthridine-6(5H)-ones, benzo[c]chromenes, and the N-methyl carbazole.Optimization of reaction conditions revealed that for phenanthridine-6(5H)-ones synthesis the best results were achieved with 3 equivalents of K2CO3 as a base, 1–5 mol% of Pd-PVP NPs in a 1:1 mixture of H2O:DMA solvent, at 100 °C for 24 hours under air atmosphere. The scope of the reaction was explored with several substituted substrates, and both electron-withdrawing and electron-donating groups were found to be well tolerated. This methodology provides ten derivatives of phenanthridine-6(5H)-ones with high yields (up to 98%) without side reactions. The N–H protection in the benzamide group was found to be crucial for the success of the intramolecular C–H bond activation reaction.
Remarkably, the preformation of Pd-PVP NPs was found to be necessary for catalytic activity, highlighting the importance of the nanocatalyst in achieving these transformations.
Finally, the study also explored the extension of the synthetic applications of Pd-PVP NPs to the cyclization of 2-halobenzyl aryl ethers, resulting in the formation of eight benzo[c]chromenes. The reactivity of aryl bromides and iodides was investigated, and the results demonstrated that aryl bromides exhibited better selectivity for the desired cyclization product. As an extension of this system, the synthesis of N-methyl carbazole was also carried out.
On the other hand, this is the first report of benzo[c]chromenes as a novel building block to obtain antitumor agents. The best in vitro antiproliferative activity results were found with 4-isopropyl-1-methyl-6H-benzo[c]chromene 7l, which inhibited cell proliferation with GI50 values in the range 3.6–8.6 μM. These are simple and robust molecule, which could be easily obtained in just two reaction steps from commercial and accessible substrates.
Overall, the use of Pd-PVP NPs as catalysts in these reactions offers a promising and sustainable approach for the synthesis of valuable heterocyclic compounds, with potential applications in the field of organic synthesis and medicinal chemistry.
Author contributions
EDDV: investigation, data curation, formal analysis, methodology, MAC: investigation, data curation, formal analysis, methodology, MDH: investigation, data curation, formal analysis, SMB: methodology, formal analysis, visualization, writing – review & editing; AGB: investigation; JMP: investigation, formal analysis, writing – review & editing; MEB: writing – original draft, writing – review & editing, conceptualization, methodology, project administration, resources, supervision, investigation, data curation, formal analysis; SEM: writing – review & editing, project administration, resources, supervision, conceptualization; PMU: writing – original draft, writing – review & editing, conceptualization, methodology, project administration, resources, supervision. All authors discussed and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by CONICET (PUE: 22920160100013CO and PIP 11220210100660CO), Agencia Nacional de Promoción Científica y Tecnica, ANCyT, FONCyT (PICT 2019-2184, PICT 2021-Cat-I-136, PICT 2018-3943, PICT 2021-GRFTI-376) and Secretaría de Ciencia y Tecnología, Universidad Nacional de Córdoba (SECyT). EDDV, MAC, and MDH gratefully acknowledge receipt of fellowship from CONICET.Notes and references
- X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed.
- J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC.
- R. Rossi, F. Bellina, M. Lessi and C. Manzini, Adv. Synth. Catal., 2014, 356, 17–117 CrossRef CAS.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed.
- R. S. Thombal, P. Y. M. Rubio, D. Lee, D. Maiti and Y. R. Lee, ACS Catal., 2022, 12, 5217–5230 CrossRef CAS.
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed.
- M. C. Bryan, B. Dillon, L. G. Hamann, G. J. Hughes, M. E. Kopach, E. A. Peterson, M. Pourashraf, I. Raheem, P. Richardson, D. Richter and H. F. Sneddon, J. Med. Chem., 2013, 56, 6007–6021 CrossRef CAS PubMed.
- K. Ohmori, M. Kitamura and K. Suzuki, Angew. Chem., Int. Ed., 1999, 38, 1226–1229 CrossRef CAS PubMed.
- G. Bringmann, N. Manchala, T. Büttner, B. Hertlein-Amslinger and R. Seupel, Chem.–Eur. J., 2016, 22, 9792–9796 CrossRef CAS PubMed.
- M. Tamiya, K. Ohmori, M. Kitamura, H. Kato, T. Arai, M. Oorui and K. Suzuki, Chem.–Eur. J., 2007, 13, 9791–9823 CrossRef CAS PubMed.
- Y. An, B.-S. Zhang, Z. Zhang, C. Liu, X.-Y. Gou, Y.-N. Ding and Y.-M. Liang, Chem. Commun., 2020, 56, 5933–5936 RSC.
- S. Ghosal, S. Singh, Y. Kumar and R. S. Srivastava, Phytochemistry, 1989, 28, 611–613 CrossRef CAS.
- V. Holl, D. Coelho, D. Weltin, J. W. Hyun, P. Dufour and P. Bischoff, Anticancer Res., 2000, 20, 3233–3241 CAS.
- L. Comoë, P. Jeannesson, C. Trentesaux, B. Desoize and J.-C. Jardillier, Leuk. Res., 1987, 11, 445–451 CrossRef PubMed.
- S. Patil, S. Kamath, T. Sanchez, N. Neamati, R. F. Schinazi and J. K. Buolamwini, Bioorg. Med. Chem., 2007, 15, 1212–1228 CrossRef CAS PubMed.
- J. Hu, X. Shi, J. Chen, X. Mao, L. Zhu, L. Yu and J. Shi, Food Chem., 2014, 148, 437–444 CrossRef CAS PubMed.
- A.-D. Yapi, N. Desbois, J.-M. Chezal, O. Chavignon, J.-C. Teulade, A. Valentin and Y. Blache, Eur. J. Med. Chem., 2010, 45, 2854–2859 CrossRef CAS PubMed.
- J. Hu, W.-D. Zhang, R.-H. Liu, C. Zhang, Y.-H. Shen, H.-L. Li, M.-J. Liang and X.-K. Xu, Chem. Biodiversity, 2006, 3, 990–995 CrossRef CAS PubMed.
- R. Jangir and N. P. Argade, RSC Adv., 2012, 2, 7087 RSC.
- H. R. Arthur, W. H. Hui and Y. L. Ng, J. Chem. Soc., 1959, 4007–4009 RSC.
- R. Pratap and V. J. Ram, Chem. Rev., 2014, 114, 10476–10526 CrossRef CAS PubMed.
- G. Appendino, S. Gibbons, A. Giana, A. Pagani, G. Grassi, M. Stavri, E. Smith and M. M. Rahman, J. Nat. Prod., 2008, 71, 1427–1430 CrossRef CAS PubMed.
- D. Killander and O. Sterner, Eur. J. Org. Chem., 2014, 2014, 1594–1596 CrossRef CAS.
- L. Zhi, J. D. Ringgenberg, J. P. Edwards, C. M. Tegley, S. J. West, B. Pio, M. Motamedi, T. K. Jones, K. B. Marschke, D. E. Mais and W. T. Schrader, Bioorg. Med. Chem. Lett., 2003, 13, 2075–2078 CrossRef CAS PubMed.
- G. A. Thakur, S. Bajaj, C. Paronis, Y. Peng, A. L. Bowman, L. S. Barak, M. G. Caron, D. Parrish, J. R. Deschamps and A. Makriyannis, J. Med. Chem., 2013, 56, 3904–3921 CrossRef CAS PubMed.
- F. Rafiee, Appl. Organomet. Chem., 2017, 31, e3820 CrossRef.
- R. R. Aleti, A. A. Festa, L. G. Voskressensky and E. V. Van der Eycken, Molecules, 2021, 26, 1–21 CrossRef PubMed.
- L. C. Campeau, M. Parisien, A. Jean and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 581–590 CrossRef CAS PubMed.
- L.-C. Campeau, M. Parisien, M. Leblanc and K. Fagnou, J. Am. Chem. Soc., 2004, 126, 9186–9187 CrossRef CAS PubMed.
- L. C. Campeau, P. Thansandote and K. Fagnou, Org. Lett., 2005, 7, 1857–1860 CrossRef CAS PubMed.
- N. Conde, F. Churruca, R. SanMartin, M. T. Herrero and E. Domínguez, Adv. Synth. Catal., 2015, 357, 1525–1531 CrossRef CAS.
- S. Pimparkar and M. Jeganmohan, Chem. Commun., 2014, 50, 12116–12119 RSC.
- Y. Tanji, N. Mitsutake, T. Fujihara and Y. Tsuji, Angew. Chem., Int. Ed., 2018, 57, 10314–10317 CrossRef CAS PubMed.
- D. E. Ames and A. Opalko, Tetrahedron, 1984, 40, 1919–1925 CrossRef CAS.
- T. Harayama, T. Akiyama and Y. Nakano, Chem. Pharm. Bull., 1997, 45, 1723–1725 CrossRef CAS.
- R. Bernini, S. Cacchi, G. Fabrizi and A. Sferrazza, Synthesis, 2008, 729–738 CAS.
- J. V. Suárez-Meneses, A. Oukhrib, M. Gouygou, M. Urrutigoïty, J.-C. Daran, A. Cordero-Vargas, M. C. Ortega-Alfaro and J. G. López-Cortés, Dalton Trans., 2016, 45, 9621–9630 RSC.
- Q.-F. Hu, T.-T. Gao, Y.-J. Shi, Q. Lei and L.-T. Yu, RSC Adv., 2018, 8, 13879–13890 RSC.
- D. Liang, Z. Hu, J. Peng, J. Huang and Q. Zhu, Chem. Commun., 2013, 49, 173–175 RSC.
- M. Feng, B. Tang, N. Wang, H.-X. Xu and X. Jiang, Angew. Chem., Int. Ed., 2015, 54, 14960–14964 CrossRef CAS PubMed.
- G. W. Wang, T. T. Yuan and D. D. Li, Angew. Chem., Int. Ed., 2011, 50, 1380–1383 CrossRef CAS PubMed.
- J. Karthikeyan and C.-H. Cheng, Angew. Chem., Int. Ed., 2011, 50, 9880–9883 CrossRef CAS PubMed.
- M. Lafrance, D. Lapointe and K. Fagnou, Tetrahedron, 2008, 64, 6015–6020 CrossRef CAS.
- M. C. Ortiz Villamizar, F. I. Zubkov, C. E. Puerto Galvis, L. Y. Vargas Méndez and V. V. Kouznetsov, Org. Chem. Front., 2017, 4, 1736–1744 RSC.
- M. Parisien, D. Valette and K. Fagnou, J. Org. Chem., 2005, 70, 7578–7584 CrossRef CAS PubMed.
- Y. Li, Y.-J. Ding, J.-Y. Wang, Y.-M. Su and X.-S. Wang, Org. Lett., 2013, 15, 2574–2577 CrossRef CAS PubMed.
- S. Santoro, S. I. Kozhushkov, L. Ackermann and L. Vaccaro, Green Chem., 2016, 18, 3471–3493 RSC.
- I. J. S. Fairlamb and N. W. J. Scott, in Nanoparticles in Catalysis, ed. S. Kobayashi, Springer International Publishing, Cham, 2020, pp. 171–205 Search PubMed.
- A. J. Reay and I. J. S. Fairlamb, Chem. Commun., 2015, 51, 16289–16307 RSC.
- K. Philippot and P. Serp, in Nanomaterials in Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2012, pp. 1–54 Search PubMed.
- N. Salarinejad, M. Dabiri and S. K. Movahed, Colloid Interface Sci. Commun., 2022, 47, 100606 CrossRef CAS.
- A. Tyagi, T. Matsumoto, T. Kato and H. Yoshida, Catal. Sci. Technol., 2016, 6, 4577–4583 RSC.
- M. Mokhtar, M. Mostafa, T. S. Saleh, S. M. Bawaked, K. S. Alghamdi and K. Narasimharao, Catalysts, 2022, 12, 1000 CrossRef.
- C. Perez-Rizquez, O. Abian and J. M. Palomo, Chem. Commun., 2019, 55, 12928–12931 RSC.
- V. Kandathil, M. Kempasiddaiah, S. B. Sasidhar and S. A. Patil, Carbohydr. Polym., 2019, 223, 115060 CrossRef CAS PubMed.
- P. Bhattacharjee, A. Dewan, P. K. Boruah, M. R. Das and U. Bora, Sustainable Chem. Pharm., 2023, 33, 101087 CrossRef CAS.
- Y. Zhang, Y. Zhao, Y. Luo, L. Xiao, Y. Huang, X. Li, Q. Peng, Y. Liu, B. Yang, C. Zhu, X. Zhou and J. Zhang, Org. Lett., 2017, 19, 6470–6473 CrossRef CAS PubMed.
- N. Losada-Garcia, A. S. Santos, M. M. B. Marques and J. M. Palomo, Nanoscale Adv., 2023, 5, 513–521 RSC.
- C. Yue, Q. Xing, P. Sun, Z. Zhao, H. Lv and F. Li, Nat. Commun., 2021, 12, 1875 CrossRef CAS PubMed.
- T. J. Williams, A. J. Reay, A. C. Whitwood and I. J. S. Fairlamb, Chem. Commun., 2014, 50, 3052–3054 RSC.
- C. G. Baumann, S. De Ornellas, J. P. Reeds, T. E. Storr, T. J. Williams and I. J. S. Fairlamb, Tetrahedron, 2014, 70, 6174–6187 CrossRef CAS.
- S.-M. Lu and H. Alper, J. Am. Chem. Soc., 2005, 127, 14776–14784 CrossRef CAS PubMed.
- N. Parveen and G. Sekar, J. Org. Chem., 2020, 85, 10514–10524 CrossRef CAS PubMed.
- H. Azizollahi, H. Eshghi and J.-A. García-López, Appl. Organomet. Chem., 2021, 35, e6020 CrossRef CAS.
- F. Ferlin, I. Anastasiou, N. Salameh, T. Miyakoshi, O. Baudoin and L. Vaccaro, ChemSusChem, 2022, 15, e202102736 CrossRef CAS PubMed.
- R. Saha and G. Sekar, J. Catal., 2018, 366, 176–188 CrossRef CAS.
- R. Cano, J. M. Pérez, D. J. Ramón and G. P. McGlacken, Tetrahedron, 2016, 72, 1043–1050 CrossRef CAS.
- E. D. Díaz-Vázquez, S. M. Soria-Castro, I. Della-Cagnoletta, S. E. Martín, G. Oksdath-Mansilla and P. M. Uberman, React. Chem. Eng., 2022, 7, 957–967 RSC.
- P. M. Uberman, L. A. Pérez, G. I. Lacconi and S. E. Martín, J. Mol. Catal. A: Chem., 2012, 363–364, 245–253 CrossRef CAS.
- P. M. Uberman, L. A. Pérez, S. E. Martín and G. I. Lacconi, RSC Adv., 2014, 4, 12330–12341 RSC.
- P. M. Uberman, C. S. García, J. R. Rodríguez and S. E. Martín, Green Chem., 2017, 19, 739–748 RSC.
- C. S. García, P. M. Uberman and S. E. Martín, Beilstein J. Org. Chem., 2017, 13, 1717–1727 CrossRef PubMed.
- C. Biglione, A. L. Cappelletti, M. C. Strumia, S. E. Martín and P. M. Uberman, J. Nanopart. Res., 2018, 20, 127 CrossRef.
- G. Altenhoff and F. Glorius, Angew. Chem., Int. Ed., 2004, 346, 1661–1664 CAS.
- G. Evindar and R. A. Batey, Org. Lett., 2003, 5, 133–136 CrossRef CAS PubMed.
- M. D. Heredia, M. Puiatti, R. A. Rossi and M. E. Budén, Org. Biomol. Chem., 2022, 20, 228–239 RSC.
- A. M. Trzeciak and A. W. Augustyniak, Coord. Chem. Rev., 2019, 384, 1–20 CrossRef CAS.
- A. Bej, K. Ghosh, A. Sarkar and D. W. Knight, RSC Adv., 2016, 6, 11446–11453 RSC.
- A. J. Reay, L. K. Neumann and I. J. S. Fairlamb, Synlett, 2016, 27, 1211–1216 CrossRef CAS.
- M. V. Polynski and V. P. Ananikov, ACS Catal., 2019, 9, 3991–4005 CrossRef CAS.
- A. F. Lee, P. J. Ellis, I. J. S. Fairlamb and K. Wilson, Dalton Trans., 2010, 39, 10473–10482 RSC.
- N. W. J. Scott, M. J. Ford, N. Jeddi, A. Eyles, L. Simon, A. C. Whitwood, T. Tanner, C. E. Willans and I. J. S. Fairlamb, J. Am. Chem. Soc., 2021, 143, 9682–9693 CrossRef CAS PubMed.
- D. O. Prima, N. S. Kulikovskaya, A. S. Galushko, R. M. Mironenko and V. P. Ananikov, Curr. Opin. Green Sustainable Chem., 2021, 31, 100502 CrossRef CAS.
- S. Campisi, M. Schiavoni, C. E. Chan-Thaw and A. Villa, Catalysts, 2016, 6, 185 CrossRef.
- G.-H. Han, S.-H. Lee, M. Seo and K.-Y. Lee, RSC Adv., 2020, 10, 19952–19960 RSC.
- R. Narayanan and M. A. El-Sayed, J. Phys. Chem. B, 2004, 108, 8572–8580 CrossRef CAS.
- R. Narayanan and M. A. El-Sayed, J. Am. Chem. Soc., 2003, 125, 8340–8347 CrossRef CAS PubMed.
- L. Zhao, G. Shen, T. Zhang, Z. Wang and Y. Liang, SynOpen, 2017, 01, 129–137 CrossRef CAS.
- S.-Q. Qin, L.-C. Li, J.-R. Song, H.-Y. Li and D.-P. Li, Molecules, 2019, 24, 437 CrossRef PubMed.
- A. Chaudhary, K. Singh, N. Verma, S. Kumar, D. Kumar and P. P. Sharma, Mini-Rev. Med. Chem., 2022, 22, 2736–2751 CrossRef CAS PubMed.
- J. Barluenga, A. M. Bayón and G. Asensio, J. Chem. Soc., Chem. Commun., 1984, 1334–1335 RSC.
- E. A. Orellana and A. L. Kasinski, Bio-Protoc., 2016, 6, e1984 Search PubMed.
- A. Monks, D. Scudiero, P. Skehan, R. Shoemaker, K. Paull, D. Vistica, C. Hose, J. Langley, P. Cronise, A. Vaigro-Wolff, M. Gray-Goodrich, H. Campbell, J. Mayo and M. Boyd, J. Natl. Cancer Inst., 1991, 83, 757–766 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02835j |
| This journal is © The Royal Society of Chemistry 2024 |