
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn easy and simple method for the immobilization of dyes through click reactions: activated alkyne, copper not needed
Julia Sánchez-Bodóna,
Ane García-Garcíaab,
Maria Diaz-Galbarriatua,
José Luis Vilas-Vilela *ab and
Isabel Moreno-Benítez*c
*ab and
Isabel Moreno-Benítez*c
aGrupo de Química Macromolecular (LABQUIMAC), Departamento de Química Física, Facultad de Ciencia y Tecnología, Universidad del País Vasco UPV/EHU, 48940 Leioa, Spain
bBCMaterials, Basque Center for Materials, Applications and Nanostructures, UPV/EHU Science Park, 48940 Leioa, Spain
cGrupo de Química Macromolecular (LABQUIMAC), Departamento de Química Orgánica e Inorgánica, Facultad de Ciencia y Tecnología, Universidad del País Vasco UPV/EHU, 48940 Leioa, Spain. E-mail: mariaisabel.moreno@ehu.eus
First published on 30th April 2024
Abstract
The copper-free azide–alkyne click reaction has shown to be a successful alternative to immobilize covalently a fluorescente compound onto poly(-L-lactic) acid (PLLA) surfaces. Proceded by basic hydrolysis and amidation reaction, typical surface characterization techniques have validated each functionaliztion step and the success of the conjugation. This method offers a catalyst-free option for various surface conjugations, extremely demanded in biomedical and biosensory fields.
Introduction
The term “click chemistry”, first coined by Barry Sharpless and coworkers in 2001, describes the formation of new C–C or C–heteroatom bonds by chemical reactions, which provide products in high yields and excellent selectivities.1,2 The click reaction allows the fabrication of complex structures from a series of smaller molecules by a select number of very efficient reactions. These building blocks contain a high-energy content that drives a spontaneous and irreversible linkage reaction. A reaction must fulfil certain requirements to be considered as a click reaction. So, products must be obtained with high yields and high selectivities from easily accessible compounds and reagents. In addition, the secondary products generated must be able to be eliminated without using chromatographic techniques, as well as they can not be harmful or toxic. Moreover, employed reagents and reaction conditions must tolerate oxygen and water media.3,4The cycloaddition between azides and alkynes to give rise to 1,2,3-triazoles, firstly described by Huisgen and coworkers in the late 1960s, can be considered the prototype among all click reactions.5,6 In this first version of the methodology, the need for high temperatures and pressures, mainly due to the poor ability of azides to act as 1,3-dipolar acceptors, constituted the most important drawbacks, which limited its application in diverse fields. Years later, in 2001, Meldal and coworkers successfully employed the first version of copper catalyst, CuI, and DIPEA as base for the click reaction. Additionally, the reaction was conducted in organic solvents and the use of copper catalyst allowed the reaction to be carried out at room temperatures, making it as one of the pioneering successful attempts at this type of reaction.5,6 This methodology was later modified by Sharpless and co-workers, who incorporated copper sulphate pentahydrate along with a reductant, sodium ascorbate, allowing the reaction to be performed in water, which overcome previously mentioned incomes and became a major scientific breakthrough. This discovery not only made C–C or C–heteroatom bond formation reactions a reality, but also transformed chemistry into a limitless tool.7,8 By addition of a catalyst, the reaction conducted a single regioisomer, particularly, 1,4-regioisomer of 1,2,3-triazole when copper(I) is employed.9,10 Since the introduction of Cu(I), azide–alkyne cycloaddition reaction has been advanced remarkably over the last decade, and has now spread to almost all areas of chemistry, applied sciences and even biomedical fields.11–15 Indeed, it was a breakthrough in drug discovery, such as the generation of lead compounds by combinatorial methods, or applications such as proteomics or nucleomics, where a new term, bioconjugation, appeared. In recent years, a primary focus of our research group has been the bioconjugation of drugs to various polymeric surfaces through click reactions, aiming to enhance their biocompatibility and thereby improve their suitability for biomedical applications such as implants or stents.16 However, the scope of this methodology has not been limited to the bioconjugation of drugs, but has been extended to other domains, including (bio)sensory or conductor materials. In fact, the resultant surface must not only deliver selective, repeatable, and sensitive electrochemical responses but also enable stable immobilization of the recognition element.17 Despite its exceptional features, the CuAAC methodology presents a major drawback regarding the complete removal of the metallic catalyst, in fact, traces of metals ions, at ppm levels, have been found after product purification. Indeed, in some cases related to biological or biomedical applications the CuAAC is not a suitable choice due to the cytotoxicity of the metal, which limits these reactions in living systems.18 Additionally, regarding luminescence properties, the CuAAC reaction presents challenges when applied to the conjugation of semiconductor QDs like CdSe due to the quenching of QDs' photoluminescence by Cu ions.19 While some studies demonstrate the utility of CuAAC for QDs conjugation, this method needs precise adjustment of Cu catalyst concentration and other reaction parameters to suit different QDs samples.20 Currently, copper-free versions of the reaction represent promising alternatives for QDs conjugation due to its relative simplicity and the absence of Cu ions.
In 2004, Bertozzi et al. introduced the term strain promoted [3 + 2] azide alkyne cycloaddition (SPAAC) reaction, which presented a novel strategy excluding the use of copper catalyst. Unlike linear acetylenes, the deformation of the carbon–carbon triple bond of a cyclooctyne (163°) allows the spontaneous reaction with azide compound despite its poor ability to act as 1,3-dipolar acceptor.21,22 This methodology was used by the same authors to label different biomolecules in cellular environments, demonstrating not only the complete bioorthogonality of the process but also the survival of the cells in the reaction media.23,24 This reaction has established itself as a robust and adaptable chemical process with broad applications in both academic and commercial ventures. Its versatility is evident in various fields, including bioconjugation processes,25 the development of hybrid and block polymers, as well as the production of high-performance and self-healing materials. Additionally, it finds utility in metabolic engineering of biological systems and the fabrication of conductor materials, among others.8,26–28 However, due to the high cost of these materials and complexity in synthesis, alternative strategies are paramount in order to address the commented challenges effectively.
In this context, this work presents the use of activated alkynes that favour the reaction between alkyne and azide avoiding the need of complex alkyne chemical structures or copper catalyst. In this proposal, the previously mentioned poor ability of azide compounds to participate as dipolarophiles would be compensated, not by strain as in Bertozzi's proposal, but by electronic effects.29 Therefore, modifying alkyne groups and becoming more reactive can be a promising alternative. The use of alkyne derived compounds with electron-attracting groups has been shown to be a rapid, efficient and cost-effective method for facilitating the reaction between azide and alkynes, reducing the activation energy required for the reaction. This novel methodology was firstly studied by Ju et al.30 in solution, where they reported the first 1,3-dipolar cycloaddition reaction between azides and electron-deficient alkynes in water. When the alkyne (either terminal or internal) had at least one neighbouring electron-withdrawing functional group, such as propiolic acid, the triazole formation was achieved even in water at room temperature without any catalysts. Despite the great versatility of this promising alternative to SPAAC, there are only few examples where this type of copper free reactions are used.30–32 For instance, it has been employed in the synthesis of hydrogels, foams or polymers among others.33–36 Although its great potential, the use of alkyne derived compound with electron-attracting groups in bioconjugation or immobilization of compounds on materials is still not widely known. While it has been shown to be a similar strategy to other copper-free click reactions, such as SPAAC, its application in these fields is still relatively new and unexplored. However, as more research is conducted and the methodology is further optimized, it has the potential to become valuable tool in the modification of polymeric surfaces for biomedical use or (bio)sensory,37–39 among others.
That is why, in this article we propose the use of an activated acetylene with an electron-attracting group. For this purpose, propiolic acid was employed as activated alkyne, which is commercially available and easily modifiable. The aim of the current study was to develop a method for the conjugation of fluorophore molecule onto polymer surfaces by employing alkyne activated-azide “click reaction”. Poly-L-lactic acid (PLLA) was employed and synthesized as substrate, where different chemical functionalization were performed in order to facilitate the covalent attachment of propiolic derivative. These alkyne activated substrate was reported to react with derivatized fluorophore compound offering a convenient way to immobilize any type of molecules by a simple modification. The surface modification procedure was confirmed employing different known techniques such as ATR-FTIR, XPS, water contact angle and fluorescence microscopy and spectroscopy.
Results and discussion
Fluorophore compound Dns-N3 was synthesized following a procedure similar to that described by Demillo et al.40 The chemical reaction mainly involved in the sulfonyl moiety of dansyl chloride and the amino group of previously synthesized 3-azidopropan-1-amine to form a sulfonamide, which contained a terminal azido group for further reaction with the terminal alkyne of modified PLLA surfaces, as shown in Scheme 1. Although, in this work dansyl derivative has been used only for the control of the conjugation owing to its fluorescence. It is worth mentioning that sulfonamides are known as broad spectrum antimicrobial agents, which can inhibit the growth of both Gram-positive and Gram-negative bacteria. Thus, the proposed methodology would be a suitable alternative to carry out the immobilization of this type of compounds.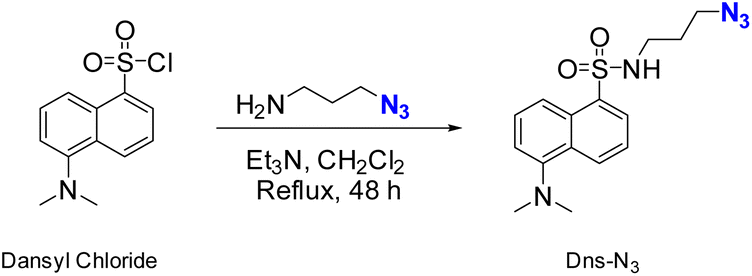 | ||
| Scheme 1 Synthesis of dansyl deritivative Dns-N3. | ||
Hydrolysis reaction of PLLA was carried out in basic media, employing NaOH 1 M during 30 min at 50 °C. As a result, carboxylic acids were generated on the surface of PLLA and were then amidated using ethylendiamine (ETDA), obtaining an amide with NH2 as ending group (PLLA-NH2 as can be seen in Scheme 2 after 1. amidation). Then, further functionalization of the terminal amine was performed in order to incorporate the alkyne group on PLLA surface, which is essential for the conjugation of the fluorophore compound. This is, in fact, the main difference between this work and the previous one carried out in our research group.16 Indeed, the first amidation of PLLA with a diamine, allowed carrying out a second amidation with propiolic acid, thus achieving an electron-deficient alkyne, which could be used to accomplish the 1,3-dipolar cycloaddition with the azide moiety of the fluorophore compound in the absence of catalyst. Finally, previously synthesized dansyl derivative was immobilized onto PLLA by copper-free azide alkyne cycloaddition reaction (Scheme 2).
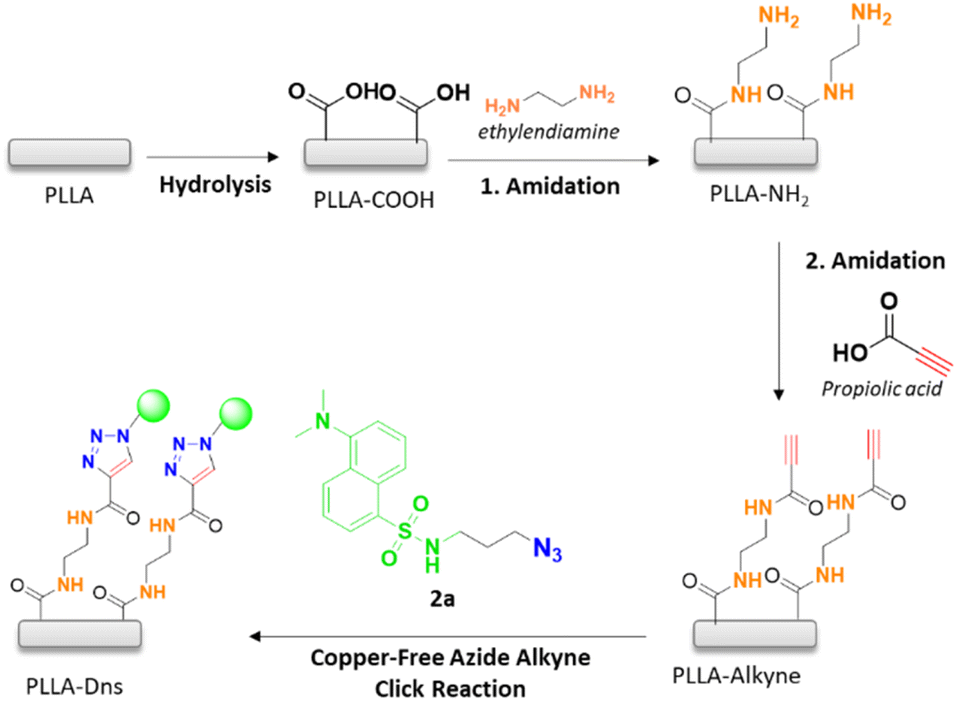 | ||
| Scheme 2 PLLA functionalization steps and dansyl derivative conjugation. | ||
The ATR-FTIR spectra of pristine PLLA, hydrolyzed PLLA, both amidated PLLA (PLLA-NH2 and PLLA-alkyne) and clicked PLLA (PLLA-Dns) are shown in Fig. 1. After 30 min of being submerged on NaOH solution, hydrolyzed PLLA samples presented a band at 3250 cm−1, corresponding to the stretching vibrations of O–H bonds, as well as the peak at 2900 cm−1 related to stretching Csp3–H bond slightly increased. These changes in ATR-FTIR spectrum evidenced the success of hydrolysis reaction on PLLA surface. After further grafting with ethylendiamine, the intensity of the band around 3250–3300 cm−1 enhanced due to the presence of NH and NH2 on the surface, which indicated that amidation reaction was carried out successfully. The incorporation of alkyne group through a second amidation reaction with propiolic acid was validated by observing two distinctive stretching bands at 2090 cm−1 and 3350 cm−1 in PLLA-alkyne samples. These bands directly corresponded to the stretching vibration of Csp–Csp and Csp–H bonds, respectively. Additionally, the presence of a peak at 1700 cm−1 corresponding to the stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O was slightly shifted to 1650 cm−1. This signal aligned with the common appearance of CO in amides, providing strong evidence that the second amidation reaction occurred successfully. Finally, after dansyl derivative (Dns-N3) immobilization via copper-free azide-yne click reaction, the Csp–Csp and Csp–H stretching signals disappeared, concluding that dansyl derivative was effectively immobilized onto PLLA.
O was slightly shifted to 1650 cm−1. This signal aligned with the common appearance of CO in amides, providing strong evidence that the second amidation reaction occurred successfully. Finally, after dansyl derivative (Dns-N3) immobilization via copper-free azide-yne click reaction, the Csp–Csp and Csp–H stretching signals disappeared, concluding that dansyl derivative was effectively immobilized onto PLLA.
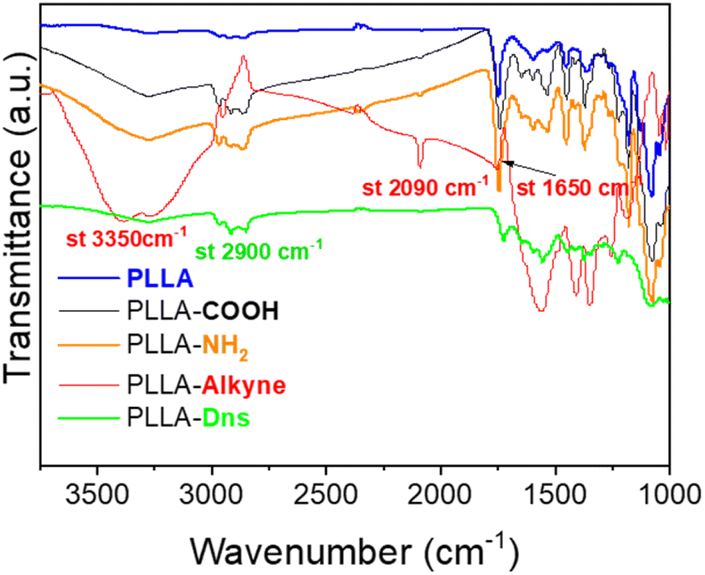 | ||
| Fig. 1 ATR-FTIR spectra of pristine PLLA and functionalized PLLA samples. | ||
Chemical compositions of functionalized surfaces were measured by XPS and the results are resumed in Fig. 2 and Table 1. As expected, PLLA-COOH only exhibited two major peaks at 284 eV and 535 eV attributing to C 1s and O 1s, respectively. In addition, the element composition of C and O were 69.9% and 29.8%, respectively. Meanwhile, high resolution spectra of C 1s showed three peaks corresponding to C–C/C–H (284.6 eV), C–O (286.3 eV) and CO (288.8 eV) bonds as it is an ester based polymer and also agreed with literature.41–43 After amidation with ethylendiamine and propiolic acid, a new peak at 400 eV attributing to N 1s appeared in the spectrum of PLLA-alkyne. Deconvolution spectra of N 1s presented only a major contribution related to C–N species. Meanwhile in the same sample, high resolution spectra of carbon exhibited that the peak at 285.8 eV (C–O/C–N) enhanced compared to the other peaks due to the introduction of amines/amides on the surface. After further functionalization with dansyl derivative, the presence of new peak around 169 eV was observed, corresponding to S 2p element. Additionally, high resolution spectra of sulphur exhibited two contributions at 164 eV and 168 eV, corresponding to C–NH/S–NH and SO species, which agreed with literature.44,45 Furthermore, high resolution spectra of nitrogen also confirmed the presence of sulphur at 402 eV.46 These results confirmed again the immobilization of dansyl derivative onto PLLA surface.
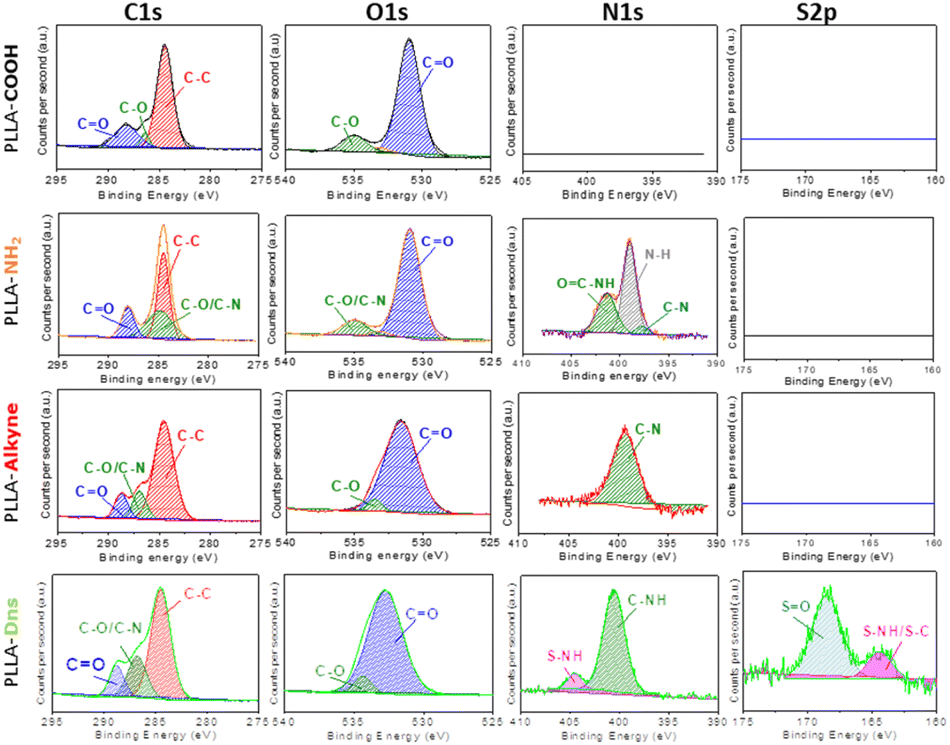 | ||
| Fig. 2 C, O, N and S deconvolution XPS spectra of PLLA-COOH, PLLA-NH2, PLLA-alkyne and PLLA-Dns samples. | ||
| Sample | C/O | C (%) | O (%) | N (%) | S (%) |
|---|---|---|---|---|---|
| PLLA | 2.4 | 69.9 | 29.8 | — | — |
| PLLA-COOH | 2.6 | 71.6 | 27.5 | — | — |
| PLLA-NH2 | 2.0 | 63.1 | 31.5 | 1.8 | — |
| PLLA-alkyne | 2.3 | 61.0 | 26.5 | 3.2 | — |
| PLLA-Dns | 2.6 | 67.2 | 26.0 | 3.8 | 0.5 |
The morphology and wettability of the surface were determined by SEM and water contact angle measurements respectively. As can be seen in Fig. 3a, the hydrolysis and subsequent functionalization reactions resulted in notable changes in the hydrophobicity of the surfaces which is reflected in the changes in the water contact angles. Pristine PLLA presented high static water contact angle value (111 ± 4.7°) due to its lack of polar functional groups on the top of the surface, which implied that the surface was hydrophobic. After hydrolysis, the water contact angle decreased to 72.4 ± 13.5°, which indicated that carboxylic acid and hydroxyl groups were successfully formed on the surface. The amidation reaction with ETDA supposed only a slight change in the contact angle (54.2 + 8.7°). In fact, similar polarities of involved moieties, such as amine or hydroxyl group, implied similar interactions with water. Nevertheless, the second amidation, once the alkyne group is incorporated, resulted in a drastic decrease in the water contact angle (18.7 ± 4.6°). Although the polarity of the attached functional groups diminished, the significantly important roughening of the surface could explain the decline in the contact angle value in this case.47,48 Moreover, after the click reaction, once the fluorophore compound was conjugated onto PLLA, the water contact angle increased again to 100 ± 8.7°, this result could be connected to the increase in hydrophobicity due to the presence of aromatic ring of dansyl derivative, specially, the naftyl group.
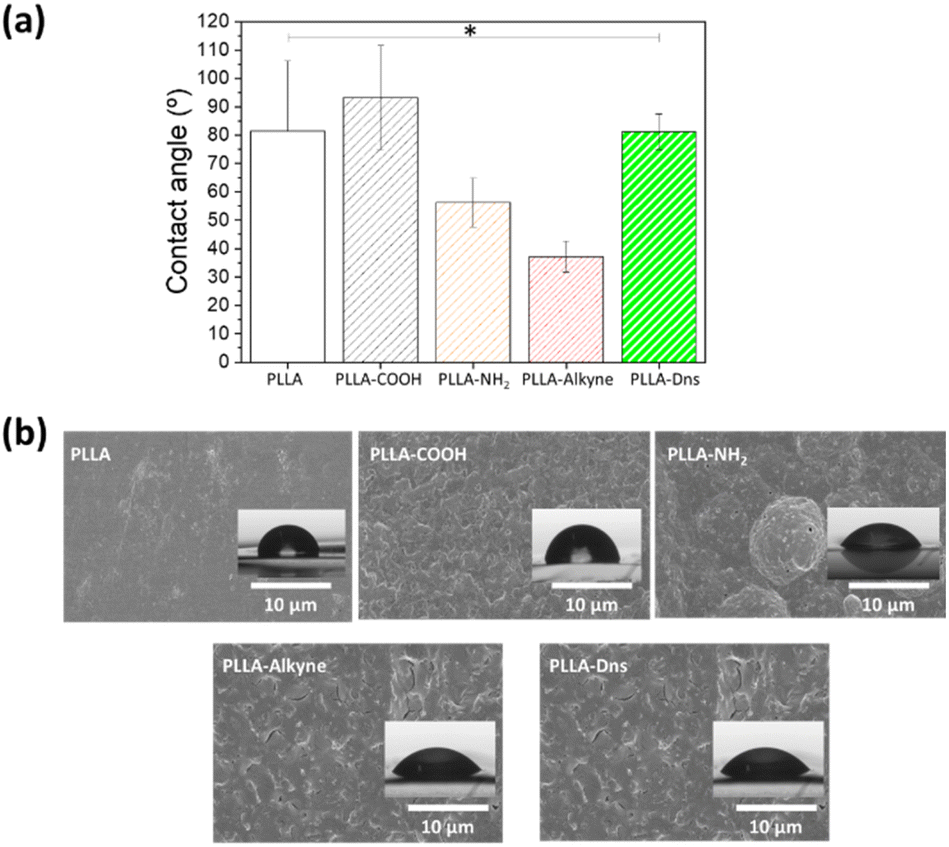 | ||
| Fig. 3 (a) Contact angle values of functionalized PLLA surfaces. Statistical significance *(p < 0.05), (b) SEM images of pristine PLLA, PLLA-COOH, PLLA-NH2, PLLA-alkyne, PLLA-Dns. | ||
SEM images indicated the surface morphologies of different PLLA samples, as can be seen in Fig. 3b. For pristine PLLA, the surface was smooth with no obvious defect. Nevertheless, after hydrolysis, PLLA-COOH sample showed some clear scratches, even flaking, which can be related to the degradation of the surface on the first step of functionalization. After first amidation, the morphology continued changing, becoming more heterogeneous with round areas. Once the alkyne was incorporated, the surface roughness increased, which is closely related with the above mentioned water contact results, since a rougher surface with a larger area is more hydrophilic.47,48
Finally, verification of dansyl derivative immobilization via copper-free azide–alkyne click reactions was conducted using both a fluorescent microscope and fluorescence spectroscopy, focusing in the characteristic emission peak at 500 nm of the fluorophore. Pristine PLLA and dansyl derivative clicked PLLA (PLLA-Dns) were compared in this study. As can be seen in Fig. 4a, as expected, PLLA-Dns surface exhibited a fluorescence maximum peak at 495 nm, specifically attributed to the dansyl compound. Meanwhile, pristine PLLA did not present any maximum at the same emission wavelength. On the other hand, confocal microscopy analysis (Fig. 4b) indicated that pristine PLLA presented no fluorescence emission across various wavelengths, even when excited at dansyl derivative's optimal excitation point around 340 nm. However, PLLA-Dns displayed a green fluorescence, attributing this emission to the fluorescence region of the dansyl derivative. These results definitively confirmed the successful immobilization of the fluorophore compound onto PLLA surfaces.
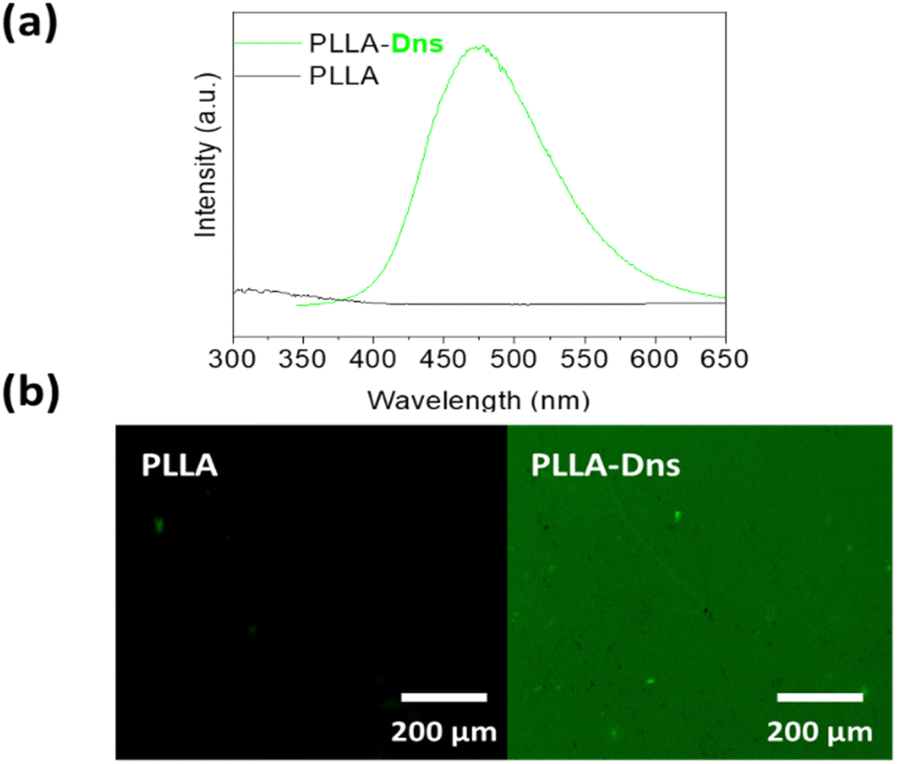 | ||
| Fig. 4 (a) Steady state fluorescence of PLLA and PLLA-Dns; (b) fluorescence microscopy images of pristine PLLA and PLLa-Dns. | ||
Materials and methods
Poly-L-lactide (PLLA) (Corbion, Amsterdam, The Netherlands) were employed as substrates. Chloroform (CHCl3, >98% Macron Fine Chemicals, Gliwice, Poland) and methanol (MeOH, destilled from Panreac) were used for the preparation of PLLA films. Sodium hydroxide (99%, Panreac, Barcelona), hydrochloridric acid (HCl, 37%, Panreac, Darmstad, Germany), N-(3-dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (EDC·HCl, 98%, Sigma Aldrich, USA), N-hydroxysuccinimide (NHS, 98%, Sigma Aldrich, USA), ethylendiamine (99%, Sigma Aldrich, USA), propiolic acid (95%, Sigma Aldrich, USA) acetic acid (99%, Sigma Aldrich, USA) and MiliQ water were used as solvent and reagents for the process of prefunctionalization. For the synthesis of the fluorophore, dansyl chloride (99%, Sigma Aldrich, Switzerland), trimethylamine (Et3N, 98%, Sigma Aldrich, USA), sodium azide (NaN3, 99%, Sigma Aldrich, USA), 3-bromopropylamine hydrobromide (98%, Sigma Aldrich, USA), acetonitrile (99%, Panreac, Darmstadt, Germany), dichloromethane (CH2Cl2, 98%, Macron Fine Chemicals, Poland), sodium hydroxide (NaOH, 99%, Panreac, Darmstadt, Germany) and sodium sulfate anhydrous (Na2SO4, 96%, Panreac, Darmstadt, Germany) were employed. Ethanol (EtOH, 98%, Panreac, Barcelona) was used as solvent in copper free click reaction. For the characterization of synthesized dansyl derivative and 3-azidopropan-1-amino, deuterated chloroform (CDCl3, 99.8%, Sigma Aldrich, USA) was employed as solvent.General procedure for the synthesis of 3-azidopropan-1-amine
The synthesis of 3-azidopropan-1-amine precursor was carried out employing reaction conditions similar to those outlined by Carboni et al.49 A detailed version of the methodology can be found in our previous work.16Synthesis of dansyl derivative (Dns-N3)
Dansyl chloride (2.0 g, 7.4 mmol) was dissolved in CH2Cl2 (20 mL) and 3-azidopropan-1-amine (0.75 g, 7.4 mmol) and Et3N (0.75 g, 7.4 mmol) were added. The reaction was refluxed for 48 h under stirring. After cooling to room temperature, water was added and then the solution was extracted with CH2Cl2 (3 × 10 mL) and washed with a saturated solution of NaCl (2 × 10 mL). Organic layers were collected, dried over Na2SO4 anhydrous and the solvent was evaporated under vacuum to afford N-(3-azidopropyl)-5-(dimethylamino)naphthalene-1-sulfonamide (Dns-N3) as a brownish oil (1.8 g, 94%). 1H-NMR (CDCl3) 2.86 (s, 6H, 3xCH3), 3.05 (t, J = 5.7 Hz, 2H, CH2), 3.27 (t, J = 5.7 Hz, 2H, CH2), 4.15 (s, 2H, NH2), 7.16 (d, J = 7.5 Hz, 1H, CHarom), 7.52 (dd, J = 7.5 Hz, J = 7.8 Hz, 2H, CHarom), 8.27 (dd, J = 8.5 Hz, J = 7.5 Hz, 2H, CHarom), 8.53 (d, J = 8.5 Hz, 1H, CHarom); 13C-RMN (CDCl3) 26.8 (CH2), 39.4 (CH2), 44.1 (CH3), 48.4 (CH2), 115.3 (Carom–H), 118.7 (Carom–C), 123.2 (Carom–H), 128.5 (Carom-H), 129.5 (Carom-H), 129.9 (Carom-C), 130.6 (Carom-H), 134.5 (Carom-S, C4), 152.0 (Carom-N).Functionalization of PLLA films
The fabrication and hydrolysis of PLLA was carried out under the same conditions as in our previous work.16 Hydrolyzed PLLA (PLLA-COOH) substrates were submitted to amidation reaction following two steps. Firstly, EDC·HCl (0.025 g, 0.13 mmol) and NHS (0.015 g, 0.13 mmol) were dissolved in acetic/acetate buffer solution of pH 5. Then, hydrolyzed PLLA films were introduced into the prepared solution for 4 h under constant stirring at room temperature. After, they were immersed in a new EDC·HCl (0.028 g, 0.15 mmol) acetic/acetate buffer solution for 2 h at 40 °C. Then, films were left to react at room temperature for 24 h. After 24 h, films were introduced in ethylendiamine (88 μL, 1.32 mmol) PBS buffer solution for 24 h at room temperature. Finally, samples were dried under vacuum for 24 h before second step. For the incorporation of the alkyne group, a previous modification of propiolic acid is required. Propiolic acid (83 μL, 1.33 mmol) is mixed with EDC·HCl (0.29 g, 1.50 mmol) and NHS (0.17 g, 1.50 mmol) in water (10 mL) for 4 h. Then, functionalized PLLA films were introduced into the solution for 24 h. Finally, samples were washed with water and dried under vacuum for 24 h.Dansyl derivative immobilization by copper-free azide–alkyne click reaction
Dansyl derivative (0.20 g, 0.61 mmol) was dissolved in EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:1 vol/vol, 10 mL) and prefunctionalized PLLA films were introduced onto the solution under constant stirring for 124 h at room temperature. Finally, dansylated PLLA (PLLA-Dns) films were washed with EtOH and dried under vacuum for 24 h.
:H2O (1:1 vol/vol, 10 mL) and prefunctionalized PLLA films were introduced onto the solution under constant stirring for 124 h at room temperature. Finally, dansylated PLLA (PLLA-Dns) films were washed with EtOH and dried under vacuum for 24 h.
Nuclear magnetic resonance (NMR)
The characterization of synthesized 3-azidopropan-1-amino and dansyl derivative was performed by proton (1H-NMR) and carbon thirteen (13C-NMR) nuclear magnetic resonance. The analysis was carried out at room temperature in an AV-300 spectrometer (300 MHz for 1H and 75.4 MHz for 13C) (Bruker, Rheinstetten, Germany) using deuterated chloroform as solvent. Chemical shifts (δ) are expressed in parts per million (ppm) relative to TMS using the residual signal of the solvent [7.26 ppm (1H) and 77.0 (13C)] as internal reference. Additionally, coupling constants (J) are expressed in Hertz (Hz).Attenuated total reflectance-Fourier transform infrared (ATR-FTIR)
ATR spectroscopy, combined with Fourier transform, has been used to analyze the vibrational bands of different bonds of all the modified PLLA surfaces, in order to visualize the functional groups generated after each functionalization. For this purpose, NICOLET Nexus FT-IR spectrophotometer (Thermo Electron Corporation) has been used and the conditions used were as follows: 32 scans, between 4000 and 500 cm−1 wavenumber, y 4 cm−1 resolution.Scanning electron microscopy (SEM)
SEM images of the surface of pristine and functionalized PLLA substrates were obtained by HITACHI S-4800 Scanning Electron Microscope. The microscope worked at 5 kV with an electron optical magnification of 100 × 100.X-ray photoelectron spectroscopy (XPS)
Elemental analysis of modified PLLA films was carried out by X-ray photoelectron spectroscopy SPECS system (XPS, SPECS Surface Nano Analysis, Berlin, Germany) using focus monochromatic radiation source 500 with dual anode Al/Ag and it is equipped with a 150 1D-DLD analyzer (Phoibos, Berlin, Germany). PLLA samples were fixed with stainless steel holders and carbon tape during de measurements. Moreover, a carbon reference was used to do the measurements.Confocal fluorescence microscopy
An Olympus FluoView FV500 confocal laser scanning with 20 times magnification (20×) was used to analyze the fluorescence of dansylated PLLA surfaces (for Dansyl λexcitation≈ 335 nm and λemission≈ 518 nm). Confocal fluorescence microscopy micrographs were taken without any previous special sample preparation.Fluorescence spectroscopy
Emission spectra of the PLLA films were recorded exciting in the UV region using an Edinburgh Instruments spectrofluorimeter (FLSP920 model, Livingston, U.K.) in front-face configuration. Samples were positioned at 40° and 50° angles to the excitation and emission beams, respectively, and were tilted at a 30° angle relative to the plane formed by the direction of incidence and detection.Conclusions
This study developed a surface treatment strategy for the conjugation of fluorescent dyes or biological compounds onto polymer surface using copper free click chemistry. The success of surface prefunctionalization was confirmed through ATR-FTIR analysis, which showed the presence of alkyne on the surface. Additionally, XPS, water contact angles, SEM and fluorescence analysis were employed to confirm that the proposed methodology was carried out successfully and the fluorescent compound was effectively immobilized. This work provides a new route to conjugate biological compounds by copper free azide alkyne click reaction, which overcome the drawbacks of the cytotoxic metal. This new and versatile approach for bioconjugation using latest copper-free click chemistry could have great potential in the biomedical field. By combining simple amidation reactions and advanced copper-free click reactions, it is possible to create bioactivated materials that can be used for a wide range of applications, such as implants coatings, biosensors or tissue engineering. In fact, this study highlights the potential of copper-free click chemistry in developing novel conjugation strategies and paves the way for future research to explore the practical applications of this technique in biomedical research.Author contributions
Conceptualization, J. S.-B. and I. M.-B.; methodology, J. S.-B., A. G.-G. and M. D.-G.; investigation, J. S.-B., A. G.-G., M. D.-G. and I. M.-B.; writing—original draft, J. S.-B.; writing—review and editing, I. M.-B.; supervision, I. M.-B. and J. L. V.-V.; funding acquisition, J. L. V.-V. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funding by Basque Government (ELKARTEK program, Department of Development and Infrastructures of the Basque Country, KK-2021-00040, KK-2021-00082 and KK-2022-00057; Grupos Consolidados IT1756-22). Moreover, authors thank for technical and human support provided by SGIker (UPV/EHU/ERDF, EU).References
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed
.
- J. Lutz, Angew. Chem., Int. Ed., 2008, 47, 2182–2184 CrossRef CAS PubMed
- Z.-R. Chen and T. Yu, Click Chemistry: Approaches, Applications, and Challenges, Nova Science Publishers, Inc, Hauppauge, New York, 2017 Search PubMed
- N. K. Devaraj and M. G. Finn, Chem. Rev., 2021, 121, 6697–6698 CrossRef CAS PubMed
- R. Huisgen, Angew. Chem., Int. Ed., 1963, 2, 565–598 CrossRef
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed
- D. Pasini, Molecules, 2013, 18, 9512–9530 CrossRef CAS PubMed
- X. Li and Y. Xiong, ACS Omega, 2022, 7, 36918–36928 CrossRef CAS PubMed
- A. Suárez, An. Quím., 2012, 108, 306–313 Search PubMed
- C. Wang, D. Ikhlef, S. Kahlal, J. Y. Saillard and D. Astruc, Coord. Chem. Rev., 2016, 316, 1–20 CrossRef CAS
- J. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS PubMed
- M. Barbosa, C. Martins and P. Gomes, U.Porto J. Eng., 2015, 1, 22–34 CrossRef
- M. Arslan and M. A. Tasdelen, Chem. Afr., 2019, 2, 195–214 CrossRef
- W. Xi, T. F. Scott, C. J. Kloxin and C. N. Bowman, Adv. Funct. Mater., 2014, 24, 2572–2590 CrossRef CAS
- J. Lahann, Click Chemistry for Biotechnology and Materials Science, John Wiley & Sons Ltd, The Atrium, Southern Gate, Chichester, West Sussex, PO19 8SQ, United Kingdom, 2009 Search PubMed
- J. Sánchez-Bodón, L. Ruiz-Rubio, E. Hernáez-Laviña, J. L. Vilas-Vilela and M. I. Moreno-Benítez, Polymers, 2021, 13, 1–15 Search PubMed
- P. Yáñez-Sedeño, A. González-Cortés, S. Campuzano and J. M. Pingarrón, Sensors, 2019, 19, 2379 CrossRef PubMed
- C. I. Z. O'Hern and K. Y. Djoko, mBio, 2022, 13, 1–4 CrossRef
- B. Ranishenka, E. Ulashchik, A. Radchanka, V. Shmanai and M. Artemyev, ChemNanoMat, 2020, 6, 292–297 CrossRef CAS
- A. R. Maity and D. Stepensky, ACS Appl. Mater. Interfaces, 2016, 8, 2001–2009 CrossRef CAS
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed
- J. Dommerholt, F. P. J. T. Rutjes and F. L. van Delft, Top. Curr. Chem., 2016, 374, 1–20 CrossRef CAS PubMed
- E. Kim and H. Koo, Chem. Sci., 2019, 10, 7835–7851 RSC
- H. I. Yoon, J. Y. Yhee, J. H. Na, S. Lee, H. Lee, S. W. Kang, H. Chang, J. H. Ryu, S. Lee, I. C. Kwon, Y. W. Cho and K. Kim, Bioconjugate Chem., 2016, 27, 927–936 CrossRef CAS PubMed
- C. J. Pickens, S. N. Johnson, M. M. Pressnall, M. A. Leon and C. J. Berkland, Bioconjugate Chem., 2018, 29, 686–701 CrossRef CAS PubMed
- H. Y. Yoon, D. Lee, D. K. Lim, H. Koo and K. Kim, Adv. Mater., 2022, 34, 1–24 Search PubMed
- R. Manova, T. A. Vanbeek and H. Zuilhof, Angew. Chem., Int. Ed., 2011, 50, 5428–5430 CrossRef CAS
- M. A. Azagarsamy and K. S. Anseth, ACS Macro Lett., 2013, 2, 5–9 CrossRef CAS PubMed
- C. Remzi Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef PubMed
- Z. Li, T. S. Seo and J. Ju, Tetrahedron Lett., 2004, 45, 3143–3146 CrossRef CAS
- M. Cetin, C. Esen, O. Daglar, S. Luleburgaz, G. Hizal, H. Durmaz and U. Tunca, Polym. Chem., 2016, 7, 7094–7100 RSC
- H. Martin-Gómez, L. Oliver-Cervelló, I. Sánchez-Campillo, V. Marchán, M. P. Ginebra and C. Mas-Moruno, Chem. Commun., 2021, 57, 982–985 RSC
- V. X. Truong, M. P. Ablett, H. T. J. Gilbert, J. Bowen, S. M. Richardson, J. A. Hoyland and A. P. Dove, Biomater. Sci., 2014, 2, 167–175 RSC
- C. Chin-Yun, L. Shu-Man, H. Jia-Qi, X. Yu-Ming, C. Tatsuya, K. Tiann-Ying, W. Ya-Ru, C. Chao-Wei and H. Hsien-Yeh, Polymers, 2021, 13, 786–797 CrossRef
- M. Jang, H. Y. Shin, D. Jang, S. M. Jo, S. Kim and S. S. Kim, ACS Appl. Polym. Mater., 2022, 4, 2712–2723 CrossRef CAS
- L. Lao, H. Tan, Y. Wang and C. Gao, Colloids Surf., B, 2008, 66, 218–225 CrossRef CAS PubMed
- A. Mukhopadhyay, K. Liu, V. Paulino, C. L. Donley and J. H. Olivier, ACS Appl. Nano Mater., 2021, 4, 8813–8822 CrossRef CAS
- I. Tsironi, J. A. Maleszka, R. S. Wilson-Kovacs, V. A. Paulino, O. Acevedo, A. Mukhopadhyay and J. H. Olivier, Chem. Mater., 2023, 35, 6877–6888 CrossRef CAS
- A. Mukhopadhyay, V. Paulino, K. Liu, C. L. Donley, B. Bernard, A. Shomar, C. Liu and J. H. Olivier, ACS Appl. Mater. Interfaces, 2021, 13, 4665–4675 CrossRef CAS PubMed
- V. G. Demillo, F. Goulinet-Mateo, J. Kim, D. Schols, K. Vermeire and T. W. Bell, J. Med. Chem., 2011, 54, 5712–5721 CrossRef CAS PubMed
- F. Renò, D. D'Angelo, G. Gottardi, M. Rizzi, D. Aragno, G. Piacenza, F. Cartasegna, M. Biasizzo, F. Trotta and M. Cannas, Plasma Processes Polym., 2012, 9, 491–502 CrossRef
- G. Sourkouni, C. Kalogirou, P. Moritz, A. Gödde, P. K. Pandis, O. Höfft, S. Vouyiouka, A. A. Zorpas and C. Argirusis, Ultrason. Sonochem., 2021, 76, 105627 CrossRef CAS PubMed
- N. De Geyter, R. Morent, T. Desmet, M. Trentesaux, L. Gengembre, P. Dubruel, C. Leys and E. Payen, Surf. Coat. Technol., 2010, 204, 3272–3279 CrossRef CAS
- A. Bunge, L. Magerusan, I. Morjan, R. Turcu, G. Borodi and J. Liebscher, J. Nanopart. Res., 2015, 17, 379–395 CrossRef
- I. Herrmann, U. I. Kramm, J. Radnik, S. Fiechter and P. Bogdanoff, J. Electrochem. Soc., 2009, 156, B1283 CrossRef CAS
- M. He, T. Yuan, W. Dong, P. Li, Q. Jason Niu and J. Meng, J. Mater. Sci., 2019, 54, 886–900 CrossRef CAS
- Y. Xiao, J. Zheng, Y. He and L. Wang, Colloids Surf., A, 2022, 653, 130008 CrossRef CAS
- E. E. Ubuo, I. A. Udoetok, A. T. Tyowua, I. O. Ekwere and H. S. Al-Shehri, J. Compos. Sci., 2021, 5, 1–9 Search PubMed
- B. Carboni, A. Benalil and M. Vaultier, J. Org. Chem., 1993, 58, 3736–3741 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2024 |