
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigning novel anti-plasmodial quinoline–furanone hybrids: computational insights, synthesis, and biological evaluation targeting Plasmodium falciparum lactate dehydrogenase†
Deepika Choudhary a,
Poonam Ranib,
Naresh Kumar Rangrac,
Girish Kumar Guptad,
Sukhbir Lal Khokra*e,
Richie R. Bhandare*fg and
Afzal B. Shaik*h
a,
Poonam Ranib,
Naresh Kumar Rangrac,
Girish Kumar Guptad,
Sukhbir Lal Khokra*e,
Richie R. Bhandare*fg and
Afzal B. Shaik*h
aDepartment of Pharmaceutical Sciences and Natural Products, Central University of Punjab, Bathinda-151401, Punjab, India. E-mail: ms_deepika86@yahoo.com
bDepartment of Pharmaceutical Sciences, Guru Jambheshwar University of Science and Technology, Hisar-125001, Haryana, India
cChitkara School of Pharmacy, Chitkara University, Baddi, Himachal Pradesh, India. E-mail: nareshrangra@gmail.com
dDepartment of Pharmaceutical Chemistry, Sri Sai College of Pharmacy, Badhani, Pathankot-145001, Punjab, India
eInstitute of Pharmaceutical Sciences, Kurukshetra University, Kurukshetra-136119, Haryana, India. E-mail: slkhokra@kuk.ac.in
fDepartment of Pharmaceutical Sciences, College of Pharmacy and Health Sciences, Ajman University, P. O. Box 346, Ajman, United Arab Emirates. E-mail: r.bhandareh@ajman.ac.ae
gCentre of Medical and Bio-allied Health Sciences Research, Ajman University, P. O. Box 346, Ajman, United Arab Emirates
hCenter for Global Health Research, Saveetha Medical College, Saveetha Institute of Medical and Technical Sciences, India. E-mail: bashafoye@gmail.com
First published on 12th June 2024
Abstract
To combat resistance against current antimalarials, modifying key pharmacophores and exploring novel parasite-specific drug targets remained one of the key drug design strategies. The resistance to quinoline-based antimalarials arises often due to the efflux of the drug. Hence, the development of newer agents containing bulkier pharmacophores will enable medicinal chemists to counteract drug resistance. In view of this, herein we designed bulkier quinoline–furanone hybrids. Initially, virtual drug-likeness and ADMET screening were conducted to optimize physicochemical properties followed by docking of the hybrids against the Plasmodium falciparum lactate dehydrogenase (PfLDH) enzyme. The most potent hybrids that emerged from the computational screening were synthesized and screened for their bioactivity against the resistant strain of Plasmodium through Schizont Maturation Inhibition assays. Among the compounds tested, 5g and 6e demonstrated the best activity, with IC50 values similar to chloroquine (CQ), and 5g exhibited superior LDH inhibition compared to CQ. Compounds 5f, 7a, and 7f showed IC50 values comparable to CQ and moderate LDH inhibition. Structure–activity relationship (SAR) analysis revealed that halogen substitutions, particularly Br and Cl, enhanced antimalarial activity, while strong electron-withdrawing (–NO2) or -donating (–OH) groups led to diminished activity. Additionally, bulkier aromatic substitutions were favoured for antimalarial activity and LDH inhibition. The investigation successfully found potent anti-plasmodial quinoline–furanone hybrids, demonstrating promising prospects for combating malaria.
1. Introduction
Malaria is mainly a disease of parasitic infection which is considered to affect almost half of the world's population. As per the WHO, 247 million malaria cases and 0.619 million deaths were reported in the World Malaria Report 2022.1 To combat this situation, the World Health Organization (WHO) recommended some preventive measures like vaccination for children and vector control.2 Malaria has become one of the deadliest infections when caused by P. falciparum particularly in the case of children under 5 years. To reduce the risk of death in children living in malaria-endemic areas, the WHO recommended the use of the malaria vaccine RTS, S/AS01. Other than that the WHO also emphasizes the use of preventive chemotherapies and vector control. However, despite the various preventive measures the increasing resistance in plasmodium is worldwide, which in turn is a matter of concern for the WHO to meet the target of 2030 proposed for the malaria strategy. Furthermore, the current antimalarial drugs are associated with reduced effectiveness and resistance. Thus among the different strategies, the search for new effective medicine as well as new drug targets should be given priority to combat malaria.3Quinoline-based antimalarial drugs mainly chloroquine (CQ) have been used to treat infection and even complicated cases of malaria caused by Plasmodium falciparum. However as mentioned above, the major problem associated with CQ is resistance, making the drug ineffective.4 One of the main reasons for the resistance and reduced effectiveness of the drug CQ is associated with the increased level of drug efflux from the receptor protein in plasmodium, as a result, resistant strains of parasite exhibit faster removal of the drug as compared to the chloroquine-sensitive parasites.5 It has been reported that CQ efflux can be reduced by designing the bulkier molecules, as these were reported to be extruded with difficulty from the receptor protein.6 The researchers have explored the potency of various bulky quinoline hybrids and found them effective even against resistant P. falciparum strains.7 Another strategy that can play an important role in drug discovery and designing of new molecules is, hybridization and the use of computational tools, many hybrid compounds comprising of quinoline–triazole, quinoline–artemisinin, quinoline–chalcone, quinoline–pyrazole, quinoline–ferrocene and bis or tris quinoline (Fig. 1), etc. have also shown promising antimalarial activities in both drug-sensitive and resistant strains.8–11
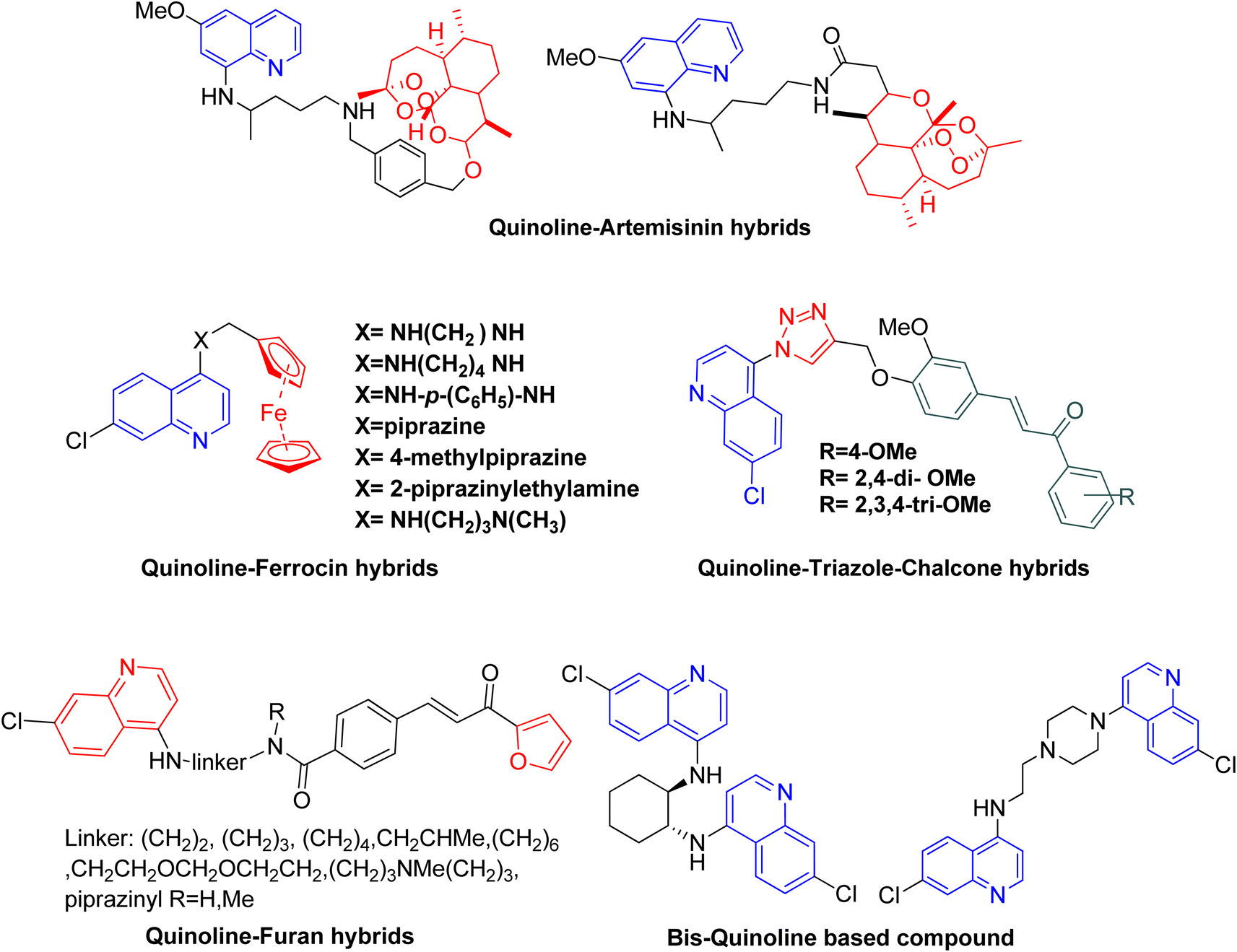 | ||
| Fig. 1 Quinoline based hybrids as antimalarial agent. | ||
The literature shows that even today the quinoline can be considered as the most versatile nucleus for the derivatization to get better potential antimalarial candidates. The concept of molecular hybridization and computational tools can further add benefits to the search for new drug targets and better chemotherapeutic agents for malaria. In consideration of all these points, various quinoline–furanone-based hybrids were designed (Fig. 2) and studied for their antimalarial potential against the resistant strain of P. falciparum. In context to explore the new drug target, Pf lactate dehydrogenase (PfLDH) was selected as the target protein. Because PfLDH is different from human LDH structurally,12 therefore, the new compounds will be more specific for the parasite. The binding affinity of the designed ligands with the receptor has been evaluated by molecular docking using the 3D crystalline structure of the enzyme PfLDH. The results were further confirmed by in vitro LDH assay. Furthermore, as we are designing some bulkier molecules, it is essential to screen them for their physio-chemical or drug-likeliness properties, therefore their ADME-T profile was also accessed virtually using in silico tools.
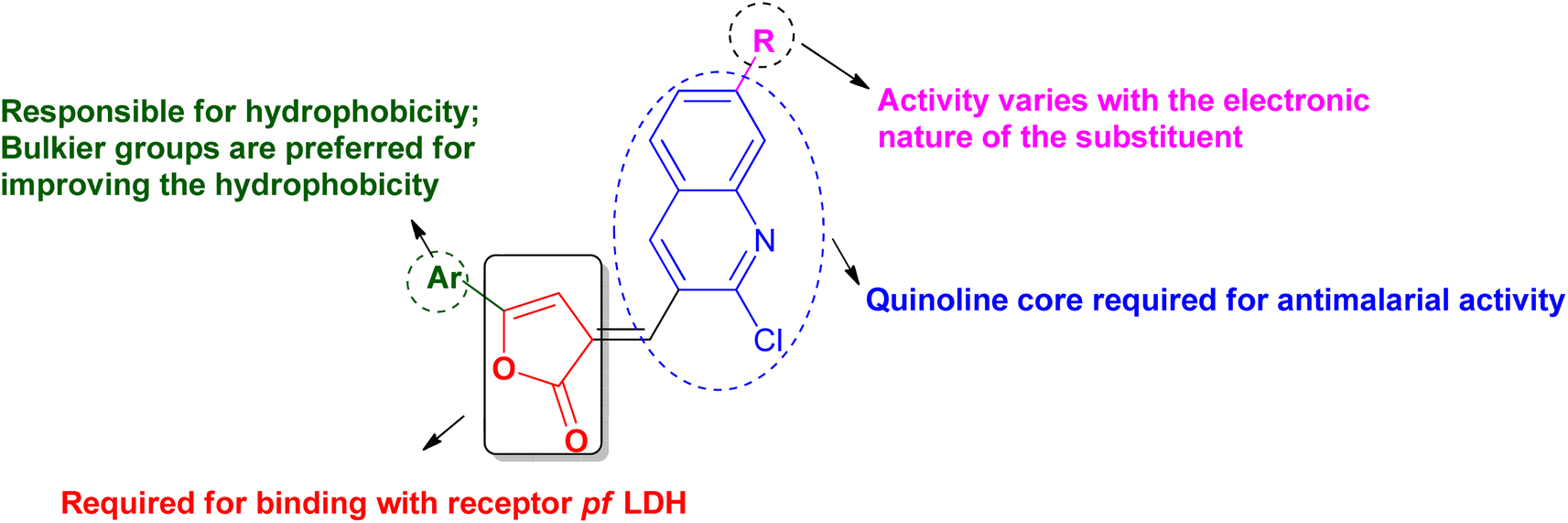 | ||
| Fig. 2 Drug design and SAR of quinoline–furanone hybrids. | ||
2. Results and discussions
2.1 Chemistry and synthesis
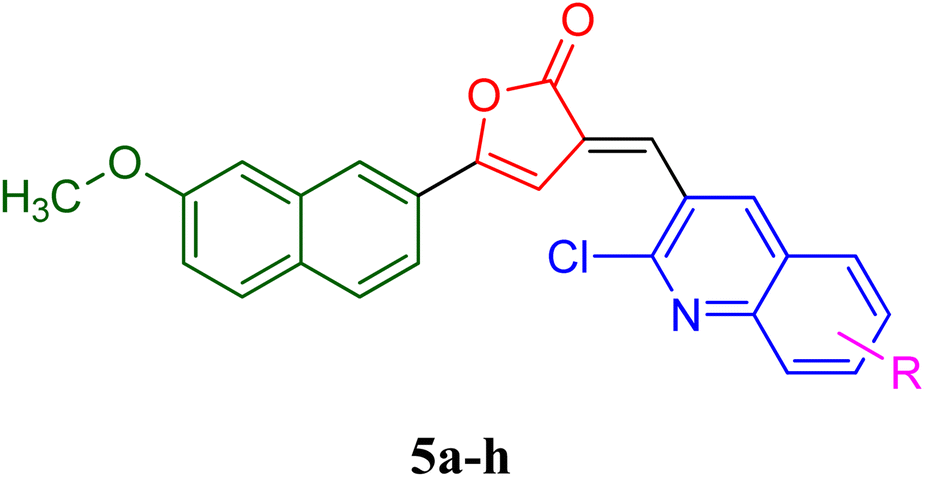
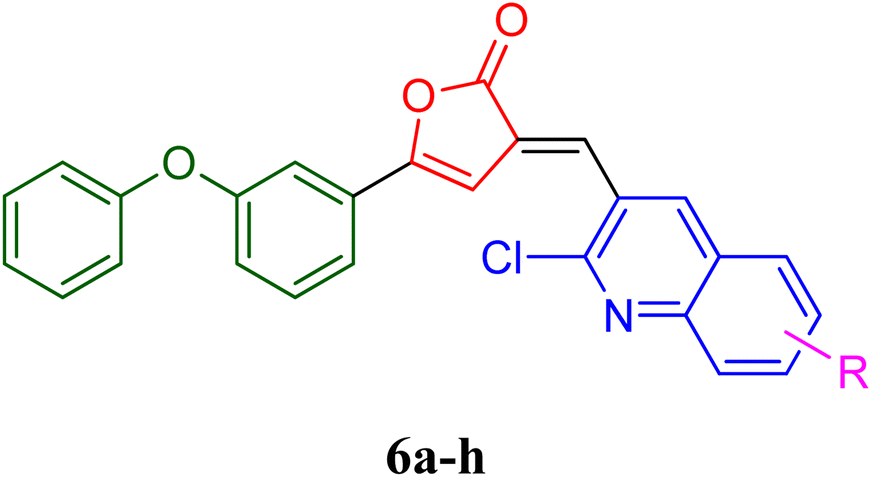
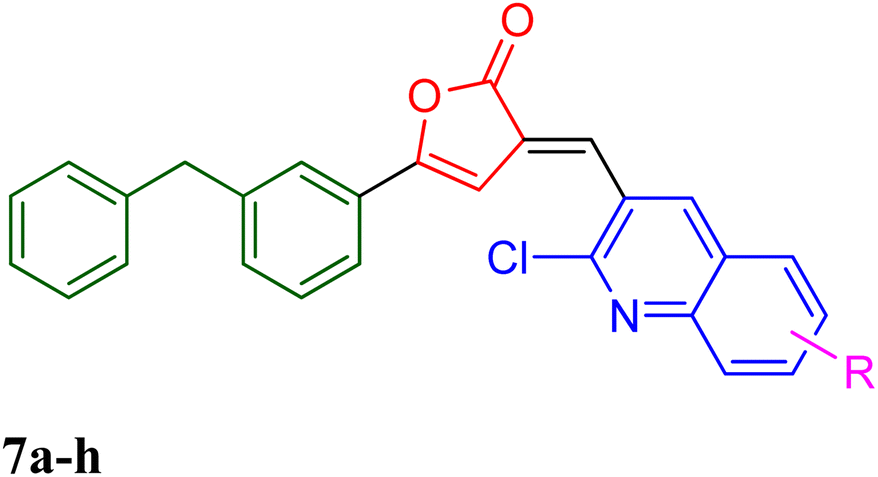
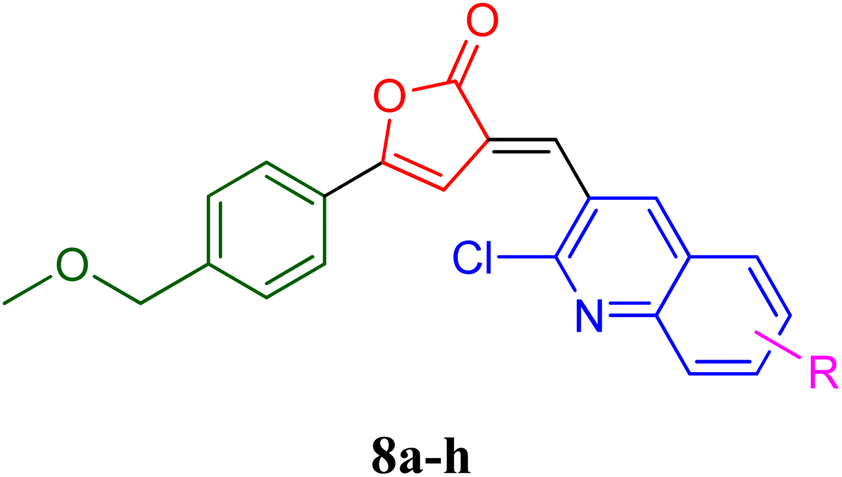
PfLDH can serve as a potential drug target for the drug discovery of new and effective antiplasmodial agents. Furthermore, the amalgamation of two different pharmacophores in one molecule and the use of computational tools to optimize the designed library of compounds can be a useful strategy in the search for new antiplasmodials.12–17 With the use of some important computational tools, strategies, and the reported SAR and available literature, libraries of furanone–quinoline hybrid ligands were generated.18–20 This library was screened for its ADME-Toxicity profile and molecular docking. The 3D crystalline structure of PfLDH was used as a target protein to screen the best hits. The whole virtual screening process with in silico ADME-T prediction and molecular docking, helped us to screen out the 11 best ligands from the library of a total of 24 designed hybrids. The synthetic scheme to prepare the best potent targeted compounds based on quinoline–furanone hybrids has been illustrated in Synthetic Scheme 1. The general structure of designed derivatives and the physical data of the synthesized compounds have been represented in Tables 1 and 2 respectively.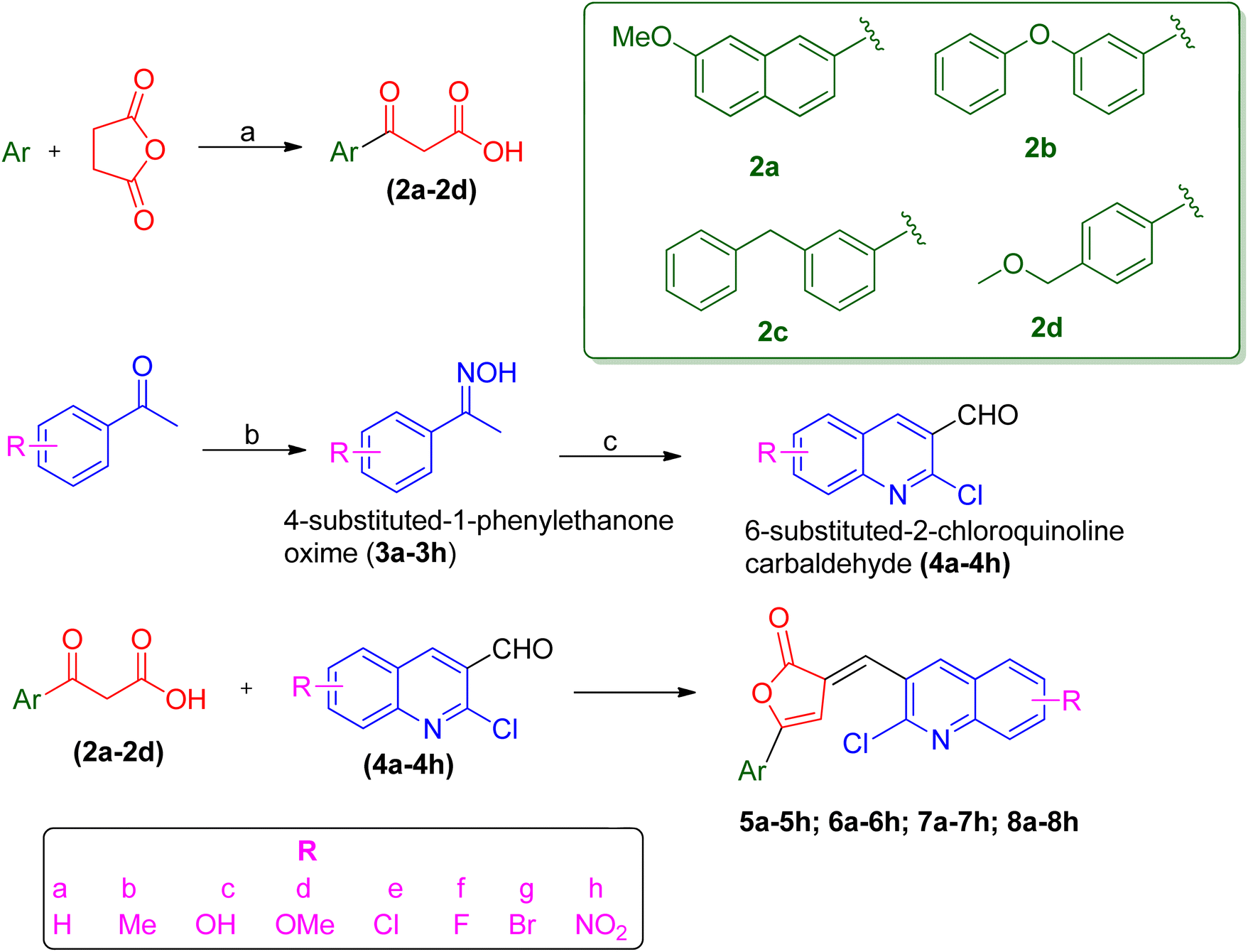 | ||
| Scheme 1 Reagents and conditions: (a) anhydrous AlCl3, reflux, 2–4 h (b) NH2OH·HCl, CH3COONa, ethanol, water, reflux for 1 h at 100 °C (c) DMF, POCl3 stirring for 16 h at 50–60 °C at anhydrous condition (d) acetic anhydride, triethylamine, heat, 5 min, anhydrous condition. | ||
 |
 |
||||||||
 |
 |
||||||||
| a | b | c | d | e | f | g | h | ||
| Where | R= | H | CH3 | OH | OMe | Cl | F | Br | NO2 |
| S. no. | Compound | Rf value | Melting point (°C) | Yield (%) |
|---|---|---|---|---|
| 1 | 5e | 0.88 | 245–246 | 42 |
| 2 | 5f | 0.88 | 263–265 | 37 |
| 3 | 5g | 0.79 | 172–174 | 22 |
| 4 | 6a | 0.81 | 216–218 | 78 |
| 5 | 6c | 0.87 | 248–250 | 85 |
| 6 | 6e | 0.82 | 215–218 | 68 |
| 7 | 6f | 0.83 | 235–238 | 42 |
| 8 | 6h | 0.84 | 286–288 | 84 |
| 9 | 7a | 0.86 | 110–112 | 73 |
| 10 | 7f | 0.76 | 150–152 | 76 |
| 11 | 7h | 0.71 | 184–186 | 28 |
The synthetic methodology was the same as the previously reported procedure,9,10 however, we have utilized some different aryl group-based β-benzoyl propionic acid. The overall synthetic scheme involves three different steps (1) the synthesis of β-benzoyl propionic acid (2) the synthesis of quinoline-3-carbaldehyde derivatives (3) the condensation of quinoline-3-carbaldehyde with β-aroyl propionic acid to yield the target compounds. The Friedel–Crafts acylation was done in the presence of anhydrous aluminum chloride as a catalyst using 4 types of aromatic groups. The different substituted quinoline-3-carbaldehyde were prepared by Vilsmeier–Haack formylation using N,N-dimethylformamide (DMF) and phosphorus oxychloride (POCl3) with a different acetophenone oxime.21 Then the condensation of β-aroyl propionic acid with quinoline-3-carbaldehyde in the presence of acetic anhydride and triethylamine gives us the proposed compounds. The characterization of synthesized compounds is done by spectral techniques such as mass, FT-IR, 1H-NMR, and elemental analysis. The IR spectral data of synthesized titled quinoline–furanone hybrids shows the characteristic bands of lactone at 1762–1687 (lactone, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) along with other bands corresponding to values like 1220–1136 (C–N), 1522–1435 (CC) and 1567–1549 (CN). The peak in 1H NMR spectra at δ value 6.5–7.5 confirm the cyclization to form furan ring. The presence of an alkene proton in place of an aldehydic proton also confirm the formation of the targeted compound.
O) along with other bands corresponding to values like 1220–1136 (C–N), 1522–1435 (CC) and 1567–1549 (CN). The peak in 1H NMR spectra at δ value 6.5–7.5 confirm the cyclization to form furan ring. The presence of an alkene proton in place of an aldehydic proton also confirm the formation of the targeted compound.
2.2 Anti-malarial activity
Evaluation of antiplasmodial activity was done with all the synthesized derivatives by in vitro methods using chloroquine-resistant (K1) strains of P. falciparum procured from the Parasite Bank of the National Institute of Malaria Research (NIMR), New Delhi. The estimation of antiplasmodial activity was also carried out in the laboratory of the same institute. The standardized WHO Schizont Maturation Test (micro-test) and the colorimetric method mainly for parasite Lactate dehydrogenase inhibition (pLDH) were employed in the current studies for the estimation of the antiplasmodial potential of synthesized hybrid compounds. In the SMI assay, a thin smear from each well was prepared according to the reported method22 and as the parameter of antiplasmodial activity assessment was the number of schizonts, the compounds in which a lesser number of mature schizonts appear in comparison with control well, were considered active as they affect the formation of schizont from ring stage. As a consequence, it was reported that the greater the number of mature schizonts, the lesser will be the %inhibition of parasitemia, hence very low activity. The images of the microscopic view of parasitized erythrocytes of culture from standard as well as test compounds are shown below in Fig. 3A–D.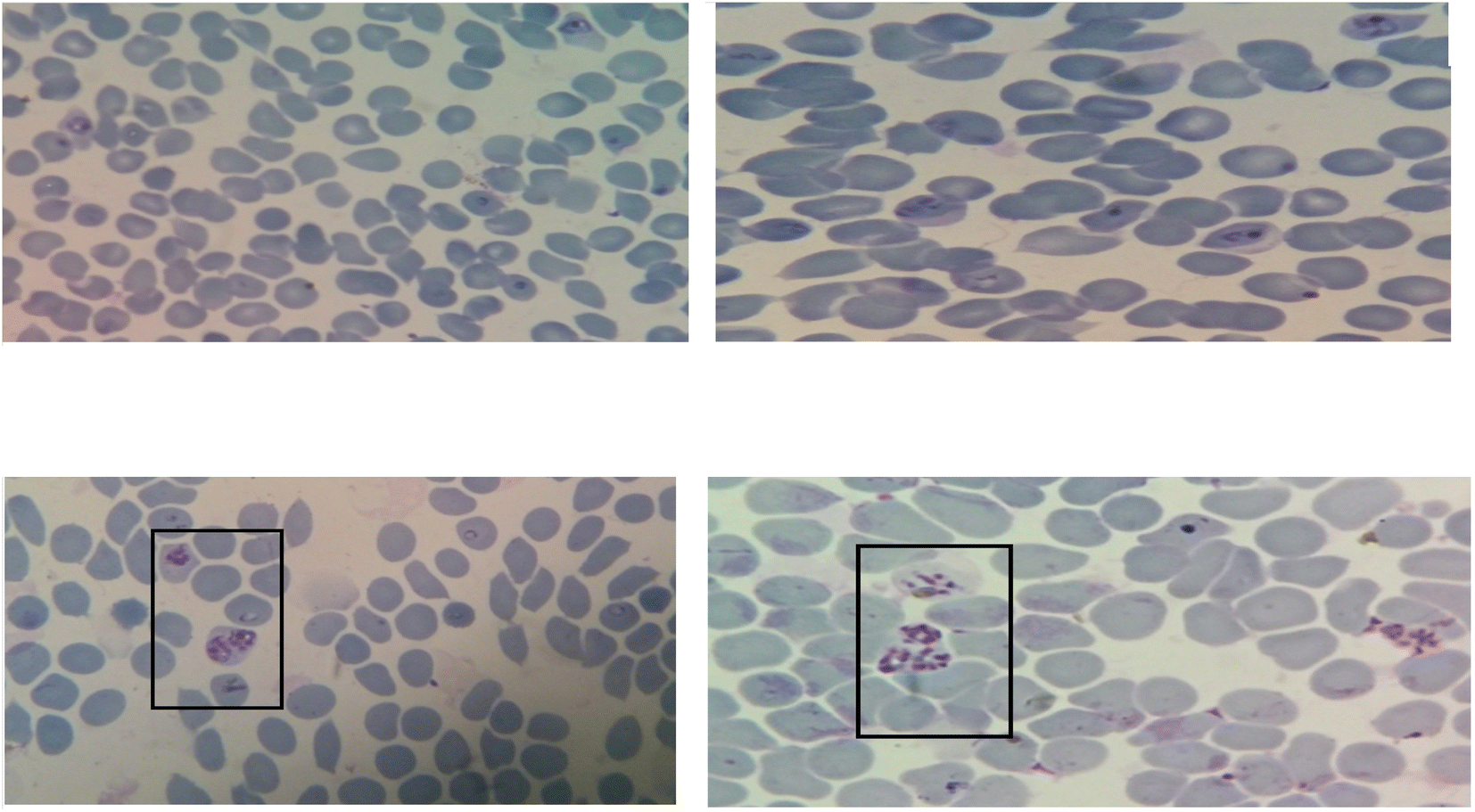 | ||
| Fig. 3 (A) The microscopic view of parasitized erythrocytes from culture having standard drug, reported with no mature schizont. (B) The microscopic view of parasitized erythrocytes from the culture of the potent test compound, reported only with some early schizont, but no mature schizont appear. (C) The microscopic view of parasitized erythrocytes from the culture of the moderately potent compound, reported with a few no. of mature schizonts (shown inside the black rectangle). (D) The microscopic view of parasitized erythrocytes from the culture of less active compound, having comparatively more number of mature dividing schizonts (shown inside the black rectangle). | ||
On the other hand in the pLDH assay, the measurement of activity of pLDH activity was the criteria, which in turn depends on the value of absorbance given by the plate reader.23 During this assay, it was observed that the enzymatic activity detected in infected erythrocytes from the control well was at least twice that of standard CQ. Moreover, the value of absorbance given by the control well was considered as 100% pLDH activity. Therefore based on the above specifications the value of absorbance for the test compounds was analyzed and results revealed that as the drug concentration increased, the corresponding value of absorbance was decreasing. The corresponding IC50 was calculated from the number of schizonts in SMI and the value of absorbance in pLDH. The potency criteria referred in both methods was based on the WHO criteria such as the compounds, those having IC50 value ≥10 exhibited poor antiplasmodial activity, those having IC50 value ≥5 were considered to possess moderate antiplasmodial activity, while the compounds having IC50 value less than 5 were considered the most active compounds among all. The data for the in vitro test is shown in Table 3.
| Compound | Antiplasmodial activity by SMI method | Antiplasmodial activity by PfLDH inhibition method | ||||
|---|---|---|---|---|---|---|
| IC50 | IC90 | IC99 | IC50 | IC90 | IC99 | |
| 5e | 3.251 | 24.143 | 66.153 | 4.283 | 24.431 | 122.192 |
| 5f | 2.825 | 15.125 | 54.156 | 3.014 | 12.474 | 28.163 |
| 5g | 2.083 | 7.794 | 32.114 | 2.314 | 12.162 | 23.118 |
| 6a | 3.125 | 16.425 | 105.134 | 6.567 | 79.142 | 179.784 |
| 6c | 23.792 | 382.468 | 453.024 | 16.710 | 276.112 | 295.828 |
| 6e | 2.168 | 5.952 | 25.112 | 4.738 | 16.129 | 23.061 |
| 6f | 3.960 | 27.479 | 124.499 | 5.379 | 77.479 | 128.428 |
| 6h | 16.837 | 252.719 | 312.148 | 14.294 | 216.952 | 228.113 |
| 7a | 2.473 | 18.129 | 39.115 | 3.202 | 12.836 | 18.184 |
| 7f | 2.569 | 12.129 | 28.591 | 5.393 | 17.425 | 22.402 |
| 7h | 8.216 | 67.126 | 125.362 | 5.539 | 88.586 | 203.628 |
| CQ (standard) | 1.953 | 2.118 | 9.862 | 2.873 | 3.102 | 10.046 |
The in vitro antiplasmodial activity data depicted that the compounds 5g and 6e were exhibiting maximum activity with IC50 value almost similar to the CQ. A total of 6 compounds 5e, 5f, 6a, 6f, 7a, and 7f, showed IC50 values comparable to CQ (<5) and after them, the compound 7h was found comparatively less potent, and the other two compounds 6c and 6h were found to have very poor antiplasmodial potential with IC50 >10 as shown in Table 3. The results of in vitro activity data give some important information regarding SAR of designed derivatives such as derivatives with either H or halogen as R will lead to an increase in antiplasmodial activity but substitution either with the strong electronic withdrawing group like –NO2 or strong electronic donating group like OH, result in almost loss or very less activity. The overall observation from in vitro antiplasmodial analysis concluded that both the quinoline and furanone can be considered important pharmacophores to design newer antiplasmodial agents which may have act as inhibitors of specific targets such as PfLDH.
2.3 Computational studies
In the process of rational drug discovery, the toxicity and bad pharmacokinetics (ADME-T) profiles are the main reason behind the failure of drugs at the later stages of drug development.24 The in silico methods like prediction of ADME-T and study of binding interaction of ligands within the protein receptor by molecular docking studies play very crucial roles in the optimization of designed hits and can reduce the overall cost as well as time of drug discovery.25 The virtual screening and insilico analysis of the designed derivatives using computational tools like QikProp for ADME prediction, TOPKAT for toxicity studies, and Molegro Virtual Docker (MVD) for molecular docking are discussed here.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logPoct) ranged 18–25, predicted water/gas partition coefficient (QPlogPw) ranged 8–17, predicted octanol/water partition coefficient (QPlogPo/w) ranged 5–7, predicted aqueous solubility, logS (QPlogS) ranged −9 to 4, predicted brain/blood partition coefficient (QPlogBB) ranged −1.5 to −0.8, and Lipinski violations were ≤1. Toxicity prediction was done by TOPKAT, in this prediction the designed library was screened for carcinogenicity and mutagenicity filter in rat model, and the results are represented in Table 5. The predictions in TOPKAT were performed through a search program based on similarity along with a check that weather the test structure prediction lies in the optimum prediction space (OPS) or not. The OPS also permits the user to determine if structures are within the model descriptor space, but in some cases, the toxicity can be predicted of those compounds that lie outside OPS. This current analysis revealed that the 11 screened analogs may have good ADME-T properties and can be successful drugs in the future.
logPoct) ranged 18–25, predicted water/gas partition coefficient (QPlogPw) ranged 8–17, predicted octanol/water partition coefficient (QPlogPo/w) ranged 5–7, predicted aqueous solubility, logS (QPlogS) ranged −9 to 4, predicted brain/blood partition coefficient (QPlogBB) ranged −1.5 to −0.8, and Lipinski violations were ≤1. Toxicity prediction was done by TOPKAT, in this prediction the designed library was screened for carcinogenicity and mutagenicity filter in rat model, and the results are represented in Table 5. The predictions in TOPKAT were performed through a search program based on similarity along with a check that weather the test structure prediction lies in the optimum prediction space (OPS) or not. The OPS also permits the user to determine if structures are within the model descriptor space, but in some cases, the toxicity can be predicted of those compounds that lie outside OPS. This current analysis revealed that the 11 screened analogs may have good ADME-T properties and can be successful drugs in the future.
| Compound | MW | SASA | HBD | HBA | QplogPo/w |
QPlogS |
QPlogKhsa |
%HOA | PSA | AffyMDCK | Affy Caco-2 | QplogBB |
QPlogPoct |
QPlogPW |
Rule of 5 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a The descriptor was calculated from QIKProp 3.6. where each descriptor represent one physicochemical property for “drug likeliness” such as MW – molecular weight; SASA – solvent assessable surface area; HBD – no. of H bond donor; HBA – no. of H bond acceptor; QPlogPo/w – predicted octanol/water partition coefficient; QPlogS – predicted aqueous solubility; QPlogKhsa – predicted human serum albumin binding; %HOA – percentage human oral absorption; PSA – total polar surface area; AffyMDCK – predicted apparent MDCK cell permeability in nm s−1, Affy Caco-2 – predicted apparent Caco-2 cell permeability in nm s−1, QplogBB – predicted blood–brain partition coefficient, QPlogPoct – predicted octanol/gas partition coefficient, QPlogPW – predicted water/gas partition coefficient rule of 5-Lipinski violations. |
|||||||||||||||
| 5e | 448.301 | 738.215 | 0 | 4 | 6.67 | −6.534 | 1.038 | 100 | 52.34 | 2832.427 | 2437.056 | −0.426 | 22.741 | 11.472 | 1 |
| 5f | 431.842 | 759.438 | 0 | 4 | 5.18 | −6.287 | 1.283 | 100 | 52.34 | 2743.324 | 2463.728 | −1.257 | 24.042 | 11.037 | 1 |
| 5g | 492.752 | 749.358 | 0 | 4 | 6.80 | −7.539 | 1.047 | 100 | 52.34 | 2853.320 | 2353.132 | −0.238 | 24.846 | 12.571 | 1 |
| 6a | 425.87 | 725.503 | 0 | 4 | 6.157 | −7.463 | 1.097 | 100 | 56.75 | 2627.307 | 2441.144 | −0.232 | 18.752 | 8.954 | 0 |
| 6c | 439.897 | 757.677 | 0 | 4 | 6.483 | −8.073 | 1.27 | 100 | 56.749 | 2628.822 | 2442.458 | −0.25 | 19.26 | 8.651 | 0 |
| 6e | 441.87 | 737.753 | 1 | 4.75 | 5.498 | −7.382 | 1.014 | 100 | 79.283 | 7242.81 | 7412.54 | −0.894 | 20.937 | 11.05 | 1 |
| 6h | 460.315 | 749.448 | 0 | 4 | 6.662 | −8.229 | 1.225 | 100 | 56.752 | 6484.175 | 2441.959 | −0.07 | 19.446 | 8.713 | 1 |
| 6f | 443.861 | 734.438 | 0 | 4 | 6.397 | −7.838 | 1.143 | 100 | 56.753 | 4715.541 | 2441.398 | −0.122 | 19.006 | 8.733 | 1 |
| 7a | 470.868 | 763.748 | 0 | 5 | 5.404 | −7.502 | 0.998 | 89.77 | 101.629 | 2651.75 | 2932.023 | −1.391 | 21.195 | 10.07 | 0 |
| 7f | 423.898 | 736.238 | 0 | 3.5 | 8.292 | −7.929 | 1.32 | 100 | 48.904 | 2627.1 | 2440.781 | −0.231 | 18.88 | 8.292 | 1 |
| 7h | 437.92 | 767.937 | 0 | 3.5 | 6.891 | −8.53 | 1.493 | 100 | 48.903 | 2628.787 | 2442.182 | −0.248 | 19.383 | 7.984 | 1 |
| Compound | ops1 | ops2 | Fragment | Carcinogenicity | Mutagenicity |
|---|---|---|---|---|---|
| a Toxicity descriptors calculated from TOPKAT. Ops1 and Ops2 – optimum prediction state. | |||||
| 5e | TRUE | TRUE | TRUE | 0.316 | X |
| 5f | TRUE | TRUE | TRUE | 0.368 | X |
| 5g | TRUE | TRUE | TRUE | 0.374 | X |
| 6a | TRUE | TRUE | TRUE | 0.011 | X |
| 6c | TRUE | TRUE | TRUE | 0.012 | X |
| 6e | TRUE | TRUE | TRUE | 0.019 | X |
| 6h | TRUE | TRUE | TRUE | 0.023 | X |
| 6f | TRUE | TRUE | TRUE | 0.024 | X |
| 7a | TRUE | TRUE | TRUE | 0.241 | X |
| 7f | TRUE | TRUE | TRUE | 0.006 | X |
| 7h | TRUE | TRUE | TRUE | 0.009 | X |
2.3.2.1 Prediction of the binding site. The 3D crystalline structure of receptor protein PfLDH having with NADH and the substrate Oxamate (co-crystalized ligands of 1LDG) was imported from the Protein Data Bank (https://www.rcsb.org) (Fig. 4) into the workspace of the software and different steps of protein preparation were done which include the deselection of all water molecules having specific distance more than 5 Å, selection of suitable cavity, or binding site prediction in PfLDH receptor. The search algorithm in MVD showed a total of five cavities of different volumes in protein. It was observed that NADH, Oxamate, and CQ were going in Cavity-1, with the largest volume and size, approximately 136.096 Å. Therefore this particular cavity was selected and others were rejected.
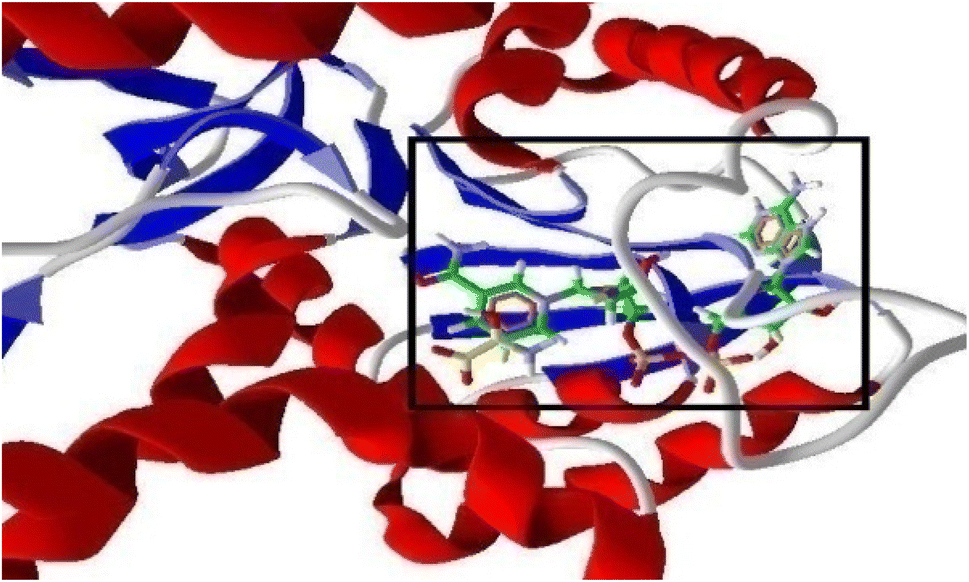 | ||
| Fig. 4 3D structure of the protein (1LDG) showing binding of NADH and Oxamate (highlighted in a window) within the pocket of PfLDH. | ||
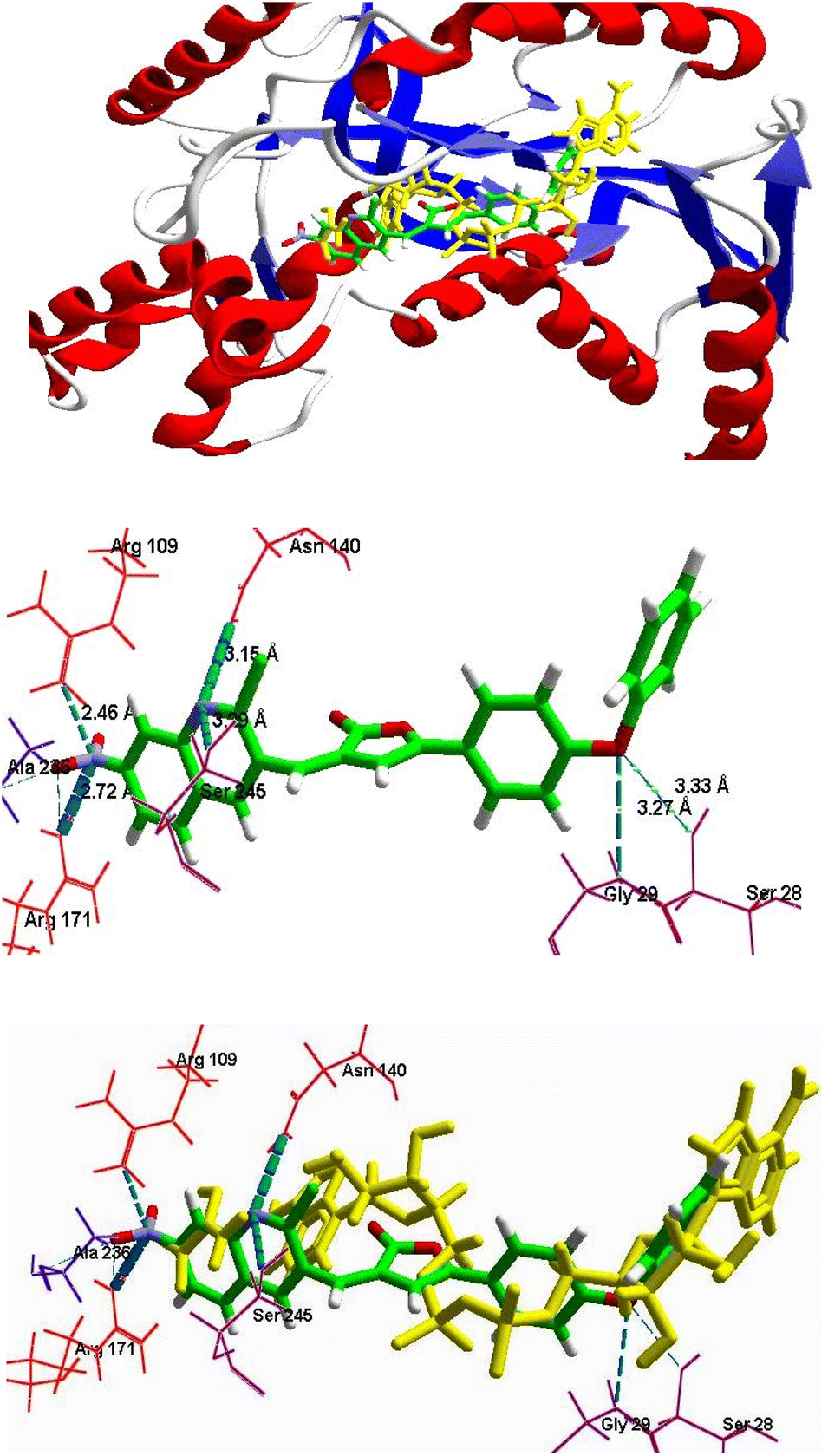
2.3.2.2 Binding mode analysis. Two major binding sites were predicted on the target protein (1LDG). One of which was the NADH binding site and another was the substrate binding domain.26 NADH binding site was identified on the N-terminal end of the enzyme with Gly 29, Ile31, Asp53, Ile54, Thr97, Gly99, Phe100, and Asn140 as key amino acids. The adenine end of the cofactor side of the binding pocket is formed by these amino acid residues and goes deep into the protein towards the nicotinamide end of the cofactor site, the substrate binding domain or the second site was constituted by amino acid residues on the C-terminal end of enzymes such as Arg109, Asn140, Arg171, His195, Val233, Ala236 and Ser245. The amino acid residues formed a binding groove on the back side of the substrate active site lying approximately 10 Ådeep within the protein structure, adjacent to the nicotinamide end of the cofactor site. All the filtered hits were successfully docked to the receptor PfLDH. After the visual binding mode analysis of the standard drug CQ, the bound ligand NADH and other ADME-T filtered hits, it was observed that all the screened hits (quinoline–furanone hybrids) were exhibited binding within the same cavity along with NADH and CQ. Moreover, it was also revealed that the screened designed hybrids not only showed better mol dock scores but having optimum interactions with the specific amino acid residues including Gly 29, Ile31, Asp53, Ile33, Thr97, Gly99, Phe100, Asn140, Arg171, His195, and Ser245. Along with that, their binding conformations were well superimposed with NADH (Fig. 5A). Molecular docking analysis concluded that these hits may exhibit competitive inhibition with of NADH which is considered essential for the enzyme PfLDH.
 | ||
| Fig. 5 (A) Superimposition in the binding mode of 6c (dark pink) with NADH, (shown in yellow color) in the pocket of protein receptor PfLDH (1LDG). (B) The binding mode of compound 6c in the pocket of protein receptor PfLDH (1LDG), having H-bond Interactions (green dotted lines) with the key amino acids. | ||
2.3.2.3 Calculation of binding energy and docking scores. The molecular docking data revealed that all the nontoxic druglike, compounds 5e–g, 6a, 6c, 6e, 6f, 6h, 7a, 7f and 7h exhibited comparative to good binding score with strong H-bond interaction with the receptor PfLDH, as represented in Table 6. The compounds 6c, 6e, 6h, 7a, and 7h showed the best moldock score with maximum binding energy, best binding conformation, and good interactions within the receptor. All ligands have good moldock score which was more than −100 except 6a and 7f in MVD (Table 6). Besides, it was also found that each ligand's binding pose was much closer to NADH and CQ with good superimposition (Fig. 4–6). It was observed that each filtered hit made consistent hydrogen bond interactions with the key amino acids such as Met30, Ile31, Asp53, Gly99, Try85, Arg109, Asn140, Arg171, Ser245, Ala236, and Thr232, which were the same as in case of the binding of reported PfLDH inhibitors. The results elucidated that CQ as well as screened hits may have a similar inhibitory mechanism against enzyme PfLDH as they can act as competitive inhibitors of NADH for binding within the receptor protein.
| S. no. | Ligands | Moldock score | Interacting residues and H-bond interaction with distance (Å) |
|---|---|---|---|
| 1 | NADH | −217.75 | Asn140 (2.65), Asn140 (3.42), Asn140 (3.13), Gly99 (3.20), Thr97 (3.38), Thr97 (2.17), Arg109 (3.09), Arg109 (2.67), Ile31 (3.13), Ser245 (2.75), Pro246 (2.41), Phe100 (2.99), Thr139 (3.25), Arg116 (3.02) |
| 2 | CQ | −153.55 | Ser245 (3.21), Asn140 (2.99) |
| 3 | 5e | −120.36 | Arg109 (2.64), Asn140 (3.36), Ser245 (3.28) |
| 4 | 5f | −114.52 | His195 (3.62), Asn140 (3.47), Arg109 (2.81) |
| 5 | 5g | −106.37 | Ala236 (3.61), Gly29 (3.74) |
| 6 | 6a | −99.23 | Gly29 (3.03), Ser245 (2.73), Asn140 (3.01) |
| 7 | 6c | −122.07 | Arg109 (3.03), Asn140 (2.78), Ser245 (3.13), Met30 (3.34), Gly29 (3.29), Ile31 (3.47), Val233 (2.96), Thr232 (3.55) |
| 8 | 6e | −122.57 | Ser245 (2.87), Asn140 (2.97), Gly29 (3.02) |
| 9 | 6f | −119.09 | Ala236 (3.07), Arg109 (2.84), Asn140 (3.12) |
| 10 | 6h | −132.77 | Arg109 (2.46), Arg171 (2.72), Arg171 (2.38), Ala236 (3.45), Gly29 (3.27), Ser245 (3.29), Asn140 (3.12), Ser28 (3.33) |
| 11 | 7a | −148.01 | Asn140 (2.97), Ser245 (3.33) |
| 12 | 7f | −97.723 | Ser245 (3.38), Asn140 (2.96), Arg109 (3.13) |
| 13 | 7h | −150.98 | Ala236 (3.17), Arg171 (2.58) |
 | ||
| Fig. 6 (A) Binding mode of 6h (green color) with NADH and substrate Oxamate (shown in yellow color) in the pocket of protein receptor PfLDH (1LDG). (B) Binding mode of 6h in the pocket of protein receptor PfLDH, having H-bond interaction with key amino acids. (C) Superimposition in the binding mode of 6h (green) with NADH (shown in yellow color) in the pocket of protein receptor PfLDH (1LDG). | ||
3. Material and methods
The compounds were synthesized using commercially available analytical grade chemicals without purification from E. Merck (Germany) and S. D. Fine Chem. Lmt (India). Melting points (MPs) were taken on slides using Labindia electrical visual melting range apparatus and are uncorrected. Benzoic acid was used as reference for the calibration of melting point apparatus. IR spectra were recorded on a PerkinElmer 1800 FT-IR spectrophotometer. 1H NMR spectra were recorded on a Bruker 300 & 400 MHz instrument using tetramethylsilane as an internal standard. Mass spectra were recorded on 2500 eV (ESI Source) using a water's Q-TOF microinstrument and elemental analysis on the PerkinElmer 2400 instrument. Progress of the chemical reaction and the purity of the synthesized compound were checked on silica gel G-coated thin-layer chromatography plates in solvent systems; petroleum ether:toluene:ethyl acetate (5:4:1, v/v/v). The visualization of spots on TLC was carried out in either in an iodine chamber or in UV cabinet at a long wavelength under a UV lamp.
3.1 Chemical synthesis of virtually screened ligands
The whole synthetic scheme was done in the following steps.3.1.2.1 Synthesis of 3-(7-methoxy-2-naphthoyl) propionic acid (2a). Dry nitrobenzene was added to the mixture of succinic anhydride (0.1 mol) and 2-methoxy-naphthalene (0.1 mol) in a round bottom flask. The mixture was heated with stirring to dissolve the content and then cooled to room temperature. Anhydrous aluminum chloride was added to a well-stirred mixture over 20 minutes, and the stirring was continued for 2–3 hours under anhydrous conditions. The reaction was allowed to stand for 2 days at room temperature while maintaining an anhydrous condition. The completion of the reaction was checked from time to time using TLC. After that excess nitrobenzene was evaporated under vacuum at 110–140 °C. A 10% sodium hydroxide solution was added to dissolve the desired reaction product and filtered to remove undissolved and unwanted material. The above solution was neutralized by the addition of dilute hydrochloric acid. Finally, precipitates of aroylpropionic acid were filtered and washed with cold water, and recrystallized with ethanol. The melting point of the compound was determined.
Note: Nitrobenzene was dried by adding 20 g anhydrous magnesium sulfate in 250 ml nitrobenzene and kept overnight.
3.1.2.2 General procedure for the synthesis of 3-(3-phenoxy-2-phenoyl)propionic acid; 2b, 3-(3-benzyl-2-phenoyl)propionic acid; 2c and 3-(2-(methoxymethyl)-1-phenoyl)propionic acid; 2d. Succinic anhydride (0.1 mol) was dissolved in 50 ml of dried starting material (diphenyl ether for 2b, diphenylmethane for 2c, and methoxy methyl benzene for 2d) under the anhydrous condition in the presence of anhydrous aluminum chloride (0.2 mol). The reaction mixture was refluxed for 2–4 hours. The reaction product was purified by dissolving in sodium hydroxide solution and filtered to remove undissolved and unwanted material. The above solution was neutralized by the addition of dilute hydrochloric acid. Finally, precipitates of aroylpropionic acid were filtered and washed with cold water, and recrystallized with ethanol. The melting point of the compound was determined.
3.1.5.1 (3-(2,6-Dichloroquinolin-3-yl)methylene)-5-(7-methoxynaphthalen-2-yl)furan-2(3H)-one 5e. Yield 42%; m.p. 245–246 °C: brown colour, Rf 0.88, IR(KBr) cm−1: 1767 (C
O), 1560 (CN str), 1203 (C–N str), 1497 (CC str), 1H NMR (300 MHz, CDCl3): 6.81 (s, 1H, βH), 7.28 (s, 1H, olefinic H), 7.45–8.18 (complex m, 11H, arylprotons). 13C NMR (100 MHz, CDCl3) δ/ppm: 167.4 (1C, C-2), 148.4 (1C, C-3), 136.3 (1C, C-5), 125.9 (1C, CH, C-4), 98.4 (1C, CH, ethylene), 131.4 (1C, C′-naphthelene), 127.2 (1C, C′, CH-naphthelene), 139.2 (1C, C′-naphthelene), 129.4 (1C, C′-naphthelene), 124.2 (1C, C′, CH-naphthelene), 119.7 (1C, C′-CH-naphthelene), 105.3 (1C, C′, CH-naphthelene), 158.2 (1C, C′-naphthelene), 118.5 (1C, C′-CH-naphthelene), 126.3 (1C, C′, CH-naphthelene), 55.9 (1C, CH3), 130.5 (1C, C-quinoline), 135.6 (1C, CH-quinoline), 126.4 (1C, C-quinoline), 128.4 (1C, CH-quinoline), 127.4 (1C, CH-quinoline), 130.8 (1C, -CH-quinoline), 127.5 (1C, CH-quinoline), 147.0 (1C, C-quinoline), 149.5 (1C, C-quinoline), MS: m/z: 449 (M+, 100%). Anal. calcd for C25H15Cl2NO3: C, 66.98; H, 3.37; N, 3.12; found: C, 67.28; H, 3.11; N, 3.52.
3.1.5.2 (3-(2-Chloro-6-fluoroquinolin-3-yl)methylene)-5-(7-methoxynaphthalen-2-yl)furan-2(3H)-one 5f. Yield 37%; m.p. 263–265 °C, yellow colour, Rf 0.88, IR (KBr) cm−1: 1758 (C
O), 1556 (CN str), 1210 (C–N str), 1428 (CC str), 1H NMR (300 MHz, CDCl3): 6.72 (s, 1H, βH), 7.47 (s, 1H, olefinic H), 4.28 (s, 3H, OCH3), 7.53 (d, 2H, ArH), 7.12 (d, 1H, ArH), 7.18 (d, 1H, ArH), 6.82 (d, 1H, ArH), 6.41 (d, 1H, ArH), 8.23 (s, 1H, quinoline-H), 7.59 (s, 1H, quinoline-H), 7.50 (d, 1H, quinoline-H), 8.42 (d, 1H, quinoline-H), 13C NMR (100 MHz, CDCl3) δ/ppm: 52.7 (1C, CH3), 133.2 (1C, C-quinoline), 134.7 (1C, CH-quinoline), 127.7 (1C, C-quinoline), 125.2 (1C, CH-quinoline), 126.8 (1C, CH-quinoline), 131.6 (1C, -CH-quinoline), 128.7 (1C, CH-quinoline), 148.3 (1C, C-quinoline), 149.2 (1C, C-quinoline), 168.3 (1C, C-2), 142.7 (1C, C-3), 135.2 (1C, C-5), 127.9 (1C, CH, C-4), 97.8 (1C, CH, ethylene), 130.3 (1C, C′-naphthelene), 125.8 (1C, C′, CH-naphthelene), 137.9 (1C, C′-naphthelene), 122.3 (1C, C′-naphthelene), 129.3 (1C, C′, CH-naphthelene), 118.2 (1C, C′-CH-naphthelene), 107.5 (1C, C′, CH-naphthelene), 157.4 (1C, C′-naphthelene), 115.2 (1C, C′-CH-naphthelene), 123.8 (1C, C′, CH-naphthelene), MS: m/z: 432 (M+, 100%). Anal. calcd for C25H15ClFNO3: C, 69.53; H, 3.50; N, 3.24; found: C, 68.98; H, 3.27; N, 3.49.
3.1.5.3 (3-(6-Bromo-2-chloroquinolin-3-yl)methylene)-5-(7-methoxynaphthalen-2-yl)furan-2(3H)-one; 5g. Yield 22%; m.p. 172–174 °C: light pink colour, Rf 0.79, IR (KBr) cm−1: 1688 (C
O), 1564 (CN str), 1190 (C–N str), 1489 (CC str), 1H NMR (300 MHz, CDCl3): 6.88 (s, 1H, βH), 7.58 (s, 1H, olefinic H), 4.12 (s, 3H, OCH3), 7.45 (d, 2H, ArH), 7.08 (d, 1H, ArH), 7.23 (d, 1H, ArH), 6.79 (d, 1H, ArH), 6.46 (d, 1H, ArH), 8.13 (s, 1H, quinoline-H), 7.85 (s, 1H, quinoline-H), 7.52 (d, 1H, quinoline-H), 8.37 (d, 1H, quinoline-H), 13C NMR (100 MHz, CDCl3) δ/ppm: 161.8 (1C, C-2), 140.5 (1C, C-3), 126.6 (1C, CH, C-4), 136.4 (1C, C-5), 98.9 (1C, CH, ethylene), 134.3 (1C, C′-naphthelene), 138.3 (1C, C′-naphthelene), 128.6 (1C, C′-naphthelene), 124.9 (1C, C′, CH-naphthelene), 123.6 (1C, C′, CH-naphthelene), 119.7 (1C, C′-CH-naphthelene), 115.2 (1C, C′, CH-naphthelene), 155.9 (1C, C′-naphthelene), 127.2 (1C, C′, CH-naphthelene), 114.8 (1C, C′-CH-naphthelene), 56.8 (1C, CH3), 134.1 (1C, C-quinoline), 135.8 (1C, CH-quinoline), 128.5 (1C, C-quinoline), 126.7 (1C, CH-quinoline), 127.4 (1C, CH-quinoline), 130.6 (1C, -CH-quinoline), 127.3 (1C, CH-quinoline), 144.9 (1C, C-quinoline), 147.4 (1C, C-quinoline), MS: m/z: 493 (M+, 100%). Anal. calcd for C25H15BrClNO3: C, 60.94; H, 3.07; N, 2.84; found: C, 60.59; H, 3.02; N, 2.08.
3.1.5.4 (3-(2-Chloroquinolin-3-yl)methylene)-5-(3-phenoxyphenyl)furan-2(3H)-one, 6a. Dark yellow, yield 78%, m.p. 216–218 °C, Rf 0.81, IR (cm−1, νmax, KBr) 1755 (C
O), 1623 (Aromatic CC), 1215 (C–O), 1066 (ArC–N). 1H NMR (CDCl3, δ ppm) 6.89 (s, 1H, furanone ring), 7.34 (s, 1H, olefinic H), 7.89 (s, 1H, quinoline ring), 7.83 (s, 1H, quinoline ring), 6.97 (d, 1H, quinoline ring), 6.28 (d, 1H, quinoline ring), 6.47 (m, 1H, quinoline ring), 6.72 (s, 1H, ArH), 6.59 (s, 1H, ArH), 6.68 (s, 1H, ArH), 6.36 (s, 1H, ArH), 6.64 (d, 2H, ArH), 6.53 (d, 2H, ArH)and 6.13 (d, 1H, ArH), 13C NMR (100 MHz, CDCl3) δ/ppm: 166.4 (1C, C-2), 137.2 (1C, C-3), 98.6 (1C, CH, C-4), 143.8 (1C, C-5), 139.6 (1C, CH, ethylene), 130.5 (1C, C-quinoline), 135.6 (1C, CH-quinoline), 126.4 (1C, C-quinoline), 128.4 (1C, CH-quinoline), 127.4 (1C, CH-quinoline), 130.8 (1C, -CH-quinoline), 127.5 (1C, CH-quinoline), 147.0 (1C, C-quinoline), 149.5 (1C, C-quinoline), 130.1 (1C, C-benzene), 119.5 (1C, CH-benzene), 128.2 (2C, CH-benzene), 116.7 (1C, CH-benzene), 159.9 (1C, C-benzene), 157.0 (1C, C-benzene), 117.5 (2C, CH-benzene), 128.6 (2C, CH-benzene), 121.9 (1C, CH-benzene) MS: m/z: 426 (M+, 100%). Anal. calcd for C26H16ClNO3: C, 73.33; H, 3.79; N, 3.38; found: C, 72.78; H, 4.19; N, 3.43.
3.1.5.5 (3-(2-Chloro-6-hydroxyquinolin-3-yl)methylene)-5-(3-phenoxyphenyl)furan-2(3H)-one, 6c. Light orange colour, yield 85%, m.p. 248–250 °C, Rf 0.87, IR (cm−1, νmax, KBr), 1760 (C
O), 1625 (Aromatic CC), 1210 (C–O), 1063 (ArC–N). 1H NMR (DMSO, δ ppm): 6.59 (s, 1H, furanone ring), 6.81 (s, 1H, olefinic H), 3.334 (broad s, 1H, OH), 9.125 (s, 1H, quinoline ring), 8.92 (s, 1H, quinoline ring), 8.00–8.02 (d, 1H, quinoline ring), 8.14–8.16 (d, 1H, quinoline ring), 7.36–7.40 (t, 3H, ArH), 7.84–7.87 (d, 2H, ArH), 7.40–7.47 (d, 2H, ArH), 7.53–7.58 (t, 2H, ArH), 13C NMR (100 MHz, DMSO) δ/ppm: 142.6 (1C, C-benzene), 108.7 (2C, CH-benzene), 120.4 (2C, CH-benzene), 125.2 (1C, CH-benzene), 141.8 (1C, CH, ethylene), 134.9 (1C, C-quinoline), 130.6 (1C, CH-quinoline), 129.3 (1C, CH-quinoline), 108.9 (1C, CH-quinoline), 122.1 (1C, CH-quinoline), 130.8 (1C, -CH-quinoline), 147.4 (1C, C-quinoline), 156.2 (1C, C-quinoline), 142.5 (1C, CH-quinoline), 163.8 (1C, C-2), 133.8 (1C, C-3), 94.9 (1C, CH, C-4), 147.4 (1C, C-5), 132.3 (1C, C-benzene), 117.4 (1C, CH-benzene), 125.6 (1C, CH-benzene), 114.8 (1C, CH-benzene), 156.9 (2C, C-benzene), MS: m/z: 442 (M+, 100%). Anal. calcd for C26H16ClNO4: C, 70.67; H, 3.65; N, 3.17; found: C, 70.36; H, 3.49; N, 3.02.
3.1.5.6 (3-(2,6-Dichloroquinolin-3-yl)methylene)-5-(3-phenoxyphenyl)furan-2(3H)-one, 6e. Brown colour, yield 68%, m.p. 225–228 °C, Rf 0.82, IR (cm−1, νmax, KBr)1720 (C
O), 1628 (Aromatic CC), 1225 (C–O), 1058 (ArC–N). 1H NMR (CDCl3, δ ppm): 6.86 (s, 1H, furanone ring), 7.59 (s, 1H, olefinic H), 7.81 (s, 1H, quinoline ring), 6.69 (s, 1H, quinoline ring), 6.62 (s, 1H, quinoline ring), 6.99 (s, 1H, quinoline ring), 6.73 (s, 1H, ArH), 6.54 (s, 1H, ArH), 6.98 (m, 2H, ArH), 6.74 (d, 1H, ArH), 7.22 (d, 2H, ArH), 6.92 (m, 1H, ArH) and 6.94 (d, 1H, ArH), 13C NMR (100 MHz, CDCl3) δ/ppm: 131.4 (1C, C-quinoline), 133.7 (1C, CH-quinoline), 126.5 (1C, CH-quinoline), 128.4 (1C, CH-quinoline), 132.2 (1C, CH-quinoline), 134.3 (1C, -CH-quinoline), 129.2 (1C, CH-quinoline), 145.6 (1C, C-quinoline), 148.6 (1C, CH-quinoline), 140.3 (1C, CH, ethylene), 165.5 (1C, C-2), 136.4 (1C, C-3), 99.8 (1C, CH, C-4), 145.9 (1C, C-5), 132.3 (1C, C-benzene), 118.2 (2C, CH-benzene), 126.3 (2C, CH-benzene), 125.2 (1C, CH-benzene), 152.8 (2C, C-benzene), 113.8 (1C, CH-benzene), 128.2 (1C, CH-benzene), 119.6 (1C, CH-benzene), 131.1 (1C, C-benzene), MS: m/z: 462 (M+, 100%). Anal. calcd for C26H15Cl2NO3: C, 67.84; H, 3.28; N, 3.04; found: C, 65.98; H, 4.19; N, 3.11.
3.1.5.7 (3-(2-Chloro-6-fluoroquinolin-3-yl)methylene)-5-(3-phenoxyphenyl)furan-2(3H)-one, 6f. Yellow colour, yield 42%, m.p. 235–238 °C, Rf 0.83, IR (cm−1, νmax, KBr), 1722 (C
O), 1625 (Aromatic CC), 1217 (C–O), 1025 (ArC–N). 1H NMR (CDCl3, δ ppm): 6.82 (s, 1H, furanone ring), 7.64 (s, 1H, olefinic H), 7.92 (s, 1H, quinoline ring), 6.78 (s, 1H, quinoline ring), 6.53 (s, 1H, quinoline ring), 6.90 (s, 1H, quinoline ring), 6.72 (s, 1H, ArH), 6.47 (s, 1H, ArH), 6.53 (d, 2H, ArH), 6.77 (d, 1H, ArH), 7.19 (d, 2H, ArH), 6.95 (d, 1H, ArH) and 6.85 (d, 1H, ArH), 13C NMR (100 MHz, CDCl3) δ/ppm: 130.2 (1C, C-quinoline), 132.4 (1C, CH-quinoline), 127.3 (1C, CH-quinoline), 129.7 (1C, CH-quinoline), 130.9 (1C, CH-quinoline), 131.7 (1C, CH-quinoline), 126.1 (1C, CH-quinoline), 144.8 (1C, C-quinoline), 147.1 (1C, CH-quinoline), 141.5 (1C, CH, ethylene), 167.9 (1C, C-2), 138.9 (1C, C-3), 97.5 (1C, CH, C-4), 140.6 (1C, C-5), 134.6 (1C, C-benzene), 117.5 (2C, CH-benzene), 121.8 (2C, CH-benzene), 122.7 (1C, CH-benzene), 154.2 (2C, C-benzene), 115.7 (1C, CH-benzene), 124.3 (1C, CH-benzene), 118.2 (1C, CH-benzene), 133.4 (1C, C-benzene), MS: m/z: 446 (M+, 100%). Anal. calcd for C26H15ClFNO3: C, 68.83; H, 3.24; N, 3.06; found: C, 68.48; H, 3.29; N, 3.02.
3.1.5.8 (3-(2-Chloro-6-nitroquinolin-3-yl)methylene)-5-(3-phenoxyphenyl)furan-2(3H)-one, 6h. Yellowish orange crystals, yield 84%, m.p. 286–288 °C, Rf 0.84, IR (cm−1, νmax, KBr) 1765 (C
O), 1623 (Aromatic CC), 1228 (C–O), 1065 (ArC–N). 1H NMR (CDCl3, δ ppm): 6.83 (s, 1H, furanone ring), 7.48 (s, 1H, olefinic H), 8.97 (s, 1H, quinoline ring), 8.46 (s, 1H, quinoline ring), 8.33 (s, 1H, quinoline ring), 8.24 (d, 1H, quinoline ring), 7.02 (d, 1H, ArH), 7.17 (s, 1H, ArH), 6.80 (s, 1H, ArH), 6.27 (s, 1H, ArH), 7.23 (d, 2H, ArH), 6.57 (m, 2H, ArH) and 6.97 (d, 1H, ArH), 13C NMR (100 MHz, CDCl3) δ/ppm: 166.3 (1C, C-2), 134.8 (1C, C-3), 96.4 (1C, CH, C-4), 146.1 (1C, C-5), 130.8 (1C, C-quinoline), 136.4 (1C, CH-quinoline), 124.8 (1C, CH-quinoline), 123.6 (1C, CH-quinoline), 147.3 (1C, CH-quinoline), 123.6 (1C, -CH-quinoline), 129.6 (1C, CH-quinoline), 148.4 (1C, C-quinoline), 152.9 (1C, CH-quinoline), 138.7 (1C, CH, ethylene), 131.3 (1C, C-benzene), 114.8 (2C, CH-benzene), 125.7 (2C, CH-benzene), 120.4 (1C, CH-benzene), 157.6 (1C, C-benzene), 156.9 (1C, C-benzene), 112.3 (1C, CH-benzene), 127.5 (1C, CH-benzene), 115.8 (1C, CH-benzene), 133.7 (1C, C-benzene), MS: m/z: 471 (M+, 100%). Anal. calcd for C26H15ClN2O5: C, 66.32; H, 3.27; N, 5.78; found: C, 65.88; H, 3.20; N, 5.19.
3.1.5.9 5-(3-Benzylphenyl)-3-((2-chloroquinolin-3-yl)methylene)furan-2(3H)-one, 7a. Yield 73%; m.p. 110–112 °C, Rf 0.86, IR (KBr) cm−1 1767 (C
O), 1563 (ArCC), 1059 (ArC–N), 865 (ArC–H), 1215 (C–O), 1H NMR (CDCl3, δ ppm) 6.89 (s, 1H, furanone ring), 7.34 (s, 1H, olefinic H), 7.97 (s, 1H, quinoline ring), 7.88 (s, 1H, quinoline ring), 6.68 (d, 1H, quinoline ring), 6.61 (d, 1H, quinoline ring), 8.01 (m, 1H, quinoline ring), 3.82 (s, 2H, CH2), 7.11 (m, 3H, ArH), 7.21 (d, 1H, ArH), 7.07 (t, 3H, ArH) and 7.06 (dd, 2H, ArH), 13C NMR (100 MHz, CDCl3) δ/ppm: 46.8 (1C, CH2), 139.4 (1C, CH, ethylene), 141.9 (1C, C-benzene), 124.7 (2C, CH-benzene), 129.3 (2C, CH-benzene), 126.3 (1C, CH-benzene), 147.5 (1C, C-benzene), 136.4 (1C, C-benzene), 122.8 (1C, CH-benzene), 123.7 (1C, CH-benzene), 125.4 (1C, CH-benzene), 127.2 (1C, CH-benzene), 160.8 (1C, C-2), 138.2 (1C, C-3), 92.7 (1C, CH, C-4), 141.4 (1C, C-5), 138.1 (1C, C-quinoline), 140.2 (1C, CH-quinoline), 122.7 (1C, CH-quinoline), 121.9 (1C, CH-quinoline), 120.4 (1C, CH-quinoline), 123.2 (1C, -CH-quinoline), 130.4 (1C, CH-quinoline), 145.0 (1C, C-quinoline), 150.4 (1C, CH-quinoline), MS: m/z: 437 (M+, 100%). Anal. calcd for C27H18ClNO2: C, 76.50; H, 4.28; N, 3.30; found: C, 75.48; H, 4.15; N, 3.49.
3.1.5.10 5-(3-Benzylphenyl)-3-((2-chloro-6-fluoroquinolin-3-yl)methylene)furan-2(3H)-one, 7f. Yield 76%; m.p. 150–152 °C, Rf 0.76, IR (KBr) cm−1 1741 (C
O), 1558 (ArCC), 1038 (ArC–N), 824 (ArC–H). 1H NMR (CDCl3): 6.91 (s, 1H, furanone ring),7.39 (s, 1H, olefinic proton), 7.81 (s, 1H, quinoline ring), 7.62 (s, 1H, quinoline ring), 7.58 (s, 1H, quinoline ring), 7.94 (s, 1H, quinoline ring), 7.16 (m, 5H, ArH), 3.69 (s, 2H, CH2), 7.08 (d, 2H, ArH), 7.11 (s, 1H, ArH), 7.10 (s, 1H, ArH), 13C NMR (100 MHz, CDCl3) δ/ppm: 162.9 (1C, C-2), 142.7 (1C, C-3), 99.1 (1C, CH, C-4), 147.2 (1C, C-5), 38.3 (1C, CH2), 135.8 (1C, CH, ethylene), 140.8 (2C, C-benzene), 128.1 (1C, CH-benzene), 119.6 (1C, CH-benzene), 117.4 (2C, CH-benzene), 124.9 (1C, CH-benzene), 132.3 (1C, C-benzene), 122.4 (1C, CH-benzene), 123.5 (1C, CH-benzene), 126.1 (1C, CH-benzene), 127.4 (1C, CH-benzene), 131.5 (1C, C-quinoline), 139.2 (1C, CH-quinoline), 127.1 (1C, CH-quinoline), 109.9 (1C, CH-quinoline), 161.1 (1C, C-quinoline), 120.8 (1C, -CH-quinoline), 129.3 (1C, CH-quinoline), 144.3 (1C, C-quinoline), 148.7 (1C, CH-quinoline), MS: m/z: 442 (M+, 100%). Anal. calcd for C27H17ClFNO2: C, 73.99; H, 3.88; N, 3.17; found: C, 72.78; H, 4.11; N, 3.28.
3.1.5.11 5-(3-Benzylphenyl)-3-((2-chloro-6-nitroquinolin-3-yl)methylene)furan-2(3H)-one, 7h. Yield 28%; m.p. 184–186 °C, Rf 0.71, IR (KBr) cm−1 1767 (C
O), 1553 (ArCC), 1051 (ArC–N), 838 (ArC–H), 1H NMR (CDCl3): 6.89 (s, 1H, furanone ring), 7.31 (s, 1H, olefinic proton), 8.23 (s, 1H, quinoline ring), 8.68 (s, 1H, quinoline ring), 8.46 (s, 1H, quinoline ring), 8.83 (s, 1H, quinoline ring), 2.84 (s, 2H, CH2), 7.09 (m, 2H, ArH), 7.12 (s, 1H, ArH), 7.11 (s, 1H, ArH), 7.25 (m, 3H, ArH), 7.18 (m, 2H, ArH), 13C NMR (100 MHz, CDCl3) δ/ppm: 132.8 (1C, C-quinoline), 137.7 (1C, CH-quinoline), 126.9 (1C, CH-quinoline), 123.8 (1C, CH-quinoline), 146.7 (1C, C-quinoline), 124.2 (1C, -CH-quinoline), 128.1 (1C, CH-quinoline), 149.4 (1C, C-quinoline), 152.4 (1C, CH-quinoline), 167.3 (1C, C-2), 135.2 (1C, C-3), 91.4 (1C, CH, C-4), 144.1 (1C, C-5), 48.4 (1C, CH2), 137.2 (1C, CH, ethylene), 141.6 (2C, C-benzene), 128.3 (2C, CH-benzene), 129.3 (2C, CH-benzene), 130.2 (2C, CH-benzene), 121.7 (1C, CH-benzene), 122.9 (1C, C-benzene), 120.1 (1C, CH-benzene), 113.8 (1C, CH-benzene), MS: m/z: 469 (M+, 100%). Anal. calcd for C27H17ClN2O4: C, 69.19; H, 3.65; N, 5.97; found: C, 68.98; H, 3.53; N, 5.78.
3.2 In vitro test for antiplasmodial activity
3.3 Computational studies
P(o/w)], number of rotatable bonds (NRB), number of hydrogen bond acceptors(HBA), number of hydrogen bond donors (HBD), total polar surface area (TPSA), solvent-accessible surface area (SASA), blood–brain partition coefficient (logBB), skin permeability (logKp), solubility (logs), percentage human oral absorption, binding to human serum albumin (logKhsa) apparent MDCK cell permeability (affyPMDCK), apparent Caco-2 cell permeability (affyPCaco) along with the Lipinski rule of 5.28 A series of steps were performed for toxicity prediction in TOPKAT. The chemical structures were entered as the molfiles code and the relevant prediction module was selected. The specific chemical class sub-model is automatically chosen from the different chemical sub-models. TOPKAT informs the user whether the prediction is within the optimum prediction space (OPS) or not, reflecting the degree of confidence to assign to that prediction. The parameter OPS, enables the user to ascertain whether the test structure is contained in the model descriptor space. TOPKAT includes various toxicity modules, but carcinogenicity, and mutagenicity are considered the most important for preliminary toxicity prediction. In the current in silico studies of quinoline–furanone derivatives, toxicity predictions were made using the same module of carcinogenicity and mutagenicity.4. Conclusions
This studies revealed that all the synthesized hybrids showed antiplasmodial activity, but the compounds 5g and 6e showed maximum activity with IC50 value almost equal to the standard drug CQ and the compounds 5e, 5f, 6a, 6b, 7a and 7f, having IC50 value comparable to CQ(<5). However, two compounds 6c and 7h were exhibiting least antiplasmodial activity with IC50 >10.The designed derivatives were made bulky to target the problem of drug efflux but at the same time, the compounds were exhibiting the drug like behavior with good ADME-T profile, confirmed by different in silico techniques. Moreover, binding mode analysis revealed that the ligands exhibited binding with the same active site residues, as reported for the cofactor NADH, which is reported essential for PfLDH enzyme, therefore our designed compounds may have competition with NADH for binding with enzyme. Moreover, similarity in binding mode with CQ which was reported as inhibitor of PfLDH, revealed that these new hybrids can also act as PfLDH inhibitors. Furthermore, the compounds with lower IC50 can be used as lead compounds for the designing and development of more potent and safe antiplasmodial agents as PfLDH inhibitors.
Data availability
Data is contained within the article.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The Financial assistance was provided by the University Grant Commission [Grant-ID-F. No. 43-381/2013(SR)]. The authors acknowledge the Institute of Pharmaceutical Sciences, Kurukshetra University, Kurukshetra, for collaboration and supporting during the research work and the University Grant Commission for providing financial support to carry out this research project. We also acknowledge Dr C. R. Pillai, Emeritus Scientist, NIMR, New Delhi, for providing all the facilities to carry out in vitro antiplamodial activity. RRB would like to thank Deanship of Graduate Studies and Research, Ajman University, UAE for their support in providing assistance in article processing charges of this manuscript.References
- World Health Organization, WHO Guidelines for Malaria, 3 June 2022 (No. WHO/UCN/GMP/2022.01 Rev. 2), World Health Organization, 2019 Search PubMed.
- M. Loeffel and A. Ross, The relative impact of interventions on sympatric Plasmodium vivax and Plasmodium falciparum malaria: a systematic review, PLoS Neglected Trop. Dis., 2022, 16, 0010541, DOI:10.1371/journal.pntd.0010541.
- D. J. Kucharski, M. K. Jaszczak and P. J. Boratyński, A review of modifications of quinoline antimalarials: mefloquine and (hydroxy) chloroquine, Molecules, 2022, 27, 1003, DOI:10.3390/molecules27031003.
- D. U. J. P. N'guessan, S. Coulibaly, A. E. M. Adouko and M. Ouattara, Antimalarial Drugs with Quinoline Nucleus and Analogs, 2023, DOI:10.5772/intechopen.113193.
- Y. Q. Hu, C. Gao, S. Zhang, L. Xu, Z. Xu, L. S. Feng and F. Zhao, Quinoline hybrids and their antiplasmodial and antimalarial activities, Eur. J. Med. Chem., 2017, 139, 22–47, DOI:10.1016/j.ejmech.2017.07.061.
- O. O. Ajani, K. T. Iyaye and O. T. Ademosun, Recent advances in chemistry and therapeutic potential of functionalized quinoline motifs–a review, RSC Adv., 2022, 12, 18594–18614, 10.1039/D2RA02896D.
- L. S. Feng, Z. Xu, L. Chang, C. Li, X. F. Yan, C. Gao and X. Wu, Hybrid molecules with potential in vitro antiplasmodial and in vivo antimalarial activity against drug-resistant Plasmodium falciparum, Med. Res. Rev., 2020, 40, 931–971, DOI:10.1002/med.21643.
- J. A. Marinho, D. S. M. Guimarães, N. Glanzmann, G. de Almeida Pimentel, I. K. da Costa Nunes, H. M. G. Pereira and C. Abramo, In vitro and in vivo antiplasmodial activity of novel quinoline derivative compounds by molecular hybridization, Eur. J. Med. Chem., 2021, 215, 113271, DOI:10.1016/j.ejmech.2021.113271.
- D. Choudhary, I. Rani, J. Monga, R. Goyal, A. Husain, P. Garg and S. L. Khokra, Pyrazole based furanone hybrids as novel antimalarial: A combined experimental, pharmacological and computational study, Cent. Nerv. Syst. Agents Med. Chem., 2022, 22, 39–56, DOI:10.2174/1871524922666220301121811.
- M. Akhter, R. Saha, O. Tanwar, M. Mumtaz Alam and M. S. Zaman, Synthesis and antimalarial activity of quinoline-substituted furanone derivatives and their identification as selective falcipain-2 inhibitors, Med. Chem. Res., 2015, 24, 879–890, DOI:10.1007/s00044-014-1139-1.
- R. Singh, V. Bhardwaj and R. Purohit, Identification of a novel binding mechanism of Quinoline based molecules with lactate dehydrogenase of Plasmodium falciparum, J. Biomol. Struct. Dyn., 2021, 39, 348–356, DOI:10.1080/07391102.2020.1711809.
- D. Choudhary, G. K. Gupta and S. L. Khokra, Structure based designing and ADME-T studies of butenolide derivatives as potential agents against receptor ICAM-1: a drug target for cerebral malaria, J. Comput. Sci., 2015, 10, 156–165, DOI:10.1016/j.jocs.2015.05.004.
- S. L Khokra, S. A Khan, D. Choudhary, S. M Hasan, A. Ahmad and A. Husain, Rational Design and Synthesis of Biologically Active Disubstituted 2 (3H) Furanones and Pyrrolone Derivatives as Potent and Safer NonSteroidal Anti-inflammatory Agents, Anti-Inflammatory Anti-Allergy Agents Med. Chem., 2016, 15, 54–71 CrossRef PubMed.
- S. L. Khokra, P. Kaushik, M. M. Alam, M. S. Zaman, A. Ahmad, S. A. Khan and A. Husain, Quinoline based furanones and their nitrogen analogues: Docking, synthesis and biological evaluation, Saudi Pharm. J., 2016, 24, 705–717, DOI:10.1016/j.jsps.2015.05.002.
- S. L. Khokra, S. A. Khan, P. Thakur, D. Chowdhary, A. Ahmad and A. Husain, Synthesis, molecular docking and potential antioxidant activity of di/trisubstituted pyridazinone derivatives, J. Chin. Chem. Soc., 2016, 63, 739–750, DOI:10.1002/jccs.201600051.
- A. Husain, S. Khokra, P. Thakur, D. Choudhary, S. Kohli, A. Ahmad and S. Khan, Molecular modeling and in silico evaluation of novel pyridazinones derivatives as anticonvulsant agents, In Silico Pharmacol., 2015, 1, 7 Search PubMed.
- A. Husain, S. A. Khan, F. Iram, M. A. Iqbal and M. Asif, Insights into the chemistry and therapeutic potential of furanones: aa versatile pharmacophore, Eur. J. Med. Chem., 2019, 171, 66–92, DOI:10.1016/j.ejmech.2019.03.021.
- F. Kayamba, T. Malimabe, I. K. Ademola, O. J. Pooe, N. D. Kushwaha, M. Mahlalela and R. Karpoormath, Design and synthesis of quinoline-pyrimidine inspired hybrids as potential plasmodial inhibitors, Eur. J. Med. Chem., 2021, 217, 113330, DOI:10.1016/j.ejmech.2021.113330.
- L. F. Moor, T. R. Vasconcelos, R. da R Reis, L. S. Pinto and T. M. da Costa, Quinoline: an attractive scaffold in drug design, Mini-Rev. Med. Chem., 2021, 21, 2209–2226, DOI:10.2174/1389557521666210210155908.
- R. Sharma, S. Patil and P. Maurya, Drug discovery studies on quinoline-based derivatives as potential antimalarial agents, SAR QSAR Environ. Res., 2014, 25, 189–203, DOI:10.1080/1062936X.2013.875484.
- M. A. Salem, M. A. Gouda and G. G. El-Bana, Chemistry of 2-(piperazin-1-yl) quinoline-3-carbaldehydes, Mini-Rev. Org. Chem., 2022, 19, 480–495, DOI:10.2174/1570193X18666211001124510.
- P. Sharma, C. R. Pillai and J. D. Sharma, In vitro schizontocidal activity of standard antimalarial drugs on chloroquine-sensitive and chloroquine-resistant isolates of Plasmodium falciparum, Indian J. Exp. Biol., 2000, 38, 1129–1133 CAS.
- S. M. Smith, M. B. Wunder, D. A. Norris and Y. G. Shellman, A simple protocol for using a LDH-based cytotoxicity assay to assess the effects of death and growth inhibition at the same time, PLoS One, 2011, 6, e26908, DOI:10.1371/journal.pone.0026908.
- S. S. Muttaqin and J. S. Maji, Screening of oxamic acid similar 3D structures as candidate inhibitor Plasmodium falciparum L-lactate dehydrogenase of malaria through molecular docking, in 2018 1st International Conference on Bioinformatics, Biotechnology, and Biomedical Engineering-Bioinformatics and Biomedical Engineering, 2018, vol. 1, pp. 1–6, DOI:10.1109/BIOMIC.2018.8610537.
- B. Chandrasekaran, S. N. Abed, O. Al-Attraqchi, K. Kuche and R. K. Tekade, Computer-aided prediction of pharmacokinetic (ADMET) properties, in Dosage Form Design Parameters, Academic Press, 2018, pp. 731–755, DOI:10.1016/B978-0-12-814421-3.00021-X.
- I. A. Shehadi, H. R. Rashdan and A. H. Abdelmonsef, Homology modeling and virtual screening studies of antigen MLAA-42 protein: identification of novel drug candidates against leukemia—an in silico approach, Comput. Math. Methods Med., 2020, 2022(1), 1–12, DOI:10.1155/2020/8196147.
- Z. Y. U. Ibrahim, A. Uzairu, G. Shallangwa and S. Abechi, Molecular docking studies, drug-likeness and in-silico ADMET prediction of some novel β-Amino alcohol grafted 1, 4, 5-trisubstituted 1, 2, 3-triazoles derivatives as elevators of p53 protein levels, Sci. Afr., 2020, 10, e00570, DOI:10.1016/j.sciaf.2020.e00570.
- M. Sinha, A. Dhawan and R. Parthasarathi, In silico approaches in predictive genetic toxicology, Methods Protoc., 2019, 351–373 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01804d |
| This journal is © The Royal Society of Chemistry 2024 |