
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChitosan-azo dye bioplastics that are reversibly resoluble and recoverable under visible light irradiation†
Mikhail Kim‡
a,
Coral Hillel‡b,
Kayrel Edwardsa,
William Pietro c,
Ozzy Mermutbc and
Christopher J. Barrett*ab
c,
Ozzy Mermutbc and
Christopher J. Barrett*ab
aDepartment of Chemistry, McGill University, Montreal, QC, Canada. E-mail: christopher.barrett@mcgill.ca
bDepartment of Physics and Astronomy, York University, Toronto, ON, Canada
cDepartment of Chemistry, York University, Toronto, ON, Canada
First published on 16th August 2024
Abstract
Biopolymer composite materials were prepared by combining bio-sourced cationic water-soluble chitosan with bi-functional water-soluble anionic azo food dyes amaranth (AMA) or allura red (ALR) as ionic cross-linkers, mixing well in water, and then slow-drying in air. The electrostatically-assembled ionically-paired films showed good long-term stability to dissolution, with no re-solubility in water, and competitive mechanical properties as plastic materials. However, upon exposure of the bioplastics to low power light at sunlight wavelengths and intensities stirring in water, the stable materials photo-disassembled back to their water-soluble and low-toxicity (edible) constituent components, via structural photo-isomerization of the azo ionic crosslinkers. XRD, UV-vis, and IR spectroscopy confirmed that these assemblies are reversibly recoverable and so can in principle represent fully recyclable, environmentally degradable materials triggered by exposure to sunlight and water after use, with full recovery of starting components ready for re-use. A density functional theory treatment of the amaranth azo dye identified a tautomeric equilibrium favouring the hydrazone form and rationalized geometrical isomerization as a mechanism for photo-disassembly. The proof-of-principle suitability of films of these biomaterial composites as food industry packaging was assessed via measurement of mechanical, water and vapour barrier properties, and stability to solvent tests. Tensile strength of the composite materials was found to be 25–30 MPa, with elongation at break 3–5%, in a range acceptable as competitive for some applications to replace oil-based permanently insoluble non-recyclable artificial plastics, as fully recyclable, recoverable, and reusable low-toxicity green biomaterials in natural environmental conditions.
1 Introduction
Heavy reliance globally on traditional disposable plastics has emerged as a pressing worldwide environmental issue due to their high stability and inability to recycle efficiently which poses significant ecological1–3 and economic challenges.4 The design of truly recyclable plastics such as for packaging faces fundamental challenges from a chemical and engineering standpoint: ideally, these materials need to be strong, flexible, water resistant, and for packaging films, capable of controlling vapour transmission rates.5 Yet maximizing these desired properties can render these materials too strong to recycle efficiently or effectively after use. Current oil-based plastics used for packaging are typically recycled at a rate of 10% or less,6 with limitations on the variety of recyclable polymers available.An alternative strategy involves designing polymer materials that can be assembled from non-toxic and readily recyclable components that are capable of reversible disassembly under specific conditions or stimuli. Such a disassembly process for example could selectively release soft bonds holding the constitutional parts together with desired properties for use, while keeping the covalent bonds intact. Some such previously reported polymer systems have been shown to disassemble on exposure to stimuli including temperature change,7 pH change,8 exposure to metal ions with chelating agents,9 mechanical force,10 or UV light.10–13 Exposure to these relatively harsh conditions however also risks the chemical or mechanical degradation of the polymer components covalently, potentially reducing recycling efficiency and re-use properties. Additionally, pH-responsive systems and those needing metal ions and chelating agents often require substantial water-based solutions of acids or bases, complicating the disassembly process and recovery of constitutional parts. Furthermore, these approaches often involve complex synthesis and result in materials mainly existing in the form of hydrogels, limiting their practical use, for instance as packaging materials.
In our work, visible light was chosen as a relatively clean, abundant, and gentle stimulus to induce photo-disassembly of water-insoluble bioplastic materials multi-layered on surfaces from water-soluble polyelectrolytes and water-soluble azo dyes as photo-reversible ionic crosslinkers, depicted schematically in Fig. 1.14 The azo dyes isomerize upon light irradiation, changing their geometry significantly from a planar trans isomer to a bent, metastable cis isomer.15 This isomerization affects structural interactions between polymer and azo dyes, gently ‘shaking’ the assemblies apart and recovering the water-solubility of each part.14 This concept of structural photo-release with azo dyes has been reported previously in some specialized systems, providing light-control over structure or solubility. For example, it was demonstrated that trans-to-cis isomerization enhanced the aqueous solubility of an anti-cancer candidate drug in aqueous environment,16 and a drug from an azobenzene–polyethylene glycol-nanoparticles micelle system was similarly shown to be released with light.17 In other previous work,18 co-crystallization of a fluorinated azobenzene with volatile components yielded halogen-bonded cocrystals that could photo-disassemble to be cut, carved, or engraved with low-power visible laser light.
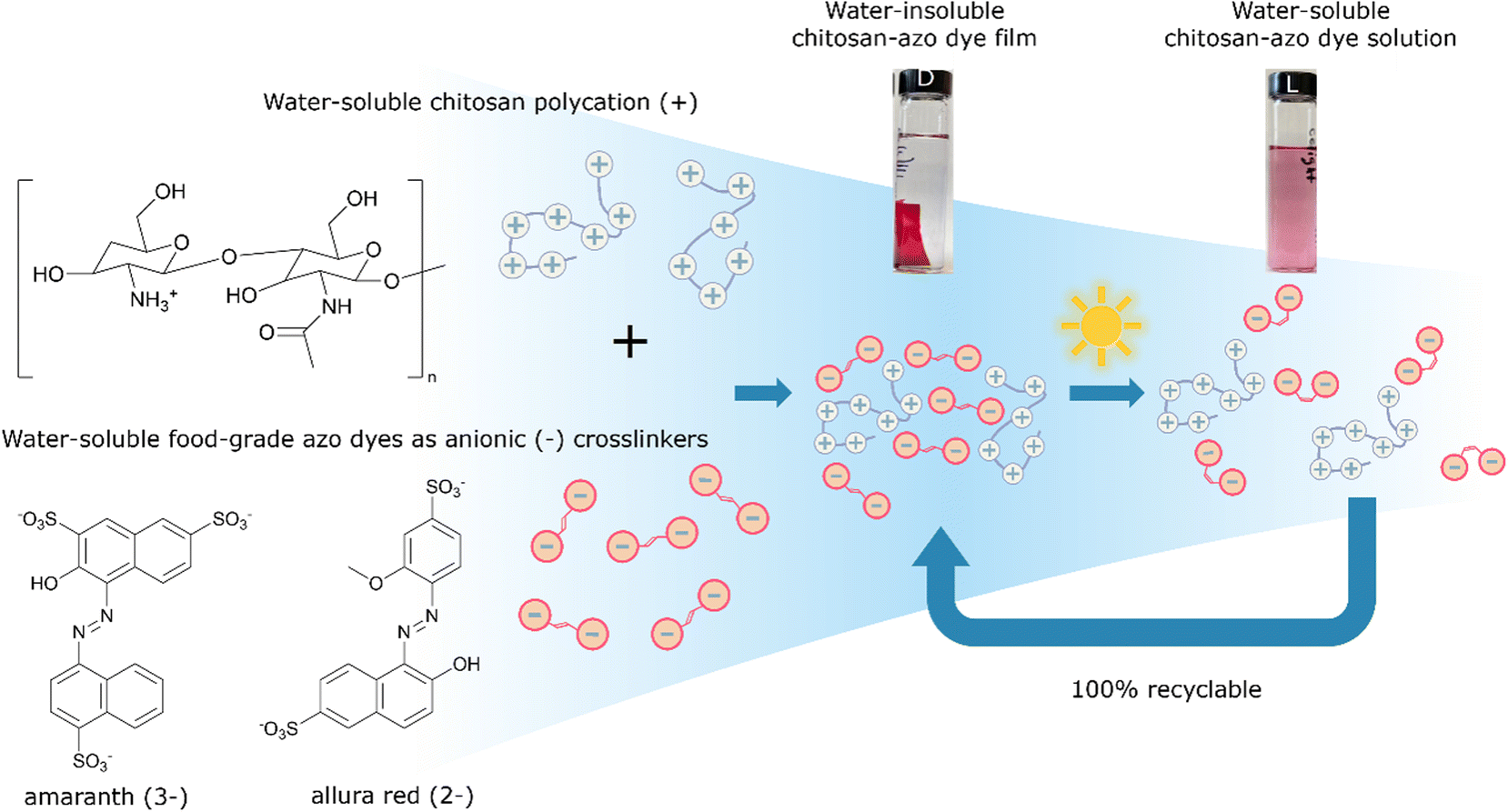 | ||
| Fig. 1 Chemical structures of the chitosan polymer and ionic crosslinker azo dyes ALR and AMA, and schematic of the self-assembly of these water-soluble components to prepare water-resistant thick film materials, until their photo-triggered disassembly to recover and recycle the aqueous components. | ||
We propose here to extend this light-triggered disassembly to low-toxicity and fully recyclable bioplastics, by combining relatively harmless anionic azo food dyes with positively charged biopolymers such as chitosan,19 now used widely in toxicity-sensitive materials applications, such as cosmetics, medicine, agriculture, and the food industry.20–22
Previously, our group has reported the aqueous assembly of materials from water solutions of polyelectrolytes such as sulphonated cellulose and sodium alginate, and water solutions of azo dyes, such as Bismarck brown Y, using a layer-by-layer (LbL) dipping technique to produce thin multilayer films on a glass substrate.14 The photo-induced disassembly of these photo-reversible materials in water was demonstrated under exposure to low-power visible light while being gently washed with water. However, the preparation of multilayer thin films by the LbL method was tedious. Moreover, they were not strong mechanically or free-standing, and they were limited to just 10s or a few 100s of nanometres in thickness, requiring a supporting substrate. In one report,19 an attempt to make thicker LbL films approaching micrometre thickness was attempted by spray-coating LbL multilayers. However, that technique was still very time-consuming and costly. For example, the production of an 80-bilayer film with a thickness of just 100 nm took 48 h. For mass production and use of such light-disassembly materials, larger-scale methods must be applied to produce the thick, strong, free-standing films many micrometres thick required for any real plastic applications, such as for packaging.
In our present work, an aqueous solvent-casting method was developed as a facile, low-cost, and high-throughput method to make thick, strong, and free-standing photo-reversible plastic films that could dis-assemble on visible light irradiation. Materials prepared by this method were water-resistant under ambient conditions and robust, yet demonstrated light-disassembly in water under gentle shaking during irradiation with blue or green light simulating sunlight. To assess the potential of these chitosan-azo dye films as replacements for petrol-based films in packaging applications, the mechanical and barrier properties of the films were measured and compared to polyethylene.
Advancing from previously-used fabric azo dyes such as Bismarck brown Y and Bismarck brown R, to low-toxicity azo food dyes such as allura red (ALR) and amaranth (AMA) has the advantage of offering systems with lower environmental impact, yet this now complicates experimental spectroscopic confirmation of the trans-to-cis isomerization, as these dyes now contain a hydroxy-group in ortho ring position with respect to the azo-group, leading to hydrazone character and associated exceedingly rapid thermal cis-to-trans back-isomerization times, too fast to observe directly with standard spectroscopies. So lastly, in place of experimental spectroscopic confirmation of photo-switching between geometric forms, a theoretical analysis of AMA was conducted via density functional theory (DFT) approaches, as a representative system for the general class of ortho-hydroxy azobenzene food dyes.
2 Materials and methods
2.1 Preparing thick free-standing films
The structures of azo dyes ALR and AMA, and chitosan (CS) polymer are presented in Fig. 1. Solvent-casting was performed by adding 10 mL of a solution of the azo dye (AMA or ALR with concentration 0.1 mass% in distilled water) dropwise into a 10 mL solution of the CS polymer (0.5 mass% solution in 1% water solution of acetic acid) and then mixed thoroughly at room temperature. These solution concentrations (0.5 mass% for CS and 0.1 mass% for azo dyes) were found to be low enough, and the addition of azo dye to CS slow enough, to prevent immediate ion–ion interaction and precipitation of the product. The transparent coloured mixture CS-azo dye was filtered through a syringe PTFE filter with a 0.2 μm pore size to remove any solids. The mixture was then placed in a 12 cm diameter 400 mL Teflon evaporating dish and left open under fume hood conditions for slow water evaporation at room temperature for 1–2 days. After evaporating, the thick film was gently peeled from the dish with plastic tweezers and a plastic spatula. The free-standing film was placed in 250 mL beaker and additionally gently rinsed with distilled water 3 times for 5 min to remove any unbound azo dye or polymer which failed to crosslink. An indication of successful rinsing was gradual disappearance of azo dye colour from rinsing water. Finally, the films were dried at 100 °C for 30 min under vacuum before recording the initial mass and used for testing. The thickness of the films we obtained was in the range of 20–800 μm and density was measured in range 0.8–1.2 g cm−3.2.2 Light disassembly experiments
To study disassembly of the films, a setup was used as depicted schematically in Fig. 2. A sample of dried film was cut into two strips of 1 × 5 cm, both were weighed, and placed into a glass 20 mL vial with 15 mL of distilled water, plus a 1% by mass aqueous solution of either acetic acid or hydrochloric acid. One vial was wrapped with aluminium foil to shield the film from light, labelled ‘D’ for dark, while the other vial was left uncovered allowing the film to be exposed to light irradiation, labelled ‘L’ for light. Different light sources were tried with powers ranging from 10 mW to 5 W and wavelengths 380, 460, 520 nm or white light bulb (light wavelength from 250 to 800 nm). The light source was placed 15 cm from the surface of the samples, a distance that was found to provide sufficient irradiation for the dis-assembly yet without heating the samples measurably to not exceed 25 °C. The vials were placed on a platform shaker for agitation and speed of the shaker was kept constant at 200–300 rpm. At certain time intervals the samples were removed from the shaker and an aliquot of each solution was centrifuged at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm to remove any film fragments which had mechanically disintegrated, but not dissolved. Using a glass Pasteur pipette the solution was carefully removed from the centrifuge tube leaving behind any solid particles. The solution was placed into clean dry quartz cuvette and analysed in duplicate by UV-vis spectroscopy. After measurements, the aliquots were returned into the vials.
000 rpm to remove any film fragments which had mechanically disintegrated, but not dissolved. Using a glass Pasteur pipette the solution was carefully removed from the centrifuge tube leaving behind any solid particles. The solution was placed into clean dry quartz cuvette and analysed in duplicate by UV-vis spectroscopy. After measurements, the aliquots were returned into the vials.
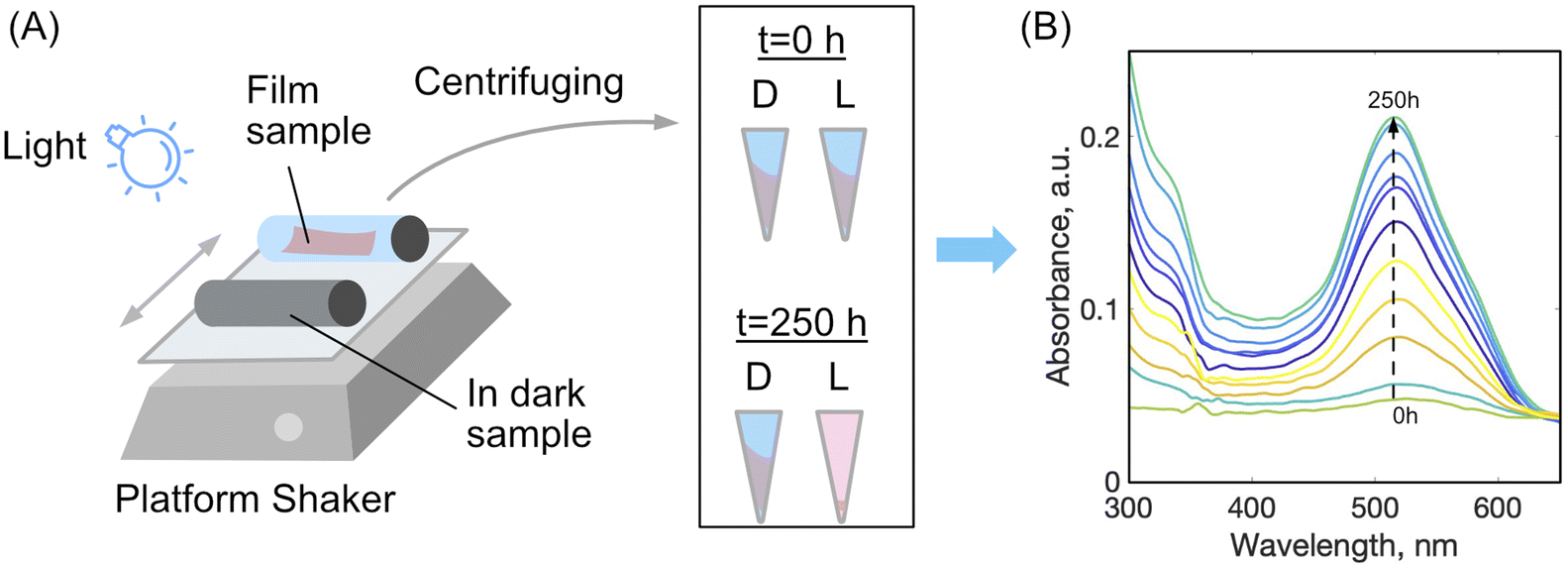 | ||
| Fig. 2 (A) The experimental setup for light-disassembly of thick films using a platform shaker under partial irradiation, with one sample kept in the dark (“D”) as a control, compared to one sample exposed to light (“L”). (B) UV-vis spectra of increasing dye release into wash water solution during a typical disassembly process. | ||
During the photo-disassembly process, the azo dye disassembled from the chitosan polymer and re-dissolved in the water solution. At time t = 0, no azo dye was present in the solution as validated with UV-vis spectroscopy. After slow re-dissolving of azo-dye and chitosan, the concentration of both components increases and can be readily detected and measured by tracking their UV-vis spectra. Since azo dyes have characteristic absorbance in the visible region, plots of dye absorbance maxima (ALR: λ = 510 nm and AMA: λ = 530 nm) versus time were used to quantify the rate of the film disassembly.
2.3 Quantum chemical calculations
Gas phase structures of the AMA azo dye were optimized with the density functional theory (DFT) method in Orca 5.0.3,23–25 with the B3LYP functional26,27 and ma-def2-TZVP basis set,28,29 including D3BJ dispersion correction.30,31 All calculations used the resolution of identity (RI) approximation with chain-of-spheres32,33 (RIJCOSX) and the def2/J auxiliary basis set.34 Calculation of thermodynamic parameters and verification of local minimum structures were carried out by vibrational analysis,35 which returned real vibrations only. Structures and vibrations were visualized with Chemcraft.363 Results and discussion
3.1 Thick film characterization by FTIR and XRD
CS-azo dye films obtained by solvent casting were elastic, and transparent with a strong colour of the incorporated dye: orange-red with ALR and red-purple with AMA. Preliminary films first cast using poly(acrylic acid) (PAA) and poly(allylamine hydrochloride) (PAH) were in contrast opaque and brittle, falling apart at the dish removal peeling step.The FTIR spectra of pure CS films, powder azo dyes, and CS-azo dye films are shown in Fig. S2.† For CS and CS-azo dye films, the bands 3200–3500 cm−1 are assigned as overlapped stretching O–H on N–H bands. The band near 2867 cm−1 is assumed to be stretching (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of the amide group CONHR of the CS.37 The presence of residual N-acetyl groups was confirmed by the bands near 1645 cm−1 (CO stretching of amide I) and 1320 cm−1 (C–N stretching of amide III), respectively. The band at 1550 cm−1 corresponds to N–H bending of amide II and is likely occluded by a band at 1548 cm−1 corresponding to the N–H bending of the primary amine.38 For both azo dyes, the two bands at 1038 and 1136 cm−1 are assigned to the coupling between naphthalene rings and stretching of SO3, and the bands at 1038, 1186, and 1200 cm−1 are linked to the SO3 stretching mode.39,40 Azo bond (NN) vibrations are identifiable between 1504 cm−1 and 1550 cm−1.41 The effect of the ionic interaction between polymer and azo dye was observed by the shifting peak of sulfonate groups of azo dyes from 1038 cm−1 to 1030 cm−1 and a shifting of the primary amine peak from 1548 cm−1 to 1559 cm−1.
O) of the amide group CONHR of the CS.37 The presence of residual N-acetyl groups was confirmed by the bands near 1645 cm−1 (CO stretching of amide I) and 1320 cm−1 (C–N stretching of amide III), respectively. The band at 1550 cm−1 corresponds to N–H bending of amide II and is likely occluded by a band at 1548 cm−1 corresponding to the N–H bending of the primary amine.38 For both azo dyes, the two bands at 1038 and 1136 cm−1 are assigned to the coupling between naphthalene rings and stretching of SO3, and the bands at 1038, 1186, and 1200 cm−1 are linked to the SO3 stretching mode.39,40 Azo bond (NN) vibrations are identifiable between 1504 cm−1 and 1550 cm−1.41 The effect of the ionic interaction between polymer and azo dye was observed by the shifting peak of sulfonate groups of azo dyes from 1038 cm−1 to 1030 cm−1 and a shifting of the primary amine peak from 1548 cm−1 to 1559 cm−1.
Fig. S3† shows XRD patterns of CS–ALR and CS–AMA films. From the pattern, the two characteristic peaks are visible at 2θ = 11° and 2θ = 20°, corresponding to count indexes (142) and (191), respectively, assigned to the CS.42 The first peak at 11° becomes wider and smaller with decreasing content of CS in the film, while the second peak at 20° becomes sharper and stronger. The weak peaks in the XRD pattern of CS reflect great disarray in chain alignment of CS,42 and the broadening of the peak at 11° toward 9° and increase in the 20° peak confirm the presence of the azo dye. In this study, the crystalline index (CrI) was determined from eqn (1) by the fitting of the diffraction profiles in Fig. S3.† This formula uses the ratio between the intensity of a crystalline reflection and intensity of the minimum peak at 2θ = 16°, which describes the diffuse halo peak (Iam). Intensity of the reflection at 20° (I200) is determined for the most frequently used hydrated form of CS.43
| CrI = (I200 − Iam)/I200 | (1) |
The lowest calculated CrI was 38% for CS films and this increased to a maximum of 56% for CS–ALR films with the mass ratio CS/ALR = 2.5/1. The higher degree of crystallinity of the CS-azo dye films can be explained by presence of crystalline azo dye and not necessarily by an increase of crystallinity of CS itself, which is also supported by the mechanical analysis of the CS-azo dye films. The higher crystallinity of CS itself would be expected to increase the tensile strength,44 however the measured strength of CS-azo dye films was in fact lower (Section 3.6).
3.2 Disassembly of the thick films
For the disassembly experiments, a vial containing chitosan/amaranth azo dye film (CS–AMA) was irradiated with 520 nm green light for 260 h, while the vial containing chitosan/allura red azo dye film (CS–ALR) was irradiated with 460 nm blue light for 130 h. The disassembly of both films was observed visually (Fig. 3). For the vials exposed to light (“L”), the films all slowly disassembled and re-dissolved, observed by the noticeable red and orange colours of the AMA and ALR increasing in the solution. For the dark (“D”) vials which were protected from light exposure, the films were sometimes fragmented into smaller pieces due to shaking but did not dissolve. Only a very faint dye colour could still be observed for the solution, likely due to the final release of a small amount of unbound excess azo dye into the solution during shaking which had not been released during the initial washing steps. To quantify the release of the azo dye into the solution, centrifuged aliquots of the solution were analyzed by UV-vis spectroscopy as a function of time. The re-dissolved compounds did not form solid insoluble material again, likely due to the high dilution. | ||
| Fig. 3 Stages of light disassembly of multilayer film (A) CS–ALR (mass ratio 1/0.125) in 1% acetic acid solution (pH = 3) at 20 °C under 460 nm blue light irradiation. (B) CS–AMA (mass ratio 1/0.125) in 1% acetic acid solution at 20 °C under 520 nm green light irradiation. For all photographs: samples “L” were exposed to light and control samples “D” were in the dark. | ||
We hypothesize that the disassembly of the CS-azo dye films occurs due to the geometric trans-to-cis isomerization of the azo dyes during irradiation with the blue or green light. Under ambient low intensity white light, both azo dyes ALR and AMA have a planar elongated orientation within the film. Fig. 1 shows structures of AMA and ALR which contain negatively charged sulfonate (SO3−) groups on opposite sides of the molecule. These negatively charged sulfonate groups can ionically bond strongly to the positively charged protonated amino groups located along the CS polymer chains. The azo dyes thus could effectively act as soft-bonded reversible crosslinkers holding the CS chains together, resulting in a water-resistant and robust material. During constant irradiation with blue or green light, the azo dyes undergo a geometric rearrangement from the trans isomer to the cis isomer, which changes the shape, orientation, and length of the crosslinker. This movement appears sufficient to disrupt or weaken the ionic bonding between the azo dyes and the CS polymer, which exposes the positive and negative ionic groups cross-linking the assemblies together, re-solubilizing each component in the moving water and, in principle, enabling them to be separated from each other. Indeed, a similar observation was made for the previously reported multilayer sulfonated cellulose-azo dye films prepared on supported substrates,14 in which the trans-to-cis geometric rearrangement of the azo dye was slow enough to be studied by pump-probe experiments and confirmed to be coincident with the photo-driven disassembly of the films.
3.3 Rate of disassembly
To estimate a relative rate of disassembly (RRD) of the films in light vs. dark, the UV-vis spectra of the solutions were analyzed to track the presence and increase in azo dye released. The spectra of food azo dyes in water (Fig. 4A) have strong absorption peaks in both the visible and UV regions; AMA has a maximum absorption peak around λmax = 530 nm, while ALR has a maximum absorption peak around λmax = 510 nm. The UV-vis spectra of the solution after disassembly together with spectra of their constitutional parts are shown in Fig. 4B. The absorbance peak in UV region 200–250 nm is a result of overlapping spectra from the azo dye, acetic acid, and CS. The sum of the absorption is higher than 1 and, hence, peaks in this region become less suitable to monitor the concentration quantitatively. Instead, the unique peaks of azo dyes in the visible region 400–800 nm were used, and with calibration curves constructed for both dyes (Fig. S4†), we could calculate a wash water dye concentration as a function of time.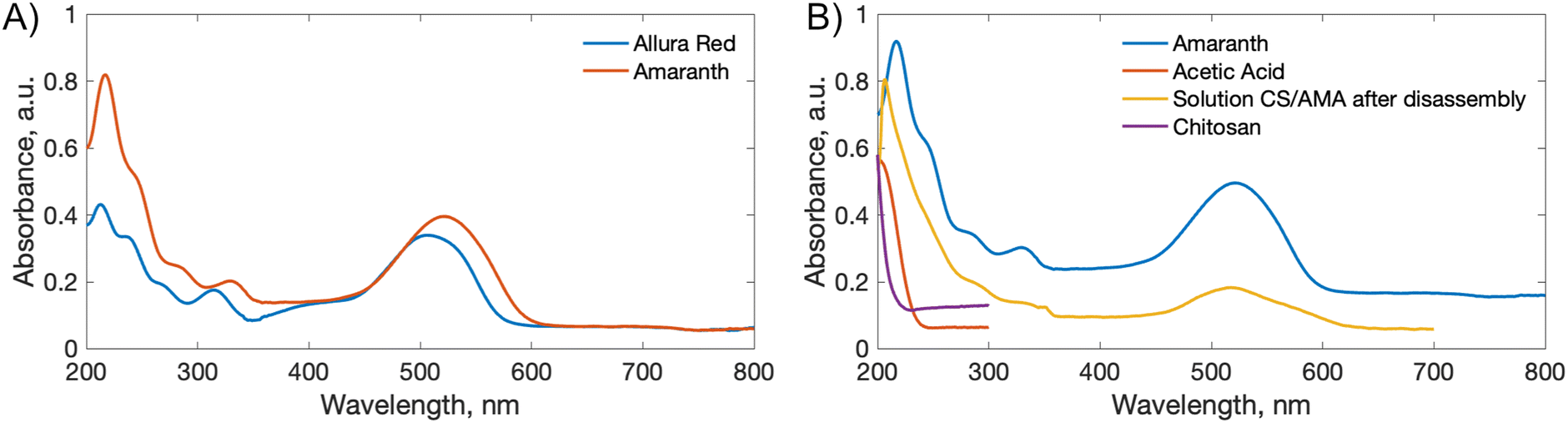 | ||
| Fig. 4 (A) UV-vis spectra of food azo dyes ALR and AMA. (B) UV-vis spectra of water solutions of AMA, CS, acetic acid, and the product of the thick film disassembly (CS absorbance peak at 200 nm and AMA peaks at 220 and 530 nm). | ||
Plotted in Fig. 5 is the absorbance vs. time profile for light-triggered disassembly of CS–AMA (Fig. 5A) and CS–ALR (Fig. 5B) compared to control vials where films were kept in the dark. In the dark, we observed the initial appearance of both the ALR and AMA dyes in the solution, which corresponded to a concentration of 0.015 mg mL−1 which, as discussed previously, we attributed to un-bound excess azo dye that wasn't completely released during the washing step. This initial concentration did not increase under dark conditions, except when exposed to light. Typical times for complete film disassembly were 150–250 h of irradiation at approximately 100 mW under both green and blue light. To determine the rate of increase in the absorbance of the azo dye released into solution (which equates to the rate of disassembly of the material), absorbance against time was further plotted (Fig. S5†) as both 1/absorbance vs. time and ln(absorbance) vs. time. We found that the R2 value was highest for the absorbance-time linear fit, which led us to conclude that the disassembly of the multilayer films can best be described by a zero-order process. The rates of disassembly in light were normalized to the weight of the samples and to the disassembly rate of samples kept in the dark. These normalized rates were termed ‘relative rates of disassembly’ (RRD) for comparison between samples and systems. For the CS–AMA material, the RRD ratio was determined to be 117:1, while the RRD for CS–ALR was determined to be 14:1. This indicated that for both CS-azo dye films, the films disassembled at a much faster rate when exposed to the light stimulus. The significantly higher RRD observed for the CS–AMA compared to CS–ALR may be rationalized by the differences in the structures of the two azo dyes with respect to charges (AMA is tri-functional while ALR is bi-functional), and perhaps also by ALR having a more favourable geometry for ionic bonding with the CS polymer which slows down the disassembly process.
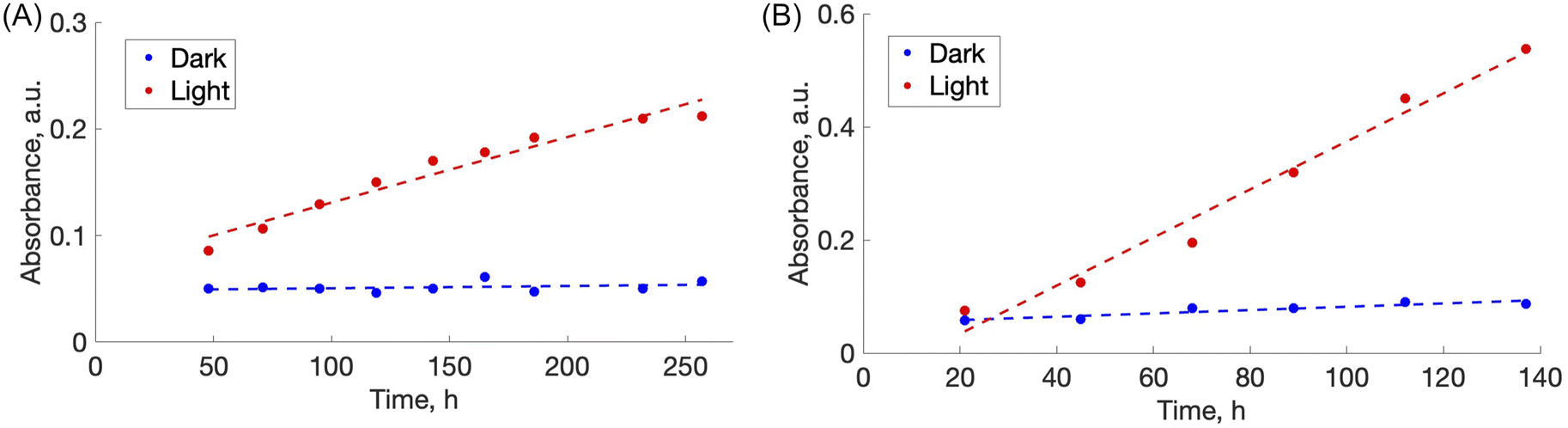 | ||
| Fig. 5 (A) Change with time of the absorbance maxima of AMA in aqueous solution during disassembly of the free-standing thick film CS–AMA. The trendline equations for “absorbance – time” fit is A = 0.0007 t + 0.0616 for light and A = 6 × 10−6 t + 0.0499 for dark. (B) Change with time of the absorbance maxima of ALR in solution during disassembly of the free-standing thick film CS–ALR. | ||
3.4 Role of the azo dye and polymer structure
Prior to assembling these thick free-standing films, the ability of water-soluble azo dyes and water-soluble polymers to form water-insoluble films was investigated. Previously used water-soluble synthetic polymers polyallylamine hydrochloride (PAH), polyacrylic acid (PAA), polyethylenimine (PEI), and polydiallyldimethylammonium chloride (PDAC) were studied to compare to the water-soluble biopolymer CS reported here. Mono-functional (methyl orange (MO)), bi-functional (ALR, sunset yellow (SY)), tri-functional (AMA, tartrazine (TZ)), and tetra-functional (Bismark Brown Y (BBY), Evans Blue (EB)) azo dyes were used as light-responsive cross-linkers for comparison. The synthetic cationic polymers PAH, PDAC, and PEI with anionic azo dyes (MO, ALR, SY, AMA, TZ, and EB), and anionic PAA with cationic BBY, form water insoluble multilayer films but all thick films were found to be water-soluble. The thick films were water soluble even with tri- and tetra-functional azo dyes (AMA, TZ, EB, BBY). On the other hand, CS-azo dye multilayered thick films were water-insoluble, except for the system CS–MO, probably because the dye is only monofunctional and thus can not crosslink multiple polymer chains to the extent to make them insoluble. One of the reasons CS forms good water-insoluble systems with azo dyes might also be the existence of many hydrogen bonds between hydroxyl groups of single chains of the polymer. CS films with tetra-functional dye EB were completely insoluble in water under any conditions tested and they did not demonstrate any light-disassembly, possibly due to the high functionality of the dye and the presence of two additional amino groups in the molecule.3.5 Effect of water acidity
CS is a polycationic polymer with a pKa = 6.3 for the amino groups. The polymer is insoluble in neutral and basic conditions, while at acidic pH values (generally less than pH = 5), the polymer becomes soluble. The protonation behaviour of CS upon dissolving in acetic acid solution is estimated by eqn (2), and the crosslinking of the protonated behaviour of CS with negatively charged AMA azo dye is described by eqn (3).| CS–NH2(s) + CH3COOH(aq) → CS–NH3+CH3COO(aq)− | (2) |
| 3CS–NH3+CH3COO(aq)− + AMA(SO3−)3(aq) → 3CS–NH3+CH3COO(aq)−AMA(SO3−)3(s) | (3) |
| CS–NH3+CH3COO−AMA(SO3−)3(s) → CS–NH3+AMA(SO3−)3(s) + CH3COO−(g) | (4) |
CS is produced commercially by deacetylation of chitin, and the proportion of amino groups in CS is related to the degree of deacetylation that describes the ratio of glucosamine to acetyl-glucosamine groups. For the CS used in the study the degree of deacetylation was 83%, determined by FTIR spectroscopy. Upon dissolving in acetic acid, the amino groups become protonated, and CS acetate solution can participate in the interaction with azo dyes. The molar ratio of amino groups of CS to sulfonate groups of azo dyes in the fabricated free-standing films varied from 15–315 to 1, i.e., from 5 to 105 times the stoichiometric amount according to eqn (3). Thus, the CS-azo dye films were made primarily of CS and contained protonated amino groups in a form of acetic acid salt. Upon drying and heating, the films release free acetic acid and some groups become unprotonated (eqn (4)), suggesting that to start the disassembly process, the washing solution should be more acidic. A series of experiments to determine the effect of water pH on the ability of the films to light-disassemble is shown in Fig. 6, where one can see that the disassembly of CS-azo dye films started only at pH conditions lower than the range of 5.5 and 6.8. Future studies will include chemical modifications of CS to increase the pH at which CS-azo dye films disassemble, such as depolymerization,45,46 or introducing into the CS structure acrylate groups.46
 | ||
| Fig. 6 Disassembly of thick film CS–AMA under 523 nm green light irradiation over 3 days duration shaking in water at different pH values. | ||
The degree of protonation of CS was regulated by conducting another series of experiments whereby 0.1 M NaOH was added to a completely dissolved (fully protonated) CS solution and the pH recorded. Using the Henderson–Hasselbalch equation (eqn (5)), we calculated the degree of protonation of CS, as eqn (5) relates the pH of the CS solution to the pKa of the weak acid CS–NH3+ (where CS- is the D-glucosamine plus N-acetyl-D-glucosamine residual of CS) with concentrations in equilibrium of deprotonated ([CS–NH2]) and protonated ([CS–NH3+]) form of CS. The acid–base equation for protonated CS is shown in eqn (6), with results provided in Table 1.
| pH = pKa + log10([CS–NH2]/[CS–NH3+]) | (5) |
| CS–NH3+ → CS–NH2 + H+ | (6) |
| CS protonation, (%) | Mass ratio CS/AMA | Disassembly rate ratio |
|---|---|---|
| 99.7 | 5 | 1.5 |
| 98.5 | 100 | 1 |
| 98.5 | 25 | 1 |
| 98.5 | 20 | 2.8 |
| 98.5 | 5 | 3.5 |
| 97.0 | 5 | 4.7 |
The ratio between disassembly rates in light and dark indicates how sufficiently the azo dye crosslinks CS and to what extent the film is light responsive. The higher the ratio, the more light was required in the disassembly process. Remarkably, this ratio was higher for films made with CS with partially unprotonated amino groups (4.7 for 97% protonated vs. 1.5 for 99.7% protonated, Table 1). The highly protonated CS films had almost equal solubility in the dark and when irradiated. With the mass ratio CS/azo dye approaching the stoichiometric ratio (for protonation degree 98.5% in Table 1), the ratio of disassembly rates increased. Thus, to achieve an optimal disassembly rate in light vs. dark, an optimal amount of protonated amino groups in the initial CS and in the film after cross-linking must be found.
3.6 Mechanical properties
To test mechanical properties, the following free-standing films were compared: (1) films made of chitosan of high molecular weight (CSH), 310000–375000 g mol−1, and of lower molecular weight (CSL), 50000–190000 g mol−1; (2) films CSH–azo dyes with mass ratio CS/azo dye = 1/0.125. CSH demonstrated the highest tensile strength. The mass ratio was the lowest ratio that permits light response and at the same time allows one to use as low amounts of azo dye as possible. Upon applying tensile stress, the films behaved as typical elastomers (Fig. 7A). CS is a competitive candidate biopolymer for use as packaging films and similar barrier materials due its relatively high tensile strength that depends on the molecular weight, measured as 45.0 ± 9.2 MPa for CSH and 38.4 ± 9.9 MPa for CSL. For the soft-bonded CS-azo dye films prepared in this study, it was shown that the cross-linking of the CS polymer with the azo dyes ALR (CSL–ALR) and AMA (CSL–AMA) did not have a significant negative effect on the tensile strength and even slightly increased elongation at break (Fig. 7C). These mechanical properties are comparable to those of widely used synthetic polymers such as HDPE (tensile strength 50 MPa, with elongation at the break 5%) and PE (tensile strength 15–40 MPa with elongation at the break 400–600%). Remarkably, the tensile stress curves of CSL, CSH, and CSH–ALR films still exhibited a yield point, after which these materials passed from reversible elastic deformation, returning to their original dimensions after the stress was released. After the yield point, the materials experience irreversible plastic deformation with changes of their shape being permanent. In contrast, CSH–AMA films behaved as a brittle material with low elongation at the break and no yield point.
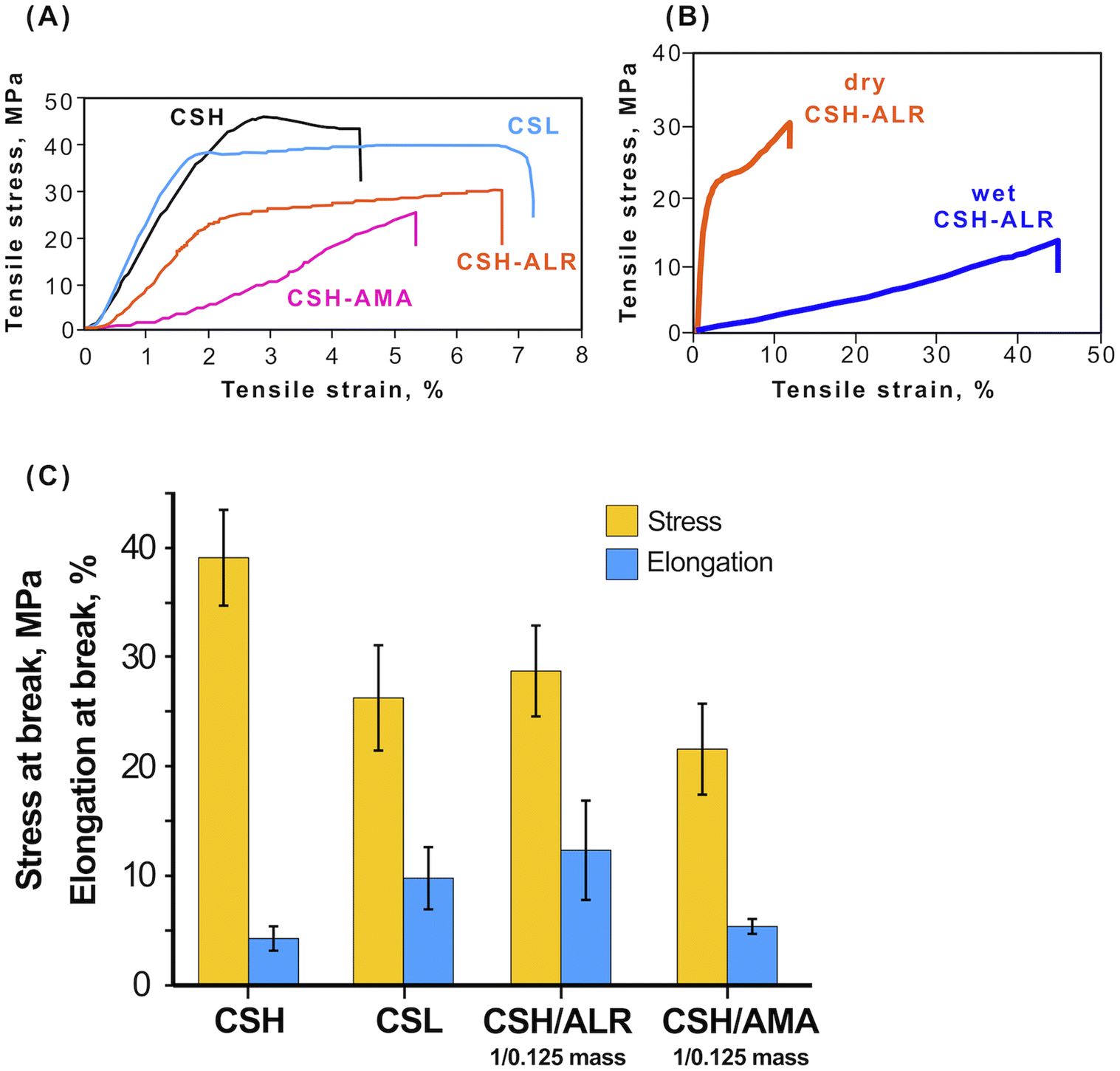 | ||
| Fig. 7 (A) Tensile stress–strain curves of CS and CS-azo dye free-standing thick films. (B) Tensile stress–strain curves of CSH–ALR film in dry and wet states. (C) Tensile strength test results for CS and CS-azo dyes thick films. | ||
Additionally, the mechanical tensile strength of CSH–ALR films (mass ratio 1:0.125) was tested in both dry and wet states (Fig. 7B). The tensile stress at break was much higher for dry films but elongation at break was 5 times larger for the wet films, indicating that properties of the films may be tunable according to the application, as for example, some biomedical applications prioritize higher elasticity of the films over toughness. The values measured for CSH–ALR (tensile strength 15–40 MPa with elongation at the break 400–600%, WVTR = 4.7–7.8 g day−1·m−2 and OTR = 2300–3100 cc (m−2 day−1)) compare reasonably well with those of standard plastic packaging.46
To demonstrate a proof-of-principle recycling ability for the CS-azo dye films, a basic recycling process was simulated as shown in Fig. 8. Pieces of the CS–AMA film were cut and mixed together with common domestic waste slurries including egg shell, organic waste, paper, polyethylene (PE), and aluminium foil. The mixture was submerged in water and under stirring was exposed to green light over 5 days. After 5 days, the water solution turned a deep red colour owing to the release of the azo dye into solution, and the liquid component was separated from the solid residue by filtration. To separate the dissolved CS from the azo dye, the pH of the solution was adjusted to basic (pH = 9) using NaOH. CS is insoluble at pH above 6.3, and therefore precipitated out of solution, leaving the azo dye dissolved. The precipitated CS was then separated by filtration and the residual azo dye removed using carbon black. The recollected CS was then redissolved in an acidic solution and solvent-cast to create a second-generation film. Fig. 9 shows the mechanical properties of the recovered CS film which had a lower tensile strength (10 MPa) and elongation at break (2%) compared to the fresh CS film (40 MPa) (12%), yet is encouraging for a first attempt. Further optimization of this recycling process is currently being studied to produce second-generation CS films with mechanical properties that are comparable to the first-generation products, such as developing a more selective filtering process (step 7) than the use of simple micro-filters used to separate CS solution from activated charcoal, which may have filtered out preferentially the high molecular weight polymer chains of CS and resulted in reduction of mechanical properties.
 | ||
| Fig. 8 Light disassembly and recovery of CS–AMA thick films in water in the presence of typical domestic waste, including PE film, eggshell and organic waste, paper, and aluminum foil. (1): Mixture of the domestic waste in water; (2): addition of CS–AMA film; (3): disassembly under white light with gentle stirring; (4): mixture on 5th day of disassembly; (5): filtrated solution of CS–AMA; (6): precipitated CS after addition of 0.1 M solution NaOH; (7): recovered CS; (8): filtrated domestic waste (paper is coloured slightly by some AMA adsorbed). | ||
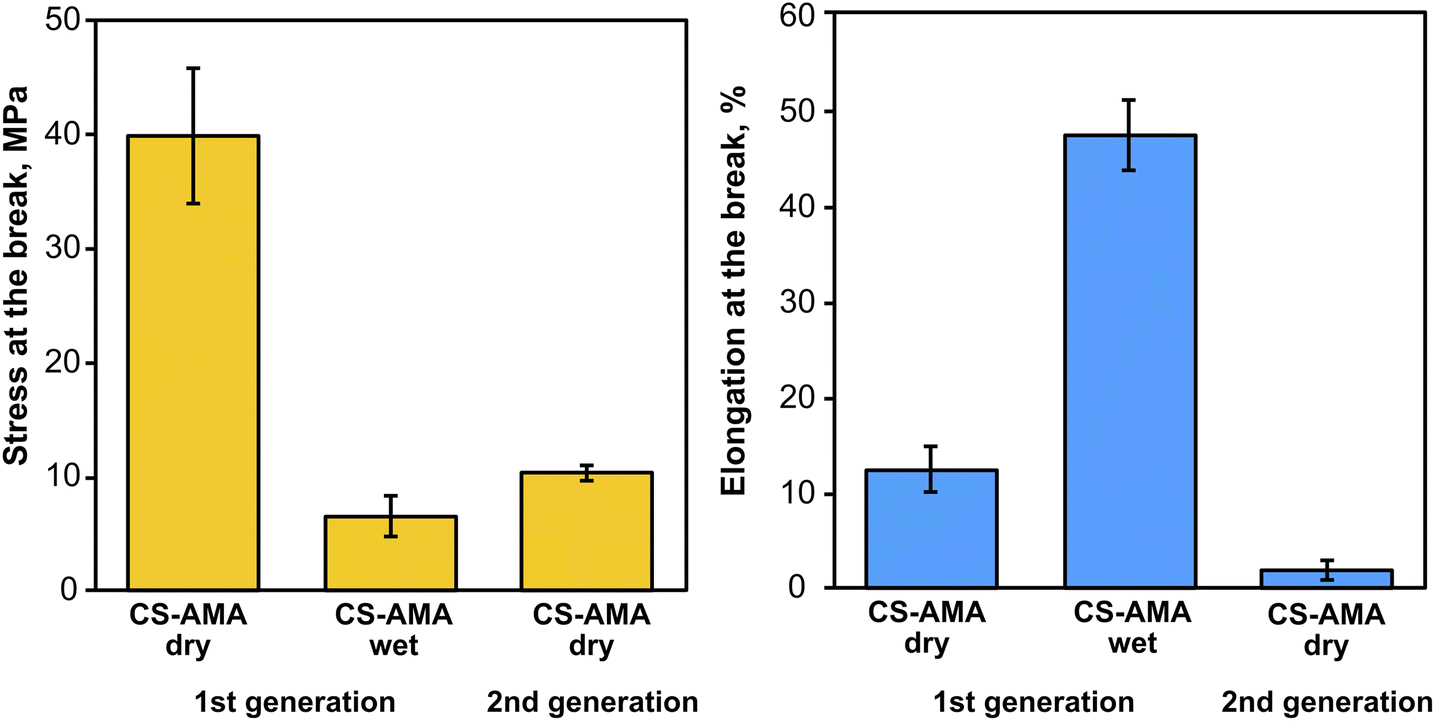 | ||
| Fig. 9 Mechanical properties of CSH–AMA thick films in dry and wet states, and after recovery (2nd generation). | ||
3.7 Interaction with water and barrier properties of the films
To be used as a packaging material, plastic must protect the enclosed goods (food, cosmetics, medication, etc.) from the environment, primarily from solvents, moisture, oxygen, and light, its so-called barrier properties. Our CS-azo dye thick films were found to be insoluble in organic solvents (ethanol, acetone, THF) as well as in the dark in water even at high temperature (70 °C). To study the interaction with water at room temperature more closely, we estimated the density of the films, tested swelling and water vapour transmission rates (WVTR), and measured contact angles. The density results of the films are presented in Table 2. The density calculated from geometric dimensions and the weight of the films were higher than density estimates obtained by gravimetric methods when the volume was measured by displacement in ethanol, suggesting that the free-standing films could be somewhat porous. The presence of azo dyes did not appear to significantly change the density, and the swelling tests showed that CS films have degree of swelling in excess of 140%. Despite the crosslinking of the chitosan films with the azo dyes, the degree of swelling of the films remained high and, surprisingly, even increased after the addition of the azo dyes to 250% (CS–ALR, mass ratio 2.5:1) and 200% (CS–AMA, mass ratio 2.5:1). The high swelling in water does not provide good barrier properties for polymer films because excess water reduces the mechanical properties of the film, although an additional partial crosslinking of CS by aldehydes could in principle potentially resolve this problem.37,47
| Film, mass ratio | Density, g cm−3 | Density, g cm−3 |
|---|---|---|
| Volume by geometry | Volume by ethanol displacement | |
| CS | 1.65 ± 0.10 | 1.54 ± 0.03 |
| CS–AMA, 2.5/1 | 1.60 ± 0.12 | 1.43 ± 0.02 |
| CS–AMA, 8/1 | 1.50 ± 0.08 | 1.38 ± 0.07 |
| CS–ALR, 2.5/1 | 1.55 ± 0.09 | 1.34 ± 0.05 |
WVTR measures the amount in grams of water vapour transmitted through a fixed area of the film per day; the lower the WVTR, the less external moisture permeates through the film, and the higher the barrier properties of the material. As shown in Table 3, the WVTR increased upon the crosslinking of the CS polymer (WVTR of pure chitosan = 59 g day−1·m−2) with azo dyes AMA and ALR, WVTR = 143–176 g day−1·m−2 for CS–AMA and WVTR = 120–162 g day−1·m−2 for CS–ALR. Such changes might be explained by an increased porosity of the films, although as described by the comparison chart on Fig. S6,† such materials can still be used as a packaging materials for some types of food such as cheese and bakery products.8 The WVTR also can be decreased by additional crosslinking of CS or by using filling materials, or by decreasing porosity by applying hot-pressing after film fabrication.
| Film | WVTR, g day−1 m−2 |
|---|---|
| CS | 59 ± 14 |
| CS–AMA mass ratio 2.5/1 | 176 ± 22 |
| CS–AMA mass ratio 5/1 | 143 ± 19 |
| CS–AMA mass ratio 8/1 | 163 ± 15 |
| CS–ALR mass ratio 2.5/1 | 146 ± 20 |
| CS–ALR mass ratio 5/1 | 162 ± 17 |
| CS–ALR mass ratio 8/1 | 120 ± 19 |
Contact angle (CA) measurements show that the CS and CS-azo dye films are strongly hydrophilic, with maximum contact angle 68±2° for CS film and lower CA for all films CS-azo dye with minimum 28±2° for CS–ALR films. To achieve more hydrophobic films, in the future a modification of CS with aldehydes (such as octanal47) might be possible.
3.8 DFT modelling of enol and keto AMA forms
Towards the development of fully recyclable bioplastics based on biodegradable polymers and food-grade azo dyes as the trigger for disassembly, mechanistic insights into the disassembly process are necessary to further tailor and optimize the material properties. Although the azo food dyes are attractive in their low toxicity, they pose a challenge for the spectroscopic verification of the isomerization process which is hypothesized to disrupt electrostatic interactions in our materials, facilitating recovery of their constituent components. Common to all the azo food dyes is an ortho-hydroxy group with respect to the azo bond which has been shown by experiment to markedly accelerate the thermal cis-to-trans isomerization rate to millisecond or sub-millisecond timescales.15,48–50 This acceleration is hypothesized to occur by a tautomeric equilibrium of azo (NN) and hydrazone (CN–N–H) forms, where the hydrazone form enables facile rotation about a weakened azo bond. Accordingly, azobenzene substitution with hydroxy groups has become one platform for the development of rapidly isomerizing azo photoswitches, especially in aqueous solution.50,51 In previous work, we mounted a DFT investigation of the cis-to-trans isomerization of ALR, suspecting isomerization rates inaccessible to our pump-probe spectroscopy equipment. We found that azo–hydrazone tautomerism weakened the azo bond and predicted a barrier of 24 kJ mol−1 for rotation at standard temperature, suggesting a rapid isomerization rate of microseconds or less.52 Theoretical investigations of the isomerization mechanisms and tautomeric equilibrium of azo food dyes are presently lacking. Therefore, we present herein quantum chemistry insights into the structure and tautomeric forms of AMA at neutral pH, where the sulphonate groups and azo bond are predicted to be unprotonated.53 This work is a first step in the characterization of AMA and its possible isomerization mechanisms.
We first investigated the enol–azo form of AMA, denoted structure 1. This structure of AMA where the azo double bond is intact is often reported in literature concerning the azo food dyes, as well as manufacturer data. All possible conformers a, b, c, and d of trans-AMA were investigated, corresponding to possible rotations of the naphthyl and naphthol rings about the azo bond as shown in Chart 1. Conformer 1c was the lowest-energy conformer, followed by 1d with a relative Gibb's free energy of 9.37 kJ mol−1 (Table 4). The orientation of the naphthol ring with respect to the azo group in both these structures enabled the formation of a pseudo-6-membered ring involving a hydrogen bond between the hydroxyl and nearby azo lone pair. In addition, this hydrogen bond tended to lock the azo group and the naphthol ring in the same plane, while the opposing naphthyl ring was twisted out of plane. This twist was commensurate with the relative energy of each conformer, where the value was 18.8° for 1c, 21.5° for 1d, and 35.5° for the highest-energy conformer 1b. In a DFT study of Ponceau 4R, an isomer of AMA, Bevziuk et al. also found that planar trans isomers were impossible, and that the lowest-energy conformer acquired a conformation which maximized the separation of the naphthyl and naphthol rings,53 as in trans-1c. Furthermore, all possible conformers of cis-1 were likewise investigated by rotating each conformer of 1 in Chart 1 about the azo bond, and then reoptimizing the resultant structure. Conformer 1a was lowest in energy, followed closely by 1c whose relative Gibb's free energy was 3.14 kJ mol−1 (Table 4). Like the trans isomer, the lowest-energy conformers were marked by the ability to form intramolecular hydrogen bonds (cis-1a), as well as the minimization of steric hindrance between the naphthol and naphthyl rings. Optimized structures of trans-1 and cis-1 minimum energy conformers are shown in Fig. 10A.
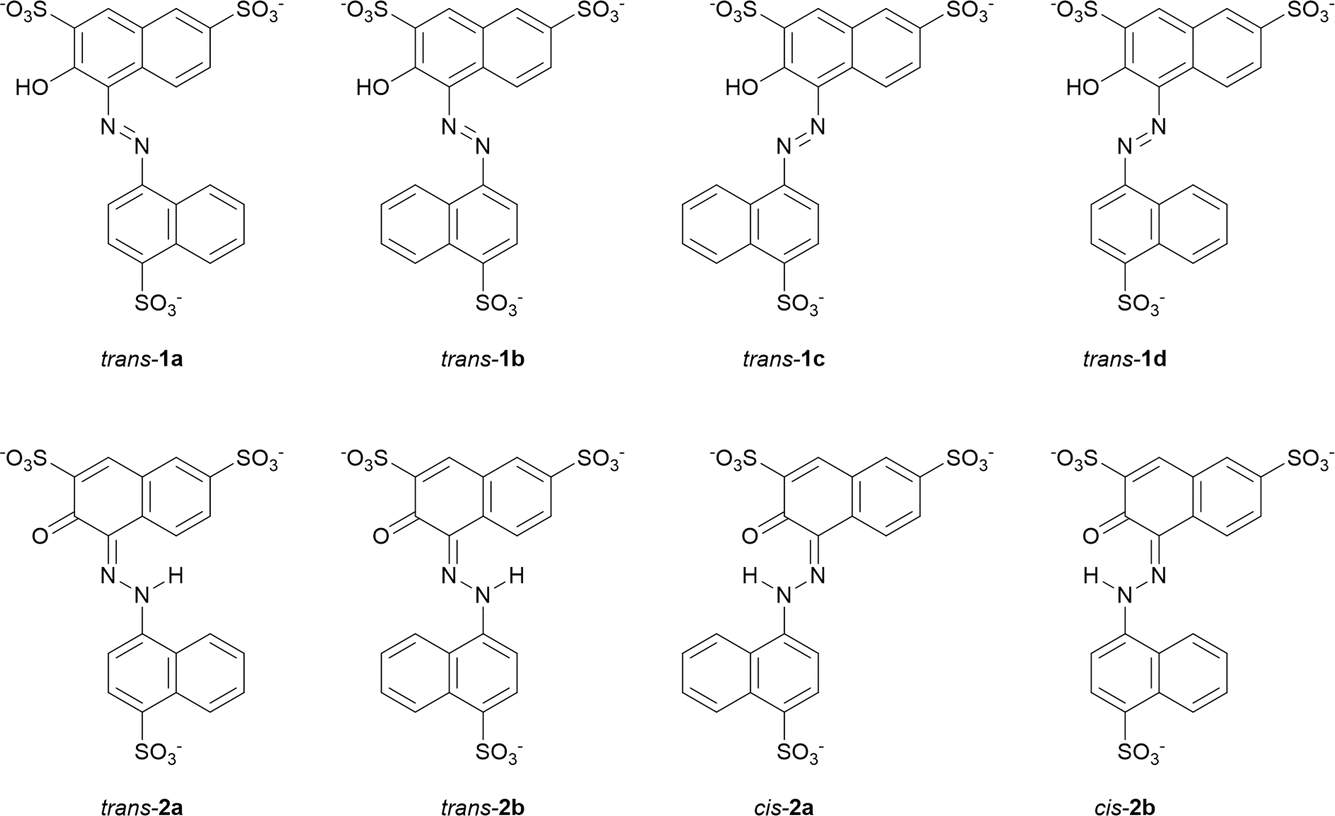 | ||
| Chart 1 Possible conformers and isomeric forms of enol–azo AMA (1) and keto–hydrazone AMA (2) investigated at the B3LYP-D3BJ/ma-def2-TZVP level. Structures for cis-1 have been omitted for simplicity. | ||
| Structure | ΔG, kJ mol−1 | |
|---|---|---|
| cis | trans | |
| 1a | 0.00 | 17.90 |
| 1b | 14.89 | 30.55 |
| 1c | 3.14 | 0.00 |
| 1d | 12.90 | 9.37 |
| 2a | 0.00 | 0.00 |
| 2b | 11.47 | 8.39 |
 | ||
| Fig. 10 DFT-optimized geometries of AMA, showing minimum-energy conformers for the trans and cis isomers of (A) the azo form 1 and (B) the hydrazone form 2. | ||
Azobenzenes ortho- or para-substituted with hydroxyl groups may exist in a tautomeric equilibrium of the enol–azo and keto–hydrazone forms. For the case of AMA, the proton is transferred from the hydroxyl group to the azo nitrogen adjacent to the naphthalene ring as shown in Scheme 1. The resonance stabilization in the hydrazone form that neutralizes the resulting charge separation concomitantly reduces the double-bond character of the azo linkage. Hydrazone dyes adopt a different naming convention from the azo dyes, where trans and cis now refer to configuration of functional groups about the CN imine bond. Moreover, literature conventions also classify the naphthol group in this case as the ‘rotor’, while the naphthyl group is the ‘stator’. For the hydrazone form, denoted structure 2, there are two possible conformers a and b for the trans and cis isomers corresponding to possible rotations of the naphthyl stator, as shown in Chart 1. In contrast to the azo form, the hydrazone form is locked into the lower energy, nearly planar cis isomer due to the formation of an intramolecular hydrogen bond between the tautomeric hydrogen and ketone oxygen in a 6-membered pseudo-ring. This hydrogen bond is lost in the trans isomer, accompanied by significant puckering of the naphthol ring due to steric interaction with the tautomeric hydrogen. Although other computational investigations tend to ignore possible rotations of the stator, our investigation explicitly verifies these assumptions, demonstrating that the rotation of the naphthyl stator in AMA is not significant. The conformers trans-2b and cis-2b presented Gibb's free energies of 8.39 kJ mol−1 and 11.47 kJ mol−1 relative to the lower energy conformers trans-2a and cis-2a, respectively (Table 4). Assuming a Boltzmann distribution of conformers, the contribution of trans-2b and cis-2b to the overall structures of the trans and cis hydrazone forms is negligible. Optimized structures of trans-2 and cis-2 minimum energy conformers are shown in Fig. 10B. Although the crystal structure of AMA has not been reported, we found agreement between our calculations and the crystallographic characterization of a similar dye, Pigment Red 40, where Heaney et al. isolated the planar cis hydrazone form and identified a covalent bond between the hydrogen atom and azo nitrogen.54 However, they found a hydrogen bond distance O⋯H of 1.85 Å, whereas our DFT calculations predicted 1.65 Å for cis-2a. This discrepancy may be attributed partly to structural differences between Pigment Red 40 and AMA, since sulphonation of the naphthol and naphthyl rings in AMA can affect the aromaticity and electronics of these rings, and hence the hydrogen bond.
 | ||
| Scheme 1 Equilibrium of enol–azo (NN) and keto–hydrazone (CN–N–H) tautomeric forms for azo dye AMA. Azo and hydrazone groups are highlighted in red. | ||
According to the relationship between the standard Gibb's free energy and the equilibrium constant for a reaction, it is possible to estimate the tautomeric equilibrium of the AMA azo and hydrazone forms. For a standard temperature and pressure of 298.15 K and 1 atm, and assuming a Boltzmann distribution of possible conformers, the standard Gibb's free energy for the unimolecular enol-to-keto (trans-1-to-cis-2) tautomerization is −6.89 kJ mol−1, corresponding to a 16:1 keto-to-enol ratio in the gas phase. Indeed, reports of other dyes based on a 1-arylhydrazone-2-naphthol skeleton have found that the keto form is more energetically favourable both in solution and the solid state, which points to an often erroneous classification of hydrazone dyes as azo dyes.54–56 In our previous work, DFT modelling of the azo food dye ALR identified an energetically favourable keto form where a nonplanar local minimum cis isomer about the azo bond could be optimized.52 Yet for AMA, this was not the case, as attempts to optimize the cis configuration about the azo bond yielded high-energy structures with respect to the hydrazone forms presented here, or simply recovered the planar hydrazone forms themselves. This observation leads us to consider possible mechanisms responsible for disassembly of the chitosan films. DFT predictions of the dipole moment show that isomerization from cis-2a to trans-2a is accompanied by a change from 8.77 D to 9.22 D, markedly less significant compared to the azo form, where isomerization from trans-1c to cis-1a results in a change from 9.89 D to 13.37 D. In addition, unlike azo isomerization where a nonplanar, disordered cis isomer is formed, hydrazone isomerization is uniquely described as an order-to-order transition57 where both isomers are nearly planar. Consequently, the typical changes associated with azobenzene isomerization—dipole moment and geometry—cannot be invoked to completely explain the mechanism of disassembly. Likely, the significant geometric change of the hydrazone isomerization process itself is responsible. This geometric change has the capability to modulate the bulk properties of soft-bonded supramolecular assemblies, which was recently shown for the chirality and polymer actuation of liquid crystals.58,59
Isomerization of the hydrazone form has been found to proceed by a keto-to-enol tautomerization which enables rotor rotation about a transient C–N bond.60–62 Other mechanisms have been reported, including in-plane inversion of the imine bond, or hybrid inversion-rotation mechanisms involving the entire hydrazone CN–N–H unit and a nonplanar intermediate.63–65 In addition, Deneva et al. used a simple explicit solvation scheme to demonstrate that water molecules present even as trace contaminants in organic solvent can catalyze the tautomerization process and lower the isomerization barrier.62 We have shown that AMA exists predominantly in the hydrazone form. Due to the loss of a favourable intramolecular hydrogen-bond upon isomerization from cis-2a to trans-2a, we suspect AMA may not exhibit bistability such as the hydrazones designed to kinetically lock supramolecular assemblies into different configurations for extended periods of time.58,59,66 Furthermore, since the presence of water in wet multilayer films can markedly accelerate the thermal isomerization process, we also suspect that, like ALR, AMA is a fast-isomerizing switch which is not easily amenable to spectroscopic verification. A thorough and comparative treatment of the azo food dyes including ALR, AMA, and others will be the subject of a forthcoming report. We plan to address the literature gap regarding the isomerization of the azo food dyes and employ multireference formulations of DFT for the correct treatment of rotational mechanisms.
4 Conclusions
We report here for the first time the fabrication of thick water-resistant films self-assembled from the water-soluble and biobased chitosan polymer, with low-toxicity food-grade azo dyes amaranth and allura red, as light-reversible crosslinkers in the material. The self-assembled dye-chitosan films possessed competitive mechanical properties (tensile strength 25–30 MPa, 3% elongation at break) and were highly water resistant. These strong, water-resistant free-standing films however underwent complete disassembly back to their water-soluble non-toxic starting materials triggered using wavelengths and intensities of visible sunlight within a few days of irradiation under running water. The chitosan/amaranth films had a relative rate of disassembly (RRD) = 117:1 in light vs. dark while the RRD for the chitosan/allura red films was 14:1. Both the polymer and photo-switchable azo dye components can be fully separated from each other and recovered to prepare second-generation smart materials in principle in a net 100% recyclable process. These new materials and their disassembly process suggest that use of sunlight for water-triggered disassembly can allow for complete recovery of reusable components for use as next-generation sustainable materials for products and recycling processes, in place of permanently insoluble oil-based plastics for some applications. DFT modeling of amaranth demonstrated that, contrary to its current classification, the dye exists predominantly in the hydrazone form, which has emerged as a class of dyes capable of significant light-triggered geometric modulation as we have shown experimentally for the films presented here. Future work on this new class of materials might include exploring other combinations of different polymers with the same dyes, to try and improve the mechanical properties. This could help address the limitations of strength of these current materials, to expand their potential application as plastics replacements beyond thin film packaging.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Mikhail Kim: conceptualization, data curation, formal analysis, methodology, validation, investigation, writing – original draft, visualization. Coral Hillel: conceptualization, data curation, formal analysis, methodology, validation, investigation, writing – original draft, writing – review & editing, visualization. Kayrel Edwards: writing – review & editing, validation, investigation. William Pietro: conceptualization, methodology, validation, project administration, funding acquisition, resources, supervision, writing – review & editing. Ozzy Mermut: conceptualization, methodology, validation, project administration, funding acquisition, resources, supervision, writing – review & editing. Christopher Barrett: conceptualization, methodology, validation, project administration, funding acquisition, resources, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the Natural Sciences and Engineering Research Council of Canada (NSERC). We also thank the Canada First Research Excellence Fund, for grants to York University through both VISTA (CFREF-2015-00013) and Connected Minds (CFREF-2022-00010) programs. The J. P. Bickell Foundation is thanked for their support.References
- D. K. A. Barnes, Nature, 2002, 416, 808–809 CrossRef CAS PubMed.
- A. L. d. F. Lacerda, L. d. S. Rodrigues, E. van Sebille, F. L. Rodrigues, L. Ribeiro, E. R. Secchi, F. Kessler and M. C. Proietti, Sci. Rep., 2019, 9, 3977 CrossRef PubMed.
- L. C. Woodall, A. Sanchez-Vidal, M. Canals, G. L. J. Paterson, R. Coppock, V. Sleight, A. Calafat, A. D. Rogers, B. E. Narayanaswamy and R. C. Thompson, R. Soc. Open Sci., 2014, 1, 140317 CrossRef PubMed.
- M. Cordier, T. Uehara, J. Baztan, B. Jorgensen and H. Yan, Ecol. Econ., 2021, 182, 106930 CrossRef.
- J. Wang, D. J. Gardner, N. M. Stark, D. W. Bousfield, M. Tajvidi and Z. Cai, ACS Sustainable Chem. Eng., 2018, 6, 49–70 CrossRef CAS.
- A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, ACS Sustainable Chem. Eng., 2020, 8, 3494–3511 CrossRef CAS.
- N. Carl, W. Müller, R. Schweins and K. Huber, Langmuir, 2019, 36, 223–231 CrossRef PubMed.
- Z.-Q. Cao and G.-J. Wang, Chem. Rec., 2016, 16, 1398–1435 CrossRef CAS PubMed.
- D. W. R. Balkenende, S. Coulibaly, S. Balog, Y. C. Simon, G. L. Fiore and C. Weder, J. Am. Chem. Soc., 2014, 136, 10493–10498 CrossRef CAS PubMed.
- M. A. Ayer, Y. C. Simon and C. Weder, Macromolecules, 2016, 49, 2917–2927 CrossRef CAS.
- Y. Ji, M. Oka and S. Honda, Polym. Chem., 2021, 12, 4621–4625 RSC.
- D. Jung, T. Rust, K. Völlmecke, T. Schoppa, K. Langer and D. Kuckling, Polym. Chem., 2021, 12, 4565–4575 RSC.
- L. Ma, R. Baumgartner, Y. Zhang, Z. Song, K. Cai and J. Cheng, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1161–1168 CrossRef CAS.
- K. E. K. Edwards, M. Kim, T. H. Borchers and C. J. Barrett, Adv. Mater., 2022, 3, 6222–6230 RSC.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- M. Ishikawa, T. Ohzono, T. Yamaguchi and Y. Norikane, Sci. Rep., 2017, 7, 6909 CrossRef PubMed.
- X. Li, D. Liu, Y. Wang, S. Xu and H. Liu, Colloids Surf., A, 2018, 555, 55–62 CrossRef CAS.
- T. Borchers, F. Topic, J.-C. Christopherson, O. S. Bushuyev, J. Vainauskas, H. M. Titi, T. Friscic and C. J. Barrett, Nat. Chem., 2022, 14, 574–581 CrossRef CAS PubMed.
- G. A. F. Roberts, Structure of Chitin and Chitosan, in Chitin Chemistry, Red Globe Press, London, ed. 1st, 1992, pp. 1–53 Search PubMed.
- Z. Yu, L. Ma, S. Ye, G. Li and M. Zhang, Carbohydr. Polym., 2020, 236, 115972 CrossRef CAS PubMed.
- T. Kean and M. Thanou, Adv. Drug Delivery Rev., 2010, 62, 3–11 CrossRef CAS PubMed.
- M. Zhang, X. H. Li, Y. D. Gong, N. M. Zhao and X. F. Zhang, Biomaterials, 2002, 23, 2641–2648 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 8, e1327 Search PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B Condens. Matter, 1988, 37, 785–789 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- J. Zheng, X. Xu and D. G. Truhlar, Theor. Chem. Acc., 2011, 128, 295–305 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2002, 4, 4285–4291 RSC.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- D. Bykov, T. Petrenko, R. Izsák, S. Kossmann, U. Becker, E. Valeev and F. Neese, Mol. Phys., 2015, 133, 1961–1977 CrossRef.
- Chemcraft – graphical software for visualization of quantum chemistry computations. Version 1.8, build 682, https://www.chemcraftprog.com.
- P. Negrea, A. Caunii, I. Sarac and M. Butnariu, Dig. J. Nanomater. Biostruct., 2015, 10, 1129–1138 Search PubMed.
- M. F. Queiroz, K. R. T. Melo, D. A. Sabry, G. L. Sassaki and H. A. O. Rocha, Mar. Drugs, 2014, 13, 141–158 CrossRef PubMed.
- C. Bauer, P. Jacques and A. Kalt, Chem. Phys. Lett., 1999, 307, 397–406 CrossRef CAS.
- R. Gong, Z. Ma, X. Wang, Y. Han, Y. Guo, G. Sun, Y. Li and J. Zhou, RSC Adv., 2019, 9, 28902–28907 RSC.
- F. Ahmed, R. Dewani, M. K. Pervez, S. Mahboob and S. A. Soomro, Bulg. Chem. Commun., 2016, 48, 71–77 Search PubMed.
- M. E. I. Badawy, T. M. R. Lotfy and S. M. S. Shawir, Bull. Natl. Res. Cent., 2019, 43, 83 CrossRef.
- E. Podgorbunskikh, T. Kuskov, D. Rychkov, O. Lomovskii and A. Bychkov, Polymers, 2022, 14, 4438 CrossRef CAS PubMed.
- N. Az-Zahra, R. Rahmi and S. Lubis, J. Phys.: Conf. Ser., 2019, 1402, 055039 CrossRef CAS.
- K. C. Krogman, R. E. Cohen, P. T. Hammond, M. F. Rubner and B. N. Wang, Bioinspiration Biomimetics, 2013, 8, 045005 CrossRef CAS PubMed.
- O. G. Piringer and A. L. Baner, Plastic Packaging, John Wiley & Sons, 2008 Search PubMed.
- Parafilm Properties, https://www.sigmaaldrich.com/deepweb/assets/sigmaaldrich/product/documents/202/568/p7668pis.pdf.
- M. Poutanen, Z. Ahmed, L. Rautkari, O. Ikkala and A. Priimagi, ACS Macro Lett., 2018, 7, 381–386 CrossRef CAS PubMed.
- J. Garcia-Amorós, A. Sánchez-Ferrer, W. A. Massad, S. Nonell and D. Velasco, Phys. Chem. Chem. Phys., 2010, 12, 13238 RSC.
- J. Garcia-Amorós and D. Velasco, Beilstein J. Org. Chem., 2012, 8, 1003–1017 CrossRef PubMed.
- J. Garcia-Amorós, M. Díaz-Lobo, S. Nonell and D. Velasco, Angew. Chem., Int. Ed., 2012, 51, 12820–12823 CrossRef PubMed.
- M. Kim, C. Hillel, K. Edwards, T. H. Borchers, O. Mermut, W. J. Pietro and C. J. Barrett, Front. Mater., 2024, 11, 1334863 CrossRef.
- K. Bevziuk, A. Chebotarev, D. Snigur, Y. Bazel, M. Fizer and V. Sidey, J. Mol. Struct., 2017, 1144, 216–224 CrossRef CAS.
- M. P. Heaney, D. K. Unruh and T. Runčevski, Cryst. Growth Des., 2022, 22, 5168–5175 CrossRef CAS.
- A. Whitaker, J. Soc. Dyers Colour., 1978, 94, 431–435 CrossRef CAS.
- A. Lyčka, Dyes Pigm., 2023, 209, 110896 CrossRef.
- B. Shao and I. Aprahamian, Chem, 2020, 6, 2162–2173 CAS.
- M. J. Moran, M. Magrini, D. M. Walba and I. Aprahamian, J. Am. Chem. Soc., 2018, 140, 13623–13627 CrossRef CAS PubMed.
- A. Ryabchun, Q. Li, F. Lancia, I. Aprahamian and N. Katsonis, J. Am. Chem. Soc., 2019, 141, 1196–1200 CrossRef CAS PubMed.
- S. M. Landge, E. Tkatchouk, D. Benítez, D. A. Lanfranchi, M. Elhabiri, W. A. Goddard III and I. Aprahamian, J. Am. Chem. Soc., 2011, 133, 9812–9823 CrossRef CAS PubMed.
- M. R. Zamanloo, R. Salmanzadeh, M. D. Esrafili, B. Seyednoruziyan, G. H. Imanzadeh and H. Eskandari, J. Phys. Org. Chem., 2023, 36, e4487 CrossRef CAS.
- V. Deneva, N. G. Vassilev, S. Hristova, D. Yordanov, Y. Hayashi, S. Kawauchi, F. Fennel, T. Völzer, S. Lochbrunner and L. Antonov, Comput. Mater. Sci., 2020, 177, 109570 CrossRef CAS.
- B. Mravec, J. Filo, K. Csicsai, V. Garaj, M. Kemka, A. Marini, M. Mantero, A. Bianco and M. Cigáň, Phys. Chem. Chem. Phys., 2019, 21, 24749–24757 RSC.
- B. Mravec, Š. Budzák, M. Medved’, L. F. Pašteka, C. Slavov, T. Saßmannshausen, J. Wachtveitl, J. Kožíšek, L. Hegedüsová, J. Filo and M. Cigáň, J. Org. Chem., 2021, 86, 11633–11646 CrossRef CAS PubMed.
- G. Wang, Y. Li, T. Song, C. Shang, J. Yang, M. Lily, Y. Fang and F. Liu, J. Phys. Chem. A, 2020, 124, 6411–6419 CrossRef CAS PubMed.
- H. Qian, S. Pramanik and I. Aprahamian, J. Am. Chem. Soc., 2017, 139, 9140–9143 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02211d |
| ‡ The first two authors MK and CH contributed equally and share first authorship. |
| This journal is © The Royal Society of Chemistry 2024 |