DOI:
10.1039/D4RA02271H
(Paper)
RSC Adv., 2024,
14, 16584-16599
Some pyrimidohexahydroquinoline candidates: synthesis, DFT, cytotoxic activity evaluation, molecular docking, and in silico studies†
Received
25th March 2024
, Accepted 8th May 2024
First published on 22nd May 2024
Abstract
Some hexahydroquinoline candidates were prepared by reacting 2-amino-3-cyano-1-cyclohexylhexahydroquinoline with oxalyl chloride and triethyl orthoformate. The computational chemical approach agreed with the product-testing results. The produced substances were examined in vitro for their antiproliferative activity against liver carcinoma (HepG2), breast adenocarcinoma (MCF7), prostate cancer (PC3), and colon cancer (HCT116) cell lines. The highest potency against the four cell lines was exhibited by hydrazide, thiosemicarbazide, and thiazolidinone derivatives. The best docking score was presented by thiosemicarbazide and thiazolidinone derivatives as they showed the highest binding to the Mcl-1 enzyme with binding energies of −8.97 and −8.90 kcal mol−1, respectively, which were higher than that of the co-crystallized ligand (LC3) with a binding energy of −8.74 kcal mol−1. Besides, the modeling pharmacokinetics disclosed their desirable drug-likeness and oral bioavailability characteristics.
Introduction
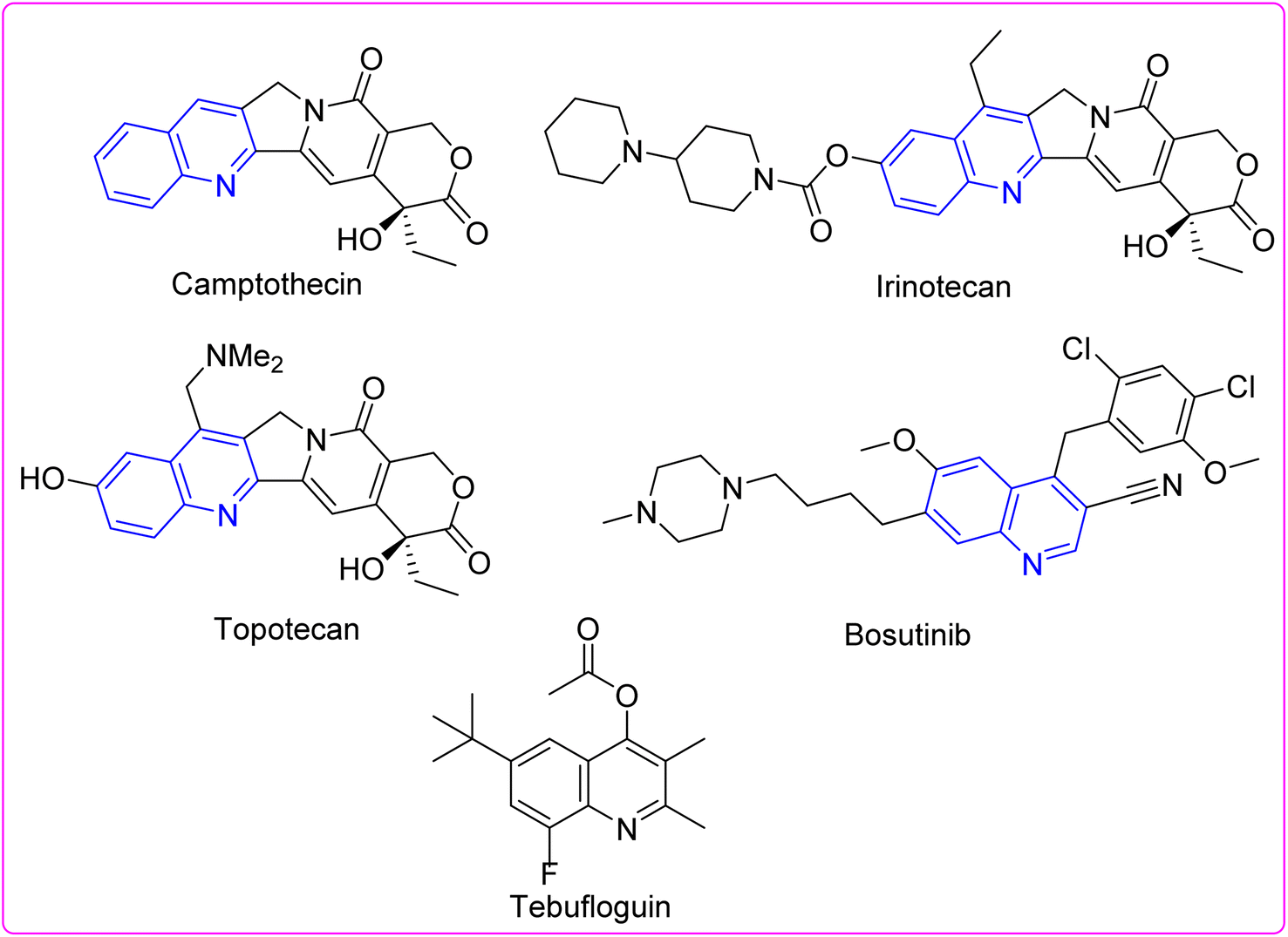
Pyrimidoquinolines have piqued the consequence of researchers due to their potential pharmaceutical applications.1–3 Pyrimidoquinolines and quinolines have demonstrated antimicrobial,4 antitumor,5,6 antiviral,7 and anti-inflammatory properties.8 Diverse quinoline structures are effective inhibitors of major microbial pathogen proteins. Some quinolines with diverse biological effects are depicted in Fig. 1.9–21 Therefore, this work describes the design and preparation of certain pyrimidohexahydroquinolines with potential antiproliferative effects, reinforced by computational chemical approach and molecular docking, which might serve as a drug development target.
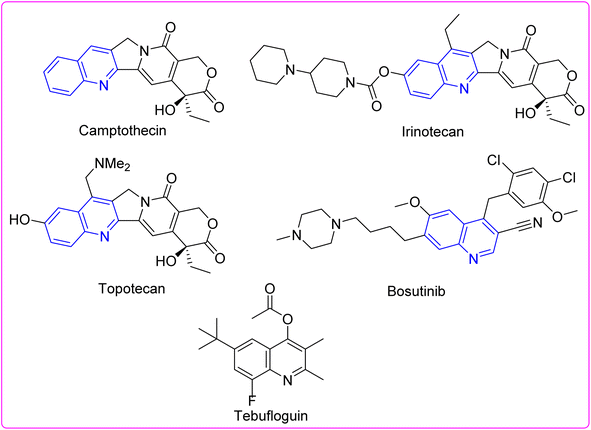 |
| | Fig. 1 Structures of some quinoline-containing drugs. | |
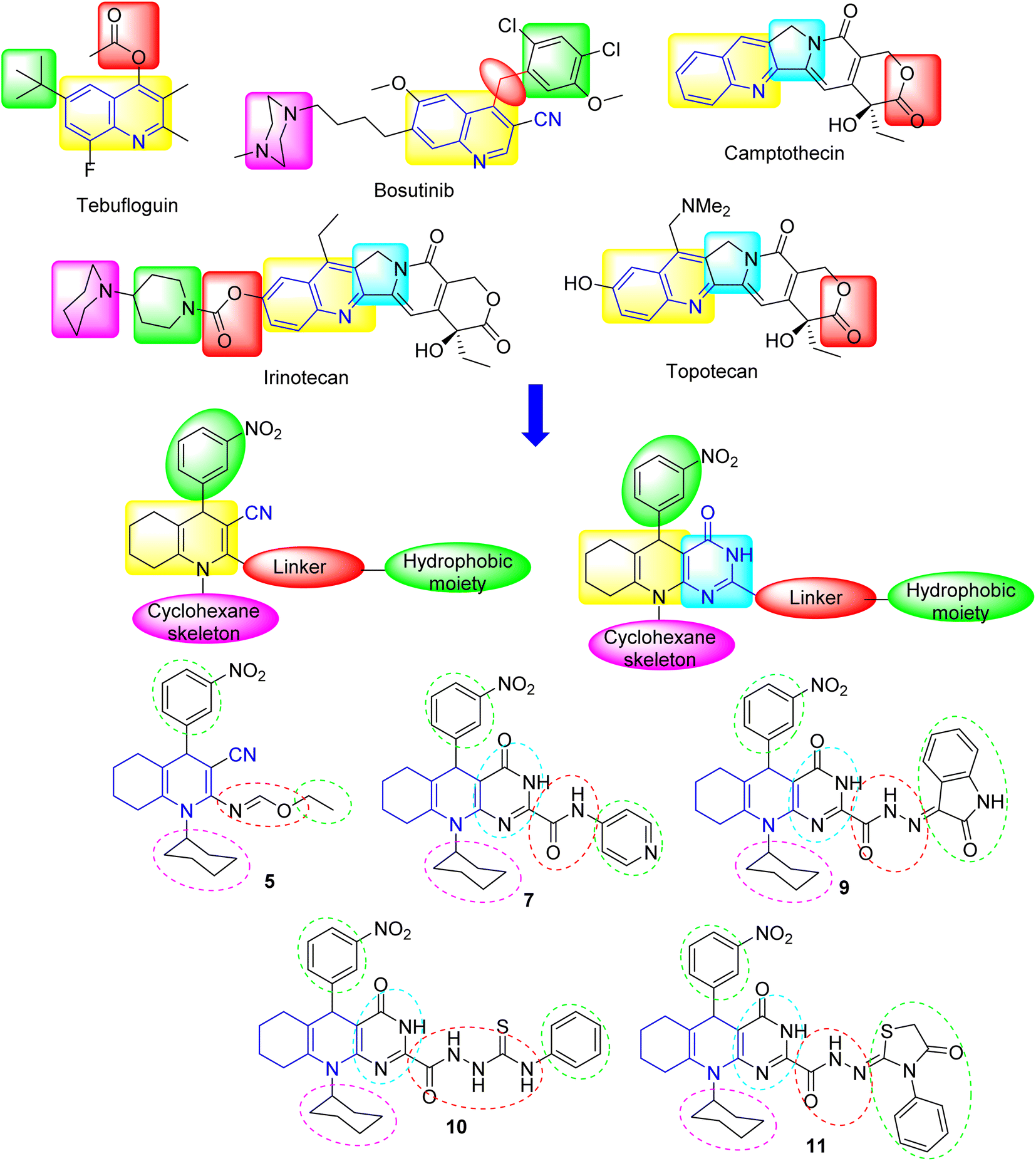
Rationale and design
Quinolines have been recognized as fundamental scaffolds owing to their existence in various pharmacologically potent compounds. In our case, to develop some antiproliferative substances, inspiration was drawn from quinoline's pharmacophoric features, which has exhibited efficacy in treating many diseases (cf. Fig. 2). Initially, using the nitro substituent in the synthesized compounds is attributed to the significant effect of the electron-withdrawing group, which improves the ability to inhibit cancer cell proliferation.22,23 This electron-withdrawing head plays a substantial role in the binding interactions with the receptor subsites via van der Waals interactions and hydrogen bonding.10 To design a new series of some hexahydroquinoline derivatives, fundamental pharmacophoric features of quinoline were retained while integrating heterocyclic and side chain moieties as follows: (i) a planar aromatic core (chromophore) associated with the active sites of enzymes was retained in their design, which displayed pi-hydrophobic or pi–pi interactions with amino acid residues; (ii) the linker was altered by adding heteroatoms like nitrogen (N), oxygen (O), and sulfur (S) to demonstrate more hydrogen bonding through interactions with tumor proteins; (iii) to enhance the antiproliferative potential, cores of pyrimidine, pyridine, indoline, thiazolidine, and phenyl were included in the design. With these features, this work intended to create some new hexahydroquinoline derivatives with enhanced antiproliferative properties.
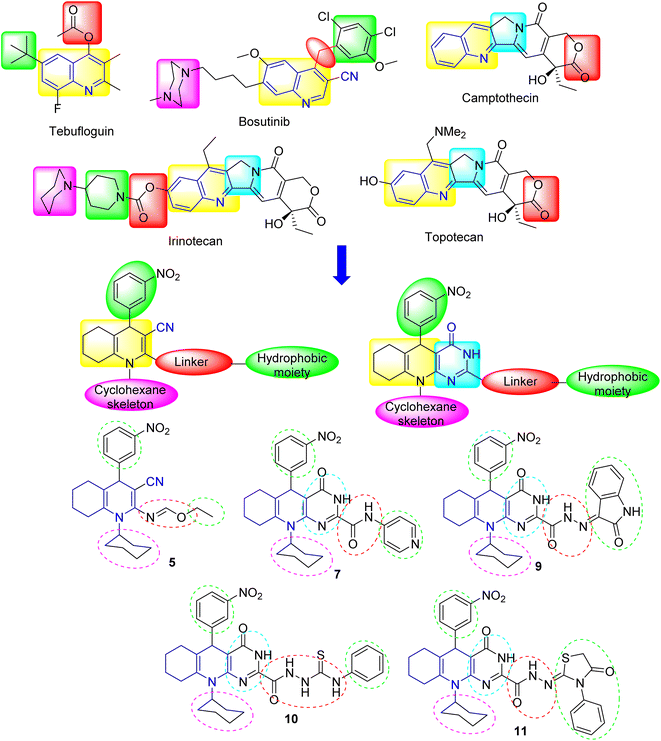 |
| | Fig. 2 Rationale and design of the structures of some anticancer agents (including quinoline core) and the target substances. | |
Results and discussion
Chemistry
A one-pot cyclo-condensation treating cyclohexanone 1 with 2-(3-nitrobenzylidine)-malononitrile 2 and cyclohexylamine 3 in boiling ethanol and triethylamine (Et3N) yielded the hexahydroquinoline-enaminonitrile derivative 4 as yellow crystals (Scheme 1). The absorption bands for primary amino and nitrile functionalities are illustrated in the IR spectra of 4. Also, its 1H NMR spectra showed exchangeable singlet signals for NH2 protons. Furthermore, its mass spectrum displayed the molecular ion peak at m/z 378 (19%). A plausible mechanistic pathway for the formation of β-enaminonitrile 4 could be visualized to occur via Scheme 2. The initial step might be the formation of the carbanion of cyclohexanone 1 using triethylamine as a base, which subsequently attacked the β-carbon of the activated arylidene malononitrile 2 to give the Michael adduct, followed by condensation with cyclohexylamine 3 to eliminate a water molecule, which underwent 1,6-exo-dig cyclization, followed by imine–amine tautomerism, to be more stabilized by conjugation.
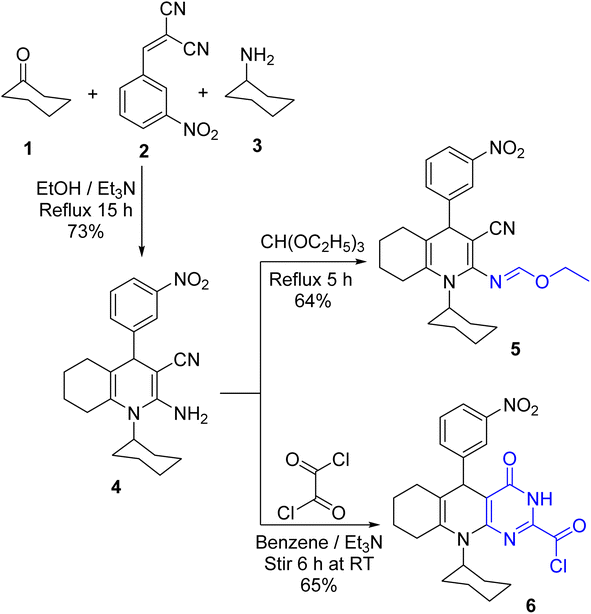 |
| | Scheme 1 Synthesis of β-enaminonitrile 4 and its reactions with triethyl orthoformate and oxalyl chloride. | |
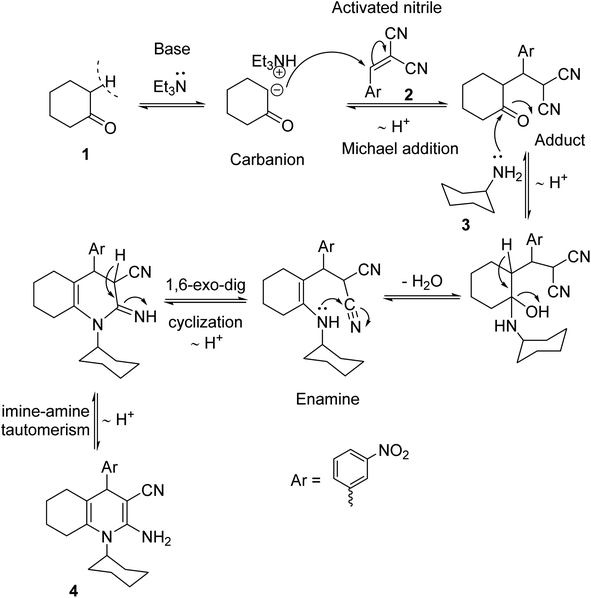 |
| | Scheme 2 A plausible mechanism for the formation of enaminonitrile 4. | |
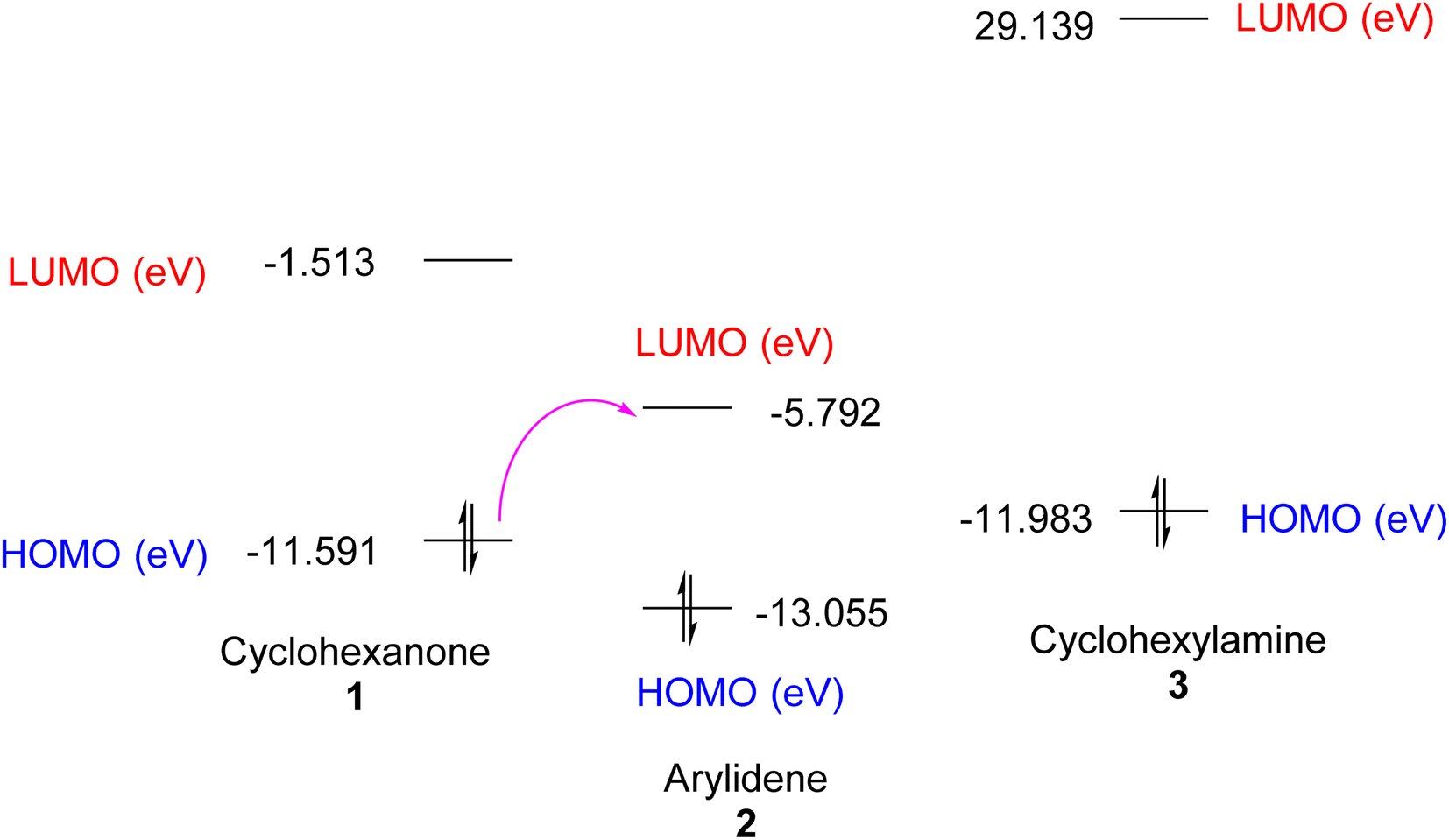
DFT simulation of the one-pot cyclocondensation reaction for the synthesis of the enaminonitrile 4 could support the plausible mechanistic pathway, as depicted in Fig. 3, which outlines the structure optimization of the intermediate that reacted to yield the final product. The computed parameters of the reacting species can be found in Table 1. The smaller energy gap between the HOMO and LUMO was found for arylidene 2, which showed good chemical reactivity.
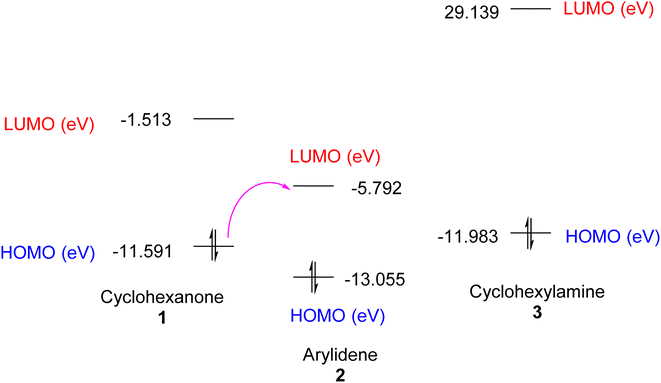 |
| | Fig. 3 EHOMO and ELUMO of reactant materials 1–3. | |
Table 1 Parameters of the reacting components to produce β-enaminonitrile 4
| Reacting components |
Stretch-bend |
Torsion |
Non-1,4 VDW |
1,4 VDW |
Dipole/dipole |
Total energy (kcal mol−1) |
 |
0.059 |
3.839 |
−1.248 |
4.486 |
−1.711 |
6.414 |
 |
0.071 |
−9.690 |
2.602 |
11.225 |
7.561 |
10.648 |
 |
0.095 |
1.841 |
−1.490 |
4.709 |
0.0 |
5.930 |
Also, the initial step of the reaction pathway could be interpreted from the charge density calculations. It was noted that charge density on the α-carbon of cyclohexanone 1 and the β-carbon of arylidene 2 was −0.101 and +0.146, respectively, which enhanced Michael addition under basic conditions (Et3N). In turn, the charge density on the amino group of cyclohexylamine 3 was −0.294, which facilitated the nucleophilic attack on the carbonyl–carbon (with charge density of +0.499) of the adduct obtained from the first step rather than the nitrile–carbon (with a charge density of +0.374).
Boiling the β-enaminonitrile 4 with triethyl orthoformate yielded the condensation product 5. The NH2 absorption was absent in the IR spectrum of 5. Further, its 1H NMR spectrum was devoid of the NH2 singlet signal and displayed quartet and triplet signals for the –CH2CH3 group. In turn, treating 4 with oxalyl chloride in benzene and Et3N at ambient temperature yielded the pyrimidoquinoline derivative 6 (cf. Scheme 1). In the IR spectrum of 6, the NH2 and CN absorptions were absent while it displayed the amide and acid chloride carbonyl absorption bands at 1675 and 1779 cm−1, respectively. Besides, its 1H NMR spectrum demonstrated an exchangeable singlet signal for the NH proton.
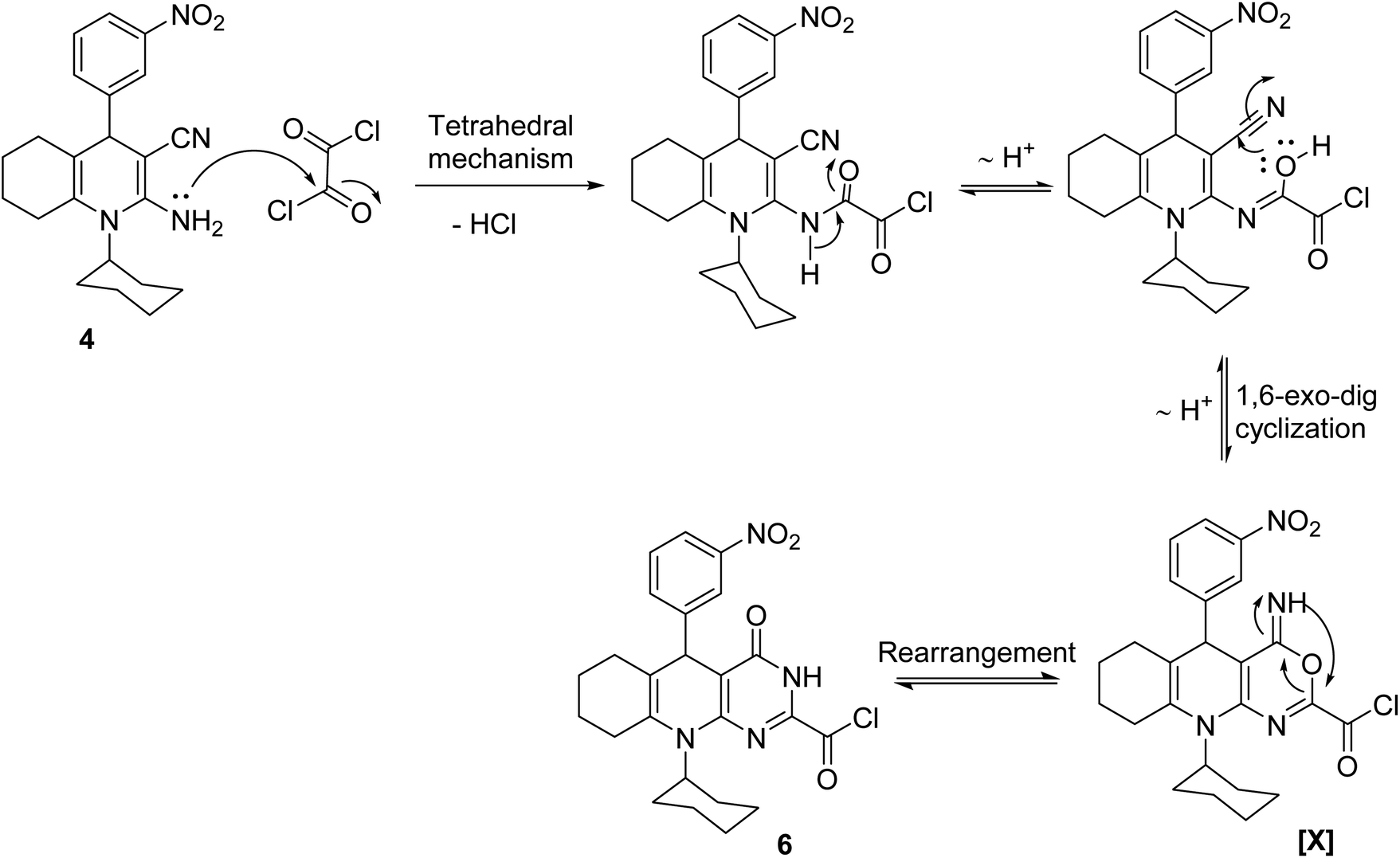
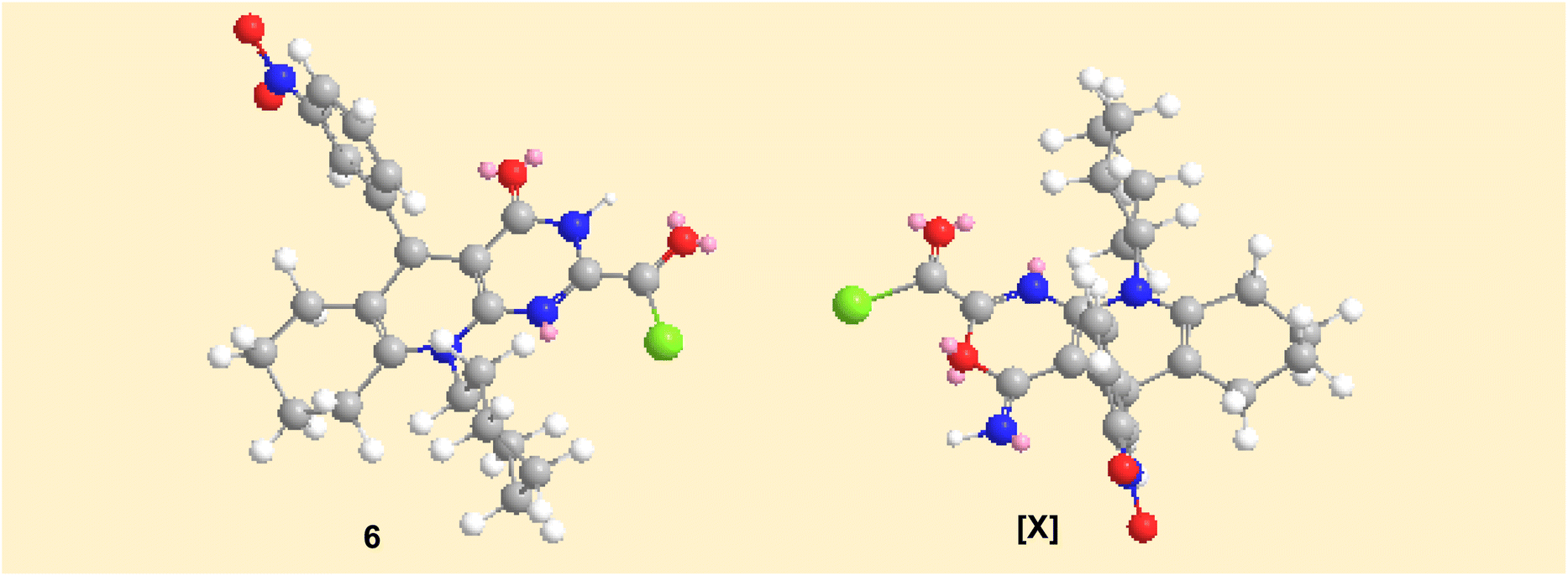
The plausible mechanistic pathway for the condensation of enaminonitrile 4 with oxalyl chloride could be described in Scheme 3. The initial step was the nucleophilic attack of the primary amino group in compound 4 on the carbonyl–carbon of oxalyl chloride to eliminate hydrogen chloride molecule via the tetrahedral mechanism, which was tautomerized into the lactim form to undergo 1,6-exo-dig cyclization, giving the intermediate [X], which subsequently rearranged into pyrimidine skeleton 6. The preferential formation of the structure of compound 6 may be interpreted by considering the stability of products. By constructing space models for pyrimidine 6 and oxazine [X] isomers using ChemBio3D Ultra 2014, it was found that the pyrimidine skeleton 6 is thermodynamically more stable than the oxazine one [X] (cf. Fig. 4) according to MM2 calculations. The total energy of the isomer [X] is 52.8448 kcal mol−1 with dipole/dipole of 10.1166, while that of pyrimidine 6 is 43.3355 kcal mol−1 with dipole/dipole of 4.3151.
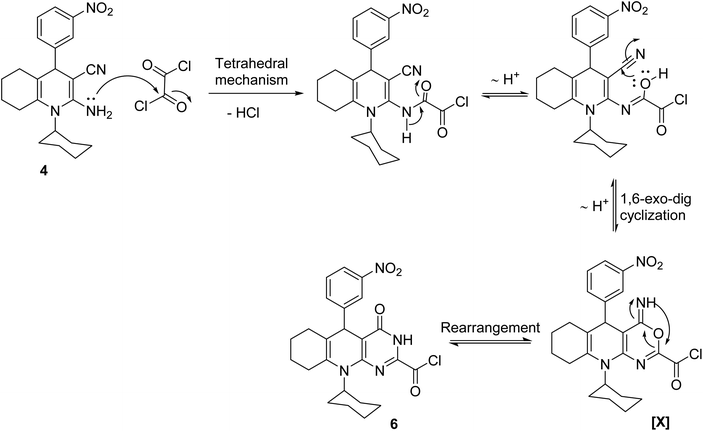 |
| | Scheme 3 A probable mechanism for the condensation of 4 with oxalyl chloride. | |
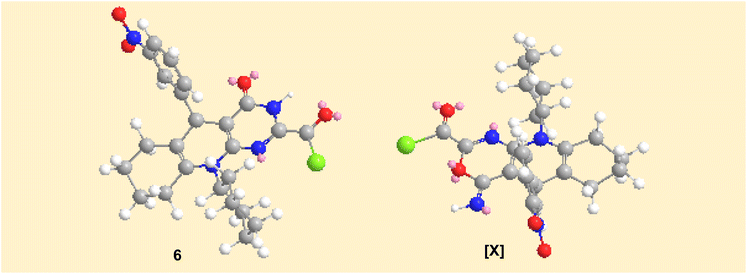 |
| | Fig. 4 Space models of both compounds obtained 6 and the intermediate [X]. | |
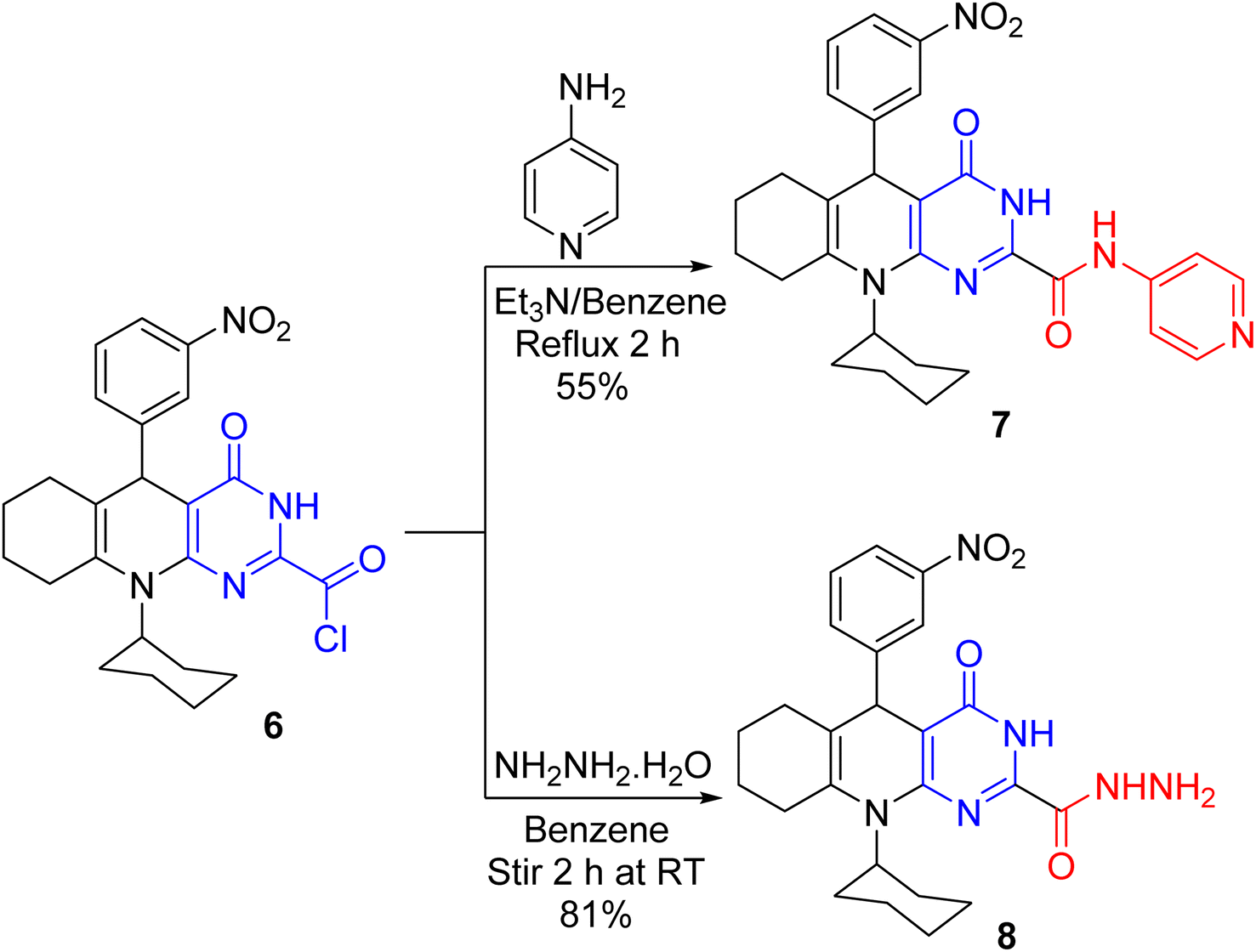
Reacting acid chloride 6 with 4-aminopyridine in benzene and Et3N furnished the carboxamide derivative 7 (Scheme 4). The absorption of acid chloride carbonyl was absent in the IR spectrum of 7 but it unveiled the absorption of amide carbonyl. Further, its 1H NMR spectrum established two exchangeable singlet signals for two NH protons. Analogously, hydrazide 8 was produced on treating 6 with hydrazine hydrate (80%) while stirring in benzene at ambient temperature. The absorption bands for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, NH, and NH2 groups are visible in the IR spectrum of hydrazide 8. Besides, its 1H NMR spectrum indicated three exchangeable singlet signals for the NH of pyrimidine, NH of hydrazide, and NH2 protons. Additionally, its mass spectrum compellingly proves structure 8 as it uncovered the molecular ion peak at m/z 464 with 23% intensity.
O, NH, and NH2 groups are visible in the IR spectrum of hydrazide 8. Besides, its 1H NMR spectrum indicated three exchangeable singlet signals for the NH of pyrimidine, NH of hydrazide, and NH2 protons. Additionally, its mass spectrum compellingly proves structure 8 as it uncovered the molecular ion peak at m/z 464 with 23% intensity.
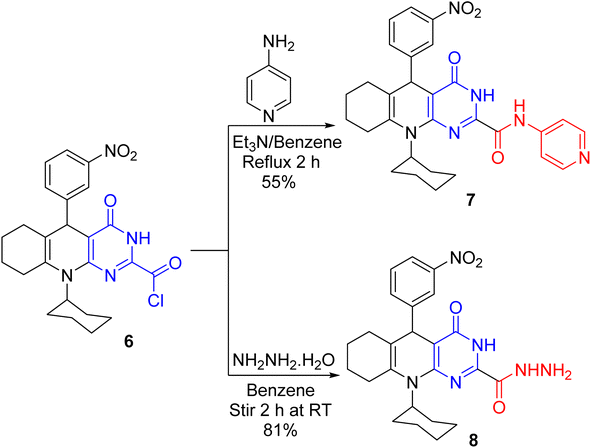 |
| | Scheme 4 Treating acid chloride 6 with 4-aminopyridine and hydrazine hydrate. | |
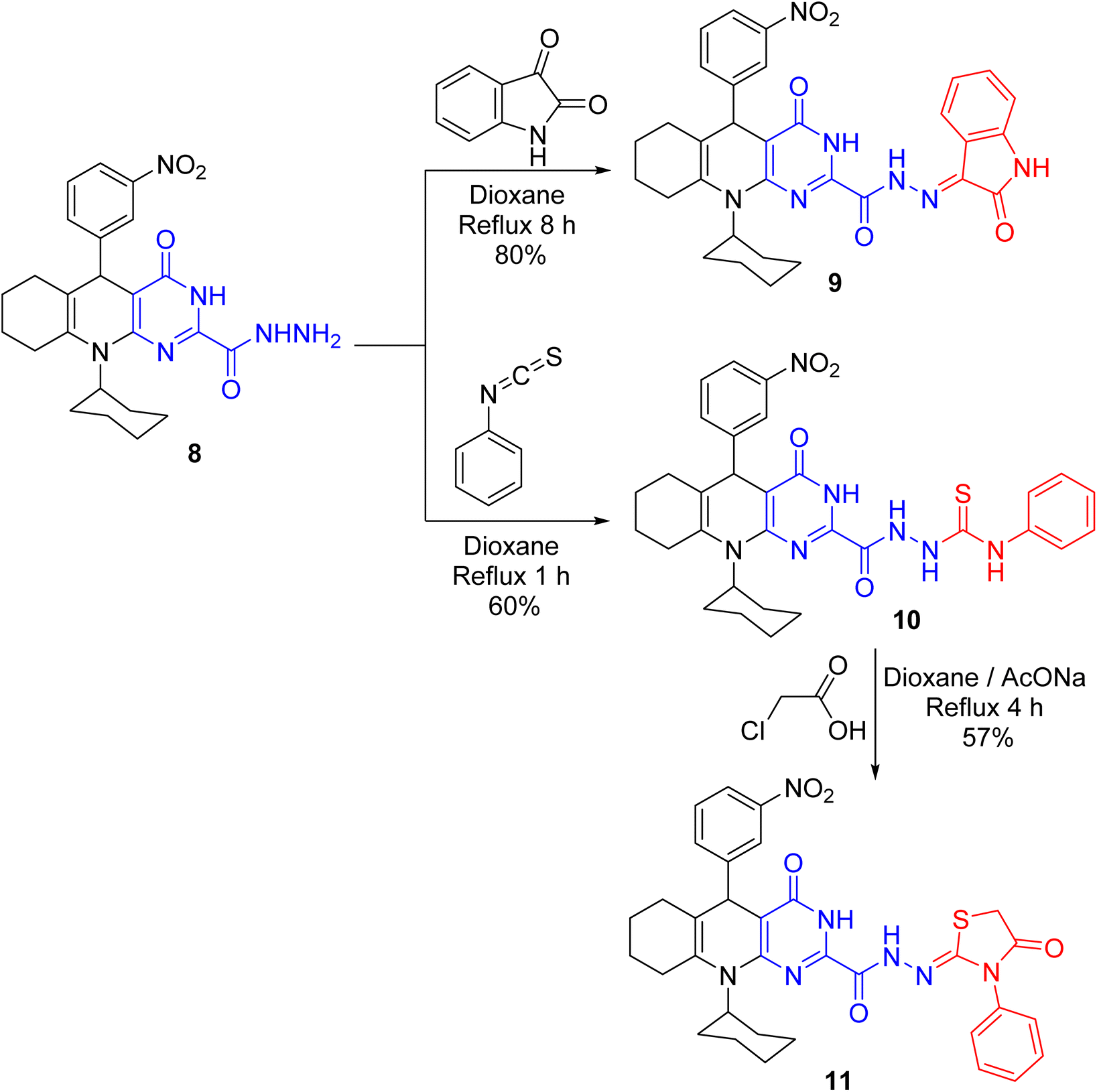
The condensation of hydrazide 8 with indolin-2,3-dione in boiling 1,4-dioxane gave hydrazone derivative 9 (Scheme 5). The IR chart of 9 unveiled the carbonyl absorption of indolinone core but no NH2 absorption. Reacting phenyl isothiocyanate with hydrazide 8 in 1,4-dioxane achieved thiosemicarbazide derivative 10. Quite a lot of pyrimidoquinoline C-nucleosides4 and thiazolidinone C-nucleosides24 demonstrate substantial antimicrobial and anticancer effects. These substances may impede the action of receptor-growth enzymes, like protein tyrosine kinase growth enzyme, in living cells.25 As a result, combining the pyrimidoquinoline backbone with the thiazolidine-C-acyclic nucleoside core in a single molecule might increase the growth-inhibitory effect of these substances. Thiazolidinone derivative 11 was produced on treating 10 with chloroacetic acid in 1,4-dioxane (Scheme 5). Thiazolidinone carbonyl absorption appeared in IR chart 11. Beyond that, a singlet signal for the CH2 of the thiazolidine core was demonstrated in its 1H NMR chart.
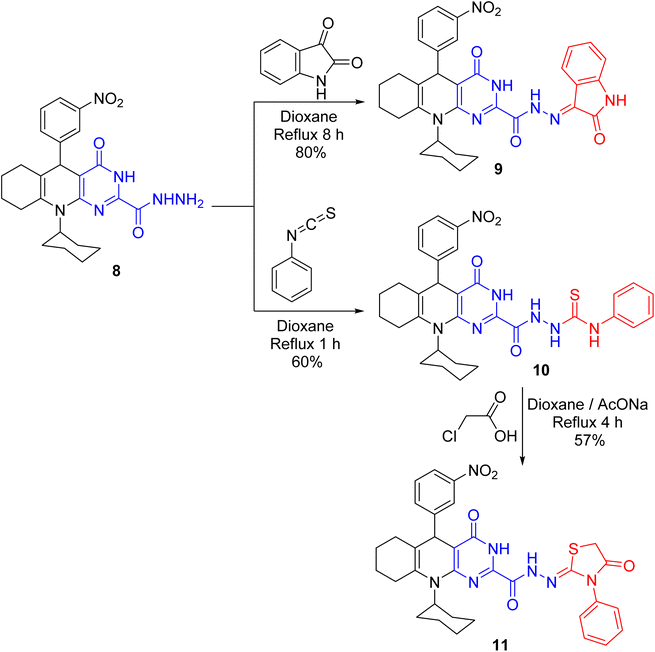 |
| | Scheme 5 Reaction of hydrazide 8 with indolin-2,3-dione and phenyl isothiocyanate. | |
Density functional theory (DFT) study
The optimization of molecular structures for the prepared substances was run with the DFT approach applying the hybrid B3LYP functional with the 3-21G basis set. DFT based on quantum chemical computation describes the structure optimization of the intermediate that reacted, furnishing the final product. The molecular structures of the substances are superior for forming a stable geometry, which is obtained through optimization. The structure's energy was reduced to a stationary point, the shape of tetrahydroquinoline derivatives was gradually optimized, and their energy was continuously reduced until the fluctuations in the molecule's energy were minimized.
The electrophilic-attacking locations are characterized by regions of highest electron density (HOMO), while the LUMO imitates the nucleophile-attacked locations. The HOMO and LUMO play strong roles in the electronic studies by quantum chemical calculations. Together, both are considered as vital tools in the computational investigations of the physicochemical, pharmacokinetic, and toxicological properties.26,27 The HOMO acts as an electron-donor, and the LUMO acts as an electron-acceptor.28 A molecule with a high EHOMO is likely to imply the strong tendency of a molecule to shift electrons.
A molecule with a smaller energy gap showed excellent chemical reactivity and such a molecule is designated as softer; meanwhile, a molecule with a larger energy gap exhibited good stability with high chemical hardness.29,30 Because the energy required to eliminate an electron from the last occupied orbital is small, the low values of the energy gap (ΔE = ELUMO − EHOMO) will lead to good inhibition efficiency.18,21 Utilizing the EHOMO and ELUMO of the synthesized compounds, we can compute such global reactivity descriptors as energy gap, ionization energy, electron affinity, chemical hardness, chemical softness, chemical potential, electrophilicity, and electronegativity.31
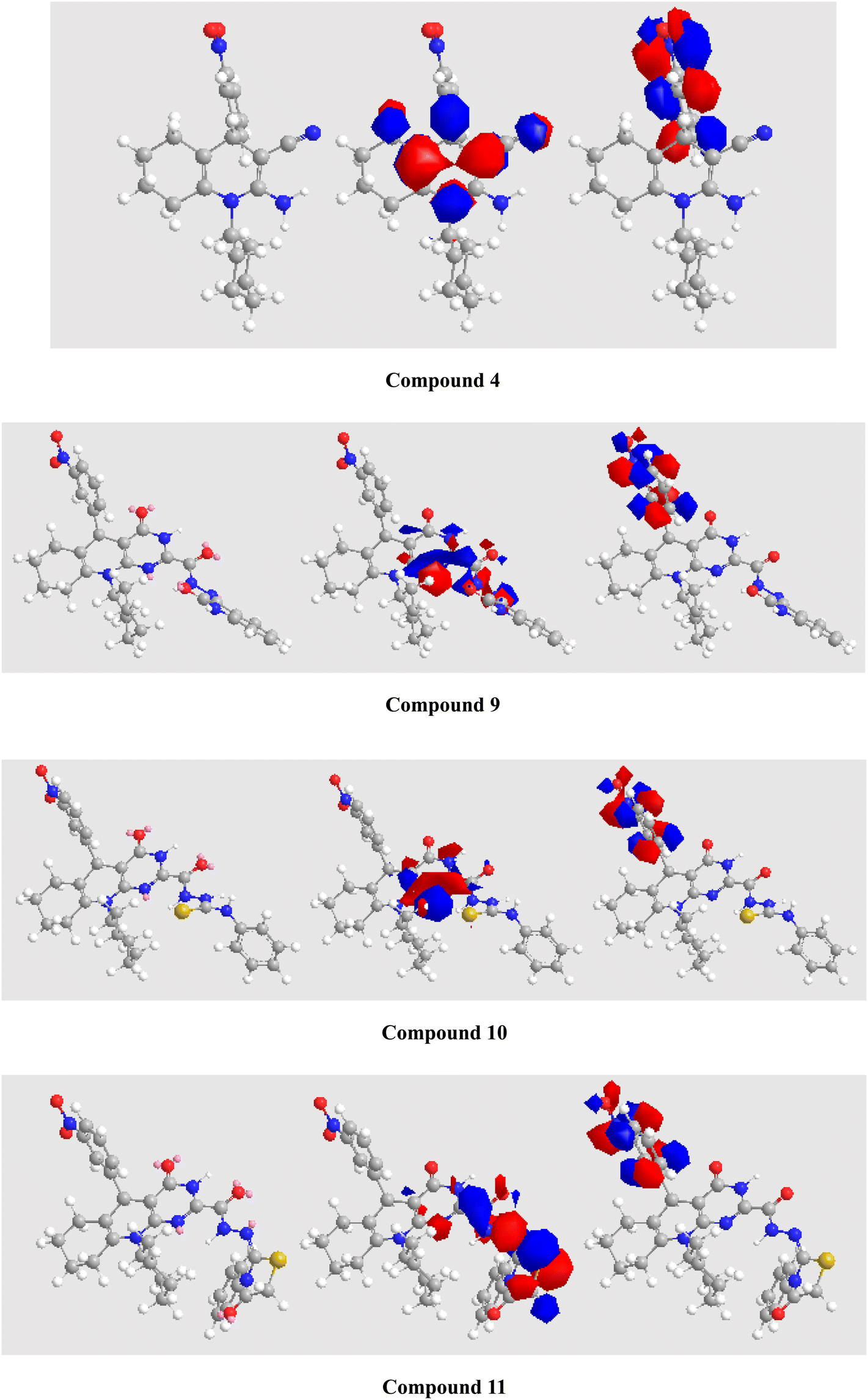
Thus, DFT simulation assisted us in demonstrating the reactions of β-enaminonitrile derivative 4 with various reagents to generate substrates 5–11, and the computed parameters are shown in Table 2. The structures were endorsed by spectral and analytical data. The dipole moments for the tetrahydroquinoline derivatives were established with a decent explanation for the substrates. Fig. 5 depicts the optimized shapes and the HOMOs and LUMOs of compounds 4–11. In compounds 4 and 5, the HOMOs are distributed over the dihydropyridine unit. In compounds 6–10, the HOMOs are spread over the pyrimidinone unit. In compound 11, the HOMO is spread over the thiazolidinone unit. However, in all the compounds, the LUMOs are localized on the nitrophenyl moiety.
Table 2 Energy level distribution and global reactivity indices of frontier orbitalsa
| Compd |
*E |
EHOMO |
ELUMO |
ΔE |
μ |
η |
ς |
μo |
ω |
n |
IP |
EA |
x |
| Dox.: doxorubicin, *E: minimized energy (kcal mol−1), μ: dipole moment, η: global hardness, ς: global softness, μo: chemical potential, ω: global electrophilicity index, n: nucleophilicity index, IP: ionization potential, EA: electron affinity, x: electronegativity. |
| 4 |
30.87 |
−7.669 |
−5.414 |
2.25 |
−2.90 |
1.12 |
0.89 |
−6.53 |
−23.87 |
−0.041 |
7.66 |
5.41 |
6.541 |
| 5 |
63.02 |
−7.705 |
−5.420 |
2.28 |
−1.07 |
1.14 |
0.87 |
−6.56 |
−24.52 |
−0.040 |
7.70 |
5.42 |
6.562 |
| 6 |
44.29 |
−7.391 |
−5.415 |
1.98 |
4.32 |
0.99 |
1.01 |
−6.40 |
−20.27 |
−0.049 |
7.39 |
5.41 |
6.403 |
| 7 |
29.88 |
−7.330 |
−5.415 |
1.92 |
−15.77 |
0.96 |
1.04 |
−6.37 |
−19.47 |
−0.051 |
7.33 |
5.41 |
6.372 |
| 8 |
24.19 |
−7.043 |
−5.413 |
1.63 |
−13.08 |
0.81 |
1.23 |
−6.22 |
−15.66 |
−0.063 |
7.04 |
5.41 |
6.228 |
| 9 |
29.25 |
−6.952 |
−5.411 |
1.54 |
−13.02 |
0.77 |
1.29 |
−6.18 |
−14.70 |
−0.068 |
6.95 |
5.41 |
6.181 |
| 10 |
22.04 |
−7.174 |
−5.417 |
1.76 |
−12.77 |
0.88 |
1.13 |
−6.29 |
−17.40 |
−0.057 |
7.17 |
5.41 |
6.295 |
| 11 |
47.52 |
−6.614 |
−5.408 |
1.21 |
−13.69 |
0.60 |
1.66 |
−6.00 |
−10.80 |
−0.092 |
6.61 |
5.40 |
6.011 |
| Dox. |
65.21 |
−6.69 |
−2.41 |
4.28 |
6.41 |
0.66 |
1.98 |
5.28 |
31.09 |
0.56 |
6.88 |
3.62 |
4.55 |
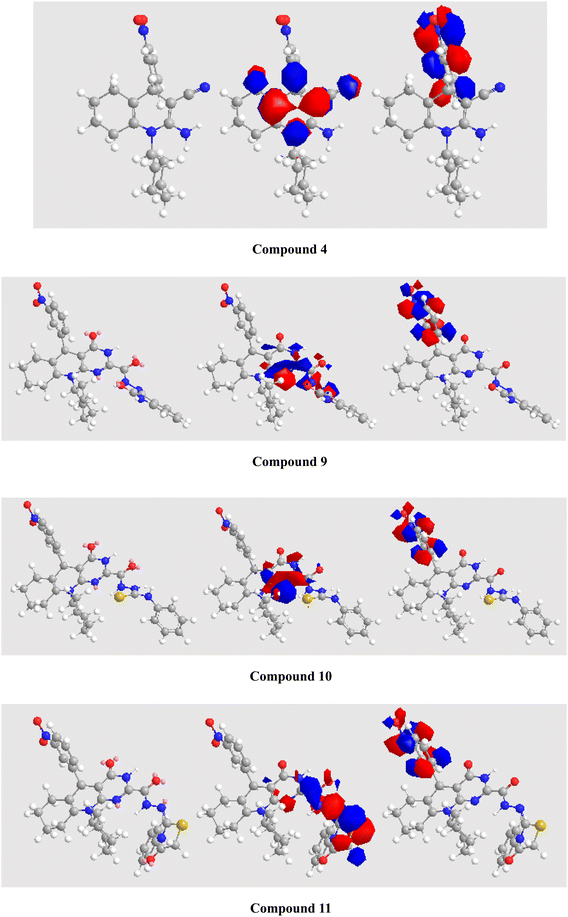 |
| | Fig. 5 Optimized configurations (left), HOMO (middle), and LUMO (right) for substrates 4–11. Atom color index: white H, grey C, blue N, red O, and green Cl (see all substrates in the ESI†). | |
With ΔE as a criterion, three most typical and popular exchange-correlation functionals like PW91 were systematically compared in terms of the typical synthesis in the gas phase upon reactions of enaminonitrile 4 with triethyl orthoformate, oxalyl chloride, 4-aminopyridine, hydrazine hydrate, indolin-2,3-dione, and phenyl isothiocyanate. In addition, for the further screening of proper functionalization, the verification of geometrical and electronic features of modeling anticancer products as well as the adsorption behaviors of typical probing reactants and solvents are suggested. This work provides general implications for how to choose a valid exchange-correlation functional in the computational solvents and catalyst on the reactant surface.
Quantum chemical parameters' calculations of the synthesized substrates are in good agreement with the anticancer efficiency (Table 2). The results indicated that the values of gap energy (ΔE) follow the sequence: heterocyclic derivatives 11 < 9 < doxorubicin. Substrates with small ΔE values are generally denoted as soft substrates, which are more reactive towards radical surface interactions, being capable of donating electrons simply to the hole surface. The scavenging ability toward positive hole, radical, tumor, and removable oxygen depend not only upon the EHOMO values but, in addition, the heteroatoms number, surface area, electron distributions, and lipophilicity must be contemplated. The dipole moment (μ, debye) and softness (σ, eV−1) for the most effective substances carrying hydrophobic groups showed decent correlation with the oxidation inhibition effectiveness. Also, the substrates with higher binding energy are of higher influence due to the strong interaction between these substrates and the receptor's active sites.
Cytotoxic activity
The cytotoxic activity of substrates was examined against four tumor cell lines, namely, liver carcinoma (HepG2), breast adenocarcinoma (MCF7), prostate cancer (PC3), and colon cancer (HCT116) cell lines, employing doxorubicin as a standard agent. The synthesized compounds are water insoluble, and they were dissolved in a mixture of water and DMSO for the biological study, and they are quite stable as they do not have cleavable bonds. The cytotoxic activity was quantified as IC50 values, as described in Table 3, applying the MTT assay.32 The results illustrate that the tested substrates unveiled varying inhibitory potency levels. The highest potency against the four cell lines was presented by the following substances: hydrazide 8 (IC50 = 8.85, 11.44, 17.39, 16.21 μM), thiosemicarbazide derivative 10 (IC50 = 7.20, 5.67, 12.46, 7.34 μM), and thiazolidinone derivative 11 (IC50 = 9.67, 8.32, 10.22, 7.85 μM). Other compounds demonstrated moderate to weak cytotoxic effects.
Table 3 Cytotoxic activity of the tested substances against the cancer cell lines
| Compound |
In vitro cytotoxicity IC50a (μM) ± S.D. |
| HepG2 |
MCF7 |
PC3 |
HCT116 |
| IC50 (μM): 1–10 (very strong). 11–20 (strong). 21–50 (moderate). 51–100 (weak) and above 100 (non-cytotoxic). S.D.: standard deviation of n = 3. |
| Doxorubicin |
4.50 ± 0.3 |
4.17 ± 0.3 |
8.87 ± 0.5 |
5.23 ± 0.3 |
| 4 |
25.34 ± 3.9 |
28.19 ± 4.3 |
24.46 ± 4.2 |
35.10 ± 3.1 |
| 5 |
61.85 ± 3.2 |
73.15 ± 3.9 |
57.11 ± 3.4 |
48.20 ± 2.1 |
| 6 |
62.10 ± 4.5 |
75.24 ± 4.8 |
66.01 ± 4.2 |
58.07 ± 3.8 |
| 7 |
37.30 ± 1.8 |
49.57 ± 1.9 |
56.30 ± 2.9 |
27.99 ± 1.4 |
| 8 |
8.85 ± 3.5 |
11.44 ± 3.1 |
17.39 ± 3.7 |
16.21 ± 2.8 |
| 9 |
33.12 ± 2.1 |
45.44 ± 3.5 |
51.91 ± 3.9 |
37.63 ± 1.9 |
| 10 |
7.20 ± 0.5 |
5.67 ± 0.3 |
12.46 ± 1.0 |
7.34 ± 0.6 |
| 11 |
9.67 ± 2.9 |
8.32 ± 2.8 |
10.22 ± 2.4 |
7.85 ± 2.2 |
Structure–activity relationship (SAR)
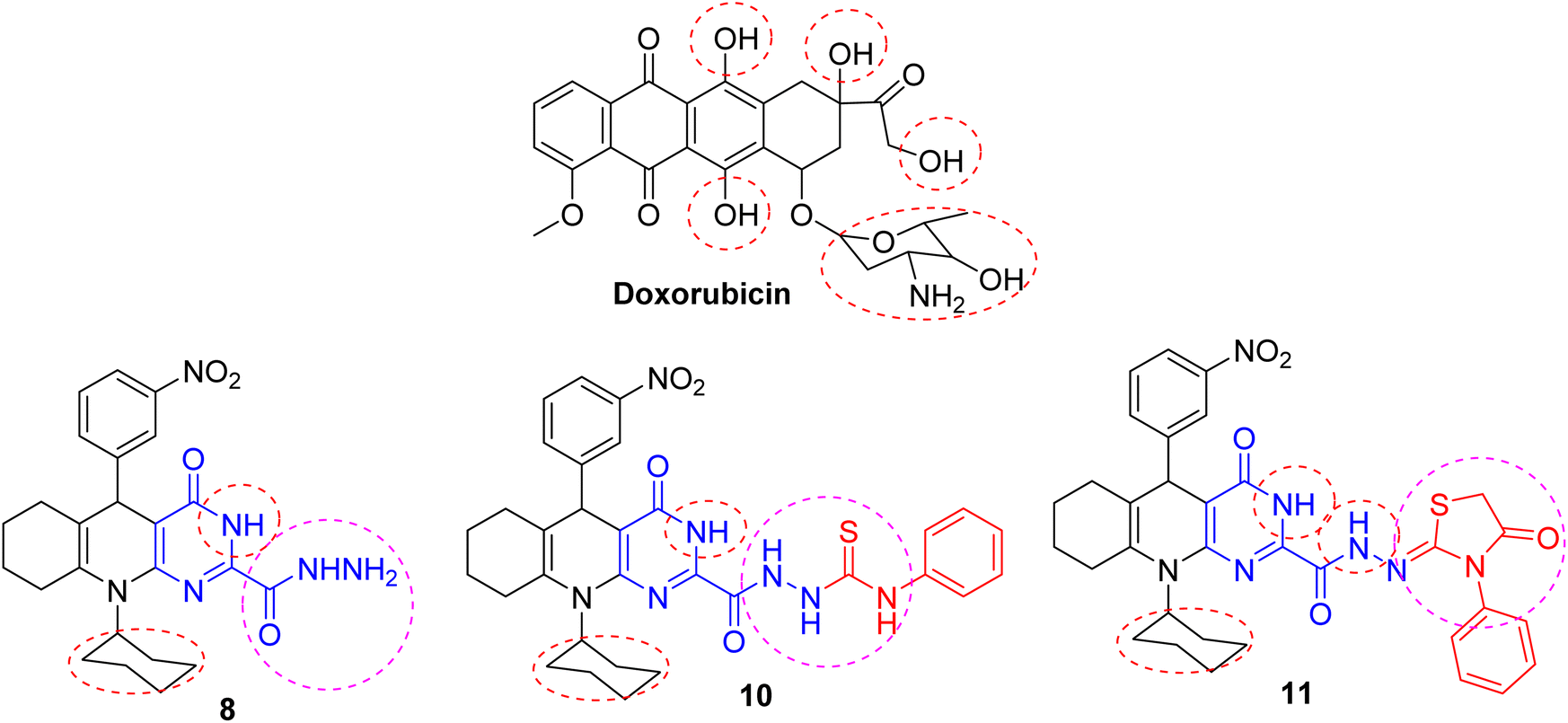
The inhibitory action of the tested substances could be correlated to structure variation and modifications compared with the doxorubicin reference. The structure of the parent substrate 4 is based on the presence of hexahydroquinoline core, which is an important scaffold of many anticancer drugs. The cytotoxic activity of the parent substrate 4 was moderate compared to the reference drug doxorubicin. The produced hexahydroquinolines with a sided electron-withdrawing head play a substantial role in binding interactions with receptor subsites via van der Waals interactions and hydrogen bonding and promote pi-stacking interactions of the loop C aromatic residue with the side chain. The existence of an aromatic core in these compounds increased the hydrophobicity, which is essential for binding with the active site of many cellular enzymes and improves their permeability into the cell membrane, therefore developing the antiproliferative activity.
Besides, the analogs with extended conjugation had higher affinity to generate a face-to-edge aromatic interaction with the receptor. Also, hydrazine and hydrazone moieties were important for bioactivity via azo-reduction into toxic amine derivatives. The hydrazone functionality is known for its antitumor properties.21,28 The introduction of another heterocyclic ring like pyrimidine, pyridine, indole, and thiazolidine rings changed the cytotoxic properties towards the cell lines. These heterocycles show many essential features as anticancer agents. Consequently, it was revealed that substituting the chlorine atom in acid chloride 6 with hydrazinyl moiety (as in compound 8) increased the inhibitory effect against the cell lines owing to hydrogen bonding with the receptor. The introduction of indolin-2-one (as in compound 9) reduced its biological profile. The addition of a thiourea side chain (as in compound 10) boosted the efficacy via extra hydrogen bonding with the receptor amino acids. The cyclization of the side chain to the thiazolidinone ring (as in compound 11) is strongly superior to the cytotoxic potency (cf. Fig. 6).
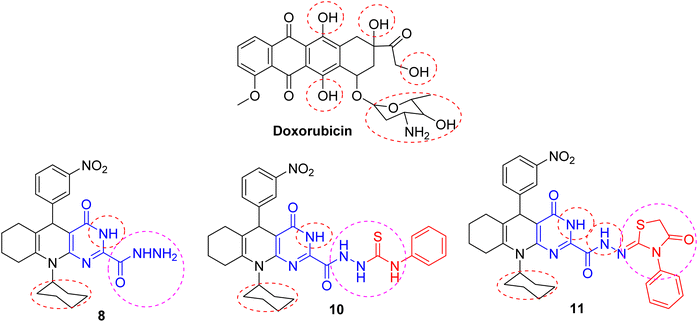 |
| | Fig. 6 SAR of the most potent substances. | |
Molecular docking studies
To grasp the pattern by which the examined compounds are bound to the active place, the most functioning candidates (8, 10, and 11) were subjected to the docking process into the Mcl-1 enzyme downloaded from the protein data bank (PDB code: 3WIX) at a resolution of 1.90 Å. Mcl-1 protein is a crucial regulator of apoptosis; therefore, inhibitors of this protein could serve as promising new anticancer drugs. To design Mcl-1 inhibitors, we performed structure-guided analyses of the corresponding selective inhibitors. Firstly, water molecules were removed from the downloaded complex. The structures of the compounds and reference ligand were drawn using ChemBio3D Ultra 14.0.
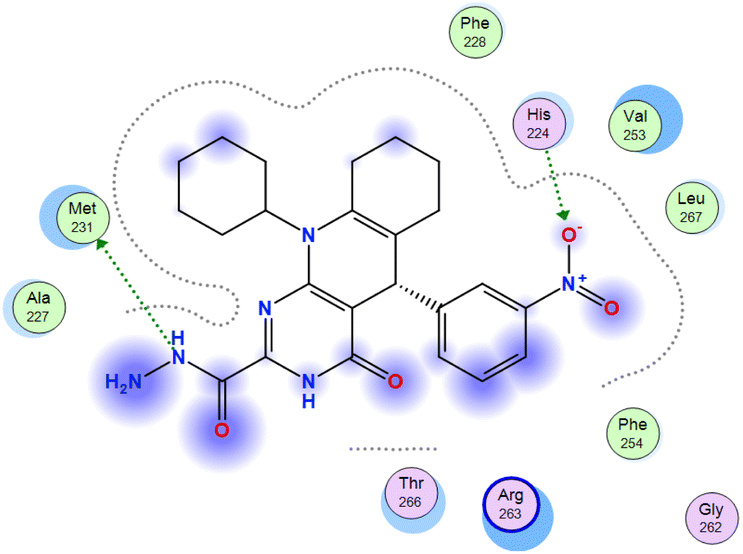
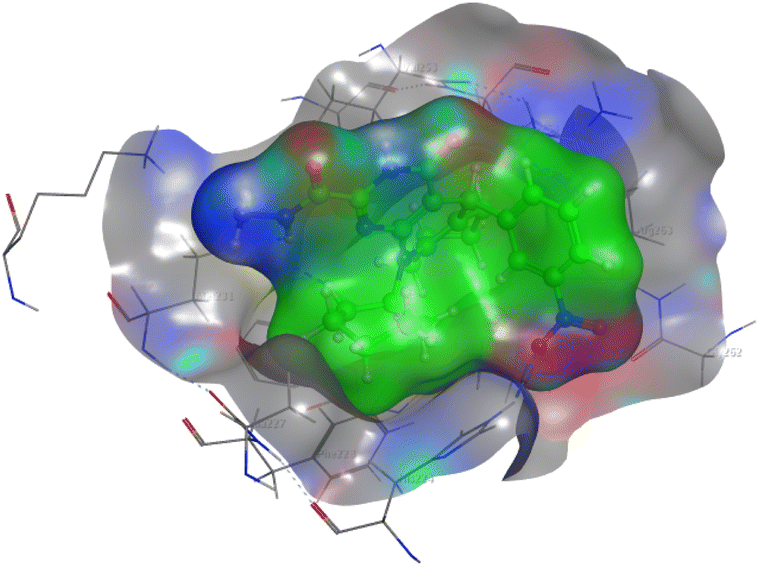
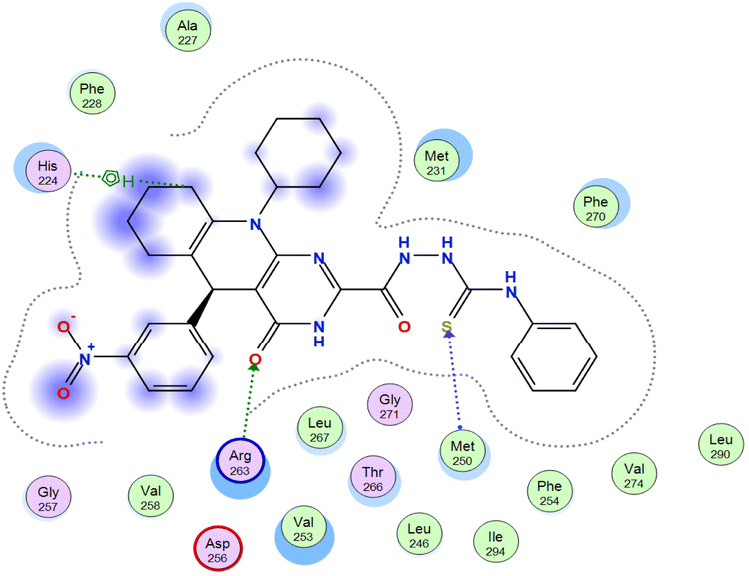
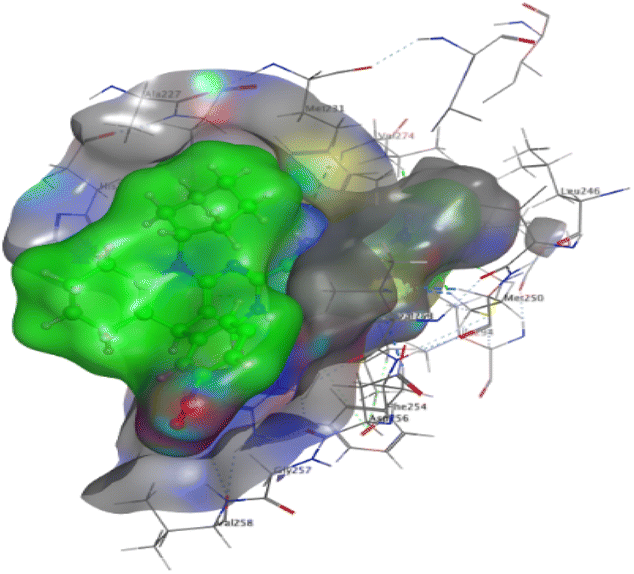
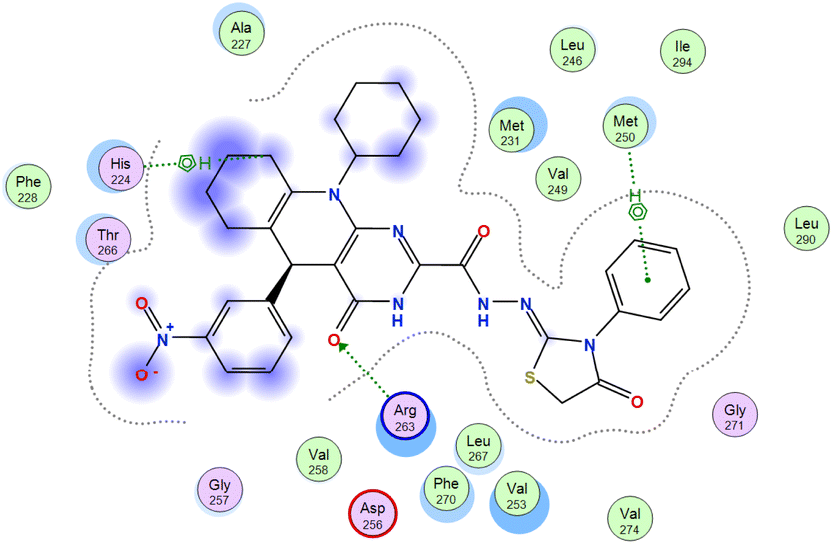
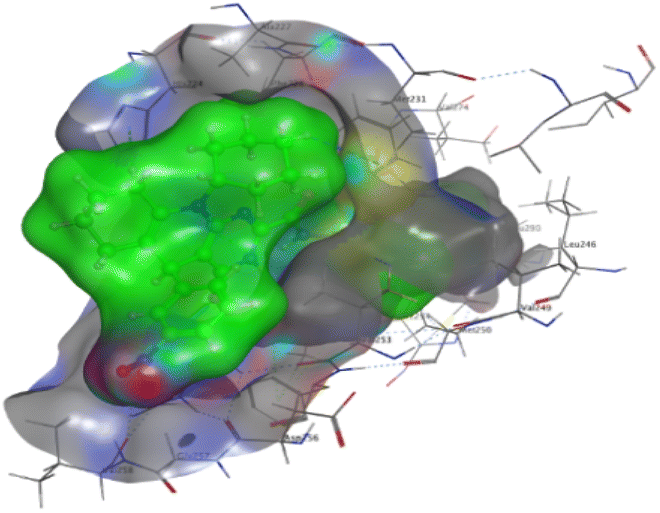
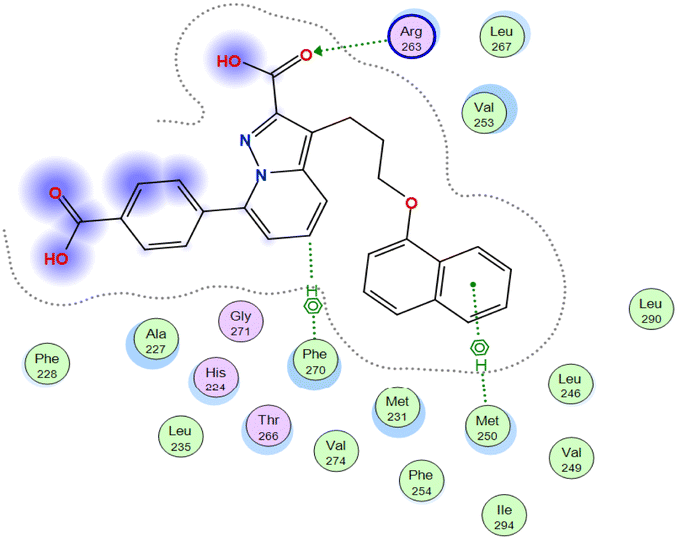
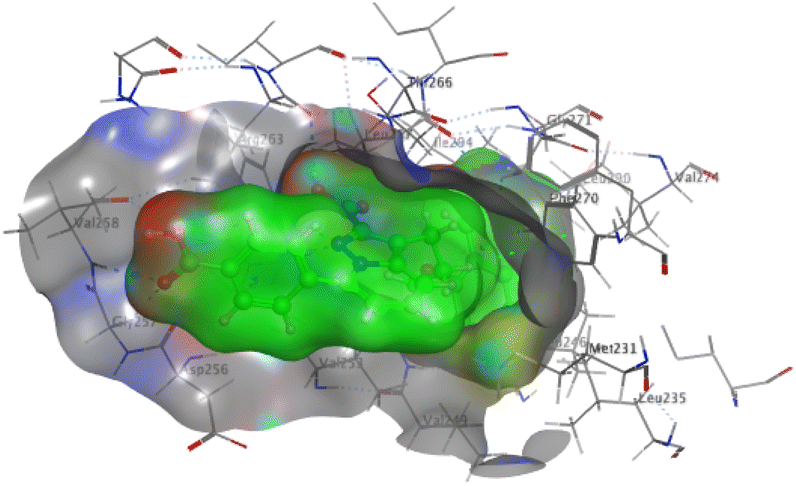
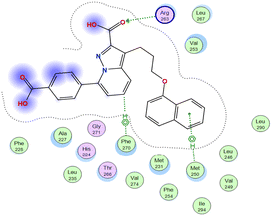
The affinities of the most active synthesized ligands (8, 10, and 11) toward the target protein (Tables 4–6) were compared according to the docking score values calculated employing MOE 2019.0102.33 The best docking score was presented by substances 10 and 11 as they unveiled the highest binding to the Mcl-1 enzyme with a binding energy of −8.97 kcal mol−1 with an RMSD of 1.41 Å and −8.90 kcal mol−1 with an RMSD of 1.99 Å, respectively, which were higher than the co-crystallized ligand (LC3) with a binding energy of −8.74 kcal mol−1 with an RMSD of 1.19 Å. The leading interactions of the co-crystallized ligand with Mcl-1 pockets were almost like that of the most effective substrates (8, 10, and 11), for example, hydrogen bonding with MET 231 and ARG 263 receptors.
Table 4 2D and 3D interactions of compounds (8, 10, and 11) with Mcl-1 binding pockets
| Compound |
2D-interaction |
3D-interaction |
| 8 |
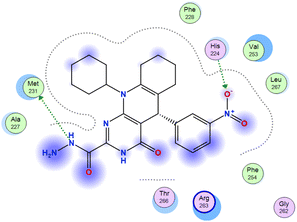 |
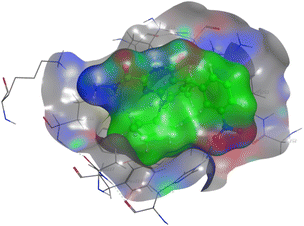 |
| 10 |
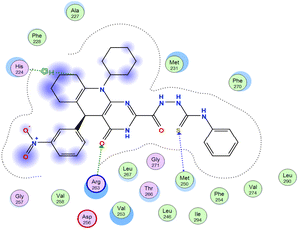 |
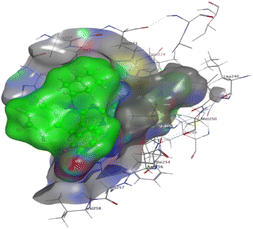 |
| 11 |
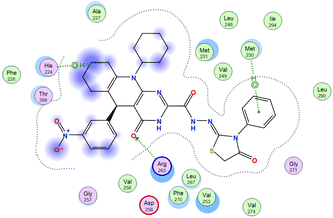 |
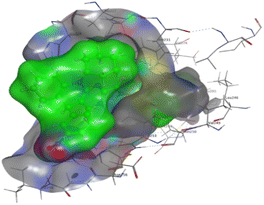 |
| Co-crystalized ligand (LC3) |
 |
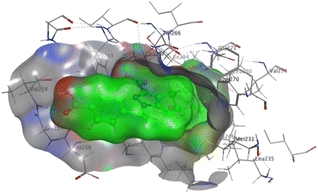 |
 |
Table 5 Ligand interactions of compounds (8, 10, and 11) with the Mcl-1 enzyme
| Compound |
Ligand |
Receptor |
Type of interaction |
Interaction distance (Å) |
E (kcal mol−1) |
| 8 |
N 7 |
SD MET 231 |
H-donor |
3.90 |
−1.8 |
| O 23 |
NE2 HIS 224 |
H-acceptor |
3.08 |
−2.5 |
| 10 |
O 1 |
NE ARG 263 |
H-acceptor |
2.95 |
−5.6 |
| S 25 |
CA MET 250 |
H-acceptor |
4.08 |
−1.1 |
| C 65 |
5-Ring HIS 224 |
H-pi |
4.34 |
−0.6 |
| 11 |
O 1 |
NE ARG 263 |
H-acceptor |
3.02 |
−5.6 |
| O 1 |
NH1 ARG 263 |
H-acceptor |
3.34 |
−0.6 |
| C 68 |
5-Ring HIS 224 |
H-pi |
4.40 |
−0.6 |
| 6-Ring |
CE MET 250 |
pi-H |
3.79 |
−1.0 |
| Co-crystalized ligand (LC3) |
O 1 |
NE ARG 263 |
H-acceptor |
3.02 |
−4.2 |
| C 23 |
6-Ring PHE 270 |
H-pi |
3.50 |
−0.7 |
| 6-Ring |
CE MET 250 |
pi-H |
3.93 |
−0.8 |
Table 6 Docking score and RMSD of compounds (8, 10, and 11) with the Mcl-1 enzyme
| Compound |
Docking score (kcal mol−1) |
RMSD (Å) |
| 8 |
−6.67 |
1.35 |
| 10 |
−8.97 |
1.41 |
| 11 |
−8.90 |
1.99 |
| Co-crystalized (LC3) |
−8.74 |
1.19 |
Modeling pharmacokinetics studies
To select substrates from a huge collection of prepared substances in the early stages of drug discovery, biological actions, and development for an effective drug, ADME profiles including physicochemical properties, lipophilicity, and drug likeness of the synthesized substances have been predicted.34–36 Substance 8, satisfying Lipinski's rule of five, demonstrates a total polar surface area (TPSA) if 149.93 Å and respectable lipophilicity, expressed by the consensus Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Po/w, which is 2.23. As seen in Table 7, it demonstrated a high GI absorption and was computed to acquire a good bioavailability score (0.55).
Po/w, which is 2.23. As seen in Table 7, it demonstrated a high GI absorption and was computed to acquire a good bioavailability score (0.55).
Table 7 ADME characteristics of compounds 8, 10, and 11
| Entry |
Compounds |
| 8 |
10 |
11 |
| Physicochemical properties/lipophilicity/druglikeness |
| Molecular weight (g mol−1) |
464.52 |
599.70 |
639.72 |
| Num. heavy atoms |
34 |
43 |
46 |
| Num. arom. heavy atoms |
12 |
18 |
18 |
| Fraction Csp3 |
0.46 |
0.35 |
0.36 |
| Num. rotatable bonds |
5 |
9 |
7 |
| Num. H-bond acceptor |
6 |
5 |
7 |
| Num. H-bond donors |
3 |
4 |
2 |
| Molar refractivity |
132.43 |
173.94 |
185.36 |
| TPSA (Å2) |
149.93 |
180.06 |
181.88 |
| Consensus LogPo/w |
2.23 |
3.98 |
4.29 |
| Lipinski's rule |
Yes |
No |
No |
| Bioavailability score |
0.55 |
0.17 |
0.17 |
|
| Pharmacokinetics |
| GI absorption |
Low |
Low |
Low |
| BBB permeant |
No |
No |
No |
| P-gp substrate |
Yes |
No |
Yes |
| CYP1A2 inhibitor |
No |
Yes |
Yes |
| CYP2C19 inhibitor |
No |
No |
No |
| CYP2C9 inhibitor |
Yes |
Yes |
Yes |
| CYP2D6 inhibitor |
No |
No |
No |
| CYP3A4 inhibitor |
Yes |
Yes |
Yes |
| LogKp (cm s−1) |
−7.10 |
−6.34 |
−6.18 |
Its skin permeation (LogKP) parameter was −7.10, thus facilitating the accessibility of the bioactive molecules through the skin. Its cyto-chrome P450 isoenzymes (CYP1A2/CYP2C19/CYP2C9/CYP2D6/CYP3A4), playing a major part in the biotransformation of drugs through O-type oxidation reactions, have been likewise anticipated (cf. Table 7). The bioavailability of the produced substances was also estimated based on their pink area on the radar chart (cf. Fig. S7–S9, cf. the ESI†). The compounds were completely included in the pink area and justified their good predicted oral bioavailability, especially compounds 8, 10, and 11.
Conclusion
Some pyrimidohexahydroquinolines were achieved by reacting the hexahydroquinoline-enaminonitrile scaffold with some carbon-centered electrophiles. The computational chemical results were in good agreement with the experimental products. The substates were screened for their in vitro antitumor activity against liver carcinoma (HepG2), breast adenocarcinoma (MCF7), prostate cancer (PC3), and colon cancer (HCT116), and unveiled that hydrazide, thiosemicarbazide, and thiazolidinone derivatives displayed strong effectiveness. The molecular docking study upheld the biological effects. In silico findings with physicochemical characteristics, lipophilicity, and drug-likeness of the potent substances have been estimated.
Experimental
General
Solvents and chemicals were purified and dried by standard techniques. Melting points (uncorrected) were assessed on a MEL-TEMP II electric melting point apparatus. The IR spectra (ν, cm−1) were recorded at the Faculty of Science, Ain Shams University, applying KBr disks on a FTIR Thermo Electron Nicolet iS10 infrared spectrometer. 1H NMR spectra (δ, ppm) were run at 300 MHz on a GEMINI NMR spectrometer employing tetramethyl silane (TMS) as an internal standard in deuterated DMSO. Mass spectra (70 eV, m/z, %) were documented at the Faculty of Pharmacy, Al-Azhar University, Egypt. Elemental analyses were done at the Faculty of Science, Ain Shams University, applying a PerkinElmer 2400 CHN elemental analyzer. The reactions were monitored by TLC on silica gel-precoated aluminum sheets 60 F254-Merck, and the spots were perceived by exposure to a UV lamp.
2-Amino-1-cyclohexyl-4-(3-nitrophenyl)-1,4,5,6,7,8-hexahydroquinoline-3-carbonitrile (4). A mixture of cyclohexanone 1 (0.98 g, 0.01 mol), 2-(3-nitrobenzylidene)malononitrile (2) (1.99 g, 0.01 mol), and cyclohexylamine 3 (0.99 g, 0.01 mol) in ethanol (30 mL) including triethylamine (0.1 mL) was refluxed for 15 h and left overnight at ambient temperature. The obtained precipitate was filtered off, washed with cold water and recrystallized from ethanol to furnish yellow crystals, mp. 95–97 °C, yield: 2.76 g (73%). IR (ν, cm−1): 2220 (CN), 3346, 3310 (NH2). 1H NMR (DMSO, δ, ppm): 1.42–1.44 (m, 2H, CH2, cyclohexyl), 1.45–1.48 (m, 4H, 2CH2, cyclohexyl), 1.64–1.67 (m, 4H, 2CH2, tetrahydroquinoline), 1.75–1.78 (m, 4H, 2CH2, cyclohexyl), 1.96–1.99 (m, 4H, 2CH2, tetrahydroquinoline), 2.54–2.58 (m, 1H, N–CH, cyclohexyl), 5.47 (s, 1H, CH tetrahydroquinoline), 6.15 (br.s, 2H, NH2, exchangeable), 6.49–6.82 (m, 4H, Ar–H). EIMS (m/z, %): 378 (M+˙, 19). Anal. calcd for C22H26N4O2 (378.48): C, 69.82; H, 6.92; N, 14.80; found: C, 69.81; H, 6.94; N, 14.82%.
Ethyl-N-(3-cyano-1-cyclohexyl-4-(3-nitrophenyl)-1,4,5,6,7,8-hexahydroquinolin-2-yl)formimidate (5). A suspension of compound 4 (0.76 g, 0.002 mol) in triethyl orthoformate (5 mL) was refluxed for 5 h; then, the reaction mixture was poured onto ice/cold water. The precipitate was filtered off, dried, and crystallized from ethanol to achieve yellow crystals, mp. 80–82 °C. yield: 0.55 g (64%). IR (ν, cm−1): 2214 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1640 (CN). 1H NMR (DMSO, δ, ppm): 1.20 (t, 3H, C
N), 1640 (CN). 1H NMR (DMSO, δ, ppm): 1.20 (t, 3H, C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3CH2, J = 6.5 Hz), 1.41–1.44 (m, 2H, CH2 cyclohexyl), 1.45–1.48 (m, 4H, 2CH2 cyclohexyl), 1.65 (m, 4H, 2CH2, tetrahydroquinoline), 1.76 (m, 4H, 2CH2 cyclohexyl), 1.98 (m, 4H, 2CH2, tetrahydroquinoline), 2.55 (m, 1H, CH cyclohexyl), 3.57 (q, 2H, CH3C2, J = 6.5 Hz), 5.42 (s, 1H, CH tetrahydroquinoline), 6.80–7.30 (m, 4H, Ar–H), 8.50 (s, 1H, CH = N). EIMS (m/z, %): 434 (M+˙, 15). Anal. calcd for C25H30N4O3 (434.54): C, 69.10; H, 6.96; N, 12.89; found: C, 69.01; H, 6.90; N, 12.91%.
3CH2, J = 6.5 Hz), 1.41–1.44 (m, 2H, CH2 cyclohexyl), 1.45–1.48 (m, 4H, 2CH2 cyclohexyl), 1.65 (m, 4H, 2CH2, tetrahydroquinoline), 1.76 (m, 4H, 2CH2 cyclohexyl), 1.98 (m, 4H, 2CH2, tetrahydroquinoline), 2.55 (m, 1H, CH cyclohexyl), 3.57 (q, 2H, CH3C2, J = 6.5 Hz), 5.42 (s, 1H, CH tetrahydroquinoline), 6.80–7.30 (m, 4H, Ar–H), 8.50 (s, 1H, CH = N). EIMS (m/z, %): 434 (M+˙, 15). Anal. calcd for C25H30N4O3 (434.54): C, 69.10; H, 6.96; N, 12.89; found: C, 69.01; H, 6.90; N, 12.91%.
10-Cyclohexyl-5-(3-nitrophenyl)-4-oxo-3,4,5,6,7,8,9,10-octahydropyrimido[4,5-b]quinoline-2-carbonyl chloride (6). To a solution of 4 (3.78 g, 0.01 mol) in benzene (20 mL) including triethylamine (0.1 mL), oxalyl chloride (1.89 g, 0.015 mol) was added dropwise at ambient temperature with stirring. The reaction mixture was stirred for 6 h. The precipitate obtained was collected to give a brown solid, yield: 3.04 g (65%). mp 151–153 °C. IR (ν, cm−1): 3352 (NH), 1779 (CO acid chloride), 1675 (CO amide). 1H NMR (DMSO, δ, ppm): 1.35–1.38 (m, 4H, 2CH2, cyclohexyl), 1.40–1.43 (m, 2H, CH2, cyclohexyl), 1.51–1.53 (m, 4H, 2CH2, cyclohexyl), 1.62–1.65 (m, 4H, 2CH2, tetrahydroquinoline), 1.94–1.98 (m, 4H, 2CH2, tetrahydroquinoline), 2.59–2.62 (m, 1H, N–CH, cyclohexyl), 5.43 (s, 1H, CH tetrahydroquinoline), 6.54–6.99 (m, 4H, Ar–H), 10.03 (br.s, 1H, NH, exchangeable). EIMS (m/z, %): 470 (M2+, 10), 468 (M+˙, 30). Anal. calcd for C24H25ClN4O4 (468.94): C, 61.47; H, 5.37; N, 11.95; found: C, 61.39; H, 5.32; N, 11.94%.
10-Cyclohexyl-5-(3-nitrophenyl)-4-oxo-N-(pyridin-4-yl)-3,4,5,6,7,8,9,10-octahydropyrimido[4,5-b]quinoline-2-carboxamide (7). A solution of 6 (4.69 g, 0.01 mol) and 4-aminopyridine (0.94 g, 0.01 mol) in benzene (20 mL) containing triethylamine (0.1 mL) was refluxed for 2 h. The reaction mixture was concentrated and then cooled. The solid obtained was filtered off and crystallized from ethanol to furnish yellow crystals, mp. 100–102 °C, yield: 2.89 g (55%). IR (ν, cm−1): 3355 (NH), 1665, 1643 (CO). 1H NMR (DMSO, δ, ppm): 1.37–1.40 (m, 4H, 2CH2, cyclohexyl), 1.43–1.46 (m, 2H, CH2, cyclohexyl), 1.52–1.55 (m, 4H, 2CH2, cyclohexyl), 1.65–1.67 (m, 4H, 2CH2, tetrahydroquinoline), 1.95–1.99 (m, 4H, 2CH2, tetrahydroquinoline), 2.59–2.62 (m, 1H, N–CH, cyclohexyl), 5.40 (s, 1H, CH tetrahydroquinoline), 6.40–6.64 (m, 8H, Ar–H), 8.89 (br.s, 1H, NH, exchangeable), 9.79 (br.s, 1H, NH, exchangeable). EIMS (m/z, %): 526 (M+˙, 21). Anal. calcd for C29H30N6O4 (526.60): C, 66.15; H, 5.74; N, 15.96; found: C, 66.07; H, 5.69; N, 15.98%.
10-Cyclohexyl-5-(3-nitrophenyl)-4-oxo-3,4,5,6,7,8,9,10-octahydropyrimido[4,5-b]quinoline-2-carbohydrazide (8). To a solution of 6 (4.69 g, 0.01 mol) in benzene (20 mL), hydrazine hydrate (0.55 mL, 0.011 mmol, 98%) was added dropwise at ambient temperature with stirring. The reaction mixture was stirred at room temperature for 2 h. The solid obtained was collected and recrystallized from ethanol to give pale-yellow crystals, mp. 130–132 °C, yield: 3.75 g (81%). IR (ν, cm−1): 1668, 1655 (CO), 3354, 3325, 3245 (NH, NH2). 1H NMR (DMSO, δ, ppm): 1.42–1.45 (m, 4H, 2CH2, cyclohexyl), 1.46–1.48 (m, 2H, CH2, cyclohexyl), 1.52–1.55 (m, 4H, 2CH2, cyclohexyl), 1.65–1.69 (m, 4H, 2CH2, tetrahydroquinoline), 2.15–2.22 (m, 4H, 2CH2, tetrahydroquinoline), 2.58–2.61 (m, 1H, N–CH, cyclohexyl), 5.44 (s, 1H, CH tetrahydroquinoline), 5.70 (br.s, 2H, NH2, exchangeable), 6.57–7.06 (m, 4H, Ar–H), 8.58 (br.s, 1H, NH, exchangeable), 8.85 (br.s, 1H, NH, exchangeable). EIMS (m/z, %): 464 (M+˙, 23). Anal. calcd for C24H28N6O4 (464.53): C, 62.06; H, 6.08; N, 18.09; found: C, 61.97; H, 6.00; N, 18.10%.
10-Cyclohexyl-5-(3-nitrophenyl)-4-oxo-N′-(2-oxoindolin-3-ylidene)-3,4,5,6,7,8,9,10-octahydropyrimido[4,5-b]quinoline-2-carbohydrazide (9). A mixture of hydrazide 8 (4.64 g, 0.01 mol) and isatin (1.47 g, 0.01 mol) in dioxane (20 mL) was refluxed for 8 h. The solid obtained was separated by filtration and crystallized from dioxane to give red crystals, mp 179–181 °C, yield: 4.74 g (80%). IR (ν, cm−1): 1665 (CO pyrimidinone), 1704 (CO indolinone), 3350, 3240 (NH). 1H NMR (DMSO, δ, ppm): 1.36–1.43 (m, 4H, 2CH2, cyclohexyl), 1.42–1.46 (m, 2H, CH2, cyclohexyl), 1.52–1.56 (m, 4H, 2CH2, cyclohexyl), 1.63–1.66 (m, 4H, 2CH2, tetrahydroquinoline), 1.99–2.33 (m, 4H, 2CH2, tetrahydroquinoline), 2.58–2.87 (m, 1H, N–CH, cyclohexyl), 5.45 (s, 1H, CH tetrahydroquinoline), 6.45 (br.s, 1H, NH indolinone, exchangeable), 6.55–7.58 (m, 8H, Ar–H), 8.59 (br.s, 1H, NH, exchangeable), 9.01 (br.s, 1H, NH, exchangeable). EIMS (m/z, %): 593 (M+˙, 25). Anal. calcd for C32H31N7O5 (593.64): C, 64.74; H, 5.26; N, 16.52; found: C, 64.68; H, 5.20; N, 15.50%.
2-(10-Cyclohexyl-5-(3-nitrophenyl)-4-oxo-3,4,5,6,7,8,9,10-octahydropyrimido[4,5-b]quinoline-2-carbonyl)-N-phenylhydrazine-1-carbothioamide (10). A mixture of hydrazide 8 (4.64 g, 0.01 mol) and phenyl isothiocyanate (1.35 g, 0.01 mol) in dioxane (20 mL) was refluxed for 1 h. The reaction mixture was evaporated under reduced pressure and the residue was triturated with ethanol. The solid obtained was collected by filtration, washed with ethanol, and recrystallized from ethanol to achieve brown crystals, mp. 140–142 °C, yield: 3.59 g (60%). IR (ν, cm−1): 1258 (CS), 1669, 1649 (CO), 3351, 3240 (NH), 3182 (NH). 1H NMR (DMSO, δ, ppm): 1.43–1.46 (m, 4H, 2CH2, cyclohexyl), 1.47–1.50 (m, 2H, CH2, cyclohexyl), 1.53–1.57 (m, 4H, 2CH2, cyclohexyl), 1.68–1.73 (m, 4H, 2CH2, tetrahydroquinoline), 2.02–2.05 (m, 4H, 2CH2, tetrahydroquinoline), 2.61–2.64 (m, 1H, N–CH, cyclohexyl), 5.44 (s, 1H, CH tetrahydroquinoline), 6.59–7.49 (m, 9H, Ar–H), 8.97 (br.s, 1H, NH, exchangeable), 9.33 (br.s, 1H, NH, exchangeable), 9.95 (br.s, 1H, NH, exchangeable), 10.10 (br.s, 1H, NH, exchangeable). EIMS (m/z, %): 599 (M+˙, 21). Anal. calcd for C31H33N7O4S (599.71): C, 62.09; H, 5.55; N, 16.35; found: C, 61.97; H, 5.50; N, 16.37%.
10-Cyclohexyl-5-(3-nitrophenyl)-4-oxo-N′-(4-oxo-3-phenylthiazolidin-2-ylidene)-3,4,5,6,7,8,9,10-octahydropyrimido[4,5-b]quinoline-2-carbohydrazide (11). A mixture of 10 (3.00 g, 0.005 mol), chloroacetic acid (0.47 g, 0.005 mol) and anhydrous sodium acetate (0.01 mmol) in dioxane (20 mL) was refluxed for 4 h. The solid formed was separated by filtration and crystallized from dioxane to produce brown crystals, mp. 161–163 °C, yield: 1.82 g (57%). IR (ν, cm−1): 1675, 1655 (CO amide), 1705 (CO thiazolidinone), 3360, 3275 (NH). 1H NMR (DMSO, δ, ppm): 1.38–1.43 (m, 4H, 2CH2, cyclohexyl), 1.44–1.47 (m, 2H, CH2, cyclohexyl), 1.53–1.57 (m, 4H, 2CH2, cyclohexyl), 1.66–1.69 (m, 4H, 2CH2, tetrahydroquinoline), 1.97–2.01 (m, 4H, 2CH2, tetrahydroquinoline), 2.59–2.62 (m, 1H, N–CH, cyclohexyl), 3.79 (s, 2H, CH2, thiazolidinone), 5.41 (s, 1H, CH tetrahydroquinoline), 6.59–7.23 (m, 9H, Ar–H), 8.39 (br.s, 1H, NH, exchangeable), 9.37 (br.s, 1H, NH, exchangeable). EIMS (m/z, %): 639 (M+˙, 21). Anal. calcd for C33H33N7O5S (639.73): C, 61.96; H, 5.20; N, 15.33; found: C, 61.87; H, 5.14; N, 15.30%.
Molecular docking
Molecular docking studies are considered an influential method for the interpretation of molecular interactions between the produced substances and the main amino acid residues at the specific binding site of the target receptor.37,38 The affinities of the produced ligands toward the target protein Mcl-1 were compared according to the docking score values calculated using the MOE 2014.0901.33 The most active derivatives (8, 10, and 11) were drawn employing ChemBio3D Ultra 14.0 and transferred to the MOE window, optimized for partial charges, and energy minimized, as previously reported.39 Then, the target protein (Mcl-1) was extracted from the PDB database (PDB ID: 3WIX40), corrected, 3D hydrogenated, and energy minimized. Finally, a docking process was run to evaluate the binding affinities of candidates toward the Mcl-1 receptor pockets. The co-crystallized inhibitor (LC3) was inserted as the reference standard and the program specifications were adjusted, as previously mentioned.41
Data availability
Full tables, figures, and all data generated or analyzed during this study are included in this article and its ESI† files.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors extend their appreciation to the Deanship of Research and Graduate Studies at King Khalid University for funding this work through Large Groups Project under grant number (RGP.2/282/45).
References
- M. Sankaran, C. Kumarasamy, U. Chokkalingam and P. S. Mohan, Bioorg. Med. Chem. Lett., 2010, 20, 7147 CrossRef CAS PubMed.
- R. S. Gouhar, W. S. I. Abou-Elmagd, M. I. El-Zahar, M. M. Kamel and D. H. El-Ghonamy, Synthesis of novel 5,6,7,8,9,10-hexahydropyrimido[4,5-b]quinoline derivatives for antimicrobial and anti-oxidant evaluation, Res. Chem. Intermed., 2017, 43, 1301 CrossRef CAS.
- M. M. Ghorab, M. A. Shaaban, H. I. Heiba, A. Zaher and A. A. Hamed, Res. Chem. Intermed., 2015, 41, 647 CrossRef CAS.
- A. B. El-Gazzar, H. N. Hafez and G. A. Nawwar, J. Med. Chem., 2009, 44, 1427 CAS.
- S. I. Alqasoumi, A. M. Al-Taweel, A. M. Alafeefy, E. Noaman and M. M. Ghorab, Eur. J. Med. Chem., 2010, 45, 738 CrossRef CAS PubMed.
- A. M. El-Naggar and S. K. Ramadan, Efficient synthesis of some pyrimidine and thiazolidine derivatives bearing quinoline scaffold under microwave irradiation, Synth. Commun., 2020, 50(14), 2188 CrossRef CAS.
- A. R. Morsy, S. K. Ramadan and M. M. Elsafty, Synthesis and antiviral activity of some pyrrolonyl substituted heterocycles as additives to enhance inactivated Newcastle disease vaccine, Med. Chem. Res., 2020, 29(6), 979 CrossRef CAS.
- B. A. El-Gazzar, H. N. Hafez, A. A. Abu-Hashem and A. S. Aly, Phosphorus, Sulfur Silicon Relat. Elem., 2009, 184, 379 CrossRef.
- H. Nishii, T. Chiba, K. Morikami, T. A. Fukami, H. Sakamoto, K. Ko and H. Koyano, Bioorg. Med. Chem. Lett., 2010, 20(4), 1405 CrossRef CAS PubMed.
- S. K. Ramadan, D. R. Abdel Haleem, H. S. M. Abd-Rabboh, N. M. Gad, W. S. I. Abou-Elmagd and D. S. Haneen, Synthesis, SAR studies, and insecticidal activities of certain N-heterocycles derived from 3-((2-chloroquinolin-3-yl)methylene)-5-phenylfuran-2(3H)-one against Culex pipiens L. larvae, RSC Adv., 2022, 12(22), 13628 RSC.
- S. K. Ramadan, H. S. M. Abd-Rabboh, N. M. Gad, W. S. I. Abou-Elmagd and D. S. Haneen, Synthesis and Characterization of Some Chitosan-Quinoline Nanocomposites as Potential Insecticidal Agents, Polycyclic Aromatic Compounds, 2023, 43(8), 7013 CrossRef CAS.
- M. M. Kaddah, A. R. Morsy, A. A. Fahmi, S. A. Rizk and S. K. Ramadan, Synthesis and biological activity on IBD virus of diverse heterocyclic systems derived from 2-cyano-N'-((2-oxo-1,2-dihydroquinolin-3-yl)methylene)acetohydrazide, Synth. Commun., 2021, 51(22), 3366 CrossRef CAS.
- M. K. Abou-Elregal, A. T. Mohamed, A. S. A. Youssef, M. M. Hemdan, S. S. Samir and W. S. I. Abou-Elmagd, Synthesis and antitumor activity evaluation of some 1,2,4-triazine and fused triazine derivatives, Synth. Commun., 2018, 48(18), 2347–2357 CrossRef CAS.
- M. R. Mahmoud, W. S. I. Abou-Elmagd, H. A. Derbala and M. H. Hekal, Synthesis and spectral characterisation of some phthalazinone derivatives, J. Chem. Res., 2012, 36(2), 75 CrossRef CAS.
- M. R. Mahmoud, M. M. El-Shahawi, W. S. I. Abou-Elmagd and M. H. Hekal, Novel Synthesis of Isoquinoline Derivatives Derived from (Z)-4-(1,3-Diphenylpyrazol-4-yl)isochromene-1,3-dione, Synth. Commun., 2015, 45(14), 1632 CrossRef CAS.
- M. R. Mahmoud, W. S. I. Abou-Elmagd, H. A. Derbala and M. H. Hekal, Novel synthesis of some phthalazinone derivatives, Chin. J. Chem., 2011, 29(7), 1446 CrossRef CAS.
- K. N. Halim, S. A. Rizk, M. A. El-Hashash and S. K. Ramadan, Straightforward synthesis, antiproliferative screening, and density functional theory study of some pyrazolylpyrimidine derivatives, J. Heterocycl. Chem., 2021, 58(2), 636 CrossRef CAS.
- N. M. Gad, W. S. I. Abou-Elmagd, D. S. Haneen and S. K. Ramadan, Reactivity of 5-phenyl-3-[(2-chloroquinolin-3-yl)methylene] furan-2(3H)-one towards hydrazine and benzylamine: A comparative study, Synth. Commun., 2021, 51(9), 1384 CrossRef CAS.
- E. A. E. El-Helw, M. Asran, M. E. Azab, M. H. Helal and S. K. Ramadan, Synthesis, Cytotoxic, and Antioxidant Activity of Some Benzoquinoline-Based Heterocycles, Polycyclic Aromatic Compounds, 2023 DOI:10.1080/10406638.2023.2270767.
- S. K. Ramadan, N. A. Ibrahim, S. A. El-Kaed and E. A. E. El-Helw, New potential fungicides pyrazole-based heterocycles derived from 2-cyano-3-(1,3-diphenyl-1H-pyrazol-4-yl) acryloyl isothiocyanate, J. Sulfur Chem., 2021, 42(5), 529 CrossRef CAS.
- M. M. Kaddah, A. A. Fahmi, M. M. Kamel, S. A. Rizk and S. K. Ramadan, Rodenticidal Activity of Some Quinoline-Based Heterocycles Derived from Hydrazide–Hydrazone Derivative, Polycyclic Aromatic Compounds, 2023, 43(5), 4231 CrossRef CAS.
- C. H. Tseng, et al., Discovery of 2-[2-(5-nitrofuran-2-yl)vinyl]quinoline derivatives as a novel type of antimetastatic agents, Bioorg. Med. Chem., 2015, 23(1), 141 CrossRef CAS PubMed.
- K. Miura, et al., Chemical and Chemotherapeutical Studies on the Furan Derivatives. Xxx. Syntheses and Antibacterial Activities of 2-(5-Nitro-2-furyl)vinyl Heterocyclics, Yakugaku Zasshi, 1963, 83, 771 CrossRef CAS PubMed.
- M. M. Kamel, H. I. Ali, M. M. Anwar, N. A. Mohamed and A. M. Soliman, Eur. J. Med. Chem., 2010, 45(2), 572 CrossRef CAS PubMed.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107 CrossRef CAS PubMed.
- A. M. Abdelrahman, A. A. Fahmi, S. A. Rizk and E. A. E. El-Helw, Synthesis, DFT and Antitumor Activity Screening of Some New Heterocycles Derived from 2,2'-(2-(1,3-Diphenyl-1H-pyrazol-4-yl)ethene-1,1-diyl)bis(4H-benzo[d][1,3]oxazin-4-one), Polycyclic Aromatic Compounds, 2023, 43(1), 721–739 CrossRef CAS.
- M. M. Kaddah, A. A. Fahmi, M. M. Kamel, S. K. Ramadan and S. A. Rizk, Synthesis, characterization, computational chemical studies and antiproliferative activity of some heterocyclic systems derived from 3-(3-(1,3-diphenyl-1H-pyrazol-4-yl)acryloyl)-2H-chromen-2-one, Synth. Commun., 2021, 51(12), 1798 CrossRef CAS.
- A. S. Elgubbi, E. A. E. El-Helw, A. Y. Alzahrani and S. K. Ramadan, Synthesis, computational chemical study, antiproliferative activity screening, and molecular docking of some thiophene-based oxadiazole, triazole, and thiazolidinone derivatives, RSC Adv., 2024, 14(9), 5926 RSC.
- V. Karunakaran and V. Balachandran, Spectrochim. Acta, Part A, 2012, 98, 229 CrossRef CAS PubMed.
- E. A. E. El-Helw, A. Y. Alzahrani and S. K. Ramadan, Synthesis and antimicrobial activity of thiophene-based heterocycles derived from thiophene-2-carbohydrazide, Future Med. Chem., 2024, 16(5), 439 CrossRef CAS PubMed.
- A. Hassaballah, S. K. Ramadan, S. A. Rizk, E. A. E. El-Helw and S. S. Abdelwahab, Ultrasonic promoted regioselective reactions of the novel spiro 3,1-benzoxazon-isobenzofuranone dye toward some organic base reagents, Polycyclic Aromatic Compounds, 2023, 43(4), 2973 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55 CrossRef CAS PubMed.
- CCG Inc., Molecular Operating Environment (MOE), Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite#910, Montreal, 2016 Search PubMed.
- A. Daina, O. Michielin and V. Zoete, SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- A. Daina, O. Michielin and V. Zoete, iLOGP: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach, J. Chem. Inf. Model., 2014, 54(12), 3284 CrossRef CAS PubMed.
- A. El-Sewedy, E. A. El-Bordany, N. F. H. Mahmoud, K. A. Ali and S. K. Ramadan, One-pot synthesis, computational chemical study, molecular docking, biological study, and in silico prediction ADME/pharmacokinetics properties of 5-substituted 1H-tetrazole derivatives, Sci. Rep., 2023, 13(1), 17869 CrossRef CAS PubMed.
- A. A. Elmaaty, et al., Anticoagulants as Potential SARS-CoV-2 Mpro Inhibitors for COVID-19 Patients: In Vitro, Molecular Docking, Molecular
Dynamics, DFT, and SAR Studies, Int. J. Mol. Sci., 2022, 23(20), 12235 CrossRef PubMed.
- M. Elagawany, et al., Ligand-based design, synthesis, computational insights, and in vitro studies of novel N-(5-Nitrothiazol-2-yl)-carboxamido derivatives as potent inhibitors of SARS-CoV-2 main protease, J. Enzyme Inhib. Med. Chem., 2022, 37(1), 2112 CrossRef CAS PubMed.
- S. M. Al-Muntaser, A. A. Al-Karmalawy, A. M. El-Naggar, A. K. Ali, N. E. Abd El-Sattar and E. M. Abbass, Novel 4-thiophenyl-pyrazole, pyridine, and pyrimidine derivatives as potential antitumor candidates targeting both EGFR and VEGFR-2; design, synthesis, biological evaluations, and in silico studies, RSC Adv., 2023, 13(18), 12184 RSC.
- Y. Tanaka, K. Aikawa, G. Nishida, M. Homma, S. Sogabe, S. Igaki, Y. Hayano, T. Sameshima, I. Miyahisa, T. Kawamoto and M. Tawada, Discovery of potent Mcl-1/Bcl-xL dual inhibitors by using a hybridization strategy based on structural analysis of target proteins, J. Med. Chem., 2013, 56(23), 9635 CrossRef CAS PubMed.
- E. M. Abbass, A. K. Khalil, M. M. Mohamed, I. H. Eissa and A. M. El-Naggar, Design, efficient synthesis, docking studies, and anticancer evaluation of new quinoxalines as potential intercalative Topo II inhibitors and apoptosis inducers, Bioorg. Chem., 2020, 104, 104255 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Hisham S. M. Abd-Rabboh
*a,
Hisham S. M. Abd-Rabboh