
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTungsten doped FeCoP2 nanoparticles embedded into carbon for highly efficient oxygen evolution reaction†
Xinyao Quan‡
 a,
Jiajia Ma‡a,
Qianshuo Shaoa,
Haocong Lia,
Lingxiang Suna,
Guili Huanga,
Su Yana,
Zhanglian Hongb,
Yuning Wang*a and
Xiaoqing Wang*c
a,
Jiajia Ma‡a,
Qianshuo Shaoa,
Haocong Lia,
Lingxiang Suna,
Guili Huanga,
Su Yana,
Zhanglian Hongb,
Yuning Wang*a and
Xiaoqing Wang*c
aInstitute of Agricultural Sciences in Taihu Lake District, Suzhou Academy of Agricultural Sciences, Suzhou, 215155, China. E-mail: wyn705@163.com
bState Key Laboratory of Silicon and Advanced Semiconductor Materials, School of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, China
cCollege of Materials and Chemical Engineering, Chuzhou University, 239000 Chuzhou, China. E-mail: xiaoqingwang2013@chzu.edu.cn
First published on 22nd May 2024
Abstract
Designing active and stable electrocatalysts with economic efficiency for oxygen evolution reaction (OER) is essential for developing water splitting process at an industrial scale. Herein, we rationally designed a tungsten doped iron cobalt phosphide incorporated with carbon (Wx–FeCoP2/C), prepared by a mechanochemical approach. X-ray photoelectron spectroscopy (XPS) revealed that the doping of W led to an increasing of Co3+/Co2+ and Fe3+/Fe2+ molar ratios, which contributed to the enhanced OER performance. As a result, a current density of 10 mA cm−2 was achieved in 1 M KOH at an overpotential of 264 mV on the optimized W0.1–FeCoP2/C. Moreover, at high current density of 100 mA cm−2, the overpotential value was 310 mV, and the corresponding Tafel slope was measured to be 48.5 mV dec−1, placing it among the best phosphide-based catalysts for OER. This work is expected to enlighten the design strategy of highly efficient phosphide-based OER catalysts.
1. Introduction
The energy and environmental crisis has led to widespread concern regarding sustainable and clean energy. With the advantages of high mass-specific energy density (120 MJ kg−1), environmental friendliness and renewability, hydrogen (H2) is considered as a promising energy carrier that can substitute the fossil fuels.1 Currently, nearly 95% of H2 production is still derived from the hydrocarbon cracking which is of high cost and low purity.2 Water electrolysis is one of the most efficient methods to produce H2 efficiently and sustainably, but encounters inevitable dynamic overpotentials associated with hydrogen and oxygen evolution reactions (HER and OER). However, OER is a rate-limiting step for the feasibility of the water splitting system as a sustainable technology for H2 production, due to its four proton-coupled electron transfer, which is kinetically not favored and requires more electrical energy input.3,4 Therefore, it is crucial to design tungsten-doped bimetallic phosphide OER catalysts to reduce the overpotential of water electrolyzers.It is well known that RuO2 and IrO2 are the state-of-the-art OER electrocatalysts, but they are severely limited by their high price and scarcity. To this end, people have been engaged in searching for new materials, which including transition-metal oxides/hydroxides/oxyhydroxides,5–13 transition metals nitrides,14–16 transition metals carbides17–19 and transition metals sulfides,20–22 as highly active and abundant OER catalysts. Among them, transition metal phosphides (TMPs) have gained tremendous attention in the field of OER owing to their adjustable composition, high stability and metallic properties.23–25 For instance, Ni5P4 film,26 CoP nanoneedles,27 and Co2P nanoparticles28 are gradually being reported for OER electrocatalytic processes with high activity and durability. However, the intrinsic OER catalytic performance of pure TMPs is not outstanding. Thus, the improvement of their OER catalytic activity is crucial for the realization of excellent OER catalysts.29
On the one hand, the introduction of a secondary metal, such as NiMoP,30 FeCoP,23 and (NixFe1−x)2P,31 would optimize the adsorption and desorption of key intermediates and create multiple active sites,24 resulting in higher catalytic activity. On the other hand, heteroatom doping (such as V,32 Mo,33 and W34) is an effective strategy to manipulate the electronic structure and reduce the energy barrier of the OER process.35 Especially, W doping has been proven to effectively enhance the activity of the catalyst by promoting the water adsorption and dissociation.36–39 More remarkably, a further boost in catalytic activity can be achieved by the combination of TMPs with functionalized carbon materials due to the improved conductivity, robust durability, and modifiable surface chemistry,29 i.e., CoxP@N, P-doped carbon,40 NiCoP/C nanoboxes,41 and FeP2–NiP2@P doped carbon.42
Based on the above considerations, we reported the simple and facile synthesis of innovative composite electrocatalysts, which was described as the W-doped FeCoP2 nanoparticles embedded in the carbon matrix, by ball milling and phosphorisation. The incorporated W and composite with carbon endow the catalysts with high crystallinity, greater conductivity, larger surface area, and good nanostructured features. As a result, the W0.1–FeCoP2/C electrocatalyst outperformed most of the previously reported OER TMPs-based catalysts. It required only 264 and 310 mV overpotentials to reach 10 mA cm−2 and 100 mA cm−2 current density for OER, respectively, with a Tafel slope of 48.5 mV dec−1 and durability of 30 h measured cycles in 1.0 M KOH, attaining a high current density (100 mA cm−2).
2. Experimental section
2.1 Materials
All chemical reagents were analytically pure and were used as received without further purification, including C15H21FeO6 (98%, Aladdin, China), Co(CH3COO)2·4H2O (98%, Aladdin), WCl6 (99%, Aladdin), NaH2PO2 (99%, Aladdin), NaH2PO2 (99%, Aladdin).2.2 Synthesis of Wx–FeCoP2/C electrocatalyst
Firstly, 4 mmol of C15H21FeO6, 4 mmol of Co(CH3COO)2·4H2O, and 0.1 mmol of WCl6 were mixed homogeneously by ball milling.After that, the mixture and 1.5 g NaH2PO2 were placed at two separate corundum boats, and the mixed precursor was at downstream in the tube furnace. Then, the tube furnace was heated at 350 °C with a heating rate of 2 °C min−1 keeping for 2 h and cooled down to room temperature under the flowing N2.
For the preparation of W0.15–FeCoP2/C, W0.05–FeCoP2/C, FeCoP2/C, WCl6 was added to the reactants at different amounts (59.4 mg, 19.8 mg, and 0 mg).
2.3 Materials characterization
The crystallographic patterns of the as-prepared materials were investigated by X-ray diffraction at 2° min−1 (XRD, Cu-Kα irradiation; λ = 1.5418 Å). The morphology and elemental distribution of the samples were observed by a scanning electron microscopy (SEM Hitachi S-8010) and a transmission electron microscopy (TEM; FEI Tecnai G2 F20 S-TWIN) equipped with an energy dispersive X-ray detector (EDX). X-ray photoelectron spectroscopy (XPS, Thermo Fisher ESCALAB 250Xi spectrophotometer) was used to characterize the chemical state of samples. The specific surface area and pore size distribution of the samples were characterized by the Kubo-X1000 specific surface and porosity analyzer. The atomic ratio of Wx–FeCoP2/C was measured by ICP-OES (Optima 8300DV).2.4 Electrochemical measurements
The electrochemical measurements were carried out in a three-electrode configuration in 1 M KOH alkaline electrolyte using Ivium electrochemical analyzer. 5 mg of samples were dispersed in 1 ml of mixed solution (500 μl ethanol, 440 μl H2O and 60 μl Nafion solution (Sigma-Aldrich, 5 wt%)). The mixture was sonicated for 30 min to form a homogeneous ink. Then 10 μl of the dispersion (containing 50 μg of sample) was loaded onto a glassy carbon electrode with 4 mm diameter (loading 0.4 mg cm−2). The resulting catalyst films were then dried at room temperature. The glassy carbon electrode with different catalysts served as the working electrode. A saturated calomel electrode (SCE) was used as the reference electrode and a graphite rod as the counter electrode. Prior to the tests, cyclic voltammetry (CV) was performed at 50 mV s−1 from 0.2 to 0.4 V vs. SCE for 30 cycles before LSV was recorded for each sample. Linear sweep voltammetry (LSV) was performed in 1 M KOH solution with a scan rate of 5 mV s−1. Electrode potentials were converted to the RHE scale using E(RHE) = E(measured) + 0.2415 + 0.059 × pH, and 90% IR correction was performed by using the ESR (equivalent series resistance) measured by EIS (electrochemical impedance spectroscopy).Tafel plots, which quantify the OER performances were obtained by plotting the polarization curves as overpotential (η) versus log current (log(j)). By fitting the linear portion of the Tafel plots to the equation (η = b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log(j) + a), the Tafel slope (b) can be acquired. Electrochemical impedance spectroscopy (EIS) measurements of the catalysts were performed at an overpotential of 0.4 V. The frequency range was 105 Hz to 10−2 Hz, and the amplitude of the applied voltage was 30 mV. The electrochemical surface areas (ECSA) were estimated by CV at different scan rates from 5 mV s−1 to 200 mV s−1. The long-term stability tests were carried out for 30 h by using the amperometric i–t curve at overpotential of 0.3 V.
log(j) + a), the Tafel slope (b) can be acquired. Electrochemical impedance spectroscopy (EIS) measurements of the catalysts were performed at an overpotential of 0.4 V. The frequency range was 105 Hz to 10−2 Hz, and the amplitude of the applied voltage was 30 mV. The electrochemical surface areas (ECSA) were estimated by CV at different scan rates from 5 mV s−1 to 200 mV s−1. The long-term stability tests were carried out for 30 h by using the amperometric i–t curve at overpotential of 0.3 V.
3. Results and discussion
The XRD patterns of the W0.1–FeCoP2/C, W0.15–FeCoP2/C, W0.05–FeCoP2/C and FeCoP2/C catalysts were displayed in Fig. 1. It can be observed that the peaks of four samples can be well indexed to FeCoP2 phase (ICSD #622953). The characteristic peaks positioned at 31.8°, 36.9°, 46.3°, 48.2° and 56.5° corresponding to the (011), (111), (211), (103) and (321) planes of FeCoP2, respectively. In addition, the broad peak near 25° can be ascribed to the carbon.43 Moreover, no other obvious diffraction peaks were observed in the XRD patterns, which indicated that there are no impure phases. Of note, compared to the FeCoP2/C, with the increase of W doping amount, the diffraction peaks of Wx–FeCoP2/C shifted to lower angles. Especially, the magnified characteristic diffraction peak at 31.6° showed a subtle shift to lower angle direction as shown in Fig. 1. It may be attributed to the incorporation of W element into the FeCoP2 lattice, resulting in the lattice expansion.44 | ||
| Fig. 1 XRD patterns of the W0.1–FeCoP2/C, W0.15–FeCoP2/C, W0.05–FeCoP2/C and FeCoP2/C. | ||
SEM characterizations were performed to explore the effect of tungsten dopant usage on the morphology of as-prepared Wx–FeCoP2/C catalysts. As shown in Fig. S1a,† FeCoP2/C was formed by the aggregation of nano particles (20–50 nm). Interestingly, as the doping amount of W increasing, Wx–FeCoP2/C showed more dense particles (Fig. S1b, c† and 2a). In particular, W0.1–FeCoP2/C showed more uniform nano particles (Fig. 2a). This result indicated a microstructural adjustment achieved by W doping in FeCoP2/C. Therefore, the surface area and pore size distribution of FeCoP2/C and Wx–FeCoP2/C were studied using BET analysis. Fig. S2† represented the N2 adsorption–desorption curves of the type IV isotherm, which extended up to the relative pressure (P/P0) range of 0.8–0.9 and an upward sweep near the saturation pressure confirmed the presence of a macroporous nature with multilayer adsorption (the insets of Fig. S2†).45 And the obtained results of the surface area, macro-/mesopore volume were tabulated in Table S1.† The surface area of pristine FeCoP2/C was 22.81 m2 g−1 with macro-/mesopore volume at 0.0095/0.074 cm3 g−1. While, Wx–FeCoP2/C possessed larger surface areas and pore volume than FeCoP2/C after W doping, in which W0.1–FeCoP2/C exhibited a maximum BET surface area as high as 36.94 m2 g−1. It was universally acknowledged that a high surface area with a highly porous structure of the material facilitates the more active sites for reaction leaded to better performance of electrocatalysts toward the OER.
 | ||
| Fig. 2 (a) SEM image of W0.1–FeCoP2/C in different magnification (b) TEM image of W0.1–FeCoP2/C (c) high-resolution TEM image of the W0.1–FeCoP2/C nanostructure with a corresponding fast Fourier transform pattern (d) HADDF-TEM image of the W0.1–FeCoP2/C and the corresponding elemental mapping of (e) carbon (f) phosphorus (g) cobalt (h) iron and (i) tungsten. | ||
The TEM image of W0.1–FeCoP2/C further confirmed the porous structure (the white circle in Fig. 2b). And the HRTEM image (Fig. 2c) of W0.1–FeCoP2/C showed typical lattice fringes that matched the standard FeCoP2 (011)/(211) interplanar spacing of 2.94/2.14 Å (ICSD #622953). Furthermore, three diffraction spots in the inset SAED patterns of Fig. 2c were assigned well to the FeCoP2 (011), (211) and (111) planes, respectively. The high-angle annular dark field scanning TEM (HAADF-STEM) (Fig. 2d) image and the corresponding elemental mappings (Fig. 2e–i) of W0.1–FeCoP2/C showed that C, P, Fe, Co, and W elements were uniformly distributed. The ICP-OES results in Table S2† showed that the elemental ratios of Fe and Co in Wx–FeCoP2/C were close to the stoichiometric ratio (1:1), while the ratio of the actual contents of W were slightly lower than that expected from the feed molar ratio: 2.44% in the feed resulted in 2.37% W, and both 4.76 and 6.89% feed ratios resulted in 4.43 and 5.99% W in the final material.
Based on the above results, the formation process of Wx–FeCoP2/C can be inferred as follows: the carbon source in C15H21FeO6 and Co(CH3COO)2·4H2O forms a three-dimensional network structure to create a small reactor during the heating process, which separates the metal elements and accommodate the phosphatization reaction at the time. As such a result of high temperature phosphatization and carbonization with space confinement effect, W doped FeCoP2 nanoparticles were encapsulated by porous carbon substrate and evenly dispersed. To confirm the successful coating of the carbon layer, the Raman spectrum was shown in Fig. S3.† FeCoP2/C and Wx–FeCoP2/C display two distinct peaks at 1348 cm−1 (D band) and 1607 cm−1 (G band) because of the carbon layer in Table S3.† And the calculated ID/IG values of FeCoP2/C and Wx–FeCoP2/C were all exceeding, which suggested that the catalyst have many defects, and the disordered structure was beneficial to improving the hydrogen ion diffusion and electron conduction.46
The electronic states and the surface chemical composition of W0.1–FeCoP2/C were determined by X-ray photoelectron spectroscopy (XPS) analysis. As illustrated in Fig. 3, the typical peaks representing Fe, Co, and P can be clearly detected in the survey XPS spectrum of W0.1–FeCoP2/C. Focusing on the high-resolution XPS spectrum of Fe 2p (Fig. 3a), the peaks located at 719.8 and 707.1 eV were demonstrated as the characteristic doublets of partially positively charged Fe–P bonds which were very close to the metallic state Fe0, while the peaks at 724.2 (728.9) and 710.8 (714.4) eV were ascribed to 2p1/2 and 2p3/2 for Fe2+ (Fe3+), respectively.47–49 Similarly, the Co (2p) spectrum (Fig. 3b) can also be deconvoluted into four sets of peaks, in where the two main peaks at 793.7 and 778.7 eV were assigned to partially positively charged Co–P bonding,50 and the strong peaks located at 797.8 (801.6) and 782.1 (784.9) eV belong to Co3+ (Co2+) 3d3/2 and Co3+ (Co2+) 3d5/2, respectively, while the peaks at 805.0 and 787.5 eV were assigned to satellite peaks.51 Meanwhile, in the XPS spectrum of P 2p (Fig. 3c), the peak at 134.3 eV correspond to P–O, and the other peak at the binding energies of 129.8 eV can be attributed to P in phosphide.51–54 The Fe, Co, P spectrum of W0.05–FeCoP2/C, W0.15–FeCoP2/C and FeCoP2/C were showed in Fig. S4–S6.† The above XPS characterization results can further prove the existence of FeCoP2 phase, which was consistent with XRD results.
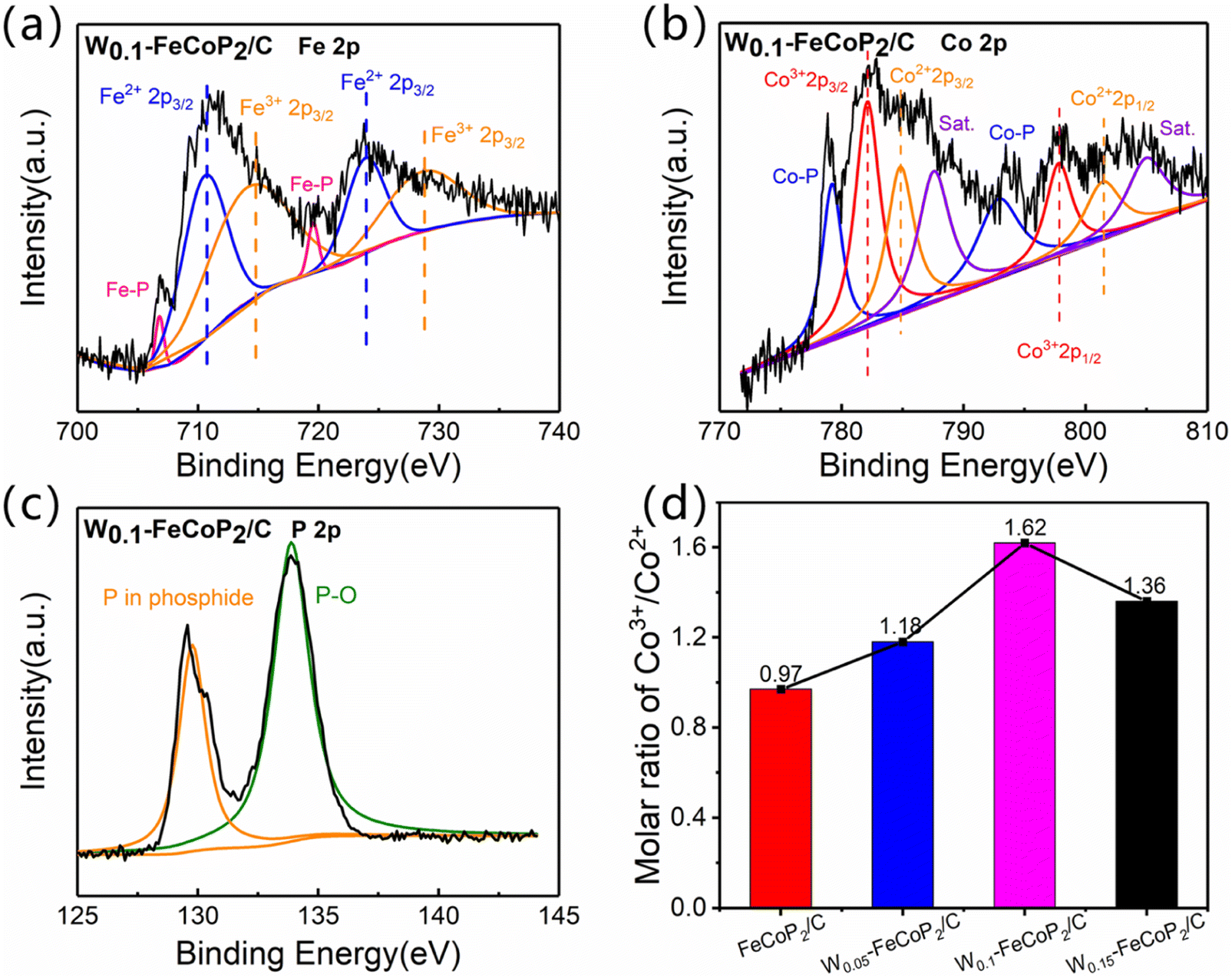 | ||
| Fig. 3 (a) Fe 2p XPS spectra for W0.1–FeCoP2/C; (b) Co 2p XPS spectra for W0.1–FeCoP2/C; (c) P 2p XPS spectra for W0.1–FeCoP2/C; (d) Co3+/Co2+ molar ratios of FeCoP2/C, W0.05–FeCoP2/C, W0.1–FeCoP2/C and W0.15–FeCoP2/C. | ||
In addition, as demonstrated by the W 4f spectrum (Fig. S7†), two weak peaks at the binding energies of 37.9 and 35.8 eV were attributed to W 4f7/2 and W 4f5/2, respectively, which suggested that W was successfully doped into FeCoP2 and mainly existed in Wx–FeCoP2 in the form of W6+.55 Notably, compared with FeCoP2/C, the peak area contents of Fe–P bond in Wx–FeCoP2 were significantly decreasing (Fig. S4†), while those of Fe3+ and Fe2+ were increasing. The above mentioned changes indicated Fe moved to the oxidation state, due to the high valence W6+ can promote the oxidation of 3d metals.56 Interestingly, similar phenomena had also occurred in the Co 2p spectra (Fig. S5†), in where the ratio of Co3+ to Co2+ content gradually changed with the change of W doping amount.
Based on the XPS results, the molar ratio of Co3+ to Co2+ (Co3+/Co2+) in each sample is quantitatively analyzed which was shown in Fig. 3d. The results showed that Co3+/Co2+ in W0.05–FeCoP2/C, W0.1–FeCoP2/C and W0.15–FeCoP2/C samples were all more than 1, and the maximum Co3+/Co2+ in W0.1–FeCoP2/C samples was 1.62. However, Co3+/Co2+ of FeCoP2/C sample was only 0.97, which indicated that the doping of tungsten atoms leaded to the increase of high price Co ions in the samples. Recently, many studies point out that high cobalt (Co3+δ, 1 ≥ δ ≥ 0) was the active center of OER catalyst,57–59 Co3+ showed higher activity than Co2+.59 The low price of Co would oxidize to the high price (Co2+ → Co3+, Co3+ → Co4+) during the catalytic reaction, while this oxidation process required a lot of energy. If Co3+ in the catalyst can be directly increased, the reaction barrier of the process will be reduced, so as to effectively improve the activity of the catalyst.60,61
The OER electrocatalytic properties of the prepared W0.1–FeCoP2/C, W0.15–FeCoP2/C, W0.05–FeCoP2/C and FeCoP2/C catalyst were measured in 1 M KOH electrolyte at room temperature. All of the potentials were calibrated against the reversible hydrogen electrode (RHE) for comparison. Fig. 4a showed the polarization curves of W0.1–FeCoP2/C, W0.15–FeCoP2/C, W0.05–FeCoP2/C and FeCoP2/C catalyst. W0.1–FeCoP2/C demonstrated a much lower onset potential and higher specific current compared to W0.15–FeCoP2/C, W0.05–FeCoP2/C and FeCoP2/C, revealing that it had excellent intrinsic OER catalytic activity. As Fig. 4c compared, to achieve J = 10 mA cm−2, W0.1–FeCoP2/C required an overpotential of 264 mV, which was significantly lower than that of W0.15–FeCoP2/C (286 mV), W0.05–FeCoP2/C (325 mV), and FeCoP2/C (389 mV). Since the current density of W0.1–FeCoP2/C achieved 100 mA cm−2, the overpotential was 310 mV which was far below FeCoP2/C (524 mV). The results indicated that sufficient doping of W atoms made catalysts behave superior to the IrO2. The performance of W0.1–FeCoP2/C was superior to those of most previously reported nanomaterials (Table S4†). The Tafel slope for the W0.1–FeCoP2/C catalyst was 48.5 mV dec−1 in Fig. 4b, confirming the OER reaction kinetics. With values of 65.5 mV dec−1 for W0.15–FeCoP2/C, 90.6 mV dec−1 for W0.05–FeCoP2/C, and 154.3 mV dec−1 for FeCoP2/C, the other catalysts had much higher Tafel slopes, indicating a lower conversion efficiency in water oxidation. The electrochemical surface areas (ECSA) were measured and compared in Fig. 4d. With measuring the capacitive current the Cdl was related to double-layer charging from the scan-rate dependence of cyclic voltammetry stripping. The corresponding CV plots at scan rates from 10 mV s−1 to 200 mV s−1, the Cdl of W0.1–FeCoP2/C was 7.15 mF cm−2 which was the highest among that of the W0.15–FeCoP2/C (6.78 mF cm−2), W0.05–FeCoP2/C (6.25 mF cm−2), and FeCoP2/C (5.78 mF cm−2). It coincided well with the polarization and Tafel slope curves. This was due to the phosphidation process, which would generate more porous structure as reported in literature. Due to the similar surface morphology of four catalysts, the values of Cdl were very similar (Table S5†). The Nyquist plots were used to analyze the charge transfer resistances in Fig. 4e. The inset of Fig. 4e showed the fitted equivalent circuit, in which Rs and Rp represented the solution resistance and charge-transfer resistance, respectively. The solution resistance was consistent for all the measurements with a value of 6 Ω. W0.1–FeCoP2/C showed a much lower interfacial charge transfer resistance of 7.86 Ω as compared to the others, which was consistent with their OER activities (FeCoP2/C < W0.05–FeCoP2/C < W0.15–FeCoP2/C < W0.1–FeCoP2/C) and signified the obviously greater reaction kinetics of W atom doped. Tungsten atoms possessed a large radius and entered into the phosphide lattice can increase the crystal surface defects, which leaded to the more active sites in the oxygen evolution reaction. Apart from the high activity, stability was also significant for OER catalyst. The electrochemical stability of the prepared W0.1–FeCoP2/C catalyst was tested by chronoamperometric (i–t) methods, as shown in Fig. 4f. The current density of W0.1–FeCoP2/C was recorded for 30 hours at an overpotential of 310 mV. The OER current remains 93.7% of its initial value after the test, which indicated the excellent durability of W0.1–FeCoP2/C. Due to carbon packed nano phosphide nanoparticles outside, W0.1–FeCoP2/C exposed more active sites used for OER reaction and simultaneously enhanced the stability.
 | ||
| Fig. 4 (a) OER polarization curves of FeCoP2/C, W0.05–FeCoP2/C, W0.1–FeCoP2/C, W0.15–FeCoP2/C and IrO2, (b) overpotential at 10 mA cm−2 and 100 mA cm−2 of FeCoP2/C, W0.05–FeCoP2/C, W0.1–FeCoP2/C and W0.15–FeCoP2/C, (c) Tafel plots for different electrocatalysts in 1 M KOH solution, (d) plots of the different current density vs. the scan rate for different electrodes, (e) Nyquist plots for different electrocatalysts recorded at 1.53 V vs. RHE, (f) the prolonged chronoamperometric response of W0.1–FeCoP2/C. | ||
In order to investigate the electron states of W0.1–FeCoP2/C after OER, XPS spectra were collected with respect to the pristine catalyst. As compared to the pristine catalyst in Fig. 5a and b, the Fe–P bond almost disappeared after the OER test and the Co 2p XPS spectrum after OER had shifted to the higher binding energy side, which indicated the high valence Co species were formed at the electrochemical oxidation process. Meanwhile, the absence of the P element was detected decreasing rapidly by XPS in Fig. 5c. However, as shown in Fig. 5d, the signal of W 4f can still be detected after OER reaction, and there was no significant change compared with that before OER reaction (Fig. S7a†). The XRD pattern of the W0.1–FeCoP2/C after OER was shown in Fig. S8,† which can be seen that there was no significant change in the overall phase of the sample. These results indicated that the surface of W0.1–FeCoP2/C has been in situ transformed into tungsten-doped Fe–Co bimetallic (hydrogen) oxides or hydroxy–hydroxide intermediates, in agreement with illustrated by previous literature, that the surface of metal phosphide materials will undergo oxidation in situ transformation into oxygen/hydroxide during OER process, and participate in the catalytic reaction as the active center.62,63 The micromorphology of W0.1–FeCoP2/C was revealed by TEM, as Fig. 5e showed, after OER W0.1–FeCoP2/C still maintained the nanoparticles embedded into carbon. That can be explained the excellent stability of W0.1–FeCoP2/C. This can be attributed to the carbon layer coated by phosphide nanoparticles, which can effectively prevent the agglomeration and deactivation of nanoparticles during the reaction process, so that tungsten-doped Fe–Co bimetallic phosphide can maintain a long time catalytic activity in the electrochemical oxygen evolution process.
 | ||
| Fig. 5 The comparison of the high-resolution XPS spectra of W0.1–FeCoP2/C before (top) and after (bottom) OER tests: (a) Fe 2p, (b) Co 2p, and (c) P 2p; (d) XPS characterization of W 4f and (e) TEM image of W0.1–FeCoP2/C after OER tests. | ||
The electrochemistry analysis, SEM, HRTEM, and X-ray analysis suggested that W0.1–FeCoP2/C possessed highly promising OER-type electrocatalytic properties with the following aspects. (1) Theoretical studies suggest that the energetics of OER intermediates can be modulated by incorporating metal elements, and that in turn these tune the catalytic activity of these materials. W6+ into FeCo bimetallic phosphides for more efficient OER catalysis is due to the modulation of the 3d energy levels of the metal atom, leading to good adsorption energies for OER intermediates. Fe, Co, W three elements coexist and synergistically make *OH energetics in an optimal position. (2) The optimal addition of tungsten results in the increasing of Co3+, which makes more electrons participate in electronic transfers for OER. (3) Phosphides are oxidized to metal oxides/hydroxides on the surface under the OER conditions in alkaline media. The metal oxides/hydroxides on the surface provide OER catalysis properties, meanwhile phosphides with metallic conductivity provide fast charge transportation. (4) The size of nano phosphide nanoparticles (W0.1–FeCoP2) is under 10 nm, which results in a large specific surface area and more active sites exposed. (5) The carbon outer layer also enables stabilization functionality toward the OER.
4. Conclusion
In conclusion, we presented a facile method for preparing W0.1–FeCoP2 nanoparticles composited with carbon which possessed outstanding OER catalytic properties. In this material, the nano phosphide nanoparticles provided abundant surface active sites and short mass transfer pathways, while the metallic conductivity facilitates charge transfer. Furthermore, the uniform distribution of Fe, Co, W three metal elements enabled adsorption energies for OER intermediates in an optimal position and lower the kinetics of OER. As expected, the W0.1–FeCoP2/C catalyst delivered a low overpotential of only 264 mV at a current density of 10 mA cm−2 and a small Tafel slope (48.5 mV dec−1), comparable with counterparts of different W atom (W0.15–FeCoP2/C, W0.05–FeCoP2/C and FeCoP2/C) catalysts, and superior to most previously reported OER catalysts. After addition of the correct amount of W into Fe–Co, highest Co3+/Co2+ molar ratio leaded to greater electronic transfers, which was the important factor in boosting the OER activity. The present work stated a promising strategy to improve OER electrocatalysis through precisely tuning the composition of multimetallic phosphides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by Grants from Subsidized Project of Suzhou Academy of Agricultural Sciences (Grant No. 22024), Agricultural Science and Technology Independent Innovation Funds of Jiangsu Province (Grant No. SCX(22)3064), Suzhou City and Tongren City Science and Technology Cooperation Project (Grant No. Tong Ke Cheng He [2020]01), the Science and Technology Foundation of Suzhou (Grant No. SNG2023002, SNG2021024).References
- G. W. Crabtree, M. S. Dresselhaus and M. V. Buchanan, The hydrogen economy, Phys. Today, 2004, 57(12), 39–44 CrossRef CAS.
- M. S. Dresselhaus and I. L. Thomas, Alternative energy technologies, Nature, 2001, 414(6861), 332–337 CrossRef CAS PubMed.
- O. Kasian, et al., The Common Intermediates of Oxygen Evolution and Dissolution Reactions during Water Electrolysis on Iridium, Angew. Chem., Int. Ed., 2018, 57(9), 2488–2491 CrossRef CAS PubMed.
- X. Y. Quan, et al., Electrospinning metal Phosphide/Carbon nanofibers from Phytic Acid for hydrogen evolution reaction catalysts, Nanotechnology, 2020, 31(41), 415602 CrossRef CAS PubMed.
- Z. P. Wu, et al., Non-Noble-Metal-Based Electrocatalysts toward the Oxygen Evolution Reaction, Adv. Funct. Mater., 2020, 30(15), 1910274 CrossRef CAS.
- S. C. Du, et al., Ni-Fe LDH/Reduced Graphene Oxide as Catalyst for Oxygen Evolution Reaction, Chem. J. Chin. Univ., 2016, 37(8), 1415–1420 CAS.
- B. L. Chen, et al., One-step construction of porous Ni/Co metal/oxide nanocubes for highly efficient oxygen evolution, Electrochem. Commun., 2018, 93, 191–196 CrossRef CAS.
- M. K. Bates, et al., Charge-Transfer Effects in Ni-Fe and Ni-Fe-Co Mixed-Metal Oxides for the Alkaline Oxygen Evolution Reaction, ACS Catal., 2016, 6(1), 155–161 CrossRef CAS.
- M. Jafarian, et al., Electrocatalytic oxidation of methane at nickel hydroxide modified nickel electrode in alkaline solution, Electrochem. Commun., 2003, 5(2), 184–188 CrossRef CAS.
- F. F. Zhang, et al., Highly dispersed ultrafine Pt nanoparticles on nickel-cobalt layered double hydroxide nanoarray for enhanced electrocatalytic methanol oxidation, Int. J. Hydrogen Energy, 2018, 43(33), 16302–16310 CrossRef CAS.
- S. Khatun and P. Roy, Cobalt chromium vanadium layered triple hydroxides as an efficient oxygen electrocatalyst for alkaline seawater splitting, Chem. Commun., 2022, 58(8), 1104–1107 RSC.
- X. Y. Chen, et al., Engineering single atomic ruthenium on defective nickel vanadium layered double hydroxide for highly efficient hydrogen evolution, Nano Res., 2023, 16, 4612 CrossRef CAS.
- N. U. Saidin, et al., One-pot gamma radiolysis synthesis of a graphene oxide-supported cobalt oxyhydroxide electrocatalyst for oxygen reduction reaction, Radiat. Phys. Chem., 2023, 205, 110680 CrossRef CAS.
- X. Peng, et al., Recent progress of transition metal nitrides for efficient electrocatalytic water splitting, Sustainable Energy Fuels, 2019, 3(2), 366–381 RSC.
- K. Xu, et al., Metallic Nickel Nitride Nanosheets Realizing Enhanced Electrochemical Water Oxidation, J. Am. Chem. Soc., 2015, 137(12), 4119–4125 CrossRef CAS PubMed.
- X. Peng, et al., In situ segregation of cobalt nanoparticles on VN nanosheets via nitriding of Co2V2O7 nanosheets as efficient oxygen evolution reaction electrocatalysts, Nano Energy, 2017, 34, 1–7 CrossRef CAS.
- H. Wang, et al., Transition metal carbides in electrocatalytic oxygen evolution reaction, Chin. Chem. Lett., 2021, 32(1), 291–298 CrossRef CAS.
- Y.-J. Tang, et al., Bimetallic Carbides-Based Nanocomposite as Superior Electrocatalyst for Oxygen Evolution Reaction, ACS Appl. Mater. Interfaces, 2017, 9(20), 16977–16985 CrossRef CAS PubMed.
- M. Li, et al., Ni Strongly Coupled with Mo2C Encapsulated in Nitrogen-Doped Carbon Nanofibers as Robust Bifunctional Catalyst for Overall Water Splitting, Adv. Energy Mater., 2019, 9(10), 1803185 CrossRef.
- R. He, X. Huang and L. Feng, Recent Progress in Transition-Metal Sulfide Catalyst Regulation for Improved Oxygen Evolution Reaction, Energy Fuels, 2022, 36(13), 6675–6694 CrossRef CAS.
- S. Wang, et al., High valence state of Ni and Mo synergism in NiS2-MoS2 hetero-nanorods catalyst with layered surface structure for urea electrocatalysis, J. Energy Chem., 2022, 66, 483–492 CrossRef CAS.
- X. Li, et al., Engineering Sulfur Vacancies in Spinel-Phase Co3S4 for Effective Electrocatalysis of the Oxygen Evolution Reaction, ACS Omega, 2022, 7(14), 12430–12441 CrossRef CAS PubMed.
- L. Kumar, et al., A Superior and Stable Electrocatalytic Oxygen Evolution Reaction by One-Dimensional FeCoP Colloidal Nanostructures, ACS Appl. Mater. Interfaces, 2022, 14(4), 5468–5477 CrossRef CAS PubMed.
- S. Li, et al., Binary Metal Phosphides with MoP and FeP Embedded in P,N-Doped Graphitic Carbon As Electrocatalysts for Oxygen Reduction, ACS Sustain. Chem. Eng., 2019, 7(13), 11872–11884 CrossRef CAS.
- C. Liu, et al., Binary nickel iron phosphide composites with oxidized surface groups as efficient electrocatalysts for the oxygen evolution reaction, Sustainable Energy Fuels, 2019, 3(12), 3518–3524 RSC.
- M. Ledendecker, et al., The Synthesis of Nanostructured Ni5P4 Films and their Use as a Non-Noble Bifunctional Electrocatalyst for Full Water Splitting, Angew. Chem., Int. Ed., 2015, 54(42), 12361–12365 CrossRef CAS PubMed.
- P. Wang, et al., Efficient Water Splitting Catalyzed by Cobalt Phosphide-Based Nanoneedle Arrays Supported on Carbon Cloth, ChemSusChem, 2016, 9(5), 472–477 CrossRef CAS PubMed.
- K. Xu, et al., Promoting Active Species Generation by Electrochemical Activation in Alkaline Media for Efficient Electrocatalytic Oxygen Evolution in Neutral Media, Nano Lett., 2017, 17(1), 578–583 CrossRef CAS PubMed.
- C.-J. Huang, et al., A review of modulation strategies for improving catalytic performance of transition metal phosphides for oxygen evolution reaction, Appl. Catal., B, 2023, 325, 122313 CrossRef CAS.
- H. Xu, et al., Hierarchical NiMo Phosphide Nanosheets Strongly Anchored on Carbon Nanotubes as Robust Electrocatalysts for Overall Water Splitting, ACS Appl. Mater. Interfaces, 2018, 10(35), 29647–29655 CrossRef CAS PubMed.
- S. Sun, et al., Tailoring the d-Band Centers Endows (NixFe1–x)2P Nanosheets with Efficient Oxygen Evolution Catalysis, ACS Catal., 2020, 10(16), 9086–9097 CrossRef CAS.
- Y. Wang, et al., Vanadium-Incorporated CoP2 with Lattice Expansion for Highly Efficient Acidic Overall Water Splitting, Angew. Chem., Int. Ed., 2022, 61(12), e202116233 CrossRef CAS PubMed.
- J. Lin, et al., Bifunctional Electrocatalysts Based on Mo-Doped NiCoP Nanosheet Arrays for Overall Water Splitting, Nano-Micro Lett., 2019, 11(1), 55 CrossRef CAS PubMed.
- X.-T. Yan, et al., Uniform W-NiCoP microneedles by molten salt decomposition as bifunctional electrocatalyst for alkaline water splitting, Appl. Mater. Today, 2021, 24, 101154 CrossRef.
- D. Thiyagarajan, et al., Nanoarchitectured porous Cu-CoP nanoplates as electrocatalysts for efficient oxygen evolution reaction, Chem. Eng. J., 2022, 432, 134303 CrossRef CAS.
- S. Ghosh, et al., W Doping in Ni12P5 as a Platform to Enhance Overall Electrochemical Water Splitting, ACS Appl. Mater. Interfaces, 2022, 14(1), 581–589 CrossRef CAS PubMed.
- J. Yan, et al., Single atom tungsten doped ultrathin α-Ni(OH)2 for enhanced electrocatalytic water oxidation, Nat. Commun., 2019, 10(1), 2149 CrossRef PubMed.
- Z. Chen, et al., Tuning electronic property and surface reconstruction of amorphous iron borides via W-P co-doping for highly efficient oxygen evolution, Appl. Catal., B, 2021, 288, 120037 CrossRef CAS.
- S.-S. Lu, et al., Tungsten-doped Ni–Co phosphides with multiple catalytic sites as efficient electrocatalysts for overall water splitting, J. Mater. Chem. A, 2019, 7(28), 16859–16866 RSC.
- Q. Shi, et al., Controllable Construction of Bifunctional CoxP@N,P-Doped Carbon Electrocatalysts for Rechargeable Zinc–Air Batteries, Energy Environ. Mater., 2022, 5(2), 515–523 CrossRef CAS.
- P. He, X.-Y. Yu and X. W. Lou, Carbon-Incorporated Nickel–Cobalt Mixed Metal Phosphide Nanoboxes with Enhanced Electrocatalytic Activity for Oxygen Evolution, Angew. Chem., Int. Ed., 2017, 56(14), 3897–3900 CrossRef CAS PubMed.
- P. Ji, et al., Double Metal Diphosphide Pair Nanocages Coupled with P-Doped Carbon for Accelerated Oxygen and Hydrogen Evolution Kinetics, ACS Appl. Mater. Interfaces, 2020, 12(1), 727–733 CrossRef CAS PubMed.
- H. Takagi, et al., XRD analysis of carbon stacking structure in coal during heat treatment, Fuel, 2004, 83(17), 2427–2433 CrossRef CAS.
- X. Q. Wang, et al., Hierarchically Porous W-Doped CoP Nanoflake Arrays as Highly Efficient and Stable Electrocatalyst for pH-Universal Hydrogen Evolution, Small, 2019, 15(37), e1902613 CrossRef PubMed.
- T. Munawar, et al., Electrochemical Performance Evaluation of Bimetallic Sulfide Nanocomposite with Fullerene (CeNdS/C60) for Efficient Oxygen Evolution Reaction (OER), Energy Fuels, 2023, 37(2), 1370–1386 CrossRef CAS.
- X. Wang, et al., Rational Design of Three-Dimensional Graphene Encapsulated with Hollow FeP@Carbon Nanocomposite as Outstanding Anode Material for Lithium Ion and Sodium Ion Batteries, ACS Nano, 2017, 11(11), 11602–11616 CrossRef CAS PubMed.
- J. Masud, et al., Iron phosphide nanoparticles as an efficient electrocatalyst for the OER in alkaline solution, J. Mater. Chem. A, 2016, 4(25), 9750–9754 RSC.
- H. Liang, et al., Amorphous NiFe-OH/NiFeP Electrocatalyst Fabricated at Low Temperature for Water Oxidation Applications, ACS Energy Lett., 2017, 2(5), 1035–1042 CrossRef CAS.
- J. Nai, et al., Formation of Ni–Fe Mixed Diselenide Nanocages as a Superior Oxygen Evolution Electrocatalyst, Adv. Mater., 2017, 29(41), 1703870 CrossRef PubMed.
- B. Deng, et al., High catalytic performance of nickel foam supported Co2P-Ni2P for overall water splitting and its structural evolutions during hydrogen/oxygen evolution reactions in alkaline solutions, J. Catal., 2019, 373, 81–92 CrossRef CAS.
- L. Chen, et al., Cobalt layered double hydroxides derived CoP/Co2P hybrids for electrocatalytic overall water splitting, Nanoscale, 2018, 10(45), 21019–21024 RSC.
- Z. Zhan, et al., Modulating Electronic Structure of NiCoP Nanoarrays by B Doping for Boosting Overall Water Splitting, Energy Fuels, 2022, 36(15), 8371–8380 CrossRef CAS.
- M. Zhi, et al., Electrochemical and microstructural analysis of nickel–yttria-stabilized zirconia electrode operated in phosphorus-containing syngas, J. Power Sources, 2008, 183(2), 485–490 CrossRef CAS.
- C. Teng, et al., Uniform NiFe phosphide nanosheets arrays on carbon cloth as high-performance oxygen evolution catalysts, Mater. Today Energy, 2019, 11, 192–198 CrossRef CAS.
- B. Zhang, et al., Homogeneously dispersed multimetal oxygen-evolving catalysts, Science, 2016, 352(6283), 333–337 CrossRef CAS PubMed.
- B. Zhang, et al., High-valence metals improve oxygen evolution reaction performance by modulating 3d metal oxidation cycle energetics, Nat. Catal., 2020, 3(12), 985–992 CrossRef CAS.
- J. Zhou, et al., Electrochemically accessing ultrathin Co (oxy)-hydroxide nanosheets and operando identifying their active phase for the oxygen evolution reaction, Energy Environ. Sci., 2019, 12(2), 739–746 RSC.
- A. Bergmann, et al., Unified structural motifs of the catalytically active state of Co(oxyhydr)oxides during the electrochemical oxygen evolution reaction, Nat. Catal., 2018, 1(9), 711–719 CrossRef CAS.
- Y. Surendranath, M. W. Kanan and D. G. Nocera, Mechanistic Studies of the Oxygen Evolution Reaction by a Cobalt-Phosphate Catalyst at Neutral pH, J. Am. Chem. Soc., 2010, 132(46), 16501–16509 CrossRef CAS PubMed.
- J. Chen, et al., Co–Fe–Cr (oxy)Hydroxides as Efficient Oxygen Evolution Reaction Catalysts, Adv. Energy Mater., 2021, 11(11), 2003412 CrossRef CAS.
- J. Chen, et al., Octahedral Coordinated Trivalent Cobalt Enriched Multimetal Oxygen-Evolution Catalysts, Adv. Energy Mater., 2020, 10(43), 2002593 CrossRef CAS.
- J. Chang, et al., Surface Oxidized Cobalt-Phosphide Nanorods As an Advanced Oxygen Evolution Catalyst in Alkaline Solution, ACS Catal., 2015, 5(11), 6874–6878 CrossRef CAS.
- S. Ding, et al., Modulating Electronic Structure of Cobalt Phosphide Precatalysts via Dual-Metal Incorporation for Highly Efficient Overall Water Splitting, ACS Appl. Energy Mater., 2019, 2(11), 8022–8030 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02326a |
| ‡ Co-first author. |
| This journal is © The Royal Society of Chemistry 2024 |