
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnprecedented iron-assisted room temperature synthesis of AgCN using acetonitrile†
Nand Kishor Gour a,
Manash J. Baruahb,
Biraj Das*c and
Mukesh Sharma*d
a,
Manash J. Baruahb,
Biraj Das*c and
Mukesh Sharma*d
aDepartment of Chemical Sciences, Tezpur University, Napaam, Tezpur – 784028, Assam, India. E-mail: nkgour1@tezu.ernet.in
bDepartment of Chemistry, D. C. B. Girls College, Jorhat, Assam 785001, India. E-mail: manashjbom@gmail.com
cDepartment of Chemistry, D. D. R. College, Chabua, Dibrugarh, Assam 786184, India. E-mail: birajdaschm@gmail.com
dDepartment of Chemistry, Suren Das College, Hajo, Kamrup, Assam 781102, India. E-mail: mcotton233@gmail.com
First published on 2nd April 2024
Abstract
A straightforward and convenient approach for producing AgCN at room temperature using acetonitrile as a source has been developed, employing various iron salts. To date, there have been no prior studies documenting the synthesis of AgCN by cleaving the C–CN bond in acetonitrile with the use of iron salts. The resulting highly crystalline material was subjected to characterization through XRD and FT-IR analysis. Additionally, the same process was used for C–CN bond breaking using Ag2S or via the formation of an AgSxOy composite. Consequently, this report is primarily dedicated to exploring the efficacy of different iron salts in breaking the C–CN bond in CH3CN. A theoretical investigation of the proposed experimental scheme has also been performed to confer the feasibility of the reaction.
Introduction
The use of non-toxic cyanide sources or cyanation agents has remained a major problem for organic chemists and the chemical industry due to its high toxicity and complex methodologies.1–4 Starting from the famous work on potassium ferrocyanide with sulfuric acid by the Swedish scientist Carl Wilhelm Scheele, numerous methodologies have been employed to date for the replacement of toxic HCN, NaCN, KCN, DDQ, malononitriles, etc. as a source of cyanating agents.1 Of particular interest is the breaking of C–CN bonds in acetonitrile (CH3CN) due to its potential applications in organic synthesis and its role as a source of cyanide ions (CN−).2 However, the high thermodynamic stability and bond dissociation energy of the C–CN bond in CH3CN limit its use as a green source of cyanide ions.5–7 To address this, various metal complexes have been developed to activate C–C bonds in aryl and alkyl cyanides.2,8,9 Henceforth, researchers have shifted their attention to the cleavage of unreactive bonds like C–C, C–O, C–N, and C–Cl particularly in the context of organic compound synthesis.10Taw et al. conducted experiments wherein they observed the displacement of dichloromethane by PhCN, leading to the formation of [Cp*(PMe3)Rh(R)(CNSiPh3)]+BAr4− when Ph3SiH was employed as a reactant.2 Subsequently, this intermediate transformed into [Cp*(PMe3)Rh(Me)(CH2Cl2)]+BAr4−. Additionally, acetonitrile cleavage was accomplished through a photoreaction utilizing the Cp(CO)2Fe(SiMe3) complex forming CpL2Fe(CN).8 In a separate study, Gou et al. reported the cleavage of the C–CN bond within CH3CN during the formation of an Ag(I) complex under solvothermal conditions.9 Furthermore, Zou et al. employed a photo-assisted process to produce metal cyanides by reacting various transition metal nitrate salts (Ag+, Zn2+, Yb2+, and Ni2+) with CH3CN.5 In a separate study, Okabayashi et al. demonstrated the formation of monomeric AgCN and AuCN through the cleavage of the C–CN bond in CH3CN in the gas phase, utilizing a sputtering reaction involving Ag sheet and CH3CN.11
In recent research endeavors, a range of vanadium-based materials were designed for the direct synthesis of AgCN from CH3CN.12–14 However, it is worth noting that, to the best of our knowledge, the synthesis of AgCN via the C–CN bond cleavage in CH3CN using iron salts has not been documented in existing literature. Due to the rapid and cost-effective synthesis of pure AgCN using iron salts, we believe this process holds great potential for various industrial and laboratory applications, as it eliminates the need for toxic alkali cyanides such as NaCN and KCN. Consequently, this report aims to investigate the potential of various iron salts in catalyzing the cleavage of the C–CN bond utilizing CH3CN as the substrate.
Experimental section
Synthesis of silver cyanide (AgCN) from silver nitrate (AgNO3)
To carry out the reaction, 100 mg of iron nitrate Fe(NO3)3 was dissolved in a 100 mL round-bottom flask containing 10 mL of CH3CN. The reaction mixture was then stirred at room temperature for 10 min after adding 2 mL of H2O2. Upon the formation of a red solution, 100 mg of AgNO3 was introduced to the solution and stirred for an additional 5 minutes, resulting in the formation of a white precipitate of AgCN. The AgCN material was thoroughly washed with distilled water and subsequently dried in an oven at 100 °C, Scheme 1.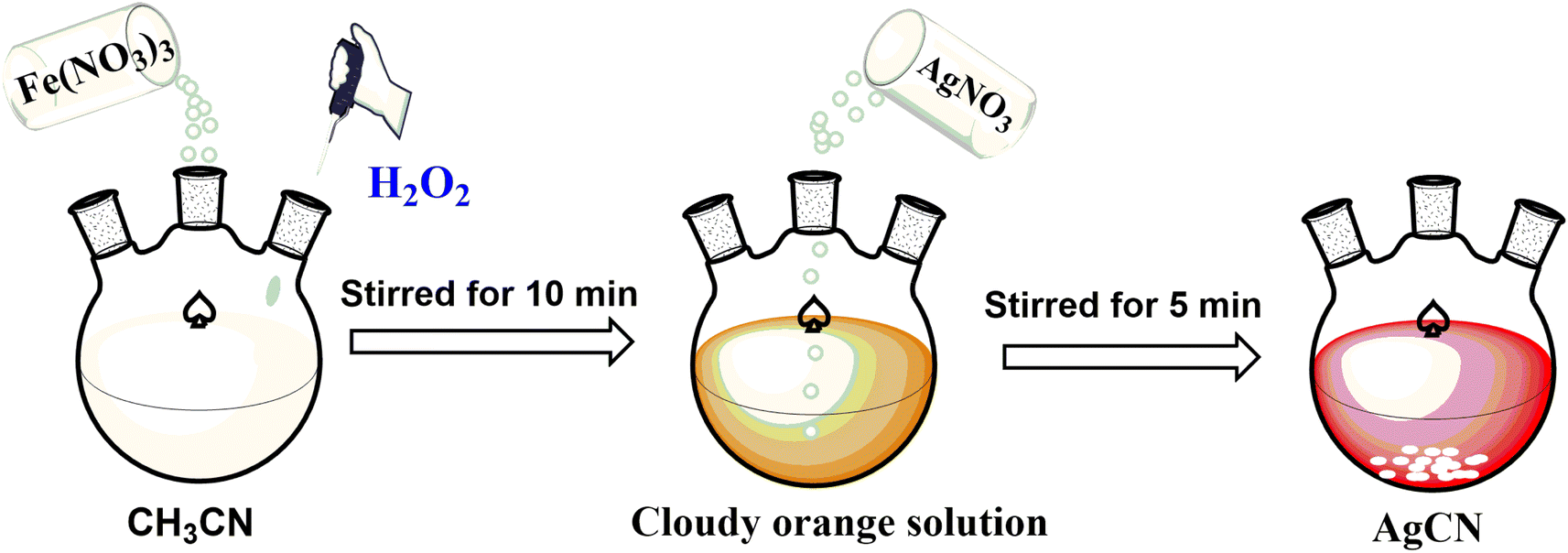 | ||
| Scheme 1 Schematic representation for the formation of AgCN. | ||
Results and discussion
The white precipitate obtained was subjected to XRD and FT-IR analysis as depicted in Fig. 1. The XRD pattern revealed sharp and well-defined peaks at 2θ values of 24, 29.7, 38.2, 49.2, 52.7, 58.4, and 61.3, which corresponded to the presence of crystalline AgCN material, Fig. 1a.14,15 Markedly, no additional peaks indicative of Ag2O or Ag NPs were observed in the XRD pattern which circuitously supports the highly crystalline nature of the formed AgCN.16,17 In the FT-IR analysis, the presence of a strong vibrational band at 2140 cm−1 and a weak band at 2163 cm−1 confirmed the existence of the –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond.18,19 Additionally, the observation of a strong band at 476 cm−1 indicated the presence of a Ag–C bond, Fig. 1b.12–14
N bond.18,19 Additionally, the observation of a strong band at 476 cm−1 indicated the presence of a Ag–C bond, Fig. 1b.12–14
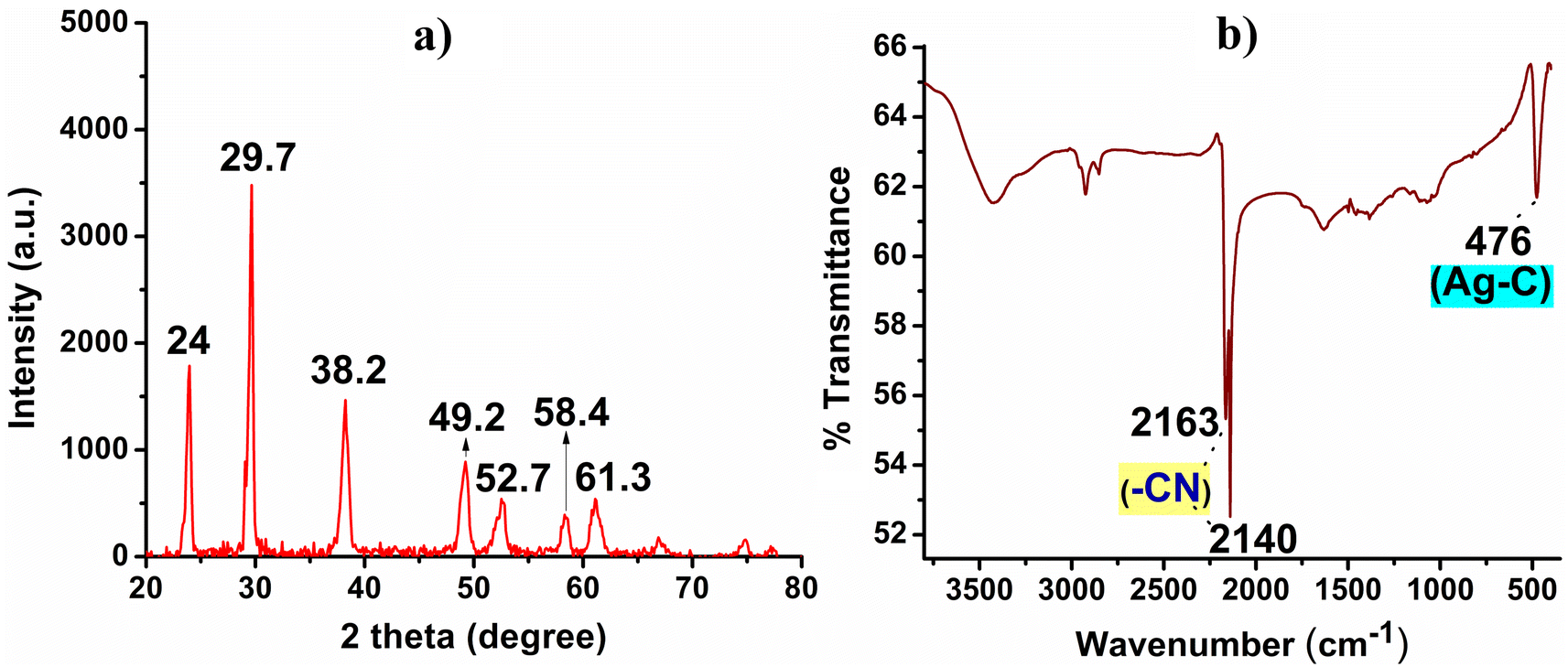 | ||
| Fig. 1 (a) XRD pattern and (b) FT-IR spectra of the synthesized AgCN. | ||
To optimize the synthesis of AgCN, we conducted experiments in which we varied the quantity of H2O2 while maintaining all other reaction parameters constant. Initially, when we employed 0.5 mL of H2O2 alongside 100 mg of Fe(NO3)3 and 10 mL of CH3CN, we did not observe the formation of AgCN precipitate. Consequently, we increased the amount of H2O2 to 1 mL, resulting in the formation of 56 mg of AgCN. To determine the ideal quantity of H2O2 needed for 100 mg of Fe(NO3)3, we conducted trials with varying amounts of H2O2, ranging up to 3 mL, as detailed in Table 1. Fortunately, we found that 2 mL of H2O2 yielded the highest conversion of AgNO3 to AgCN, making it the optimal condition. Further increases in the amount of H2O2 did not enhance AgCN formation. Therefore, we concluded that the combination of 2 mL of H2O2, 100 mg of Fe(NO3)3, and 10 mL of CH3CN represented the optimum reaction conditions.
| S. no. | Fe(NO3)3/FeSO4·7H2O (100 mg) | CH3CN (mL) | H2O2 (mL) | AgCN (mg) |
|---|---|---|---|---|
| 1 | Fe(NO3)3 | 10 | 0.5 | 0 |
| 2 | Fe(NO3)3 | 10 | 1 | 56 |
| 3 | Fe(NO3)3 | 10 | 1.5 | 64 |
| 4 | Fe(NO3)3 | 10 | 2 | 75 |
| 5 | Fe(NO3)3 | 10 | 2.5 | 76 |
| 6 | Fe(NO3)3 | 10 | 3 | 76 |
| 7 | FeSO4·7H2O | 10 | 2 | 48 |
To delve deeper into the reaction, we carried out additional investigations where we substituted iron nitrate Fe(NO3)3 with alternative iron salts while keeping the same reaction conditions. Specifically, we used ferrous sulfate (FeSO4·7H2O) as the iron salts in this experiment. Intriguingly, we observed the formation of AgCN through the cleavage of CH3CN with iron salts. However, there were subtle distinctions in their reactivity and outcomes when compared to the results obtained with Fe(NO3)3, which are summarized in Table 1. The red solution (Scheme 1) underwent recycling for up to three successive rounds in the cyanide extraction process, consistently exhibiting comparable activity and leading to the generation of silver cyanide (AgCN).
Silver sulfide (Ag2S) ores play a vital role in global silver production and the extraction of silver from these ores is imperative to satisfy the demand for this precious metal.20 The process of obtaining silver from Ag2S ores can be financially sound, given that these ores frequently contain substantial silver content. This contributes to the revenue of mining companies and has a positive impact on both local and national economies.14–16 Recognizing the substantial demand for silver extraction from Ag2S ore, we endeavored to reproduce the same reaction under standard conditions, following our successful synthesis of AgCN using AgNO3. Unfortunately, we did not observe the formation of AgCN in this instance. Given that Ag2S is highly insoluble in both acids and organic solvents, we proposed that AgSxOy material, which is soluble in hydrogen peroxide, might play a role in the reaction.20 To investigate this hypothesis, we synthesized a composite of Ag2S and Fe(NO3)3 by gradually introducing H2O2. The FT-IR analysis of this composite revealed a prominent band at 1108 cm−1, indicating the transformation of Ag2S into AgSxOy, Fig. 2.14,21 Subsequently, when we added this synthesized composite to a mixture of CH3CN and H2O2 at room temperature, AgCN was formed. To obtain pure AgCN, we washed the resulting sample with an aqueous solution of oxalic acid.
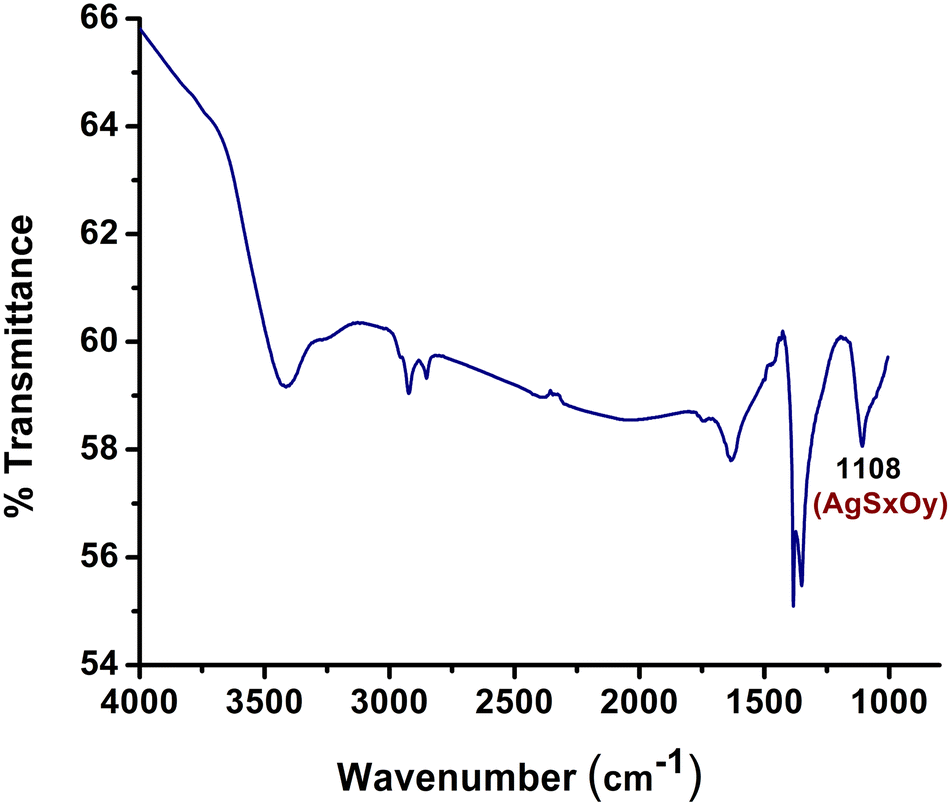 | ||
| Fig. 2 FT-IR spectra of the synthesized composites. | ||
Theoretical investigation
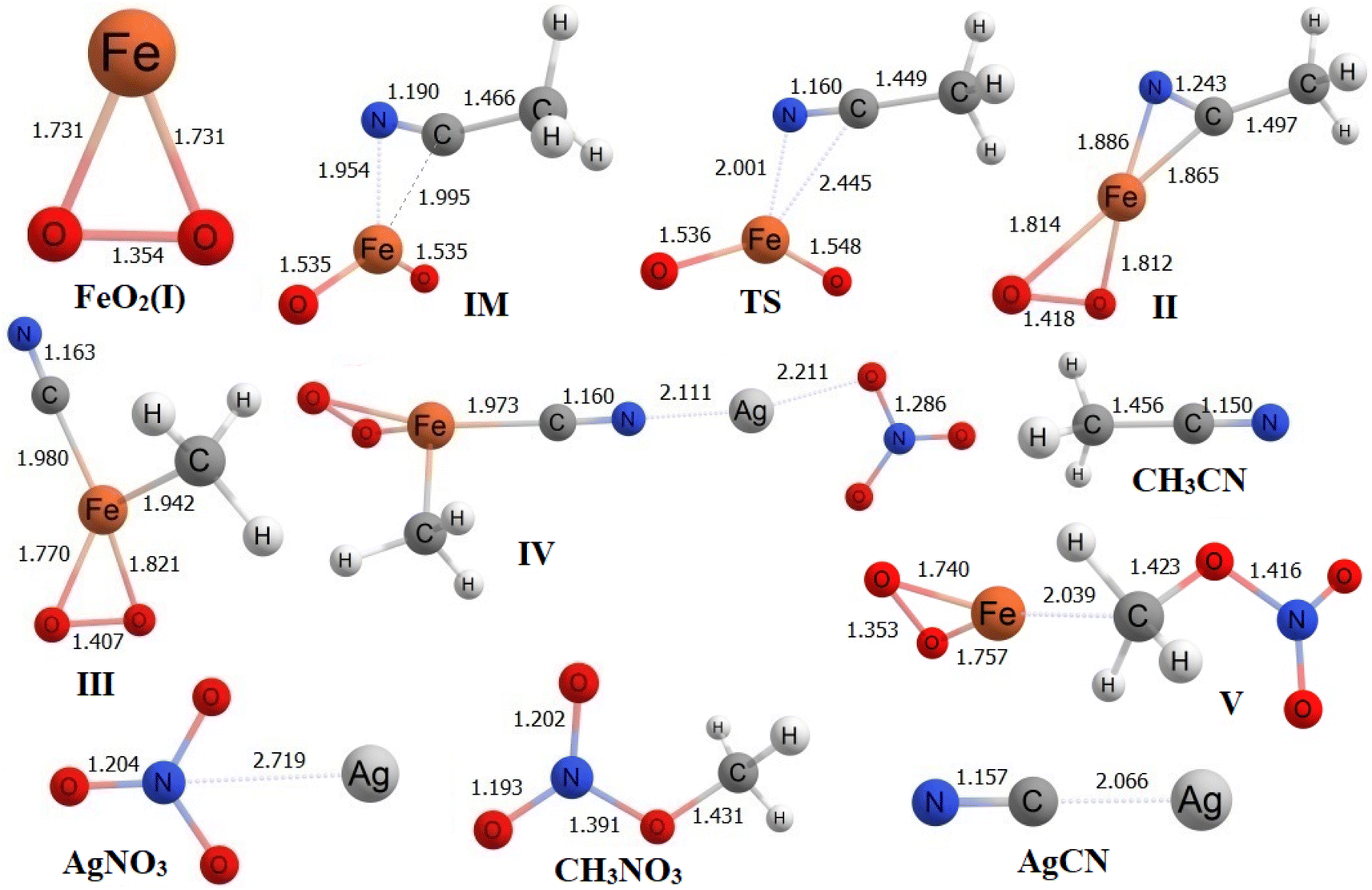 | ||
| Fig. 3 Optimized geometries of all species at ωB97XD/6-311++G(d,p)/LANL2DZ level of theory. | ||
Within structure II, the bond distances between the C atom and N atom of CH3CN with the Fe atom of FeO2 are 1.865 Å and 1.886 Å, respectively. To analyze the transformation from structure I to structure II, an IRC calculation was conducted. The IRC plot confirms that this TS effectively connects structures I and II along a reaction coordinate. In the transition from structure II to structure III, the CH3 group in CH3CN migrates toward the Fe atom, forming a bond at a distance of 1.942 Å. Simultaneously, the C atom of CN forms a bond with the Fe atom at a distance of 1.980 Å. In the subsequent step, AgNO3 is introduced to structure III, and during the conversion to structure IV, the distance between the N atom of CN in structure IV and the Ag atom of AgNO3 is measured at 2.111 Å. Meanwhile, the distance between the C atom of CN and the Fe atom is 1.973 Å. As the process proceeds from structure IV to structure V, AgCN is removed from structure IV to yield structure V. In structure V, the Fe–C and C–O distances are found to be 2.039 Å and 1.423 Å, respectively. Finally, in the last step, CH3NO3 is entirely desorbed from structure V, leading to the reformation of structure I.
Thermochemistry of the reaction
Thermo-chemical data, including total energy (E0), enthalpy (H), and Gibbs's free energy (G), for all the species were determined through frequency calculations, and these values can be found in Table S3.† We calculated the enthalpy of the reaction (ΔrHo) and Gibbs's free energy (ΔrGo) for each step in the process illustrated in Fig. 3, and these values are provided in Table 2.| Reaction steps | ΔrHo | ΔrGo |
|---|---|---|
| FeO2 (I) + CH3CN → IM | −85.96 | −74.77 |
| IM → TS | 4.38 | 3.86 |
| TS → II | −25.68 | −25.87 |
| II → III | −6.35 | −7.29 |
| III + AgNO3 → IV | −49.72 | −41.79 |
| IV → V + AgCN | 172.12 | 166.31 |
| V → FeO2+ AgCN + CH3NO3 | 14.35 | 3.38 |
In the transition from FeO2 (I) + CH3CN to IM, both ΔrHo and ΔrGo exhibit negative values, indicating that this step is exothermic (ΔrHo < 0) and spontaneous (ΔrGo < 0) at room temperature. However, the subsequent step is slightly endothermic and non-spontaneous. The following three steps are characterized as exothermic and spontaneous processes. During the transformation from structure IV to structure V, which involves the removal of AgCN and requires an input of energy, this step is highly endothermic and non-spontaneous. Finally, in the last step where structure V converts back into structure I and CH3NO3, we also observe a slightly endothermic and non-spontaneous behavior.
Energy profile diagram
Using the absolute total energy data for all the species from Table S3,† we have determined the relative energies of these species for FeO2 and explored their positions on the potential energy surface (PES), as depicted in Fig. 4. It is evident from Fig. 4 that, in addition to CH3CN within FeO2 (I), an intermediate (IM) structure is formed, resulting in a decrease in energy by 85.8 kcal mol−1. Subsequently, this IM transforms into Structure II through a transition state (TS) structure, with an energy barrier of 4.4 kcal mol−1 relative to IM. The energy of structure II is measured at −25.7 kcal mol−1. Moving forward, the energy of structure III decreases further by −6.9 kcal mol−1 relative to structure II. In the next step, the introduction of AgNO3 to structure III leads to a further decrease in the energy of structure IV relative to structure III, amounting to −50.4 kcal mol−1. Following this, AgCN is eliminated from structure IV, forming structure V. This step necessitates an input of energy equal to 173.2 kcal mol−1. Finally, structure V undergoes a conversion into FeO2 (I) with the elimination of CH3NO3. This final step also requires an input of 14.3 kcal mol−1 relative to structure V.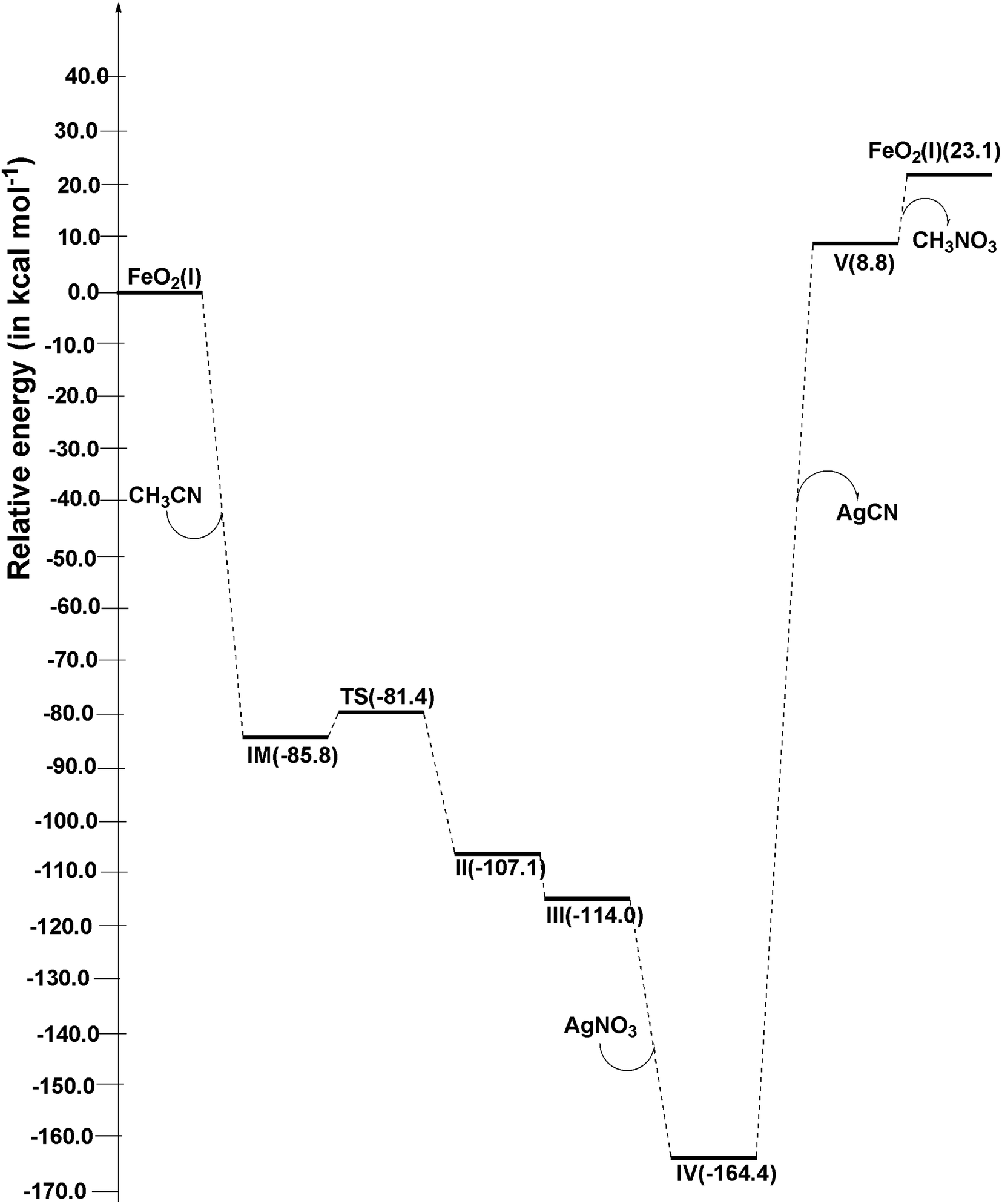 | ||
| Fig. 4 Potential energy diagram of proposed reaction at ωB97XD/6-311++G(d,p)/LANL2DZ level of theory. | ||
Catalytic study
To see the efficacy and reactivity of the freshly synthesized AgCN, the material was employed for the dye degradation process and the details of compared results with the commercially available AgCN are provided in Fig. S1.† Upon investigation, it was determined that the synthesized AgCN exhibited greater reactivity compared to its commercially available counterpart.Conclusions
This report presents a straightforward and efficient method for synthesizing AgCN using acetonitrile as a cyanide source at room temperature. What sets our work apart is the use of various iron salts, as there is no prior research demonstrating the synthesis of AgCN through the cleavage of the C–CN bond in acetonitrile with iron salts. Our study primarily focuses on investigating the potential of different iron salts to promote the cleavage of the C–CN bond in CH3CN for AgCN synthesis. Through theoretical analysis, we delve into the thermochemistry of all the involved steps, thereby discussing the feasibility of the reaction. We anticipate that this supplementary theoretical exploration will contribute to raising the quality of the experimental investigation. The freshly synthesized AgCN material showcased enhanced reactivity in the dye degradation process compared to its commercially available counterpart underlining the uncompromised reactivity of the freshly synthesized AgCN.Author contributions
Dr Gour conducted all the theoretical calculations for the experiment and provided insights into result interpretation. Dr Baruah processed the experimental data, conducted analysis, drafted the manuscript, and created the figures. Dr Sharma and Dr Das conceived and devised the experiments, contributed to the result analysis, writing, reviewing, and editing of the final manuscript. All authors engaged in result discussions and collaborated on the final manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
B. D. and M. S. acknowledge SAIC, Tezpur University for support during analysis. B. D. and M. S. also thank Prof. Kusum K. Bania for his preliminary support during the analysis.Notes and references
- A. M. Nauth and T. Opatz, Org. Biomol. Chem., 2019, 17, 11–23 RSC.
- F. L. Taw, P. S. White, R. G. Bergman and M. Brookhart, J. Am. Chem. Soc., 2002, 124, 4192–4193 CrossRef CAS PubMed.
- T. Ooi, Y. Uematsu and K. Maruoka, J. Am. Chem. Soc., 2006, 128, 2548–2549 CrossRef CAS PubMed.
- J. Schörgenhumer and M. Waser, Org. Chem. Front., 2016, 3, 1535–1540 RSC.
- S. Zou, R. Li, H. Kobayashi, J. Liu and J. Fan, Chem. Commun., 2013, 49, 1906–1908 RSC.
- Y. Zhu, M. Zhao, W. Lu, L. Li and Z. Shen, Org. Lett., 2015, 17, 2602–2605 CrossRef CAS PubMed.
- M. Tobisu and N. Chatani, Chem. Soc. Rev., 2008, 37, 300–307 RSC.
- H. Nakazawa, T. Kawasaki, K. Miyoshi, C. H. Suresh and N. Koga, Organometallics, 2004, 23, 117–126 CrossRef CAS.
- L. R. Guo, S. S. Bao, Y. Z. Li and L. M. Zheng, Chem. Commun., 2009, 20, 2893–2895 RSC.
- K. Fukumoto, T. Oya, M. Itazaki and H. Nakazawa, J. Am. Chem. Soc., 2009, 131, 38–39 CrossRef CAS PubMed.
- T. Okabayashi, E. Y. Okabayashi, F. Koto, T. Ishida and M. Tanimoto, J. Am. Chem. Soc., 2009, 131, 11712–11718 CrossRef CAS PubMed.
- B. Das, M. Sharma, C. Kashyap, A. K. Guha, A. Hazarika and K. K. Bania, Appl. Catal., 2018, 568, 191–201 CrossRef CAS.
- B. Das, M. Sharma, M. J. Baruah, K. K. Borah and K. K. Bania, Inorg. Chim. Acta, 2019, 498, 119160 CrossRef CAS.
- G. A. Bowmaker, B. J. Kennedy and J. C. Reid, Inorg. Chem., 1998, 37, 3968–3974 CrossRef CAS PubMed.
- Y. J. Lu, D. Janmanchi, T. Natarajan, Z. H. Lin, W. H. Wanna, I. J. Hsu, D. L. M. Tzou, T. Ayalew Abay and S. S. F. Yu, ChemCatChem, 2022, 14, e202200030 CrossRef CAS.
- J. Ren and Y. Zhu, RSC Adv., 2020, 10, 6114–6120 RSC.
- X. Wei, F. Cheng, Y. Yao, X. Yi, B. Wei, H. Li, Y. Wu and J. He, RSC Adv., 2021, 11, 18417–18422 RSC.
- M. C. Ríos, N. F. Bravo, C. C. Sánchez and J. Portilla, RSC Adv., 2021, 11, 34206–34234 RSC.
- G. Deschênes, J. Rajala, A. R. Pratt, H. Guo, M. Fulton and S. Mortazavi, Min., Metall., Explor., 2011, 28, 37–42 Search PubMed.
- H. Jiang, F. Xie and D. B. Dreisinger, Hydrometallurgy, 2015, 158, 149–156 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B, John Wiley & Sons, NY, 5th edn, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The details of the material used, physical measurements, computational details, cartesian coordinates of all species, harmonic vibrational frequencies, total energy, enthalpy, and Gibb's free energy of all species. Procedure for the catalytic degradation of MB dye. See DOI: https://doi.org/10.1039/d4ra01790k |
| This journal is © The Royal Society of Chemistry 2024 |