
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLipo-polymeric nano-complexes for dermal delivery of a model protein†
Abhay Tharmatta,
Deepak Kumar Sahela,
Reena Jatyana,
Anupma Kumarib,
Amit Mishrac,
Anupama Mittal a and
Deepak Chitkara*a
a and
Deepak Chitkara*a
aDepartment of Pharmacy, Birla Institute of Technology and Science Pilani, Vidya Vihar, Pilani, Rajasthan 333031, India. E-mail: deepak.chitkara@pilani.bits-pilani.ac.in; Tel: +91 9660 456 009 Tel: +91 1596 255 835
bCentre for Comparative Medicine, Institute of Liver and Biliary Sciences (ILBS), New Delhi, 110070, India
cDepartment of Bioscience & Bioengineering, Indian Institute of Technology, Jodhpur, Rajasthan, India
First published on 26th June 2024
Abstract
Delivering macromolecules across the skin poses challenges due to the barrier properties of stratum corneum. Different strategies have been reported to cross this barrier, such as chemical penetration enhancers and physical methods like microneedles, sonophoresis, electroporation, laser ablation, etc. Herein, we explored a cationic lipo-polymeric nanocarrier to deliver a model protein across the skin. A cationic amphiphilic lipo-polymer was used to prepare blank nanoplexes, which were subsequently complexed with anionic fluorescein-tagged bovine serum albumin (FITC-BSA). Blank nanoplexes and FITC-BSA complexed nanoplexes showed sizes of 93.72 ± 5.8 (PDI-0.250) and 145.9 ± 3.2 nm (PDI-0.258), respectively, and zeta potentials of 25.6 ± 7.0 mV and 9.17 ± 1.20 mV. In vitro cell culture, and toxicity studies showed optimal use of these nanocarriers, with hemocompatibility data indicating non-toxicity. Ex vivo skin permeation analysis showed a skin permeation rate of 33% after 24 h. The optimized formulation was loaded in a carbopol-based gel that exhibits non-Newtonian flow characteristics with shear-thinning behavior and variable thixotropy. The nanoplexes delivered via gel demonstrated skin permeation of 57% after 24 h in mice skin ex vivo. In vivo skin toxicity testing confirmed the low toxicity profile of these nanocarriers. These results are promising for the transdermal/dermal delivery of macromolecules, such as protein therapeutics, using nanoplexes.
1. Introduction
The skin, the largest organ of the human body, provides a physical barrier against various environmental aggressors such as pathogens, pollutants, and toxins, regulates water loss, and protects against harmful UV radiation.1 It comprises three distinct layers: the epidermis, dermis, and subcutaneous tissue. The epidermis, which is the outermost layer, is primarily responsible for mechanical strength and protection from external stimuli. The middle layer, the dermis, contains a network of blood vessels, nerves, and hair follicles, while the subcutaneous tissue acts as a cushion and insulation.2 Delivering therapeutic molecules, including drugs, proteins, and nucleic acids, across the skin is a formidable challenge due to the stratum corneum's barrier properties that mainly comprises tightly packed dead keratinocytes. The stratum corneum is both hydrophobic and negatively charged, making it particularly challenging for hydrophilic and negatively charged molecules to penetrate the skin.3,4Due to its distinct benefits, nanotechnology has emerged as a very promising option for drug administration into the skin.5,6 One of the important factors that influence the efficacy of such delivery systems is the size of the molecules being transported. The molecular size directly impacts the penetration ability through skin layers, with smaller molecules often exhibiting enhanced permeability. Thus, understanding the relationship between molecular size and skin penetration is paramount for the design and optimization of effective delivery strategies. Further, understanding and manipulating the size of formulations is paramount in developing efficient dermatological therapies, ensuring that drugs not only reach their target site within the skin but also maintain their structural integrity and functional efficacy upon administration. One significant aspect of nanotechnology in drug delivery is cationic nanocarriers, which have shown improved results due to their ability to permeate the skin.7 Better delivery of therapeutic is achieved with cationic nanocarriers due to their positive charge and ability to interact with negatively charged cells and tissues.8 These nanocarriers may protect therapeutics from breakdown by encapsulating or complexing with them. They are an attractive choice for topical administration because they improve/provide better skin penetration. However, there are significant limitations connected with the usage of cationic nanocarriers. The potential toxicity of cationic polymers is a major issue because of their ability to bind to and affect the function of negatively charged macromolecules.9 Also, they can induce inflammation and immune responses, limiting their therapeutic applications.10 Scientists have been working to find solutions to these problems by creating novel cationic polymers/lipids with enhanced biocompatibility and decreased toxicity. For example, Jin et al. developed a dermally applicable gene delivery system comprised of 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), Tween 20, and tricaprin-modified cationic lipid nanoparticles (cLNs). The cLNs were nanoscale-sized, highly positive, and demonstrated enhanced transfection potential in cells without cytotoxic effects. In vitro and in vivo experiments showed successful transfer of plasmid DNA to the skin, suggesting the promising potential of cLNs as a gene delivery system for cutaneous gene therapy and gene vaccine delivery in preclinical and clinical applications.7 To enhance the efficacy of topical tretinoin (TTN), Ebrahimi et al. created a new formulation using cationic nanocapsules. Through statistical optimization of three factors, an ideal formulation was developed, exhibiting a spherical shape, a particle size of 116.3 nm, and a high encapsulation effectiveness of 83.2%. Increased drug deposition in the epidermis, reduced skin irritation, and higher photostability were all seen in TTN-loaded nanocapsules.11 In contrast, cationic nanocarriers, especially cationic polymers, have shown encouraging outcomes in drug delivery despite these drawbacks, further can be complexed with proteins and peptides, resulting in enhancing their stability and bioavailability.10–12 The lack of stability and bioavailability of proteins and peptides, despite their importance as therapeutic agents, severely restricts their utilization.13 To prevent breakdown and facilitate transport across cell membranes, proteins and peptides may be complexed with cationic polymers. For instance, Petrilli et al. developed nanoparticles from monoolein and oleic acid, further functionalized with membrane transduction peptides TAT or PNT, and complexed with siRNA, which had positive zeta potentials, nanoscale diameters, and efficient siRNA release. In L929 cells, TAT functionalization improved the absorption efficiency.14
Moreover, the choice of polymer in these delivery systems significantly influences their performance and stability. Polymers serve as a carrier for these therapeutics and provide protection, controlled release, and targeting functionalities. Hence, elucidating the rationale behind selecting specific polymer is essential for comprehending the design principles and expected outcomes of skin delivery systems. Further, understanding and manipulating the size of formulations is paramount in developing efficient dermatological therapies, ensuring that drugs not only reach their target site within the skin but also maintain their structural integrity and functional efficacy upon administration. Lipo-polymers, a class of synthetic materials that combine lipids and polymers, hold great promise for the delivery of therapeutic proteins, miRNAs and RNPs (ribonucleoproteins).15–17 These versatile materials offer enhanced stability, controlled release, and improved penetration into target tissues. In skin diseases like psoriasis, lipo-polymers can encapsulate proteins and nucleic acid-based therapeutics, such as miRNA and RNPs, to improve their delivery. This approach enables the simultaneous delivery of multiple therapeutic agents, allowing for a synergistic effect in treating the disease. Researchers are looking at lipo-polymers as a means of transporting molecules into the skin, to create a novel and efficient platform for topical therapy that might lead to more effective treatment for psoriasis and other skin disorders.18–20 Previously, we have demonstrated that cationic lipo-polymers can efficiently complex with RNPs, enhance their stability, and enable efficient delivery.17,21,22 Herein, we investigated the potential of cationic lipo-polymers for delivering a model protein into the skin. A cationic amphiphilic lipo-polymer was utilized to form a complex with fluorescein isothiocyanate-labeled bovine serum albumin (FITC-BSA). We hypothesize that cationic nanocarrier will increase the penetration of FITC-BSA through the skin and improve its localization within the skin layers. The significance of this research lies in developing a novel drug delivery system that can potentially enhance the delivery of therapeutic molecules into the skin. This improvement could be particularly significant for treating skin diseases, such as psoriasis, eczema, and acne that require drug delivery into the skin.
2. Materials and method
2.1. Materials
Fetal Bovine Serum (FBS), OptiMEM™ reduced serum media, Snakeskin dialysis tubing™ (3.5 kD), Dulbecco's Modified Eagle's Medium (DMEM), DAPI (4′,6-diamidino-2-phenylindole), Micro BCA™ Protein Assay Kit, and Hoechst were obtained from Thermo Fischer Scientific (Massachusetts, USA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was procured from Merck (Mumbai, India). Tin(II) 2-ethyl hexanoate, cholesterol, N,N-dimethyldipropylenetriamine (DP), 4-(2-aminoethyl)morpholine, hydroxyl benzotriazole (HOBt), benzyl bromide, bis(hydroxymethyl)propionic acid, 1-ethyl-3-(3-dimethyl aminopropyl) carbodiimide hydrochloride (EDC·HCl), methoxy poly(ethylene glycol) (mPEG, 5000 Da), HEPES buffer were bought from Sigma Aldrich (St. Louis, MO). All the remaining solvents and chemicals were of analytical grade and were procured locally.2.2. Methodology
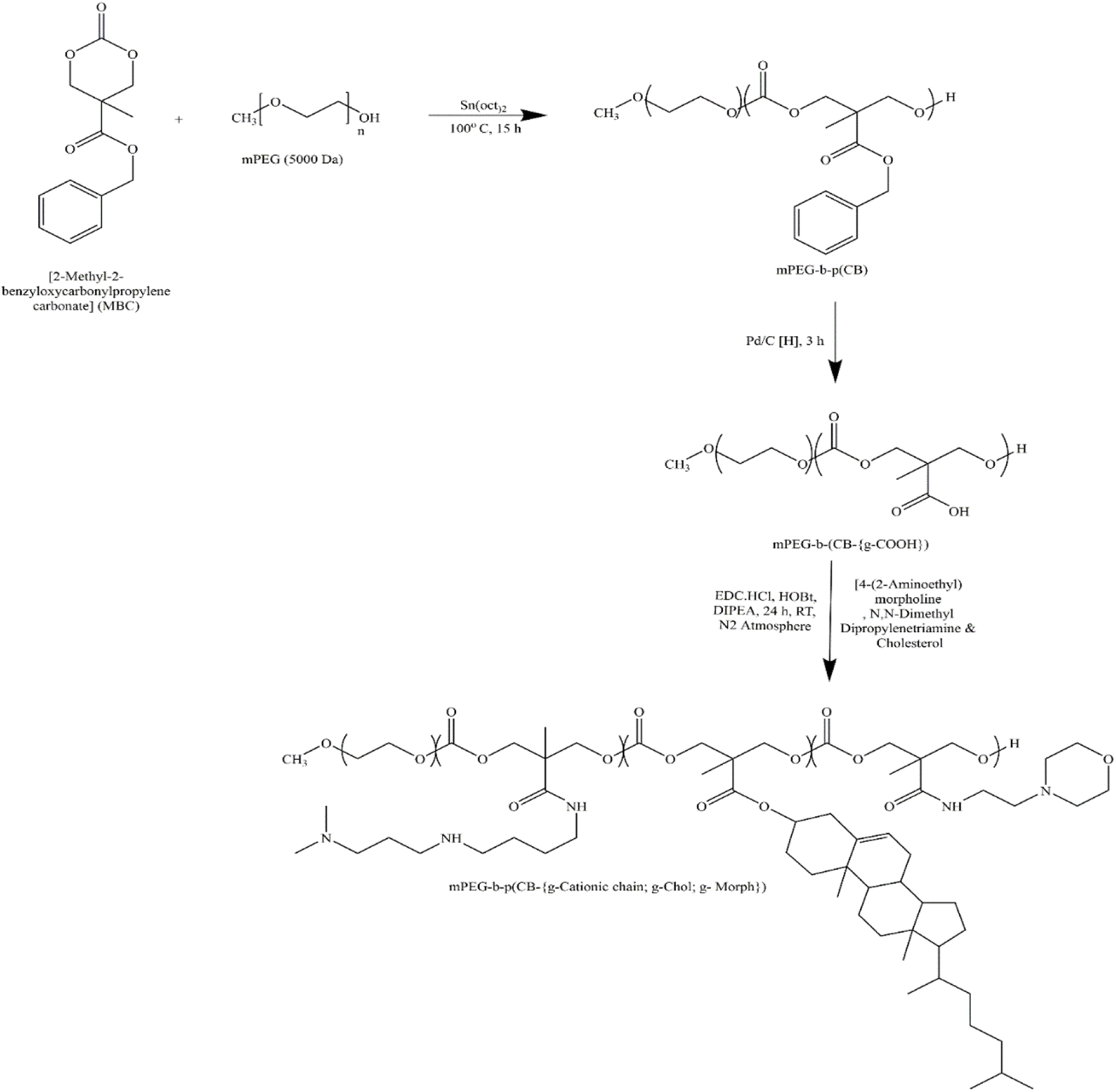 | ||
| Fig. 1 Synthetic scheme of amphiphilic cationic copolymer mPEG-b-p(CB-{g-cationic chain; g-chol; g-morph}). Reprinted with permission from ref. 17. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio as preservatives. Furthermore, propylene glycol (PG) (6.5 g) was added, and the total weight was adjusted to 50 g using Milli-Q water. The pH was carefully adjusted to 6.8 using a 1 M sodium hydroxide solution to obtaine the BSA-NPX gel.:0.5, 1:1, 1:2, 1:4, and 1:8). Before the zeta-potential measurement, the mixtures were added and kept at room temperature for 30 min, respectively.17,25:2, 1:4, and 1:8) were applied on the skin surface in the donor area. At 6, 12, and 24 h, 0.5 mL of the receptor solution was withdrawn and replaced with a fresh medium to maintain the total volume. For comparison, the same application of the BSA-NPX alone and in a gel containing BSA-NPX was tested on normal and psoriatic skin. The permeation of the loaded protein was measured using a microplate reader (BioTeK Epoch). The skin tissue was fixed in 5% paraformaldehyde, dehydrated, and prepared in paraffin blocks. 5 μm thick tissue sections were prepared, stained with DAPI and imaged using confocal microscopy.20:2, 1:4, and 1:8) to evaluate the treatment's dose-dependent effects. Skin samples were collected from the mice at specific intervals, i.e., 6, 12, and 24 h post-application to monitor temporal changes in response to the treatment regimen. Following collection, the skin samples underwent thorough preparation before undergoing in vivo bioimaging using IVIS Lumina XR (PerkinElmer, USA). The bioimaging utilized an excitation wavelength of 490 nm and an emission wavelength of 525 nm, as previously demonstrated by Pukale et al.19
:1 ratio as preservatives. Furthermore, propylene glycol (PG) (6.5 g) was added, and the total weight was adjusted to 50 g using Milli-Q water. The pH was carefully adjusted to 6.8 using a 1 M sodium hydroxide solution to obtaine the BSA-NPX gel.:0.5, 1:1, 1:2, 1:4, and 1:8). Before the zeta-potential measurement, the mixtures were added and kept at room temperature for 30 min, respectively.17,25:2, 1:4, and 1:8) were applied on the skin surface in the donor area. At 6, 12, and 24 h, 0.5 mL of the receptor solution was withdrawn and replaced with a fresh medium to maintain the total volume. For comparison, the same application of the BSA-NPX alone and in a gel containing BSA-NPX was tested on normal and psoriatic skin. The permeation of the loaded protein was measured using a microplate reader (BioTeK Epoch). The skin tissue was fixed in 5% paraformaldehyde, dehydrated, and prepared in paraffin blocks. 5 μm thick tissue sections were prepared, stained with DAPI and imaged using confocal microscopy.20:2, 1:4, and 1:8) to evaluate the treatment's dose-dependent effects. Skin samples were collected from the mice at specific intervals, i.e., 6, 12, and 24 h post-application to monitor temporal changes in response to the treatment regimen. Following collection, the skin samples underwent thorough preparation before undergoing in vivo bioimaging using IVIS Lumina XR (PerkinElmer, USA). The bioimaging utilized an excitation wavelength of 490 nm and an emission wavelength of 525 nm, as previously demonstrated by Pukale et al.192.2.10.1. Cells. Dr Munia Ganguly of the CSIR-Institute of Genomics and Integrated Biology (CSIR-IGIB) in New Delhi generously provided the Human epidermal keratinocytes (HaCat) cells. These cells were maintained at 37 °C in a humidified environment with 5% CO2 and were cultivated in Dulbecco's modified Eagle's medium (DMEM), which was supplemented with 10% fetal bovine serum (FBS) and 1% antibiotics (100 U mL−1 penicillin/streptomycin).
2.2.10.2. Uptake efficiency. HaCat cells were seeded in a 6-well cell culture plate with cell density of 1 × 105 cells per well in DMEM (10% FBS), and incubated at 37 °C in 5% CO2 overnight for cells to adhere. The following day, PBS was used to wash the cells, and old medium was changed with fresh optiMEM medium containing naked FITC-BSA or BSA-NPX. After predetermined periods (i.e., 6 h and 12 h) for nuclear staining, the cells were rinsed with PBS and stained with Hoechst dye (100 mg mL−1). They were then examined with a fluorescence microscope (Zeiss Axio Vert A1). To quantitatively measure the uptake efficiency, HaCat cells were seeded in a 6-well cell culture plate at a cell density of 2 × 105 cells per well in DMEM (10% FBS) and incubated at 37 °C in 5% CO2 for 24 h. Following incubation, the cells were treated with naked FITC-BSA or BSA-NPX for 12 h. Subsequently, the media was changed, and the cells were washed twice with PBS. Trypsinization was performed to detach the cells, which were then harvested by centrifugation at 1200 rpm for 3 min. The harvested cells were suspended in 70% ethanol (1 mL), pre-cooled to −20 °C, and incubated for 20 min. Flow cytometry was analysed using a CytoFLEX flow cytometer (Beckman Coulter) equipped with a 488 nm laser. FITC is excited at 488 nm and peaks at around 530 nm. The detection process employed filters, finely tuned to capture wavelengths between 525 and 545 nanometers, and the resulting data underwent analysis through Cyt Expert software, version 2.3.17,19
:4) and naked FITC-BSA. After 48 h of incubation, the cells were again cultured for 4 h in fresh DMEM medium supplemented with 3-(4,5-dimethyl thiazolyl-2)-2,5-diphenyl tetrazolium bromide (MTT) at a concentration of 0.5 mg mL−1. Following the incubation period, the medium containing MTT was withdrawn, and 200 μL of dry DMSO was added to dissolve the formazan crystals. Then, cell viability was determined using the following equation and quantified using a microplate reader (BioTeK Epoch) at 570 and 630 nm:17,19 :1 volume ratio after cooling in tap water. The mixture was centrifuged at 4000 rpm for 10 min to separate the organic layer. The absorbance was measured at 532 nm and 1,1,3,3-tetramethoxypropane (TMP) was used as a standard.18,30 For Catalase estimation, homogenate (0.1 mL) was mixed with phosphate buffer (0.1 mL; 0.01 M). 2 mL of potassium dichromate acetic acid reagent (5% potassium dichromate solution mixed in glacial acetic acid in 1:3), was added to the reaction and kept on boiling water bath for 15 min. The green-coloured mixture was cooled and absorbance was measured at 570 nm against blank. Catalase was assayed calorimetrically at 620 nm. The internal blank solution set was run under identical conditions without adding H2O2 (2–10 μmol mL−1). The concentration of Catalase was expressed as μmoles of catalase per mg protein.31 For Nitric oxide (NO) estimation, 0.1 M phosphate buffer (pH 7.4) was used to make a skin tissue homogenate a 10% (w/v). The skin homogenate was mixed with an equivalent amount of Greiss reagent, which included 0.1% N-1-naphthyl ethylenediamine dihydrochloride, 1% sulphanilamide, and 2.5% o-phosphoric acid. After incubating for 10 min, the absorbance at 540 nm was measured. The amount of nitrite present was calculated by reference from a standard curve established with different sodium nitrite concentrations.32
:1 volume ratio after cooling in tap water. The mixture was centrifuged at 4000 rpm for 10 min to separate the organic layer. The absorbance was measured at 532 nm and 1,1,3,3-tetramethoxypropane (TMP) was used as a standard.18,30 For Catalase estimation, homogenate (0.1 mL) was mixed with phosphate buffer (0.1 mL; 0.01 M). 2 mL of potassium dichromate acetic acid reagent (5% potassium dichromate solution mixed in glacial acetic acid in 1:3), was added to the reaction and kept on boiling water bath for 15 min. The green-coloured mixture was cooled and absorbance was measured at 570 nm against blank. Catalase was assayed calorimetrically at 620 nm. The internal blank solution set was run under identical conditions without adding H2O2 (2–10 μmol mL−1). The concentration of Catalase was expressed as μmoles of catalase per mg protein.31 For Nitric oxide (NO) estimation, 0.1 M phosphate buffer (pH 7.4) was used to make a skin tissue homogenate a 10% (w/v). The skin homogenate was mixed with an equivalent amount of Greiss reagent, which included 0.1% N-1-naphthyl ethylenediamine dihydrochloride, 1% sulphanilamide, and 2.5% o-phosphoric acid. After incubating for 10 min, the absorbance at 540 nm was measured. The amount of nitrite present was calculated by reference from a standard curve established with different sodium nitrite concentrations.323. Results
3.1. Preparation and characterization of nanoplexes (BSA-NPX) and gel loaded with BSA-NPX
As previously described, an emulsion-based approach was employed to prepare blank NPX from the synthesized cationic lipopolymer (mPEG-b-(CB-g-cation chain; g-chol; g-morph)).17 The amphiphilic cationic copolymer mPEG-b-(CB-{g-cationic chain; g-chol; g-morph}) was found to have a molecular weight of 24553 Da, comprising 22 cholesterol units, 18 cationic chain units, and 25 morpholine units as reported earlier.17 The BLK-NPX were incubated for 30 min at room temperature with naked FITC-BSA at different ratios to obtain BSA-NPX. Blank NPX and BSA-NPX showed a particle size of 93.72 ± 5.8 (PDI-0.250) and 145.9 ± 3.2 nm (PDI-0.258), and the zeta potential of 25.6 ± 7.0 mV and 9.17 ± 1.20 mV, respectively (Fig. 2(a and b)). The FITC-BSA encapsulation efficiency was determined using a bicinchoninic acid (BCA) test, which revealed an encapsulation efficiency of 75%, indicating efficient complexation FE-SEM was used to study the morphology of the resultant BSA-NPX, which revealed a spherical form (Fig. 2(c and d)).
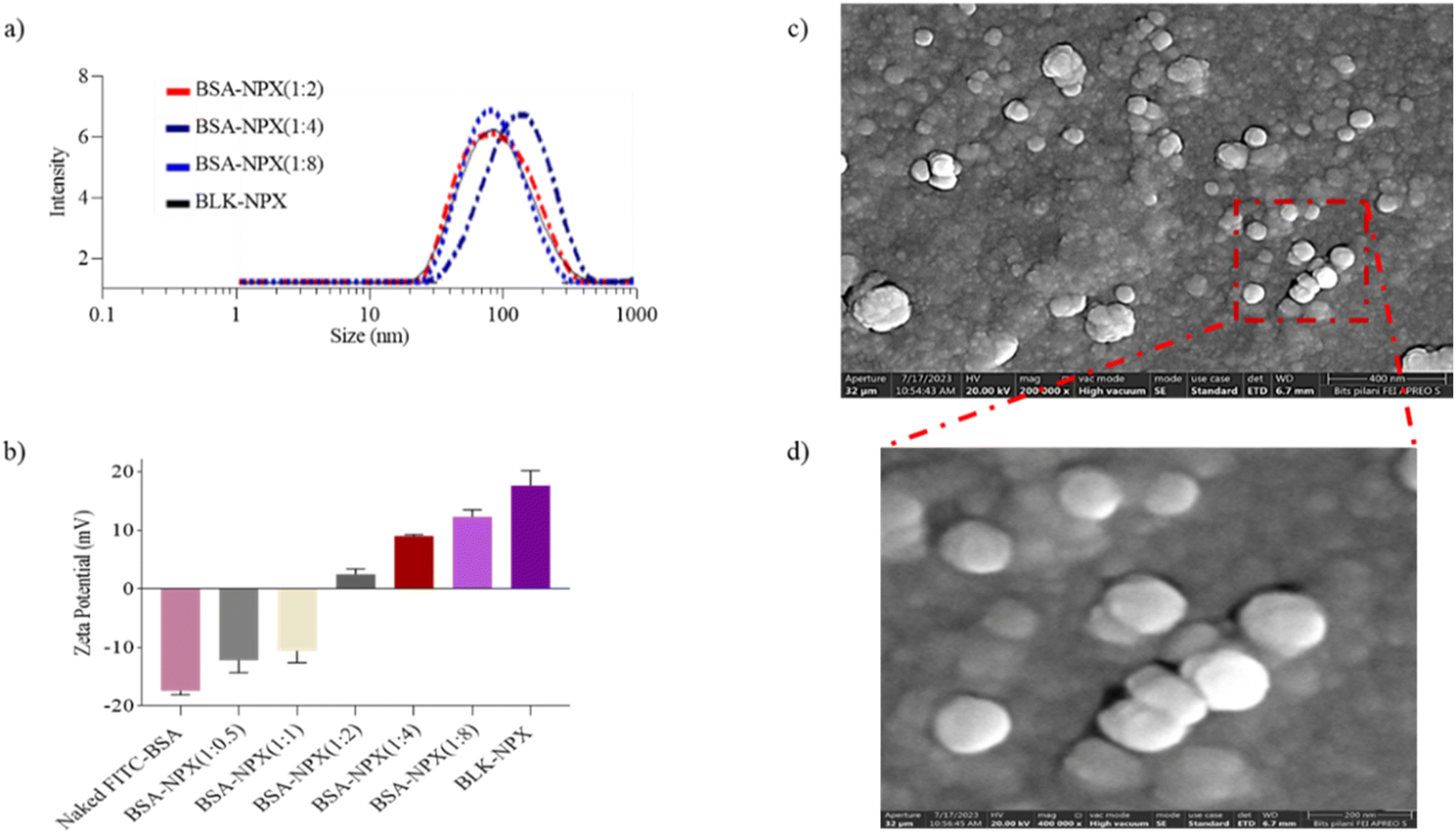 | ||
| Fig. 2 In vitro characterization of BSA-NPX formulations complexed at different ratios of BSA to NPX. (a) Illustrates the size range assessment; (b) illustrates the complexation efficiency of BSA-NPX with respect to BLK-NPX; (c and d) illustrates morphological representation of BSA-NPX. Data are represented as mean ± SD (n = 3). | ||
To evaluate the potential impact of aggregation on the therapeutic efficacy of BSA-NPX when incorporated into a topical gel, an analysis was conducted using FE-SEM of the BSA-NPX Gel. The obtained results demonstrated that the morphology of BSA-NPX remained intact, and no observable aggregation was detected upon formulation into the hydrogel (refer to Fig. 3e and f), with higher magnification (100000×) in Fig. 3f to visualise the nanoparticles embedded in a gel matrix; conversely, the lower magnification (50000×) in Fig. 3e was appropriate for depicting the overall morphology and texture of the blank gel. The viscosity (η) of the BSA-NPX gel was 100025.6 cP at a constant shear rate of 10 s−1 and a temperature of 25 °C. The relation between shear stress (mPa) and shear rate (s−1) as well as viscosity (η) and shear rate (s−1) revealed non-Newtonian flow features, including distinctive shear-thinning behavior and variable thixotropy(Fig. 3(a–d)).
 | ||
| Fig. 3 Rheological measurements and oscillatory rheological analysis of BSA-NPX. (a) Amplitude sweep test data indicating viscoelastic gel formation with G′ > G′′ within the linear viscoelastic (LVE) region wherein G′ is storage modulus and G′′ is loss modulus (s−1); (b) shear stress against shear rate (s−1) graphs; (c) viscosity (η) against shear rate, (d) stability of gel 3D matrix evident from the graph of viscosity (η) against temperature (°C), (e–f) scanning electron microscope image of blank gel and BSA-NPX gel. | ||
3.2. Ex vivo characterization of BSA-NPX and BSA-NPX gel
Skin samples treated with BSA-NPX complexes in ratios of 1:2, 1:4, and 1:8 exhibited a significant increase in fluorescence intensity compared to skin samples treated with FITC-BSA alone at all-time points (6, 12, and 24 h). This indicates a notable enhancement in the penetration of FITC-BSA into the deeper skin layers, namely the viable epidermis and dermis when complexed with NPX. Confocal analysis was performed on these skin tissues for each complexed ratio, and protein quantification was conducted using an ELISA plate reader. The results showed protein quantities ranging from 30–70% in all complexed ratios after 24 h (Fig. 4). To evaluate the permeation of BSA-NPX gel, an ex vivo permeation study was conducted, demonstrating that less than 25% of FITC-BSA reached the receiver compartment in all instances. Furthermore, in psoriatic skin, less than 45% FITC-BSA permeated to the receiver compartment within 24 h (Fig. 5).
 | ||
| Fig. 4 Ex vivo skin permeation study of the developed NPX in normal swiss albino mice skin; (a–d) illustrates quantitative permeation of naked FITC-BSA after treatment with naked FITC-BSA (control), BSA-NPX (1:2), BSA-NPX (1:4) and BSA-NPX (1:8), respectively, (e–h) illustrates the confocal microscopic images of the skin at 6, 12 and 24 h for naked FITC-BSA (control), BSA-NPX (1:2), BSA-NPX (1:4) and BSA-NPX (1:8), with DAPI staining (blue) and FITC-BSA (green), respectively; Data are represented as mean ± SD (n = 3). | ||
 | ||
| Fig. 5 Comparative ex vivo skin permeation study of the developed NPX gel Swiss albino mice skin; (a and b) Illustrates quantitative permeation of naked FITC-BSA after treatment with naked FITC-BSA gel (control), BSA-NPX (1:4) gel in normal skin, (c and d) illustrates the confocal microscopic images of the skin at 6, 12 and 24 h for naked FITC-BSA gel (control), BSA-NPX (1:4) gel in normal skin, respectively; (e and f) illustrates quantitative permeation of naked FITC-BSA after treatment with naked FITC-BSA gel (control), BSA-NPX (1:4) gel in psoriatic skin, (g and h) illustrates the confocal microscopic images of the skin at 6, 12 and 24 h for naked FITC-BSA gel (control), BSA-NPX (1:4) gel in psoriatic skin, respectively. Data are represented as mean ± SD (n = 3). | ||
3.3. In vivo bioimaging
The in vivo bioimaging analysis conducted using an in vivo imaging system revealed that BSA-NPX complexes with higher ratios of BSA to NPX exhibited an increased fluorescence intensity in the whole skin tissue, indicating improved uptake and retention within the target tissue. These findings highlight the importance of optimizing the BSA-NPX ratio for efficient fluorescence-based imaging applications (Fig. 6). | ||
| Fig. 6 In vivo bioimaging of skin tissue excised from Swiss albino mice treated with naked FITC-BSA, BSA-NPX (1:2, 1:4 and 1:8) after 6 h, 12 h and 24 h, compared to negative control. | ||
3.4. In vitro cell culture studies
:4) in HaCaT cells. Fig. 7(a) demonstrates that within a 6 h incubation period, HaCaT cells efficiently internalized BSA-NPX, as indicated by the prominent green signal from FITC-BSA complexed with NPX. Conversely, the control group treated with naked FITC-BSA showed no internalization. To quantitatively assess the uptake, flow cytometry analysis was performed as shown in Fig. 7(b). The results revealed that BSA-NPX (1:4) treatment led to an increase in fluorescence to 82.77% with respect to 0.77%, when cells were treated with naked FITC-BSA.
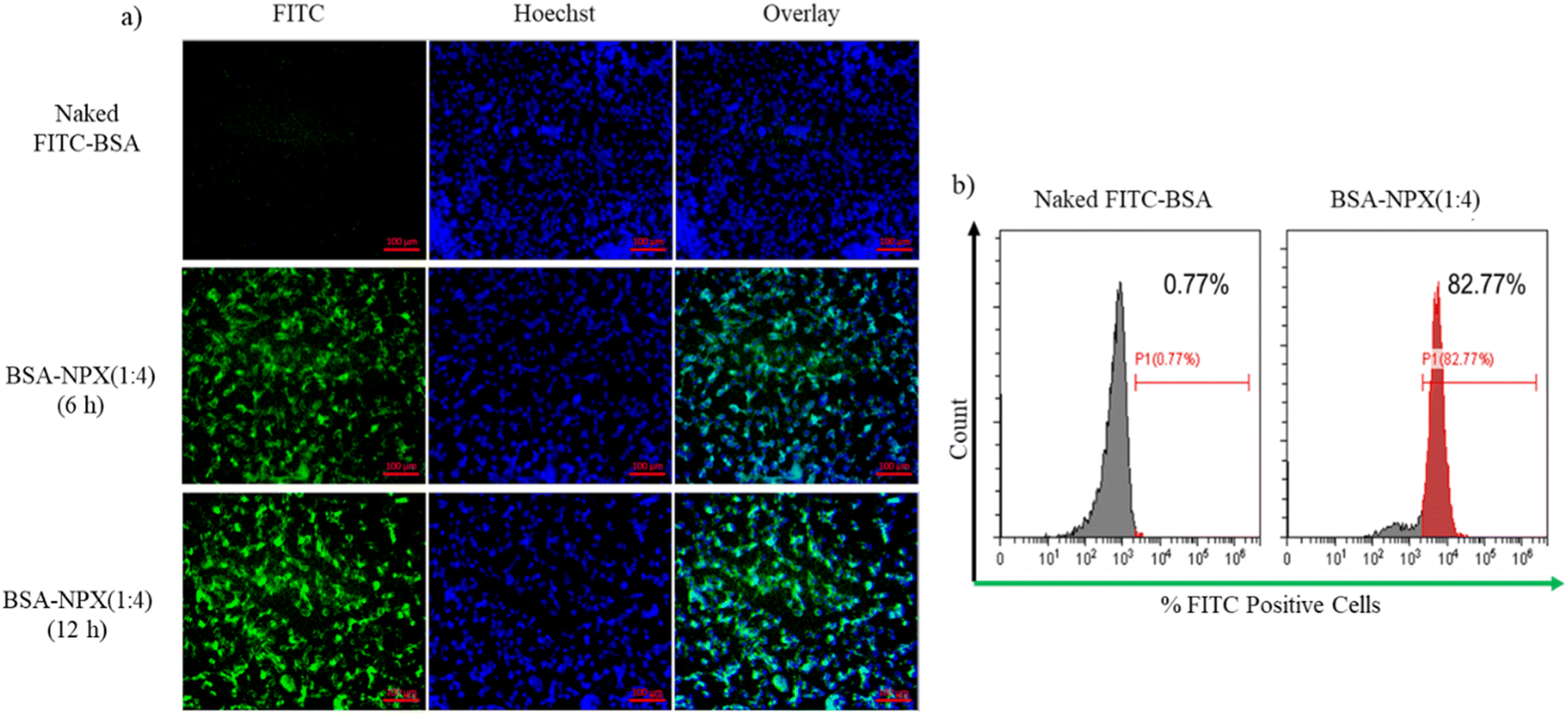 | ||
| Fig. 7 (a) Fluorescence microscopy; (b) flow cytometry analysis of cellular uptake following incubation with naked FITC-BSA and BSA-NPX (1:4) for 12 h in HaCaT cells (magnification 40×; scale bar 200 μm). | ||
:4) for 48 h. The results obtained from the assay revealed the absence of any cytotoxic effects by retaining their viability compared to the control untreated cells. Intriguingly, even at a concentration of 500 μg mL−1, reflecting the dose dependence response in all experimental groups exhibited cell viability levels in the 80–100% range, as illustrated in Fig. 8.
 | ||
| Fig. 8 Cytotoxicity of naked FITC-BSA, BLK-NPX and, BSA-NPX (1:4) in HaCaT cells till 48 h; data are represented as mean ± SD (n = 3). | ||
3.5. Hemocompatibility study
When evaluating the safety of NPX for possible biological uses, its compatibility with erythrocytes is critical. To that purpose, we performed an experiment in which naked FITC-BSA, BLK-NPX, and BSA-NPX (1:4) were incubated for 1 h with 1 mL of freshly obtained mice erythrocytes. Triton X 100 was used as a positive control since it causes complete hemolysis. Naked FITC-BSA, BLK-NPX, and BSA-NPX (1:4) caused less than 6% hemolysis at the maximum dose tested (50 μg mL−1), reflecting its safety response, which mimics realistic exposure, indicating a low degree of hemolysis. This observation was supported by visual and microscopic studies (Fig. S3†).
3.6. In vivo skin toxicity
H&E staining of a normal skin sample to a treated skin sample showed no significant changes in the skin morphology. The presence of intact keratinocytes and fibroblasts, along with the absence of hyperkeratosis, parakeratosis, and capillary proliferation, suggests that the treatment did not cause any significant damage to the skin tissue. These findings indicate that the BSA-NPX therapy may be safe for use on the skin (Fig. 9a, b, e and f).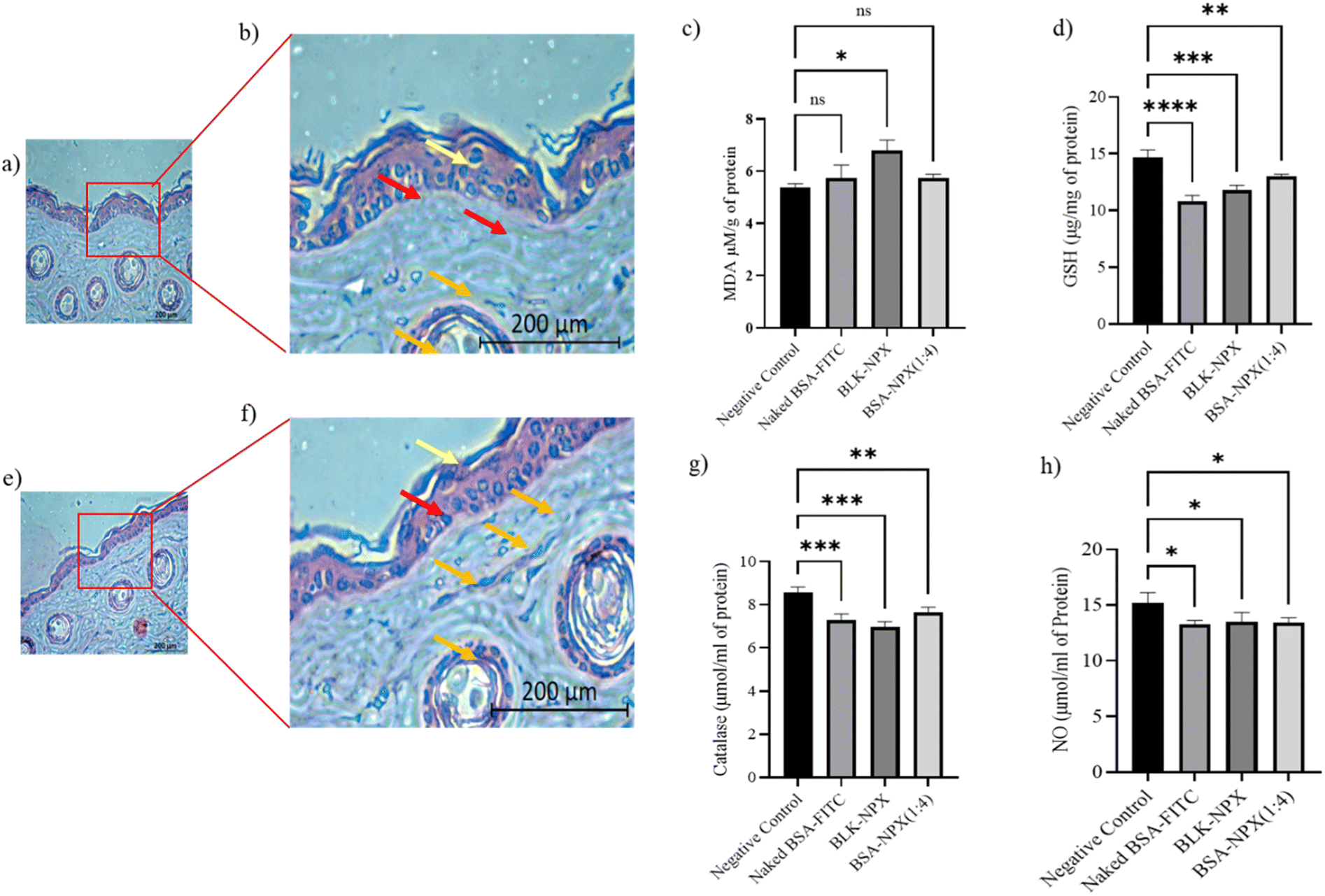 | ||
| Fig. 9 (a and b) Visual illustration of H&E staining of healthy skin of Swiss albino mice after incubation to that of (c and d) with BSA-NPX (1:4) after 24 h, respectively; (e and f) illustrated MDA, GSH, catalase and NO levels. The red arrow represents the healthy dermis in both images; the Yellow arrow represents a healthy protective stratum corneum layer; the Orange arrow represents the migrated fibroblast cells. Each value is represented as mean ± SD (n = 3). * for p < 0.05, ** for p < 0.01, *** for p < 0.001, **** for p < 0.0001, and “ns” for not significant versus negative control. | ||
3.7. ROS scavenging efficacy
In light of the correlation between oxidative stress and its pivotal involvement in the initiation and progression of skin inflammation, which is ascribed to its toxic effects,33,34 we initiated a comprehensive toxicity profiling study encompassing naked FITC-BSA, BLK-NPX, and BSA-NPX (1:4) in the context of normal skin. This assessment involved the measurement of key markers, including malondialdehyde (MDA), glutathione (GSH), Nitric Oxide (NO), and catalase. Upon topical application, we observed a significant increase in MDA levels when compared to the control group, concomitant with a decrease in the levels of GSH and NO when treated with BSA-NPX (1:4). This effect was accompanied by a corresponding decrease in catalase levels, suggesting the potential decomposition of reactive oxygen species (ROS) in comparison to the control skin (Fig. 9c, d, g and h).
4. Discussion
Proteins, with their high specificity in action and function, offer ideal properties for therapeutic molecules with minimal side effects and optimum efficacy.35 However, delivering proteins topically for conditions like psoriasis is challenging due to their instability and large size, coupled with the impermeable nature of the skin barrier.36 Innovative strategies are needed to overcome these hurdles in the delivery of proteins, and several techniques, such as iontophoresis, microneedles, and ultrasound-assisted delivery, have shown promise in enhancing protein permeation. These techniques hold great potential for improved protein delivery and therapeutic outcomes in treatment.4,37 Further, it has been reported that nanocarriers can protect proteins and enhance their penetration into the skin. These carriers can be engineered to improve stability, prolong release, and target the affected skin layers.35 For example, Choi et al. demonstrated the effectiveness of a chitosan-conjugated, Pluronic-based nanocarrier in delivering proteins, namely Insulin and BSA, through human skin. The study showed that the chemically crosslinked nanocarrier had remarkable permeability for proteins of various sizes.12 Hassan et al. devised a novel vancomycin (VCM) delivery method to combat methicillin-resistant Staphylococcus aureus (MRSA) infections. The NPX was formulated using dextran sulfate sodium salt as a polyelectrolyte complexing agent. The in vitro and in vivo studies showed promising results, with enhanced antibacterial activity and lower MRSA load compared to standard VCM.38 Cationic nanocarriers, consisting of cationic lipids/polymers and anionic biologics, have also been explored for improved stability, cellular uptake, and bioavailability.7 These systems could be further used for the topical delivery of protein and peptides; however, they may cause skin irritation and interact with the skin's natural barrier thereby reducing efficacy.39 By carefully formulating and optimizing these materials, the drawbacks of cationic nanocarriers in topical delivery can be overcomed, enhancing the efficacy of topical drug delivery.40Using cationic amphiphilic copolymers, we previously demonstrated the effective production of lipopolymeric nanoplexes. These nanoplexes successfully encapsulated and transported miRNA-34a, which has a negative charge, to MCF-7 and 4T1 cells.16,21 We have further utilized these nanoplexes for complexing CRISPR/Cas ribunucleoproteins and plasmids.17,22 These findings confirmed the importance of cationic lipopolymer in the transport of negatively charged macromolecules. To study the delivery capability of these nanoplexes for topical route, the current study utilized a cationic lipo-polymer to prepare the BLK-NPX with a positive surface charge that could complex negatively charged model protein (FITC-BSA). The copolymer mPEG-b-(CB-{g-cationic chain; g-chol; g-morph}) was successfully synthesized, which possesses a polycarbonate backbone having biodegradability and biocompatibility properties, and pendant groups to impart multifunctionality to the polymer as reported earlier.17 The cationic chain provides a positive charge, facilitating its complexation with negatively charged model protein. Earlier, we demonstrated how cationic lipopolymeric NPX may effectively distribute sgRNA/Cas9 ribonucleoproteins (RNPs) for gene editing. The nanoparticles were 117.3 ± 7.64 nm in size, with a zeta potential of +6.17 ± 1.04 mV and a high RNP entrapment efficiency. These qualities resulted in a significant increase in gene editing efficiency, both in vitro and in vivo.17 For studying the delivery route we utilized the Bovine Serum Albumin (BSA), a model protein that has been extensively studied for drug delivery applications due to its stability, biocompatibility, and low immunogenicity.12,41 Fluorescein isothiocyanate (FITC) is a fluorescent dye that can track the protein and determine its localization within the skin.42 BSA can be labelled with fluorescein isothiocyanate (FITC-BSA) to visualize and track its transport in cells and tissues. This application has been extended in the current study, where blank NPX were incubated with FITC-BSA to produce BSA-NPX. The optimal ratio for complexation was determined by evaluating the zeta potential of the NPX. Blank and FITC-BSA-loaded NPX showed a size of 93.72 ± 5.8 and 145.9 ± 3.2 nm, respectively, and the zeta potential of 25.6 ± 7.0 mV and 9.17 ± 1.20 mV. This aligns with prior findings, where an augmentation in particle size and a reduction in zeta potential were recognized as key indicators of charge-based nano-complex formation.43 Optimizing entrapment efficiency in NPX makes it possible to achieve targeted delivery of proteins to specific sites in the body, resulting in improved therapeutic outcomes and reduced side effects.44 The encapsulation efficiency in the developed formulations was more than 75%, and the morphology was spherical, as confirmed through in vitro characterization and FE-SEM. For the practical application of the formulation on the skin, a gel has proven to be a safer and more convenient. Therefore, carbopol 974P was selected as the gelling agent due to its highly cross-linked structure, resulting in a gel with high viscosity and favorable rheological properties suitable for topical use.19 BSA-NPX (1:4) formulated into a topical hydrogel utilizing carbopol 974P at 0.75% w/w. FE-SEM analysis indicated that the NPX maintained their spherical morphology without notable aggregation. The rheological behavior of the BSA-NPX gel was evaluated, since topical pharmaceutical preparations must exhibit optimal flow characteristics for achieving therapeutic benefits.18 The gel's rheograms showed a non-Newtonian, shear-thinning flow with variable thixotropy desirable for topical formulations, making them easier to process, package, apply, and stay on the skin for a prolonged time.20
In our pursuit of understanding the permeation behavior of the BSA-NPX complex across different ratios, we embarked on ex vivo skin permeation studies. These studies used healthy skin from Swiss albino mice after removing the stratum corneum, chosen for its consistent and well-characterized permeation characteristics. Despite its higher permeability relative to human or pig skin, the mouse model provides valuable initial insights into formulation behavior and permeation kinetics. The selection of mice skin for ex vivo permeation studies is pragmatic, offering accessibility for preliminary investigations. This approach enables efficient screening before transitioning to pig or human skin, ensuring a thorough understanding of skin delivery dynamics. This choice minimized the influence of extraneous factors, aligning with the focus of our current research.45 Our findings shed light on the penetration of BSA-NPX complexes into deeper skin layers, such as the viable epidermis and dermis, with varying protein quantities detected in the receptor compartments (ranging from 30% to 50%). These results provide a crucial baseline for comprehending the impact of different ratios on permeation behavior. In a previous study, we reported skin permeation through in vivo bioimaging analysis for a lipid-polymer hybrid system for small molecule delivery,19 and this study employed the same protocol. Notably, BSA-NPX complexes at higher ratios (1:4 and 1:8) displayed increased fluorescence, indicating enhanced uptake and retention in the target tissue. Incorporating these complexes into a gel matrix demonstrated a potential strategy for enhancing skin permeation by promoting adherence to the skin surface and extending contact time. The BSA-NPX (1:4) gel formulation displayed promising results, achieving up to 30% permeation within 24 h. Recognizing the importance of dermal drug delivery in diseased skin, we evaluated BSA-NPX (1:4) in a psoriatic skin model, resulting in 45% permeation. These results showcase the potential efficacy of NPX in treating skin diseases like psoriasis and pave the way for more effective therapeutic approaches. To validate these significant findings and gain deeper insights into the behavior of different NPX complexes and the BSA-NPX (1:4) gel within the skin tissue, we harnessed the power of confocal microscopy.46,47 Combining the ex vivo permeation study with confocal microscopy gave us a more comprehensive understanding of the behavior of different complexed NPX and BSA-NPX (1:4) gel in the skin tissue and optimized formulation for future applications. Confocal microscopy was also used to visualize the distribution and extent of penetration of the BSA-NPX (1:4) in psoriatic skin after ex vivo permeation.
Efficient intracellular drug delivery by nanoparticles is paramount for maximizing their therapeutic effectiveness, and extensive research has already delved deeply into this realm, encompassing various mechanisms, including endocytosis, broadly classified into phagocytosis and pinocytosis.48 We have also explored the internalization pattern of nanocarrier by employing HaCaT cells, an immortal keratinocyte cell derived from adult human skin known to mimic normal skin pathophysiology in in vitro cultures.19 We observed no cytotoxicity in HaCaT cells after treatment with BSA-NPX (1:4), possibly attributed to the biodegradable and biocompatible nature of the developed cationic lipo-polymer as reported earlier.17 Furthermore, to assess the potential toxicity of the NPX, particularly in terms of hemolysis, considering the possibility of topical application leading to systemic exposure, hemocompatibility studies were also performed. A model protein was chosen to mimic the delivery of large molecules using nanoplexes. Herein, we focused on their therapeutic benefit in skin diseases such as psoriasis, prioritizing their skin penetration across stratum corneum, local effects, skin delivery efficacy and toxicity. Though quantitative estimation of protein delivery into the blood is warranted.49 Hemocompatibility assays demonstrated the non-toxicity of NPX towards erythrocytes, indicating their potential for safe biomedical applications. In preclinical toxicity assessment, it's imperative to consider the morphological characteristics and carefully document any noteworthy observations related to the skin. This thorough evaluation ensures a comprehensive understanding of the effects of the drug delivery system on the skin.50 In vivo skin toxicity was assessed by comparing formulation-treated skin to control skin and checked for H&E staining for possible toxicity.51 We did the histological examination of the skin tissue treated with BSA-NPX (1:4) and revealed no discernible changes in the cells compared to the control skin after 24 h of application. In light of the correlation between oxidative stress and its pivotal involvement in initiating and progressing skin inflammation, which is ascribed to its toxic effects.33,34 We initiated a comprehensive toxicity profiling study encompassing naked FITC-BSA, BLK-NPX, and BSA-NPX (1:4) in the context of normal skin, mainly by checking MDA, reduced glutathione (GSH), nitric oxide (NO), and catalase levels. We observed a notable increase in MDA levels, albeit remaining within a range indicative of minimal lipid peroxidation and oxidative damage, suggesting a favorable outcome. In contrast, GSH levels slightly decreased within the expected range, reflecting its antioxidant activity in mitigating oxidative stress without severe depletion.52 Furthermore, NO levels displayed a modest reduction, aligning with the prevention of oxidative damage and inflammation.53 Catalase levels remained relatively constant or marginally decreased, indicating its protective role against oxidative damage. These findings underscore the significance of ROS scavenging in mitigating skin inflammation, highlighting the potential utility of targeted interventions in managing oxidative stress-related skin conditions, and also collectively suggest that our application, specifically BSA-NPX (1:4), did not show toxicity, which is crucial for a drug delivery system for topical use in treating skin diseases.
5. Conclusions
To summarize, the current study assessed the use of cationic polymeric NPX for effective topical delivery of a model protein (FITC-BSA). The emulsion-based method used to prepare blank NPX that were subsequently complexed with FITC-BSA, which proved beneficial in various in vitro assays. In vitro cell culture and toxicity studies showed optimal use of these nanocarriers, with initial hemocompatibility data suggesting non-toxicity. In vivo skin toxicity testing also confirmed the low toxicity profile of these nanocarriers. Overall, these nanocarriers have the potential to be used as a versatile platform for delivering macromolecular therapeutics for the treatment of various skin diseases, such as skin cancer, psoriasis, eczema, acne, and skin infections. These findings provide a promising basis for further clinical evaluation and potential commercial use.Conflicts of interest
The authors show no conflict of interest.Acknowledgements
We acknowledge BITS Pilani, Pilani campus, Rajasthan, for their support throughout the research.References
- C. Parrado, S. Mercado-Saenz, A. Perez-Davo, Y. Gilaberte, S. Gonzalez and A. Juarranz, Front. Pharmacol, 2019, 10, 759 CrossRef CAS PubMed.
- N. N. Schommer and R. L. Gallo, Trends Microbiol., 2013, 21, 660–668 CrossRef CAS PubMed.
- S. Coulman, C. Allender and J. Birchall, Crit. Rev. Ther. Drug Carrier Syst., 2006, 23, 205–258 CrossRef CAS PubMed.
- Y. Zhang, J. Yu, A. R. Kahkoska, J. Wang, J. B. Buse and Z. Gu, Adv. Drug Delivery Rev., 2019, 139, 51–70 CrossRef CAS PubMed.
- W. H. De Jong and P. J. Borm, Int. J. Nanomed., 2008, 3, 133–149 CrossRef CAS PubMed.
- A. Tharmatt, D. K. Sahel, K. Raza, M. M. Pandey, A. Mittal and D. Chitkara, J. Drug Delivery Sci. Technol., 2023, 83, 104365 CrossRef CAS.
- S. E. Jin and C. K. Kim, Colloids Surf., B, 2014, 116, 582–590 CrossRef CAS PubMed.
- S. Adepu and S. Ramakrishna, Molecules, 2021, 26, 5905 CrossRef CAS PubMed.
- T. Merdan, J. Kopecek and T. Kissel, Adv. Drug Delivery Rev., 2002, 54, 715–758 CrossRef CAS PubMed.
- B. Xie, K. Du, F. Huang, Z. Lin and L. Wu, Front. Pharmacol, 2021, 12, 762362 CrossRef CAS PubMed.
- S. Ebrahimi, R. Mahjub, R. Haddadi and S. Y. Vafaei, BioMed Res. Int., 2021, 2021, 4603545 Search PubMed.
- W. I. Choi, J. H. Lee, J. Y. Kim, J. C. Kim, Y. H. Kim and G. Tae, J. Controlled Release, 2012, 157, 272–278 CrossRef CAS PubMed.
- B. J. Bruno, G. D. Miller and C. S. Lim, Ther. Delivery, 2013, 4, 1443–1467 CrossRef CAS PubMed.
- R. Petrilli, J. O. Eloy, F. S. Praça, J. O. Del Ciampo, M. A. Fantini, M. J. Fonseca and M. V. Bentley, J. Biomed. Nanotechnol., 2016, 12, 1063–1075 CrossRef CAS PubMed.
- J. Kuhn, Y. Lin, A. Krhac Levacic, N. Al Danaf, L. Peng, M. Höhn, D. C. Lamb, E. Wagner and U. Lächelt, Bioconjugate Chem., 2020, 31, 729–742 CrossRef CAS PubMed.
- S. Sharma, S. Pukale, D. K. Sahel, P. Singh, A. Mittal and D. Chitkara, Mater. Sci. Eng., C, 2021, 128, 112305 CrossRef CAS PubMed.
- D. K. Sahel, M. Salman, M. Azhar, S. G. Goswami, V. Singh, M. Dalela, S. Mohanty, A. Mittal, S. Ramalingam and D. Chitkara, J. Mater. Chem. B, 2022, 10, 7634–7649 RSC.
- S. S. Pukale, D. K. Sahel, A. Mittal and D. Chitkara, J. Drug Delivery Sci. Technol., 2022, 76, 103672 CrossRef CAS.
- S. S. Pukale, S. Sharma, M. Dalela, A. K. Singh, S. Mohanty, A. Mittal and D. Chitkara, Acta Biomater., 2020, 115, 393–409 CrossRef CAS PubMed.
- S. S. Pukale, A. Mittal and D. Chitkara, AAPS PharmSciTech, 2021, 22, 238 CrossRef CAS PubMed.
- S. Sharma, S. Mazumdar, K. S. Italiya, T. Date, R. I. Mahato, A. Mittal and D. Chitkara, Mol. Pharm., 2018, 15, 2391–2402 CrossRef CAS PubMed.
- D. K. Sahel, S. G. Goswami, R. Jatyan, A. Kumari, A. Mittal, S. Ramalingam and D. Chitkara, Macromol. Rapid Commun., 2023, 2300101 CrossRef CAS PubMed.
- N. Barbero, C. Barolo and G. Viscardi, World J. Chem. Educ., 2016, 4, 80–85 CAS.
- S. Gebrekidan, B. H. Woo and P. P. DeLuca, AAPS PharmSciTech, 2000, 1, 17–25 CrossRef PubMed.
- T. Asimakopoulos and G. Staikos, Eur. Polym. J., 2015, 71, 567–574 CrossRef CAS.
- K. S. Prayag, A. T. Paul, S. K. Ghorui and A. B. Jindal, J. Drug Delivery Sci. Technol., 2023, 81, 104215 CrossRef CAS.
- M. Korani, S. M. Rezayat, K. Gilani, S. Arbabi Bidgoli and S. Adeli, Int. J. Nanomed., 2011, 6, 855–862 CrossRef CAS PubMed.
- I. C. Roseboom, H. Rosing, J. H. Beijnen and T. P. C. Dorlo, J. Pharm. Biomed. Anal., 2020, 191, 113590 CrossRef CAS PubMed.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS PubMed.
- H. Ohkawa, N. Ohishi and K. Yagi, Anal. Biochem., 1979, 95, 351–358 CrossRef CAS PubMed.
- C. J. Weydert and J. J. Cullen, Nat. Protoc., 2010, 5, 51–66 CrossRef CAS PubMed.
- L. C. Green, D. A. Wagner, J. Glogowski, P. L. Skipper, J. S. Wishnok and S. R. Tannenbaum, Anal. Biochem., 1982, 126, 131–138 CrossRef CAS PubMed.
- S. R. Pinnell, J. Am. Acad. Dermatol., 2003, 48, 1–22 CrossRef PubMed.
- J. Chen, Y. Liu, Z. Zhao and J. Qiu, Int. J. Cosmet. Sci., 2021, 43, 495–509 CrossRef CAS PubMed.
- A. Varanko, S. Saha and A. Chilkoti, Adv. Drug Delivery Rev., 2020, 156, 133–187 CrossRef CAS PubMed.
- Y. Huang, Y. S. Park, C. Moon, A. E. David, H. S. Chung and V. C. Yang, Angew. Chem., Int. Ed., 2010, 49, 2724–2727 CrossRef CAS PubMed.
- M. A. Oberli, C. M. Schoellhammer, R. Langer and D. Blankschtein, Ther. Delivery, 2014, 5, 843–857 CrossRef CAS PubMed.
- D. Hassan, C. A. Omolo, R. Gannimani, A. Y. Waddad, C. Mocktar, S. Rambharose, N. Agrawal and T. Govender, Int. J. Pharm., 2019, 558, 143–156 CrossRef CAS PubMed.
- K. S. Paudel, M. Milewski, C. L. Swadley, N. K. Brogden, P. Ghosh and A. L. Stinchcomb, Ther. Delivery, 2010, 1, 109–131 CrossRef CAS PubMed.
- K. Yadav, A. Soni, D. Singh and M. R. Singh, Prog. Biomater., 2021, 10, 1–17 CrossRef CAS PubMed.
- M. Karimi, S. Bahrami, S. B. Ravari, P. S. Zangabad, H. Mirshekari, M. Bozorgomid, S. Shahreza, M. Sori and M. R. Hamblin, Expert Opin. Drug Delivery, 2016, 13, 1609–1623 CrossRef CAS PubMed.
- R. Natarajan, N. Northrop and B. Yamamoto, Curr. Protoc. Neurosci., 2017, 79, 9–59 Search PubMed.
- G. Chen, A. A. Abdeen, Y. Wang, P. K. Shahi, S. Robertson, R. Xie, M. Suzuki, B. R. Pattnaik, K. Saha and S. Gong, Nat. Nanotechnol., 2019, 14, 974–980 CrossRef CAS PubMed.
- R. Solaro, F. Chiellini and A. Battisti, Materials, 2010, 3, 1928–1980 CrossRef CAS.
- A. K. Indra and M. Leid, Permeability Barrier: Methods and Protocols, 2011, pp. 73–81 Search PubMed.
- A. M. Pena, X. Chen, I. J. Pence, T. Bornschlögl, S. Jeong, S. Grégoire, G. S. Luengo, P. Hallegot, P. Obeidy, A. Feizpour, K. F. Chan and C. L. Evans, Adv. Drug Delivery Rev., 2020, 153, 147–168 CrossRef CAS PubMed.
- A. D. Elliott, Curr. Protoc. Cytom., 2020, 92, e68 CrossRef PubMed.
- J. Mosquera, I. García and L. M. Liz-Marzán, Acc. Chem. Res., 2018, 51, 2305–2313 CrossRef CAS PubMed.
- W. I. Choi, J. H. Lee, J. Y. Kim, J. C. Kim, Y. H. Kim and G. Tae, J. Controlled Release, 2012, 157, 272–278 CrossRef CAS PubMed.
- C. Herkenne, I. Alberti, A. Naik, Y. N. Kalia, F. X. Mathy, V. Préat and R. H. Guy, Pharm. Res., 2008, 25, 87–103 CrossRef CAS PubMed.
- F. F. Schmidt, S. Nowakowski and P. J. Kluger, Front. Bioeng. Biotechnol., 2020, 8, 388 CrossRef PubMed.
- T. R. Schalcher, J. L. F. Vieira, C. G. Salgado, R. d. S. Borges and M. C. Monteiro, Rev. Soc. Bras. Med. Trop., 2013, 46, 645–649 CrossRef PubMed.
- U. Karaman, T. Celik, T. R. Kiran, C. Colak and N. U. Daldal, Kor. J. Parasitol., 2008, 46, 293 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02337d |
| This journal is © The Royal Society of Chemistry 2024 |