
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sodium halide solid state electrolyte of Na3YBr6 with low activation energy
Xiang-Yu Niua,
Xin-Yi Doua,
Cheng-Yu Fua,
Yi-Chun Xu*b and
Xu-Yong Feng *a
*a
aSchool of Materials Science and Engineering, Anhui Provincial Key Laboratory of Advanced Functional Materials and Devices, Hefei University of Technology, Hefei 230009, Anhui, P. R. China. E-mail: 2021800026@hfut.edu.cn
bKey Laboratory of Materials Physics, Institute of Solid-State Physics, Chinese Academy of Sciences, P. O. Box 1129, Hefei 230031, P. R. China. E-mail: xuyichun@issp.ac.cn
First published on 7th May 2024
Abstract
Halide solid-state electrolytes (SSEs) are considered promising candidates for practical applications in all-solid-state batteries (ASSBs), due to their outstanding high voltage stability and compatibility with electrode materials. However, Na+ halide SSEs suffer from low ionic conductivity and high activation energy, which limit their applications in sodium all-solid-state batteries. Here, sodium yttrium bromide solid-state electrolytes (Na3YBr6) with a low activation energy of 0.15 eV is prepared via solid state reaction. Structure characterization using X-ray diffraction reveals a monoclinic structure (P21/c) of Na3YBr6. First principle calculations reveal that the low migration activation energy comes from the larger size and vibration of Br− anions, both of which expand the Na+ ion migration channel and reduce its activation energy. The electrochemical window of Na3YBr6 is determined to be 1.43 to 3.35 V vs. Na/Na+, which is slightly narrower than chlorides. This work indicates bromides are a good catholyte candidate for sodium all solid-state batteries, due to their low ion migration activation energy and relatively high oxidation stability.
1. Introduction
All solid-state batteries (ASSBs) have attracted much attention in the past decades, due to their high safety and high energy density.1–6 The main difference between traditional batteries and ASSBs is the use of solid-state electrolytes (SSEs) to replace organic liquid electrolytes, which brings higher safety and better stability. However, SSEs also have new technique and economic issues, which hinder the commercialization of ASSBs. For example, the contact between SSEs and solid electrodes is poor, leading to high interface resistance. Furthermore, this contact would be even worse with volume change of electrodes during cycling, making ASSBs decay fast.2,7–9 Also, the Li+ ion concentration is always ten times higher than in liquid electrolytes, which increases the cost of ASSBs obviously.Sodium has a similar ionic radius and potential, but is more abundant than lithium. The replacement of Li with Na can significantly reduce the cost while the electrochemical performance can be maintained to a large extent.10–14 However, similar technical issues still exist in Na based ASSBs. Since these issues are caused by the use of SSEs, developing high-performance SSEs is expected to solve these issues. In the past decades, several types Na ion SSEs have been developed and applied in Na ASSBs, such as oxides (NASICON, etc.),15–19 sulfides (Na3PS4, etc.),20–23 halides (Na2ZrCl6),13,24,25 borides26–30 and polymers.31 However, these electrolytes all have certain issues. For example, polymers are soft and easy to process, posing good compatibility to electrode materials and low cost, but the ionic conductivity is poor at room temperature.31 Sulfides have high ionic conductivity up to 10−2 S cm−1, which is comparable or even higher than liquid electrolytes, but the chemical and electrochemical stability is poor, leading to poor cycle performance when combining with high voltage cathodes.14 Oxides and halides are both stable against high voltage cathodes, but the rigid feature of oxides makes the poor contact to electrodes,32 while the ionic conductivity of halides is poor.33,34 The most reported sodium halide SSEs are chlorides, such as Na3YCl6,35 Na2ZrCl6,13,25 NaAlCl4 (ref. 33) etc. In such materials, the diffusion bottleneck is too small for Na+ ion diffusion, leading to high activation energy and low ionic conductivity at room temperature. To broaden the diffusion bottleneck and enhance the ion conduction, anions with bigger size can be applied, such as Br−, I− etc. Considering the oxidation stability, Br− would be a good option for Na halide SSEs. Several bromides have been predicted to have higher ionic conductivity, such as Na3YBr6, Na3MBr6 etc.35–37 But little experimental results have proved this.
In this paper, Na3YBr6 with space group of P21/c was synthesized via solid state reaction, which shows different ion conduction characteristics to previous simulation results. A much lower ionic conductivity (4.57 × 10−8 S cm−1) combined relatively low activation energy (0.15 eV) was achieved. This indicates the presence of fast ion migration paths in Na3YBr6, but the mobility of Na+ ions is poor. Theoretical simulations indicate this low migration activation energy is due to the vibration of Br− ions during Na+ ion migration, which further broadens the size of diffusion bottleneck and enables a fast Na ion migration path. Further enhancement in ionic conductivity can be achieved by increasing the mobility of Na+ ions, such as local structure rearrangement. Cyclic voltammetry test proves a relatively high oxidation stability up to 3.3 V vs. Na/Na+ for Na3YBr6.
2. Experiments
The raw materials of NaBr, YBr3 was purchased from Aladdin and used without further purification. Stoichiometric NaBr and YBr3 were weighed and hand milled for around 30 min to obtain an even mixture. This mixture was further sintered in a sealed quartz tube at different temperatures for 10 h to obtain sintered Na3YBr6. Then the sample with highest ionic conductivity was further ball milled at 500 rpm for another 10 h to optimize the structure and ionic conductivity.Na2.9PS3.9Cl0.1 was prepared according to our previous report.23 The raw materials of Na2S, P2S5 and NaCl purchased from SINOPHARM was mixed according to stoichiometric ratio and ball milled at 500 rpm for 10 h. Then the mixture was sintered at 420 °C for 10 h to obtain final products.
The structure and phase purity were characterized with powder X-ray diffraction (PXRD). The as-sintered powders were placed on a glass holder, flatted and covered by a thin Kapton film to ensure an inert environment. The PXRD patterns were acquired on a PANalytical X'Pert PRO powder X-ray diffractometer at 45 kV and 40 mA at room temperature by using a Cu Kα radiation (λ = 1.5406 Å) with a fixed scanning speed of 2° min−1 from 10° to 80°.
The ionic conductivities and activation energies of Na3YBr6 samples were measured with Ac impedance spectroscopy (EIS) at variable temperatures, in the frequency range from 1 MHz to 1 Hz and a potential perturbation of 50 mV (Biologic SP-200). The as-prepared Na3YBr6 samples were cold pressed under a pressure of 300 MPa into pellets with a diameter of 12 mm and thickness of around 1 mm. The pellets were sandwiched by two steel rods for EIS measurements. The electronic conductivity was also measured using the same cell, with applying a voltage of 0.5 V and collecting current till it is stable.
The electrochemical stability of Na3YBr6 was tested with linear scanning voltammetry. The Na3YBr6 sample was hand milled with 30 wt% Super P and used as the cathode, Na2.9PS3.9Cl0.1 (ref. 23 and 38) was used as solid-state electrolyte and Na was used as anode. These three layers were pressed under 300 MPa in a cylinder cell respectively, with a diameter of 12 mm. The mass of different layers was 10 mg (Na3YBr6), 100 mg (Na2.9PS3.9Cl0.1) and 200 mg (Na) respectively. The cells were charged to 5 V or discharged to 0 V vs. Na/Na+, with a scan rate of 1 mV s−1.
To get insights into the migration of ion migration in atomic level, density functional theory based first-principles calculations were performed by the Vienna Ab initio Simulation Package (VASP) code.39 The interaction between ion and electron is described by the projector augmented wave (PAW) method.40 The generalized gradient approximation (GGA) with Perdew–Burke–Ernzerhol (PBE) function41 was used for the exchange correlation energy. A plane wave energy cutoff of 520 eV with 8 × 6 × 4 k-point meshes are applied to optimized the structure of Na3YBr6 until the force convergence to less than 0.01 eV Å−1. Based on the optimized structure, the transition states and energy barriers of diffusion paths were calculated by the Climbing Image Nudged Elastic Band (CINEB) in the VTST package.42,43
3. Results and discussions
After high-temperature sintering, pure Na3YBr6 can be obtained, as shown in Fig. 1a. The XRD pattern for Na3YBr6 samples sintered between 400 °C and 500 °C are similar, all can be indexed to the pure Na3YBr6 with monoclinic structure (P21/c). The lattice parameters are 7.17 Å (a), 7.67 Å (b), 12.84 Å (c) and 123.16° (β) (PDF# 47-1223). Comparing to Na3YCl6 (504.51 Å3, Z = 2),44 the unit cell volume of Na3YBr6 is bigger (591.22 Å3, Z = 2), indicating a larger size of Na+ ion migration path in Na3YBr6. When the temperature increases to 550 °C, the XRD changes significantly. For example, peaks around 27° emerge together and peak at 50° shifts and increases significantly, which indicates a structure change or decomposition of Na3YBr6.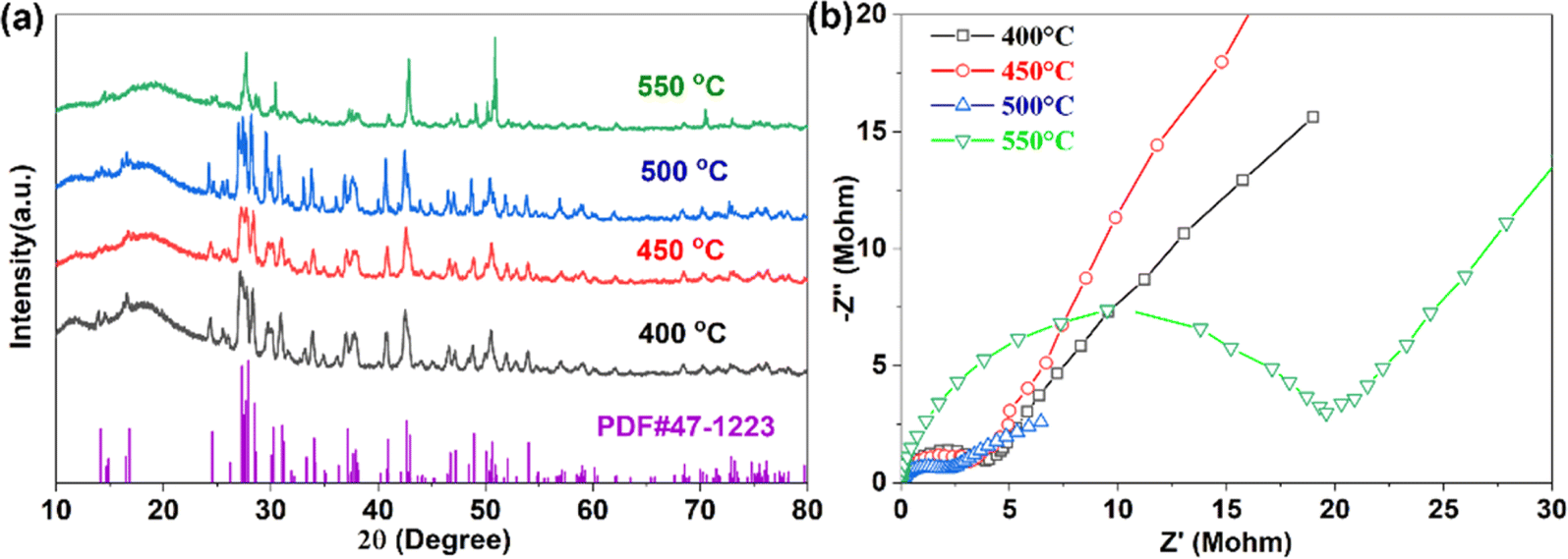 | ||
| Fig. 1 XRD pattern (a) and EIS (b) of Na3YBr6 sintered at different temperatures. | ||
The ionic conductivities of Na3YBr6 samples are calculated based on EIS (Fig. 1b), which shows only one clear semicircle. The grain boundary resistance maybe small and negligible. So we only consider the bulk resistance here. When temperature increases from 400 °C to 500 °C, the resistance of Na3YBr6 continuously decreases from 4 MΩ to about 2 MΩ, the corresponding ionic conductivity increases from 2.38 × 10−8 S cm−1 to 4.57 × 10−8 S cm−1. This indicates that Na3YBr6 powders are nonionic conductive, which is contradicting to the simulation results.36 As the temperature increases to 550 °C, the decomposition of Na3YBr6 (XRD pattern in Fig. 1a) leads to a significant decrease in its ionic conductivity to 5.10 × 10−9 S cm−1.
SEM images of different samples are shown in Fig. 2. When sintered at 450 °C, the particles aggregate tightly. The irregular particle shape indicates the low crystallinity (Fig. 2a), which is consistent to the XRD pattern. When temperature increases, the particle size grows to hundreds of nanometers (Fig. 2b and c). When the temperature continues to rise to 550 °C, the decomposition of the product reduces the particle size, and the fast ion diffusion at high temperatures further aggregates the particles together (Fig. 2d).
 | ||
| Fig. 2 SEM images of Na3YBr6 sintered at 400 °C (a), 450 °C (b), 500 °C (c), 550 °C (d), and Na3YBr6 ball milled at 500 rpm after sintering at 500 °C (e and f). | ||
For crystal materials with low ionic conductivity, local structure disordering after ball milling can significantly enhance the ionic conductivity, by changing the site energy and distribution of charge carriers at different sites.5,45 Here, the 500 °C sintered sample was further ball milled at 500 rpm for 10 h. After ball milling at 500 rpm for 10 h, the XRD pattern shows that the sample still maintains the Na3YBr6 structure, as shown in Fig. 3a. The difference is that the diffraction peaks become wider after ball milling, resulting in the adjacent peaks overlapping together. The broad peaks also indicate a decrease in crystal size and an increase in disordering in the Na3YBr6 sample. As shown in Fig. 2d and e, the particle size of Na3YBr6 significantly decreases after ball milling and these particles significantly agglomerate under pressure. Meanwhile, cracks between aggregates form, as shown in Fig. 2d and e. The tightly contact between small particles may help to reduce the grain boundary resistance and enhance the total ionic conductivity. Ionic conductivity can be significantly enhanced, from 4.57 × 10−8 S cm−1 to 2.84 × 10−6 S cm−1 (Fig. 3b), which is even higher than Na3YCl6 from literature.
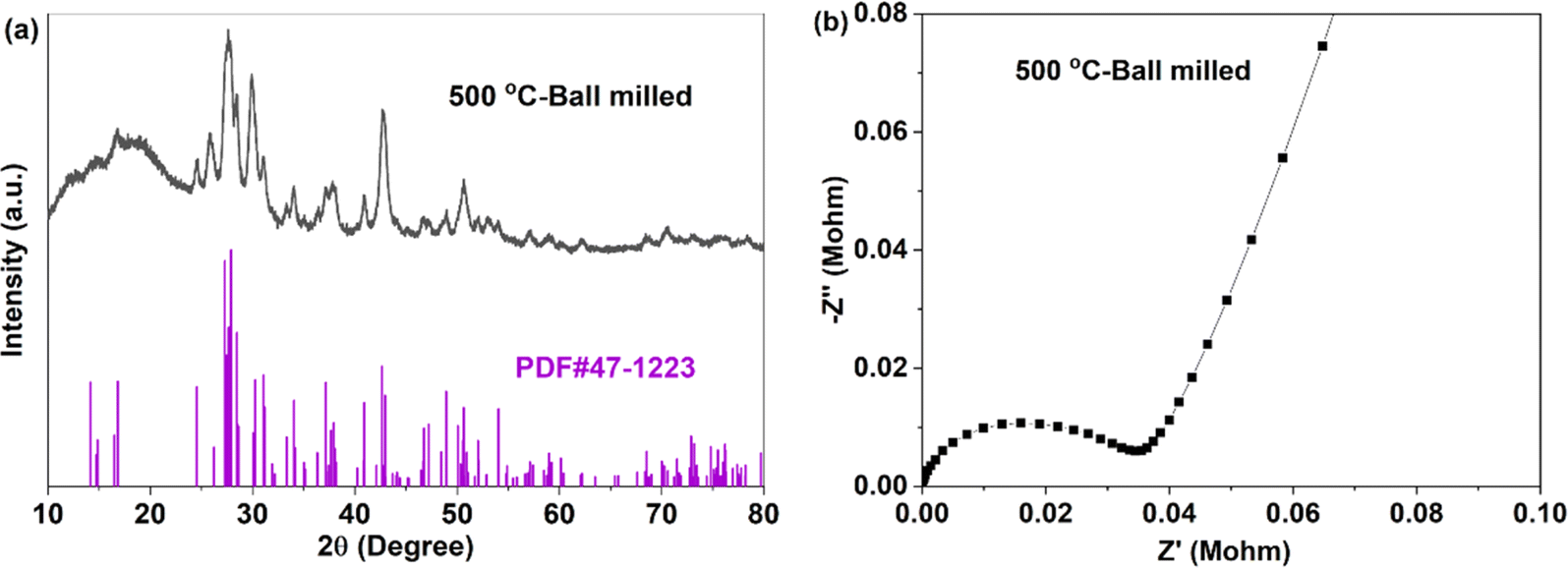 | ||
| Fig. 3 XRD pattern (a) and EIS (b) of Na3YBr6 sintered at 500 °C and ball milled at 500 rpm for 10 h. | ||
Variable temperature EIS measurements were further conducted to obtain ion migration activation energy of different samples, as shown in Fig. 4. When temperature increases, the impedance decreases (Fig. 4a and b), indicating an increase of ionic conductivity. The relationship between ionic conductivity and temperature follows the Arrhenius formula 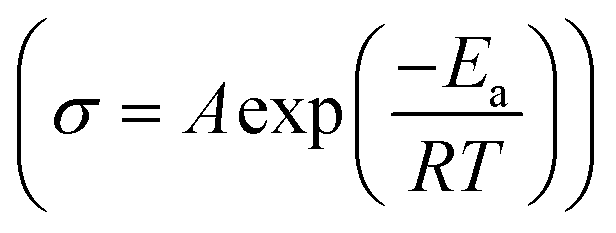 within a certain temperature range. That is, the logarithm of ion conductivity is proportional to the reciprocal of temperature, and the slope is related to the migration activation energy. The activation energy can be calculated based on the Arrhenius plots (Fig. 4c), which is 0.15 eV for the 500 °C sample and 0.36 eV after ball milling. Although the activation energy of Na3YBr6 is low and comparable to other fast ion conductors. The pre-exponential factor A is much lower than other fast ion conductors, which is 3.7 × 10−5 for the 500 °C sample and 2.8 for the 500 °C sample after ball milling (Fig. 4c). This means Na+ ions in the Na3YBr6 sample are quite stable and have a low frequency to migrate to surrounding sites. As a result, the ionic conductivity of Na3YBr6 is still low (4.57 × 10−8 S cm−1) with such low activation energy (0.15 eV). According to the Arrhenius formula, the ionic conductivity is affected with two factors, pre-exponential factor A and activation energy Ea. Here, the pre factor A is positively correlated with the concentration of charge carriers and the jumping frequency of charge carrier to the surrounding sites. The migration activation energy is related to the structure change along the migration path. The ball milling would change the local structure around Na+ ions, which reduces the stability of Na+ ions in Na3YBr6 and increase their jumping frequency to adjacent sites (A). Meanwhile, the distortion may increase the migration activation energy (Ea). Here, the combined effect of both is an increase in ion conductivity, similar to the Ca2+ doped Na3PS4.46 Further enhancement in ionic conductivity can increase the site energy and thus migration frequency of Na+ ions through microstructure optimization, such as anion doping or lattice parameter adjustment. As solid-state electrolytes, low electronic conductivity is necessary. Here, the electronic conductivity was measured and the result is shown in Fig. 4d. It is calculated that the electronic conductivity of 500 °C sample after ball milling is about 4.2 × 10−10 S cm−1, which is four orders of magnitude lower than the ionic conductivity.
within a certain temperature range. That is, the logarithm of ion conductivity is proportional to the reciprocal of temperature, and the slope is related to the migration activation energy. The activation energy can be calculated based on the Arrhenius plots (Fig. 4c), which is 0.15 eV for the 500 °C sample and 0.36 eV after ball milling. Although the activation energy of Na3YBr6 is low and comparable to other fast ion conductors. The pre-exponential factor A is much lower than other fast ion conductors, which is 3.7 × 10−5 for the 500 °C sample and 2.8 for the 500 °C sample after ball milling (Fig. 4c). This means Na+ ions in the Na3YBr6 sample are quite stable and have a low frequency to migrate to surrounding sites. As a result, the ionic conductivity of Na3YBr6 is still low (4.57 × 10−8 S cm−1) with such low activation energy (0.15 eV). According to the Arrhenius formula, the ionic conductivity is affected with two factors, pre-exponential factor A and activation energy Ea. Here, the pre factor A is positively correlated with the concentration of charge carriers and the jumping frequency of charge carrier to the surrounding sites. The migration activation energy is related to the structure change along the migration path. The ball milling would change the local structure around Na+ ions, which reduces the stability of Na+ ions in Na3YBr6 and increase their jumping frequency to adjacent sites (A). Meanwhile, the distortion may increase the migration activation energy (Ea). Here, the combined effect of both is an increase in ion conductivity, similar to the Ca2+ doped Na3PS4.46 Further enhancement in ionic conductivity can increase the site energy and thus migration frequency of Na+ ions through microstructure optimization, such as anion doping or lattice parameter adjustment. As solid-state electrolytes, low electronic conductivity is necessary. Here, the electronic conductivity was measured and the result is shown in Fig. 4d. It is calculated that the electronic conductivity of 500 °C sample after ball milling is about 4.2 × 10−10 S cm−1, which is four orders of magnitude lower than the ionic conductivity.
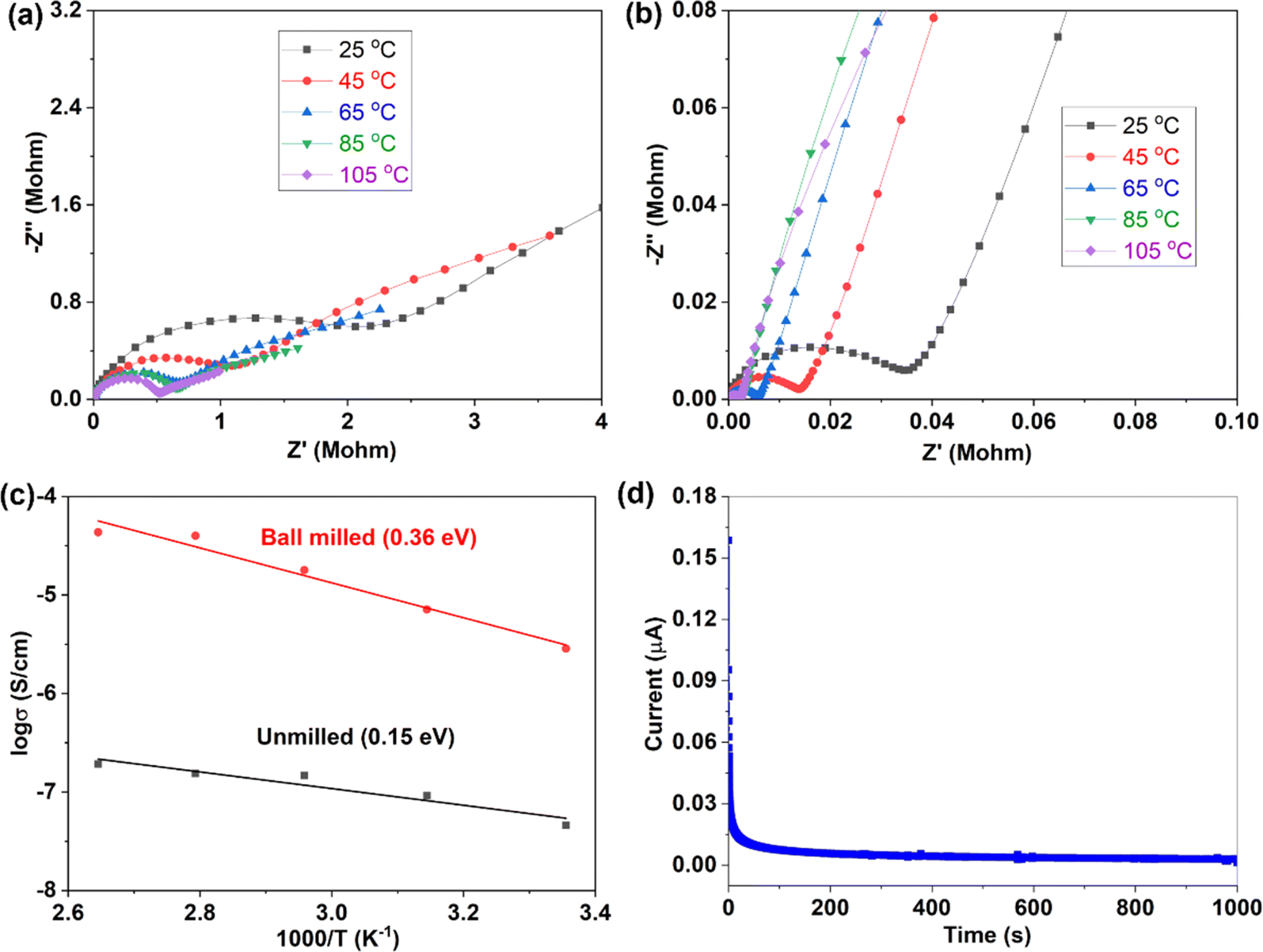 | ||
| Fig. 4 Variable temperature EIS of 500 °C samples before (a) and after (b) ball milling, (c) Arrhenius plots of 500 °C samples before and after ball milling, (d) DC polarization curve of 500 °C sample after ball milling. | ||
Since the activation energy of Na3YBr6 measured here is much lower than the predicted value (∼0.43 eV) from BVSE method, which is much higher than the experimental results here (0.15 eV).36 Here, the relaxation of atoms next to Na+ during migration is considered, which should be closer to the actual migration process. The crystal structure of Na3YBr6 (monoclinic P21/n) was built according to the previous work36 and optimized to obtain the possible migration path way as shown in Fig. 5a. The energy barriers for Na+ migrating along the Na1–Na2–Na1′–Na2′–Na1 vary from 0.22 eV to 0.28 eV as shown in Fig. 5b, which is more consistent with the experimental results. Theoretical simulation results indicate that the relaxation of Br− ions in bromide solid electrolytes plays an important role in reducing the activation energy of sodium ion migration and promoting the conduction of Na+ ions, which cannot be ignored in theoretical simulations.
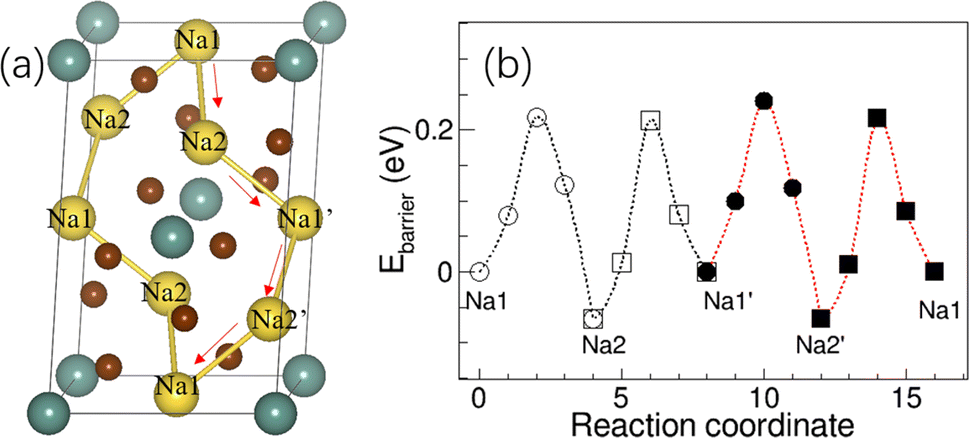 | ||
| Fig. 5 (a) The structure of Na3YBr6 and diffusion path of Na+, (b) the migration energy barrier of Na+ in the Na3YBr6. | ||
The electrochemical stability of Na3YBr6 is also tested with linear scanning voltammetry and the result is shown in Fig. 6. To improve the activity of the Na3YBr6 sample and more accurately reflect its electrochemical stability, conductive carbon is added to Na3YBr6. When the anion changes from Cl− to Br− (Na3YCl6 vs. Na3YBr6), the oxidation potential is supposed to be reduced. For Na3YBr6, it starts to oxidize at around 3.35 V vs. Na/Na+, which is comparable to theoretical results and lower than that of Na3YCl6.35 This is because in halides, oxidation reactions usually occur on anions, so Br− with poor oxidation stability gives bromide (Na3YBr6) a lower oxidation potential to chlorides. The reduction potential of Na3YBr6 is simulated to be 0.57 V vs. Na/Na+.35,36 However, it is as high as 1.43 V vs. Na/Na+ according to our experimental results. This means the Na3YBr6 should be suitable for catholyte but not for anolyte.
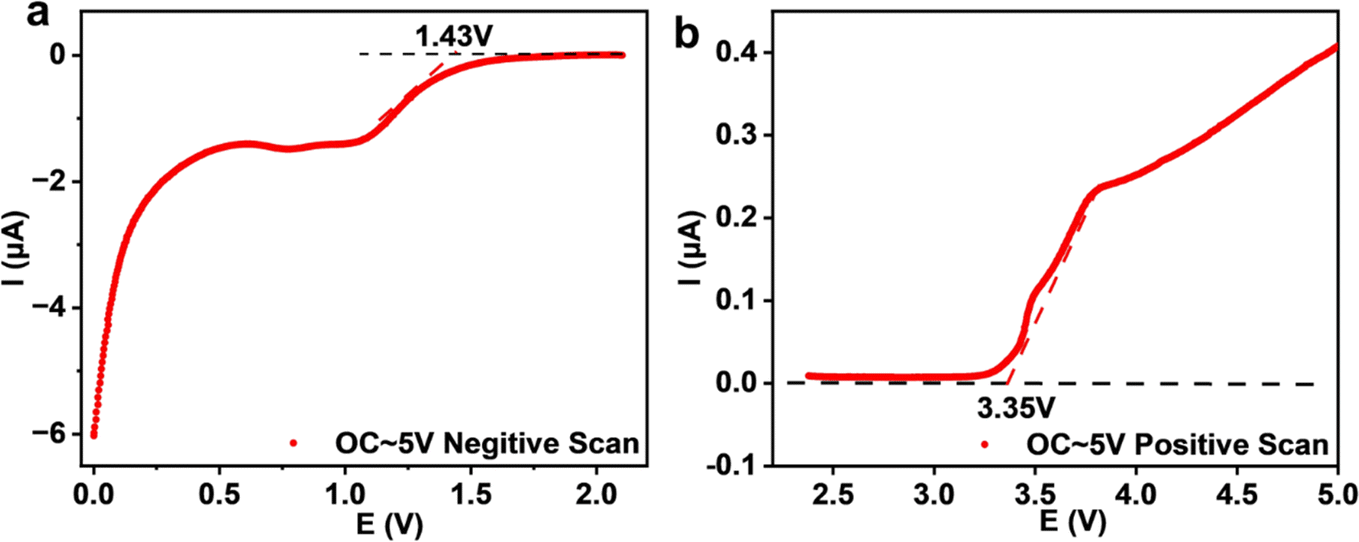 | ||
| Fig. 6 Linear scanning voltammetry of a Na|Na2.9PS3.9Cl0.1|Na3YBr6 + C cell. | ||
4. Conclusions
In summary, Na3YBr6 with a low activation energy (0.15 eV) is prepared via solid state reaction. Structure characterizations reveal a monoclinic structure (P21/c) of Na3YBr6, which has a bigger unit cell volume comparing to that of Na3YCl6. Experimental results show relatively low activation energy of 0.15 eV in sintered Na3YBr6 and 0.36 eV in ball milled Na3YBr6. Theoretical simulation results reveal that this low ion migration activation energy is due to the larger size of Br− anion and the relaxation of Br− anion during Na+ migration. The ionic conductivities of Na3YBr6 are measured to be 4.57 × 10−8 S cm−1 and 2.84 × 10−6 S cm−1 before and after ball milling, due to the low pre-exponential factor. Further enhancement in ionic conductivity can be achieved by local structure arrangement to increase the site energy and mobility of Na+ ions. The electronic conductivity of Na3YBr6 is measured to be 4.2 × 10−10 S cm−1, which is typically electronic insulator. Finally, the electrochemical stable window of Na3YBr6 is determined to be 1.43–3.35 V vs. Na/Na+, which should be stable against most cathode materials.Author contributions
X. F. and Y. X. conceived the research. X. N., X. D. and C. F. performed the materials synthesis, conductivity measurement, structural characterization and electrochemical tests. Y. X. conducted theoretical simulations. X. F. wrote the manuscript under the assistance of X. N., X. D., Y. X. and C. F.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Major Science and Technology Projects in Anhui Province (202203a05020032 and 2022e03020004), the Fundamental Research Funds for the Central Universities (JZ2022HGTB0251) and Innovation and Entrepreneurship Project for College Students (202210359011).References
- V. Thangadurai and B. Chen, Chem. Mater., 2022, 34, 6637–6658 CrossRef CAS.
- T. Famprikis, P. Canepa, J. A. Dawson, M. S. Islam and C. Masquelier, Nat. Mater., 2019, 18, 1278–1291 CrossRef CAS PubMed.
- T. Krauskopf, F. H. Richter, W. G. Zeier and J. Janek, Chem. Rev., 2020, 120, 7745–7794 CrossRef CAS PubMed.
- X. Zhan, J. F. Bonnett, M. H. Engelhard, D. M. Reed, V. L. Sprenkle and G. Li, Adv. Energy Mater., 2020, 10, 2001378 CrossRef CAS.
- J. Zhang, C. Lou, L. Dong, W. Ping, H. Xiang, M. Tang and X. Feng, Small Struct., 2024, 2300565 CrossRef.
- X. Zhao, Z. Zhao-Karger, M. Fichtner and X. Shen, Angew. Chem., Int. Ed., 2020, 59, 5902–5949 CrossRef CAS PubMed.
- J. Janek and W. G. Zeier, Nat. Energy, 2023, 8, 230–240 CrossRef.
- N. Zhao, W. Khokhar, Z. Bi, C. Shi, X. Guo, L.-Z. Fan and C.-W. Nan, Joule, 2019, 3, 1190–1199 CrossRef CAS.
- Y. Gao, A. M. Nolan, P. Du, Y. Wu, C. Yang, Q. Chen, Y. Mo and S.-H. Bo, Chem. Rev., 2020, 120, 5954–6008 CrossRef CAS PubMed.
- Z. Liu, J. Shen, S. Feng, Y. Huang, D. Wu, F. Li, Y. Zhu, M. Gu, Q. Liu, J. Liu and M. Zhu, Angew. Chem., Int. Ed., 2021, 133, 21128–21137 CrossRef.
- Z. Wu, Y. Ni, S. Tan, E. Hu, L. He, J. Liu, M. Hou, P. Jiao, K. Zhang, F. Cheng and J. Chen, J. Am. Chem. Soc., 2023, 145, 9596–9606 CrossRef CAS PubMed.
- F. Ding, C. Zhao, D. Xiao, X. Rong, H. Wang, Y. Li, Y. Yang, Y. Lu and Y.-S. Hu, J. Am. Chem. Soc., 2022, 144, 8286–8295 CrossRef CAS PubMed.
- E. A. Wu, S. Banerjee, H. Tang, P. M. Richardson, J.-M. Doux, J. Qi, Z. Zhu, A. Grenier, Y. Li, E. Zhao, G. Deysher, E. Sebti, H. Nguyen, R. Stephens, G. Verbist, K. W. Chapman, R. J. Clément, A. Banerjee, Y. S. Meng and S. P. Ong, Nat. Commun., 2021, 12, 1256 CrossRef CAS PubMed.
- X. Feng, H. Fang, P. Liu, N. Wu, E. C. Self, L. Yin, P. Wang, X. Li, P. Jena, J. Nanda and D. Mitlin, Angew. Chem., Int. Ed., 2021, 60, 26158–26166 CrossRef CAS PubMed.
- R. Li, D. Jiang, P. Du, C. Yuan, X. Cui, Q. Tang, J. Zheng, Y. Li, K. Lu, X. Ren, S. Gao and X. Zhan, Chem. Sci., 2022, 13, 14132–14140 RSC.
- Q. Ma, M. Guin, S. Naqash, C.-L. Tsai, F. Tietz and O. Guillon, Chem. Mater., 2016, 28, 4821–4828 CrossRef CAS.
- Q. Ma, T. Ortmann, A. Yang, D. Sebold, S. Burkhardt, M. Rohnke, F. Tietz, D. Fattakhova-Rohlfing, J. Janek and O. Guillon, Adv. Energy Mater., 2022, 12, 2201680 CrossRef CAS.
- Z. Deng, T. P. Mishra, E. Mahayoni, Q. Ma, A. J. K. Tieu, O. Guillon, J.-N. Chotard, V. Seznec, A. K. Cheetham, C. Masquelier, G. S. Gautam and P. Canepa, Nat. Commun., 2022, 13, 4470 CrossRef CAS PubMed.
- J. A. S. Oh, X. Xu, Z. Zeng, K. Wang, N. Y. J. Tan, E. Kok, J. Huang and L. Lu, Energy Environ. Mater., 2023, 6, e12472 CrossRef CAS.
- T. Famprikis, J. A. Dawson, F. Fauth, O. Clemens, E. Suard, B. Fleutot, M. Courty, J.-N. Chotard, M. S. Islam and C. Masquelier, ACS Mater. Lett., 2019, 1, 641–646 CrossRef CAS.
- N. Tanibata, M. Deguchi, A. Hayashi and M. Tatsumisago, Chem. Mater., 2017, 29, 5232–5238 CrossRef CAS.
- S. Takeuchi, K. Suzuki, M. Hirayama and R. Kanno, J. Solid State Chem., 2018, 265, 353–358 CrossRef CAS.
- X. Feng, P. Chien, Z. Zhu, I. Chu, P. Wang, M. Immediato-Scuotto, H. Arabzadeh, S. P. Ong and Y. Hu, Adv. Funct. Mater., 2019, 29, 1807951 CrossRef.
- R. Schlem, A. Banik, M. Eckardt, M. Zobel and W. G. Zeier, ACS Appl. Energy Mater., 2020, 3, 10164–10173 CrossRef CAS.
- H. Kwak, J. Lyoo, J. Park, Y. Han, R. Asakura, A. Remhof, C. Battaglia, H. Kim, S.-T. Hong and Y. S. Jung, Energy Storage Mater., 2021, 37, 47–54 CrossRef.
- T. J. Udovic, M. Matsuo, A. Unemoto, N. Verdal, V. Stavila, A. V. Skripov, J. J. Rush, H. Takamura and S. Orimo, Chem. Commun., 2014, 50, 3750–3752 RSC.
- K. Yoshida, T. Sato, A. Unemoto, M. Matsuo, T. Ikeshoji, T. J. Udovic and S. Orimo, Appl. Phys. Lett., 2017, 110, 103901 CrossRef.
- A. Unemoto, M. Matsuo and S. Orimo, Adv. Funct. Mater., 2014, 24, 2267–2279 CrossRef CAS.
- J. Cuan, Y. Zhou, T. Zhou, S. Ling, K. Rui, Z. Guo, H. Liu and X. Yu, Adv. Mater., 2019, 31, 1803533 CrossRef PubMed.
- Y. Sadikin, M. Brighi, P. Schouwink and R. Černý, Adv. Energy Mater., 2015, 5, 1501016 CrossRef.
- X. Wang, C. Zhang, M. Sawczyk and J. Sun, Nat. Mater., 2022, 21, 1057–1065 CrossRef CAS PubMed.
- Q. Ma, C.-L. Tsai, X.-K. Wei, M. Heggen, F. Tietz and J. T. S. Irvine, J. Mater. Chem. A, 2019, 7, 7766–7776 RSC.
- J. Park, J. P. Son, W. Ko, J.-S. Kim, Y. Choi, H. Kim, H. Kwak, D.-H. Seo, J. Kim and Y. S. Jung, ACS Energy Lett., 2022, 7, 3293–3301 CrossRef CAS.
- H. Kwak, J.-S. Kim, D. Han, J. S. Kim, J. Park, G. Kwon, S.-M. Bak, U. Heo, C. Park, H.-W. Lee, K.-W. Nam, D.-H. Seo and Y. S. Jung, Nat. Commun., 2023, 14, 2459 CrossRef CAS PubMed.
- Y. Qie, S. Wang, S. Fu, H. Xie, Q. Sun and P. Jena, J. Phys. Chem. Lett., 2020, 11, 3376–3383 CrossRef PubMed.
- R. Li, K. Xu, K. Liu, R. Si and Z. Zhang, Chem. Mater., 2022, 34, 8356–8365 CrossRef CAS.
- H. Huang, C. Chi, J. Zhang, X. Zheng, Y. Wu, J. Shen, X. Wang and S. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 36864–36874 CrossRef CAS PubMed.
- I.-H. Chu, C. S. Kompella, H. Nguyen, Z. Zhu, S. Hy, Z. Deng, Y. S. Meng and S. P. Ong, Sci. Rep., 2016, 6, 33733 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- W. Liao and R. Dronskowski, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, i72–i73 CrossRef CAS.
- N. Flores-González, N. Minafra, G. Dewald, H. Reardon, R. I. Smith, S. Adams, W. G. Zeier and D. H. Gregory, ACS Mater. Lett., 2021, 3, 652–657 CrossRef PubMed.
- C. K. Moon, H.-J. Lee, K. H. Park, H. Kwak, J. W. Heo, K. Choi, H. Yang, M.-S. Kim, S.-T. Hong, J. H. Lee and Y. S. Jung, ACS Energy Lett., 2018, 3, 2504–2512 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |