
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInnovation of 6-sulfonamide-2H-chromene derivatives as antidiabetic agents targeting α-amylase, α-glycosidase, and PPAR-γ inhibitors with in silico molecular docking simulation†
Hamdy Khamees Thabet *a,
Ahmed Ragab*b,
Mohd Imranc,
Mohamed Hamdy Helala,
Saleh Ibrahim Alaqelc,
Ahmed Alshehride,
Abida Ash Mohdc,
Saleh Saad Alshammaria,
Yousry A. Ammarb and
Moustafa S. Abusaifb
*a,
Ahmed Ragab*b,
Mohd Imranc,
Mohamed Hamdy Helala,
Saleh Ibrahim Alaqelc,
Ahmed Alshehride,
Abida Ash Mohdc,
Saleh Saad Alshammaria,
Yousry A. Ammarb and
Moustafa S. Abusaifb
aDepartment of Chemistry, College of Sciences and Arts, Northern Border University, Rafha, 91911, Saudi Arabia. E-mail: Hamdy.salem@nbu.edu.sa
bDepartment of Chemistry, Faculty of Science (Boys), Al-Azhar University, Nasr City, 11884, Cairo, Egypt. E-mail: ahmed_ragab@azhar.edu.eg; Ahmed_ragab7@ymail.com
cDepartment of Pharmaceutical Chemistry, College of Pharmacy, Northern Border University, Rafha 91911, Saudi Arabia
dDepartment of Pharmacology and Toxicology, College of Pharmacy, Northern Border University, Rafha 91911, Saudi Arabia
eDepartment of Pharmacology, College of Clinical Pharmacy, Imam Abdulrahman Bin Faisal University, King Faisal Road, Dammam 31441, Saudi Arabia
First published on 14th May 2024
Abstract
A new series of 2-imino or 2-oxo-2H-chromene-6-sulfonamide derivatives 2–9 with potential anti-diabetic activity were designed and synthesized. The new 6-sulfonamide chromenes were synthesized by reacting 3-formyl-4-hydroxybenzenesulfonyl chloride with activated methylene derivatives in the presence of ammonium acetate as a catalyst. The structure of the products was confirmed by spectroscopic analysis. All the designed derivatives 2–9 were evaluated for their activity against α-amylase and exhibited inhibitory percentage values higher than 93% at 100 μg mL−1. Additionally, the IC50 values represented a variable degree of activity with two derivatives 2 and 9 exhibiting the most promising derivative results with IC50 values of 1.76 ± 0.01 and 1.08 ± 0.02 μM, respectively, compared to Acarbose (IC50 = 0.43 ± 0.01 μM). Additionally, these derivatives showed potency against the α-glucosidase enzyme with IC50 values of 0.548 ± 0.02 and 2.44 ± 0.09 μg mL−1, compared to Acarbose (0.604 ± 0.02 μg mL−1). Moreover, the in vitro PPAR-γ transactivation assay revealed that chromene-6-sulfonamide derivatives 2 and 9 exhibited potential PPAR-γ activity with IC50 values of 3.152 ± 0.03 and 3.706 ± 0.32 μg mL−1, respectively, compared to Pioglitazone (4.884 ± 0.29 μg mL−1). This indicates that these derivatives have insulin sensitivity and glucose metabolism activity. The in silico ADMET prediction showed that these derivatives have an acceptable range of oral bioavailability, drug-likeness, and a safe toxicity profile, including being non-cytotoxic, non-mutagenic, non-immunotoxic, and non-carcinogenic. Finally, computational docking analysis demonstrated the ability of these derivatives to interact with α-amylase, α-glucosidase, and PPAR-γ enzymes, with confirmed successful placement due to good binding energy values and various interactions within the pocket.
1. Introduction
One of the most prevalent metabolic diseases is diabetic mellitus, which causes hyperglycemia due to defective insulin action or secretion, and this disease affects 451 million people worldwide (aged 18–99 years), according to the World Health Organization.1,2 The number of patients with diabetes is expected to increase to 693 million by 2045, according to the International Diabetes Federation (IDF).3 It has been reported that diabetes complications are associated with alterations in the body's antioxidant defense system, increased oxidative stress, and dyslipidemia.3 Many studies have shown that diabetes complications affect 20 to 90.5% of the population.4,5 Diabetes mellitus can be divided into four types: Type I diabetes mellitus, Type II diabetes mellitus, gestational diabetes mellitus (GDM), and finally, specific DM (cystic fibrosis diabetes, drug-induced diabetes, monogenic diabetes, and latent autoimmune diabetes in adults).6 It has been shown that diabetes mellitus can cause ketoacidosis and hyperosmolarity7 and chronic complications, such as peripheral and cerebrovascular disease, foot ulcers, retinopathy, and renal impairment.8 It is still unknown how to cure diabetes mellitus, but there are ways to manage it, including taking oral anti-diabetic drugs and insulin and modifying your diet.9 Moreover, in some cases, medicinal plants may provide an alternative method of treating diabetes mellitus.10 The most common long-term health complications associated with diabetes include renal failure, neurologic complications, cerebrovascular disease, limb amputation, coronary artery disease, blindness, and premature death.11,12 Most patients who take oral hypoglycemic medications suffer from gastrointestinal abnormalities, which affect more than half of patients when they are administered α-glucosidase and α-amylase inhibitors. Unabsorbed carbohydrates produce intestinal gas (colon gases), resulting in abdominal bloating, cramps, flatulence, and diarrhea.13 These chemotherapeutic medications are not recommended for people with gastrointestinal problems, whether acute or chronic, hepatic dysfunction, and renal impairment.14 Besides, to develop effective antidiabetic agents, we need to understand the adverse effects of these agents.15Coumarin is a heterocyclic compound containing one oxygen atom in its bicyclic ring and exhibited extensive application in designing new potent therapeutic molecules.16 Coumarine naturally occurs in several plants, including green tea, tonka beans, yellow sweet clover, chicory, lavender, and cassia.17 Researchers and drug developers prefer coumarin compounds due to their advantages of multiple targets and low toxicity. Coumarin and its derivatives have antimutagenic, antiviral, antioxidant,18 antibacterial,19 and anticancer activities.18,20 Moreover, the coumarin derivatives exhibited many bioactive potentials against monoamine oxidase, carbonic anhydrase, cholinesterase, urease, topoisomerase, lipoxygenase, alkaline phosphatase, histone deacetylase, α-glucosidase, and cyclooxygenase.21 Additionally, many coumarin derivatives inhibit the α-glucosidase and α-amylase.22,23 The extraction of Anacardiumouest Linn plant containing coumarins and terpenoids was shown to lower blood glucose levels in diabetic rats.24 Besides, umbelliferone (7-hydroxycoumarin) is used to reduce blood glucose levels.25,26 H. Sun et al., synthesized new coumarin–flavonoid hybrids, as compound I that displayed glucosidase inhibitory activity with IC50 value of 1.47 ± 0.07 μM (Acarbose = 224.70 ± 14.14) and amylase with IC50 value of 6.89 ± 1.17 μM (Acarbose = 2.72 ± 0.30 μM).27 Asgari et al., developed a new bis-coumarin derivatives conjugated with 1,2,3-triazole nucleus to exhibit IC50 values of compound II (IC50 = 13.0 ± 1.5 μM) and compound III (IC50 = 16.4 ± 1.7 μM) against α-glucosidase inhibitory activity.28 In addition, Elahabaadi et al., modified coumarin with dithiocarbamate and found that the introduction of a nitro group at meta or para position at the phenyl ring of carbamate moiety led to high activity against α-glucosidase enzyme with IC50 values of for compound IV (IC50 = 101.6 ± 4.7 μM) and compound V (IC50 = 85.0 ± 4.0 μM).29 Channabasappa et al., synthesized new chorine with triazole fragment compound VI, demonstrating inhibitory activity to a-amylase with IC50 = 4.11 μM compared to Acarbose = 2.66 μM (ref. 23) Fig. 1.
 | ||
| Fig. 1 Structure of the previously reported chromone derivatives as anti-diabetic agents as α-glucosidase and α-amylase inhibitors. | ||
Furthermore, the scientific community's interest became more attractive to the sulfonamide (–SO2NH2) core due to the wide variety of biological activity, such as anti-HIV, antimalarial, antithyroid, anticancer, high ceiling diuretic, insulin-releasing antidiabetic, carbonic anhydrase activity, and antimicrobial activity.30–36 Recently, in 2022, the FDA approved some new sulphonamides as Mitapivat is a pyruvate kinase activator (used to treat hemolytic anemia),37 Baricitinib as a selective Janus Kinase 1 (JAK 1) inhibitor (used to treat atopic dermatitis and severe rheumatoid arthritis),38 Vonoprazan is an acid blocker for potassium-competitive (used to treat H. pylori), and Omidenepag isopropyl used to reduce the intraocular pressure for glaucoma patients.28
As a result of all the findings above, we urgently need additional drugs to compensate for the shortcomings of current drugs. Moreover, modification of the bioactive core to design a new heterocyclic for a specific target is our research group goal.39–41 Herein, this article synthesized a new 2H-chromene 6-sulfonamide. Moreover, all the designed derivatives are chosen to investigate the in vitro α-amylase inhibitory activity. The most active members were also used for further evaluation against α-glycosidase and PPAR-γ. Finally, the in silico ADMET and docking simulation was undertaken to determine the oral bioavailability, toxicity, and binding mechanism, including the interacting groups inside the active site of the pocket.
2. Results and discussion
2.1. Chemistry
Due to the high simplicity, low molecular weight, and significant reactivity of 3-formyl-4-hydroxybenzenesulfonyl chloride (1), the authors planned to use it as a starting material to construct and build up a novel bioactive chromene sulphonamide derivative. As a highly pure compound, the starting material (1) was obtained from ChemSpider with ID: 74821554. Also, the starting material included two electrophile active centers, known as the carboxaldehyde and the sulfonyl chloride groups. Meanwhile, the sulfonyl chloride group is more highly reactive toward some nucleophiles than the carboxaldehyde function, but this reactivity is not controlled in most cases. Moreover, the preparation of 3-formyl-4-hydroxybenzenesulfonamide is the main target for most chemists and biologists due to the high use of substituted benzene sulphonamide moiety (Ar–SO2NH2) that exhibited many biological activities, such as anti-bacterial agent,42 anti-cancer agent,43 anti-Alzheimer disease,44 anti-diabetics,45 and anti-inflammatory.46 Unluckily, the synthetic pathways for preparing 3-formyl-4-hydroxybenzenesulfonamide as an intermediate are tricky and incompletely successful due to impurities and low yield percentage. This reason is explained through the reaction of 3-formyl-4-hydroxybenzenesulfonyl chloride (1) with ammonia (NH3) which produced mixtures of the desired 3-formyl-4-hydroxybenzenesulfonamide (low yield) and undesired 4-hydroxy-3-(iminomethyl)benzene-sulfonamide (high yield). Therefore, the authors planned to produce the target bioactive materials via a one-pot three components reaction: 3-formyl-4-hydroxybenzenesulfonyl chloride (1), different activated methylene, and ammonium acetate (CH3COONH4). Additionally, the ammonium acetate used in this reaction act as a reagent to produce the sulphonamide moiety (–SO2NH2) and as a catalytic base to produce the cyclic chromene moiety in the presence of active methylene derivatives. The synthetic routes to prepare novel eight compounds are depicted in Schemes 1–3. | ||
| Scheme 1 Illustrate synthesis of 3-substituted-2-imino-2H-chromene-6-sulfonamide 2 & 3 and 3-substituted-2-oxo-2H-chromene-6-sulfonamide 4 & 5. | ||
 | ||
| Scheme 2 Mechanism illustrated the synthetic approach of 3-cyano-2-imino-2H-chromene-6-sulfonamide 2. | ||
 | ||
| Scheme 3 Synthesis of the target N-(substitutedphenyl)-2-imino-6-sulfamoyl-2H-chromene-3-carboxamide 6–9. | ||
Firstly, the sulfonyl chloride derivative 1 is allowed to react with an equimolar amount of malononitrile in the presence of ammonium acetate via one-pot synthesis, yielding the corresponding 3-cyano-2-imino-2H-chromene-6-sulfonamide (2) as a high pure biomaterial (Scheme 1). The authors examined the physical properties of this product as a light-orange powder, in an excellent yield, with recorded melting point at 190–192 °C.
The chemical structure of the designed derivatives was confirmed using elemental analysis and spectral data. So, the IR data of compound 2 displayed the lack of absorption band at around 1680 cm−1 related to the carboxaldehyde group (–CHO). Also, the IR chart revealed new sharp stretching frequencies at ν 3414, 3335, 3233 cm−1 related to the NH2 sulfonamide moiety and NH of the 2-imino chromene (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH), besides absorption bands at ν 2203, 1620, 1592, 1399, and 1185 cm−1 assignable to CN, CNH, CC, and SO2 functions, respectively. Moreover, its 1H NMR spectra showed two singlet signals in the down-field region at δ 8.76 and 9.39 ppm due to 4H-pyran and NH proton. Also, other singlet proton signals at δ 8.10 ppm due to the NH2 function of sulfonamide moiety, aromatic protons as doublet signal at δ 7.32 ppm corresponding to the proton of H8-chromene with coupling constant (J = 8.8 Hz), doublet of doublet signal equivalent to H7-chromene at δ 7.86 ppm with coupling constant (J = 8.4, 4.0 Hz), and singlet signal at 8.39 ppm for the H5-chromene. Furthermore, the 13C NMR spectrum represented only ten signals equivalent to the compound's total carbon atoms. The chart displayed signals at δ 96.01, 111.45, and 116.21 ppm related to carbon attached to the cyano group, C4 of pyran moiety, and cyano group, respectively. Although, in the downfield, other signals appeared at δ 138.12, 157.90, and 160.53 ppm related to carbon attached to sulfonamide moiety, carbon of (O–CC), and carbon attached to imino function (CNH), respectively. Besides signals of aromatic carbons in the middle region ranging between δ 118.25–131.91 ppm equivalent to four aromatic carbons.
NH), besides absorption bands at ν 2203, 1620, 1592, 1399, and 1185 cm−1 assignable to CN, CNH, CC, and SO2 functions, respectively. Moreover, its 1H NMR spectra showed two singlet signals in the down-field region at δ 8.76 and 9.39 ppm due to 4H-pyran and NH proton. Also, other singlet proton signals at δ 8.10 ppm due to the NH2 function of sulfonamide moiety, aromatic protons as doublet signal at δ 7.32 ppm corresponding to the proton of H8-chromene with coupling constant (J = 8.8 Hz), doublet of doublet signal equivalent to H7-chromene at δ 7.86 ppm with coupling constant (J = 8.4, 4.0 Hz), and singlet signal at 8.39 ppm for the H5-chromene. Furthermore, the 13C NMR spectrum represented only ten signals equivalent to the compound's total carbon atoms. The chart displayed signals at δ 96.01, 111.45, and 116.21 ppm related to carbon attached to the cyano group, C4 of pyran moiety, and cyano group, respectively. Although, in the downfield, other signals appeared at δ 138.12, 157.90, and 160.53 ppm related to carbon attached to sulfonamide moiety, carbon of (O–CC), and carbon attached to imino function (CNH), respectively. Besides signals of aromatic carbons in the middle region ranging between δ 118.25–131.91 ppm equivalent to four aromatic carbons.
In the same way, the other aliphatic activated methylene, such as 2-amino-1,1,3-propenetricarbonitrile, ethyl acetoacetate, and diethyl malonic acid is reacted with sulfonyl chloride salicylaldehyde 1 in the presence of ammonium acetate as a reagent and catalyst, afforded the corresponding 3-(1-amino-2,2-dicyanovinyl)-2-imino-2H-chromene-6-sulfonamide (3), 3-acetyl-2-oxo-2H-chromene-6-sulfonamide (4), and ethyl 2-oxo-6-sulfamoyl-2H-chromene-3-carboxylate (5), respectively (Scheme 1). The physical properties of the three bioactive materials were examined as pure materials and recorded in the experimental section. Also, the chemical structures of the same compounds were confirmed using elemental analysis and spectral data (IR, 1H NMR, and 13C NMR). For example, the IR analysis of 2-oxo-chromene derivative 5 confirmed the presence of strong absorption frequencies at ν 3183, 3064, 1765, and 1710 cm−1 related to the NH2 of sulfonamide function, sp2-CH group, and two carbonyl functions, respectively. Also, two frequencies of sulfonyl moiety (SO2) at ν 1366, 1180 cm−1. Continuously, 1H NMR spectral of the same compound revealed new signals shielded in the up-field region as triplet signal at δ 1.29 ppm due to methyl protons of ester and quartet signals for methylene protons of ester at δ 4.27 ppm, as well as two significant singlet signals at δ 6.78 and 8.96 ppm related to the amino protons (NH2) and 4H-pyran, respectively. Meanwhile, the aromatic protons appeared as two doublet signals at δ 7.61 and 7.92 ppm with the same coupling constant (J = 7.2 Hz) identified to H8 and H7-chromene, respectively. In addition, a singlet signal appears at δ 8.40 ppm H5-chromene. Furthermore, its 13C NMR showed two signals at δ 13.99 and 60.58 ppm owing to carbons of the ester group, respectively. In addition, signals at δ 140.33, 156.58, 161.62, and 168.14 attributed to carbon attached to the sulfonyl group, carbon attached to (O–CC), carbonyl of 2-oxo chromene, and carbonyl of the ester group, respectively.
As described in Scheme 2, the mechanistic pathway illustrated the synthetic pathway for producing the target bioactive 2-imino-2H-chromene derivative 2 as an example. Firstly, the mechanistic pathway moves forward via the nucleophilic addition of NH3 (liberated from ammonium acetate) to the sulfonyl chloride group (SO2Cl) to produce the sulfonamide moiety (intermediate i) by liberating the HCl that reacted with an excess of ammonia and formed ammonium chloride (NH4Cl) as a by-product. Secondly, the ammonium acetate works as a source of ammonia that pulls a proton from malononitrile to form an active carbanion malononitrile, which is allowed to react with the formyl group in (intermediate i) via condensation reaction to yield the non-isolable (intermediate ii) that subsequently underwent intramolecular cyclization through nucleophilic attack of the phenolic hydroxyl (OH) group to the nitrile function to furnish the desired product 2.
Furthermore, the authors planned to produce some bioactive chromene containing carboxamide group via only a simple one-pot reaction process. According to the previous strategy, the starting material 1 is heated under reflux with different active cyano-acetanilide derivatives A1–A4 in the presence of ammonium acetate and yielding the corresponding N-(substitutedphenyl)-2-imino-6-sulfamoyl-2H-chromene-3-carboxamide 6–8 (Scheme 3). The IR spectra of the carboxamide derivatives 6–8 revealed characteristic frequencies around ν 3431–3430 cm−1 related to the NH2 and NH groups, besides two distinct bands related to the carbonyl groups of 2-oxo-chromene and the carbonyl of the amide function at ν 1727–1726, and 1655–1654 cm−1, respectively. The 1H NMR data of compound 6 exhibited four sharp singlet signals at δ 6.90, 8.43, and 10.46 ppm due to the amino protons, H4-chromene, and NH proton, respectively. The 19F NMR spectra of compounds 6 and 7 revealed signals at δ −118.03 and −61.28 ppm related to C–F and CF3, respectively, and these values fall within the range of the reported values.47,48
Finally, the bioactive material ethyl 4-(2-oxo-6-sulfamoyl-2H-chromene-3-carboxamido)benzoate (9) was isolated through the reaction of compound 1, ethyl 4-(2-cyanoacetamido)benzoate (A4), and ammonium acetate in ethanolic solution under reflux condition for 5 hs and in the presence of ammonium acetate as a catalyst. In an acceptable yield, the compound is isolated as a pale-orange powder with melting point = 233–235 °C. The IR spectra viewed bands at ν 3430, 1726, and 1655 cm−1 due to NH2, carbonyl of 2-oxo chromene, and the carbonyl of the amide function, respectively. Its 1H NMR data displayed the two signals in the up-field region at δ 1.24 and 4.18 ppm corresponding to the ester protons as a triplet and quartet splitting and signals de-shielded at δ 5.91, 7.99, and 10.23 ppm assignable to the amino of sulfonamide, H4-chromene, and NH amide protons, respectively. Besides, the aromatic protons ranged from δ 6.92–7.60 ppm. The 13C NMR spectra of the same compound displayed significant signals at δ 14.76, 61.03, 157.58, 163.46, and 166.20 ppm, equivalent to methyl carbon, methylene carbon, carbonyl of 2-oxo chromene, amide carbonyl carbon, and carbonyl of ester group respectively.
2.2. Antidiabetic activity
| Cpd no. | The α-amylase activity | |
|---|---|---|
| Inhibitory percentage at 100 μg mL−1 | IC50 ± SD (μM) DF = 100 | |
| 2 | 96.1 | 1.76 ± 0.01 |
| 3 | 96 | 14.62 ± 0.16 |
| 4 | 94.2 | 8.55 ± 0.31 |
| 5 | 93.4 | 32.58 ± 0.33 |
| 6 | 95.4 | 2.11 ± 0.03 |
| 7 | 96.2 | 4.39 ± 0.06 |
| 8 | 95.4 | 4.23 ± 0.06 |
| 9 | 96.6 | 1.08 ± 0.02 |
| Acarbose | 96.5 | 0.43 ± 0.01 |
As represented in Table 1, the 2-oxo-6-sulfamoyl-2H-chromene-3-carboxamide derivative 9 emerged the most active hypoglycemic agent by inhibiting the α-amylase enzyme with IC50 value of 1.08 ± 0.02 μM. In addition, the 3-cyano-2-imino-2H-chromene-6-sulfonamide (2) revealed the second active member among the synthesized derivatives with IC50 value of 1.76 ± 0.01 μM compared to Acarbose (IC50 = 0.43 ± 0.01 μM). The activity of the synthesized derivatives was displayed due to two positions only in the chromene nucleus: the imino or carbonyl at position two and the fragments at position three (cyano, vinyl, ethyl acetate, acetyl, and carboxamide). Firstly, for the imino group, grafting the cyano as compound 2 increases the inhibitory potency by nearly 8.31-fold when replaced with 1-amino-2,2-dicyanovinayl derivative 3, indicating that an increase in the conjugation system at position three decreases the activity.
On the other hand, the IC50 values displayed introducing the carboxamide moiety with different substituents at position three of chromenes 6–9 demonstrated inhibitory activity with IC50 values ranging from 1.08 ± 0.02 to 4.39 ± 0.06 μM (i.e., below 5 μM). Generally, introducing the electron-withdrawing groups at the para position is more effective than the meta position. Additionally, introducing N-(4-fluorophenyl)-3-carboxamide fragment showed IC50 value of 2.11 ± 0.03 μM, while replacing the fluoro with chloro atom decreased the α-amylase activity enzyme with IC50 value of 4.23 ± 0.06 μM. Further, the replacement of N-(4-fluorophenyl)-3-carboxamide in compound 6 with -N-(3-(trifluoromethyl)phenyl), as showed in compound 7 to increase the number of fluorine atoms, reduces the activity with nearly two folds and demonstrated IC50 value of 4.39 ± 0.06 μM. To our surprise, replacing the halogenated atoms (4-F and 4-Cl) or 3-trifluoromethyl with ethyl carboxylate group (COOEt) causes an increase in the activity with IC50 value of 1.08 ± 0.02 μM. Furthermore, replacing the carboxamide on 2-oxo-2H-chromene pharmacophore with ethyl carboxylate or acetyl group causes reduction in the activity with IC50 values for compound 4 (IC50 = 8.55 ± 0.31 μM) and compound 5 (IC50 = 32.58 ± 0.33 μM) indicating the presence of amide group and its aromatic moiety is important in activity Fig. 2.
 | ||
| Fig. 2 General structure with different substituents that exhibited the activity and SAR study of the newly designed chromene derivatives 2–9. | ||
Finally, the designed chromene demonstrated α-amylase inhibitory activity. Additionally, the SAR study showed that the carboxamide in position three and the carbonyl group in position two are essential in reducing glucose levels by inhibiting the α-amylase enzyme. Moreover, any change in these two groups by introducing esters or acetyl instead of carboxamide doesn't enhance the α-amylase activity, except for inserting a cyano group at position three and replacing carbonyl with an imino group.
| Cpd no. | α-Glucosidase activity | |
|---|---|---|
| Inhibitory potential at 10 μg mL−1 | IC50 ± SD (μg mL−1) DF = 100 | |
| 2 | 90.7 | 0.548 ± 0.02 |
| 9 | 83.8 | 2.44 ± 0.09 |
| Acarbose | 90.1 | 0.604 ± 0.02 |
| Cpd No | PPAR-γ | |
|---|---|---|
| IC50 ± SD (μg mL−1) | Folds | |
| 2 | 3.152 ± 0.03 | 1.55 |
| 9 | 3.706 ± 0.32 | 1.32 |
| Pioglitazone | 4.884 ± 0.29 | 1 |
2.3. In silico toxicity and drug likeness prediction
Recently, there has been great concern about the toxicity of developed molecules to be used as candidate drugs,49,50 so in this section, we study the toxicity prediction of the most active derivatives 2 and 9, as well as positive controls (Acarbose and Pioglitazone). The toxicity prediction was performed by constructing the structure using the previously reported method,51,52 where the structure was exported to the ProTox-II prediction web server tool (https://tox-new.charite.de/protox_II/last access 28/4/2024).The most active 6-sulfamoyl-2H-chromene derivatives (2 and 9) and positive controls (Acarbose and Pioglitazone) exhibited a safe profile to non-cytotoxic, non-mutagenic, non-immunotoxic (except for Acarbose that showed activity with a probability value of 0.99), and non-carcinogenic. Fortunately, 3-cyano-2-imino-2H-chromene-6-sulfonamide (2) revealed non-hepatotoxic activity compared to other tested compound 9 and positive controls. Moreover, 6-sulfamoyl-2H-chromene derivatives 9 and Pioglitazone belong to class IV in the toxicity class with LD50 expected values of 1000 mg kg−1, while the 2-imino-2H-chromene-6-sulfonamide derivative 2 (class V with LD50 = 4870 mg kg−1) and Acarbose (class VI with LD50 = 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mg kg−1). The tested derivatives were predicted to be inactive members to in term of heat shock factor response element (HSE) (increase in temperature), mitochondrial membrane potential (MMP), and phosphoprotein p53 gene (Table 4).
000 mg kg−1). The tested derivatives were predicted to be inactive members to in term of heat shock factor response element (HSE) (increase in temperature), mitochondrial membrane potential (MMP), and phosphoprotein p53 gene (Table 4).
| Oral toxicity prediction | Most active 6-sulfamoyl-2H-chromene derivatives (2 and 9) and positive controls Acarbose and Pioglitazone | ||||
|---|---|---|---|---|---|
| 2 | 9 | Acarbose | Pioglitazone | ||
| a For ADMETlab2.0 the values described as: −3 = (high safe with probability ∼ 0.0–0.1); −2 = (moderate safe with probability ∼ 0.1–0.3); −1 = (safe with probability ∼ 0.3–0.7); +1 = (low risk with probability ∼ 0.5–0.7); +2 = (moderate risk with probability ∼ 0.7–0.9); and +3 = (high risk with probability ∼ 0.9–1.0). | |||||
| Oral toxicity prediction | |||||
| ProTox-II prediction | LD50 mg kg−1 | 4870 | 1000 | 24000 |
1000 |
| Toxicity class | V | IV | VI | IV | |
| Hepatotoxicity | Inactive | Active | Active | Active | |
| 0.66 | 0.53 | 0.65 | 0.51 | ||
| Carcinogenicity | Inactive | Inactive | Inactive | Inactive | |
| 0.50 | 0.62 | 0.84 | 0.52 | ||
| Immunotoxicity | Inactive | Inactive | Active | Inactive | |
| 0.99 | 0.99 | 0.99 | 0.89 | ||
| Mutagenicity | Inactive | Inactive | Inactive | Inactive | |
| 0.72 | 0.65 | 0.76 | 0.71 | ||
| Cytotoxicity | Inactive | Inactive | Inactive | Inactive | |
| 0.80 | 0.74 | 0.70 | 0.73 | ||
| Neurotoxicity | Inactive | Inactive | Inactive | Active | |
| 0.75 | 0.84 | 0.60 | 0.83 | ||
| Nephrotoxicity | Inactive | Active | Active | Inactive | |
| 0.52 | 0.59 | 0.80 | 0.50 | ||
| Cardiotoxicity | Inactive | Inactive | Active | Inactive | |
| 0.74 | 0.54 | 0.60 | 0.52 | ||
| Ecotoxicity | Inactive | Inactive | Inactive | Inactive | |
| 0.76 | 0.81 | 0.66 | 0.58 | ||
| Clinical toxicity | Inactive | Active | Active | Active | |
| 0.57 | 0.55 | 0.65 | 0.86 | ||
| Nutritional toxicity | Inactive | Inactive | Inactive | Inactive | |
| 0.70 | 0.60 | 0.52 | 0.56 | ||
| Heat shock factor response element (HSE) | Inactive | Inactive | Inactive | Inactive | |
| 0.97 | 0.97 | 0.98 | 0.98 | ||
| Mitochondrial membrane potential (MMP) | Inactive | Inactive | Inactive | Inactive | |
| 0.78 | 0.78 | 0.97 | 0.57 | ||
| Phosphoprotein (tumor suppressor) p53 | Inactive | Inactive | Inactive | Inactive | |
| 0.90 | 0.93 | 0.97 | 0.91 | ||
| ADMETLab2.0 | Eye corrosion | −3 | −3 | −3 | −3 |
| Eye irritation | −2 | −3 | −3 | −3 | |
| Respiratory toxicity | +2 | −3 | −3 | −2 | |
| Skin sensitization | −3 | −3 | −3 | −1 | |
| Half life time (t1/2 = h) | 0.669 | 0.154 | 0.835 | 0.311 | |
| Clearance (mL min−1 kg−1) | 1.737 | 0.639 | 0.681 | 8.271 | |
Moreover, our work was extended to study the prediction of additional toxicity items based on ProTox-3.0 – prediction of toxicity of chemicals web tools. The results showed that the most active 6-sulfamoyl-2H-chromene derivatives 2 and 9 exhibited inactive properties for neurotoxicity, cardiotoxicity, ecotoxicity, clinical toxicity, and nutritional toxicity, except for ethyl 4-(2-oxo-6-sulfamoyl-2H-chromene-3-carboxamido)benzoate (9) which showed activity for neurotoxicity and clinical toxicity with probability values of 0.59 and 0.55, respectively. Additionally, Acarbose and Pioglitazone demonstrated safe profiles for ecotoxicity and nutritional toxicity with high probability values. In contrast, Pioglitazone showed active properties for neurotoxicity (P ∼ 0.83) and clinical toxicity (P ∼ 0.86). On the other hand, Acarbose exhibited active properties for nephrotoxicity (P ∼ 0.80), cardiotoxicity (P ∼ 0.60), and clinical toxicity (P ∼ 0.65).
Furthermore, some other important toxicity prediction items and excretion properties such as [half lifetime (t1/2 = h) and clearance (mL min−1 kg−1)] were predicted using ADMETLab2.0 web server (https://admetmesh.scbdd.com/service/evaluation/index last accessed 26/1/2024). The tested derivatives and drugs showed non-corrosive and non-irritant for the eye, non-sensitizer for the skin, and non-respiratory toxic with probability properties ranging from moderate to high safe, except for 3-cyano-2-imino-2H-chromene-6-sulfonamide (2) that demonstrated respiratory toxicity with probability value between 0.7-0.9 indicating moderate risk. The Pioglitazone exhibited the lowest safety profile with non-sensitizer to the skin (probability values ∼ 0.3–0.7) and an alarming pharmacophore related to the thiazole ring. Additionally, all tested compounds featured short half-time values from 0.154 to 0.835 h, as well as low clearance values ranging between 0.639 and 1.737 mL min−1 kg−1, except for Pioglitazone which displayed a moderate clearance value of ∼8.271 mL min−1 kg−1 (Table 4).
The designed 6-sulfamoyl-2H-chromene derivatives and Pioglitazone do not violate the Lipinski rule (RO5), indicating that these derivatives in clinical trials tend to be shorter and, therefore, have a better chance of success. However, Acarbose doesn't obey RO5 due to three violations violations: MW > 500, NorO > 10, NHorOH > 5. Also, 2H-chromene derivatives 2 and Pioglitazone passed with the Veber rule, while 2H-chromene derivatives 9 and Acarbose failed due to TPSA > 140 Å. Additionally, the 6-sulfamoyl-2H-chromene derivatives 2 and 9 exhibited soluble properties with logS (ESOL) values of −2.06 and 3.46, respectively. On the other hand, they showed highly soluble properties logS (ESOL) = −2.13, while Pioglitazone showed moderate solubility logS (ESOL) = −4.31. For synthetic accessibility, the 2H-chromene derivatives (2 and 9) and Pioglitazone exhibited easy synthesis with values of 2.89, 3.21, and 3.46, respectively. Acarbose represented difficult synthetic accessibility with a probability of 7.34. All derivatives demonstrated good bioavailability scores with probability values ranging from 0.17 to 0.55 (Table 5).
| Test items | Most active 6-sulfamoyl-2H-chromene derivatives (2 and 9) and positive controls Acarbose and Pioglitazone | ||||
|---|---|---|---|---|---|
| 2 | 9 | Acarbose | Pioglitazone | ||
| Molecular properties | |||||
| a M. Soluble = moderately soluble; H. Soluble = highly soluble. | |||||
| SwissADME | MLogP | −0.57 | 1.13 | −6.94 | 2.01 |
| TPSA (Å2) | 129.32 | 154.15 | 321.17 | 93.59 | |
| MW | 249.25 | 416.40 | 645.60 | 356.44 | |
| nHBA (NO) | 6 | 8 | 19 | 4 | |
| nHBD (OHNH) | 2 | 2 | 14 | 1 | |
| NRB | 1 | 7 | 9 | 7 | |
| Fraction Csp3 | 0.00 | 0.11 | 0.92 | 0.32 | |
| Drug likeness and medicinal chemistry prediction | |||||
| LogS (ESOL) |
−2.06 | 3.46 | −2.13 | −4.31 | |
| Solubility | Soluble | Soluble | H. Soluble | M. Soluble | |
| PAINS | 0 | 0 | 0 | 0 | |
| Lead-likeness | No (1) | No (1) | No (2) | No (2) | |
| Synthetic accessibility | 2.89 | 3.21 | 7.34 | 3.46 | |
| Bioavailability score | 0.55 | 0.55 | 0.17 | 0.55 | |
| Lipinski rule (violation) | Yes (0) | Yes (0) | No (3) | Yes (0) | |
| Veber rule (violation) | Yes (0) | No (1) | No (1) | Yes (0) | |
2.4. In silico molecular docking simulation
To determine the conformation and ligand–protein interaction of the most active derivatives 2 and 9 that exhibited the activity with lower IC50 values on α-amylase, α-glucosidase, and PPAR-γ, the docking simulation carried out compared to the positive controls used in each in vitro study (Acarbose and Pioglitazone), as well as antidiabetic drugs have sulphonamide moiety e.g., Chlorpropamide, Glibenclamide. The docking simulation was performed using Molecular Operating Environmental (MOE) 10.2009 software53,54 inside the active site of α-amylase (PDB: 2QV4), α-glucosidase (PDB: 3w37), and PPAR-γ (PDB: 3SZ1).55–57 | ||
| Fig. 3 3D interaction of (A) co-crystallized Acarbose as superimposable (green is the original pose, while turquoise is our pose), (B) compound 2, and (C) compound 9 inside the active site of α-amylase (PDB: 2QV4). | ||
The 3-cyano-2-imino-2H-chromene-6-sulfonamide (2) showed binding energy S = −8.88 kcal mol−1 and interacted with pocket through two sidechain H-bonds acceptor with Tyr151 with nitrogen of cyano group (3.3 Å and strength of 13%) and His201 with nitrogen of imino group at position two of chromene (2.1 Å and strength of 12%). Moreover, the amino acid residue Asp300 located on the sidechain formed H-bond donor with the amino group of sulfonamide moiety with a distance of 2.5 Å and strength of 13% (Fig. 3b). Additionally, this compound exhibited hydrophobic interactions with Glu233, Tyr58, and Tyr62. Meanwhile, the 2-oxo-6-sulfamoyl-2H-chromene-3-carboxamide 9 displayed the highest binding energy S = −9.29 kcal mol−1, confirming the in vitro α-amylase's IC50 = 1.08 ± 0.02 μM. Compound 9 demonstrated two H-bonds interaction with His201 (sidechain acceptor) and Asp300 (sidechain donor) as the interaction of co-crystallized ligands with bond lengths of 2.0 Å and strength of (27 and 32%), respectively. In addition, the compound's center is located over the phenyl ring of Tyr151, exhibiting arene–arene interaction and hydrophobic interaction inside the pocket (Fig. 3c). Moreover, the docking simulation of Chlorpropamide and Glibenclamide, as antidiabetic drugs with a sulphonamide moiety, exhibited higher binding energy than the designed derivatives 2 and 9. Chlorpropamide showed a binding energy of S = −2.99 kcal mol−1, with one hydrogen bond sidechain donor between the residue Asp300 and NH of the sulfonamide group (NH–SO2−), with a bond length of 2.0 Å and a strength of 14% Fig. S1 and S2.† On the other hand, Glibenclamide demonstrated a binding energy of S = −4.98 kcal mol−1 through an arene–arene interaction between the m-chloro phenyl group and Tro59 Fig. S3 and S4 (see ESI for figures).†
O] and the sulfonamide group [–NH–SO2−] in the Glibenclamide exhibited ligand exposure with the pocket. They appeared blue color through the 2D diagram Fig. S7 and S8 (see ESI for figures).†
 | ||
| Fig. 4 3D interaction of (A) co-crystallized Acarbose as superimposable (green is the original pose, while turquoise is our pose), (B) compound 2, and (C) compound 9 inside the active site of α-glucosidase (PDB: 3w37). | ||
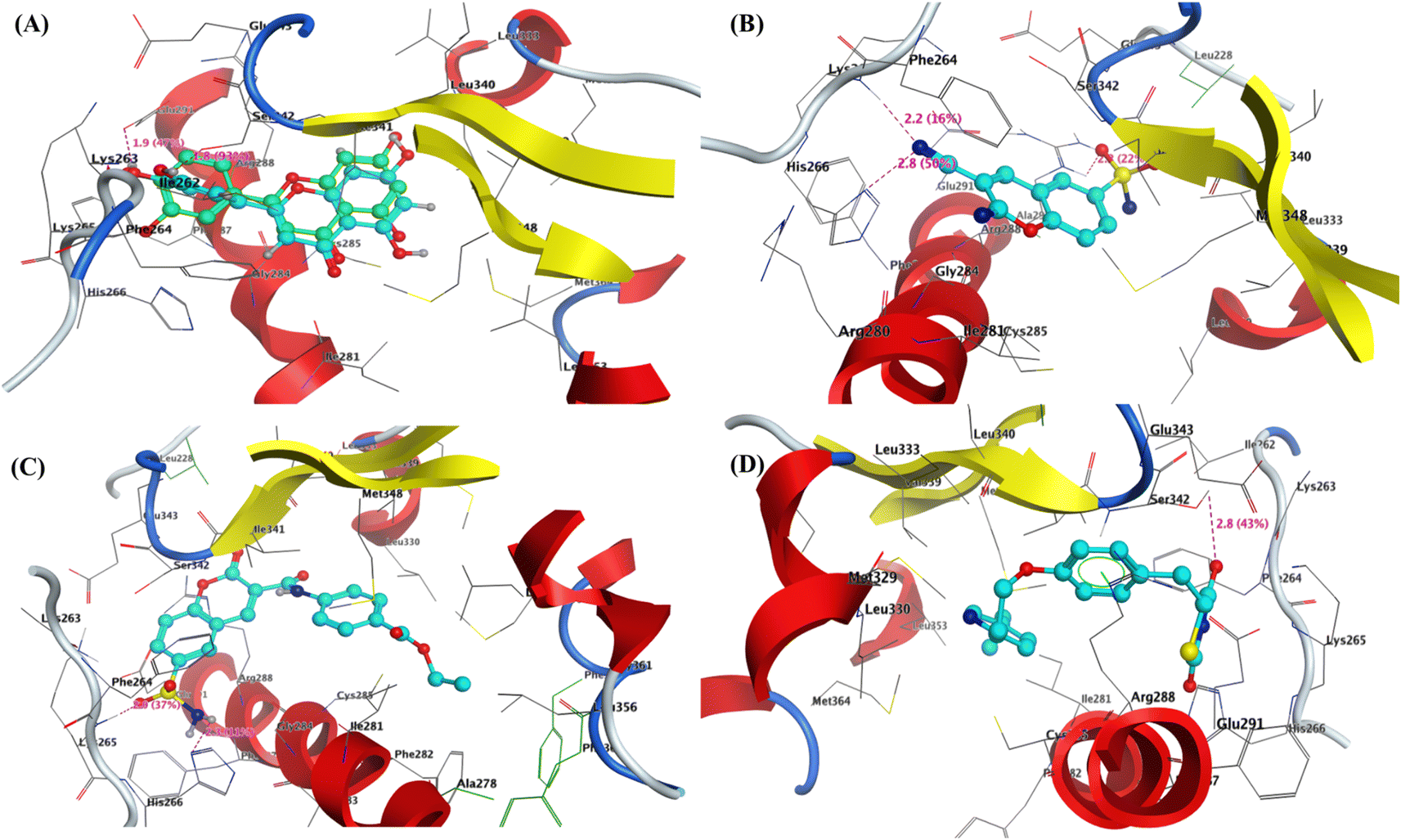 | ||
| Fig. 5 3D interaction of (A) co-crystallized Ligand as superimposable (green is the original pose, while turquoise is our pose), (B) compound 2, and (C) compound 9, and (D) Pioglitazone inside the active site of PPAR-γ (PDB: 3SZ1). | ||
Furthermore, the most active 6-sulfamoyl-2H-chromene derivatives 2 and 9 showed the lowest binding energy (S = −13.64 and −12.67 kcal mol−1) than Pioglitazone as positive control (−12.43 kcal mol−1). The 3-cyano-2-imino-2H-chromene-6-sulfonamide (2) interacted with PPAR-γ with three hydrogen bond acceptors with residues Lys262 and His266 that interacted with the nitrogen of cyano group with a distance of 2.2 and 2.8 Å, while the third one displayed between the oxygen of sulfonyl group with Arg288 with a bond length of 2.2 Å and strength of 22%. The structure of compound 2 is located inside the pocket and formed hydrophobic interactions with (Leu228, Leu333, Leu340, Glu343, Ser342, Ile341, Phe264, and Gly284) Fig. 5b. Similarly, the 2-oxo-6-sulfamoyl-2H-chromene-3-carboxamide 9 exhibited two hydrogen bonds with residues Lys265 and His266 that bounded to oxygen and amino of sulfonamide (–SO2NH2) with bond lengths of 2.0 and 2.3 Å, respectively Fig. 5c. Finally, the Pioglitazone bounded to the common amino acid residue Ser342 with H-bond sidechain acceptor with the oxygen of carbonyl at C4 of thiadiazol-2,4-dione with bond length of 2.8 Å and strength of 43%. Besides, the Arg288 could interact with phenyl ring of Pioglitazone through arene–cation interaction Fig. 5d. For Chlorpropamide and Glibenclamide, it was found that the binding energy of these antidiabetic sulfonamide drugs exhibited binding energy S = −4.44 and −7.81 kcal mol−1, respectively. Chlorpropamide displayed one hydrogen bond backbone acceptor between the residue Ser289 and oxygen of SO2 with a bond length of 2.2 Å and strength of 38% Fig. S9 and S10.† On the other hand, Glibenclamide showed one hydrogen bond sidechain donor between the residue Ser289 and NH of sulfonamide with a bond length of 2.7 Å and strength of 32% Fig. S11 and S12 (see ESI figures).†
Finally, it can be concluded that the most active 6-sulfamoyl-2H-chromene derivatives 2 and 9 showed lower binding energy than Chlorpropamide and Glibenclamide as antidiabetic drugs have sulphonamide moiety against α-amylase, α-glucosidase, and PPAR-γ enzymes.
3. Conclusion
Herein, this article synthesized new 2H-chromene 6-sulfonamide by reaction of 5-sulfonyl salicylaldehyde with different reagents containing active methylene group in the presence of ammonium acetate as a basic catalyst, where the reaction proceeded by condensation followed by intramolecular cyclization to afford the target compounds. Moreover, the in vitro α-amylase inhibitory activity was evaluated for all the designed derivatives. The 2-oxo-6-sulfamoyl-2H-chromene-3-carboxamide derivative 9 emerged as the most active hypoglycemic agent by inhibiting the α-amylase enzyme with IC50 value of 1.08 ± 0.02 μM, while the 3-cyano-2-imino-2H-chromene-6-sulfonamide (2) revealed the second active member among the synthesized derivatives with IC50 value of 1.76 ± 0.01 μM compared to Acarbose (IC50 = 0.43 ± 0.01 μM). Among the tested compounds, two derivatives exhibited good inhibitory activity and were used for further evaluation against α-glycosidase and PPAR-γ. Surprisingly, compounds 2 and 9 showed inhibitory potential to α-glucosidase with IC50 values of 0.548 ± 0.02 and 2.44 ± 0.09 μg mL−1 compared to Acarbose as a positive FDA-approved drug (IC50 = 0.604 ± 0.02 μg mL−1). Further, the most active 6-sulfonamide-2H-chromenes 2 and 9 were potent agonists against PPAR-γ receptors with IC50 values of 3.152 ± 0.03 and 3.706 ± 0.32 μg mL−1 compared to Pioglitazone (IC50 = 4.884 ± 0.29 μg mL−1). The in silico ADMET, including molecular properties, medicinal chemistry, drug-likeness properties, and toxicity prediction, were also studied. Finally, the docking simulation was performed on the active site of α-amylase, α-glycosidase, and PPAR-γ and determined the best conformation and, therefore, determined the binding energy and interaction types inside the pockets. The promising outcomes of most active derivatives encourage us to investigate further several enzymatic tests, including lipase, aldose reductase, DPP4, and assess more in vivo studies to improve efficacy, reduce potential side effects, etc.4. Experimental
4.1. Chemistry
4.1.3.1. 3-Cyano-2-imino-2H-chromene-6-sulfonamide (2). Light-orange powder; m. p. = 190–192 °C, 91% yield; anal. calcd (%) for C10H7N3O3S (249.24 g mol−1): C, 48.19; H, 2.83; N, 16.86; found (%): C, 48.48; H, 2.70; N, 16.99; IR (KBr): νmax = 3414, 3335, 3233 (NH & NH2), 3080, (sp2-CH), 2203 (CN), 1620 (C
NH), 1592 (CC), 1399, 1185 (SO2); 1H NMR (δ, ppm) = 7.32 (d, J = 8.8 Hz, 1H, H8-phenyl), 7.86 (dd, J = 8.4, 4.0 Hz, 1H, H7-phenyl), 8.10 (s, 2H, NH2), 8.39 (s, 1H, H5-phenyl), 8.76 (s, 1H, H4-pyran), 9.39 (s, 1H, NH); 13C NMR (ppm) δ 96.01 (C–CN, C3-pyran), 111.45 (C4-pyran), 116.21 (CN), 118.25 (C-sp2), 122.61(C-sp2), 127.71(C-sp2), 131.91(C-sp2), 138.12 (C–SO2), 157.90 (O–CC), 160.53 (CNH).
4.1.3.2. 3-(1-Amino-2,2-dicyanovinyl)-2-imino-2H-chromene-6-sulfonamide (3). Deep-orange powder; m. p. = 176–178 °C, 84% yield; anal. calcd (%) for C13H9N5O3S (315.31 g mol−1): C, 49.52; H, 2.88; N, 22.21; found (%): C, 49.23; H, 2.91; N, 22.11; IR (KBr): νmax = 3418, 3336, 3227 (NH & 2NH2), 3040, (sp2-CH), 2203 (CN), 1621 (C
NH), 1568 (CC), 1400, 1185 (SO2); 1H NMR (δ, ppm) = 4.99 (s, 2H, NH2), 6.94 (d, J = 8.0 Hz, 1H, H8-phenyl), 7.40 (d, J = 8.4 Hz, 1H, H7-phenyl), 7.53 (br.s, 2H, NH2), 7.66 (s, 1H, H5-phenyl), 7.95 (s, 1H, H4-pyran), 8.07 (s, 1H, NH); 13C NMR (ppm) δ 52.65 (C-(CN)2), 113.48 (CN), 118.25 (C4-pyran), 122.61 (C-sp2), 127.71 (C-sp2), 131.91 (C-sp2), 136.68 (C-sp2), 138.12 (C-sp2), 140.05 (C–SO2), 156.69 (O–CC), 160.08 (CNH), 178.95 (NH2–CC).
4.1.3.3. 3-Acetyl-2-oxo-2H-chromene-6-sulfonamide (4). Orange powder; m. p. = 151–153 °C, 78% yield; anal. calcd (%) for C11H9NO5S (267.26 g mol−1): C, 49.44; H, 3.39; N, 5.24; found (%): C, 49.78; H, 3.15; N, 5.55; IR (KBr): νmax = 3394, 3300 (NH2), 3110 (sp2-CH), 2980 (sp3-CH), 1716 (2CO), 1563 (C
C), 1367, 1270 (SO2); 1H NMR (δ, ppm) = 2.31 (s, 3H, COCH3), 6.70 (br.s, 2H, NH2), 7.33 (d, J = 8.8 Hz, 1H, H8-phenyl), 7.87 (d, J = 8.4 Hz, 1H, H7-phenyl), 8.40 (s, 1H, H5-phenyl), 8.74 (s, 1H, H4-pyran); 13C NMR (ppm) δ 25.90 (CO–CH3), 119.54 (C-sp2), 119.91 (C-sp2), 122.06 (C-sp2), 127.30 (C-sp2), 131.65 (C-sp2), 138.55 (C-sp2), 144.90 (C–SO2), 157.46 (O–CC), 161.15 (CO), 183.43 (CO–CH3).
4.1.3.4. Ethyl 2-oxo-6-sulfamoyl-2H-chromene-3-carboxylate (5). Red powder; m. p. = 165–167 °C, 76% yield; anal. calcd (%) for C12H11NO6S (297.28 g mol−1): C, 48.48; H, 3.73; N, 4.71; found (%): C, 48.43; H, 3.82; N, 4.75; IR (KBr): νmax = 3183 (br.NH2), 3064 (sp2-CH), 1765, 1710 (2CO), 1570 (C
C), 1366, 1180 (SO2); 1H NMR (δ, ppm) = 1.29 (t, J = 6.0 Hz, 3H, CH3CH2O-), 4.27 (q, J = 5.6 Hz, 2H, CH3CH2O-), 6.78 (s, 2H, NH2), 7.61 (d, J = 7.2 Hz, 1H, H8-phenyl), 7.92 (d, J = 7.2 Hz, 1H, H7-phenyl), 8.40 (s, 1H, H5-phenyl), 8.96 (s, 1H, H4-pyran); 13C NMR (ppm) δ 13.99 (CH3CH2O-), 60.58 (CH3CH2O-), 118.26 (C-sp2), 122.61 (C-sp2), 125.61 (C-sp2), 127.71 (C-sp2), 131.91 (C-sp2), 138.12 (C-sp2), 140.33 (C–SO2), 156.58 (O–CC), 161.62 (CO), 168.14 (CO–OEt).
4.1.4.1. N-(4-fluorophenyl)-2-oxo-6-sulfamoyl-2H-chromene-3-carboxamide (6). Light-red powder; m. p. = 215–217 °C, 67% yield; anal. calcd (%) for C16H11FN2O5S (362.33 g mol−1): C, 53.04; H, 3.06; N, 7.73; found (%): C, 53.24; H, 2.91; N, 7.66; IR (KBr): νmax = 3431 (br.NH2 & NH), 3097 (sp2-CH), 1727 (CO), 1654 (CO–NH), 1605 (C
C), 1359, 1191 (SO2); 1H NMR (δ, ppm) = 6.90 (s, 2H, NH2), 7.21 (d, J = 8.4 Hz, 2H, Ar–H), 7.69 (d, J = 9.2 Hz, 1H, H8-phenyl), 7.72 (d, J = 8.8 Hz, 2H, Ar–H), 7.76 (d, J = 8.4 Hz, 1H, H7-phenyl), 8.34 (s, 1H, H5-phenyl), 8.43 (s, 1H, H4-pyran), 10.46 (s, 1H, CO–NH); 13C NMR (ppm) δ 115.30 (C-sp2), 121.49 (C-sp2), 124.45 (C-sp2), 126.66 (C-sp2), 127.33 (C-sp2), 128.56 (C-sp2), 129.43 (C-sp2), 129.74 (C-sp2), 130.60 (C-sp2), 134.71 (C-sp2), 146.68 (C-sp2), 152.44 (C–SO2), 156.92 (O–CC), 161.85 (C–F), 162.61 (CO), 166.15 (CO–NH); 19F NMR (376 MHz, DMSO) δ −118.03 (s, 1F).
4.1.4.2. 2-Oxo-6-sulfamoyl-N-(3-(trifluoromethyl)phenyl)-2H-chromene-3-carboxamide (7). Deep-red powder; m. p. = 223–225 °C, 83% yield; anal. calcd (%) for C17H11F3N2O5S (412.34 g mol−1): C, 49.52; H, 2.69; N, 7.79; found (%): C, 49.74; H, 2,79; N, 7.54; IR (KBr): νmax = 3431 (br.NH2 & NH), 3098 (sp2-CH), 1727 (CO), 1654 (CO–NH), 1573 (C
C), 1360, 1191 (SO2); 1H NMR (δ, ppm) = 6.73 (s, 2H, NH2), 7.23 (d, J = 6.8 Hz, 1H, Ar–H), 7.29–7.32 (m, 2H, Ar–H + H8-phenyl), 7.48 (d, J = 6.8 Hz, 1H, Ar–H), 7.79 (d, J = 6.8 Hz, 1H, H7-phenyl), 7.83–7.87 (m, 1H, Ar–H), 8.18 (s, 1H, H5-phenyl), 8.36 (s, 1H, H4-pyran), 9.37 (s, 1H, CO–NH); 13C NMR (ppm) δ 111.70 (C-sp2), 115.34 (C-sp2), 115.62 (C-sp2), 118.63 (C-sp2), 119.07 (C-sp2), 121.37 (C-sp2), 123.20 (C-sp2), 124.25 (C–F3), 125.82 (C-sp2), 127.49 (C-sp2), 129.64 (C-sp2), 133.33 (C-sp2), 136.44 (C-sp2), 144.24 (C–SO2), 155.30 (O–CC), 159.38 (CO), 164.34 (CO–NH); 19F NMR (376 MHz, DMSO) δ/ppm −61.29 (s, 3F).
4.1.4.3. N-(4-chlorophenyl)-2-oxo-6-sulfamoyl-2H-chromene-3-carboxamide (8). Deep-yellow powder; m. p. = 209–211 °C, 77% yield; anal. calcd (%) for C16H11ClN2O5S (378.78 g mol−1): C, 50.74; H, 2.93; N, 7.40; found (%): C, 50.82; H, 2.86; N, 7.45; IR (KBr): νmax = 3430 (br.NH2 & NH), 3104 (sp2-CH), 1726 (CO), 1654 (CO–NH), 1573 (C
C), 1380, 1137 (SO2); 1H NMR (δ, ppm) = 6.72 (s, 2H, NH2), 7.32 (d, J = 8.8 Hz, 2H, Ar–H), 7.51 (d, J = 8.4 Hz, 1H, H8-phenyl), 7.81 (d, J = 10.4 Hz, 1H, Ar–H), 7.86 (d, J = 6.4 Hz, 1H, Ar–H), 7.98 (d, J = 8.8 Hz, 1H, H7-phenyl), 8.10 (s, 1H, H5-phenyl), 8.39 (s, 1H, H4-pyran), 9.38 (s, 1H, CO–NH); 13C NMR (ppm) δ 114.19 (C-sp2), 116.83 (C-sp2), 117.77 (C-sp2), 118.61 (C-sp2), 119.57 (C-sp2), 122.40 (C-sp2), 126.33 (C-sp2), 128.50 (C-sp2), 130.57 (C-sp2), 131.16 (C-sp2), 133.84 (C-sp2), 134.88 (C-sp2), 142.41 (C–SO2), 153.98 (O–CC), 159.44 (CO), 164.09 (CO–NH).
4.1.4.4. Ethyl 4-(2-oxo-6-sulfamoyl-2H-chromene-3-carboxamido)benzoate (9). Pale-orange powder; m. p. = 233–235 °C, 86% yield; anal. calcd (%) for C19H16N2O7S (416.40 g mol−1): C, 54.80; H, 3.87; N, 6.73; found (%): C, 54.82; H, 3.88; N, 6.68; IR (KBr): νmax = 3430 (br.NH2 & NH), 3073 (sp2-CH), 1726 (2CO), 1655 (CO–NH), 1572 (C
C), 1361, 1192 (SO2); 1H NMR (δ, ppm) = 1.24 (t, J = 7.2 Hz, 3H, CH3CH2O-), 4.18 (q, J = 5.6 Hz, 2H, CH3CH2O-), 5.91 (s, 2H, NH2), 6.53 (d, J = 8.4 Hz, 2H, Ar–H), 6.92 (d, J = 6.4 Hz, 1H, H8-phenyl), 7.51 (d, J = 6.8 Hz, 1H, H7-phenyl), 7.60 (d, J = 6.8 Hz, 2H, Ar–H), 7.86 (s, 1H, H5-phenyl), 7.99 (s, 1H, H4-pyran), 10.23 (s, 1H, CO–NH); 13C NMR (ppm) δ 14.76 (CH3CH2O-), 61.03 (CH3CH2O-), 115.07 (C-sp2), 115.52 (C-sp2), 116.76 (C-sp2), 120.77 (C-sp2), 124.50 (C-sp2), 127.35 (C-sp2), 128.87 (C-sp2), 130.59 (C-sp2), 132.14 (C-sp2), 134.05 (C-sp2), 138.30 (C-sp2), 140.18 (C-sp2), 144.61 (C–SO2), 155.12 (O–CC), 157.58 (CO), 163.46 (CO–NH), 166.20 (CO–OEt).
4.2. Biological evaluation
4.3. Molecular docking simulation
To determine the binding mode pattern for the most active chromene derivatives 2 and 9, the docking simulation conducted inside the active site of α-amylase (PDB: 2QV4, chain A), α-glucosidase (PDB: 3w37, chain A), and PPAR-γ (PDB: 3SZ1, chain B) as described previously.55–57 The 3D structure of proteins was obtained from the protein data bank (https://www.rcsb.org/), and each protein was prepared by default protocol where the water molecules were removed, and only one chain was selected for docking purposes. The active site was protonated, then the energy was minimized using MMFF94X with a gradient of 0.05 kcal mol−1 and saved as an mdb file.63,64 The structure of most active 6-sulfamoyl-2H-chromene derivatives 2 and 9, as well as Pioglitazone were drawn and constructed using chembiodraw14, then prepared by protonation and minimized using standard protocol for MOE as described previously.65–67Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at Northern Border University for funding this work through research group no. (RG-NBU-2022-1705).References
- N. H. Cho, J. E. Shaw, S. Karuranga, Y. Huang, J. D. da Rocha Fernandes, A. W. Ohlrogge and B. Malanda, Diabetes Res. Clin. Pract., 2018, 138, 271–281 CrossRef CAS PubMed.
- A. D. Association, Diabetes Care, 2013, 37, S81–S90 CrossRef PubMed.
- Z. D. Kifle, M. Abdelwuhab, A. D. Melak, G. Genet, T. Meseret and M. Adugna, Metab. Open, 2022, 13, 100174 CrossRef CAS PubMed.
- P. O. U. Adogu, H. N. Chineke, M. U. Ewuzie, O. O. Enwere and N. B. Egenti, J. Diabetes Mellitus, 2015, 5, 49 CrossRef.
- A. Ali, F. Iqbal, A. Taj, Z. Iqbal, M. J. Amin and Q. Z. Iqbal, Pak. J. Med. Sci., 2013, 29, 899–902 Search PubMed.
- D. Care, Diabetes Care, 2017, 40, S11–S24 CrossRef PubMed.
- A. E. Kitabchi, G. E. Umpierrez, M. B. Murphy and R. A. Kreisberg, Diabetes Care, 2006, 29, 2739–2748 CrossRef CAS PubMed.
- S. S. S. Surya, A. D. Salam, D. V Tomy, B. Carla, R. A. Kumar and C. S. C. Sunil, Int. J. Pharm. Biomed. Sci., 2014, 4, 337–347 Search PubMed.
- D. K. Patel, R. Kumar, D. Laloo and S. Hemalatha, Asian Pac. J. Trop. Biomed., 2012, 2, 411–420 CrossRef CAS PubMed.
- M. Przeor, Pharmaceuticals, 2022, 15, 65 CrossRef CAS PubMed.
- P. Subash Babu, S. Prabuseenivasan and S. Ignacimuthu, Phytomedicine, 2007, 14, 15–22 CrossRef CAS PubMed.
- L. Pari and N. Rajarajeswari, Chem.-Biol. Interact., 2009, 181, 292–296 CrossRef CAS PubMed.
- S. M. Patil, R. M. Martiz, A. M. Satish, A. M. Shbeer, M. Ageel, M. Al-Ghorbani, L. Ranganatha, S. Parameswaran and R. Ramu, Molecules, 2022, 27, 3888 CrossRef CAS PubMed.
- S. M. Patil, R. M. Martiz, R. Ramu, P. S. Shirahatti, A. Prakash, B. R. P. Kumar and N. Kumar, J. Biomol. Struct. Dyn., 2022, 40, 12491–12505 CrossRef CAS PubMed.
- A. J. Scheen, Diabet. Epidemiol. Manag., 2023, 11, 100145 CrossRef.
- M. S. Asgari, M. Mohammadi-Khanaposhtani, M. Kiani, P. R. Ranjbar, E. Zabihi, R. Pourbagher, R. Rahimi, M. A. Faramarzi, M. Biglar, B. Larijani, M. Mahdavi, H. Hamedifar and M. H. Hajimiri, Bioorg. Chem., 2019, 92, 103206 CrossRef CAS PubMed.
- D. Egan, R. O'kennedy, E. Moran, D. Cox, E. Prosser and R. D. Thornes, Drug Metab. Rev., 1990, 22, 503–529 CrossRef CAS PubMed.
- J. V Patil, S. S. Soman, S. Umar, P. Girase and S. Balakrishnan, ChemistrySelect, 2024, 9, e202303391 CrossRef.
- E. A. Fayed, M. Mohsen, S. M. A. El-Gilil, D. S. Aboul-Magd and A. Ragab, J. Mol. Struct., 2022, 1262, 133028 CrossRef CAS.
- N. Q. Khai and T. K. Vu, Anti-Cancer Agents Med. Chem., 2024, 24, 18–29 CrossRef CAS PubMed.
- A. Ibrar, S. A. Shehzadi, F. Saeed and I. Khan, Bioorg. Med. Chem., 2018, 26, 3731–3762 CrossRef CAS PubMed.
- Q. Shen, J. Shao, Q. Peng, W. Zhang, L. Ma, A. S. C. Chan and L. Gu, J. Med. Chem., 2010, 53, 8252–8259 CrossRef CAS PubMed.
- V. Channabasappa, K. Kumara and A. K. Kariyappa, J. Chem. Sci., 2021, 133, 130 CrossRef CAS.
- S. Gorain, J. Pal and S. J. Biswas, in Antidiabetic Medicinal Plants, ed. M. Naeem and T. Aftab, Academic Press, 2024, pp. 141–166 Search PubMed.
- S. Shahdadi, F. Hamidi and B. Fathi, Heliyon, 2024, 10, e24436 CrossRef CAS PubMed.
- V. Kumar, D. Ahmed, A. Verma, F. Anwar, M. Ali and M. Mujeeb, BMC Complementary Altern. Med., 2013, 13, 273 CrossRef PubMed.
- H. Sun, X. Song, Y. Tao, M. Li, K. Yang, H. Zheng, Z. Jin, R. H. Dodd, G. Pan, K. Lu and P. Yu, Future Med. Chem., 2018, 10, 1055–1066 CrossRef CAS PubMed.
- M. S. Asgari, M. Mohammadi-Khanaposhtani, M. Kiani, P. Rashidi Ranjbar, E. Zabihi, R. Pourbagher, R. Rahimi, M. A. Faramarzi, M. Biglar, B. Larijani, M. Mahdavi, H. Hamedifar and M. H. Hajimiri, Bioorg. Chem., 2018, 92, 103206 CrossRef PubMed.
- E. Elahabaadi, A. A. Salarian and E. Nassireslami, Polycyclic Aromat. Compd., 2022, 42, 4317–4327 CrossRef CAS.
- E. L. R. Stokstad and T. H. Jukes, J. Nutr., 1987, 117, 1335–1341 CrossRef CAS PubMed.
- M. N. L. Nalam, A. Peeters, T. H. M. Jonckers, I. Dierynck and C. A. Schiffer, J. Virol., 2007, 81, 9512–9518 CrossRef CAS PubMed.
- Y. L. Hong, P. A. Hossler, D. H. Calhoun and S. R. Meshnick, Antimicrob. Agents Chemother., 1995, 39, 1756–1763 CrossRef CAS PubMed.
- A. Ragab, S. A. Fouad, O. A. A. Ali, E. M. Ahmed, A. M. Ali, A. A. Askar and Y. A. Ammar, Antibiotics, 2021, 10, 162 CrossRef CAS PubMed.
- R. R. Raslan, Y. A. Ammar, S. A. Fouad, S. A. Hessein, N. A. M. Shmiess and A. Ragab, RSC Adv., 2023, 13, 10440–10458 RSC.
- M. A. Ismail, M. S. Abusaif, M. S. A. El-Gaby, Y. A. Ammar and A. Ragab, RSC Adv., 2023, 13, 12589–12608 RSC.
- F. Carta, C. T. Supuran and A. Scozzafava, Future Med. Chem., 2014, 6, 1149–1165 CrossRef CAS PubMed.
- M. A. Gouda and B. H. M. Hussein, Lett. Drug Des. Discovery, 2017, 14, 1425–1432 CrossRef CAS.
- A. Markham, Drugs, 2017, 77, 697–704 CrossRef CAS PubMed.
- H. A. Mohamed, Y. A. Ammar, G. A. M. Elhagali, H. A. Eyada, D. S. Aboul-Magd and A. Ragab, J. Mol. Struct., 2023, 1287, 135671 CrossRef CAS.
- M. S. A. El-Gaby, Y. A. Ammar, M. A. Ismail, A. Ragab and M. S. Abusaif, Heterocycl. Commun., 2023, 29, 20220170 CrossRef CAS.
- Y. A. Ammar, A. Ragab, M. A. Migahed, S. Al-Sharbasy, M. A. Salem, O. K. M. Riad, H. M. R. M. Selim, G. A. Abd-elmaksoud and M. S. Abusaif, RSC Adv., 2023, 13, 27363–27384 RSC.
- K. Zessel, S. Mohring, G. Hamscher, M. Kietzmann and J. Stahl, Chemosphere, 2014, 100, 167–174 CrossRef CAS PubMed.
- V. Garaj, L. Puccetti, G. Fasolis, J.-Y. Winum, J.-L. Montero, A. Scozzafava, D. Vullo, A. Innocenti and C. T. Supuran, Bioorg. Med. Chem. Lett., 2005, 15, 3102–3108 CrossRef CAS PubMed.
- U. Košak, B. Brus, D. Knez, R. Šink, S. Žakelj, J. Trontelj, A. Pišlar, J. Šlenc, M. Gobec, M. Živin, L. Tratnjek, M. Perše, K. Sałat, A. Podkowa, B. Filipek, F. Nachon, X. Brazzolotto, A. Więckowska, B. Malawska, J. Stojan, I. M. Raščan, J. Kos, N. Coquelle, J.-P. Colletier and S. Gobec, Sci. Rep., 2016, 6, 39495 CrossRef PubMed.
- M. Bavadi, K. Niknam and O. Shahraki, J. Mol. Struct., 2017, 1146, 242–253 CrossRef CAS.
- R. Schobert, R. Stehle and H. Walter, Tetrahedron, 2008, 64, 9401–9407 CrossRef CAS.
- D. Borah, P. N. Bhattacharyya, B. C. Nath, R. Chetia, N. F. Islam, B. Sarma and J. Fluor, Chem, 2024, 274, 110259 CAS.
- M. Kou, J. Zheng, T. Chen, Y. Wu, L. Huang, D. Cao, D. Chen and J. Fluor, Chem, 2024, 276, 110279 CAS.
- Y. A. Ammar, J. A. Micky, D. S. Aboul-Magd, S. M. A. Abd El-Hafez, S. A. Hessein, A. M. Ali and A. Ragab, Chem. Biol. Drug Des., 2023, 101, 245–270 CrossRef CAS PubMed.
- M. M. Abdelgalil, Y. A. Ammar, G. A. M. Elhag Ali, A. K. Ali and A. Ragab, J. Mol. Struct., 2023, 1274, 134443 CrossRef CAS.
- A. Ragab, M. S. Abusaif, N. A. Gohar, D. S. Aboul-Magd, E. A. Fayed and Y. A. Ammar, Bioorg. Chem., 2023, 131, 106307 CrossRef CAS PubMed.
- A. Ragab, S. A. Ibrahim, D. S. Aboul-Magd and M. H. Baren, RSC Adv., 2023, 13, 34756–34771 RSC.
- A. Ragab, M. S. Abusaif, D. S. Aboul-Magd, M. M. S. Wassel, G. A. M. Elhagali and Y. A. Ammar, Drug Dev. Res., 2022, 1305–1330 CrossRef CAS PubMed.
- R. Ayman, M. S. Abusaif, A. M. Radwan, A. M. Elmetwally and A. Ragab, Eur. J. Med. Chem., 2023, 249, 115138 CrossRef CAS PubMed.
- Y. M. Abdel-Baky, A. M. Omer, E. M. El-Fakharany, Y. A. Ammar, M. S. Abusaif and A. Ragab, Sci. Rep., 2023, 13, 22792 CrossRef CAS PubMed.
- A. S. Hassan, N. M. Morsy, W. M. Aboulthana and A. Ragab, Drug Dev. Res., 2023, 84, 3–24 CrossRef CAS PubMed.
- V. M. Patil, K. N. Tilekar, N. M. Upadhyay and C. S. Ramaa, ChemistrySelect, 2022, 7, e202103848 CrossRef CAS.
- K. E. Saadon, N. M. H. Taha, N. A. Mahmoud, G. A. M. Elhagali and A. Ragab, J. Iran. Chem. Soc., 2022, 19, 3899–3917 CrossRef CAS.
- A. Ragab, S. A. Fouad, Y. A. Ammar, D. S. Aboul-Magd and M. S. Abusaif, Antibiotics, 2023, 12, 128 CrossRef CAS PubMed.
- M. J. Mphahlele, N. M. Magwaza, S. T. Malindisa and Y. S. Choong, Chem. Biol. Drug Des., 2021, 98, 234–247 CrossRef CAS PubMed.
- S. H. Aly, A. M. Elissawy, M. A. El Hassab, T. A. Majrashi, F. E. Hassan, E. B. Elkaeed, W. M. Eldehna and A. N. B. Singab, J. Enzyme Inhib. Med. Chem., 2024, 39, 2292482 CrossRef PubMed.
- E. A. Fayed, A. Thabet, S. M. A. El-Gilil, H. M. A. Elsanhory and Y. A. Ammar, J. Mol. Struct., 2024, 1301, 137374 CrossRef CAS.
- R. Ayman, A. M. Radwan, A. M. Elmetwally, Y. A. Ammar and A. Ragab, Arch. Pharm., 2023, 356, e2200395 CrossRef PubMed.
- M. Basseem, A. A. Emam, F. H. Kamal, A. M. Gamal and S. A. Abo Faraha, Appl. Organomet. Chem., 2023, 37, 1–23 CrossRef.
- H. F. Rizk, M. A. El-Borai, A. Ragab, S. A. Ibrahim and M. E. Sadek, Polycyclic Aromat. Compd., 2023, 43, 500–522 CrossRef CAS.
- S. A. Ibrahim, A. Ragab and H. A. El-Ghamry, Appl. Organomet. Chem., 2022, 36, e6508 CrossRef CAS.
- E. S. A. E. H. Khattab, A. Ragab, M. A. Abol-Ftouh and A. A. Elhenawy, J. Biomol. Struct. Dyn., 2022, 40, 13291–13309 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02143f |
| This journal is © The Royal Society of Chemistry 2024 |