
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntiviral activity of sulphated specialized metabolites from sea urchin Clypeaster humilis: in vitro and in silico studies†
Fahd M. Abdelkarem a,
Hamdy K. Assafa,
Yaser A. Mostafabc,
Aldoushy Mahdyd,
Modather F. Husseinef,
Samir A. Rossgh and
Nesma M. Mohamed*ij
a,
Hamdy K. Assafa,
Yaser A. Mostafabc,
Aldoushy Mahdyd,
Modather F. Husseinef,
Samir A. Rossgh and
Nesma M. Mohamed*ij
aDepartment of Pharmacognosy, Faculty of Pharmacy, Al-Azhar University, Assiut 71524, Egypt
bPharmaceutical Organic Chemistry Department, Faculty of Pharmacy, Assiut University, Assiut 71526, Egypt
cPharmaceutical Chemistry Department, Faculty of Pharmacy, Badr University, Assiut 77771, Egypt
dDepartment of Zoology, Faculty of Science, Al-Azhar University, Assiut, 71524, Egypt
eChemistry Department, Collage of Science, Jouf University, P.O. Box 2014, Sakaka 72388, Saudi Arabia
fChemistry Department, Faculty of Science, Al-Azhar University, Asyut Branch, Assiut 71524, Egypt
gNational Center for Natural Products Research, Research Institute of Pharmaceutical Sciences, School of Pharmacy, The University of Mississippi, Mississippi 38677, USA
hDepartment of BioMolecular Sciences, Division of Pharmacognosy, School of Pharmacy, University of Mississippi, Mississippi 38677, USA
iDepartment of Pharmacognosy, Faculty of Pharmacy, Assiut University, Assiut 71526, Egypt. E-mail: nesma.Mohamed269@pharm.aun.edu.eg
jDepartment of Pharmacognosy, Faculty of Pharmacy, Badr University, Assiut 77771, Egypt
First published on 30th April 2024
Abstract
Chemical investigations of the sea urchin Clypeaster humilis has led to separation of twelve compounds including one new sulfonic acid derivative (7R) tridec-1-en-7-yl hydrogen sulphate (1), first isolated from natural source, pyridine-3-yl methane sulfonate (2), and first isolated from marine organisms, boldine (12), in addition to nine known compounds (3–11), which were isolated for the first time from the genus Clypeaster. Their structures were elucidated based on spectroscopic analyses (1D and 2D NMR), HR-ESI-MS as well as comparison with the previously reported data. The antiviral activity of the crude extract and sulphated compounds were evaluated using MTT colorimetric assay against Coxsackie B4 virus. The crude extract and compound 1 showed very potent antiviral activity with a percentage of inhibition equal to 89.7 ± 0.53% and 86.1 ± 0.92%, respectively. Results of the molecular docking analysis of the isolated compounds within Coxsackie Virus B4 (COX-B4) X-ray crystal structure and quantum chemical calculation for three sulphated compounds are in a consistent adaptation with the in vitro antiviral results. The pharmacokinetic properties (ADME) of isolated compounds were determined.
Introduction
Natural products have become ever more significant in the discovery and development of pharmaceuticals during the past few decades. Many natural remedies are thought to be useful alternatives for treating a variety of diseases and disorders.1 One of the world's least investigated environments is the marine ecosystem. Additionally, marine ecosystem is known for its higher taxonomic diversity composition than its terrestrial counterpart. This provides a tremendous source of innovative compounds with diverse biological activity.2Sea urchins (Echinoidae) are widely distributed marine organisms which belong to the phylum Echinodermata.3 Clypeaster is one of the most widely distributed irregular echinoids genera in the Red Sea. Three species of clypestaroids were founds in the Egyptian coasts: Clypeaster humilis, Clypeaster fervens and Clypeaster reticulatus.4,5 Clypeaster humilis is distributed mainly in a shallow water sediment reaching lengths of 5–8 cm with a flattened test and two aboral and oral sides. Several metabolites were isolated from sea urchins as polyhydroxylated naphthaquinones, nucleosides, fatty acids, glycerol derivatives, diterpenes and steroids, in addition to, sulphated compounds, polysaccharides, acid polysaccharide, sphingolipid, glycolipids, and phospholipids6 with diverse biological activities as anticancer, antimicrobial, anti-inflammatory, antioxidant, anticoagulant, anti-fungal, anti-parasitic, hepatoprotective, anti-viral, anti-diabetic, anti-lipidemic, gastro-protective and cytotoxic properties.7–12
Coxsackie B virus (CVB) is a significant human pathogen belonging to the Picornavirus family, with six serotypes of group B are recognized CV-B1 to CV-B6. CVB causes symptoms such as fever, rash, and upper respiratory illness. In some cases, the virus can cause a variety of diseases, including gastrointestinal illness, myocarditis, pneumonia, aseptic meningitis, encephalitis, and hepatitis.13–16 CVB may persist in pancreatic ductal and β-cells, also persistence in other sites, such as the intestine, blood cells and thymus, these sites could serve as a reservoir for infection.17 Also, it may cause pancreatitis leading to beta-cell destruction and type-1 diabetes mellitus.18 Since 1987, several outbreaks caused by CVB were reported in different parts of the world. Repeated outbreaks of aseptic meningitis, myocarditis, and pancreatitis caused by CVB have been frequently reported. Furthermore, the epidemiological data and incidence of CVB in infants and children have also been investigated in China and India.19–22
The mechanism of CVB, which cause functional impairment and β-cell death characterized by nuclear pyknosis. Apoptosis appears to play a minor role during a productive infection in β-cells.23 There is no specific recommended treatment for CVB, symptomatic and supportive care for the associated syndromes is appropriate. Patients with neurological complications may need antiepileptics for seizures and sedation for delirium.24 The current used medications that, inhibit virus uptake by binding to the virus capsid (e.g., pleconaril) or inactivate viral proteins (e.g., NO-metoprolol and ribavirin) or inhibit cellular proteins which are essential for viral replication (e.g., ubiquitination inhibitors), or immunoglobulin therapy, immunoabsorption or specific antibody therapy, such as interferons that inhibit the immune system.25 It is necessary to continue researching possible cures for CVB through preventing viral attachment to cells for proliferation, viral replication, and complications of the virus in different organs of the body.
Our research aimed to isolate and characterize specialized metabolites from the marine organisms in the Red Sea. Hence, our study dealt with a detailed isolation and structural elucidation of compounds from the sea urchin C. humilis followed by evaluation of the antiviral activity of crude extract and sulphated compounds against COX-B4 virus at the maximum non-toxic concentration (MNTC).
Materials and methods
General experimental procedures
Optical rotations were recorded on Rudolph AutoPol IV automatic polarimeter (Rudolph Research Analytical, Hackettstown, NJ, USA) at room temperature; UV spectra were recorded by a Hewlett-Packard 8452A UV-Vis spectrometer (American Laboratory Trading, CT, USA); nuclear magnetic resonance (NMR) spectra were recorded in deuterated solvents and acquired on Bruker Avance DRX spectrometer (Bruker Biospin Gmbh, Rheistetten, Germany) at 400 MHz (1H) and 100 MHz (13C), 500 MHz (1H) and 125 MHz (13C). The chemical shifts are given in δ (ppm) and were calibrated using the residual solvent signals; coupling constants (J) are reported in hertz (Hz). The signals in the spectra are described as s (singlet), d (doublet), t (triplet), m (multiplet) and br (broad resonances). High-resolution mass spectra were obtained using an HR-ESI-TOF-MS spectrometer with the Analyst QS software for data acquisition and processing (Agilent Series 1100 SL, ESI source model #G1969A, Agilent Technologies, Palo Alto, CA, USA). Solvents used for extraction and isolation were analytical grade (Fisher Scientific, Fair Lawn, NJ, USA). Column chromatography was carried out on Diaion HP-20 resin (Mitsubishi Chemical Corporation, Tokyo, Japan), normal silica gel 60 F254 (40–65 μm, Merck, Darmstadt, Germany), Sephadex LH-20 (0.25–0.1 mm, Mitsubishi Kagaku, Tokyo, Japan), and reversed-phase C18 silica gel (Polarbond, JT Baker), with analytical grade solvents from Fisher Scientific. Thin layer chromatography (TLC) was conducted on silica 60 F254 (0.2 mm, Merck, Darmstadt, Germany) pre-coated aluminium sheets and spot detection was done by spraying with 5% vanillin solution in conc. H2SO4–EtOH (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :95) followed by heating. All solvents used for extractions and separations were analytical grade (Merck, Darmstadt, Germany).
:95) followed by heating. All solvents used for extractions and separations were analytical grade (Merck, Darmstadt, Germany).
Experimental
Collection of marine organism
The sea urchin Clypeaster humilis was collected and identified by Dr Aldoushy Mahdy (Faculty of Science, Al-Azhar University, Assiut branch, Egypt) from the shallow coarse sand areas of Red Sea in front of the National Institute of Oceanography and Fisheries, Hurghada, Egypt, at geographical coordinates 27°17′02′′N 33°46′18′′E. A voucher sample (CH-22) has been deposited at the Pharmacognosy Department, Faculty of Pharmacy, Al-Azhar University, Assiut Branch, Egypt.Extraction and isolation of compounds
The collected samples of Clypeaster humilis (n = 42) were first dissected to separate the outer shell from the gonads, and then all samples were extracted by maceration technique using 90% methanol (4 × 2 L) under room temperature for 72 h each time. After filtration, the methanolic extract was evaporated at low pressure at 45 °C using a rotary evaporator, resulting in dry viscous greenish brown extract (27 g). The obtained crude extract was subjected to column chromatography (CC) using the polymeric adsorbent Diaion (HP-20) as adsorbent (3 × 150 cm) and eluted using mixtures of H2O–MeOH in the following order: 100% H2O, 25% MeOH, 50% MeOH, 75% MeOH, and 100% MeOH. The column was finally washed with acetone to afford six main fractions I–VI, respectively.Fraction II (0.212 g) was purified using sephadex LH-20 CC (2 × 60 cm) using solvent system H2O–MeOH (20:80) to afford compound 2 (7 mg) colourless residue.
Fraction III (0.664 g) was subjected to normal silica gel CC (2 × 70 cm) using DCM–MeOH mixtures in order of increasing polarities (90:10 to 75:25) to afford three main sub-fractions M-2 (37 mg), M-3 (28 mg), and M-4 (56 mg). Sub-fraction M-2, eluted with 15% MeOH, was further purified on reversed phase silica CC (1.5 × 60 cm) using H2O as eluent to afford compound 3 (16.2 mg) and compound 5 (3.7 mg) as creamy amorphous powder. Subfraction M-4, eluted with 25% MeOH, was subjected to reversed phase silica CC (1.5 × 60 cm) using H2O–MeOH as solvent system and subfractions eluted with system 95:5 was collected together to afford a residue of 25 mg weight, which was further purified using sephadex LH-20 CC (1 × 50 cm) and MeOH as eluent to obtain compound 4 (5.1 mg) as creamy amorphous powder.
Fraction IV (0.160 g) showed on the TLC one major spot which was isolated using normal silica gel CC (1.5 × 60 cm) and DCM–MeOH (85:15) as solvent system to afford compound 6 (8.9 mg) needle crystals.
Fraction V (0.187 g) was fractionated on sephadex LH-20 CC (2 × 60 cm) using DCM–MeOH (30:70) to afford two main subfractions M-5 (31 mg) and M-6 (35 mg). Subfraction M-5 was subjected to reversed phase silica CC using H2O–MeOH (25:75) to get compound 1 (4.9 mg) white amorphous powder. Subfraction M-6 was subjected to silica gel CC (1.5 × 60 cm) using DCM–acetone (90:10) to obtain compound 7 (3.9 mg) colourless residue.
Fraction VI (0.447 g) was chromatographed on normal silica gel CC (2 × 70 cm) using n-hexane–acetone mixtures to increase polarity to afford four main subfractions (M-7 to M-10). Subfraction M-7 (87 mg), eluted with 5% acetone, was subjected to reversed phase silica CC using H2O–MeOH (10:90) as solvent system to afford compound 8 (3.2 mg) as colourless residue and compound 9 (7.5 mg) oily residue. Subfraction M-8 (24 mg) and M-9 (21 mg) were eluted with 10% and 30% acetone, respectively. These fractions were further purified by crystallization from MeOH to afford compound 10 (12 mg) as white amorphous powder. Subfraction M-10 (47 mg), which was eluted with 40% acetone, was subjected to sephadex LH-20 CC using MeOH as eluent to afford compound 11 (3.5 mg) as amorphous powder and compound 12 (3.8 mg) as colourless residue.
Antiviral assay
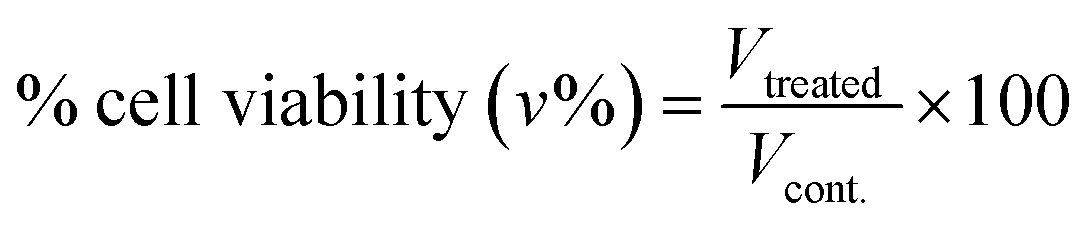 :1 v/v) of non-lethal dilution of tested sample and the virus suspension for one hour. Add 100 μL from viral/sample suspension. Place on a shaking table, 150 rpm for 5 minutes. Incubate (37 °C, 5% CO2) for 24 h cell viability was also assessed using MTT assay. The antiviral activity expressed as % of inhibition using the following formula:
:1 v/v) of non-lethal dilution of tested sample and the virus suspension for one hour. Add 100 μL from viral/sample suspension. Place on a shaking table, 150 rpm for 5 minutes. Incubate (37 °C, 5% CO2) for 24 h cell viability was also assessed using MTT assay. The antiviral activity expressed as % of inhibition using the following formula:| Antiviral activity (%) = (Atv − Acv)/(Acd − Acv) × 100 |
Molecular docking study
Generally, docking simulations were used to study the molecular structure and structure–activity relationship of various compounds. In our study, in silico simulations of the compounds were performed using Molecular Operating Environment (MOE® 2014 software) within crystal structure of Coxsackie Virus B4 (COX B4) X-ray crystal structure (PDB ID: 6ZCK) revealed from protein data bank (RCSB protein data bank; https://www.rcsb.org/). Proteins structures were also prepared according to MOE LigX protocol and their structures were protonated at a cutoff value of 15 Å. Crystal structure of protein was validated by re-docking of co-crystallized ligand and its docking score and RMSD (Å) were in the acceptable range. Structure of the tested compounds was drawn using Chem®Draw program and were energy minimized using MOE ligand preparation tool.Validation of docking process of co-crystallized ligand was carried out by triangular placement method and London δG for rescoring 1 for only 10 retained docking poses of each compound and its RMSD was within acceptable value for docking protocols (RMSD = 1.34 Å). The resultant docking poses for each compound were examined and arranged according to their free binding energy value (kcal mol−1). Finally, 2D interactions for each pose were inspected individually for binding interactions.
Quantum chemical calculation
Quantum-chemical calculations of the sulphated compounds (1, 2 and 11) were made to find molecular properties using the Gauss View 06 and Gaussian 09 program package.30,31 Frontier molecular orbital (EHOMO and ELUMO) analysis, molecular structure, and Surface Electrostatic Potential (SEP) of sulphated compounds (1, 2 and 11) were determined by using Becke–3–Lee–Yang Parr (B3LYP)32 using the DFT/B3LYP method with 6-311++G(d,p) basis set, and their 3D plots were verified with density functional theory (DFT) methods with 6-311++G(d,p) in the ground state. From the data obtained from the Gauss view, the theoretical values for EHOMO, ELUMO, ΔE, electron affinity, ionization potential, softness, hardness, electronegativity, and hydrophobicity were calculated according to Oyewole et al., 2020.33 The quantum-chemical descriptors were calculated using eqn (1)–(8) (Table S1).† All methods were carried out in accordance with relevant guidelines.ADME analysis of the isolated compounds
Absorption, distribution, metabolism, and excretion (ADME) for determination of Lipinski's parameter and pharmacokinetic properties were investigated using SwissADME predictor software (http://www.swissadme.ch/).Results and discussion
A combination of chromatographic techniques resulted in the isolation of twelve compounds (1–12) from the crude extract of the sea urchin Clypeaster humilis (Fig. 1). This study represents the separation of two new compounds (1 and 2) from natural source and one compound (12) reported for the first time from marine organisms. Their structures were elucidated based on 1D, 2D-NMR and HR-ESI-MS spectral analyses. The 1H and DEPT-Q NMR data of 1 and 2 were shown in Tables 1 and 2. The key HMBC and 1H–1H COSY correlations were clarified in Fig. 2. The spectral data were given in the ESI (Fig. S1–S13†).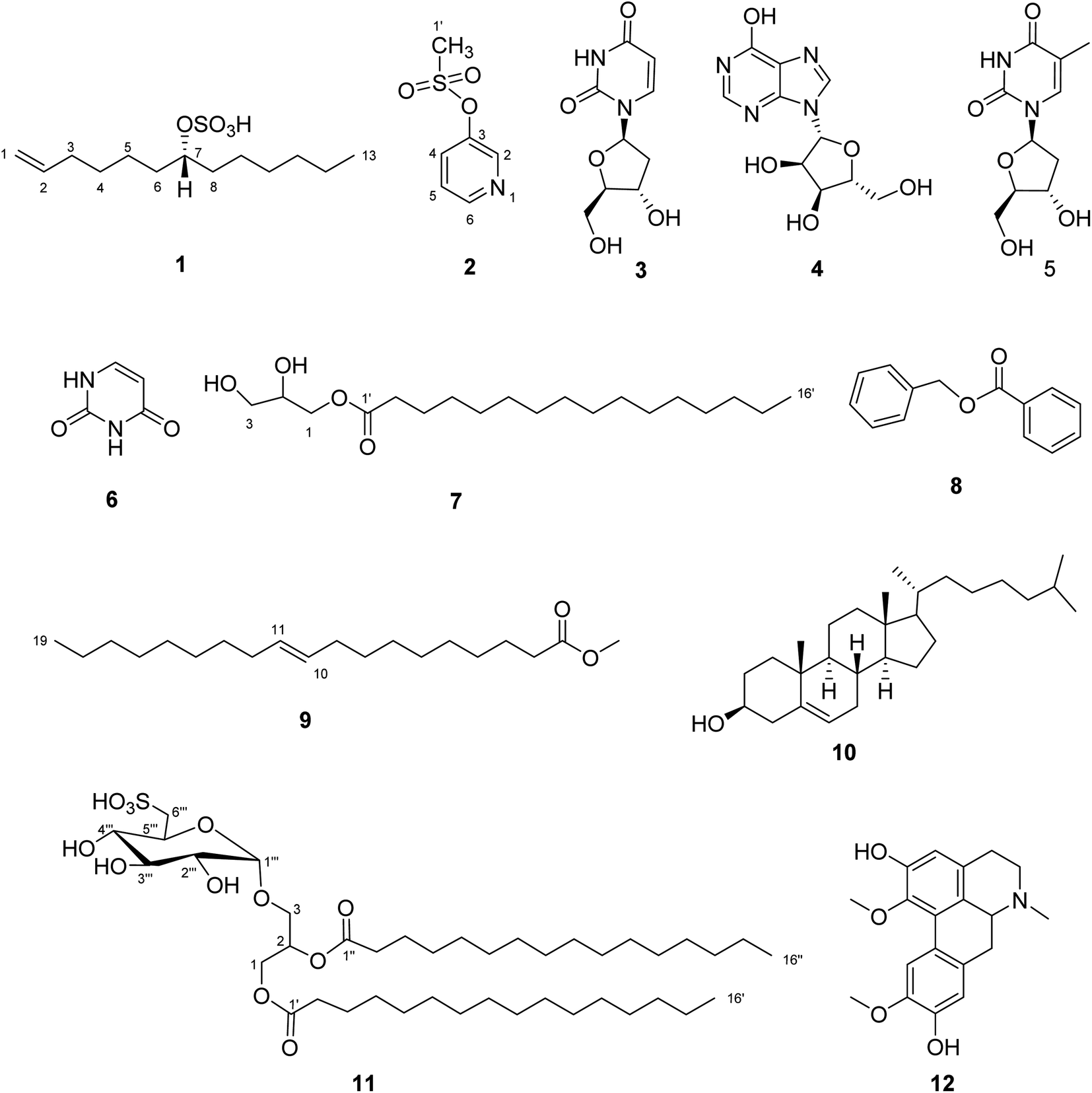 | ||
| Fig. 1 Structure of the isolated compounds (1–12). | ||
| Position | δHa | δCa | ||
|---|---|---|---|---|
| Chemical shift (ppm) | Multiplicity | J in (Hz) | ||
| a Measured in CD3OD, 1H-NMR (400 MHz) and DEPT-Q (100 MHz). | ||||
| 1 | a = 4.91 and b = 4.97 | dd | 2, 10.4 | 114.71 |
| dd | 2, 17.2 | |||
| 2 | 5.80 | m | — | 140.10 |
| 3 | 2.04 | m | — | 35.21 |
| 4 | 1.47 | m | — | 30.55 |
| 5 | 1.60 | m | — | 25.91 |
| 6 | 1.61 | m | — | 34.81 |
| 7 | 4.32 | tt | 6, 12 | 80.93 |
| 8 | 1.63 | m | — | 34.93 |
| 9 | 1.42 | m | — | 29.99 |
| 10 | 1.47 | m | — | 30.07 |
| 11 | 1.35 | m | — | 28.19 |
| 12 | 1.33 | m | — | 23.73 |
| 13 | 0.91 | t | 6.8 | 14.39 |
 | ||
Fig. 2 Key HMBC ( ) and 1H–1H COSY ( ) and 1H–1H COSY ( ) correlations of compounds 1 and 2. ) correlations of compounds 1 and 2. | ||
Structural elucidation of the isolated compounds
Compound 1 was obtained as white amorphous powder (4.9 mg) with [α]D25 − 18.77° (c = 0.1, MeOH). The negative HR-ESI-MS analysis showed a characteristic molecular ion peak [M − H]+ at m/z 277.1479 (calcd for C13H25O4S, 277.1474) (Fig. S1†). The DEPT-Q spectrum revealed characteristic signals at δC 140.10 (CH) and δC 114.71 (CH2) assigned to terminal olefinic double bond (Table 1), this was further approved from 1H-NMR signals at δH H-1a = 4.91 (1H, dd, J = 2, 10.4 Hz), H-1b = 4.97 (1H, dd, J = 2, 17.2 Hz), and H-2 (1H, 5.80, m) assigned to three olefinic protons, which were clarified by HSQC spectrum. Moreover, 1H-NMR spectrum (Fig. S3†) showed the presence of characteristic oxymethine proton at δH 4.32 (tt, J = 6, 12 Hz, H-7) attached to C-7 (δC 80.93), which confirmed from DEPT-Q and HSQC spectra (Fig. S4 and S5†). Moreover, the more downfield chemical shift of C-7 (δC 80.93) indicated the presence of sulphated moiety.8,34 The DEPT-Q spectrum showed terminal methyl group C-13 (δC 14.39), in addition to a group of signals at (δC 23.73 to 34.93) assigned to CH2 cluster revealing the existence of terminal chain moiety.28,35 The HMBC spectrum (Fig. 2 and S6†) showed significant correlations between (H-1a, H-1b, and C-3) and (H-3, C-1, C-2, and C-4), which confirmed the presence of terminal olefinic double bond, in addition to, (H-13, C-10, C-11, and C-12), that confirmed the existence of terminal chain moiety. The attachment of sulphate moiety to C-7 was confirmed from the NMR (1D and 2D) spectroscopy together with the characteristic fragments at m/z 193.0015 and 194.0680 (M+–C6H13) and (M+–C6H12) in the HR-ESI-MS (Fig. S2†). The 1H–1H COSY spectrum (Fig. 2 and S7†) illustrated the presence of correlations between the following (H-1/H-2), (H-2/H-3), (H-6/H-7), and (H-7/H-8), which confirmed the structure of compound 1. The configuration of the only chiral center was deduced from coupling constant of H-7 (δH 4.32, tt, J = 6, 12 Hz), which indicated the β orientation of H-7, on the other hand the sulphated moiety is on the opposite direction which is α oriented.28,35 From the previous data, compound 1 was new compound and identified as (7R) tridec-1-en-7-yl hydrogen sulphate.Compound 2 was obtained as colourless residue (7 mg), the negative HR-ESI-MS analysis showed a characteristic molecular ion peak [M − H]− at m/z 172.0013 (calcd for C6H6NO5S, 172.0069) (Fig. S8†). The DEPT-Q spectrum revealed characteristic five signals at δC 145.63 (CH), δC 155.20 (C), δC 125.81 (CH), δC 125.73 (CH) and δC 145.09 (CH) assigned to pyridine moiety (Table 2). This was further approved by 1H-NMR (Fig. S9†) with four signals at δH 8.90 (1H, d, J = 5.2 Hz), δH 7.91 (1H, d, J = 8.0 Hz), δH 7.91 (1H, d, J = 8.0 Hz) and δH 8.48 (1H, t, J = 7.6 Hz), respectively, which were clarified by HSQC spectrum (Fig. S11†), this confirmed the presence of pyridine moiety.36 Additionally, a singlet signal integrated with three protons at δH 4.29 (3H, s) was appeared in the 1H-NMR spectrum, and correlated with its corresponding carbon atom (δC 46.26) in the HSQC spectrum revealed the existence of terminal methyl moiety.37 Moreover, the HMBC spectrum (Fig. 2 and S12†) showed significant correlations between (H-2 and C-6), (H-4, C-3 and C-5), (H-5, C-3 and C-4) and (H-6, C-2 and C-3) which confirmed the presence of pyridine moiety, in addition to, (H-1′, C-2 and C-3) that confirmed the existence of terminal methyl moiety. The 1H–1H COSY spectrum (Fig. 2 and S13†) illustrated the presence of correlations between (H-5 and H-6) which confirmed the structure of compound 2. From the previous data, compound 2 was first isolated from natural source and identified as pyridine-3-yl methane sulfonate.38,39
The structures of the known compounds were elucidated as 2′-deoxyuridine (3),40 inosine (4),41 thymidine (5),42 uracil (6),7 glyceryl monopalmitate (7),43 benzyl benzoate (8),44,45 methyl-(E)-nonadec-10-enoate (9),46 cholesterol (10),47,48 1,2-di-O-palmitoyl-3-O-(6′′′-sulfo-α-D-quinovopyranosyl)-glycerol (11),49 boldine (12)50 based on comparisons of their 1D (1H and DEPT-Q) NMR spectroscopic data with previously reported data, in addition to mass analysis (Fig. S1–S36†).
Antiviral assay
Marine organisms are known for specialized metabolites with pivotal antiviral activities. These metabolites have diverse structures; however, they are common in having a sulphate moiety. The previously reported sulphated polysaccharides, sulphated steroids and sulfo-lipids may exert their action via inhibiting the different stages of the viral infection process, which involves blocking the initial entry of the virus or inhibiting its transcription and translation by modulating the immune response of the host cell. The possible mechanism of action may be attributed to the fact that negatively charged sulphate group may interact with positively charged protein sites and consequently explain their antiviral activity.1,2,51,53
| Molecule # | Docking score (S; kcal mol−1) | Binding interactions | ||
|---|---|---|---|---|
| a.a. residues | Type | Length (Å) | ||
| 1 | −5.16 | ARG228 | H-acceptor | 3.22 |
| ARG228 | H-acceptor | 2.94 | ||
| ARG228 | H-acceptor | 3.14 | ||
| 2 | −3.69 | PHE236 | H-acceptor | 3.06 |
| PHE236 | H-pi | 3.92 | ||
| THR235 | pi-H | 3.95 | ||
| 3 | −3.92 | — | ||
| 4 | −4.53 | |||
| 5 | −4.33 | |||
| 6 | −4.31 | |||
| 9 | −4.04 | |||
| 11 | −5.36 | THR235 | H-donor | 2.85 |
| 12 | −6.11 | |||
| QFW | −5.35 | ARG228 | H-acceptor | 3.17 |
| ARG228 | H-acceptor | 3.15 | ||
| PHE236 | H-pi | 3.94 | ||
To achieve the best possible binding orientation, the target compounds and the co-bound inhibitor were docked onto the protein's probable binding site to explore their interactions and binding mechanisms with the important amino acids in the active sites. Interestingly, visual inspection of docking poses generated for each of test compound showed that sulphated molecules (1, 2, and 12) have binding interactions (in the form of H-bond and hydrophobic pi interactions) with key amino acid residues (Arg 228, THR 235, & PHE 236) with docking score as did co-crystallized ligand, QFW (Fig. 3).
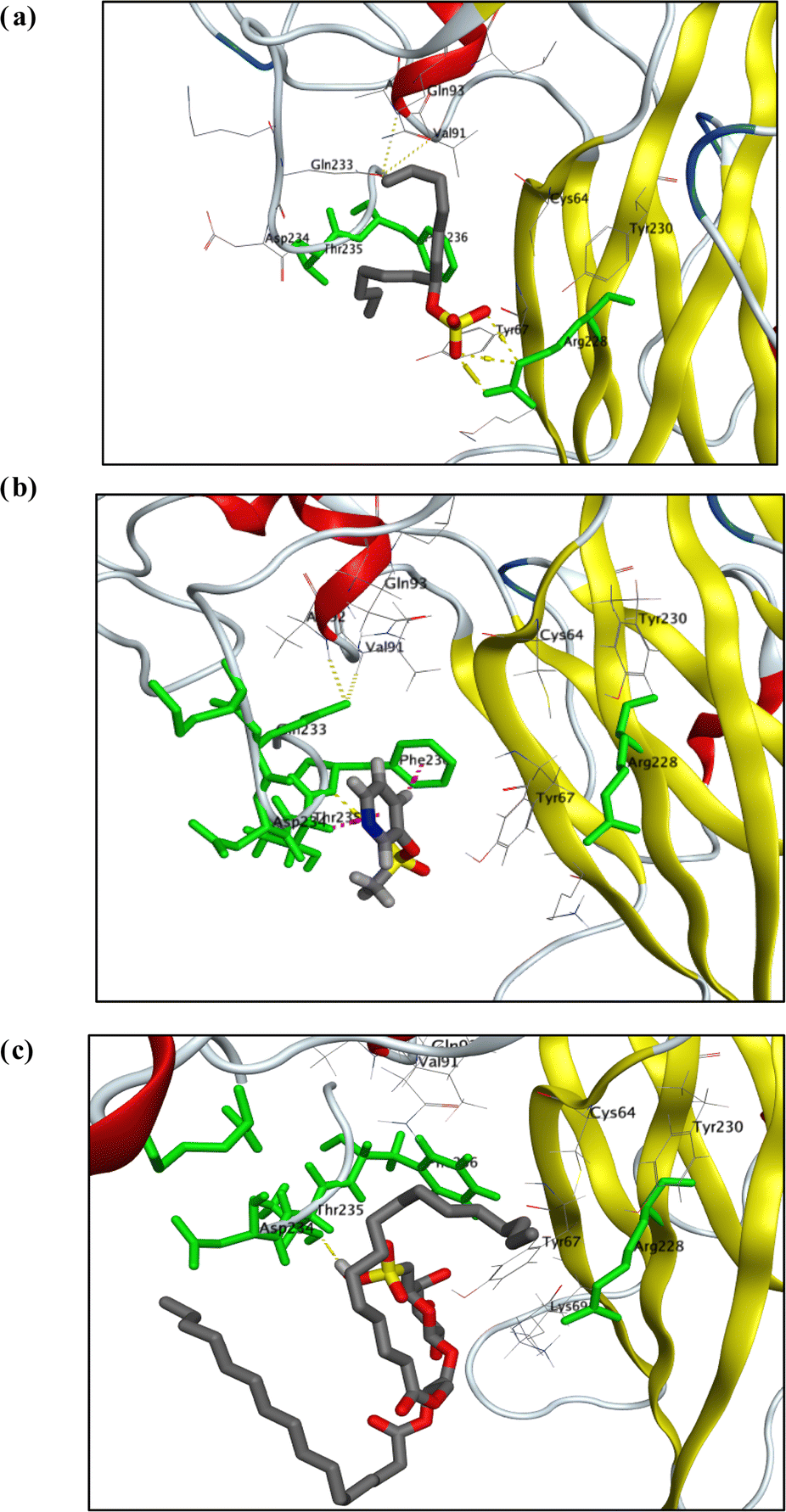 | ||
| Fig. 3 Visualization diagrams of docking poses of compound 1 (a), 2 (b) and 11 (c) within X-ray crystal structure of COX-B4 (PDB ID: 6ZCK) active site: key amino acid residues (ARG 228, THR 235, and PHE 236) were colored in light-green bold line; H-bonds (yellow-dotted lines); pi-H and/or H-pi interactions (light-purple-dotted lines). | ||
QFW (the co-crystallized ligand) is a 4-[(6-propoxynaphthalen-2-yl)sulfonylamino]benzoic acid derivative which targets the VP1 hydrophobic pocket, which has an entrance located at the base of the canyon-like depression surrounding each capsid fivefold axis. The site is normally occupied by the pocket factor; however, binding of chemically optimized compounds dislodges the lipid due to the drugs having a much higher binding affinity. Replacement of the pocket factor with capsid binders provides entropic stabilization by raising the un-coating free energy barrier against thermal or receptor-induced conformational changes. In this way, the sulphated compounds (1, 2 and 11) can prevent formation of expanded 135S intermediates or A-particles, which is a required step for genome release. These remarkable results were consistent with findings obtained with antiviral assay of these sulphated compounds (1, 2 and 11).54,55
Quantum chemical calculation
Both physical and chemical properties of sulphated compounds (1, 2 and 11) was measured using chemical quantum method as in (Table S2† and Fig. 4) based on DFT parameter. Compound 1 is the most active compound with lowest ΔE = 0.06 and ionization potential = 0.20. The stability of compounds measured by degree of softness or hardness. Low hardness and high softness values for the three sulphated compounds reflect high stability for the compounds, in which compound 1 is the most stable with softness value equal to 30.93 eV in comparison with compound 2 and 11 with values equal to 13.14 and 12.92 eV, respectively. Additionally, compound 1 showed the highest electrophilicity value equal to 0.91 in comparison with 2 and 11 with values equal to 0.69 and 0.50, respectively. All of this parameter showed high stability and reactivity of sulphated compounds.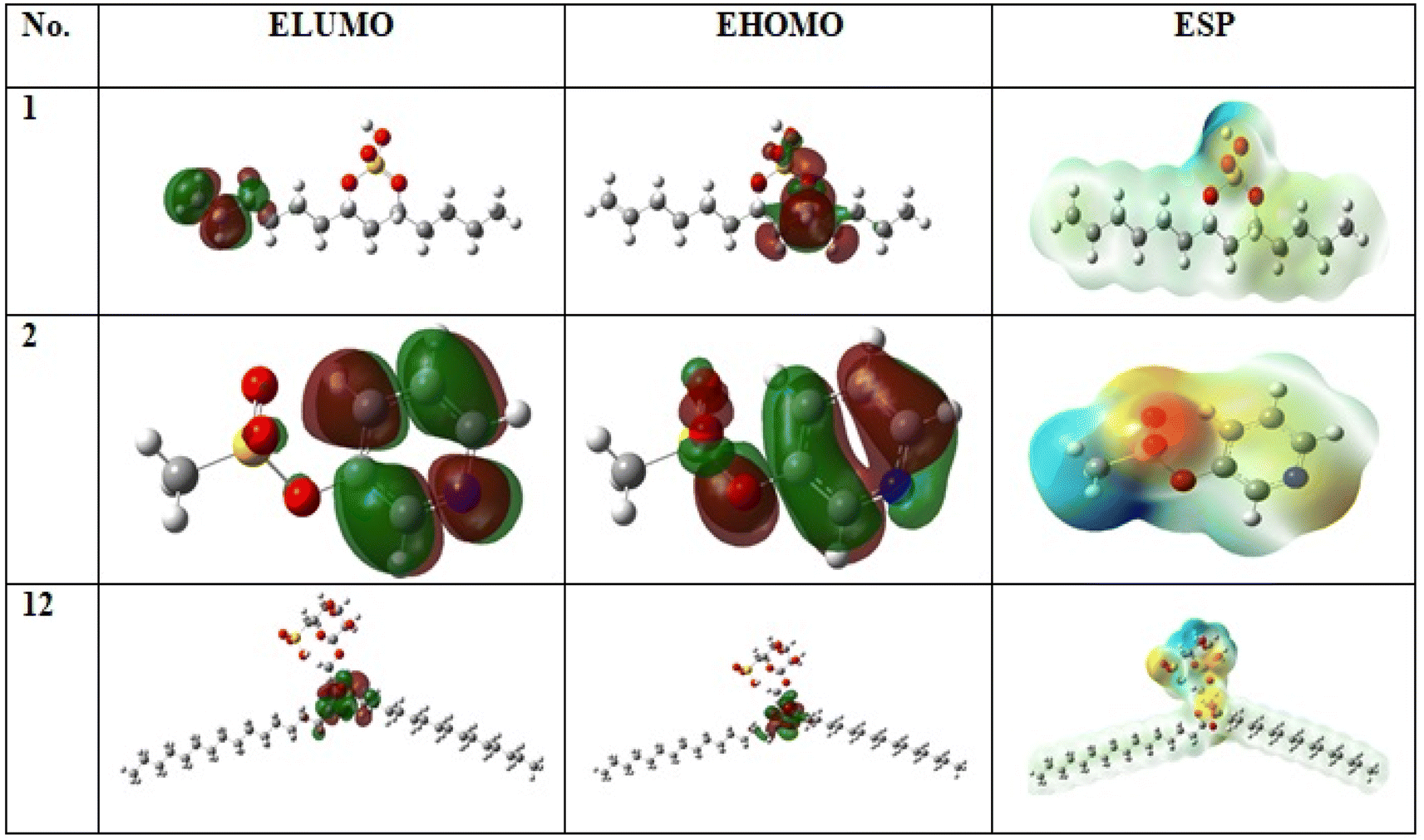 | ||
| Fig. 4 Frontier molecular orbitals (HOMOs, LUMOs) and electrostatic potential (ESP) surface of the compounds (1, 2 and 11) by using the DFT/B3LYP/6-311++G(2d,2p) method. | ||
ADME analysis of the isolated compounds
Concerning the promising antiviral activity of the tested compounds, we decided to check their obedience to Lipinski rules and their feasibility as therapeutic agents. Compounds with no more than one violation of RO5 criteria could be potential orally active drug candidate as stated by Lipinski rule: M.W. below 500 dalton, ten or less H-bond acceptor centers, five or less H-bond donor centers, ten or less rotatable bonds, and a partition coefficient ≤ 5. RO5 expansions improved drug likeness predication of compounds presented by the molar refractivity (40 to 130) and total polar surface area (not exceeding 140 Å). As seen from (Table 4) that all the compounds have values within acceptable ranges of all RO5 criteria indicating their potential as valuable therapeutic agents.| # | MFa | Mb | Nrotbc | HBAd | HBDe | MRf | TPSAg | iLogPh | Water solubility | HIA%i | BBB permeantj | Fk | PAINSl |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a MF, molecular formula.b M, molecular mass (dalton).c Nrotb, # of rotatable bonds.d HBA, hydrogen bond acceptor.e HBD, hydrogen bond donor.f MR, molar refractivity.g TPSA, total polar surface area.h iLogP, octanol/water partition coefficient.i HIA%, human gastrointestinal absorption.j BBB permeant, blood–brain barrier penetration.k F, Abbott oral bioavailability score.l PAINS, pan-assay interference compounds. | |||||||||||||
| 1 | C13H26O4S | 278.4 | 12 | 4 | 1 | 75.75 | 71.98 | 2.85 | Soluble | High | No | 0.85 | 0 |
| 2 | C6H7NO3S | 173.2 | 2 | 4 | 0 | 39.7 | 64.6 | 1.21 | Very soluble | High | Yes | 0.55 | 0 |
| 3 | C9H12N2O6 | 244.2 | 2 | 6 | 4 | 54.3 | 125 | 0.44 | Very soluble | Low | No | 0.55 | 0 |
| 4 | C10H12N4O5 | 268.2 | 2 | 8 | 4 | 60.3 | 134 | 0.88 | Very soluble | Low | No | 0.55 | 0 |
| 5 | C10H13N5O3 | 251.2 | 2 | 6 | 3 | 61.5 | 119.3 | 1.03 | Very soluble | High | No | 0.55 | 0 |
| 6 | C10H14N2O5 | 242.2 | 2 | 5 | 3 | 58.1 | 104.5 | 1.16 | Very soluble | High | No | 0.55 | 0 |
| 9 | C14H12O2 | 212.2 | 4 | 2 | 0 | 62.2 | 26.3 | 2.68 | Soluble | High | Yes | 0.55 | 0 |
| 11 | C28H48O | 400.7 | 6 | 1 | 1 | 128.4 | 20.2 | 5.13 | Poor | Low | No | 0.55 | 0 |
| 12 | C19H21NO4 | 327.4 | 2 | 5 | 2 | 96 | 62.16 | 2.93 | Soluble | High | Yes | 0.55 | 0 |
| RO5 | ≤500 | ≤10 | ≤10 | ≤5 | ≤130 | ≤140 | ≤5 | ||||||
Remarkably, the ADME study for the isolated compounds showed that estimated water solubility of all test molecules showed a poor to very high degree of water solubility, which co-inside with the estimated values for both GIT absorption, and BBB penetration, as shown in (Table 4). Finally, the calculated Abbott oral bioavailability score (F) of all tested compounds were found above zero with no violation to pan-assay interference panel, indicating their high potential as drug candidates in clinical studies.
Conclusions
This study reports isolation of twelve compounds from the sea urchin C. humilis collected from the red sea including three sulfated compounds. Two compounds of them were undescribed namely (7R) tridec-1-en-7-yl hydrogen sulfate (1), pyridine-3-yl methane sulfonate (2), and one previously reported 1,2-di-O-palmitoyl-3-O-(6′′′-sulfo-α-D-quinovopyranosyl)-glycerol (11), together with nine known compounds including boldine (12), which isolated for the first time from marine organisms. The crude extract and sulfated compounds subjected to MTT assay to evaluate their antiviral activity against COX-B4 virus. The crude extract and compound 1 showed potent antiviral activity. In silico studies supported the in vitro results by using molecular docking stimulation and chemical quantum analyses. Sulfated metabolites could be considered as promising source for a future development of antiviral drugs.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We would like to acknowledge the Egyptian Ministry of Higher Education and Scientific Research and the National Center for Natural Products Research (NCNPR), School of Pharmacy, University of Mississippi, Mississippi, USA for their financial support. We are thankful to Dr Baharthi Avula for providing the HR-ESI-MS.References
- N. Hans, A. Malik and S. Naik, Bioresour. Technol. Rep., 2021, 13, 100623 CrossRef CAS PubMed.
- F. Carvalhal, M. Correia-da-Silva, E. Sousa, M. Pinto and A. Kijjoa, J. Mol. Endocrinol., 2018, 61, T211–T231 CAS.
- F. Hu, X. Chi, M. Yang, P. Ding, D. Yin, J. Ding, X. Huang, J. Luo, Y. Chang and C. Zhao, Sci. Rep., 2021, 11, 15116 CrossRef CAS PubMed.
- A. Mancosu and J. H. Nebelsick, Acta Palaeontologica Polonica, 2017, 62, 627–646 CrossRef.
- J. H. Nebelsick and A. Mancosu, Contribution from museum of planteology, 2022, vol. 34, pp. 157–148 Search PubMed.
- A. Sibiya, J. Jeyavani, J. Sivakamavalli, C. Ravi, M. Divya and B. Vaseeharan, Regional Studies in Marine Science, 2021, 44, 101760 CrossRef.
- S. Kan, G. Chen, C. Han, Z. Chen, X. Song, M. Ren and H. Jiang, Nat. Prod. Res., 2011, 25, 1243–1249 CrossRef CAS PubMed.
- F. M. Abdelkarem, E.-E. K. Desoky, A. M. Nafady, A. E. Allam, A. Mahdy, A. Ashour and K. Shimizu, Nat. Prod. Res., 2022, 36, 1118–1122 CrossRef CAS PubMed.
- Y. Liu, H. Yan, K. Wen, J. Zhang, T. Xu, L. Wang, X. Zhou and X. Yang, J. Food Biochem., 2011, 35, 932–938 CrossRef CAS.
- D.-S. Lee, X. Cui, W. Ko, K.-S. Kim, I. C. Kim, J. H. Yim, R.-B. An, Y.-C. Kim and H. Oh, Arch. Pharmacal Res., 2014, 37, 983–991 CrossRef CAS PubMed.
- P. Francis and K. Chakraborty, Med. Chem. Res., 2020, 29, 656–664 CrossRef CAS.
- D. M. Moreno-García, M. Salas-Rojas, E. Fernández-Martínez, M. del Rocío López-Cuellar, C. G. Sosa-Gutierrez, A. Peláez-Acero, N. Rivero-Perez, A. Zaragoza-Bastida and D. Ojeda-Ramírez, PeerJ, 2022, 10, e13606 CrossRef PubMed.
- A. Wilder-Smith, Tropical Diseases, Travel Medicine and Vaccines, 2021, 7, 1–11 CrossRef PubMed.
- M. A. AL-Zobaei and Z. R. AL-Ani, Egyptian Academic Journal of Biological Sciences. G, Microbiology, 2019, 11, 1–12 Search PubMed.
- C. C. Kemball, M. Alirezaei and J. L. Whitton, Future Microbiol., 2010, 5, 1329–1347 CrossRef PubMed.
- U. Kühl, M. Pauschinger, M. Noutsias, B. Seeberg, T. Bock, D. Lassner, W. Poller, R. Kandolf and H.-P. Schultheiss, Circulation, 2005, 111, 887–893 CrossRef PubMed.
- M. P. Nekoua, E. K. Alidjinou and D. Hober, Nat. Rev. Endocrinol., 2022, 18, 503–516 CrossRef CAS PubMed.
- A. Carré, F. Vecchio, M. Flodström-Tullberg, S. You and R. Mallone, Endocr. Rev., 2023, bnad007 Search PubMed.
- Z. Han, Y. Zhang, K. Huang, J. Wang, H. Tian, Y. Song, Q. Yang, D. Yan, S. Zhu and M. Yao, BMC Infect. Dis., 2019, 19, 1–10 CrossRef PubMed.
- K. Payne, P. Kenny, J. M. Scovell, K. Khodamoradi and R. Ramasamy, Sexual Medicine Reviews, 2020, 8, 518–530 CrossRef PubMed.
- O. T. Ebob, S. B. Babiaka and F. Ntie-Kang, Nat. Prod. Bioprospect., 2021, 11, 611–628 CrossRef CAS PubMed.
- A. G. Atanasov, S. B. Zotchev, V. M. Dirsch and C. T. Supuran, Nat. Rev. Drug Discovery, 2021, 20, 200–216 CrossRef CAS PubMed.
- M. Roivainen, S. Rasilainen, P. Ylipaasto, R. Nissinen, J. Ustinov, L. Bouwens, D. c. L. Eizirik, T. Hovi and T. Otonkoski, J. Clin. Endocrinol. Metab., 2000, 85, 432–440 CAS.
- N. Tariq and C. Kyriakopoulos, Group B coxsackie virus, 2020 Search PubMed.
- H. Fechner, S. Pinkert, A. Geisler, W. Poller and J. Kurreck, Molecules, 2011, 16, 8475–8503 CrossRef CAS PubMed.
- P. Jadhav, N. Kapoor, B. Thomas, H. Lal, N. Kshirsagar and N. Am, J. Med. Sci., 2012, 4, 641 Search PubMed.
- S. A. Zidan, M. A. Orabi, M. A. Mustafa, M. AAl-Hammady, M. S. Kamel and J. Pharmacogn, Phytochemistry, 2016, 5, 247–251 Search PubMed.
- H. K. Assaf, A. M. Nafady, M. S. Abdelkader, A. E. Allam, M. S. Kamel and J. Pharmacogn, Phytochemistry, 2015, 4, 282–290 CAS.
- A. Karimi, M.-T. Moradi, M. Rabiei and S. Alidadi, Antiviral Chem. Chemother., 2020, 28, 2040206620916571 CAS.
- D. G. El-Hosari, W. M. Hussein, M. O. Elgendy, S. O. Elgendy, A. R. Ibrahim, A. M. Fahmy, A. Hassan, F. A. Mokhtar, M. F. Hussein and M. E. Abdelrahim, Pharmaceuticals, 2023, 16, 1378 CrossRef CAS PubMed.
- P. Houghton, R. Fang, I. Techatanawat, G. Steventon, P. J. Hylands and C. Lee, Methods, 2007, 42, 377–387 CrossRef CAS PubMed.
- J. Tirado-Rives and W. L. Jorgensen, J. Chem. Theory Comput., 2008, 4, 297–306 CrossRef CAS PubMed.
- R. O. Oyewole, A. K. Oyebamiji and B. Semire, Heliyon, 2020, 6(5), e03926 CrossRef PubMed.
- H.-J. Zhang, J.-B. Sun, H.-W. Lin, Z.-L. Wang, H. Tang, P. Cheng, W.-S. Chen and Y.-H. Yi, Nat. Prod. Res., 2007, 21, 953–958 CrossRef CAS PubMed.
- N. A. Chira, A. Nicolescu, R. Stan and S. Rosca, Rev. Chim., 2016, 67, 1257–1263 CAS.
- A. Ito, R. Kasai, K. Yamasaki and H. Sugimoto, Phytochemistry, 1993, 33, 1133–1137 CrossRef CAS.
- H.-Q. Wang, C.-Z. Peng and Y.-G. Chen, Chem. Nat. Compd., 2015, 51, 1167–1168 CrossRef CAS.
- M. Del Giudice, G. Settimj and M. Delfini, Tetrahedron, 1984, 40, 4067–4080 CrossRef CAS.
- P. Leowanawat, N. Zhang, A.-M. Resmerita, B. M. Rosen and V. Percec, J. Org. Chem., 2011, 76, 9946–9955 CrossRef CAS PubMed.
- D. R. Abou-Hussein, J. M. Badr and D. T. Youssef, Nat. Prod. Sci., 2007, 13, 229–233 CAS.
- H. Wu, Z. Su, H. Aisa, A. Yili and B. Hang, Chem. Nat. Compd., 2007, 43, 472–473 CrossRef CAS.
- P. T. Huyen, T. Van Loc, T. Van Sung and T. T. P. Thao, Chem. Nat. Compd., 2019, 55, 141–143 CrossRef CAS.
- H. Wang, R. Zhang, K. Zhang, X. Chen and Y. Zhang, Molecules, 2022, 27, 2858 CrossRef CAS PubMed.
- A. R. Rosandy, L. B. Din, W. Yaacob, N. I. Yusoff, I. Sahidin, J. Latip, S. Nataqain and N. M. Noor, Malays. J. Anal. Sci., 2013, 17, 50–58 Search PubMed.
- H. Shen, X. Lu, K.-Z. Jiang, K.-F. Yang, Y. Lu, Z.-J. Zheng, G.-Q. Lai and L.-W. Xu, Tetrahedron, 2012, 68, 8916–8923 CrossRef CAS.
- J. E. Patterson, I. R. Ollmann, B. F. Cravatt, D. L. Boger, C.-H. Wong and R. A. Lerner, J. Am. Chem. Soc., 1996, 118, 5938–5945 CrossRef CAS.
- Q. Dong, Z. Wang, H. Liu, C. Zhang, D. He, G. Wu and L. Zhang, Chem. Nat. Compd., 2011, 47, 114–115 CrossRef CAS.
- W. S. Grainger and E. J. Parish, Steroids, 2015, 101, 103–109 CrossRef CAS PubMed.
- Z. Cantillo-Ciau, R. Moo-Puc, L. Quijano and Y. Freile-Pelegrín, Mar. Drugs, 2010, 8, 1292–1304 CrossRef CAS PubMed.
- W. M. N. H. W. Salleh, N. M. Shakri, M. A. Nafiah, N. Ab Ghani, N. E. Rasol, N. S. Rezali and M. H. Jauri, Bull. Chem. Soc. Ethiop., 2022, 36, 963–969 CrossRef CAS.
- E. Plouguerné, L. M. De Souza, G. L. Sassaki, J. F. Cavalcanti, M. T. Villela Romanos, B. A. Da Gama, R. Crespo Pereira and E. Barreto-Bergter, Mar. Drugs, 2013, 11, 4628–4640 CrossRef PubMed.
- K. R. Gustafson, J. H. Cardellina, R. W. Fuller, O. S. Weislow, R. F. Kiser, K. M. Snader, G. M. Patterson and M. R. Boyd, J. Natl. Cancer Inst., 1989, 81, 1254–1258 CrossRef CAS PubMed.
- P. G. Spear, Cell. Microbiol., 2004, 6, 401–410 CrossRef CAS PubMed.
- J. W. Flatt, A. Domanska, A. L. Seppälä and S. J. Butcher, Commun. Biol., 2021, 4, 250 CrossRef CAS PubMed.
- T.-C. Chen, K.-F. Weng, S.-C. Chang, J.-Y. Lin, P.-N. Huang and S.-R. Shih, J. Antimicrob. Chemother., 2008, 62, 1169–1173 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01966k |
| This journal is © The Royal Society of Chemistry 2024 |