
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBase mediated aza-[2 + 1] annulation and regioselective aziridine ring-opening cascade: mild synthesis of functionalized β-amino ketones from cyclic N-sulfonyl aldimines and α-carbonyl sulfonium salts†
Xiaoqiang Lei *,
Yanyan Sun,
Qinglan Guo and
Jiangong Shi*
*,
Yanyan Sun,
Qinglan Guo and
Jiangong Shi*
State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, China. E-mail: xqlei@imm.ac.cn; shijg@imm.ac.cn
First published on 28th May 2024
Abstract
Cyclic N-sulfonyl aldimines are well-known aza-[2C]-synthons for various [2 + n] annulation reactions. Herein we describe a novel base mediated [2 + 1] annulation and a regioselective aziridine ring-opening reaction cascade, which provides an efficient and distinct synthetic strategy from readily available cyclic N-sulfonyl aldimines and α-carbonyl sulfonium salts leading to β-amino ketone derivatives through the corresponding fused tri-substituted aziridines. This one-pot, two-step process involves formation of C–C and C–N bonds and subsequent cleavage of a C–N bond. The features of the developed reaction include the use of mild reaction conditions, broad substrate scope, and excellent yields. The synthetic utility of this approach was demonstrated by gram-scale operation and further product derivatizations.
Introduction
β-Amino ketones (β-AKs) and related derivatives are ubiquitous substructures of many natural products and biologically active molecules (Fig. 1).1 For example, jasplakinolide is an unusual cyclodepsipeptide natural molecule isolated from Marine Sponge, which can be used as an actin polymerization inducer.1a Ondansetron is a known drug for the treatment of vomiting caused by chemotherapy.1b Eperisone hydrochloride is commonly used to treat spastic paralysis disease.1c Taxine B shows potent antiarrhythmic activity.1d Furthermore, β-Aks have been also identified as versatile building blocks in organic synthesis. For example, they have been widely used for the synthesis of β-amino alcohols, β-amino acids, β-lactams, 1,3-alkamines, and complex nitrogen-containing natural molecules.2 | ||
| Fig. 1 Representative β-amino ketones-containing compounds. | ||
Given their importance and utility in drug discovery and organic synthesis, many methods have been developed for the construction of such scaffolds over the past decades (Scheme 1a). Among them, Mannich-type reactions stand as the most conventional and classical approaches to access β-AKs.3 In addition, aza-Michael addition reaction of α,β-unsaturated ketones with amine-based nucleophiles represent another reliable protocol to access β-AKs motifs.4 In parallel, transition metal-catalyzed reactions, such as coper-catalyzed decarboxylative Mannich reaction,5 palladium-catalyzed aminocarbonylation,6 rhodium-catalyzed hydrogenation of β-ketoenamides,7 and palladium-catalyzed Tsuji–Wacker oxidation of internal allylamines,8 have been also successfully employed in preparing structurally diverse and synthetically useful β-AKs and their related analogues. Despite great advances have been made for such molecule synthesis, most of those methodologies usually required the use of corresponding pre-functionalized substrates, expensive catalysts, and harsh reaction conditions. From a synthetic standpoint, the development of a novel and mild synthetic strategy for the rapid assembly of β-AKs from easily accessible starting materials is still highly desirable and significant.
 | ||
| Scheme 1 Strategies for the synthesis of β-amino ketones. | ||
Because of their versatile reactivities, sulfamate-derived cyclic aldimines have attracted considerable attention from the synthetic community. Recently, they have been extensively used as electrophiles in various transformations, including alkynylation,9 allylation,10 and arylation,11 etc.12 Additionally, sulfamate-derived cyclic aldimines are also useful building blocks for synthesizing β-Aks.5,13 For example, Ma and co-workers described a novel Cu-catalyzed asymmetric decarboxylative Mannich reaction of sulfamate-derived cyclic aldimines with β-ketoacids to synthesize β-Aks (Scheme 1b).5a The group of Wang reported another asymmetric version of the Mannich reaction to achieve chiral β-Aks from cyclic aldimines using cinchona alkaloid as catalyst (Scheme 1c).13c With an electrophilic center (C1) and one nucleophilic center (N2) arranged consecutively in the same molecule, sulfamate-derived cyclic aldimines often serve as ideal aza-[2C]-synthons in a variety of [2 + 2],14 [2 + 3],15 [2 + 4],16 and [2 + 5]17 annulations for constructing diverse four-to seven-membered bicyclic N heterocycles. However, its potential for fused bicyclic aziridine formation has not been fully explored.18,13b The traditional strategies for the synthesis of such type of bicyclic aziridine frameworks mainly rely on the aza-Darzens reactions,19 copper-catalyzed intramolecular aziridination of unsaturated iminoiodinanes,20 and Rh-catalyzed intramolecular aziridinations of alkenes with sulfamate esters.21 On the other hand, owing to the intrinsic ring strain, aziridines can be further converted to functionalized linear compounds via nucleophilic ring-opening reactions to release the ring strain.22 In this context, we envisioned that the expected β-AKs could be prepared from the corresponding benzobicyclic sulfamidate-fused α-carbonyl aziridines through hydrogen-mediated reductive ring-opening reactions, whereas the latter in turn could be obtained via a novel base mediated [2 + 1] annulations of cyclic aldimines with sulfonium salts (Scheme 1d). However, this cascade synthetic strategy, namely, a “sew-and-cut” approach, is much more challenging and which has not been utilized in the synthesis of β-AKs so far. The majority ring-opening reactions for cyclic sulfamidate-fused aziridines typically employ organometallic reagents (e.g., grignard reagents, organolithium reagents) or heteroatom-contained reagents as nucleophiles.23 Comparably, hydrogen-promoted ring-opening processes are still scarce. Furthermore, different types of ring-opening products (7-membered ring vs. 6-membered ring) might be obtained in theory due to regioselectivity of fused aziridines to ring opening.23b Therefore, control of the selectivity of this ring-opening reaction is another challenge. Coincidently, while our research was underway, Lin disclosed a method toward fused aziridines via saccharin-derived cyclic ketimines and sulfur ylides under reflux conditions.24 Herein we describe a cascade [2 + 1] annulation/hydrogen-mediated reductive ring-opening reaction between sulfamate-derived cyclic aldimines and α-carbonyl sulfonium salts to access β-AKs derivatives under mild conditions.
Results and discussion
We initially studied the designed [2 + 1] annulation reaction using sulfamate-derived cyclic aldimine 1a and sulfur ylides precursors dimethyl(2-oxo-2-phenylethyl)sulfonium bromide 2a as the model substrates (Table 1). Treatment of 1a with 2a with potassium carbonate (2.0 equiv.) in dichloromethane at room temperature for 1.5 h, the expected fused aziridine 3a was obtained smoothly in 96% yield with excellent diastereoselectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) (entry 1). Subsequent various inorganic bases screening demonstrated that sodium carbonate exhibited excellent activity, affording the product in 98% yield (entries 2 and 3). Other inorganic bases, including KOH and NaOH shown comparable catalytic activities to give the [2 + 1] product 3a in moderate yields (entries 4 and 5). We then investigated a series of organic bases. To our delight, the commonly used base Et3N delivered the expected 3a in nearly quantitative yield with a significant reaction time decrease (entry 8). Finally, the solvent effect on the reaction was examined. It was revealed that the reaction yield was not improved when polar solvents such as THF and CH3CN were used as the reaction medium (entries 9 and 10).
:1 dr) (entry 1). Subsequent various inorganic bases screening demonstrated that sodium carbonate exhibited excellent activity, affording the product in 98% yield (entries 2 and 3). Other inorganic bases, including KOH and NaOH shown comparable catalytic activities to give the [2 + 1] product 3a in moderate yields (entries 4 and 5). We then investigated a series of organic bases. To our delight, the commonly used base Et3N delivered the expected 3a in nearly quantitative yield with a significant reaction time decrease (entry 8). Finally, the solvent effect on the reaction was examined. It was revealed that the reaction yield was not improved when polar solvents such as THF and CH3CN were used as the reaction medium (entries 9 and 10).
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent | Time (h) | Yield (%) |
| a Reaction conditions: 1a (0.40 mmol), 2a (0.6 mmol) and base (0.80 mmol) in solvent (3.0 mL).b Isolated yield. DMAP = 4-dimethylaminopyridine. DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene. THF = tetrahydrofuran. | ||||
| 1 | K2CO3 | CH2Cl2 | 15 | 96 |
| 2 | Cs2CO3 | CH2Cl2 | 15 | 82 |
| 3 | Na2CO3 | CH2Cl2 | 24 | 98 |
| 4 | KOH | CH2Cl2 | 3 | 75 |
| 5 | NaOH | CH2Cl2 | 3 | 77 |
| 6 | DMAP | CH2Cl2 | 24 | 41 |
| 7 | DBU | CH2Cl2 | 0.5 | Trace |
| 8 | Et3N | CH2Cl2 | 1.5 | 99 |
| 9 | Et3N | THF | 1.5 | 83 |
| 10 | Et3N | CH3CN | 1.5 | 86 |
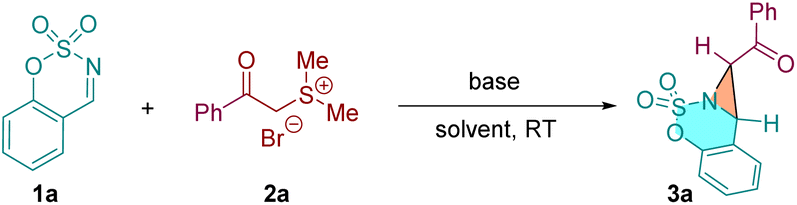
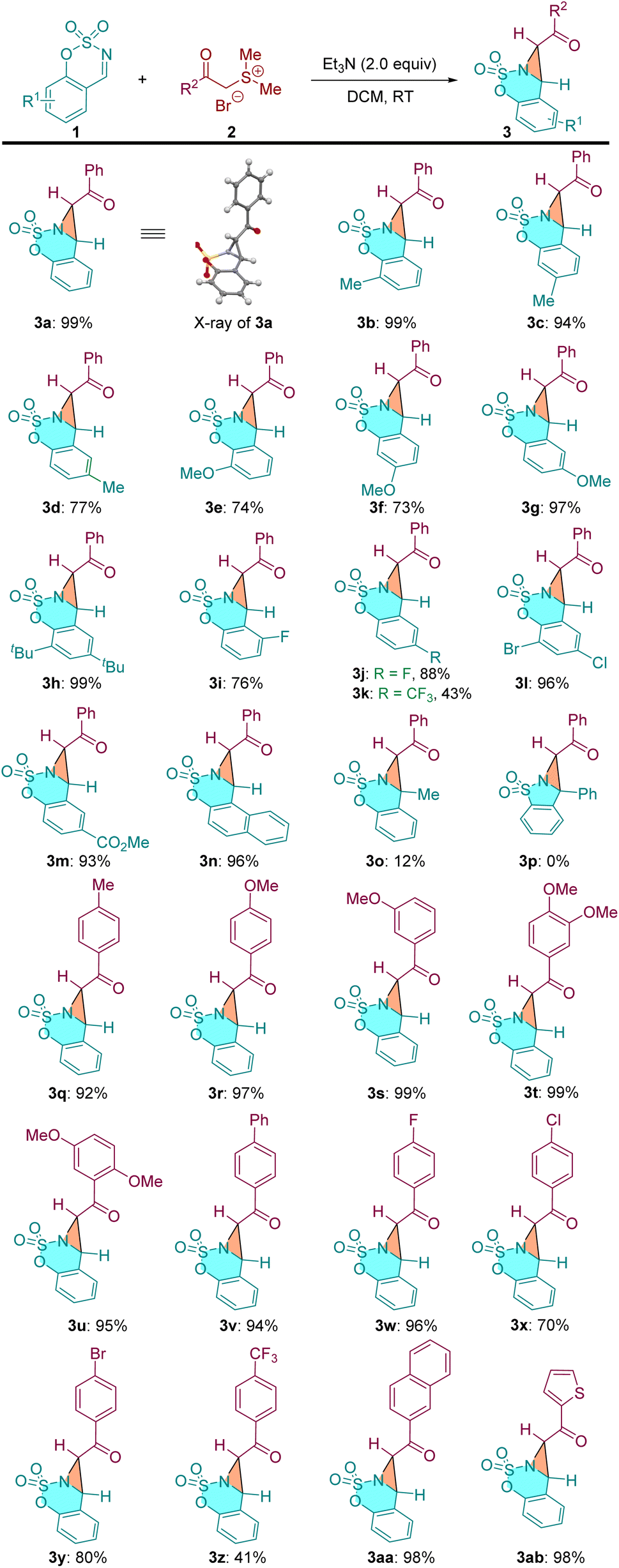
With the optimized reaction conditions in hand, the scope of this [2 + 1] annulation reaction was examined. As shown in Table 2, a variety of cyclic aldimines reacted smoothly with α-carbonyl sulfonium salt 2a to furnish the corresponding products in excellent yields (3a–3n, 73–99% yields). Cyclic aldimines bearing both electron-donating groups (–Me, –OMe, –tBu) and electron-withdrawing groups (–F, –CF3, –Cl, –Br, –CO2Me) on the phenyl ring were all well tolerated under identified conditions. It was found that the substitution position of the substituents on the cyclic aldimines has notable influence on the efficiency of this [2 + 1] annulation. For example, the ortho- and meta-substituted aldimines delivered the desired products 3e and 3f in moderate yields (74% and 73%), while the para-substituted substrate gave 3g in 97% yield. The methylated ketimine provided 3o only in 12% yield, and the phenyl group substituted ketimine did not afford the desired bicyclic aziridine probably due to the steric effect (structure not shown). Notably, the saccharin-derived imine substrate 3p, which has proven to be suitable reactant in various annulations,24 did not give the expected product under our conditions. We then investigated the scope of the α-carbonyl sulfonium salts. A range of substrates bearing diverse substaitutes on the phenyl ring were all compatible to the current reaction conditions (3q–3y). The CF3 substituted substrate gave a slightly lower yield (3z). Beyond phenyl, 1−naphthyl and 1−thienyl derived sulfonium salts were also applicable to this reaction (3aa and 3ab).
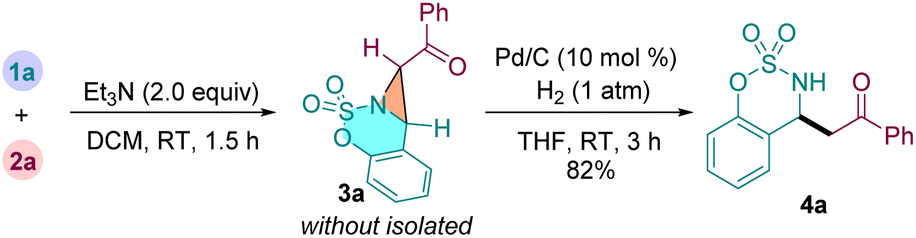
We then turned our attention to explore our designed cascade reactions to synthesize β-AKs from 1a and 2a by one-pot procedure (eqn (1)). Thus, treatment of cyclic aldimine 1a with α-carbonyl sulfonium salt 2a under standard conditions afforded the fused aziridine 3a. Then, the solvent dichloromethane was evaporated and the crude product 3a was directly redissolved in tetrahydrofuran without isolation. Finally, the reaction mixture further stirred at room temperature for 3 h in the presence of Pd/C (10 mol%) under hydrogen atmosphere (1 atm). Gratifyingly, the expected β-amino ketone 4a was isolated in 82% yield based on 1a. This result shows that our designed tandem strategy was feasible for preparing β-AKs.
 | (1) |
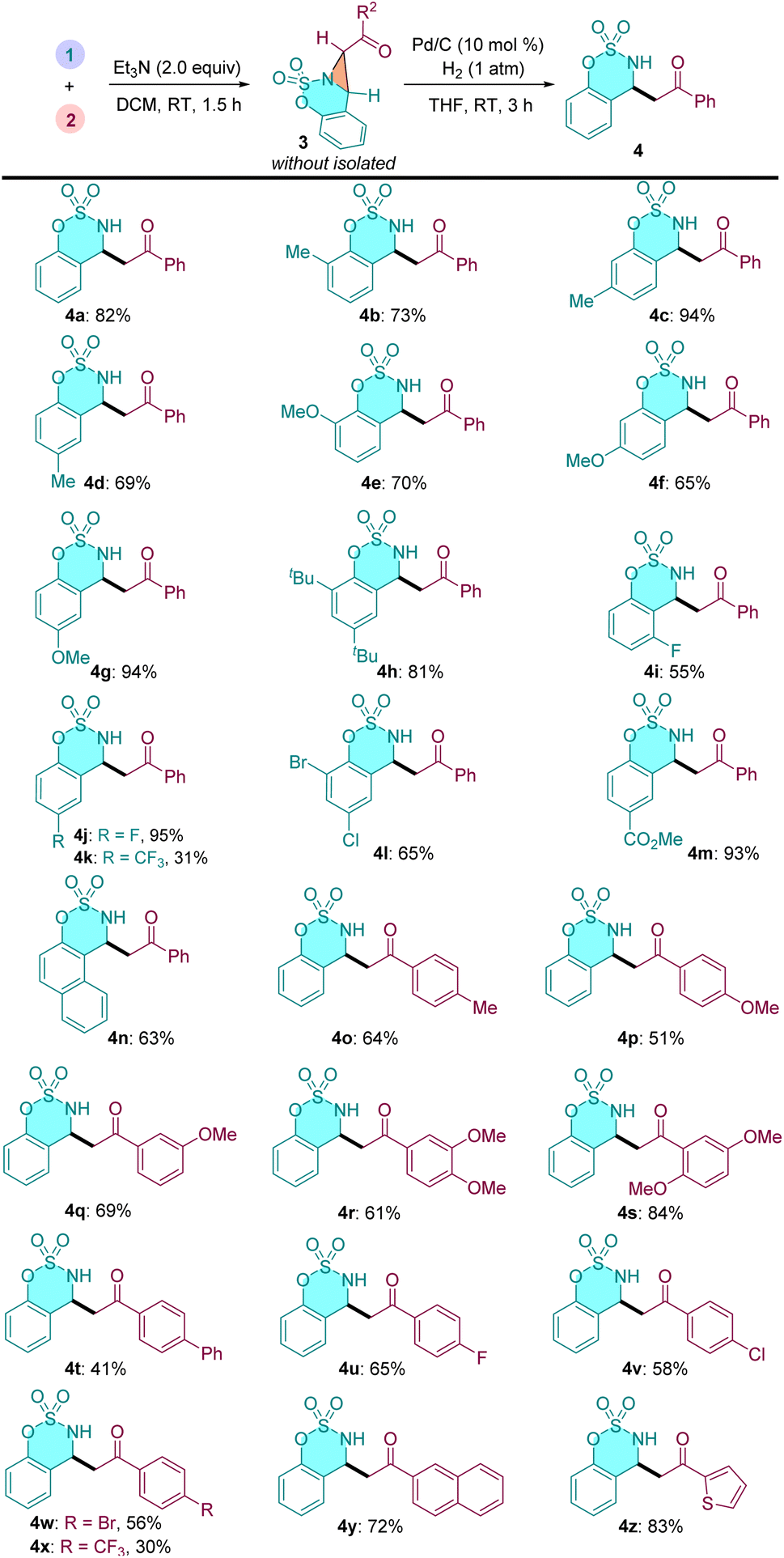
Next, we explored the substrate scope and generality of this one-pot sequential reaction (Table 3). In general, a series of cyclic aldimines and α-carbonyl sulfonium salt smoothly participated in this cascade process under the standard conditions, affording the desired β-amino ketones in moderate to good yields. The electronic property of the substituent on the cyclic aldimine moieties had some impact on this reaction efficiency. For example, cyclic aldimines bearing electron-donating substituents generally gave higher yields than those with electron-withdrawing groups (4b–4h vs. 4i–4l). Meta-methoxy substituted aldimine 4f resulted a slightly lower yield because of an over-reduction byproduct was observed. Similarly, α-carbonyl sulfonium salts with diverse functional groups led to the corresponding β-AKs 4o–4x in moderate to good yields. Some other aromatic rings, such as 4y–4z also can be incorporated into the products.
To demonstrate the synthetic utility of this approach, several transformations of products were carried out (Scheme 2). A 3.0 mmol scale reactions were conducted, 4a (0.69 g) and 4g (0.85 g) were obtained in good yields (Scheme 2a). Subjecting β-amino ketone 4a with hydroxylamine hydrochloride delivered the corresponding ketoxime 5 in 91% yield. The carbonyl group of 4a was reduced by sodium borohydride to generate alcohols 6 and 6' in 99% yield (1.6:1 dr). Treatment of 4a with iodomethane under basic condition afforded methylated 7 in nearly quantitative yield. The free amino group of the 4g was protected by treatment with allyl bromide.
 | ||
| Scheme 2 Synthetic applications. | ||
Based on the previous reports and the above results, a plausible mechanism is described in Scheme 3. In the presence of base, the α-carbonyl sulfonium salt 2 undergoes one molecule of hydrobromic acid elimination to form the sulfur ylide 2′. Then, the ylide 2′ attacks the electron-deficient cyclic aldimine 1a to give the zwitterion intermediates IM1 and IM2 through two possible pathways (path a and b). Subsequent intramolecular SN2 nucleophilic substitution reaction proceeds through the less steric effect intermediate IM1 leads to the trans-trisubstituted fused-aziridine product 3.
 | ||
| Scheme 3 Plausible mechanism. | ||
Conclusions
In summary, we have developed a novel base-promoted formal [2 + 1] annulation reaction of cyclic N-sulfonyl aldimines with α-carbonyl sulfonium salts for the efficient synthesis of bicyclic fused aziridines. With hydrogen as nucleophile, these fused tri-substituted aziridines can be readily transformed to β-amino ketone derivatives in good to excellent yields through ring-opening reactions. Different from the conventional strategies, the developed protocol provides a distinct “sew-and-cut” strategy for the rapid access of β-amino ketones by the strategic utilization of the cascade reactions. This one-pot, two-steps process involves creation of C–C and C–N bonds and subsequent selectivity cleavage of a C–N bond. The mild reaction conditions enabled such reaction to compatible with a broad range of functional groups. The synthetic utility of this chemistry was demonstrated by gram-scale operation and further products derivatizations.Author contributions
X. Lei and J. Shi conceived and directed the project. X. Lei designed experiments and X. Lei and Y. Sun performed experiments, analysed the experimental data. X. Lei wrote the manuscript and J. Shi revised the manuscript. Q. Guo discussed the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial supports from the National Science Foundation of China [82293685, 82293682 (82293680), and 22201306] and CAMS Innovation Fund for Medical Sciences (CIFMS) (2021-I2M-1-028).Notes and references
-
(a) S. J. Robinson, B. I. Morinaka, T. Amagata, K. Tenney, W. M. Bray, N. C. Gassner, R. S. Lokey and P. Crews, J. Med. Chem., 2010, 53, 1651–1661 CrossRef CAS PubMed
; (b) R. J. Milne and R. C. Heel, Drugs, 1991, 41, 574–595 CrossRef CAS PubMed
-
(a) J. Barluenga, E. Aguilar, S. Fustero, B. Olano and A. L. Viado, J. Org. Chem., 1992, 57, 1219–1223 CrossRef CAS
- For selected reviews, see:
(a) M. Arend, B. Westermann and N. Risch, Angew. Chem., Int. Ed., 1998, 37, 1044–1070 CrossRef
- For selected reviews and examples, see:
(a) D. Colantoni, S. Fioravanti, L. Pellacani and P. A. Tardella, Org. Lett., 2004, 6, 197–200 CrossRef CAS PubMed
-
(a) H.-X. Zhang, J. Nie, H. Cai and J.-A. Ma, Org. Lett., 2014, 16, 2542–2545 CrossRef CAS PubMed
- J. Cheng, X. Qi, M. Li, P. Chen and G. Liu, J. Am. Chem. Soc., 2015, 137, 2480–2483 CrossRef CAS PubMed
-
(a) H. Geng, K. Huang, T. Sun, W. Li, X. Zhang, L. Zhou, W. Wu and X. Zhang, J. Org. Chem., 2011, 76, 332–334 CrossRef CAS PubMed
- M. Garbacz and S. Stecko, Org. Biomol. Chem., 2023, 21, 115–126 RSC
- Y.-L. Li, J.-X. Liu, X.-P. Chen, Y. Zhou, Y.-C. Xiao and F.-E. Chen, Adv. Synth. Catal., 2020, 362, 3202–3207 CrossRef CAS
-
(a) Y. Luo, A. J. Carnell and H. W. Lam, Angew. Chem., Int. Ed., 2012, 51, 6762–6766 CrossRef CAS PubMed
-
(a) T. Nishimura, A. Noishiki, C. C. Tsui and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 5056–5059 CrossRef CAS PubMed
-
(a) A.-J. Xia, T.-R. Kang, L. He, L.-M. Chen, W.-T. Li, J.-L. Yang and Q.-Z. Liu, Angew. Chem., Int. Ed., 2016, 55, 1441–1444 CrossRef CAS PubMed
-
(a) S. Zhang, L. Li, Y. Hu, Z. Zha, Z. Wang and T.-P. Loh, Org. Lett., 2015, 17, 1050–1053 CrossRef CAS PubMed
- R.-R. Liu, J.-P. Hu, J.-J. Hong, C.-J. Lu, J.-R. Gao and Y.-X. Jia, Chem. Sci., 2017, 8, 2811–2815 RSC
- For selected examples, see:
(a) M. Rommel, T. Fukuzumi and J. W. Bode, J. Am. Chem. Soc., 2008, 130, 17266–17267 CrossRef CAS PubMed
- For selected examples, see:
(a) P.-C. Zheng, J. Cheng, S. Su, Z. Jin, T.-H. Wang, S. Yang, L.-H. Jin, B.-A. Song and Y. R. Chi, Chem.–Eur. J., 2015, 21, 9984–9987 CrossRef CAS PubMed
- Y. Wu, C. Yuan, C. Wang, B. Mao, H. Jia, X. Gao, J. Liao, F. Jiang, L. Zhou, Q. Wang and H. Guo, Org. Lett., 2017, 19, 6268–6271 CrossRef CAS PubMed
- B.-H. Zhu, J.-C. Zheng, C.-B. Yu, X.-L. Sun, Y.-G. Zhou, Q. Shen and Y. Tang, Org. Lett., 2010, 12, 504–507 CrossRef CAS PubMed
-
(a) L. Degennaro, P. Trinchera and R. Luisi, Chem. Rev., 2014, 114, 7881–7929 CrossRef CAS PubMed
-
(a) P. Dauban and R. H. Dodd, Org. Lett., 2000, 2, 2327–2329 CrossRef CAS PubMed
- K. Guthikonda, P. M. Wehn, B. J. Caliando and J. Du Bois, Tetrahedron, 2006, 62, 11331–11342 CrossRef CAS
- S. Stanković, M. D’hooghe, S. Catak, H. Eum, M. Waroquier, V. V. Speybroeck, N. D. Kimpe and H.-J. Ha, Chem. Soc. Rev., 2012, 41, 643–665 RSC
-
(a) F. Duran, L. Leman, A. Ghini, G. Burton, P. Dauban and R. H. Dodd, Org. Lett., 2002, 4, 2481–2483 CrossRef CAS PubMed
- C. Lin, M.-C. Wang, P. Wie, Q. Xing and C.-H. Liu, Eur. J. Org Chem., 2023, 26, e202300300 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data of new compounds, and NMR spectra. CCDC 2334209. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra02817a |
| This journal is © The Royal Society of Chemistry 2024 |