
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdsorption effects of acetone and acetonitrile on defected penta-PdSe2 nanoribbons: a DFT study
Nguyen Thanh Tien a,
Nguyen Hai Danga,
Pham Thi Bich Thaoa,
K. Dien Vobc,
D. M. Hoatde and
Duy Khanh Nguyen*fg
a,
Nguyen Hai Danga,
Pham Thi Bich Thaoa,
K. Dien Vobc,
D. M. Hoatde and
Duy Khanh Nguyen*fg
aCollege of Natural Sciences, Can Tho University, 3-2 Road, Can Tho City 900000, Vietnam
bDivision of Applied Physics, Dong Nai Technology University, Bien Hoa City, Vietnam
cFaculty of Engineering, Dong Nai Technology University, Bien Hoa City, Vietnam
dInstitute of Theoretical and Applied Research, Duy Tan University, Ha Noi 100000, Vietnam
eFaculty of Natural Sciences, Duy Tan University, Da Nang 550000, Vietnam
fLaboratory for Computational Physics, Institute for Computational Science and Artificial Intelligence, Van Lang University, Ho Chi Minh City, Vietnam
gFaculty of Mechanical – Electrical and Computer Engineering, School of Technology, Van Lang University, Ho Chi Minh City, Vietnam. E-mail: khanh.nguyenduy@vlu.edu.vn
First published on 21st May 2024
Abstract
Using DFT calculations, the structural and electronic properties of the ZZ7 p-PdSe2 nanoribbons (ZZ7) with the four kinds of vacancy defects, including ZZ7-VPd, ZZ7-VSe, ZZ7-VPd+Se, and ZZ7-V2Se are studied, in which their stability, diverse geometries, and altered electronic properties are determined through the formation energies, optimal structural parameters, electronic band structures, and DOSs. Specifically, the formation energies of all studied systems show significant negative values around −3.9 eV, evidencing their good thermal stability. The geometries of four defective structures exhibit different diversification, whereas only the ZZ7-V2Se structure possesses the highly enhanced feature, identified as the most effective substrate for the acetone and acetonitrile adsorption. On the electronic behaviors, the ZZ7 band structure displays the nonmagnetic metallic characteristics that become the ferromagnetic half-metallic band structures for the ZZ7-VPd and ZZ7-VSe and the ferromagnetic semi-metallic band structures for the ZZ7-VPd+Se and ZZ7-V2Se. For adsorption of the acetone and acetonitrile on the ZZ7-V2Se structure, the energetic stability, adsorption sites, adsorption distances, charge transfers, and electronic characteristics of the adsorbed systems are determined by the adsorption energies, optimal adsorption sites, adsorption distances, Mulliken populations, and DOSs. The adsorption energies of the acetone- and acetonitrile-adsorbed ZZ7-V2Se systems display significant values at −1.2 eV and −0.86 eV at the preferable sites of 8 and 11, respectively, indicating their great adsorption ability. The adsorption mechanism of the acetone- and acetonitrile-adsorbed systems belongs to the physisorption owing to absence of chemical bonds, in which the bond lengths of the ZZ7-V2Se substrate show a very small deviation. Under the acetone and acetonitrile adsorptions, the ferromagnetic semi-metallic DOSs of the ZZ7-V2Se become the ferromagnetic half-metallic DOSs for the ZZ7-V2Se-acetone-8 and the ferromagnetic semiconducting DOSs for the ZZ7-V2Se-acetonitrile-11. Our systematic results can provide a complete understanding of the acetone- and acetonitrile adsorptions on the potential ZZ7-V2Se structure, which is very useful for nanosensor application.
1 Introduction
Two-dimensional (2D) materials have attracted considerable attention in recent years due to their unique physical and chemical properties, such as high surface area, tunable band gap, and diverse electronic structures.1–10 Among various 2D materials, transition metal dichalcogenides (TMDs) are promising candidates for nanoelectronic and nanosensor applications, as they exhibit a wide range of electronic behaviors, from metallic to semiconducting, depending on their composition and structure.11–13 One of the ways to tune the electronic properties of 2D materials is to create nanostructures, including nanoribbons, nanotubes, and quantum dots that result in edge terminations and quantum confinement effects.14–17 Specifically, nanoribbons are particularly interesting, as they can exhibit different edge configurations and edge states, which can significantly affect their electronic and magnetic properties. For example, zigzag-edged nanoribbons usually have metallic edge states, while armchair-edged nanoribbons can have either metallic or semiconducting behavior depending on their width.18–22 Another way to modify the electronic properties of 2D materials is to adsorb molecules or atoms on their surfaces, which can induce charge transfer, doping, or band gap engineering.23–27 The adsorption of organic molecules on 2D materials is especially relevant for the development of nanosensors, as the interaction between the molecules and the 2D material can alter the electrical conductivity, optical absorption, or surface plasmon resonance of the 2D material, which can be detected by various sensing techniques.28 Moreover, the adsorption of organic molecules on 2D materials can also provide insight into the nature and strength of the intermolecular forces, such as van der Waals and electrostatic that govern the adsorption process.29,30 Regarding adsorption effects of organic molecules on the 2D materials, it has been widely reported in recent studies. Using the van der Waals DFT calculations, the structural and electronic properties of the acetone, acetonitrile, ammonia, benzene, methane, methanol, ethanol, and toluene adsorbed 2D silicene have been studied by Thaneshwor P. Kaloni et al., in which they have indicated that the adsorption energies of the adsorbed silicene systems show the significant values from −0.11 to −0.95 eV from acetone to toluene, respectively, confirming the good adsorption ability of the silcene substrate.31 Very recently, Chen-Hao Yeh has systematically investigated the sensitivity of the acetone, methanol, ethanol, and formyl aldehyde adsorbed pristine 2D transition metal dichalcogenides (TMDs) and 2D Janus TMD monolayers under DFT calculations. This study has resulted in the calculated adsorption energies of the adsorbed systems around −0.25 eV to −0.34 eV and determined that the MoS2, Janus MoSSe, and Janus MoSTe showed the greater sensitivity toward acetone than other ones.32Pentagonal palladium diselenide (p-PdSe2) is a novel 2D material that has attracted attention for its potential applications in nanoelectronics, optoelectronics, and catalysis.33,34 It has a unique pentagonal structure that exhibits high stability, tunable band gap, and strong anisotropy. In addition, the electronic properties of p-PdSe2 can be significantly affected by the presence of defects, such as vacancies, which can introduce localized states, spin polarization, and band gap opening. Therefore, it is essential to understand how defects influence the electronic structure and transport properties of p-PdSe2, as well as its interaction with organic molecules that can act as adsorbates, dopants, or sensors.35,36 Using DFT calculations, Yiping Ma et al., have studied the adsorption of the NH3, N2O, NO2, and NO gases on the PdSe2 monolayer, in which the calculated adsorption energies of the adsorbed PdSe2 systems showed the significant values of −0.287 eV, −0.228 eV, −0.579 eV, and −0.481 eV for NH3, N2O, NO2, and nd NO adsorptions, respectively, indicating that NH3 and N2O display the physisorption, while NO2 and NO belong to the chemisorption.37
Moreover, the 2D p-PdSe2 structure can be existed in a quasi one-dimensional structure so-called 1D p-PdSe2 nanoribbons that can trigger many extraordinary properties owing to its 1D quantum confinements and edge configurations.38 Very recently, the 1D p-PdSe2 nanoribbons has be successfully synthesized by Giang. D Nguyen et al., using a facile hybrid approach of top-down and bottom-up processes. The experimental results of this study has shown that the 1D p-PdSe2 nanoribbons was formed in a dimer shape on the PdSe2 substrate, and the formed 1D p-PdSe2 nanoribbons displayed the metallic behavior, which has been further confirmed by their DFT results.39 However, the results reported by Giang. D Nguyen et al., have not yet specified various edge structures of the formed 1D p-PdSe2 nanoribbons, in which the different edge structures can lead to different fundamental properties, as determined by well-known graphene nanoribbons.19,20 Using the density functional theory (DFT) calculations, the structural, magneto-electronic, electronic transport properties of the p-PdSe2 nanoribbons within the specific edge structures of the sawtooth–sawtooth (SS), armchair–armchair (AA), zigzag–armchair (ZA), and zigzag–zigzag (ZZ) have been reported in detail by N. T. Tien et al., whereas the DFT results have showed that the SS, AA, ZA, and ZZ structures present the high buckling and diverse geometries.40 On the electronic properties, the AA, ZZ, ZA p-PdSe2 nanoribbons exhibit the ferromagnetic metallic characteristics, while the SS p-PdSe2 nanoribbon shows the non-magnetic semiconducting characteristics. It is worthwhile to note that the p-PdSe2 nanoribbons possess more diverse edge shapes than the only two unique edge configurations of the graphene nanoribbons.19,20 With rich edge structures, 1D quantum confinements, and puckered 1D pentagonal layered structure, the p-PdSe2 nanoribbons can serve as good platform for adsorption of organic molecules. Especially, creating different vacancy defects in the p-PdSe2 nanoribbons is expecting to result in more diverse geometries and enriched essential properties as well as greatly enhancing its adsorption ability. Thus, a complete investigation in the p-PdSe2 nanoribbons within different vacancy defects and adsorption of acetone and acetonitrile molecules on the defected p-PdSe2 nanoribbons is an interesting topic, which has not yet reported up to date.
In this paper, using the DFT calculations, the structural and electronic properties of the p-PdSe2 nanoribbons containing in four different kinds of vacancy defects, including single Pd vacancy, single Se vacancy, double vacancies of one Pd and one Se atom, and double Se vacancies are investigated by the formation energies, optimal structural parameters, electronic band structures, and density of states (DOSs). Furthermore, adsorption effects of acetone and acetonitrile molecules on the most effective defected structure of the double Se vacancies are taken into account, whereas the adsorption energies, adsorption sites, change in bond lengths under adsorption, adsorption distances, Mulliken populations, electronic band structures, and DOSs of the adsorbed systems are calculated to determine the adsorption mechanisms, adsorption profile, and adsorption effects on the geometric and electronic characteristics.
2 Methodology
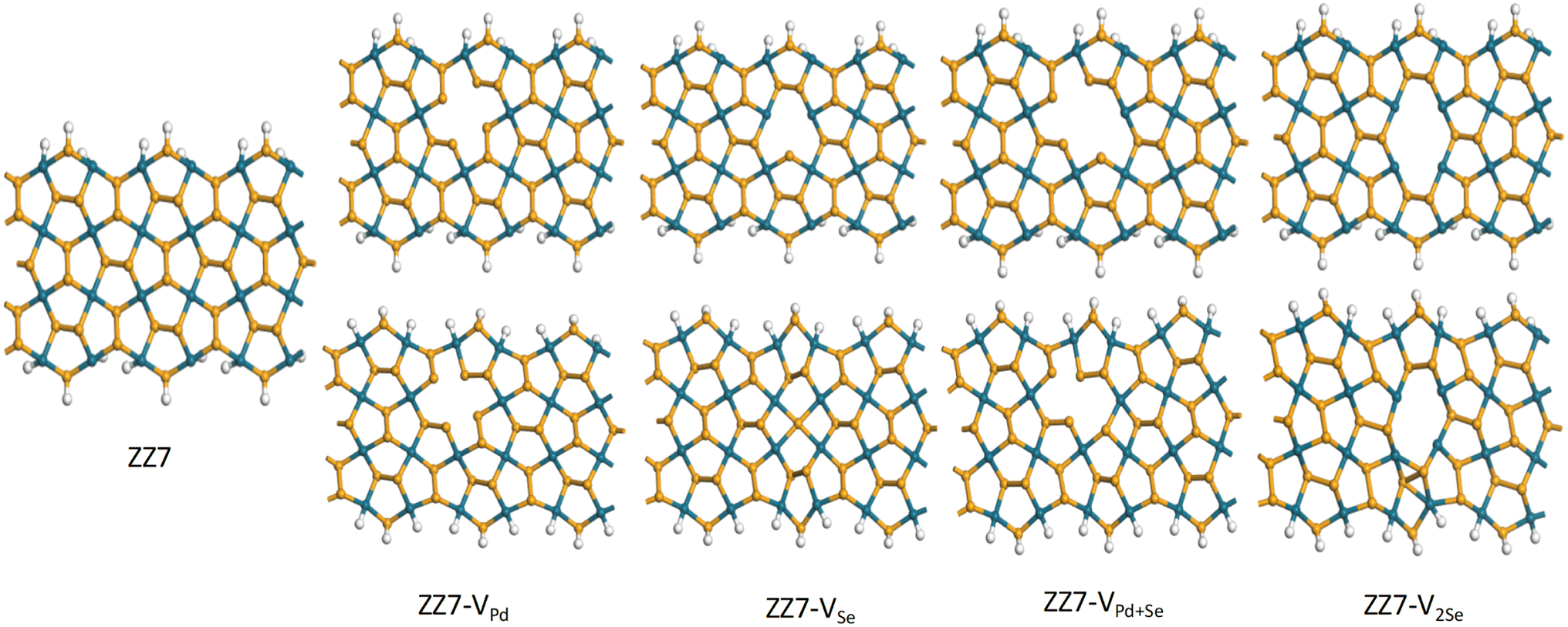
The defective vacancies can be created by removing one Se atom, one Pd atom, two atoms consisting of one Pd atom and one Se atom, or two Se atoms. With ribbon structures, vacancies can form at the edges or inside the structure. Unlike penta graphene nanoribbon, the SS edge structure is considered to be the most stable,14 the ZZ edge structure of penta palladium diselenide nanoribbon displays the most stable energy.40 However, edge-defected ZZ p-PdSe2NRs are unstable. The ZZ7 p-PdSe2NR samples were investigated, including pristine ZZ7 p-PdSe2NR, ZZ7 p-PdSe2NR with single Pd vacancy, ZZ7 p-PdSe2NR with single Se vacancy, ZZ7 p-PdSe2NR with double vacancies of Pd and Se, ZZ7 p-PdSe2NR with double Se vacancies are denoted as ZZ7, ZZ7-VPd, ZZ7-VSe, ZZ7-VPd+Se, ZZ7-V2Se, respectively, whereas the edges of these structures are passivated by hydrogen atoms (Fig. 1). | ||
| Fig. 1 (a) ZZ7 p-PdSe2NR without vacancy (ZZ7), (b) ZZ7 p-PdSe2NR with single Pd vacancy (ZZ7-VPd), (c) ZZ7 p-PdSe2NR with single Se vacancy (ZZ7-VSe), (d) ZZ7 p-PdSe2NR with double vacancies of Pd and Se (ZZ7-VPd+Se), (e) ZZ7 p-PdSe2NR with double Se vacancies (ZZ7-V2Se). | ||
To investigate the structural stability and electronic properties of defective ZZ7 p-PdSe2NR samples, the density functional theory (DFT) calculations are utilized, in which the ZZ7 p-PdSe2NR samples with different vacancies are optimized via the Perdew–Burke–Ernzerhof (PBE) potential within the gradient generalized approximation (GGA) as included in the CASTEP software, while the ATK package is used to further calculate formation energy and electronic structure.41–43 The number of k-points in the Brillouin zone is chosen to be 1 × 1 × 3 and 1 × 1 × 400 in the x, y, and z directions, respectively, for structure optimization and electronic structure calculation with a cutoff energy of 1000 eV, and the kinetic temperature of the electron system is 300 K. During the optimization process, the energy convergence is 10−6 eV, the maximum Hellman Feynman force acting on each atom is less than 0.01 eV Å−1. The van der Waals (vdW) interaction is also considered under the DFT-D2 framework in long-range dispersion correction.44
3 Results and discussion
3.1. Effect of vacancy types on the structural and electronic properties of ZZ p-PdSe2
 | (1) |
In eqn (1), EC (eV per atom) is the formation energy of system. Etotal, EPd, Se, H (eV), and nPd, Se, H are the total energy in ground-state energies, the energies of isolated Pd, Se, H atoms, and the numbers of atoms in a unit cell, respectively.
 | ||
| Fig. 2 The studied structures before and after optimization. | ||
As a result, the calculated formation energies are presented in Table 1, whereas the significant negative values evidence the good structural stability of all investigated samples that can be fully feasible for experimental synthesis approach.45 Among the four defected samples, the defected sample of ZZ7-VPd achieves the most stability due to its lowest formation energy at −3.96 eV; however, difference in the EC value of the ZZ7-VPd and other samples is insignificant as identified from Table 1. From the optimal defected structures shown at the bottom part of Fig. 2, it can be identified that only the three vacancy samples can form polygons around the defect position with introduced new bonds,46 including the VSe, VPd+Se, and V2Se. Specifically, ZZ7-VSe, ZZ7-V2Se exhibit two new bonds, while the ZZ7-VPd+Se only displays one new bond. In contrast, the ZZ7-VPd dot not form a polygon around the vacancy position, leading to its negligible structural changes. Critically, the buckling height of ZZ7-V2Se is significantly enhanced around the two new bonds compared to other samples. This enhanced buckling feature will increase the possible adsorption sites around the vacancy region of the ZZ7-V2Se structure due to the broken symmetry. Thus, the ZZ7-V2Se structure are identified as an effective substrate for organic molecules adsorption that will be fully explored in the latter part.
| Sample | EC (eV) |
|---|---|
| ZZ7 | −3.98 |
| ZZ7-VPd | −3.96 |
| ZZ7-VSe | −3.95 |
| ZZ7-VPd+Se | −3.95 |
| ZZ7-V2Se | −3.93 |
Furthermore, it is worthy mentioning that p-PdSe2 structure is formed by pentagonal rings, whereas each Pd atom is bonded to four Se atoms, while two adjacent Se atoms form covalent bonds in a single layer. When a Pd atom is lost, the optimized structure does not change significantly because the distance between Se atoms is too large to allow the creation of new bonds. For the remaining three vacancy samples, new bonds are formed because both Pd and Se have electrons in the d subshell, which can form bonds when the distance between two atoms is satisfied.47 The results of the optimal structural parameters of the vacancy samples show that the bond length in all four vacancy samples are slightly modified (maximum is 0.263 Å), in which the positions towards the edge have more variation.
As compared with its two-dimensional (2D) counterpart, the bond lengths of defective PdSe2 nanoribbon show more variation. Specifically, for the 2D PdSe2 system with Pd vacancy, the bond lengths increase by about 0.010 Å, whereas the bond lengths of ZZ7-VPd variate between 0.011 and 0.156 Å. In addition, as compared to the defective sawtooth penta-graphene nanoriibons (D-SSPGNR) with C1, C2 and 2C vacancies, which form polygons without new bonds,48 the ZZ p-PdSe2 NR with Pd vacancy do not form polygons, leading to no new bonds appeared. In contrast, ZZ7-VSe, ZZ7-VPd+Se, ZZ7-V2Se can form polygons with appearing of some new bonds. Thus, the optimal structural parameters of defective ZZ p-PdSe2 NR change much less than those of the D-SSPNR structures. Specifically, the largest bond length change of D-SSPNRs is 1.344 Å, while that of defective ZZ p-PdSe2NRs is 0.286 Å. In particular, for the ZZ7 p-PdSe2NR structure with 2Se vacancies, there is a significant change in buckling compared to the D-SSPGNR structures.
 | ||
| Fig. 3 Band structure of (a) ZZ7, (b) ZZ7-VPd, (c) ZZ7-VSe, (d) ZZ7-VPd+Se, and (e) ZZ7-V2Se. | ||
The 1D band structures of the pristine and defective ZZ7 p-PdSe2NR samples discussed in Fig. 3 can be fully confirmed by their spin-projected density of states (DOSs) as shown in Fig. 4, in which the black and green solid curves display the DOSs of the pristine and defective systems, respectively, the spin-up and spin-down DOSs are illustrated via the positive and negative DOSs. Meanwhile, the Fermi level are marked using the black dashed lines. To identifying the vacancies-induced new states, combining the DOSs of the pristine and defective systems into a single plot is made as shown in Fig. 4(a–c) for the ZZ7 and ZZ7-VPd, ZZ7 and ZZ7-VSe, ZZ7 and ZZ7-VPd+Se, and ZZ7 and ZZ7-V2Se, respectively. As a result, the higher spin-splitting bands of the defective structures compared to the pristine one are confirmed by higher spin asymmetric peaks in the DOSs components of the defective systems, and vice versa. The negligible spin-splitting metallic energy bands of the pristine system is verified by the negligible spin asymmetric peaks and existing of DOSs at the Fermi level. The half-metallic band structures of the ZZ7-VPd and ZZ7-VSe structures discussed in Fig. 3(b and c) are verified by absence of the spin-up DOSs and existence of the spin-down DOSs at the Fermi level as observed by the green curves in Fig. 4(a and b), respectively. On the other hand, the semi-metallic band structures of the ZZ7-VPd+Se and ZZ7 and ZZ7-V2Se discussed in Fig. 3(d and e) are consistent with remaining the DOSs at the Fermi level in both spin-up and spin-down components. Particularly, in the vacancies-induced DOSs, new linear electronic states appear more frequently in the conduction band near the Fermi level, mainly contributed by atoms surrounding or adjacent to the defect ring, demonstrating the localized electron confinement effect around the defect. Therefore, in the energy range near the Fermi energy, the electronic dynamic properties of defected structures are pseudo-zero-dimensional. The above characteristic is expected to create some new physical properties of the defected structures.
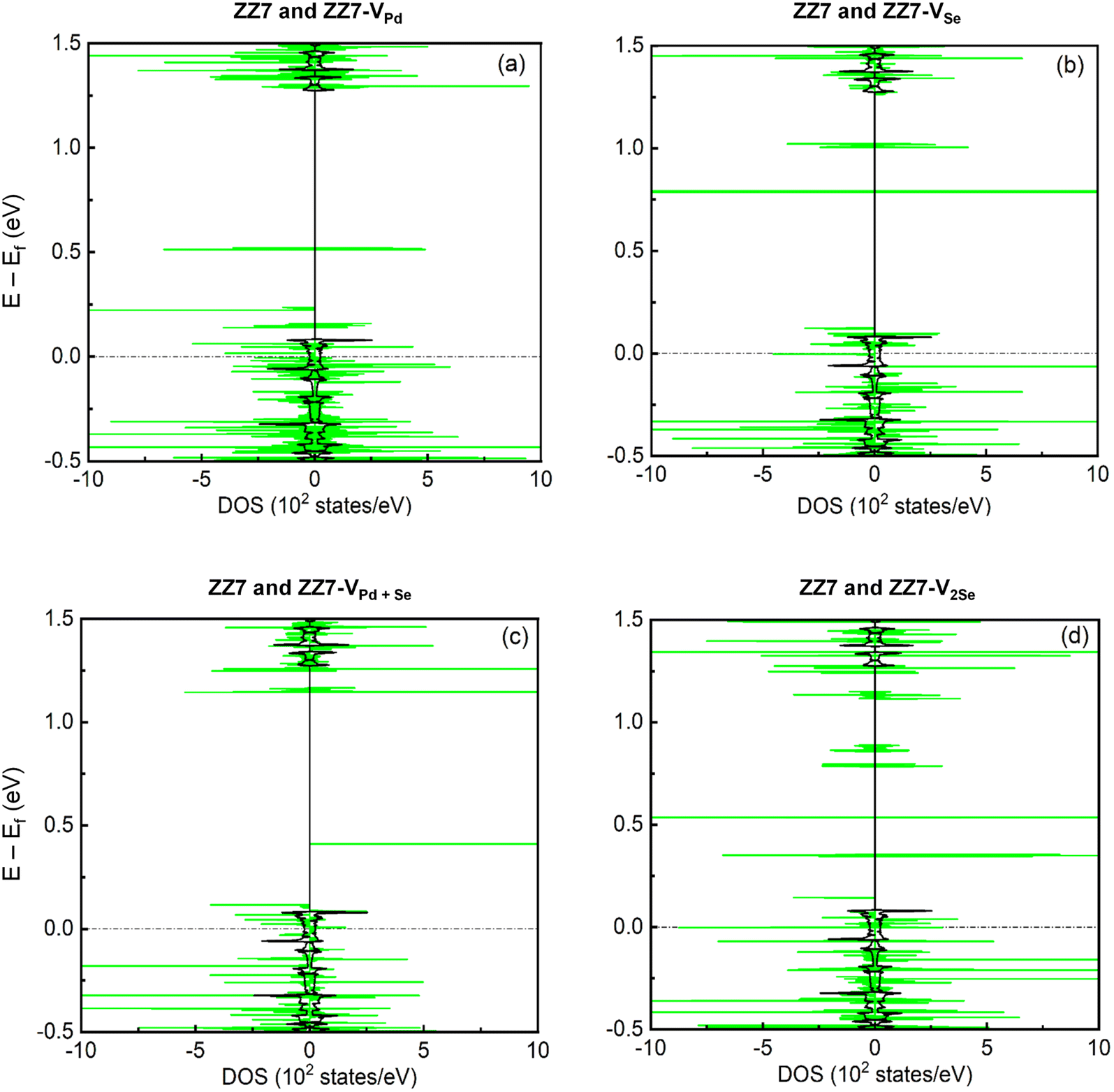 | ||
| Fig. 4 Total density of state of (a) ZZ7 and ZZ7-VPd, (b) ZZ7 and ZZ7-VSe, (c) ZZ7 and ZZ7-VPd+Se, (d) ZZ7 and ZZ7-V2Se. The black solid line depicts the ZZ7 structure and the black dashed line indicates the Fermi level. | ||
To fully determine the vacancies-created new states and vacancies-related spin-polarized peaks, the spin-projected density of states (spin-DOSs) of vacancy ZZ7 p-PdSe2NR samples are taken into account, in which the spin-DOSs of the pristine one is also performed for comparison in vacancy-induced reshape. As a result, the spin-DOSs of the pristine ZZ7, ZZ7-VPd, ZZZ7-VSe, ZZ7-VPd+Se, and ZZ7-V2Se are shown in Fig. 5(a–f), respectively. Fig. 5 shows that the vacancy patterns create new states near the Fermi level, which is similar to the Se vacancy and Pd vacancy pattern of the 2D PdSe2 structure.43 Additionally, the spin-splitting DOSs of the vacancy samples show stronger spin-polarized peaks than that of the pristine one. Specifically, in the pristine sample, the energy levels with spin-up and spin-down electronic states are symmetrical, and have small differences in intensity. In the vacancy samples, the spin-up and spin-down states near the Fermi level are asymmetric and have obvious intensity differences. Compared with the 2D defect structures, respectively VPd, VSe, VPd+Se, V2Se, the ribbon structure also changes DOS and forms new states in the conduction band, but there are some fundamental difference. Specifically, ZZ7-VPd appears new states in the conduction band, while the defective 2D PdSe2–VPd structure does not have. In addition, the defective structures with a single or double vacancies show the strong spin polarization, while the defective 2D PdSe2 only exhibits the spin polarization when the five-atom vacancies (VPd+4Se) are made. Besides, the number of new states formed in the conduction band near the Fermi level in the defective samples of the ZZ7-VSe, ZZ7-VPd+Se, ZZ7-V2Se is more than in their corresponding 2D defective structure. To further evaluate the influence of vacancies on the electronic properties of the ZZ7 p-PdSe2NR structure, the contributions of atoms were statistically calculated through the density of eigenstates. The investigated states are numbered in green in the Fig. 5. The results show that the total density of states (DOSs) with new energy levels in the vicinity of the Fermi level is mainly contributed by the local atoms around the defective regions, while the contribution of the remaining atoms is negligible.
 | ||
| Fig. 5 Spin-projected density of states of (a and d) ZZ7, (b) ZZ7-VPd, (c) ZZZ7-VSe, (e) ZZ7-VPd+Se, and (f) ZZ7-V2Se. The blue and red solid lines depict the spin-up and spin-down states, respectively. The black dashed line indicates the Fermi level. The green numbers display vacancies-induced prominent peaks. | ||
3.2. Adsorption effects of acetone and acetonitrile molecules on the pristine ZZ7 and ZZ7-V2Se 1D structures
As formerly identified, the ZZ7-V2Se system with the enriched structural features shows high feasibility for adsorption of the acetone and acetonitrile molecules as compared with the other systems. In this section, we investigate the adsorption of the acetone and acetonitrile molecules on both pristine ZZ7 structure at 3 marked sites as shown Fig. 6(a) and the ZZ7-V2Se structure at 11 marked sites in Fig. 6(b), respectively. The structural stability, electronic structure, and molecular adsorption profile of the acetone and acetonitrile molecules absorbed on the pristine ZZ7 and ZZ7-V2Se samples are calculated using the DFT calculations, in which the adsorbed systems are optimized using DFT Perdew Burke–Ernzerhof (PBE) pseudopotential within Generalized Gradient Approximation (GGA) as implemented in the CASTEP package. The number of k-points in the Brillouin zone is chosen to be 1 × 1 × 3 with a cutoff energy of 850 eV, and a kinetic temperature of the electronic system is set at 300 K. During the optimization process, the maximum force and stress concentrations are set at 0.03 eV Å−1 and 0.05 Gpa, respectively. To determine the stability of the adsorbed systems, the adsorption energy (Ead) are determined according to the following formula49| Ead = EZZ7-V_2Se-molecule − EZZ7-V_2Se − Emolecule | (2) |
 | ||
| Fig. 6 Acetone/acetonitrile adsorption sites on (a) ZZ7 and (b) ZZ7-V2Se substrates. | ||
As a result, the negative and positive Ead values are respectively responsible for the stability and instability of the adsorbed systems. The calculated Ead values and shortest distance from the acetone molecule to the ZZ7-V2Se substrate are presented in Table 2, in which except for positions of 6, 7, 9 showing the unstable structures owing to their positive Ead values, the remaining positions displaying the significant negative (Ead) values, indicating the possible adsorption of the acetone molecule on the substrate. The shortest distance between the acetone molecule and the ZZ7-V2Se substrate is larger than the sum of the covalent radii of the nearest atoms in the acetone molecule and the Pd/Se atoms in the substrate. This implies that no chemical bonds are formed between the acetone molecule and the ZZ7-V2Se substrate, evidencing that the physical adsorption mechanism is dominant for the acetone adsorption onto the ZZ7-V2Se substrate at sites of 1, 2, 3, 4, 5, 8, 10 and 11, in which their shortest distances are 2.226 Å, 3.312 Å, 2.901 Å, 2.941 Å, 3.172 Å, 2.567 Å, 2.519 Å, 2.571 Å, respectively.44 Critically, the adsorption energy of the acetone on the ZZ7-V2Se substrate at the site of 8 is smallest with the lowest Ead value of −1.20 eV among other adsorption sites. This proves that the adsorption site of 8 is the most preferable adsorption site among the investigated 11 adsorption sites as evidenced in Table 2 and illustrated Fig. 7. As compared with the adsorption of acetone on boron nitride nanotube (BNNTs),50 the obtained adsorption energy of the acetone adsorbed BNNTs system at the most optimal configuration is about −0.93 eV, which is larger than that of −1.20 eV obtained in the acetone adsorbed ZZ7-V2Se system at its most optimal configuration, while the shortest adsorption distance between the BNNTs and the acetone adsorbent were found at 1.654 Å that is very shorter than that of 2.567 Å presented in the acetone-adsorbed ZZ7-V2Se system, confirming the acetone adsorbed BNNTs and the acetone adsorbed ZZ7-V2Se belong to the chemisorption and physisorption, respectively.
| Acetone | d (Å) | Ead (eV) | Acetonitrile | d (Å) | Ead (eV) |
|---|---|---|---|---|---|
| 1 | 2.226 | −0.65 | 1 | 2.097 | −0.20 |
| 2 | 3.312 | −0.67 | 2 | 3.031 | −0.50 |
| 3 | 2.901 | −0.25 | 3 | 2.546 | −0.60 |
| 4 | 2.941 | −0.82 | 4 | 3.663 | +1.10 |
| 5 | 3.172 | −0.01 | 5 | 3.027 | −0.40 |
| 6 | 2.992 | +0.11 | 6 | 3.576 | −0.10 |
| 7 | 3.477 | +0.14 | 7 | 1.891 | +0.30 |
| 8 | 2.567 | −1.20 | 8 | 3.077 | +0.12 |
| 9 | 3.318 | +0.16 | 9 | 3.255 | −0.05 |
| 10 | 2.519 | −0.58 | 10 | 2.084 | −0.70 |
| 11 | 2.571 | −0.70 | 11 | 2.149 | −0.86 |
 | ||
| Fig. 7 Side view and top view of acetone/acetonitrile adsorbed on the ZZ7-V2Se substrate at the 8 and 11 sites, respectively. | ||
As for the acetonitrile-adsorbed ZZ7-V2Se substrate, their calculated Ead values and the shortest distances from the acetonitrile molecule to the ZZ7-V2Se substrate are also shown in the Table 2. Except for the sites of 4, 7, 8 showing the unstable structures due to their positive Ead values, the remaining sites present the significant negative Ead values, indicating for the possible adsorption of the acetonitrile molecule on the ZZ7-V2Se substrate. The results presented in Table 2 show that the shortest distances between the acetonitrile molecule and the ZZ7-V2Se substrate are larger than the sum of the covalent radii of the nearest atoms in the acetonitrile molecule and the Pd/Se atoms in the substrate. This means that no chemical bonds are formed between the acetonitrile molecule and the ZZ7-V2Se substrate. Therefore, the physical adsorption mechanism is also considered as the dominant mechanism in the acetonitrile-adsorbed ZZ7-V2Se system. Among the 11 adsorption sites (Table 2), the acetonitrile adsorbed at the site of 11 displays the lowest adsorption energy of −0.86 eV, evidencing that the site of 11 is its most optimal adsorption site as illustrated in Fig. 7
As for the acetone/acetonitrile-adsorbed pristine ZZ-PdSe2 substrate at the investigated 3 sites of 1, 2, and 3 as shown in Fig. 6, the obtained adsorption energies are at −0.02 eV, −0.03 eV, and −0.01 eV, respectively. The negative Ead values are insignificant as compared with that of the acetone/acetonitrile-adsorbed ZZ7-V2Se system (Table 2), showing that the acetone/acetonitrile molecules adsorbing on the ZZ7-V2Se substrate are more stable than the acetone/acetonitrile molecules adsorbing on the pristine ZZ-PdSe2 substrate. The dominant adsorption of the ZZ7-V2Se substrate is since the vacancy defects trigger free electrons, which can easily interact with the adsorbed molecules, increasing their adsorption capacity as compared to the non-vacancy structure. This firmly confirms the great potential of the ZZ7-V2Se structure for nanosensor applications.
Furthermore, bond angles and bond lengths of the acetone adsorbing on the ZZ7-V2Se substrate at the most stable site of 8 are presented in Table 3 (after adsorption), in which these bond angles and bond lengths show a small variation compared to that before adsorption. Specifically, the detailed changes in the bond lengths of the ZZ7-V2Se substrate with and without acetone/acetonitrile adsorption are presented in Table 4. For the bonds forming the polygon, significant changes are observed in the bond lengths of d46, d20, d39 and d40 as shown Fig. 8(b), the bonds around the Pd site corresponding to the optimal acetone adsorption site of 8, proving that there is an interaction between the adsorbent molecule and the substrate, causing the small change in the bond lengths. For bond lengths outside the defect ring, symmetric bond pairs including the d8 and d3, d5 and d12, d18 and d26, and d43 and d29 (Fig. 8(b)) show a significant variation in bond lengths compared to before adsorption (Fig. 8(a)). This affects the width and buckling of the defect ring.
| Bond length | d (Å) | Bond angle | α (°) |
|---|---|---|---|
| Acetone-8 | |||
| C92–O91 | 1.249 | C83–C92–C87 | 118.151 |
| C92–C83 | 1.495 | H84–C83–H85 | 110.650 |
| C92–C87 | 1.494 | H84–C83–H86 | 109.445 |
| C83–H84 | 1.098 | H85–C83–H86 | 106.362 |
| C83–H85 | 1.099 | H88–C87–H89 | 110.911 |
| C83–H86 | 1.103 | H88–C87–H90 | 106.752 |
| C87–H88 | 1.100 | H89–C87–H90 | 108.275 |
| C87–H89 | 1.096 | O91–C92–C83 | 121.102 |
| C87–H90 | 1.104 | O91–C92–C87 | 120.725 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Acetonitrile-11 | |||
| N83–C84 | 1.166 | N83–C84–C85 | 176.185 |
| C84–C85 | 1.449 | H86–C85–H87 | 109.419 |
| C85–H86 | 1.098 | H86–C85–H88 | 109.414 |
| C85–H87 | 1.100 | H87–C85–H88 | 108.905 |
| C85–H88 | 1.100 | ||
| Bond | ZZ7-V2Se-acetone | ZZ7-V2Se | Bond | ZZ7-V2Se-acetone | ZZ-7-DVSe |
|---|---|---|---|---|---|
| d9 | 2.555 | 2.498 | d12 | 2.543 | 2.475 |
| d13 | 2.455 | 2.466 | d26 | 2.388 | 2.541 |
| d23 | 2.436 | 2.455 | d29 | 2.417 | 2.472 |
| d24 | 2.499 | 2.455 | d34 | 2.627 | 2.691 |
| d35 | 2.446 | 2.435 | d39 | 2.488 | 2.659 |
| d46 | 2.513 | 2.601 | d40 | 2.452 | 2.523 |
| d20 | 2.447 | 2.492 | d43 | 2.474 | 2.529 |
| d19 | 2.452 | 2.482 | d18 | 2.389 | 2.543 |
| d4 | 2.438 | 2.432 | d5 | 2.404 | 2.478 |
| d8 | 2.549 | 2.603 | d3 | 2.575 | 2.560 |
| Bond | ZZ7-V2Se-acetonitrile | ZZ7-V2Se | Bond | ZZ7-V2Se-acetonitrile | ZZ-7-DVSe |
|---|---|---|---|---|---|
| d9 | 2.346 | 2.498 | d12 | 2.401 | 2.475 |
| d13 | 2.467 | 2.466 | d26 | 2.365 | 2.541 |
| d23 | 2.470 | 2.455 | d29 | 2.399 | 2.472 |
| d24 | 2.452 | 2.455 | d34 | 2.534 | 2.691 |
| d35 | 2.410 | 2.435 | d39 | 2.349 | 2.659 |
| d46 | 3.415 | 2.601 | d40 | 2.422 | 2.523 |
| d20 | 2.454 | 2.492 | d43 | 2.411 | 2.529 |
| d19 | 2.479 | 2.482 | d18 | 2.371 | 2.543 |
| d4 | 2.430 | 2.432 | d5 | 2.401 | 2.478 |
| d8 | 2.547 | 2.603 | d3 | 2.496 | 2.560 |
 | ||
| Fig. 8 (a) ZZ7, (b) ZZ7-V2Se, (c) acetone, and (d) acetonitrile. | ||
As for the acetonitrile adsorbing on the ZZ7-V2Se substrate at the most optimal site of 11, their bond angles and bond lengths are also shown in Table 3, whereas their bond angles and bond lengths display a small variation compared to that before adsorption (similar identification in the former acetone-absorbed cases). In addition, the change in the bond lengths of the ZZ7-V2Se substrate after and before adsorption are shown in Table 4. Similar to the case of acetone adsorption onto ZZ7-V2Se at the site of 8, for the bonds forming the defect ring, significant changes are recorded in the neighboring bonds of d46 and d47 as marked by red symbol in Fig. 8(b). Specifically, bond lengths of d46, d39 and d40 are situated around the Pd position corresponding to the position of the hydrogen atoms of the acetonitrile molecule after adsorption onto ZZ7-V2Se at the optimal adsorption site of 11. Considering the bonds adjacent to the defect ring, the symmetric bond pairs are as follows d8 and d3, d5 and d12, d18 and d26, and d43 and d29, which show a significant variations in bond lengths compared to before adsorption. The calculated results in the structural parameters of the substrate and the molecule after adsorption (Fig. 8(b)) indicate the great impact on the ZZ7-V2Se substrate under the acetonitrile adsorption at the site of 11, in which the acetonitrile molecule tended to be pulled to the end of the defect ring, whereas two new bonds (46 and 47) are formed as compared to the pristine structure, causing a significant change in the bond lengths adjacent to these two bonds as displayed in Fig. 8(b). In addition, effects are also observed for the strong stretching of symmetric bond pairs around the defect ring.
The changes in the structural parameters owing to the acetone/acetonitrile adsorption result in changes in the electronic properties of the adsorbed systems that are determined by their electric density of states (DOSs). Using the DFT calculations, DOSs of the ZZ7-V2Se, ZZ7-V2Se-acetone-8, and ZZ7-V2Se-acetonitrile-11 are presented in Fig. 9(a–c), respectively, in which the Fermi level (dashed line) is set at zero energy, and the spin-up and spin-down components are marked by blue and red curves, respectively. As for the DOSs of the ZZ7-V2Se in Fig. 9(a), it shows many spin-asymmetric peaks, whereas there are both spin-up and spin-down states localized at the Fermi level, regarding as the ferromagnetic semi-metal as early identified in Fig. 3(e) and 5(f). Under the acetone-8 and acetonitrile-11 adsorptions, the DOSs of the non-adsorbed system in Fig. 9(a) is significantly reshaped. For the DOSs of the ZZ7-V2Se-acetone-8 in Fig. 9(b), it displays many new peaks with higher spin polarization, in which its spin-up component (blue curve) presents a vacant region centered from the Fermi level, while its spin-down component (red curve) remains the electronic states localized the Fermi level. The identified DOSs feature in Fig. 9(b) can be regarded as the ferromagnetic half-metal. In contrast, the vacant states appearing in both spin-up and spin-down components (blue and red curves) in the DOSs of the ZZ7-V2Se-acetonitrile-11 as shown in Fig. 9(c), while its newly formed peaks also shows high spin polarization. The DOSs feature identified in Fig. 9(c) results in its ferromagnetic semiconducting characteristics. Obviously, under the acetone-8 and acetonitrile-11 adsorptions, the significant transition from the ferromagnetic semi-metal to ferromagnetic half-metal to ferromagnetic semiconductor is occurred. To determine the specific contributions of each components in the DOSs of the adsorbed systems, the partial density of states (PDOSs) of the ZZ7-V2Se-acetone-8 and ZZ7-V2Se-acetonitrile-11 systems are presented in Fig. 10(a and b), respectively, in which the contribution of the ZZ7-V2Se and acetone components are situated at the middle and bottom parts. As observed in Fig. 10(a), the contribution of the ZZ7-V2Se component is mostly contributed in the valence and low-lying energies, while the contribution of the acetone component is highly dominated in the highest conduction energies. In contrast, the contribution of the acetonitrile component is separately contributed in conduction energies from 0.5 eV to 1.25 eV as identified in Fig. 10(b). In addition, the acetone/acetonitrile-related prominent peaks show minor spin polarization, indicating that the magnetism of the adsorbed systems is mainly due to the ZZ7-V2Se component.
 | ||
| Fig. 9 Total density of states of (a) ZZ7-V2Se, (b) ZZ7-V2Se-acetone-8, and (c) ZZ7-V2Se-acetonitrile-11. | ||
 | ||
| Fig. 10 Partial density of states of (a) ZZ7-V2Se-acetone-8 and (b) ZZ7-V2Se-acetonitrile-11. | ||
To determine the charge distribution in molecules and the substrate as well as the charge transfer, the Mulliken populations of the ZZ7-V2Se-acetone-8 and ZZ7-V2Se-actonitrile-11 systems are presented in Table 5, in which the atoms in the acetone/acetonitrile molecules and defective ring atoms are illustrated in Fig. 11(a and b), respectively. Observed from Table 5, it shows that the hydrogen (H) has a positive charge, while palladium (Pd) has a negative charge after the acetone adsorption. Specifically, H84 has a charge of 0.099e and Pd48 has a charge of −0.139e; H88 has a charge of 0.066e and Pd45 has a charge of −0.004e. These opposite charge values evidence the Coulomb interaction between these pairs of atoms. Besides, it shows the charge changes of the atoms of the acetone molecule and the ZZ7-V2Se substrate (Table 5), while the charge changes on the adsorbed molecule are strongly observed on hydrogen atoms near Pd, and all hydrogen atoms have a positive charge. Meanwhile, the charge change on the ZZ7-V2Se substrate before and after adsorption mainly occurs on Pd48, Pd45 and neighboring atoms. As a result, the strong Coulomb interaction occurs at hydrogen atoms of the acetone molecule and Pd48 and Pd45 atoms, leading to the physical adsorption of acetone molecules on the ZZ7-V2Se substrate. As for the ZZ7-V2Se-acetonitrile-11, its Mulliken population shown in Table 5 indicates that the change in charge on the ZZ7-V2Se substrate before and after adsorption mainly occurs at the atoms at the end of the defect ring, whereas the two newly formed bonds appear (d46 and d47). After the acetonitrile-11 adsorption, hydrogen has a positive charge and palladium has a negative charge. Specifically, H86 has a charge of 0.084e and Pd54 has a charge of −0.115e; H88 has a charge of 0.083e and Pd48 has a charge of −0.092e. This opposite charge values result in the Coulomb interaction between these pairs of atoms. Critically, the C84 has the clearest charge transfer before and after the acetonitrile adsorption, in which the C84 atom becomes positively charged after the adsorption, and the bond between C84 and N83 is a triple bond with a bond length of about 1.166 Å. On the other hand, the shortest distance from the acetonitrile molecule to the ZZ7-V2Se substrate is 2.149 Å, corresponding to the distance from the N83 to P63 atom. From Mulliken analysis, it confirms that the Coulomb interaction exists at C84–N83–P63. As a result, the strong Coulomb interactions occurred at the pairs H86–P54, H88–P48 and C84–N83–P63, leading to the physical adsorption of acetonitrile molecules on the ZZ7-V2Se substrate.
| Atom | Before adsorption | After adsorption | Atom | Before adsorption | After adsorption |
|---|---|---|---|---|---|
| Charge | Charge | Charge | Charge | ||
| Acetone | |||||
| C83 | −0.098 | −0.102 | Se16 | 0.058 | 0.042 |
| H84 | 0.029 | 0.099 | Se28 | 0.083 | 0.075 |
| H85 | 0.047 | 0.038 | Pd63 | −0.167 | −0.158 |
| H86 | 0.031 | 0.033 | Se19 | 0.082 | 0.056 |
| C87 | −0.097 | −0.115 | Pd48 | −0.169 | −0.139 |
| H88 | 0.046 | 0.066 | Se25 | 0.061 | 0.088 |
| H89 | 0.031 | 0.034 | Se13 | 0.087 | 0.073 |
| H90 | 0.030 | 0.049 | Pd54 | −0.160 | −0.103 |
| O91 | −0.366 | −0.351 | Se22 | 0.081 | 0.020 |
| C92 | 0.349 | 0.390 | Pd45 | −0.044 | −0.004 |
|
|||||
| Acetonitrile | |||||
| N83 | −0.216 | −0.209 | Se16 | 0.058 | 0.045 |
| C84 | −0.027 | 0.147 | Se28 | 0.083 | 0.065 |
| C85 | −0.076 | −0.082 | Pd63 | −0.167 | −0.081 |
| H86 | 0.106 | 0.084 | Se19 | 0.082 | 0.061 |
| H87 | 0.106 | 0.119 | Pd48 | −0.169 | −0.092 |
| H88 | 0.106 | 0.093 | Se25 | 0.061 | 0.000 |
| Se13 | 0.087 | 0.069 | |||
| Pd54 | −0.160 | −0.115 | |||
| Se22 | 0.081 | −0.022 | |||
| Pd45 | −0.044 | −0.066 | |||
 | ||
| Fig. 11 (a) acetone adsorbed on the ZZ7-V2Se substrate at the 8 site, (b) acetonitrile adsorbed on the ZZ7-V2Se substrate at the 11 site. | ||
4 Conclusion
Using the DFT calculations, the diverse geometric and electronic properties of the ZZ7 p-PdSe2 nanoribbons (ZZ7) with the four different kinds of vacancy defects, including the ZZ7-VPd, ZZ7-VSe, ZZ7-VPd+Se, and ZZ7-V2Se are determined by the formation energies, optimal structural parameters, electronic band structures, and density of states (DOSs). The formation energies (EC) of the ZZ7 structure and the four defective ZZ7 structures show the significant negative values around −3.9 eV, indicating their good stability that can be fully reached out by experimental approaches. For the four defective structures, their optimal geometries display the significantly different diversification, as compared with the pristine one: e.g., the ZZ7-VSe and ZZ7-V2Se exhibit two new bonds, the ZZ7-VPd+Se only displays one new bond, and conversely the ZZ7-VPd dot not form a polygon around the vacancy position, in which only the buckling feature of the ZZ7-V2Se structure is critically enhanced around the two new bonds that greatly increase its adsorption ability, rendering the ZZ7-V2Se structure is most effective substrate for adsorption of the acetone and acetonitrile molecules. On the electronic characteristics, the electronic band structure of the pristine ZZ7 structure displays the non spin-splitting energy bands and the highest occupied valence bands intersecting with the Fermi level, regarded as the nonmagnetic metallic characteristics, which are verified by existing of electronic states at the Fermi level and negligible spin asymmetric peaks in its corresponding DOSs. Under various vacancies, the nonmagnetic metallic band structure of the pristine structure are fully modified, depending on different kinds of vacancies. For the ZZ7-VPd and ZZ7-VSe structures, their band structures display highly spin-splitting energy bands with the half-metallic behavior, regarded as the ferromagnetic half-metallic characteristic that can be confirmed by concurrently existing of the spin-up electronic states and spin-down vacant states at the Fermi level and their highly spin-asymmetric peaks in their corresponding DOSs. As for the ZZ7-VPd+Se and ZZ7-V2Se structures, their band structures show the ferromagnetic semi-metallic characteristics, as identified by highly spin-splitting bands and concurrently intersecting of one highest occupied valence spin-up band and one lowest unoccupied conduction spin-down band with the Fermi level, which are verified by the highly spin-asymmetric peaks and co-existing of the spin-up and spin-down electronic states with the Fermi level in their corresponding DOSs.As for the adsorption of the acetone and acetonitrile molecules on the central ZZ7-V2Se structure, the energetic stability, adsorption sites, adsorption distances, bond lengths, charge distributions, charge transfers, and electronic characteristics of the adsorbed systems are fully determined by the adsorption energies, optimal adsorption sites, optimal distances, optimal bond lengths, Mulliken populations, and density of states (DOSs). Meanwhile, the adsorption energies of the acetone- and acetonitrile-adsorbed pristine ZZ7 structure are also calculated to compare its adsorption ability with that of the adsorbed ZZ7-V2Se structure, whereas the pristine ZZ7 structure only has 3 possible adsorption sites that increases to 11 possible adsorption sites in the ZZ7-V2Se structure owing to its formed vacancy ring. As a result, the adsorption energies of the acetone/acetonitrile on the pristine ZZ7 structure at the possible sites of 1, 2, and 3 show insignificant values without any obviously preferable absorption site that indicates the low adsorption ability and instability of the adsorbed-pristine ZZ7 systems: e.g., −0.05/−0.02 eV, −0.07/−0.03 eV, and −0.01/−0.01 eV, respectively. As expected, the adsorption energies of the acetone- and acetonitrile-adsorbed ZZ7-V2Se systems display the greatly significant values with the obviously preferable adsorption sites: e.g., −1.20 eV and −0.86 eV for the acetone adsorption at the site of 8 and the acetonitrile adsorption at the site of 11, respectively, indicating that the greater adsorption ability and stability of the ZZ7-V2Se substrate. Owing to the large adsorption distances leading to the absence of chemical bonds between the adsorbent molecules and the ZZ7-V2Se substrate, the adsorption mechanism of the acetone- and acetonitrile-adsorbed systems is regarded as the physical adsorption (physisorption), in which the good stability of the adsorbed systems are mainly due to the charge transfer between the adsorbent molecules (loss charge) and the substrate (gain charge) as identified from their Mulliken populations. As a result of the physisorption, the bond lengths of the ZZ7-V2Se substrate show a very small deviation under the acetone and acetonitrile adsorptions as identified by their optimal bond lengths before and after adsorptions. For adsorption effects of the acetone and acetonitrile on the electronic properties, changes in electronic behaviors of the adsorbed ZZ7-V2Se systems compared with that of the ZZ7-V2Se substrate are determined by the reshaped DOSs of the adsorbed systems. Specifically, the ferromagnetic semi-metallic DOSs features of the ZZ7-V2Se substrate become the ferromagnetic half-metallic and ferromagnetic semiconducting characteristics in the DOSs spectrum of the ZZ7-V2Se-acetone-8 and ZZ7-V2Se-acetonitrile-11, respectively. With the complete results and analyses, this study provides a complete understanding of the acetone and acetonitrile adsorptions on the ZZ7-V2Se structure, in which the ZZ7-V2Se structure show its great potential as the effective substrate for adsorption of small organic molecules of the acetone and acetonitrile. Furthermore, such ZZ7-V2Se structure can be expected to effectively adsorb other small organic molecules, such as aldehyde31,32 that can be fully investigated using the DFT theoretical framework developed in this study.
Data availability
The data that supports the findings of this study are available within the article.Author contributions
N. T. Tien did numerical calculations with the assistance of N. H. Dang, P. B. Thao, V. K. Dien, D. M. Hoat, N. D. Khanh, in which the manuscript wrote by N. D. Khanh and N. T. Tien.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research is supported by Vietnam Ministry of Education and Training under grant number B2023-TCT-03. The calculations were carried out on the Can Tho University (MOMA Lab) high-performance computing facilities.References
- K. Khan, A. K. Tareen, M. Aslam, R. Wang, Y. Zhang, A. Mahmood and Z. Guo, J. Mater. Chem. C, 2020, 8, 387–440 RSC.
- V. K. Dien, P. T. B. Thao, N. T. Han, N. D. Khanh, M. F. Lin and N. T. Tien, Phys. Rev. B, 2023, 108, 205406 CrossRef.
- A. Chaves, J. G. Azadani, H. Alsalman, D. R. Da Costa, R. Frisenda, A. J. Chaves and T. Low, npj 2D Mater. Appl., 2020, 4, 29 CrossRef CAS.
- N. D. Khanh, J. Guerrero-Sanchez, V. V. Tuan, R. Ponce-Pérez and D. M. Hoat, Surf. Interfaces, 2022, 32, 102114 CrossRef.
- N. D. Khanh, H. D. Quang and D. M. Hoat, RSC Adv., 2022, 12, 9828–9835 RSC.
- D. M. Hoat, N. D. Khanh, J. Guerrero-Sanchez, R. Ponce-Pérez, V. V. On, J. F. Rivas-Silva and G. H. Cocoletzi, Appl. Surf. Sci., 2021, 566, 150711 CrossRef CAS.
- N. T. T. Tran, G. Gumbs, N. D. Khanh and M. F. Lin, ACS Omega, 2020, 5, 13760–13769 CrossRef CAS PubMed.
- N. T. T. Tran, N. D. Khanh, S. Y. Lin, G. Gumbs and M. F. Lin, ChemPhysChem, 2019, 20, 2473–2481 CrossRef CAS PubMed.
- V. V. On, D. M. Hoat, N. D. Khanh, R. Ponce-Pérez, V. V. Tuan, J. F. Rivas-Silva and G. H. Cocoletzi, Phys. Scr., 2020, 95, 105806 CrossRef CAS.
- N. D. Khanh, D. M. Hoat, A. Bafekry, V. V. On, J. F. Rivas-Silva, M. Naseri and G. H. Cocoletzi, Physica E Low Dimens. Syst. Nanostruct., 2021, 131, 114732 CrossRef.
- T. Chowdhury, E. C. Sadler and T. J. Kempa, Chem. Rev., 2020, 120, 12563–12591 CrossRef CAS PubMed.
- Y. Hu, W. Zheng, S. Fan, J. Zhang and X. Liu, Appl. Phys. Rev., 2023, 10, 031306 CAS.
- T. M. D. Huynh, N. D. Khanh, N. T. D. Hien, V. K. Dien, H. D. Pham and M. F. Lin, Front. Mater., 2021, 7, 569756 CrossRef.
- N. T. Tien, P. T. B. Thao, N. H. Dang, N. D. Khanh and V. K. Dien, Nanomaterials, 2023, 13, 1728 CrossRef CAS PubMed.
- N. T. Tien, P. T. B. Thao, V. T. Phuc and R. Ahuja, J. Phys. Chem. Solids, 2020, 146, 109528 CrossRef CAS.
- M. T. Dang, P. T. B. Thao, T. T. N. Thao and N. T. Tien, AIP Adv., 2022, 12, 065008 CrossRef CAS.
- H. D. Pham, S. Y. Lin, G. Gumbs, N. D. Khanh and M. F. Lin, Front. Phys., 2020, 8, 561350 CrossRef.
- X. Jia, M. Jofmann, V. Meunier, B. G. Sumpter, J. Campos-Delgado, J. M. Romo-Herrera and M. S. Dresselhaus, Science, 2009, 323, 1701–1705 CrossRef CAS PubMed.
- N. D. Khanh, N. T. T. Tran, N. T. Tien and M. F. Lin, Sci. Rep., 2018, 8, 17859 CrossRef PubMed.
- N. D. Khanh, Y. T. Lin, S. Y. Lin, Y. H. Chiu, N. T. T. Tran and M. F. Lin, Phys. Chem. Chem. Phys., 2017, 19, 20667–20676 RSC.
- N. T. Tien, N. V. Ut, B. T. Hoc, T. T. N. Thao and N. D. Khanh, Adv. Condens. Matter Phys., 2019, 2019 Search PubMed.
- T. Y. Mi, N. D. Khanh, R. Ahuja and N. T. Tien, Mater. Today Commun., 2021, 26, 102047 CrossRef CAS.
- N. T. T. Tran, D. K. Nguyen, O. E. Glukhova and M. F. Lin, Coverage-dependent essential properties of halogenated graphene: A DFT study, Sci. Rep., 2017, 7(1), 17858 CrossRef PubMed.
- N. D. Khanh, N. T. T. Tran, Y. H. Chiu and M. F. Lin, Sci. Rep., 2020, 10, 12051 CrossRef.
- N. D. Khanh, V. D. Dat, V. V. On, and M. F. Lin, Feature-rich quasiparticle properties of halogen-adsorbed silicene nanoribbons, CRC Press, 2022, pp. 97–121 Search PubMed.
- N. D. Khanh, N. T. T. Tran, Y. H. Chiu and M. F. Lin, Sci. Rep., 2019, 9, 13746 CrossRef PubMed.
- D. M. Hoat, N. D. Khanh, J. Guerrero-Sanchez, R. Ponce-Pérez, V. V. On, J. F. Rivas-Silva and G. H. Cocoletzi, Appl. Surf. Sci., 2021, 566, 150711 CrossRef CAS.
- Z. Huang, H. Liu, R. Hu, H. Qiao, H. Wang, Y. Liu and H. Zhang, Nano Today, 2020, 35, 100906 CrossRef CAS.
- W. Y. Chen, X. Jiang, S. N. Lai, D. Peroulis and L. Stanciu, Nat. Commun., 2020, 11, 1302 CrossRef CAS PubMed.
- N. Rohaizad, C. C. Mayorga-Martinez, M. Fojtu, B. M. Latiff and M. Pumera, Chem. Soc. Rev., 2021, 50, 619–657 RSC.
- T. P. Kaloni, G. Schreckenbach and M. S. Freund, J. Phys. Chem. C, 2014, 118, 23361–23367 CrossRef CAS.
- C. H. Yeh, ACS Omega, 2020, 5, 31398–31406 CrossRef CAS PubMed.
- G. Zhang, M. Amani, A. Chaturvedi, C. Tan, J. Bullock, X. Song and A. Javey, Appl. Phys. Lett., 2019, 114(25), 253102 CrossRef.
- A. D. Oyedele, et al., J. Am. Chem. Soc., 2017, 139, 14090–14097 CrossRef CAS PubMed.
- J. D. Correa, L. Chico, V. Nunez, S. Bravo and M. Pacheco, Sens. Actuators, A, 2024, 115113 CrossRef CAS.
- J. Huang and J. Kang, J. Phys. Chem. C, 2022, 126, 20678–20685 CrossRef CAS.
- Y. Ma, X. Gong, F. Xiao, Y. Liu and X. Ming, ACS Appl. Nano Mater., 2022, 5, 11519–11528 CrossRef CAS.
- C. Long, Y. Liang, H. Jin, B. Huang and Y. Dai, ACS Appl. Energy Mater., 2018, 2, 513–520 CrossRef.
- G. D. Nguyen, A. D. Oyedele, A. Haglund, W. Ko, L. Liang, A. A. Puretzky and A. P. Li, ACS Nano, 2020, 14, 1951–1957 CrossRef CAS PubMed.
- N. T. Tien, P. T. B. Thao and N. D. Khanh, Surf. Sci., 2023, 728, 122206 CrossRef CAS.
- S. J. Clark, et al., Z. für Kristallogr.–Cryst. Mater., 2005, 220, 567–570 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- K. Stokbro, et al., Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 075420 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- C. Gonzalez, B. Biel and Y. J. Dappe, Nanotechnology, 2016, 27, 105702 CrossRef CAS PubMed.
- A. V. Kuklin, L. V. Begunovich, L. Gao, H. Zhang and H. Agren, Phys. Rev. B, 2021, 104, 134109 CrossRef CAS.
- N. T. Tien, P. T. B. Thao and D. H. Chuong, Comput. Mater. Sci., 2022, 203, 111065 CrossRef CAS.
- M. Fu, L. Liang, Q. Zou, G. D. Nguyen, K. Xiao, A. P. Li and Z. Gai, J. Phys. Chem. Lett., 2019, 11, 740–746 CrossRef PubMed.
- X. Xia, S. Guo, L. Xu, T. Guo, Z. Wu and S. Zhang, IEEE Electron Device Lett., 2021, 42, 573–576 CAS.
- M. D. Ganji and M. Rezvani, J. Mol. Model., 2013, 19, 1259–1265 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |