
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition-metal ions intercalation chemistry enabled the manganese oxides-based cathode with enhanced capacity and cycle life for high-performance aqueous zinc-ion batteries†
Hongyu Zhao *,
Li Wang and
Meiling Li
*,
Li Wang and
Meiling Li
School of Electronic Engineering, Lanzhou City University, Lanzhou 730000, Gansu, People's Republic of China. E-mail: ruofan_1007@126.com
First published on 27th March 2024
Abstract
Aqueous zinc-ion batteries (AZIBs) employing mild aqueous electrolytes are recognized for their high safety, cost-effectiveness, and scalability, rendering them promising candidates for large-scale energy storage infrastructure. However, the practical viability of AZIBs is notably impeded by their limited capacity and cycling stability, primarily attributed to sluggish cathode kinetics during electrochemical charge–discharge processes. This study proposes a transition-metal ion intercalation chemistry approach to augment the Zn2+ (de)intercalation dynamics using copper ions as prototypes. Electrochemical assessments reveal that the incorporation of Cu2+ into the host MnO2 lattice (denoted as MnO2–Cu) not only enhances the capacity performance owing to the additional redox activity of Cu2+ but also facilitates the kinetics of Zn2+ ion transport during charge–discharge cycles. Remarkably, the resulting AZIB employing the MnO2–Cu cathode exhibits a superior capacity of 429.4 mA h g−1 (at 0.1 A g−1) and maintains 50% capacity retention after 50 cycles, surpassing both pristine MnO2 (146.8 mA h g−1) and non-transition-metal ion-intercalated MnO2 (MnO2–Na, 198.5 mA h g−1). Through comprehensive electrochemical kinetics investigations, we elucidate that intercalated Cu2+ ions serve as mediators for interlayer stabilization and redox centers within the MnO2 host, enhancing capacity and cycling performance. The successful outcomes of this study underscore the potential of transition-metal ion intercalation strategies in advancing the development of high-performance cathodes for AZIBs.
1. Introduction
The ever-increasing demand for the utilization of renewable energy, such as solar, wind, tidal energy, etc., has triggered the development of energy storage devices because of the uneven distribution nature in time and space of renewable energy.1–3 Thus, developing efficient and affordable energy storage devices/systems shows tremendous research potential for humanity's journey toward a sustainable society. Aqueous zinc ion batteries (AZIBs) have been considered some of the most promising candidates for next-generation energy storage devices/systems due to their unique merits of high safety, affordable cost, and high scalability, in which the charge carrier Zn2+ ions migrate between the Zn metal anode and cathode materials capable of reversible Zn2+ insertion/extraction.4–7 However, the practical applications of AZIBs are greatly hindered by the unexpected capacity and cycling performance of the used cathode.8–10 Therefore, it is a critical and challenging issue to develop a suitable cathode material for the realization of high-performance AZIBs.In the past years, extensive research efforts have been devoted to exploring and developing different cathode materials for the AZIBs, which mainly include manganese oxides, vanadium oxides, Prussian blue analogs, etc. Among them, manganese oxide (MnO2) shows promising potential as the cathode material because of its superior electrochemical properties with a high specific capacity of about 308 mA h g−1 and wide operation voltage of 1.2 V at the mild electrolyte (i.e., ZnSO4).11–13 Its versatile crystal forms of α-MnO2, β-MnO2, δ-MnO2, and γ-MnO2 enable the ubiquitous electrolyte ions Zn2+ diffusion pathways due to well-defined interlayered or tunnel structure.14 It is worth noting that the MnO2-based electrode suffers from structure variation during the electrochemical charge–discharge operations, in which the hydrated ions (Zn2+ or H+ ions) occur insertion/extraction in the host of MnO2 materials leading to the shrinkage and expansion of structures. Such a continued structure variation often results in undesired long-term instability during electrochemical cycling until the structure collapses and releases MnO2 due to the Jahn–Teller effect.15 To stabilize the host structures of MnO2, an ions-intercalated (such as K+, Mg2+, and Al3+, etc.) strategy has been introduced to the interlayered or tunnel regimes, and the electrochemical performance of the MnO2 has been enhanced to some extent.16–22 For example, Liu et al. reported that K+ ions-intercalated MnO2 (i.e., MnO2–K) exhibited a capacity of 266 mA h g−1 and an excellent capacity retention percentage of 92% after 200 cycles.23 Therefore, it is reasonable that the ion-intercalation of MnO2 may be effective in enhancing the cyclic stability of MnO2-based cathodes for high-performance AZIBs. However, the introduction of foreign ions reported in previous studies may obstruct the electrolyte ions Zn2+ insertion/extraction during the electrochemical charge–discharge process, impeding the improvement of the capacity properties due to the limited accessible active sites.
In this work, we have proposed a transition-metal ions intercalation chemistry strategy to enhance the electrochemical performance of MnO2-based electrodes with the prototypical model of copper ions (Cu2+), in which the Cu2+ ions were in situ pre-embedded in the electrodeposited MnO2. Introducing Cu2+ ions into the host MnO2 material (i.e., MnO2–Cu) enhances the capacity performance of the MnO2-based electrode because of the extra capacity contribution of the redox nature of Cu2+. Meanwhile, the Cu2+ ion intercalation and the accompanying structural water molecules in the interlayered regime play a role as the pillars among [MnO6] polymorph blocks, which promotes the Zn2+ ion kinetics and stabilizes the MnO2 structure during the electrochemical charge–discharge process. The resultant AZIB configured with MnO2–Cu cathode delivers the highest capacity of 429.4 mA h g−1 (0.1 A g−1) and capacity retention of 50% after 50 cycles, which is far superior to that of pristine MnO2 and non-transition-metal ions intercalated MnO2 (i.e., MnO2–Na). This work may provide significant guidance for the development of high-performance AZIBs.
2. Experimental
Fabrication of MnO2-based electrodes
The MnO2-based electrodes were prepared using the electrodeposition technique on the carbon cloth (CC). Before the electrodeposition process, the CC was cut into a small disc with a 1-inch diameter, cleaned with acetone and deionized water several times, following a drying process in the vacuum oven at 60 °C for 6 h. To enhance the hydrophilicity of the obtained CC disc, additional plasma treatment was performed for 5 min in an oxygen atmosphere. The MnO2 materials were then electrodeposited on the treated CC discs. The electrodeposition process was performed in a three-electrode electrochemical system with the treated CC disc as the working electrode, Pt-foil as the counter electrode, and saturated calomel electrode (SCE) as the reference electrode. For a typical electrodeposition process, the seed layer of the MnO2 was first deposited by the cyclic voltammetry (CV) method with a scan rate of 5 mV s−1 under a potential window of 0–1.2 V. And galvanostatic charging–discharging (GCD) operations with 100-cycle were further performed with the electrochemical parameters of the current density of 5 mA cm−2 and the potential of 0–1.2 V to complete the electrodeposition process of MnO2-based electrode. Here the electrolyte recipes for the electrodeposition of pristine MnO2, MnO2–Na, and MnO2–Cu were 0.02 M Mn(NO3)2, a mixture of 0.02 M Mn(NO3)2 + 0.1 M NaNO3, and a mixture of 0.02 M Mn(NO3)2 + 0.1 M Cu(NO3)2, respectively. Note that, for the MnO2–Cu electrodepositing process, the deposition potential window was set at 0.6–1.2 V to avoid the Cu2+/Cu redox potential.Materials characterization
The morphologies and microstructure of the obtained samples (i.e., MnO2, MnO2–Na and MnO2–Cu) were examined by field-emission scanning electron microscopy (FE-SEM, CIQTEK SEM4000). Transmission electron microscopy (TEM, Thermo-fisher Talos 200S) equipped with energy-dispersive X-ray spectroscopy (EDS) was used to characterize the microstructure and elemental distribution of the obtained MnO2–Cu. X-ray photoelectron spectroscopy (XPS, KRATOS AXIS-Ultra DLD) was employed to understand the surface chemistry of the samples. The crystal structure of the obtained samples was analyzed by X-ray diffraction (XRD, Philips, X'pert Pro, Cu K-alpha, 0.154056 nm). To quantify the mass-specific capacity of the electrode, the mass loading of active materials (i.e., MnO2-based materials) in the different electrodes was weighted by the Mettler microbalance (Mettler Toledo, XS105DU).Electrochemical performance measurements
The electrochemical properties of the different samples (i.e., MnO2, MnO2–Na, MnO2–Cu) were investigated under the three-electrode system, which is the same as the electrodeposition system. And typical CV curves and electrochemical impedance spectroscopy (EIS, with the frequency ranging from 0.01 Hz to 100 kHz at an alternating current voltage disturbance amplitude of 5 mV) were recorded in the electrochemical working station (Chenhua CHI660). To better evaluate the electrochemical performance of the obtained electrode, the CR232 coin-cell was further assembled with the MnO2-based electrodes (i.e., MnO2@CC, MnO2–Na@CC and MnO2–Cu@CC) as the freestanding cathode and a zinc metal foil as the anode with the same size as the cathode. The glass-fiber film with a pore size of about 100 nm was directly used as the separator and the aqueous mixture of 2 M ZnSO4 with 0.1 M MnSO4 as the electrolyte. During the coin-cell tests, the charge–discharge operations at different current densities were done on a LAND battery testing system (LANHE CT3002A) to study the rate capability and cycling performance of the assembled AZIBs.3. Results and discussion
The MnO2 reveals versatile crystal forms, such as α-MnO2, β-MnO2, δ-MnO2, and γ-MnO2. Typically, δ-MnO2 manifests superior electrochemical performance due to its layered architecture with an interlayer spacing of 0.7 nm, which secures the facilitated electrolyte ions kinetics. Thus, we adopt δ-MnO2 as the host material to demonstrate the effectiveness of the transition-metal ions intercalation chemistry strategy. The Cu2+ ions intercalated δ-MnO2 (i.e., MnO2–Cu) have been successfully fabricated via in situ electrochemical deposition method. To better illustrate the performance-enhancing effect of transition-metal ions (i.e., Cu2+) intercalation, we have also synthesized pristine δ-MnO2 (i.e., MnO2) and non-transition-metal ions intercalated δ-MnO2 (MnO2–Na) as the comparison. It can be found that all three samples display a loose and porous structure (Fig. 1a–c), and the magnified micrograph (insets of Fig. 1a–c) reveals the MnO2 sheets aggregating nature of the obtained samples. And the ions' intercalation operations did not induce the observable morphological changes. Note that the vertically located MnO2 sheets in the samples may effectively increase the contact between the electrode material and the electrolyte, allowing more electrolyte ions to participate in the redox reaction with well-defined ionic diffusion pathways. Fig. 1d confirms the nature of δ-MnO2 of the electrodeposited samples, and the diffraction peaks in the XRD spectrum match well with the standard phase of δ-MnO2 (JCPDS #18-0802). The intercalated ions did not induce the impurity phase during electrodeposition, indicating the existence of ions form of sodium ions and copper ions in the host material MnO2. The presence and valence state of copper ions in the MnO2–Cu were studied with the XPS core-level spectra of Cu 2p (Fig. S1†), which fully demonstrates the divalent copper ion properties (i.e., Cu2+). To further verify the intercalation of Cu in the MnO2–Cu sample, element mapping experiments have been conducted using EDS mapping, and the results are shown in Fig. 1e. An even distribution of elements Mn, O and Cu can be found in the sample-located profile, revealing the uniform Cu2+ intercalation in the host material MnO2. The same conclusion can also derived for the electrodeposited MnO2–Na sample, in which the elements Mn, O, and Na are distributed evenly in the EDS signal-captured region (Fig. S2†). Note that the ions intercalation (both for the Na+ and Cu2+ ions) induces the interlayer spacing shrinkage, and the interlay spacing of the sample MnO2, MnO2–Na, and MnO2–Cu are measured to be 0.72 nm, 0.69 nm, and 0.65 nm, respectively, as shown in Fig. S3.† After the ions' intercalation, the interlayer spacing shrinkage should be attributed to the strong interlayer interaction due to the strong bonding between the ions (with the positive valence) and the terminated O of [MnO6] octahedron. And the smaller interlayer spacing of MnO2–Cu was considered the most bonding strength because of the +2 valence value compared to the MnO2–Na. The interlayered structure of all three samples indicates the delta-phase (i.e., δ-MnO2) nature of the MnO2-based electrodes, agreeing well with that of XRD results (Fig. 1e). | ||
| Fig. 1 Morphological and structural characterization of the obtained MnO2, MnO2–Na and MnO2–Cu. Representative SEM images of the sample (a) MnO2, (b) MnO2–Na, (c) MnO2–Cu, and the corresponding inset shows the local view of the samples, respectively; (d) XRD spectrum of the obtained samples; (e) elemental maps of the sample MnO2–Cu. | ||
The valence and chemical bond information of the obtained MnO2, MnO2–Na and MnO2–Cu were further investigated with the XPS spectrum, as shown in Fig. 2. Fig. 2a shows the high-resolution XPS spectra of Mn 2p. Generally, the typical Mn 2p edge can be fitted and deconvoluted into two main valence states (Mn3+, and Mn4+).24 It can be found that the ions intercalation process will lead to the improved percentage of Mn3+ in the sample, and the MnO2–Cu exhibits the highest proportion of Mn3+ among the three samples. The nominal valence states of Mn in the three samples were calculated from the energy level splitting of Mn 3s (Fig. 2b),25 indicating the lowest valence states of Mn in the sample MnO2–Cu. The core level spectra of O 1s in Fig. 2c were fitted with three peak positions: Mn–O–Mn (529.9 eV), Mn–OH (531.4 eV) and H–O–H (532.4 eV).26 The Mn–O–Mn was consistent with the MnO2-dominated nature of the three samples, the Mn–O–H comes from the hydrated nature of MnO2 obtained in the aqueous system, and the signal of H–O–H corresponds to interlayer-located water molecules. It is worth noting that the MnO2–Cu possesses the lowest percentage of Mn–O–H, which may be attributed to the electron cloud interaction between the Mn and Cu in the interlayer regime. Considering the structure observations and chemical property analysis, it is evident that Cu2+ ions intercalated MnO2 were obtained successfully in the experiments.
 | ||
| Fig. 2 The valence and chemical bond information of the obtained MnO2, MnO2–Na and MnO2–Cu. (a) XPS core-level spectra of Mn 2p; (b) XPS core-level spectra of Mn 3s; (c) XPS core-level spectra of O 1s. | ||
The electrochemical properties of the obtained MnO2, MnO2–Na and MnO2–Cu electrodes were evaluated as the cathode in the assembled AZIBs (in the form of CR2032 coin-cell) with an aqueous mixture of 2.0 M ZnSO4 and 0.1 M MnSO4, and the results were shown in Fig. 3 and S4.† Fig. 3a shows the CV curves of the different samples at a scan rate of 0.1 mV s−1. Two oxidation peaks located at 1.55 V and 1.6 V can be observed, which correspond to the typical two-step oxidation reaction of MnO2-based electrodes. And two reduction peaks can be identified at 1.38 V and 1.27 V in the discharging part of the CV curves, which should be considered as the two-step reduction reaction of MnO2-based electrodes. These two pairs of oxidation and reduction peaks were mainly related to the H+ and Zn2+ insertion/extraction behaviors during the electrochemical charge–discharge process, which was also reported by many other works. Note that the typical oxidation peak (Peak-2) displays the shifting toward lower potential, but the reduction peak (Peak-3) shifts to higher potential with the sample sequence of MnO2, MnO2–Na, and MnO2–Cu, indicating the lower overpotential with the ion intercalation in the host material MnO2. It is fully evident that the MnO2–Cu earns improved electrochemical reversibility. The MnO2–Cu exhibits the most pronounced capacity performance since it has the biggest CV area among the curves. Quantitative studies based on the b-values were performed, where the b-values can be derived from the oxidation/reduction peak in the CV curves recorded at various scan rates according to the equation i = avb (where i is the peak current, and v is the scan rate).27 In principle, the b-value can be used to reflect the electrochemical kinetics during electrochemical reactions, which varies from 0.5 to 1.0. The value of “0.5” reveals that the redox reaction is limited by the diffusion-controlled kinetics, while the redox reaction is dominated by the capacitive-controlled behavior for b = 1.28 By simple mathematical processing (i.e., linearly fitting operations), the b-values of MnO2–Cu are derived to be 0.62, 0.71 and 0.68 for the Peak-1, Peak-2 and Peak-3, respectively (Fig. 3b). The b-values of the MnO2–Cu are higher than those of MnO2 and MnO2–Na, indicating excellent electrochemical kinetics of MnO2–Cu among the three samples. Moreover, the capacity contribution of the different electrodes was deconvoluted into diffusion-controlled and capacitive-controlled capacity by Dunn's method,28,29 as shown in Fig. 3c–e. It can be found that the MnO2–Cu possesses the highest capacitive-controlled capacity portion, indicating the fastest electrochemical kinetics. Here the enhanced electrochemical kinetics of MnO2–Cu electrode should be considered as two aspects. One is that interlayer intercalated Cu2+ benefits the electrochemical kinetics of electrolyte ions in the interlayer regimes. And the other is that the defective MnO2 (with the reduced nominal valence of MnO2–Cu) shows enhanced electrochemical activity, especially for the surface or near-surface regimes of MnO2. Both the b-value and capacity contribution studies fully demonstrate that the Cu2+ intercalation of MnO2 (i.e., MnO2–Cu) owns the enhanced fast ion transportation and charge transfer behavior in the MnO2–Cu electrode.
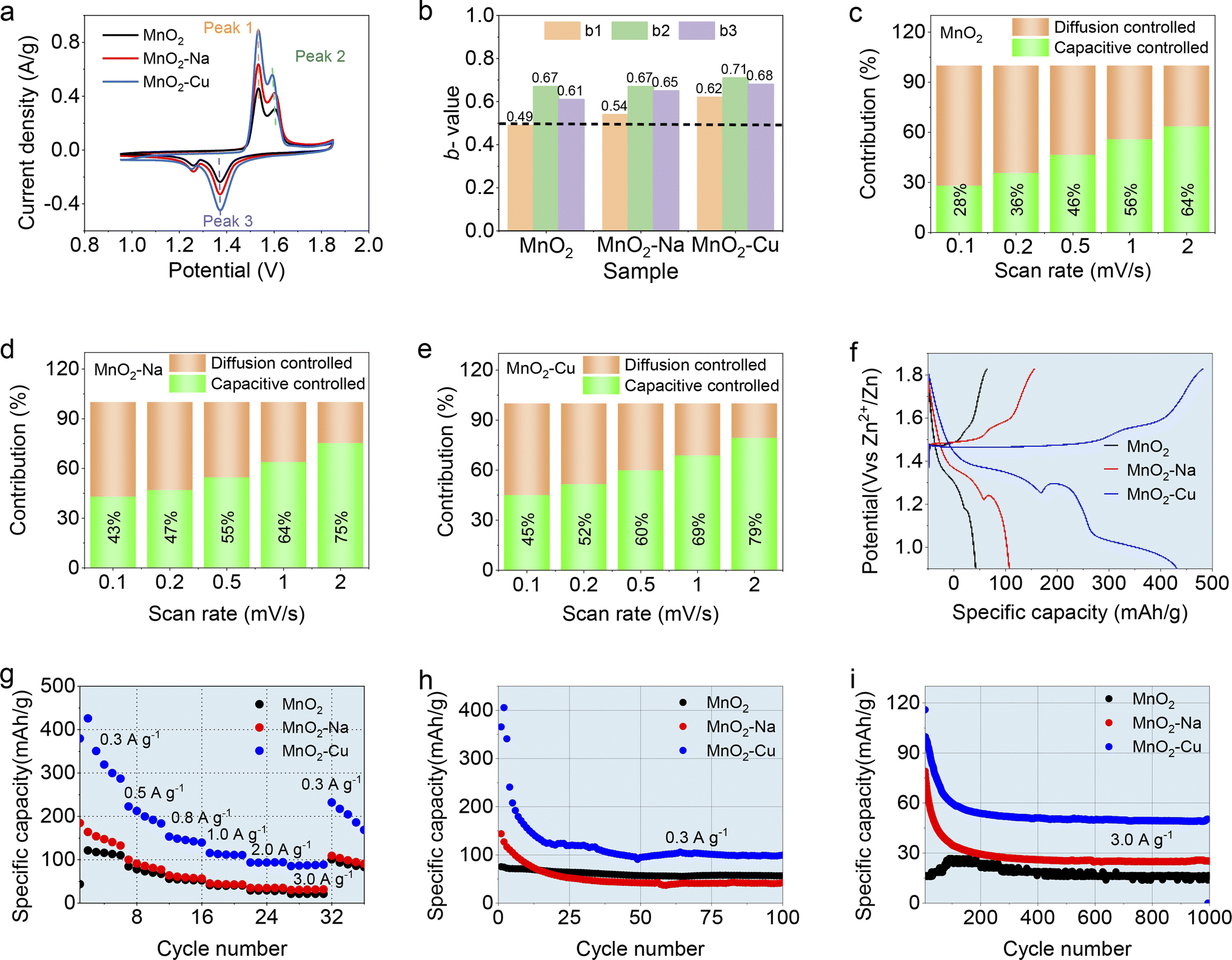 | ||
| Fig. 3 Electrochemical performance of configured AZIBs with the obtained MnO2, MnO2–Na and MnO2–Cu electrodes. (a) CV curves recorded at the scan rate of 0.1 mV s−1; (b) b-value derived from the CV curves at different scan rates; (c)–(e) deconvoluted capacity contribution of the diffusion-controlled and capacitive-controlled process; (f) representative GCD plots collected at 0.3 A g−1; (g) specific capacity at different current densities ranging from 0.3 A g−1 to 3 A g−1; (h) and (i) cycling performance at a current density of 0.3 A g−1 and 3 A g−1. | ||
Fig. 3f shows the GCD plots collected at the current density of 0.3 A g−1. Apart from the two common discharge platforms for all the samples, the MnO2–Cu reveals an extra discharge platform at the potential of 0.95 V, which should be considered as the redox reaction of Cu2+. And the common two discharge platforms of 1.34 V and 1.2 V for all three samples are considered as the intercalation of H+ and Zn2+, respectively, which is consistent with the previous reports. Such an extra redox reaction in the GCD plot was responsible for the capacity improvement of MnO2–Cu. Fig. S5† shows the ex situ XPS analysis of MnO2–Cu at different charge–discharge states. It can be seen that the Cu2+ satellite peaks change from strong to weak and then become strong again with the charging and discharging process, confirming the valence change of the intercalated Cu2+ in host material MnO2. Fig. 3g displays the specific capacity of the three samples recorded at different current densities. It can be observed that the MnO2–Cu delivers the highest capacity of 429.4 mA h g−1 (0.1 A g−1), which is much superior to that of pristine MnO2 (146.8 mA h g−1) and MnO2–Na (198.5 mA h g−1). And the rate capability of the samples can be obtained from the specific capacity at different current densities. The MnO2–Cu reveals the highest capacity retention among the three samples with the current densities increasing from 0.1 A g−1 to 3 A g−1. Further cycling stability studies were performed at the current densities of 0.3 A g−1 and 3 A g−1, as shown in Fig. 3h and i. The MnO2–Cu at the initial cycling stage exhibits the highest specific capacity up to about 400 mA h g−1, and it decreases till to a stable performance as the cycling process proceeds, which should be attributed to the dissolution of Mn2+ due to the Jahn–Teller effect. Such a capacity trend is consistent with the results of the CV curves. Note that the Jahn–Teller effect (i.e., Jahn–Teller distortion), derived from high spin states of Mn3+ (t2g3–eg1), will cause an abnormal change of O–Mn–O bond length in the [MnO6] octahedra, and a large lattice strain and anisotropic volume will be induced accordingly when the MnO2-based electrodes used during the electrochemical charge–discharge processes. Eventually, the MnO2 electrodes reveal obvious structural degradation with the cycling operations. In principle, the Jahn–Teller effect occurs in all the MnO2 electrodes because of the redox reaction of Mn4+/Mn3+ during the charge–discharge process, which shows a significant relation to the redox level of MnO2 materials. Since the deficient capacity performance of the unmodified MnO2 electrode during cycling tests, the Jahn–Teller effect shows a less significant impact on the capacity decay of the MnO2 electrode compared to that of MnO2–Cu and MnO2–Na electrodes. Even though a substantial capacity decline occurs in the MnO2–Cu, it still shows the best capacity performance among the three samples. Till the cycling operations to 1000-cycle, the MnO2–Cu electrode retains the capacity of 50 mA h g−1 and MnO2–Na with 30 mA h g−1, corresponding to the capacity retention of 50% and 30% for the MnO2–Na and MnO2–Cu electrodes, respectively. Note that the MnO2–Cu electrode shows a more rapid capacity decay in the initial cycles, especially at the low charge–discharge current density, as shown in Fig. 3h. The underlying reason should be considered as two aspects. One is that the fact of ions intercalation contributes to the capacity fading due to the robust Jahn–Teller effect, especially at the low charge–discharge current density, and the same trend can be found in the MnO2–Cu and MnO2–Na electrodes (Fig. 3h and i). The other aspect should be attributed to the more significant content of Mn3+ in the MnO2–Cu, which may induce the more significant disproportionation reaction (Mn3+ → Mn4+ + Mn2+) with more soluble Mn2+ generated and then robust MnO2 collapsing.
To get insight into the electrochemical kinetics of the obtained MnO2, MnO2–Na and MnO2–Cu electrodes, the EIS spectrum was collected at different cycling stages (Fig. 4a–c). It can be found that the charge transfer resistance (Rct) of the three samples increases exceptionally rapidly during the initial cycling stage, which is due to the electrochemical activation effect. As the cycling process proceeds, the charge transfer resistance (Rct) tends to be stabilized with similar values among the three samples. Note that the Warburg impedance shows significant differences for the three samples, and the Warburg impedance of MnO2–Cu is significantly lower compared with that of MnO2 and MnO2–Na within the initial 100-cycle stage, indicating that the blocked ion diffusion inside the sample. Fig. 4d shows the GITT curve during the discharge process of the different electrodes. It can be observed that a discharge platform was achieved at a relatively stable voltage of 1.38 V, which should be considered as the regular insertion of Zn2+ and H+ at this stage. The GITT-derived diffusivity coefficient reveals two separation stages, the high diffusion coefficient stage of H+ and the relative-low diffusion coefficient stage of Zn2+, agreeing well with the previous reports. Accordingly, a stable insertion platform for Zn2+ with a diffusion coefficient of 2.23 × 10−10 cm2 s−1, 5.68 × 10−10 cm2 s−1, and 2.56 × 10−9 cm2 s−1 can be calculated for the MnO2, MnO2–Na and MnO2–Cu electrodes, respectively. The highest diffusion coefficient of MnO2–Cu reveals superior electrochemical kinetics, consistent with the electrochemical performance. More impressively, such facilitated electrochemical kinetics of MnO2–Cu enable outstanding electrochemical performance at the condition of mass loading of MnO2. Fig. S6† displays the electrochemical performance of the MnO2–Cu electrodes with a mass loading of 9.6 mg cm−2, exhibiting the highest capacity of 3.45 mA h cm−2 with the calculated specific mass capacity of 359.3 mA h g−1. About 50% capacity is retained after the charge–discharge density increases ten times (from 2 mA cm−2 to 20 mA cm−2).
 | ||
| Fig. 4 Electrochemical kinetics configured AZIBs with the obtained MnO2, MnO2–Na and MnO2–Cu electrodes. (a) MnO2; (b) MnO2–Na; (c) MnO2–Cu; (d) GITT profiles and the derived corresponding diffusivity coefficient. | ||
4. Conclusion
In summary, we have proposed a transition-metal ions intercalation chemistry strategy to enhance electrochemical performance with the prototype of copper ions. Due to the redox nature of transition-metal ions, introducing Cu2+ ions on the host MnO2 (i.e., MnO2–Cu) enhances the capacity performance of the MnO2-based electrode because of the extra capacity contribution of the redox nature of Cu2+. Meanwhile, the Cu2+ ion intercalation and the accompanying structural water molecules in the interlayered regime play a role as the pillars among [MnO6] polymorph blocks, which promotes the Zn2+ ion kinetics and stabilizes the MnO2 structure during the electrochemical charge–discharge process. The resultant AZIBs configured with MnO2–Cu cathode deliver the highest capacity of 429.4 mA h g−1 (0.1 A g−1), which is much superior to that of pristine MnO2 (146.8 mA h g−1) and non-transition-metal ions intercalated MnO2 (i.e., MnO2–Na, 198.5 mA h g−1). The success achieved in this work suggests the transition-metal ions intercalation strategy may aid in the future development of advanced cathodes for high-performance AZIBs.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the Innovation Foundation Program of Gansu Province Colleges and Universities (No. 2022B-219), Doctoral Scientific Research Foundation of Lanzhou City University (No. LZCU-BS2020-02).References
- A. A. Kebede, T. Kalogiannis, J. Van Mierlo and M. Berecibar, Renewable Sustainable Energy Rev., 2022, 159, 112213 CrossRef CAS.
- O. S. Vasilkov, V. S. Dobysh and IEEE, in IEEE Conference of Russian Young Researchers in Electrical and Electronic Engineering (EIConRus), Saint Petersburg Electrotechnical University, Russia, 2019, pp. 728–730 Search PubMed.
- L. Wen, Y. Shi, J. Chen, B. Yan and F. Li, Chin. Phys. B, 2016, 25, 018207 CrossRef.
- W. Du, E. H. Ang, Y. Yang, Y. Zhang, M. Ye and C. C. Li, Energy Environ. Sci., 2020, 13, 3330–3360 RSC.
- S. Huang, J. Zhu, J. Tian and Z. Niu, Chem.–Eur. J., 2019, 25, 14480–14494 CrossRef CAS PubMed.
- D. Selvakumaran, A. Pan, S. Liang and G. Cao, J. Mater. Chem. A, 2019, 7, 18209–18236 RSC.
- I. R. Tay, J. Xue and W. S. V. Lee, Adv. Sci., 2023, 10, 2303211 CrossRef CAS PubMed.
- P. He, Y. Quan, X. Xu, M. Yan, W. Yang, Q. An, L. He and L. Mai, Small, 2017, 13, 1702551 CrossRef PubMed.
- L. Wang and J. Zheng, Mater. Today Adv., 2020, 7, 100078 CrossRef.
- S. Zuo, X. Xu, S. Ji, Z. Wang, Z. Liu and J. Liu, Chem.–Eur. J., 2021, 27, 830–860 CrossRef CAS PubMed.
- T. Xiong, Z. G. Yu, H. Wu, Y. Du, Q. Xie, J. Chen, Y.-W. Zhang, S. J. Pennycook, W. S. V. Lee and J. Xue, Adv. Energy Mater., 2019, 9, 1803815 CrossRef.
- G. G. Yadav, D. Turney, J. Huang, X. Wei and S. Banerjee, ACS Energy Lett., 2019, 4, 2144–2146 CrossRef CAS.
- N. Zhang, Y.-R. Ji, J.-C. Wang, P.-F. Wang, Y.-R. Zhu and T.-F. Yi, J. Energy Chem., 2023, 82, 423–463 CrossRef CAS.
- X. S. Cui, Y. X. Zhang, J. L. Zhang, E. Xie and J. C. Fu, Adv. Mater. Technol., 2023, 8, 2300321 CrossRef CAS.
- J. Heo, S. Chong, S. Kim, R. Kim, K. Shin, J. Kim and H.-T. Kim, Batteries Supercaps, 2021, 4, 1881–1888 CrossRef CAS.
- P. Hu, T. Zhu, X. Wang, X. Wei, M. Yan, J. Li, W. Luo, W. Yang, W. Zhang, L. Zhou, Z. Zhou and L. Mai, Nano Lett., 2018, 18, 1758–1763 CrossRef CAS PubMed.
- F. Jing, Y. Liu, Y. Shang, C. Lv, L. Xu, J. Pei, J. Liu, G. Chen and C. Yan, Energy Storage Mater., 2022, 49, 164–171 CrossRef.
- K. Sada, B. Senthilkumar and P. Barpanda, J. Mater. Chem. A, 2019, 7, 23981–23988 RSC.
- Q. Xie, G. Cheng, T. Xue, L. Huang, S. Chen, Y. Sun, M. Sun, H. Wang and L. Yu, Mater. Today Energy, 2022, 24, 100934 CrossRef CAS.
- P. Xu, H. Yi, G. Shi, Z. Xiong, Y. Hu, R. Wang, H. Zhang and B. Wang, Dalton Trans., 2022, 51, 4695–4703 RSC.
- L. Yan, B. Liu, J. Hao, Y. Han, C. Zhu, F. Liu, X. Zou, Y. Zhou and B. Xiang, J. Energy Chem., 2023, 82, 88–102 CrossRef CAS.
- J. Zhang, W. Li, J. Wang, X. Pu, G. Zhang, S. Wang, N. Wang and X. Li, Angew. Chem., Int. Ed., 2023, 62, e202215654 CrossRef CAS PubMed.
- G. Liu, H. Huang, R. Bi, X. Xiao, T. Ma and L. Zhang, J. Mater. Chem. A, 2019, 7, 20806–20812 RSC.
- H. W. Nesbitt and D. Banerjee, Am. Mineral., 1998, 83, 305–315 CrossRef CAS.
- M. Fujiwara, T. Matsushita and S. Ikeda, J. Electron Spectrosc., 1995, 74, 201–206 CrossRef CAS.
- M. Han, J. Huang, S. Liang, L. Shan, X. Xie, Z. Yi, Y. Wang, S. Guo and J. Zhou, iScience, 2020, 23, 100797 CrossRef CAS PubMed.
- P. He, Y. Quan, X. Xu, M. Yan, W. Yang, Q. An, L. He and L. Mai, Small, 2017, 13, 1702551 CrossRef PubMed.
- S. Khamsanga, R. Pornprasertsuk, T. Yonezawa, A. A. Mohamad and S. Kheawhom, Sci. Rep., 2019, 9, 8441 CrossRef PubMed.
- T. Brousse, D. Bélanger and J. W. Long, J. Electrochem. Soc., 2015, 162, A5185 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data and related descriptions, including XPS spectra, elemental mapping, TEM images and CV curves. See DOI: https://doi.org/10.1039/d4ra01815j |
| This journal is © The Royal Society of Chemistry 2024 |