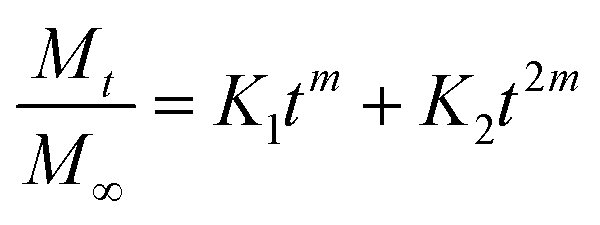DOI:
10.1039/D4RA02439G
(Paper)
RSC Adv., 2024,
14, 23886-23901
FericipXT-coated PEGylated rutile TiO2 nanoparticles in drug delivery: in vitro assessment of imatinib release†
Received
31st March 2024
, Accepted 3rd July 2024
First published on 30th July 2024
Abstract
This study presents a facile synthesis strategy for magnetic field-responsive PEGylated iron-supplement-coated rutile titanium dioxide (TiO2) nanoparticles (NPs) for stimulus-responsive drug delivery. Imatinib, an anticancer drug, was successfully loaded into NPs, and its in vitro release was investigated under different pH conditions. XRD analysis confirmed the successful synthesis of PEGylated iron supplement-coated rutile titania NPs. HR-TEM studies revealed an increased NP size due to the coating, PEGylation, and drug loading, which was corroborated by FTIR spectra, confirming the drug loading into the NPs. DLS provided a hydrodynamic diameter of 642.2 nm and polydispersity index of 0.277 for PEGylated NPs, indicating their enhanced biodistribution and narrow size distribution. PEGylated NPs exhibited a negative zeta potential of −32.89 mV, indicating high stability. In vitro drug-release studies demonstrated controlled release with maximum efficiency under acidic conditions. Hemolysis assay confirmed the safety and biocompatibility of PEGylated NPs. All drug-loaded nanoformulations followed the Peppas–Sahlin model, suggesting Fickian diffusion and Case II relaxation mechanism of drug release. These NPs have the potential for the targeted delivery and controlled release of chemotherapeutics, thereby minimizing side effects.
1 Introduction
1.1 Challenges in cancer therapy and its solution
Cancers, commonly referred to as malignant tumors, are characterized by uncontrolled growth of cells on account of mutation in their genetic material. The adverse effects of cancer and the dire prognosis of patients include high mortality, poor quality of life and expensive treatments.1 Much technological development and research have been conducted to combat cancer, including chemotherapy, immunotherapy and radiation therapy. However, these techniques are associated with certain flaws, such as low drug concentrations at the tumor site, non-selectivity to target cells or tissues and low drug efficacy due to their short half-life. The solution to these issues lies in the usage of targeted drug delivery systems. In such systems, the targeted specificity and selectivity of the drug are greatly enhanced. The main advantage of the targeted therapy is that the drug concentration of chemotherapy in the tumor environment can be maximized and toxicity to other non-target organs can be minimized.2 In addition, if the drug is locally delivered, this can improve drug effectiveness by avoiding the long journey required to reach the target site through the hostile environment when the drug is administered via the systemic route. To better understand tumorigenesis and increase drug availability, several vectors have been explored that can deliver drugs in a controlled manner without causing side effects. To date, the use of versatile nanomaterials, including polymers, lipids, inorganic molecules, hydrogels and other macromolecular scaffolds, has led to the development of systems to deliver chemotherapy drugs directly to tumor sites with improved treatment efficiency.3
1.2 Enhanced permeation effect in tumors
General features of a tumor include leaky vasculature and poor lymphatic drainage. Several approaches have been proposed to target tumors, for example, the enhanced permeability and retention effect, where leaky blood vessels promote small-molecule permeation and poor lymphatic drainage allow storage and accumulation of chemotherapy drugs in the tumor cells.4 Unlike freely diffusing drugs, a nanocarrier can enter the tumor tissue through leaky vasculature due to its enhanced permeability and retention effects. In the enhanced permeation effect (EPR), the delivered agent is locally released and rapidly absorbed by the cancer cell to perform its function. Targeted tumor cells that express or overexpress specific receptors or antigens can be targeted using ligands such as antibodies or small peptides that recognize and bind to them.5 Smaller molecules accumulate in the tumor faster than larger molecules, but larger molecules can be retained in the tumor longer. Therefore, size is a dominant factor in controlling drug accumulation at the tumor site.6
1.3 Nanocarriers for targeted therapy
Active targeting refers to the interaction of ligand-receptors after the nanoparticles (NPs) reach the target site through systemic circulation. Ligand–receptor interaction is possible only if the two components are in close proximity (0.5 nm). Active targeting of tumors can be achieved by functionalizing NPs with proteins, peptides, nucleic acid aptamers, carbohydrates, and other small molecules.7 To date, several classes of materials for targeted therapy have been developed, including biodegradable polymers, liposomes, dendrimers, nanoshells, and NPs based on nucleic acids. Biodegradable nanoparticles are widely used in cancer therapy due to their high biocompatibility. For targeted delivery, sustained release and site-specific delivery are key requirements. Another important factor is the stability of the NPs to stay longer in the blood and eventually accumulate in tumors.8 Drug release from these NPs can be controlled by diffusion or swelling followed by mass diffusion or erosion in a time-dependent manner. The release rate can be adjusted by modifying the polymer, developing new polymers, or synthesizing copolymers. The main advantage of these biodegradable polymers is to maintain drug concentrations within the optimal range for longer periods of time, thus increasing drug efficacy and improving patient compliance. Thus, targeted therapy is a combination of targeted administration with continuous drug release, allowing maximum use of the drug delivered to the tumor site.
Titanium dioxide nanoparticles (TiO2 NPs) are being studied extensively for their drug delivery applications. Their properties can be modified by carefully controlling the synthesis parameters. They can be effectively functionalized using different coatings and surfactants. TiO2 NPs can be tweaked to respond to different stimuli such as light, sound, pH, and temperature and initiate the drug release as has been seen in different studies.9,10 The extensive study of TiO2 nanoparticles is not solely due to their ease of functionalization, flexibility and usability they offer. Rather, it is the highly encouraging results obtained from their application in drug delivery that have led to such extensive research. Recently, Uribe-Robles et al.11 functionalized TiO2 nanospheres with folic acid for the targeted delivery of temozolomide for the treatment of glioblastoma. The nanocarriers exhibited high loading capacity and retained the drug for 48 h, which was successfully internalized into the cancer cells resulting in high cytotoxicity. Abdel-Megeed et al.12 found that TiO2 nanostructures loaded with adriamycin showed a significant upregulation of ATP-binding cassette transporter membrane proteins and downregulation of the C-myc gene, which make them a promising candidate for addressing the drug resistance issue associated with adriamycin and for enhancing its therapeutic efficacy against breast cancer.
In developing a smart drug delivery system for targeted drug delivery, it is essential for the nanocarrier loaded with the drug to effectively navigate to the intended site without premature release. Once at the target site, the drug should be released. While recent studies have focused on drug delivery, there is limited exploration into the transportation of drug-carrying nanocarriers to the target location to minimize unwanted side effects caused by the drug circulating throughout the body and being released at undesired sites. In this study, we investigated the pH-responsive release profile of the chemotherapeutic drug imatinib from PEGylated iron supplement-coated rutile TiO2 NPs. The coating with iron supplement was performed to induce a magnetic response in the NPs so that they respond to the applied magnetic field. The formulation used to synthesize the final version of NPs ensures that the PEG chains will rupture and the NPs will degrade to release the drug only around acidic pH (considering the fact that the pH around the tumors is mostly acidic). The magnetic properties of bare and drug-loaded NPs were studied and in vitro drug release was inspected over acidic, neutral and basic pH levels. Moreover, an attempt to understand the drug release mechanism from different formulations was also made using different mathematical models.
2 Experimental section
2.1 Materials
Materials used were titanium tetraisopropoxide (TTIP), ethanol, nitric acid (HNO3), PEG 4000 solution, phosphate buffer saline (PBS) solution of pH 7.4, acetate buffer solution of pH 4.4, borate buffer solution of pH 9.0 and deionized (DI) water. These were obtained from Sigma Aldrich Pvt. Ltd, Bangalore, India, and were used without any further purification. Ferrous ascorbate with folic acid tablets under the brand name FericipXT manufactured by Cipla Ltd. were purchased from pharmaceuticals in Chandigarh, India. Dialysis tubing and clips were purchased from Himedia. IKA-RCT basic IKAMAG safety control magnetic stirrer with a hot plate from Merck, India was used. pH testing kit by Insif, India was used to determine the final pH of the solution. For calcination, a muffle furnace supplied by Insif, India was used. Conical high alumina crucibles of volume 30 ml supplied by Thomas Scientific were used for high-temperature calcination.
2.2 Methods
2.2.1 Synthesis of PEGylated iron supplement-coated rutile TiO2 NPs. Rutile TiO2 NPs were synthesized following the sol–gel method. The procedure for synthesis was the same as adopted by us in our previous study with the only difference being that the quantity of the nitric acid was chosen to obtain the pH of the final solution 1.0.13 Further, TiO2 NPs were calcined at 800 °C to obtain the desired rutile phase. To enhance the magnetic behaviour of the rutile TiO2 NPs, they were coated with FericipXT tablets (iron supplement) with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 core:shell ratio. The ratio of 1:1 was decided based on our previously obtained results, which served as preliminary data for this study.14 1:1 core:shell ratio ensured significant coating of the shell material over the host material while also keeping the properties of the host intact. For coating, the procedure mentioned by Mehr et al.15 was followed. Briefly, an optimal amount of FericipXT tablets was added to 250 ml of DI water. Next, TiO2 NPs were added followed by dropwise addition of 1 M NaOH solution. Finally, the solution was heated to reflux at 90 °C. The resulting solution was centrifuged, washed with DI water and calcined at 800 °C. The characteristic white color of TiO2 NPs was changed to brown, indicating the formation of Fe2+ coating on them. For PEGylation, the synthesized iron supplement-coated rutile TiO2 NPs were added to a 1% concentration of PEG 4000 solution and stirred for one day at 750 rpm. A lower PEG concentration was chosen to avoid the growth of a thicker PEG layer on the NPs. For the final step, PEG-coated NPs were centrifuged at 9000 rpm for 30 min at room temperature, washed with distilled water and dried.
:1 core:shell ratio. The ratio of 1:1 was decided based on our previously obtained results, which served as preliminary data for this study.14 1:1 core:shell ratio ensured significant coating of the shell material over the host material while also keeping the properties of the host intact. For coating, the procedure mentioned by Mehr et al.15 was followed. Briefly, an optimal amount of FericipXT tablets was added to 250 ml of DI water. Next, TiO2 NPs were added followed by dropwise addition of 1 M NaOH solution. Finally, the solution was heated to reflux at 90 °C. The resulting solution was centrifuged, washed with DI water and calcined at 800 °C. The characteristic white color of TiO2 NPs was changed to brown, indicating the formation of Fe2+ coating on them. For PEGylation, the synthesized iron supplement-coated rutile TiO2 NPs were added to a 1% concentration of PEG 4000 solution and stirred for one day at 750 rpm. A lower PEG concentration was chosen to avoid the growth of a thicker PEG layer on the NPs. For the final step, PEG-coated NPs were centrifuged at 9000 rpm for 30 min at room temperature, washed with distilled water and dried.
2.2.2 Synthesis of drug-loaded PEG/Fe@TiO2 NPs. To encapsulate the NPs with the drug (imatinib), the procedure performed by Akram et al.16 was chosen with minor modifications. A fixed amount of the drug was reconstituted in 40 ml of DI water along with the addition of 500 mg of the synthesized NPs. The solution was stirred for 2 days and then stored for one day. It was then centrifuged and filtered. In this way, three samples were collected, one with the drug-loaded on the rutile TiO2 NPs (TD), the other with the drug loaded on the FericipXT-coated rutile TiO2 NPs (FD) and the third sample was the drug loaded onto the PEGylated FericipXT-coated rutile TiO2 NPs (PD). To measure the amount of the drug in the yield obtained, the absorbance of the filtrate was measured at 254 nm. The amount of the drug successfully retained on NPs was determined as a percentage of the encapsulation efficiency in eqn (1). The drug loading percentage reflects the mass ratio of the drug to the nano-drug that was synthesized by conjugating the drug with a fixed amount of NPs. The % drug loading was calculated by finding the drug concentrations in the loaded NPs considered for the study divided by the number of NPs used, and then multiplied by 100 according to eqn (2).| | |
Encapsulation efficiency (%) = (WDrug on the NPs/WTotal drug used) × 100
| (1) |
| | |
Drug loading (%) = (concentration of the drug in the NPs/weight of the loaded NPs considered for the study) × 100
| (2) |
2.2.3 In vitro drug release by dialysis method. In the present study, an in vitro drug release study of the drug imatinib was studied over three different pH values viz. 4.4, 7.4 and 9.0. Three types of drug-loaded NP formulations used for the study were TD, FD and PD. Firstly, the calibration curve of the drug-imatinib (D) was plotted in three different pH mediums. For this, a stock solution of imatinib was prepared and dissolved in three different pH buffer solutions. The calibration curve is a plot with the concentration (μg ml−1) on the X-axis and absorbance on the Y-axis. The curve was mathematically analyzed to obtain the slope and intercept. pH-dependent release behavior at three different pH levels was investigated. All three types of drug-loaded NPs formulations were inserted into dialysis tubes having a Molecular Weight Cut Off (MWCO) of 8 kDa each. In total, 9 sets were prepared. 3 sets of dialysis tubes contained imatinib-loaded TiO2 NPs (TD), the other 3 sets of dialysis tubes contained imatinib-loaded Fe@TiO2 NPs (FD) and the final 3 sets of the dialysis tubes contained imatinib-loaded PEGylated Fe@TiO2 NPs (PD). To mimic a neutral pH environment, 3 dialysis tubes containing 5 ml of each of all the three types of drug-NPs formulations were dipped into 500 ml of 0.1 M phosphate buffer solution (pH 7.4), separately. Similarly, for the in vitro study under an acidic environment, 500 ml of 0.1 M Acetate buffer solution (pH 4.4) was used and likewise, 500 ml of 0.1 M borate buffer solution (pH 9.0) was used to achieve basicity. The dialysis tubes were hung with the help of clips in their respective beakers containing solutions to imitate different pH environments. The entire setup was stirred at 37 °C. After a fixed interval of time, 3 ml of the dialysate was withdrawn from each of the beakers to monitor the absorbance of the imatinib molecules released into the respective buffer at 254 nm with the help of a UV-visible spectrophotometer. The withdrawn amount of the dialysate was replaced with an equal quantity of the fresh buffer solution to avoid the sink conditions. In each observation, the value of the absorbance obtained was used to calculate the concentration of the drug released into the media using the following expression:| | |
Concentration of the drug (μg ml−1) = (absorbance − intercept)/slope
| (3) |
where slope and intercept were obtained from the calibration curve. Percentage of the drug released and the cumulative percentage of the drug released was calculated using the following methods:| | |
% drug released = (concentration of the drug/total amount of the drug used) × 100
| (4) |
| | |
Cumulative % release = (volume of the sample withdrawn (ml)/bath volume (ml)) × d(t−1) + dt
| (5) |
where dt is the percent drug released at time t and d(t−1) is the percent drug released at the time (t − 1).
2.2.4 Hemolysis assay. Fresh anticoagulated human blood treated with the citric-acid-dextrose solution was added into seven vials in equal quantities. 1 ml suspension of PEGylated iron-supplement coated TiO2 NPs (P) was added into the five vials at the concentrations of 10 μg ml−1, 50 μg ml−1, 100 μg ml−1, 300 μg ml−1 and 500 μg ml−1, respectively. 0.9% saline solution was taken as negative control and 1% Triton X-100 solution was used as a positive control. The blood samples containing PEGylated NPs, positive control and negative control, were gently shaken and incubated for 45 minutes at 37 °C. They were then centrifuged for 3 minutes at the rate of 4000 revolutions per minute. After centrifugation, the blood sample was split into two components, the red part ‘heme’ at the bottom and a transparent plasma as the top layer. The upper clearer solution was taken out from the samples for the absorbance study at 540 nm. The percentage of hemolysis occurring in the samples was calculated using the formula indicated as follows:| |
 | (6) |
2.3 Characterization
The synthesized samples were characterized by X-ray diffraction (XRD) and high-resolution transmission electron microscopy (HR-TEM). XRD was carried out using the Panalytical X'Pert Pro instrument equipped with x'Celerator solid-state detector, manufactured by Panalytical, Netherlands. The sample was firmly placed onto the glass slide, which was carefully laid down onto the sample holder. XRD was performed at the Sophisticated Analytical Instrumentation Facility (SAIF), Panjab University, Chandigarh, India. HR-TEM was carried out using JEOL 2100 Plus, Japan at CIL, Panjab University, Chandigarh, India. The powdered sample was dispersed in ethanol and ultrasonicated for about an hour. A drop of the solution was then carefully placed onto the copper grid. The grid was eventually air-dried under room temperature and was used for imaging after half an hour. Ultraviolet-visible (UV-vis) spectroscopy was used to perform the absorbance studies. A double beam UV-VIS-NIR Spectrophotometer - Lambda 750 from PerkinElmer, USA was used for the said purpose. A fixed amount of NPs (in μg) was dispersed in DI water of a specific pH. The baseline correction was performed before conducting the study. Afterwards, blanking was performed with the help of a reference sample. Since this work involved studying the behaviour of the drug release under different pH levels, the reference samples as well as the original samples were prepared in the respective pH conditions accordingly. Dynamic light scattering (DLS) verified the particle size distribution of the resulting NPs. A vibrating sample magnetometer (VSM) was used to analyze the magnetic behaviour of the bare NPs and the drug-loaded different formulations of the NPs. The samples were placed in the sample holder and all the studies were performed at room temperature.
3 Results and discussion
3.1 X-ray diffraction
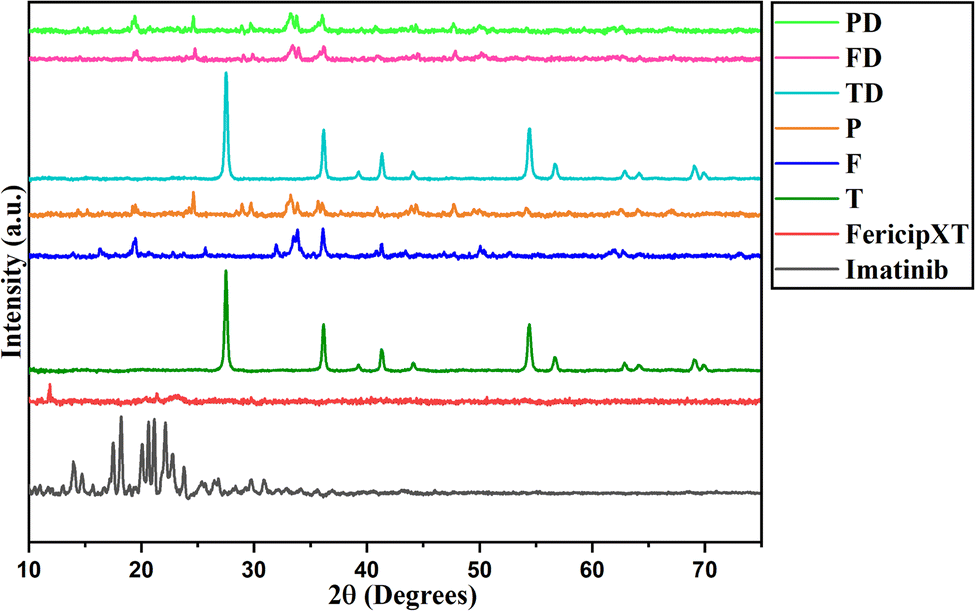
Rutile TiO2 NPs were synthesized and were then treated with FericipXT tablets to obtain a coating over them. Finally, these iron tablets-coated rutile titania NPs were PEGylated to obtain another layer on them. Fig. 1 demonstrates the XRD plot of imatinib (D), FericipXT tablet (iron supplement was used for coating rutile TiO2 NPs - FXT), rutile TiO2 NPs (T), FericipXT-coated rutile TiO2 NPs in the 1:1 ratio (F), PEGylated 1:1 FericipXT-coated rutile TiO2 NPs (P), imatinib-loaded TiO2 NPs (TD), imatinib-loaded FericipXT-coated rutile TiO2 NPs (FD) and imatinib-loaded PEGylated FericipXT-coated rutile TiO2 NPs (PD). Table 1 (ESI File S1†) demonstrates the symbols for different samples used in this paper. The XRD pattern of T belongs to the pure rutile phase (JCPDS Pattern No. 01-073-1782). The highly crystalline nature of T was lost after the coating with the FericipXT tablet. Table 2 (ESI File S1†) highlights the different phases of iron-based compounds appearing on the surface of TiO2 NPs after their coating with the iron tablet in a 1:1 core:shell ratio. The details of the different phases obtained and their quantification were performed after the analysis of the XRD data through HighScore Plus software. The XRD pattern of F is different from that of T and FXT because of the formation of iron oxides during the core–shell formation process. The ferrous ascorbate salt and folic acid were converted into different iron oxides belonging to different crystal systems. Since all the compounds are iron-based, it can be inferred that the entire surface of the TiO2 NPs was coated and the thickness of the coating was also more than 10 nm. X-rays can easily penetrate the 10 nm coating thereby displaying the structural peaks of the core beneath. However, in this case, no TiO2 phase was obtained, which only indicates that the thickness of the iron coating was > 10 nm. In our previous study,17 the thickness of the shell was ∼10 nm and the XRD pattern of the Autrin tablet-coated anatase TiO2 NPs was exactly similar to that of the bare TiO2 NPs, which was possible only because of the thin shell formation. The XRD pattern of the P was almost similar to that of the F but a little less crystalline. It can be concluded that the PEG layer lowers the crystallinity of the sample.
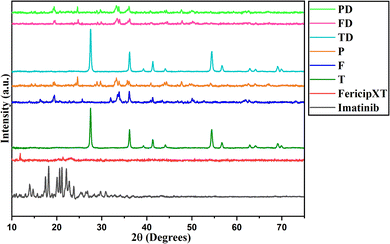 |
| | Fig. 1 XRD of imatinib (drug), FericipXT tablet (iron supplement) used for coating, rutile TiO2 NPs (T), FericipXT-coated rutile TiO2 NPs in the 1:1 ratio (F), PEGylated FericipXT-coated rutile TiO2 NPs (P), imatinib-loaded TiO2 NPs (TD), imatinib-loaded FericipXT-coated rutile TiO2 NPs (FD) and imatinib-loaded PEGylated FericipXT-coated rutile TiO2 NPs (PD) (from bottom pattern to the top). | |
The XRD plot of D shows a set of diffraction peaks between 10° and 30°. The XRD plot of FericipXT did not show any well-defined peaks, rather an almost flat plot was obtained indicating the amorphous nature of the tablet. The XRD pattern of P was similar to F with a few extra peaks of low intensity emerging at 24.686°, 28.918° and 29.726°, which correspond to the iron titanium oxide (Fe9TiO15) and orthorhombic phase of Fe2O3. Antarnusa et al.18 coated Fe3O4 NPs with PEG 4000 and observed the appearance of extra phases corresponding to α-Fe2O3, α-FeO(OH) and γ-FeO(OH). The XRD plots marked as TD, FD and PD have retained the diffraction pattern obtained for the host NPs. The drug loading did not disturb the diffraction pattern of the host NPs and thus, the diffraction peaks of the individual NPs were retained. However, no diffraction peaks related to imatinib were detected in the individual XRD patterns of TD, FD and PD. This implies that imatinib was present in an amorphous state on the NPs. Likewise, Li et al.19 synthesized a drug, 5-FU loaded chitosan-coated ZnSe/ZnS NPs (5-Fu–CS–ZnSe/ZnS NPs) and observed that the XRD plot of 5-FU displayed diffraction peaks indicating its crystalline nature, however, no such peak of the drug was obtained in the XRD of 5-Fu–CS–ZnSe/ZnS NPs depicting that the drug was present in its amorphous form onto the functionalized NPs. Raj et al.20 prepared solid-lipid NPs and loaded them with cytarabine, an anticancer drug meant for treating leukemia. The XRD plot obtained for cytarabine was crystalline in nature, but the XRD pattern of the cytarabine-loaded solid-lipid NPs did not show the characteristic peak of cytarabine. They too reported that cytarabine was present in its amorphous form in the NPs. In another study, the XRD of quercetin presented sharp diffraction peaks confirming its crystalline nature but when the same quercetin was loaded onto PEGylated CdSe/CdS and PEGylated CdSe/ZnS core/shell NPs and when their XRD characterization was performed no peak of the antioxidant molecule was detected. This confirmed that the quercetin existed in a non-crystalline, i.e. amorphous state onto the NPs.21 Patel et al.22 suggested that the crystalline behaviour of the conjugant is often suppressed when they are loaded onto the NPs resulting in an amorphous form.
3.2 FT-IR spectroscopy
Fig. 2 displays the FTIR spectra obtained for different samples and Table 3 (ESI file S1†) highlights the different functional groups associated with the samples corresponding to the peaks obtained in their relevant FTIR spectra. In the case of imatinib (D), the broad region between 3200 and 4000 cm−1 shows characteristic peaks at 3237.05 cm−1, 3414.83 cm−1, 3475.74 cm−1 and 3550.83 cm−1 corresponding to the stretching vibration of the O–H group.23 A pattern similar to D can be observed in TD, FD and PD for the said range of wavenumbers, which confirmed the presence of the drug onto these NPs along with the presence of stretching mode of the O–H group. The peak corresponding to 3550.83 cm−1 in D was observed at 3546.26 cm−1 in PD. The peak at 3475.74 cm−1 in D was shifted to 3463.61 cm−1, 3463.83 cm−1, 3462.55 cm−1, 3470.46 cm−1 in T, P, TD and PD, respectively. For the peak at 3414.83 cm−1 in D, the corresponding peaks in FXT, T, F, P, TD, FD, PD were obtained at 3429.14 cm−1, 3425.40 cm−1, 3434.15 cm−1, 3414.61 cm−1, 3431.66 cm−1, 3416.92 cm−1, 3414.12 cm−1, respectively. The peak obtained at 3237.05 cm−1 in D was only been observed in PD at 3238.17 cm−1. For the same range of wavenumbers, FericipXT showed a broad region, which was also obtained in F. With this, we can confirm the successful coating of rutile TiO2 NPs with FericipXT. The FTIR patterns of T-TD, F-FD and P-PD also matched those of T, F and P representing the host NPs, which after loading of the drug became TD, FD and PD, respectively.
 |
| | Fig. 2 FTIR spectra of imatinib, FericipXT, T, F, P, TD, FD and PD. | |
D exhibits a small peak at 2920.87 cm−1, which corresponds to the stretching mode of the C–H group. The same peak was obtained in FD and PD at 2917.30 cm−1 and 2915.98 cm−1, respectively. D showed a double-peak pattern at 1617.31 cm−1 and 1638.36 cm−1, corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching. The similar pattern was repeated in TD, FD and PD at (1622.01 cm−1 and 1637.07 cm−1), (1619.09 cm−1 and 1637.31 cm−1) and (1617.73 cm−1 and 1637.72 cm−1), respectively. This double-peak pattern was observed only in the drug-loaded NPs and was not observed in the bare NPs. Instead, the bare NPs such as T, F and P demonstrated a single peak at 1636.22 cm−1, 1634.89 cm−1, 1636.59 cm−1, respectively corresponding to CC stretching.
C stretching. The similar pattern was repeated in TD, FD and PD at (1622.01 cm−1 and 1637.07 cm−1), (1619.09 cm−1 and 1637.31 cm−1) and (1617.73 cm−1 and 1637.72 cm−1), respectively. This double-peak pattern was observed only in the drug-loaded NPs and was not observed in the bare NPs. Instead, the bare NPs such as T, F and P demonstrated a single peak at 1636.22 cm−1, 1634.89 cm−1, 1636.59 cm−1, respectively corresponding to CC stretching.
The peaks obtained between 1380 and 1467 cm−1 contribute to C–H bending and those obtained in the range 1100–1280 cm−1 belong to C–O stretching. The peaks obtained at 750–900 cm−1 correspond to out-of-plane bending of C–H aromatic hydrocarbons. The peak at 613 cm−1 in T is attributed to the Ti–O–Ti bridging stretching mode. Moreover, the peaks obtained at 620.10 cm−1, 613.00 cm−1, 619.31 cm−1, 613.76 cm−1, 621.13 cm−1, 617.87 cm−1 and 620.01 cm−1 correspond to Ti–O–Ti bridging stretching mode of D, T, F, P, TD, FD and PD, respectively. The reason that the peak at 620.10 cm−1 in D is related to Ti–O–Ti bonding is due to the fact that an imatinib tablet also has a coating of titanium dioxide. However, the peak obtained at 617.44 cm−1 in FXT can be attributed to the Fe–O bond.24 The FTIR results validate that imatinib conjugated with T, F and P.
3.3 UV-vis spectroscopy
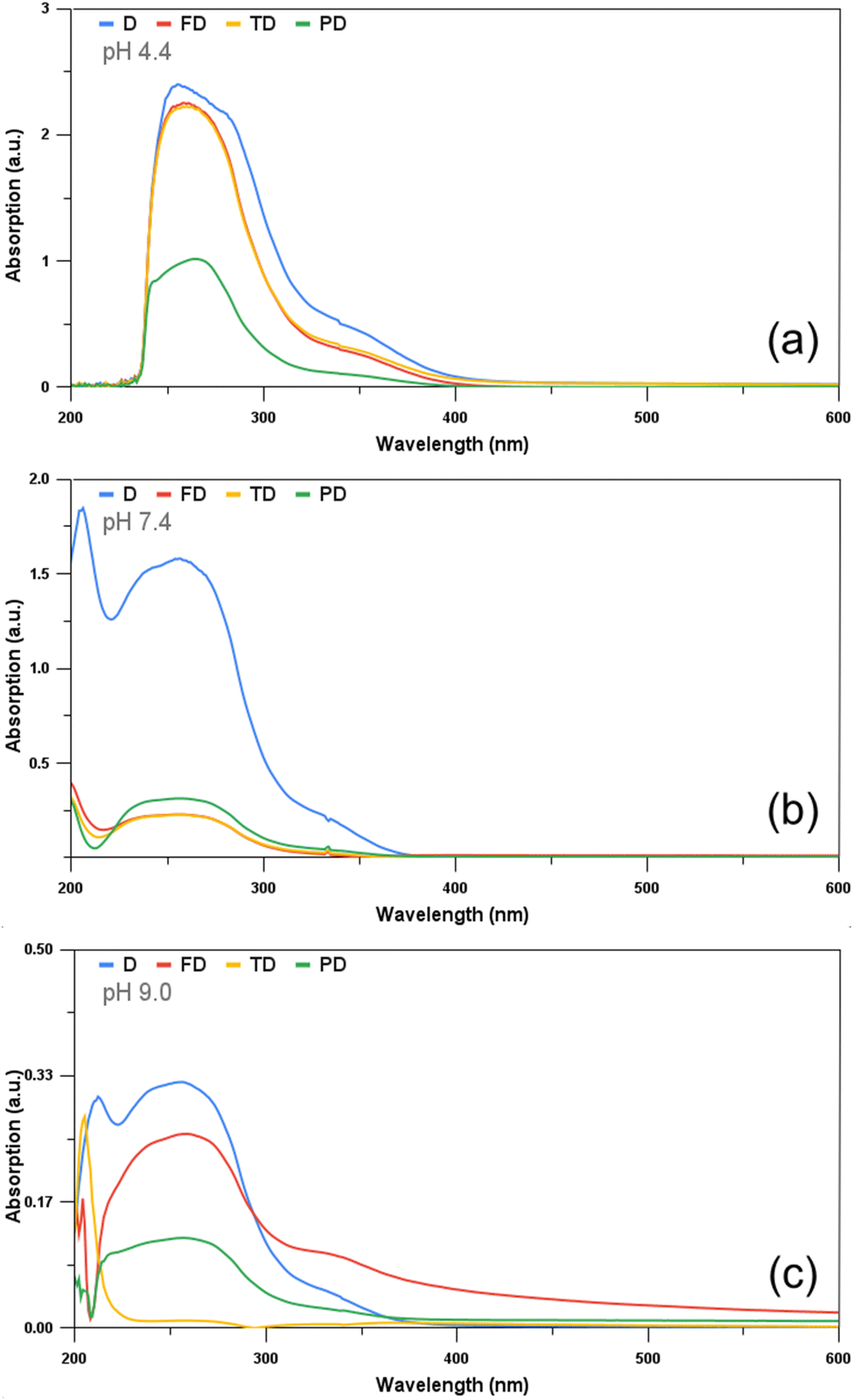
In order to obtain the percentage in vitro drug released through the absorbance of the drug (imatinib), at first, the calibration curve of imatinib was plotted in different pH mediums, viz. pH 4.4, 7.4 and 9.0, in which the drug-release study was to be conducted. The calibration curve was plotted with the concentration of imatinib in μg ml−1 along the X-axis and the absorbance value obtained along the Y-axis. For this, the stock solution of imatinib was prepared under three different pH environments. The absorption spectra of the drug in three different buffer solutions against known values of concentration were acquired and plotted. Fig. 3(a)–(c) denote the calibration curve of the drug (imatinib) obtained under pH 4.4, 7.4 and 9.0, respectively. With the help of the equation of straight line and least square fitting, the slope and intercept for all three categories were obtained. These slopes and intercepts were used to calculate the concentration of the drug released in the in vitro drug-release study.
 |
| | Fig. 3 Calibration curve obtained for imatinib under pH (a) 4.4, (b) 7.4 and (c) 9.0. | |
These curves were obtained by plotting the concentration of the drug (μg ml−1) added along the X-axis and the corresponding absorbance measured along the Y-axis. The absorbance of the drug increased with increasing concentration. imatinib and mostly the maximum percentage of chemotherapeutic drugs exhibit maximum solubility in acid which decreases with increasing pH. This can be verified from the calibration curves obtained below as the maximum value of absorbance received for 1000 μg ml−1 is the highest under pH 4.4 and the least under pH 9.0. Fig. 4(a)–(c) denote the UV-visible absorbance plots obtained for the D, T, F and P under pH 4.4, 7.4 and 9.0, respectively. The drug showed the maximum absorbance in all the pH conditions. In pH 4.4 and 7.4, TD and FD showed almost similar behaviour with them showing more absorption in the acidic medium and very less absorption in the neutral medium. Furthermore, PD showed very limited absorption in an acidic medium and even less in a neutral medium. However, the absorbance displayed by PD in a neutral medium was more than TD and FD, which can be attributed to the biocompatible nature of the PEGylated NPs. Overall, the absorbance behaviour demonstrated by TD, FD and PD ascertains their usage in pH-responsive drug delivery applications.
 |
| | Fig. 4 UV-visible absorption spectra obtained for the drugs D, TD, FD and PD at pH (a) 4.4, (b) 7.4 and (c) 9.0. | |
3.4 HR-TEM
Fig. 5(a) depicts the HR-TEM image of T. They showed a size range of 25–35 nm. Fig. 5(b) displays the image of F. Their size was approximately 60 nm. Thus, the iron coating has augmented the size of the core NPs. On comparing with our previous study, we can easily conclude that the rutile phase results in a bigger size of the NPs.17 Furthermore, the PEGylation of the NPs further increased the size of the resulting NPs to the range 120–145 nm. Fig. 5(c) shows the HR-TEM image of P. Both the iron tablet-coating and the PEG layer can be easily observed as distinct layers from the image.
 |
| | Fig. 5 HR-TEM images of (a) T, (b) F, (c) P, (d) D, (e) TD, (f) FD and (g) PD. | |
In addition, the morphology of all NPs in Fig. 5(a)–(c) were almost spherical. Liu et al.25 observed a slight increase in the size of the TiO2 NPs when they were functionalized with hyaluronic acid. The HRTEM of D is shown in Fig. 5(d) and that of TD is shown in Fig. 5(e). The characteristic patterns obtained for D in Fig. 5(d) can also be seen in Fig. 5(e) ensuring the loading of the drug onto the NPs. The inset in Fig. 5(e) explains that the shape of the particles after loading is round and rectangle-like. The longer side of the rectangular-shaped TD varied between 40 and 75 nm. Clearly, there was an increase in size as well as morphological change. Fig. 5(f) demonstrates the HRTEM image of FD. It is obvious that the drug has been loaded onto the NPs as the NPs themselves were independent spherical-shaped particles, as depicted in Fig. 5(b). However, now the particles are aggregated. Furthermore, one can observe the elongation of the NPs as well. The inset of Fig. 5(f) shows the magnified image of the NPs. The layer around the NPs is clearly visible. The dark patches can be associated with the loaded drug. The nanostructures obtained are highly agglomerated due to which their size cannot be computed by analyzing the image. The HRTEM image of PD is shown in Fig. 5(g) with the inset showing its magnified view. These NPs have somehow managed to retain their round shape, although they are not perfectly round. The layer around the NPs can be seen. However, the particle size post-drug-loading was decreased. PD appeared to be agglomerated and possessed a varied size distribution in the range of 30–80 nm. This reduction in the size of PD might be due to the lattice strain generated on account of the loading of imatinib molecules, which have restricted the growth and nucleation of the P during the drug-loading process.26
3.5 SAED
Fig. 6(a)–(c) present the SAED patterns for T, F and P, respectively. The SAED pattern for T shows very bright circular concentric dotted patterns, which is indicative of the highly crystalline nature of the NPs, as shown in Fig. 6(a). Fig. 6(b) displays the diffuse concentric rings, which show that the crystalline behaviour of T has weakened after the coating with the iron tablet. Finally, Fig. 6(c) again displays a scattered dotted pattern, which is indicative of the crystalline nature of P. A few random spots are visible due to the PEG coating on the NPs. This implies that the crystallinity decreased due to polymer coating and defect density was increased.27 These observations get along well with the results obtained from XRD analysis. Gayathri et al.28 coated Eu:Gd2O3 NPs with silica and a diffuse ring pattern was obtained, which confirmed the amorphous nature of the NPs. Initially, a well-defined ring structure was obtained in the SAED pattern for Eu:Gd2O3 NPs. The SAED of D is depicted in Fig. 6(d) which shows bright spots illustrating its crystalline nature. Fig. 6(e) demonstrates the SAED pattern for TD. Concentric rings of dim spots are visible and this image when compared with the SAED pattern of T as shown in Fig. 6(a), one can clearly see that the conjugation of the drug has dimmed the bright concentric spots pattern as obtained for rutile TiO2 NPs. Moreover, no different pattern of rings/spots have been obtained which clarifies that the drug has been deposited in amorphous form onto the rutile TiO2 NPs. This observation gets along well with the XRD pattern obtained for TD in .Fig. 1 Fig. 6(f) highlights the SAED pattern of FD. The difference is that instead of having a diffused rings pattern, two bright spots and a few random spots are visible. Here, we can say that a certain proportion of the drug is present in crystalline form. However, this is in contrast to what we observe in XRD of FD as shown in Fig. 1. Further, Fig. 6(g) presents the SAED pattern of PD. The SAED pattern for P showed random bright spots, however, here two extremely dim concentric rings are visible along with a few dim spots. Thus, the conjugation of Imatinib has brought distinct changes in the SAED patterns of the respective NPs.
 |
| | Fig. 6 SAED patterns obtained for (a) T, (b) F, (c) P, (d) D (e) TD, (f) FD and (g) PD. | |
3.6 DLS and ZP
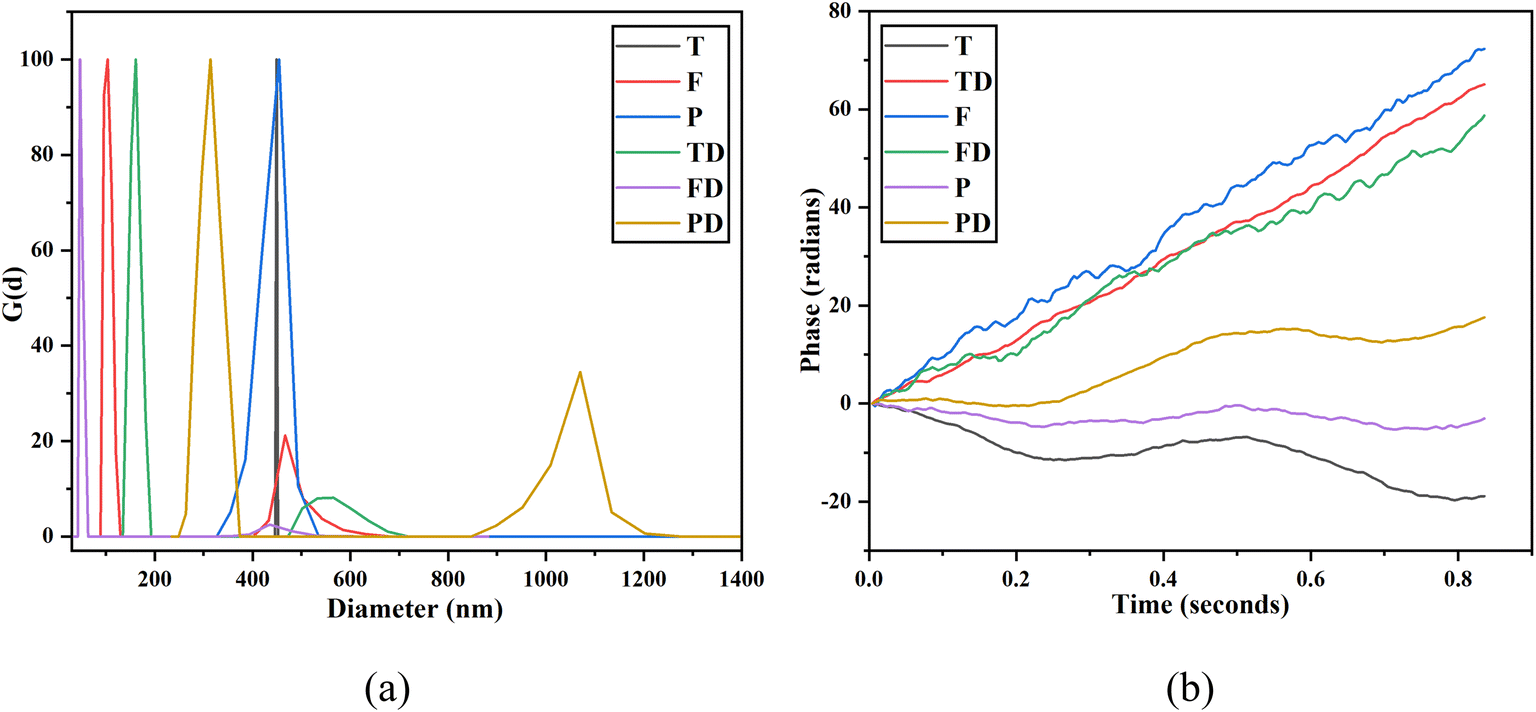
DLS is an ideal technique for performing size distribution measurements. DLS studies more than tens of thousands of particles thereby minimizing the error generated due to single particle encounters. The hydrodynamic size and polydispersity index (PDI) are both crucial parameters to optimize the performance and understand the in vitro migration of the NPs. The readings were taken by dispersing the NPs in water where 5 runs of each sample were taken for 30 seconds each. The measurements were performed at 25 °C using a wavelength of 658.0 nm. The difference between the hydrodynamic diameter and core diameter is that core diameter tells how much drug can be placed inside the NPs whereas hydrodynamic diameter is equivalent to the diameter of the particle, which would experience the same drag force in the fluid as would the dispersed nanoparticle in question. Hydrodynamic diameter comes into the picture when NPs are dispersed into some liquid medium. When NPs are dispersed in a liquid, the hydration layer gets attached over their surface due to which the apparent size of the NPs increases. Fig. 7(a) illustrates the intensity-weighted size distribution for T, F, P, TD, FD and PD. The data is plotted between the differential number G(d) and the diameter (in nm). The mean diameters obtained for T, F and P are 449.1 nm, 438.2 nm and 438.5 nm, respectively. The mean diameters obtained for TD, FD and PD are 198.3 nm, 59 nm and 432.2 nm, respectively. For T, a straight-line plot was obtained. Also, a close to zero value of PDI, i.e. 0.005 confirms that there is the least width in the distribution plot for T. A little decrease in the mean diameter was observed as T was coated with an iron supplement. Not much difference was observed in the mean diameters of F and P. A drastic decrease in the mean diameter was observed when both T and F were loaded with imatinib. Thus, drug loading modified the mean diameter of both T and F, however, PEGylated NPs did not face much difference post-drug-loading. For FD, a very narrow size distribution was obtained with a peak value of 59 nm. This observation matches with our HR-TEM results, which confirmed the size of FericipXT-coated rutile TiO2 NPs to be ∼60 nm. For the rest of the samples, the obtained size is more than what was obtained from HR-TEM. The intensity-weighted mean diameters are different from the hydrodynamic diameters of the NPs. Table 4 (ESI File S1†) summarizes the hydrodynamic diameter in nm and PDI of T, F, P, TD, FD and PD. The PDI of all the samples was below 0.3, which indicates a narrow size distribution for all the NPs. Moreover, a non-zero value of PDI is an indicator of width in the distribution. When dispersed NPs move through a fluid, a thin electric dipole layer of the solvent attaches to their surface. This layer affects the movement of the NPs through the medium. Thus, hydrodynamic diameter is the diameter comprising the core, shell, surface coating (if any) plus the solvent layer, whereas, in the analysis via HR-TEM, the so-called solvent or the hydration layer is not present. In addition, TEM is a number-based technique whereas DLS is an intensity-based technique.29,30 Hence, it is evident that the hydrodynamic size is greater than the size obtained via measurements such as the Debye–Scherrer's formula, FESEM and HR-TEM. In the present study, the sizes of the NPs are much greater than those predicted from the HR-TEM study, which can be attributed to the hydrated layer present around them. However, the sizes of the NPs and their PDIs obtained are acceptable and are indicative of the appropriate synthesis of NPs.
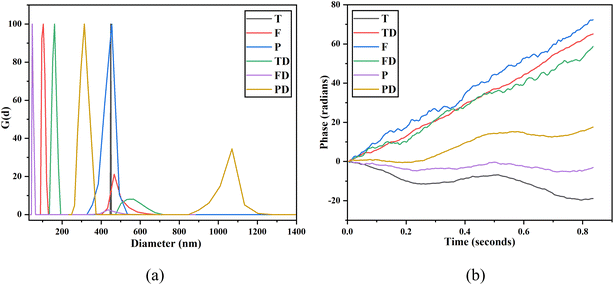 |
| | Fig. 7 (a) DLS size distribution plots obtained for T, F, P, TD, FD and PD and (b) zeta potential plots of T, F, P, TD, FD and PD. | |
Zeta potential is basically the charge that develops at the interface between a solid surface and a liquid medium. The motion of the particles is a product of Brownian motion and the attraction or repulsion occurring among the particles under the influence of an electric field. In the case the repulsive force dominates over the attractive force, the formulation becomes stable. The values of ZP indicate the interaction of the NPs with the surroundings and can be used to understand their stability, surface characteristics and adsorption phenomena. The values of zeta potential greater than +25 mV or less than −25 mV provide more stability to the sample as the particles will stay dispersed for a longer duration, dominating the van der Waals forces of attraction. Fig. 7(b) illustrates the ZP plots for T, F, P, TD, FD and PD. The mean zeta potential obtained for T, F, P, TD, FD, PD are −17.21 mV, +4.19 mV, −32.89 mV, +2.30 mV, −7.53 mV and −16.42 mV, respectively. ZP is known to vary with pH and gets more positive or negative with acidic and basic pH, respectively. ZP neither measures the charge nor the charge density, rather it tells about the surface potential. Thus, only the magnitude of ZP matters rather than the robust positive/negative charges associated with it.31 Here, PEGylated NPs show the maximum stability with −32.89 mV as the value of zeta potential. Thus, it is confirmed that PEGylation is beneficial for enhancing the stability of the NPs, which can then be effectively used in drug-delivery applications.
3.7 VSM
Undoped TiO2 NPs also demonstrated weak ferromagnetism with 0.004 emu g−1 saturation magnetization as shown in Fig. 8(a). Fig. 8(b) illustrates the VSM plot of FericipXT, F and FD. The plots of FericipXT and FD are almost superimposed. The most striking observation was that the magnetization induced in F was much greater than that induced in FericipXT alone. However, the magnetization induced in F was again reduced when it was loaded with D. In addition, the saturation magnetization of P was lower than F. A similar observation made by Tai et al.32 was that the saturation magnetization of the magnetic NPs decreased when they were coated with PEG 600 Da and the value was further decreased with the increase in the PEG content. A similar study also concluded that the saturation magnetization of the magnetite NPs decreased with increasing PEG content.18 Further, Fig. 8(c) shows the VSM plots of P and PD. Near superparamagnetic behaviour was observed for both samples. However, the magnetization induced in PD was more than that in P. Fig. 8(d) compares the VSM plots of FericipXT, FD and PD and all three plots seem to be nearly overlapping. Thus, PD can be suitably used for magnetically stimulated drug delivery applications. Liu et al.33 synthesized TiO2-coated Fe3O4 nanospheres for the delivery of Daunomycin. They observed that the coating of TiO2 onto the superparamagnetic Fe3O4 resulted in a superparamagnetic VSM plot for the obtained core–shell nanospheres but with reduced saturation magnetization, which was further reduced upon the loading of daunomycin. Thus, the drug loading often decreases the saturation magnetization of the otherwise magnetic compound. However, here, we observed that the magnetic behaviour of T was significantly improved upon coating with FericipXT and upon PEGylation. These NPs offer the notable potential to be used as magnetically guided drug-delivery vehicles. Moreover, for the second time, we demonstrated that coating with commercially available iron supplements induces superparamagnetism in the otherwise diamagnetic TiO2 NPs.17
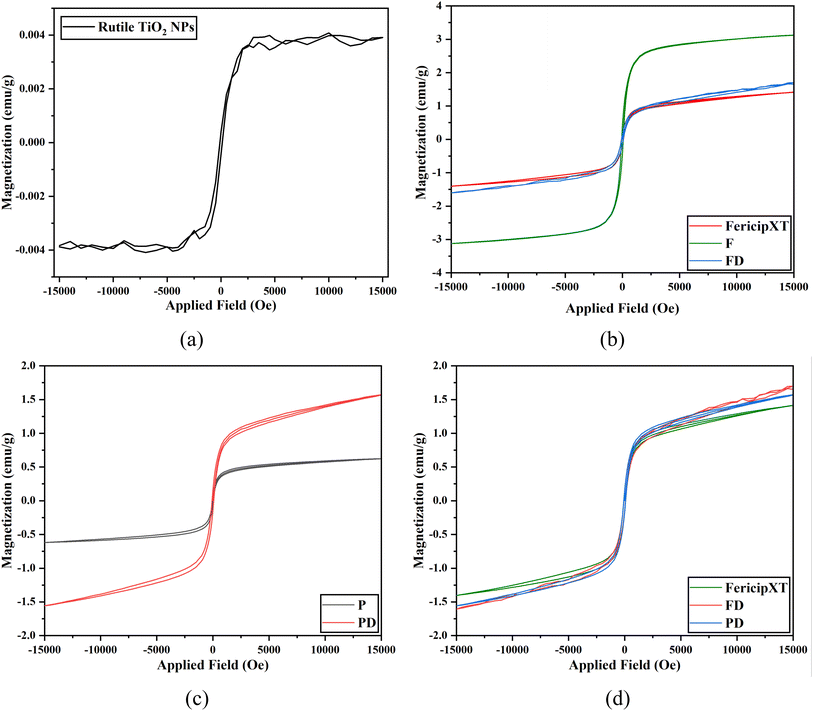 |
| | Fig. 8 VSM curves obtained for (a) T, (b) FericipXT, F and FD, (c) P and PD and (d) FericipXT, FD and PD. | |
3.8 In vitro drug release study
For the drug release studies, we investigated the release behavior at different pH levels. Various parameters such as the chemistry of the drug, drug–NP interaction, method of drug loading, percent of drug loading, solubility of the drug in the release media, surface modification and functionalization affect the rate of drug release under different conditions.34 The terminology shown in Table 5 (ESI File S1†) has been used to indicate the samples in different pH solutions.
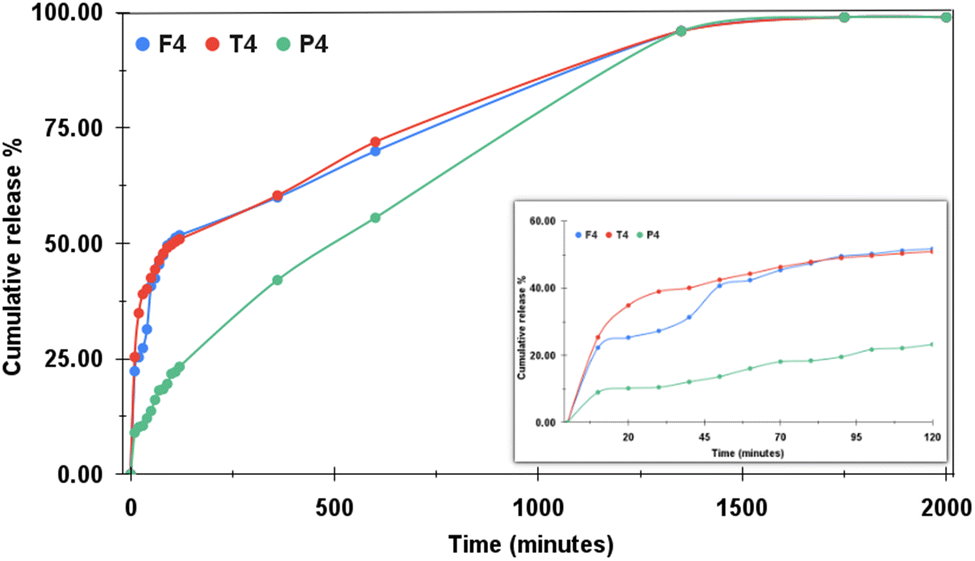
For all the pH mediums, the study was undertaken for 2000 minutes. The dialysis tube chosen for the study also influences the rapidity or retardation of the drug release. Fig. 9 shows the drug release profile from the samples T4, F4 and P4 at pH 4.4 with cumulative release percentage plotted along the Y-axis and the time (in minutes) along the X-axis. The inset displays the drug release plot obtained for the first two hours. The pH around malignant tumors is mostly acidic, which is why most of the chemo drugs are designed to release faster around acidic environments. The orally administered drugs first reach into the stomach after consumption, where the highly acidic medium breaks them down into their components from where the drug molecules are released into the bloodstream. For the acidic pH, out of all the three samples, P4 showed much-controlled drug release behaviour as it released only 23.29% of the drug in the first two hours, whereas T4 and F4 exhibited initial burst release behaviour by releasing 50.9% and 51.76% of the drug, respectively, in just 120 minutes. Thus, half of the drug was released by both T4 and F4 at the end of two hours. The drug release profiles of both T4 and F4 almost matched the entire release pattern and both released approximately 99% of the drug in the study of 2000 minutes. P4 demonstrated a very controlled drug-release behaviour. At the end of 1350 minutes, P4 also showed 96% of the drug release like the other two and from then onwards, its profile also overlapped with those of T4 and F4. Thus, in an acidic medium, P proved to be the best drug carrier showcasing a very controlled drug release. Moreover, the plot obtained for P4 exhibited no sharp ups or downs, rather it showed a very smooth linear increment for the total time period of the study. Here, we can conclude that PEGylation of the NPs improves the sustained drug release behaviour of the NPs by ensuring that the same desired quantity of the drug will be released systematically for a longer duration. This enhances drug retention but minimizes the side effects, quantity of dosages and drug release at unwanted sites.
 |
| | Fig. 9 Imatinib release profiles of TD (denoted by T4), FD (F4) and PD (P4) at pH 4.4. | |
Similarly, Fig. 10 demonstrates the drug release profile of T7, F7 and P7 at pH 7.4. The pH of normal blood generally lies between the range of 7.35 to 7.45. The NPs to be used as drug-carrying vehicles are expected to release the drug solely under acidic environments and less to no release under normal pH. With this minimal drug release can be assured during the circulation and more effective drug release at the target locations can be achieved. Under normal pH, T7 showed the maximum cumulative release percentage of 31.89% whereas F7 released 14.86% and P7 released 18.64% of the drug. Both F7 and P7 showed overly controlled drug release behaviour. The plots of both F7 and P7 ran parallelly, however, F7 outperformed P7 in the context of the least drug released under normal pH. The inset in Fig. 2 shows the initial release pattern in the first two hours. The better performance of F7 and P7 than T7 concludes that the surface modification of T improves their drug-release behaviour. It is clear from the observation that iron coating and the PEG layer are better able to hold the drug effectively for longer hours, mitigating the chances of unnecessary drug release before reaching the target site. Hettiarachchi et al.35 have reported that the pH and redox-triggered release of doxorubicin (DOX) from carbon dots was faster in acidic pH than in pH 7.4. Xu et al.36 reported in their study that more than 90% of the bare imatinib was released in the pH 5.5 in the initial 8 hours. However, when imatinib was loaded onto the liposomes, only 48.3% of the drug was released in 48 hours. PEG molecules are known to retain their helical structure in aqueous solutions.37 In the present study, the PEGylated NPs showed the most sustained drug release behaviour than T and F under acidic and neutral pH conditions. Kumskova et al.38 reported that polymers of different molecular weights affect the different parameters of the drug-loaded NPs such as percent drug loading, pattern of drug release and degradation of the NPs. Jain et al.29 reported the initial burst release of DOX from PEGylated CdSe/ZnS core–shell NPs in the first 180 minutes of the drug-release study followed by slower and sustained release of the drug. The drug release was higher in the acidic pH than in the neutral pH. The drug-release mechanism was found to be associated with Weibull with a lag-time model, which indicates the release of the drug by dissolution. Bharati et al.30 found in their study that PEG-diamine functionalized CdSe/ZnS NPs are capable of regulating the pH-dependent in vitro release of quercetin. An initial burst release took place for the first 200 minutes and more quercetin was released at neutral pH than at mildly acidic pH. Thus, different drug molecules favor different pH conditions to get released.
 |
| | Fig. 10 Imatinib release profiles of TD (denoted by T7), FD (F7) and PD (P7) at pH 7.4. | |
Additionally, the samples were also tested under the basic medium of pH 9.0. Fig. 11 illustrates the drug-release profiles for T9, F9 and P9. The inset shows the release profile for the initial two hours. F9 almost restricted the release of the drug and showed only 4.73% of the drug release in the entire study. However, P9 attained 19.1% of the drug release and T9 released 17.44% of the drug. This observation was strikingly different as P9 behaved strangely under this pH level. Normally, as the pH increases, the drug release should decrease. TD followed this notion and they released 99% of the drug at pH 4.4, 31.89% of the drug at pH 7.4 and 17.44% of the drug at pH 9.4. Similarly, FD released 99% of the drug under pH 4.4, 14.86% of the drug under pH 7.4 and just 4.73% of the drug at pH 9.4. Both TD and FD demonstrated maximum drug release under acidic conditions and accordingly limited the release of the drug as the pH was increased to neutral, further restricting the drug release when the pH turned basic. P outperformed the other two under acidic conditions by exhibiting sustained drug release still achieving 99% of the drug release in the end. In neutral pH, the cumulative drug release achieved was reduced by 80.36%. However, in the buffer solution exhibiting basicity, PD showed 19.1% of the drug release, which is more than that achieved in neutral pH by 0.46%. In other words, it can be put that the P behaved alike under both neutral and basic pH mediums. It was expected that the cumulative drug release percentage would be lesser in basic pH than that acquired in neutral pH, but no such observation was made. Similar behaviour was observed by Jain et al.39 where the swelling rate of polymethyl methacrylate (PMMA) accelerated in a slightly alkaline medium on account of hydrolytic cleavage of the polymer chains. The hydrolytic cleavage results in acidic species in the polymer network creating repulsion among positively charged polymer chains thereby increasing the swelling ratio.
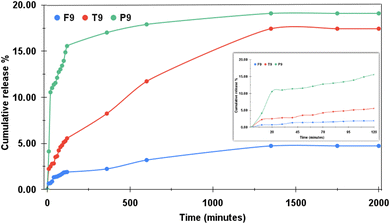 |
| | Fig. 11 Imatinib release profiles of TD (denoted by T9), FD (F9) and PD (P9) at pH 9.0. | |
Kadivar et al.40 in their study reported that 99.56% of the imatinib gets released within 20 minutes in 0.1 N HCl solution. The NPs synthesized by us demonstrated sustained drug-release behaviour, which is much needed for an effective chemotherapeutic treatment. If a drug is retained for a longer time, this would diminish the requirement of drug dosages thereby minimizing the side effects associated with drug overdose. The maximum drug release attained by the NPs synthesized by us under acidic pH conditions suggests that they undergo comparatively faster decomposition and promote faster drug release under acidic pH conditions as compared to neutral pH conditions. In a nutshell, the results revealed that the release of imatinib is strongly pH-responsive. The pH-dependent release behaviour of imatinib can help improve the efficiency of the drug-delivery system involving the nanocarriers and the drug.
3.9 Hemolysis assay
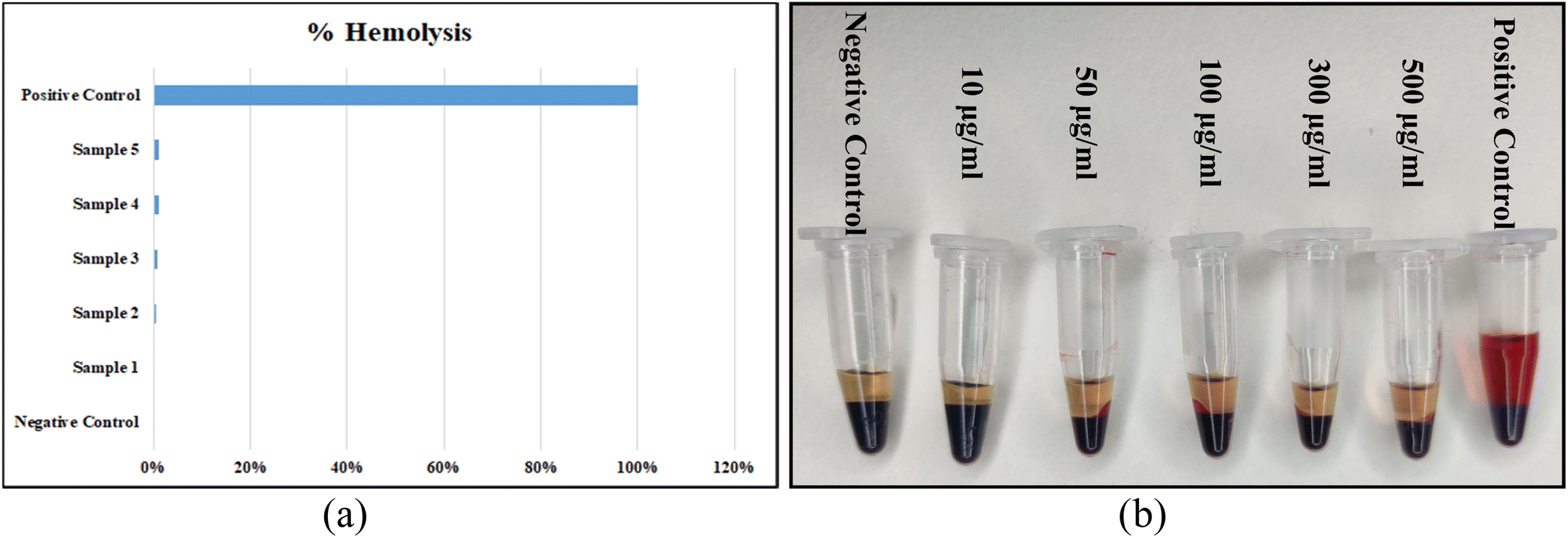
Hemolysis assay was performed to understand the blood compatibility of the PEGylated NPs. PEGylation was performed on the NPs to improve their biocompatibility and the same can be observed from the results depicted in Fig. 12(a). 0% hemolysis was observed in the blood sample obtained by centrifuging it with 0.9% saline solution and this was taken as the negative control. 100% hemolysis was obtained by centrifuging the blood sample with 1% Triton X-100 solution and this was treated as the positive control. The ratio of hemolysis of the samples was in the range between 0-1.2%, which was less than the critical safe limit of hemolysis for biomaterials as per ISO/TR 7406. Thus, the damage posed by the samples was very little. The least hemolysis that occurred in samples 2 to 5 was almost undetectable by the naked eye. Moreover, it was observed that the percentage of hemolysis was incremented slightly with the increase in the concentration of the NPs.
 |
| | Fig. 12 (a) The bar-chart representation of the %hemolysis obtained in all the five samples in comparison with the positive and negative control and (b) the images of all the five blood samples containing the NPs in the respective concentrations, positive control and negative control obtained after centrifugation. | |
The actual pictures of samples 2 to 5, positive control and negative control after centrifugation are depicted in Fig. 12(b). The percentage of hemolysis increased slightly with the increase in the concentration of the NPs. Aisaka et al.41 suggested that grave attention should be paid towards the polymorphs of the NPs when studying their toxicity. However, in their experiment, no direct evidence of the generation of any oxidative stress intracellularly by TiO2 NPs was observed. The plasma abolished any sort of hemolysis initiated by both anatase and rutile TiO2 NPs. Thus, our PEGylated FericipXT-coated rutile TiO2 NPs are a safer option for drug-delivery applications. Additionally, there is evidence that confirms that PEGylation minimizes the hemolysis induced by NPs and also the percent hemolysis is significantly reduced by increasing the PEG content. PEG coating ameliorates the systemic delivery of the therapeutic and is also helpful in overcoming extracellular barriers. Further, the biodistribution of the NPs and their clearance from the body is also promoted.42
3.10 Mathematical models and understanding the drug release mechanism
In order to understand the drug-release mechanism, the in vitro drug-release data was studied using different mathematical models. The different models proposed to assist in understanding the dissolution profile of the drug with respect to time. Different factors such as the size of the NPs, their shape, porosity, crystallinity, surfactants, polymer-coating, the drug itself, and the process of drug-loading affect the release kinetics. The drug is released in various ways such as burst release, extended release, controlled release, delayed release and sustained release.43 The first 60% of the release curve obtained is employed for the statistical analysis, which tells about the mechanism governing the drug-release pattern. The drug-release profiles obtained for imatinib release from different samples under different pH levels were analyzed against various mathematical models such as the zero-order model, first-order model, Higuchi model, Hixson-Crowell model, Weibull model, Noyes-Whitney model, Korsmeyer-Peppas model, Peppas–Sahlin model and 2nd-degree polynomial model.
The model providing the highest value of the coefficient of determination R2 will be chosen as the model describing the drug-release mechanism. Table 6 (ESI File S1†) describes the coefficient of correlation R2 values for all the samples against nine models chosen by us. Among all the models, the highest value of R2 was obtained for the Peppas–Sahlin model. Peppas and Sahlin44 proposed a release kinetics model given by eqn (7) as follows:
| |
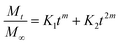 | (7) |
where
K1,
K2 and
m are constants. The highest
R2 values denote that the drug-release mechanism can be easily understood by means of the Peppas–Sahlin model, which suggests that the release of the drug from the system is governed by a combination of Fickian diffusion and Case II relaxation. Fickian diffusion release is due to the general molecular diffusion of the drug, which occurs because of the chemical potential gradient. Case II relaxation release is associated with the stress and state transition occurring in hydrophilic polymers that swell in water or biological fluids. Thus, we can conclude that the release of imatinib from the different NP-based formulations is best fitted to the Peppas–Sahlin model and the drug follows Fickian diffusion coupled with Case II relaxation.
4 Conclusions
Rutile TiO2 NPs were coated with the iron supplement, FericipXT to enhance their magnetic response for magnetically guided drug delivery application. They were further PEGylated to improve their biocompatibility and reduce toxicity. XRD, FTIR and HR-TEM confirmed the successful synthesis of NPs and loading of the chemotherapeutic. The superparamagnetic response displayed by the NPs ensures their magnetically guided targeted drug-delivery behaviour. An increased hydrodynamic size obtained for PEGylated NPs provides enhanced stability and prolonged circulation time within biological systems. The PEGylation process involves the covalent attachment of PEG chains to the nanoparticle surface. This modification not only shields the nanoparticles from rapid clearance by the immune system but also reduces their tendency to aggregate. Consequently, the larger hydrodynamic size allows for improved colloidal stability, minimizing the risk of premature particle aggregation and improving their overall performance. The larger hydrodynamic size contributes to enhanced accumulation at the target site through the EPR effect. This phenomenon occurs due to the leaky vasculature and inadequate lymphatic drainage typically found in tumors. Thus, we can conclude that the PEGylated NPs synthesized in this study can potentially improve the therapeutic efficacy of the anticancer drugs, leading to better treatment outcomes and reduced side effects.
The formulations synthesized were tested for in vitro delivery of imatinib, a chemotherapeutic in three different pH levels. A sustained release of the drug was achieved by the PEGylated NPs. Moreover, different nano-formulations were observed to release the drug following the Peppas–Sahlin model under all the pH conditions. Thus, the drug release mechanism followed both Fickian diffusion and Case II relaxation. These drug-carrying NPs offer potential applications in magnetically-guided targeted delivery of chemotherapeutics as confirmed from the findings of VSM characterization. The chances of premature drug release can be diminished and those of site-specific drug release can be greatly enhanced thereby minimizing the side effects of the dosages provided. Moreover, the PEGylated NPs appeared non-toxic as the percent hemolysis occurred belonged to a safe level.
5 Future prospects
The potential of NPs in drug delivery is promising for the future. By loading chemo drugs onto NPs and delivering them to the target site within the body, the side effects of these drugs can be minimized. Our research focuses on the in vitro release of an anticancer drug loaded onto different formulations of rutile TiO2 NPs. This study is unique as it analyzes the loading of imatinib onto TiO2 NPs, which has not been previously studied. Previous research has shown that these NPs can successfully and controllably deliver Doxorubicin and Daunorubicin. To achieve targeted drug delivery, we have modified the magnetic properties of the NPs to make them magnetically responsive. We coated TiO2 NPs with an iron supplement to enhance their magnetic character and biocompatibility. However, the research did not cover magnetically directed drug delivery in vivo. The study also includes testing the toxicity of the NPs for hemolysis and ROS generation. The scope of the study involves using TiO2 NPs modified with magnetic shells and a suitable polymer, controlling the size and shape of the NPs, loading a single drug, and checking the pH-responsive in vitro drug release. Although TiO2 NPs have shown efficiency in carrying and delivering drugs in a sustained manner, they and other similar NPs may still pose health hazards. It is important to address these concerns and explore greener and eco-friendly synthesis methods. Further research can focus on synthesizing TiO2-based nanostructures and nanocomposites, enhancing the porosity of the NPs for better drug loading, improving their biocompatibility, studying the behavior of different antitumor drugs when loaded onto the NPs, studying dual-drug delivery, designing drug-delivery vehicles that are photo and thermal-responsive, attaching target-specific receptors to the NPs, performing in vitro cytotoxicity assays for different cancer cell lines, and simulating human physiological conditions to check the targeted drug release. Combining different methods such as drug delivery via nanocarriers and photodynamic therapy (PDT) or sonodynamic therapy (SDT) can also have a significant impact on cancer treatment.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
The authors confirm their contribution to the paper as follows: Conceptualization – S. Bhullar. Data curation – S. Bhullar. Formal Analysis – S. Bhullar, N. Goyal, S. Gupta. Funding acquisition – S. Bhullar, S. Gupta. Investigation – S. Bhullar. Methodology – S. Bhullar, S. Gupta. Project administration – S. Bhullar, S. Gupta. Resources – S. Bhullar, N. Goyal, S. Gupta. Software – S. Bhullar, N. Goyal. Supervision – N. Goyal, S. Gupta. Validation – S. Bhullar, N. Goyal, S. Gupta. Visualization – S. Bhullar. Writing – original draft – S. Bhullar. Writing – review and editing – S. Bhullar, N. Goyal, S. Gupta. All authors reviewed the results and approved the final version of the manuscript.
Conflicts of interest
The authors have no Conflicts of Interest to disclose.
Acknowledgements
The authors declare that (s)he has no relevant or material financial interests that relate to the research described in this paper. We are grateful to the Sophisticated Analytical Instrumentation Facility (SAIF) and CIL, Panjab University, Chandigarh for timely characterizations.
References
- M. Yan, X. Fan, H. Si, X. Wang, Z. Wang, Z. Wang, X. Lv, H. Yin, Y. Jia, L. Jiang, Y. Xia and Y. Liu, Association between gene polymorphism and adverse effects in cancer patients receiving docetaxel treatment: a meta-analysis, Cancer Chemother. Pharmacol., 2022, 89, 173–181, DOI:10.1007/s00280-021-04374-3.
- M. T. Manzari, Y. Shamay, H. Kiguchi, N. Rosen, M. Scaltriti and D. A. Heller, Targeted drug delivery strategies for precision medicines, Nat. Rev. Mater., 2021, 6, 351–370, DOI:10.1038/s41578-020-00269-6.
- A. Yadav, S. Singh, H. Sohi and S. Dang, Advances in Delivery of Chemotherapeutic Agents for Cancer Treatment, AAPS PharmSciTech, 2022, 23, 25, DOI:10.1208/s12249-021-02174-9.
- T. A. Georgescu, R. Bohiltea, O. Munteanu, C. Grigoriu, I. Paunica and M. Sajin, A mini-review regarding the carcinogenesis and morphology of serous tumors of the ovary, fallopian tube and peritoneum, J. Mind. Med. Sci., 2021, 8(1), 7, DOI:10.22543/7674.81.P4452.
- A. D. Wong, M. Ye, M. B. Ulmschneider and P. C. Searson, Quantitative Analysis of the Enhanced Permeation and Retention (EPR) Effect, PLoS One, 2015, 10(5), e0123461, DOI:10.1371/journal.pone.0123461.
- Q. Meng, H. Meng, Y. Pan, J. Liu, J. Li, Y. Qib and Y. Huang, Influence of nanoparticle size on blood–brain barrier penetration and accumulation of anti-seizure medicines in the brain, J. Mater. Chem. B, 2022, 10, 271–281, 10.1039/D1TB02015C.
- J. Yoo, C. Park, G. Yi, D. Lee and H. Koo, Active Targeting Strategies Using Biological Ligands for Nanoparticle Drug Delivery Systems, Cancers, 2019, 11(5), 640, DOI:10.3390/cancers11050640.
- A. Tewabe, A. Abate, M. Tamrie, A. Seyfu and E. A. Siraj, Targeted Drug Delivery – From Magic Bullet to Nanomedicine: Principles, Challenges, and Future Perspectives, J. Multidiscip. Healthc., 2021, 14, 1711–1724, DOI:10.2147/JMDH.S313968.
- M. Jarosz, J. Kapusta-Kołodziej, A. Pawlik, K. Syrek and G. D. Sulka, Drug delivery systems based on titania nanostructures, in Nanostructures for Drug Delivery, Elsevier, 2017, pp. 299–326 Search PubMed.
- S. Jafari, B. Mahyad, H. Hashemzadeh, S. Janfaza, T. Gholikhani and L. Tayebi, Biomedical Applications of TiO2 Nanostructures: Recent Advances, Int. J. Nanomed., 2020, 15, 3447–3470 CrossRef CAS PubMed.
- M. Uribe-Robles, E. Ortiz-Islas, E. Rodriguez-Perez, F. F. Valverde, T. Lim and A. A. Martinez-Morales, Targeted delivery of temozolomide by nanocarriers based on folic acid-hollow TiO2 -nanospheres for the treatment of glioblastoma, Biomater. Adv., 2023, 151, 213442 CrossRef CAS PubMed.
- R. M. Abdel-Megeed, A. H. Z. Abdel-Hamid and M. O. Kadry, Titanium dioxide nanostructure-loaded Adriamycin surmounts resistance in breast cancer therapy: ABCA/P53/C-myc crosstalk, Future Sci. OA, 2024, 10(1), FSO979 CrossRef PubMed.
- S. Bhullar, S. Gupta and N. Goyal, A recipe for optimizing TiO2 nanoparticles for drug delivery applications, Open Nano, 2022, 8, 100096, DOI:10.1063/5.0001662.
- S. Bhullar, N. Goyal and S. Gupta, Synthesizing and Optimizing Rutile TiO2 Nanoparticles for Magnetically Guided Drug Delivery, Int. J. Nanomed., 2022, 21(17), 3147–3161, DOI:10.2147/IJN.S367358.
- M. E. Mehr, H. Maleki-Ghaleh, M. Yarahmadi, M. Kavanlouei and M. H. Siadati, Synthesis and characterization of photocatalytic zinc oxide/titanium oxide (core/shell) nanocomposites, J. Alloys Compd., 2021, 882, 160777, DOI:10.1016/j.jallcom.2021.160777.
- M. W. Akram, F. Raziq, M. Fakhar-e-Alam, M. H. Aziz, K. S. Alimgeer, M. Atif, M. Amir, A. Hanif and W. A. Farooq, Tailoring of Au-TiO2 nanoparticles conjugated with doxorubicin for their synergistic response and thermodynamic therapy applications, J. Photochem. Photobiol., A, 2019, 384, 112040, DOI:10.1016/j.jphotochem.2019.112040.
- S. Bhullar, N. Goyal and S. Gupta, In vitro pH-responsive release of imatinib from iron-supplement coated anatase TiO2 nanoparticles, Sci. Rep., 2022, 12, 4600, DOI:10.1038/s41598-022-08090-7.
- G. Antarnusa and E. Suharyadi, A synthesis of polyethylene glycol (PEG)–coated magnetite Fe3O4 nanoparticles and their characteristics for enhancement of biosensor, Mater. Res. Express, 2020, 7, 056103, DOI:10.1088/2053-1591/ab8bef.
- Y. Li, J. Xu, Y. Xu, L. Huang, J. Wang and X. Cheng, Synthesis and characterization of fluorescent chitosan-ZnSe/ZnS nanoparticles for potential drug carriers, RSC Adv., 2015, 5, 38810–38817, 10.1039/C5RA02933C.
- R. Raj, P. M. Raj and A. Ram, Preparation and characterization of solid lipid nanoparticles loaded with cytarabine via a micellar composition for leukemia, RSC Adv., 2016, 6, 53578–53586, 10.1039/C6RA10111A.
- S. Bharti, Interaction of Drugs with Semiconductor Core Shell Nanoparticles, PhD thesis, Panjab University, Chandigarh, 2018, https://hdl.handle.net/10603/241578, (Source: INFLIBNET Centre) Search PubMed.
- A. Patel, P. Heussen, J. Hazekamp and K. P. Velikov, Stabilisation and controlled release of silibinin from pH responsive shellac colloidal particles, Soft Matter, 2011, 7, 8549, 10.1039/C1SM05853C.
- M. Kaya, Ö. Şahin and C. Saka, Preparation and TG/DTG, FT-IR, SEM, BET Surface Area, Iodine Number and Methylene Blue Number Analysis of Activated Carbon from Pistachio Shells by Chemical Activation, Int. J. Chem. React. Eng., 2018, 16(2), 20170060, DOI:10.1515/ijcre-2017-0060.
- S. W. Hwang, A. Umar, G. N. Dar, S. H. Kim and R. I. Badran, Synthesis and Characterization of Iron Oxide Nanoparticles for Phenyl Hydrazine Sensor Applications, Sens. Lett., 2014, 12, 97–101, DOI:10.1166/sl.2014.3224.
- E. Liu, Y. Zhou, Z. Liu, J. Li, D. Zhang, J. Chen and Z. Cai, Cisplatin loaded hyaluronic acid modified TiO2 nanoparticles for neoadjuvant chemotherapy of ovarian cancer, J. Nanomater., 2015, 2015 DOI:10.1155/2015/390358.
- P. Bhattacharya and S. Neogi, Gentamicin coated iron oxide nanoparticles as novel antibacterial agents, Mater. Res. Express, 2017, 4(9), 095005, DOI:10.1088/2053-1591/aa8652.
- S. A. Zulfiqar, R. Khan, T. Zeb, M. ur Rahman, Burhanullah, S. Ali, G. Khan, Z. ur Rahman and A. Hussain, Structural, optical, dielectric and magnetic properties of PVP coated magnetite (Fe3O4) nanoparticles, J. Mater. Sci.: Mater. Electron., 2018, 29, 20040–20050, DOI:10.1007/s10854-018-0134-6.
- T. Gayathri, R. Kumar, S. Dhilipkumaran, C. K. Jayasankar, P. Saravanan and B. Devanand, Microwave-assisted combustion synthesis of silica-coated Eu:Gd2O3 nanoparticles for MRI and optical imaging of cancer cells, J. Mater. Sci.: Mater. Electron., 2019, 30, 6860–6867, DOI:10.1007/s10854-019-00999-6.
- S. Jain, S. Bharti, G. K. Bhullar and S. K. Tripathi, pH dependent drug release from drug conjugated PEGylated CdSe/ZnS nanoparticles, Mater. Chem. Phys., 2020, 240, 122162, DOI:10.1016/j.matchemphys.2019.122162.
- S. Bharti, G. Kaur, S. Gupta and S. K. Tripathi, PEGylated CdSe/ZnS core/shell nanoparticles for controlled drug release, Mater. Sci. Eng., B, 2019, 243, 115–124, DOI:10.1016/j.mseb.2019.03.015.
- S. Bhattacharjee, Block copolymer micelles for drug delivery: design, characterization and biological significance, J. Controlled Release, 2016, 235, 337–351, DOI:10.1016/j.jconrel.2016.06.017.
- M. F. Tai, C. W. Lai and S. B. A. Hamid, Facile Synthesis Polyethylene Glycol Coated Magnetite Nanoparticles for High Colloidal Stability, J. Nanomater., 2016, 8612505, DOI:10.1155/2016/8612505.
- M. C. Liu, B. Liu, X. Y. Sun, H. C. Lin, J. Z. Lu, S. F. Jin, S. Q. Yan, Y. Y. Li and P. Zhao, Core/Shell Structured Fe3O4@TiO2-DNM Nanospheres as Multifunctional Anticancer Platform: Chemotherapy and Photodynamic Therapy Research, J. Nanosci. Nanotechnol., 2018, 18, 4445–4456, DOI:10.1166/jnn.2018.15338.
- V. Chandrakala, V. Aruna and G. Angajala, Review on metal nanoparticles as nanocarriers : current challenges and perspectives in drug delivery systems, Emerg, Mater, 2022, 237, 1–23, DOI:10.1007/s42247-021-00335-x.
- S. D. Hettiarachchi, E. K. Cilingir, H. Maklouf, E. S. Seven, S. Paudyal, S. Vanni, R. M. Graham and R. M. Leblanc, pH and redox triggered doxorubicin release from covalently linked carbon dots conjugates, Nanoscale, 2021, 13, 5507–5518, 10.1039/D0NR08381J.
- H. Xu, H. Ji, Z. Li, W. Qiao, C. Wang and J. Tang, In vivo Pharmacokinetics and in vitro Release of imatinib Mesylate-Loaded Liposomes for Pulmonary Delivery, Int. J. Nanomed., 2021, 16, 1221–1229, DOI:10.2147/IJN.S294626.
- D. Q. M. Craig, Polyethyelene Glycols and Drug Release, Drug Dev. Ind. Pharm., 1990, 16(17), 2501–2526, DOI:10.3109/03639049009058544.
- Y. Ermolenko, O. N. Gorunova, V. V. Dunina, D. B. Petrenko, N. G. Novikova, A. Alekseeva, N. Osipova, K. A. Kochetkov, A. Morozov and S. Gelperina, Quantitative analysis of palladacycle-tagged PLGA nanoparticle biodistribution in rat organs by means of atomic absorption spectrometry and inductively coupled plasma mass spectrometry, J. Anal. At. Spectrom., 2021, 36(11), 2423–2430, 10.1039/D1JA00260K.
- S. Jain and A. K. Bajpai, Designing polyethylene glycol (PEG) – plasticized membranes of poly(vinyl alcohol-g-methyl methacrylate) and investigation of water sorption and blood compatibility behaviors, Des. Monomers Polym., 2013, 16(5), 436–446, DOI:10.1080/15685551.2012.747162.
- A. Kadivar, B. Kamalidehghan, H. A. Javar, E. T. Davoudi, N. D. Zaharuddin, B. Sabeti, L. Y. Chung and M. I. Noordin, Formulation and In Vitro, In Vivo Evaluation of Effervescent Floating Sustained-Release imatinib Mesylate Tablet, PLoS One, 2015, 10, e0126874, DOI:10.1371/journal.pone.0126874.
- Y. Aisaka, R. Kawaguchi, S. Watanabe, M. Ikeda and H. Igisu, Hemolysis Caused by Titanium Dioxide Particles, Inhalation Toxicol., 2008, 20, 891–893, DOI:10.1080/08958370802304123.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, PEGylation as a strategy for improving nanoparticle-based drug and gene delivery, Adv. Drug Delivery Rev., 2016, 99(Pt A), 28–51, DOI:10.1016/j.addr.2015.09.012.
- S. Jafari, M. Soleimani and M. Badinezhad, Application of different mathematical models for further investigation of in vitro drug release mechanisms based on magnetic nano-composite, Polym. Bull., 2022, 79, 1021–1038, DOI:10.1007/s00289-021-03537-9.
- N. A. Peppas and J. J. Sahlin, A simple equation for the description of solute release. III. Coupling of diffusion and relaxation, Int. J. Pharm., 1989, 57(2), 169–172, DOI:10.1016/0378-5173(89)90306-2.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Navdeep Goyala and
Shikha Guptac
*ab,
Navdeep Goyala and
Shikha Guptac