
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailoring MIL-100(Fe)-derived catalyst for controlled carbon dioxide conversion and product selectivity†
Hany E. Ahmed abc,
Mohamed K. Albolkany*d,
Mohamed E. El-Khoulya and
Ahmed Abd El-Moneim*ace
abc,
Mohamed K. Albolkany*d,
Mohamed E. El-Khoulya and
Ahmed Abd El-Moneim*ace
aNanoscience Program, Institute of Basic and Applied Sciences, Egypt-Japan University of Science and Technology, New Borg El-Arab City, Alexandria 21934, Egypt. E-mail: hany.elmenyawi@ejust.edu.eg; mohamed.elkhouly@ejust.edu.eg; ahmed.abdelmoneim@ejust.edu.eg
bNational Institute of Standards, Tersa St, El-Matbah, Haram, P. O. Box: 136, Code No 12211, Giza, Egypt
cGraphene Center of Excellence for Energy and Electronics Applications, Egypt-Japan University of Science and Technology, New Borg El-Arab, 21934, Egypt
dDepartment of Environmental Studies, Institute of Graduate Studies and Research, Alexandria University, Alexandria, Egypt. E-mail: mohamed.masoud@alexu.edu.eg
ePhysical Chemistry Department, National Research Centre, El-Dokki, Cairo, 12622, Egypt
First published on 29th April 2024
Abstract
Here in, we are reporting the effect of the catalyst particle size on the catalytic activity and product selectivity by understanding the strength of the interaction between the active catalyst and the reactants (CO2 and H2). In this regard, two catalytic systems having different active catalyst particle sizes and support surface areas were synthesized using metal–organic frameworks (MOF) (MIL-100(Fe)) having two crystal size ranges as sacrificial templates. The active catalyst having smaller nanoparticles exhibited greater chemisorption of hydrogen (Fe–H bond), resulting in heightened selectivity for paraffin due to hydrogenation of re-adsorbed olefins. Conversely, larger nanoparticles showed enhanced chemisorption of CO2 (Fe–C bond), leading to increased selectivity for olefins (O/P = 0.15). Additionally, a reduction in particle size boosts activity from 24% to 38.7% at 340 °C/20 bar. While, higher particle size enhances the selectivity towards C5+ from 11.1 to 45.6% at (300 °C/10 bar) and 9.6 to 21.3% at (340 °C/20 bar).
1. Introduction
The current levels of carbon dioxide surpass those of the preceding 800![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 years, reaching over 419 parts per million in 2023.1,2 The high CO2 level has resulted in hazardous environmental problems such as global warming and sea-level rise as a consequence, in addition to ocean acidification. These concerns called for the development of effective CO2 capture methods followed by utilization such as the catalytic conversion into value-added chemicals which would provide an extra economic motivation to support efforts in CO2 capture.3,4 In a plausible and achievable situation where green hydrogen is produced through electrolysis, the conversion of CO2 to paraffins and olefins, which are highly sought-after basic chemicals, could become a highly appealing technology.5 Nevertheless, the development of efficient and selective catalysts for this process is still needed.6–9 The process of converting CO2 to paraffins and olefins typically involves a direct pathway. This pathway begins with the transformation of CO2 into CO through a reverse water gas shift (RWGS) reaction, followed by the conversion of CO into hydrocarbons using the Fischer–Tropsch synthesis (FTS) sequentially in the same reactor.10–14 Alternatively, the transformation of CO2 into hydrocarbons could be achieved through another pathway where methanol act as an intermediary.15–17 An important benefit of this method is the ability to surpass the limitations of the Anderson–Schulz–Flory (ASF) distribution and achieve selectivity to olefins and paraffins in the hydrocarbon fraction that exceeds 80%.18–22 Nevertheless, the significant affinity for CO in the production of methanol renders this process unfavorable, as CO accounts for more than 50% of the whole output. Alternatively, by utilizing the FTS route, it is possible to maintain CO selectivity at a level lower than 40%.23–26
000 years, reaching over 419 parts per million in 2023.1,2 The high CO2 level has resulted in hazardous environmental problems such as global warming and sea-level rise as a consequence, in addition to ocean acidification. These concerns called for the development of effective CO2 capture methods followed by utilization such as the catalytic conversion into value-added chemicals which would provide an extra economic motivation to support efforts in CO2 capture.3,4 In a plausible and achievable situation where green hydrogen is produced through electrolysis, the conversion of CO2 to paraffins and olefins, which are highly sought-after basic chemicals, could become a highly appealing technology.5 Nevertheless, the development of efficient and selective catalysts for this process is still needed.6–9 The process of converting CO2 to paraffins and olefins typically involves a direct pathway. This pathway begins with the transformation of CO2 into CO through a reverse water gas shift (RWGS) reaction, followed by the conversion of CO into hydrocarbons using the Fischer–Tropsch synthesis (FTS) sequentially in the same reactor.10–14 Alternatively, the transformation of CO2 into hydrocarbons could be achieved through another pathway where methanol act as an intermediary.15–17 An important benefit of this method is the ability to surpass the limitations of the Anderson–Schulz–Flory (ASF) distribution and achieve selectivity to olefins and paraffins in the hydrocarbon fraction that exceeds 80%.18–22 Nevertheless, the significant affinity for CO in the production of methanol renders this process unfavorable, as CO accounts for more than 50% of the whole output. Alternatively, by utilizing the FTS route, it is possible to maintain CO selectivity at a level lower than 40%.23–26
The majority of catalysts outlined for the direct hydrogenation of CO2 to value-added hydrocarbons are Fe-based catalytic systems.27–30 The primary factor contributing to the exceptional performance of iron is its ability to effectively catalyze both the RWGS and FTS reactions.31 Specifically, magnetite and iron carbide serve as the active phases for these reactions, respectively.32–35 In our previous research work, we optimized the catalytic system composition through utilization of metal–organic framework (MOF) as a sacrificial template to form a carbon-supported iron nanoparticles for converting carbon dioxide into valuable hydrocarbons.36 This approach deviated from the conventional usage of inorganic supports like Al2O3, SiO2, and TiO2.37–40 By leveraging the unique properties of MOFs, we were able to significantly enhance the process. Achieving improved metal dispersion within a carbon matrix, small particle size (averaged around 3 nm), and high metal loading after the pyrolysis of MIL-100(Fe) at low temperature.41–43 As a result, CO2 conversion percentage was around 44.1% where paraffins were obtained with a selectivity of 99%, while the O/P ratio remained below 0.01.
Modifying the MOF preparation process may result in a notable alteration in the MOF crystal size, consequently affecting the particle size of the iron nanoparticles upon pyrolysis of the MOF at low temperature.44–47 The catalyst particle size is a crucial factor in heterogeneous catalysis, as well as in the process of CO2 hydrogenation.48–50 Magnetite nanoparticles were prepared with different sizes and used to examine the impact of reduction and carburization processes on the catalytic efficiency of iron-based catalysts in CO2 hydrogenation.51 Studies have demonstrated that larger Fe3O4 nanoparticles demonstrate enhanced resistance to reduction and carburization. Various iron species were formed within the catalysts during the procedure, resulting in different catalytic performances. The catalyst with the ideal proportion of magnetite and Hӓgg carbide phases demonstrated a CO2 conversion of 41.7% and an O/P ratio of 1.71.
Here, we focused on studying the correlation between the size of iron nanoparticles and carbon dioxide conversion as well as hydrocarbons selectivity. To the best of our knowledge, such studies are rare in literature. For achieving that, two catalytic systems having different Fe particle sizes were prepared by tailoring the parent MOF crystal size. Synthesizing MIL-100(Fe) following two strategies (at room temperature in a basic medium and hydrothermally in an acidic medium) led to different MOF crystal sizes. Consequently, producing two particle size ranges for the active catalyst (Fe species) after pyrolyzing the as-prepared MOF. The experimental results demonstrate that the chemisorption of hydrogen (Fe–H bond) on Fe nanoparticles with a smaller size (3 nm) derived from MIL-100(Fe) prepared at room temperature is higher than that on Fe nanoparticles with a larger size (10 nm) derived from MOF synthesized through solvothermal method. Conversely, the larger nanoparticles have a higher affinity for carbon dioxide chemisorption, particularly through the creation of Fe–C bonds. The improved chemisorption process prevents the re-adsorption of olefins, leading to a much greater level of selectivity towards olefins (with an O/P ratio of 0.15). Decreasing the size of the particles enhances the surface area accessible for contact with the fed gas, leading to a proportional rise in hydrogenation activity from 14 to 17.1% at (300 °C/10 bar) and 24 to 38.7% at (340 °C/20 bar). Conversely, the increase in particle size led to an increase in selectivity towards C5+ from 11.1 to 45.6% at (300 °C/10 bar) and 9.6 to 21.3% at (340 °C/20 bar).
2. Experimental
2.1. Materials
Trimesic acid (BTC) (98% purity), ferrous sulfate heptahydrate (FeSO4·7H2O) (>98% purity), ferric nitrate nonahydrate (Fe(NO3)3·9H2O) (>98% purity), sodium hydroxide (NaOH) (>97% purity), n-octane (≥99% purity), and ethanol (99.8% HPLC grade) were purchased from Fisher Scientific, UK with laboratory reagent grade. Nitric acid (ACS, Reag. 69%) was provided by EMSURE. Hydrogen gas of high purity (grade 4, 99.99%) was produced by the utilization of an on-demand hydrogen generation system known as H-Genie®. This process involved water electrolysis, resulting in the generation of hydrogen gas under pressures of up to 100 bar. A purification system is typically placed downstream of the electrolyzer to achieve a purity level of grade 5, with a minimum of 99.999% purity. All other gases of grade 5 (99.999% purity) are provided or imported by Air Supply Co., Egypt.2.2. Preparation methodology
The resulting dark-brown suspension was filtered then washed in hot water (80 °C) twice (each includes stirring at 80 °C for 3 h) followed by washing in ethanol once at 60 °C for 3 h. The solid material was collected by centrifuge then subjected to vacuum drying at a temperature of 70 °C. A total of 12 grams of dark brown powder was collected per run.
:0.66 H3-BTC:0.6 HNO3:166 H2O. The reaction took place in a Teflon-lined autoclave at a temperature of 150 °C for 15 hours. The solid product of a light-orange color was obtained through filtration and subsequently washed with deionized water and ethanol following the same washing protocol described above for the MIL-100(Fe) prepared at room temperature. A total of 1.5 grams of light-orange powder was collected per run.2.3. Characterization
Fourier-transform infrared spectroscopy (FTIR) was done using Bruker Vertex 70 instrument. Powder X-ray diffraction was collected using Shimadzu XRD-6100 instrument equipped with Cu Kα radiation (λ = 1.54178 Å). X-ray photoelectron spectroscopy (XPS) was employed to analyse the surface chemical and electronic properties. The analysis was conducted using K-ALPHA (Thermo Fisher Scientific, USA) with monochromatic X-ray Al Kα radiation. Morphological features, nanoparticle dispersion, crystal size distribution (CSD), and particle size distribution (PSD) of the prepared catalysts were investigated using Transmission Electron Microscopy-Energy-Dispersive X-ray Spectroscopy (TEM-EDX) (JEOL 2100 Plus) and Scanning Electron Microscopy (SEM) (JEOL JSM-6010LV). The N2 sorption isotherms were measured using BELSORP II mini (BEL Japan instruments). Subsequently, the obtained isotherms were subjected to analysis using the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) t-plot methods to determine the specific BET surface area, mean pore diameter, and total pore volume. The pristine and pyrolyzed MOFs were degassed at 150 °C for 24 hours prior to conducting measurements. The temperature programmed reduction (TPR) studies were conducted using a MICROTRAC BELCAT II catalyst analyzer. The catalyst samples (100 mg) were initially subjected to activation in an argon flow at a temperature of 400 °C for a duration of 4 hours to remove physically adsorbed water, followed by cooling down to 50 °C. The samples were subjected to a gas mixture consisting of 10% hydrogen (H2) and 90% argon (Ar), which flowed at a rate of 50 mL min−1. The temperature of the samples was incrementally raised at a constant rate of 10 °C min−1. The hydrogen consumption was consistently measured using a thermal conductivity detector. Temperature programmed desorption (TPD) measurements of CO2 and H2 were conducted at a temperature of 50 °C with a MICROTRAC BELCAT II catalyst analyzer. The samples (100 mg) utilized for the chemisorption investigation were initially subjected to reduction at a temperature of 400 °C in a flow of H2 gas for a duration of 5 hours, this was followed by cooling to 50 °C. Subsequently, a predetermined quantity of carbon dioxide (25% concentration) for CO2-TPD, and hydrogen (60% concentration) for H2-TPD was introduced into the system, utilizing helium and argon gases as a diluent for CO2-TPD and H2-TPD, respectively. Then, the samples were purged with the diluent inert gas at 50 °C for 30 minutes to remove physically adsorbed CO2 and H2 before desorption step. Then the temperature of the samples was raised at a constant rate of 10 °C min−1. The hydrogen and CO2 desorption were consistently measured using a thermal conductivity detector.2.4. Catalysis experiments
Catalyst activity measurements were conducted for CO2 hydrogenation in a fixed-bed reactor with a continuous-flow configuration and high-pressure conditions. The reactor had an internal diameter of 10 mm. The selection of stainless steel (grade 316) as the material for the reactor was based on its higher corrosion resistance compared to other alloys.53–56 Moreover, under similar reaction conditions, a control experiment demonstrated that the reactor, glass beads, glass wool, and SiC exhibited no catalytic activity. The catalyst underwent in situ reduction at a temperature of 340 °C, with a heating rate of 10 °C per minute, for a duration of 24 hours. This reduction process occurred under a continuous flow of a gas mixture consisting of hydrogen and carbon monoxide in a ratio of 2:1, with a flow rate of 45 mL per minute. To ensure the presence of iron oxide and iron carbide phases (the active phases for RWGS and FT reactions, respectively), we utilized syngas instead of pure H2 during the reduction stage. The catalysis process was performed on a catalyst weighing 1.5 grams. The catalyst was diluted with a similar weight of SiC. This was done at the catalysis temperatures of 300 and 340 °C and pressures of 10–20 bar. Throughout, a constant H2/CO2 ratio of 3 and a gas hour space velocity (GHSV) of 2400 mL g−1 h−1 were maintained. The cold trap connected to the reactor tube output was charged with a volume of 20 mL of deionized water and a mass of 2 g of n-octane, which served as the solvent for the liquid hydrocarbon products. The reactor was connected to a chromatographic system for monitoring the gaseous reaction products.
The composition of the permanent gases (carbon monoxide, methane, and carbon dioxide) was analyzed using a Shimadzu-GC-2014 gas chromatography (GC) system equipped with a 3 m ShinCarbon Restek column. The molar concentrations of the permanent gases were determined using external standards. The temperatures of the column, injection ports, and TCD detector in the GC/TCD system were set to 120 °C, 100 °C, and 180 °C, respectively. Simultaneously, the flow rate of the helium carrier gas was adjusted to 20 mL min−1. The analysis of the light hydrocarbon fraction (C1–C6) was conducted using a GC equipped with flame ionization detector (FID) with a Rt-alumina BOND/Na2SO4 column (30 m, 0.32 mm ID, 5 μm film). The GC/FID system was calibrated by employing an external secondary standard gas (C1–C6) obtained from a local petrochemical facility. Following a period of maintaining a temperature of 40 °C for six minutes, the GC oven was subsequently heated to 100 °C at a rate of 15 °C per minute for a duration of 37 minutes. The carrier gas, helium (He), flow was 20 mL min−1, while the samples were injected at a split ratio of 150. The liquid product was analyzed after mixing with n-dodecane as internal standard through direct injection on an MXT-1 Restek column (60 m length, 0.53 mm ID, and a 5 μm film). The gas chromatography (GC) equipment employed for this purpose was the SRI-GC-8610 C. The temperature of the column oven was initially set at 35 °C and kept for a duration of 3 minutes. Subsequently, the temperature increased gradually to 240 °C at a rate of 5 °C min−1 followed by a further increase to 300 °C at a rate of 10 °C min−1. The temperature was then held at 300 °C for 60 minutes. Helium was utilized as the carrier gas at a flow rate of 5 mL min−1. The conversion rates of CO2 and H2, as well as the selectivity of the resulting products, were determined using the following mathematical equations.



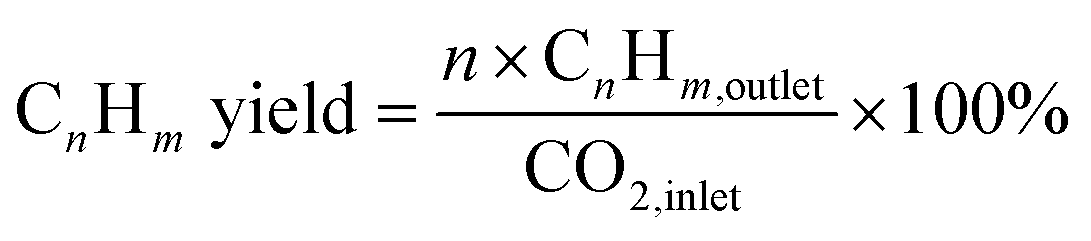


3. Results and discussion
3.1. Characterization of MIL-100(Fe) frameworks and derived catalytic systems
For synthesizing MIL-100(Fe) MOF, solvothermal (ST) and room temperature-based (RT) methods were utilized to form two different particle sizes of the MOF. The morphological structures of the prepared MOFs were confirmed through TEM analysis (Fig. 1(a) and (b)). The crystal size distribution (CSD) histogram inset revealed a crystal size of 249 nm and 210 nm for the ST-based MOF and RT-based MOF, respectively (Table S1†). The discernible variation in crystal size is expected to give rise to a disparity in the nanoparticle size of iron in the Fe-based catalytic system after the pyrolysis of the MOF at low temperature (500 °C). Consequently, this discrepancy will also inevitably impact the resultant support surface area. These hypotheses will be proved stepwise in the characterization of the catalytic system.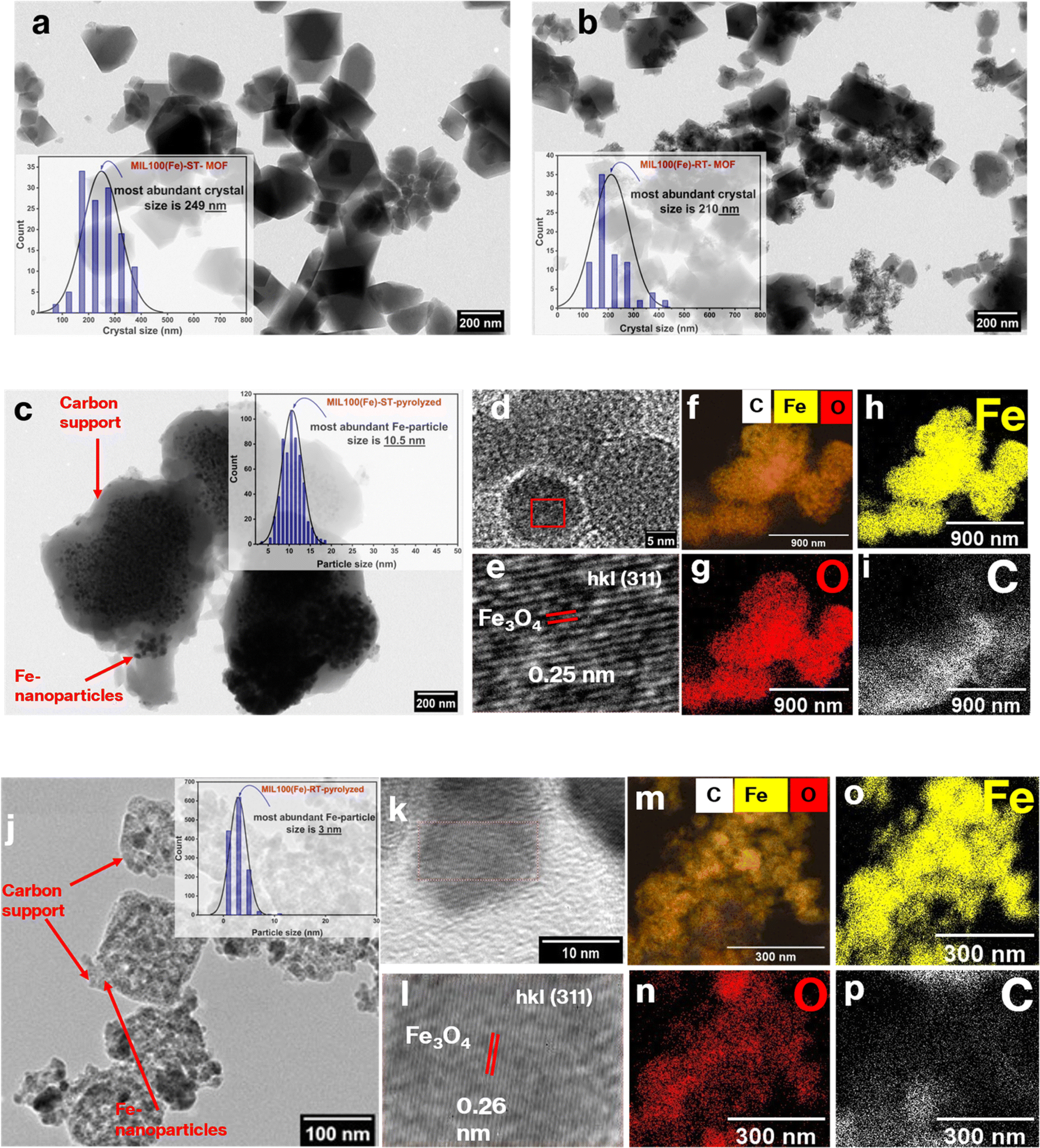 | ||
| Fig. 1 TEM images for (a) MIL-100(Fe)-ST (inset crystal size distribution (CSD)), (b) MIL-100(Fe)-RT (inset crystal size distribution (CSD)), (c) MIL-100(Fe)-ST after pyrolysis, and PSD (inset), (d and e) HRTEM images, and (f–i) TEM-EDS elemental mapping; (j) TEM image for MIL-100(Fe)-RT after pyrolysis, and PSD (inset), (k and l) HRTEM images, and (m–p) TEM-EDS elemental mapping. | ||
The powder X-ray diffraction (PXRD) patterns of the pristine MOFs refered to high crystallinity and complete matching with the simulated pattern (Fig. 2(a)). These results were further substantiated by the TEM images, which effectively captured the octahedral morphology of the MOF (Fig. 1(a) and (b)). These findings confirm that both strategies successfully achieve the intended MOF structure.52 After successful synthesis of the MIL-100(Fe) samples, they were pyrolyzed at low temperature (500 °C) under N2 atmosphere to prepare the Fe-based catalytic system. Pyrolysis at such low temperature led to framework collapse into iron oxide nanoparticles dispersed on carbon framework. The carbon support inherited the MOF porous structure which endows the catalyst with high surface area and more exposed active sites. The PXRD patterns revealed that magnetite emerged as the primary iron phase, exhibiting a face-centered cubic (fcc) structure in accordance with the JCPDS file number 39-1346 (Fig. 2(b)). Furthermore, the confirmation of magnetite as an active phase in the pyrolyzed MOF for both catalysts was achieved through the high-resolution TEM (HR-TEM) images of the pyrolyzed catalysts, as depicted in Fig. 1(d, e) and (k, l) The interplanar spacing values of 0.26 nm and 0.256 nm observed in the RT and ST catalyst samples, respectively, can be attributed to the (311) lattice planes inside the face-centered cubic (fcc) structure of magnetite. Based on the TEM analysis, the particle size distribution provides further validation of the X-ray diffraction (XRD) findings.
 | ||
| Fig. 2 PXRD patterns of (a) the simulated, ST-based, and RT-based MIL-100(Fe) and (b) ST-based, and RT-based pyrolyzed sample, and magnetite reference. | ||
Specifically, the results indicate that the most prevalent particle size for the iron oxide derived from MIL-100(Fe)-RT particles was 3 nm, whereas for MIL-100(Fe)-ST-based iron particles, it was 10 nm Fig. 1(c, j). Additionally, the elemental mapping analysis, as depicted in Fig. 1(f–i) and (m–p), demonstrates the uniform distribution of Fe nanoparticles and their effective covering in the carbon support structure. Generally, the presence of uniformly dispersed nanoparticles of small size leads to an augmentation in the surface area of the active phase. Consequently, these cause an enhancement in the adsorptivity of the feed gas mixture on the surface of the catalyst, as well as an increase in its reducibility and reaction activity.57
Moreover, the temperature programmed reduction (TPR) analysis revealed that the reduction of larger Fe3O4 nanoparticles in ST-based catalyst is more challenging when exposed to a flow of H2 before the reaction. The effect is evident in Fig. 3, where it is observed that smaller particles exhibit a higher degree of hydrogen consumption throughout the reduction process, whilst larger particles exhibit a lower degree of consumption (Table S2†). In contrast to the catalyst based on RT-MOF (smaller particles), the first consumption peak of ST-based catalyst (larger particles) necessitates a higher temperature to initiate the reduction process which represents a positive correlation between the particle size and the catalyst reducibility.58,59
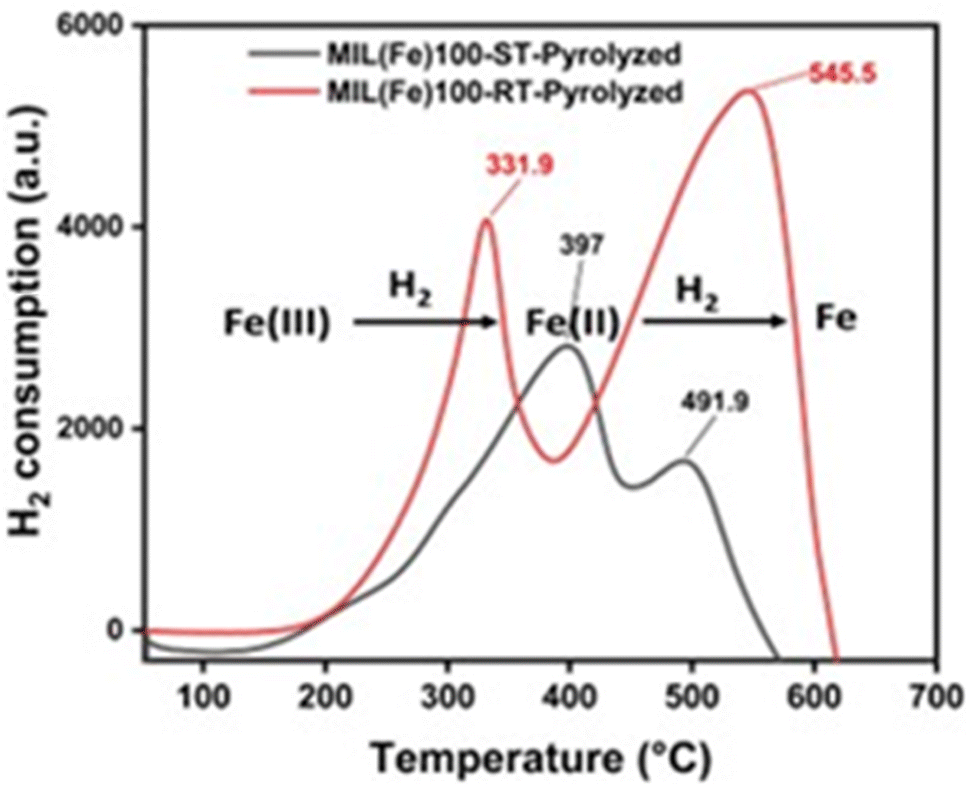 | ||
| Fig. 3 TPR profile of pyrolyzed MIL-100(Fe)-ST and RT. | ||
3.2. Catalysis experiment
The full characterization of the two MIL-100(Fe)-derived catalytic systems showed an obvious difference in the catalyst support porosity, surface area (Table S1†) and active catalyst particle size (Fig. 1). These differences are related to the different preparation methods. Because the active catalyst particle size and surface area are key factors that greatly affect the carbon dioxide hydrogenation and product selectivity, we aimed to investigate the effect of these two factors. The particle size of the catalyst derived from the MIL-100(Fe)-ST is threefold larger than that of the catalyst based on MIL-100(Fe)-RT. The present study involved doing a comparative analysis at different temperature conditions, specifically at 300 and 340 °C, as well as varying pressures of 10 and 20 bar. Additionally, the analysis was performed at a gas hourly space velocity (GHSV) of 2400 mL gcat−1 h−1, and with a hydrogen to carbon dioxide (H2/CO2) ratio of 3. In our previous research,36 a comprehensive investigation was conducted to examine the impact of temperature variations within the range of 300 to 340 °C on the conversion of carbon dioxide and the selectivity of carbon monoxide, methane, and other hydrocarbons. The findings revealed a notable enhancement in the catalyst's performance for carbon dioxide conversion when temperature increased. This improvement can be primarily attributed to the thermodynamics of the reaction responsible for converting carbon dioxide into carbon monoxide, known as the RWGS reaction, which exhibits greater reactivity at elevated temperatures.58,60 This phenomenon was verified in the present study, where the temperature was raised from 300 to 340 °C. Consequently, the carbon dioxide conversion exhibited an increase from 17 to 38.7% using the RT-based catalyst and from 14% to 24% when using the ST-based catalyst as depicted in Fig. 4(a). As previously stated, the process of CO2 hydrogenation occurs through a two-step reaction mechanism. The RWGS process involves the conversion of carbon dioxide (CO2) into carbon monoxide (CO), which serves as an intermediate product in a subsequent reaction.61,62 This intermediate, CO, is then further transformed into hydrocarbons through the Fischer–Tropsch (FT) reaction. The rate of the FT reaction showed a notable enhancement with an increase in the reaction temperature.63 The decrease in CO selectivity from 34 to 11.3 and 28 to 13.7% using the RT and ST-based catalysts, respectively, can be attributed to two main factors: the quick conversion of CO from the RWGS process into hydrocarbons, and the increase in total hydrocarbon (specially, CH4) production with rising temperature. Furthermore, it was observed that the selectivity for methane (CH4) exhibited an increase from 31.6% to 43%. However, the selectivity for heavier hydrocarbons (C5+) steadily decreased as the temperature was raised, as shown in Table 1. The process of methane generation exhibits a preference for higher temperatures, although the chain-growth probability diminishes as the reaction temperature rises, as described by α values in Table 1.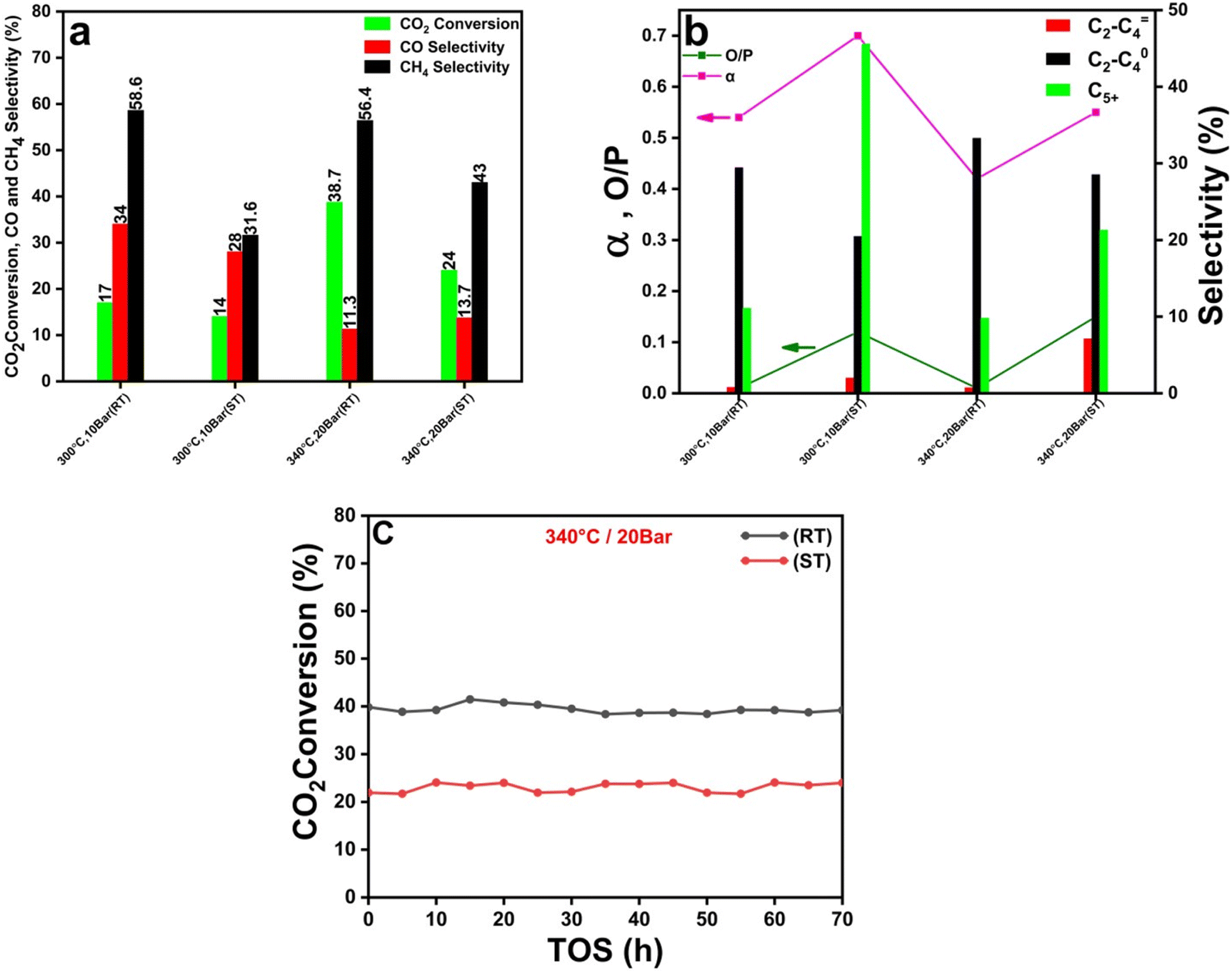 | ||
Fig. 4 (a) CO2 conversion, and CO and CH4 product selectivity at different operation conditions, (b) O/P ratio, α (chain growth probability), and C2–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) 4, C2–C04, and C5+ product selectivity of MIL-100(Fe)-RT, and MIL-100(Fe)-ST derived catalysts at different operation conditions, and (c) catalytic performance stability, carbon dioxide conversion for a reaction time of 70 h at 340 °C and 20 bar. 4, C2–C04, and C5+ product selectivity of MIL-100(Fe)-RT, and MIL-100(Fe)-ST derived catalysts at different operation conditions, and (c) catalytic performance stability, carbon dioxide conversion for a reaction time of 70 h at 340 °C and 20 bar. | ||
| Catalyst | T (°C) | P (bar) | GHSV (mL gcat−1 h−1) | H2/CO2 | CO2 conversion (%) | CO selectivity (%) | O/P | C1–C4 selectivity (%) | C5+ selectivity (%) | Hydrocarbon yield (%) | αc | Carbon balance (%) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C1 | C2–C4 | Total | ||||||||||||
| a C5–C23.b C5–C16.c Chain growth probability. (α) can be calculated from the slope of ASF plot by using single variable regression. | ||||||||||||||
| ST-based-catalyst | 340 | 20 | 2400 | 3 | 24 | 13.7 | 0.15 | 43 | 35.6 | 78.6 | 21.3a | 20.69 | 0.55 | 100 |
| 300 | 10 | 14 | 28 | 0.12 | 31.6 | 22.7 | 54.3 | 45.6a | 10.36 | 0.7 | 93 | |||
| RT-based- catalyst | 340 | 20 | 38.7 | 11.3 | 0.01 | 56.4 | 34 | 90.2 | 9.6b | 34.33 | 0.42 | 95 | ||
| 300 | 10 | 17.1 | 34 | 0.01 | 58.6 | 30.2 | 88.9 | 11.1b | 11.23 | 0.54 | 96 | |||
When the working pressure is elevated, there is a significant enhancement in CO2 conversion, accompanied by a conspicuous rise in the overall yield of the hydrocarbons, as mentioned in Table 1. Besides its elevated working pressure, the mesoporous structure of the catalyst inherited from MOFs boosts the adsorption of gas on the catalyst's surface. Consequently, this leads to an increased likelihood of both RWGS and FTS reactions taking place.62,63 The CO2 conversion % increased when pressure increased from 10 to 20 bar. However, the selectivity towards CO dropped, the selectivity towards CH4 increased. This study elucidates the combined impact of reducing temperature and pressure, namely from 340 °C and 20 bar to 300 °C and 10 bar, on the probability of chain growth. The synergistic effect of decreasing temperature and pressure resulting in enhancing the chain growth probability from 0.55 to 0.7 for the ST-based catalyst. The RT-based catalyst exhibits enhanced CO2 conversion than the ST-based catalyst, which can be explained by the smaller particle size. This reduced particle size leads to a greater exposure of active sites, hence facilitating the FTS reaction and gas adsorption.
In the case of catalysts containing iron (Fe), an increase in particle size has been observed to positively influence the chain growth probability and facilitate hydrogenation, resulting in greater selectivity towards hydrocarbons with higher molecular weights and olefins.64 Consequently, at 340 °C, the RT-based catalyst with a smaller particle size shows a lower α value of 0.42 than the ST-based catalyst with a larger particle size (α = 0.55). Similarly, the chain growth probability shows the same trend at 300 °C where the ST-based catalyst shows a higher α value of 0.7 than the RT-based catalyst (α = 0.54). The ST-based catalyst exhibited a significant rise in the proportion of C5+ compounds compared to the RT-based catalyst, as indicated in Fig. 4(b). The observed O/P ratios of the catalyst with smaller Fe particle size, as depicted in Table 1, did not exceed 0.02. This observation serves as empirical evidence that paraffin is the prevailing product across all examined settings, constituting 99% of the overall output (Fig. 5(c) and (d)). Nevertheless, the O/P ratios of the catalyst with larger Fe particle size reached a numerical value of 0.15 as visually represented in Fig. 4(b) at 340 °C and 20 bar.
 | ||
| Fig. 5 GC-FID liquid chromatogram as a function of carbon number of MIL-100(Fe)-ST at (a) 340 °C and 20 bar, (b) 300 °C and 10 bar and MIL-100(Fe)-RT at (c) 340 °C and 20 bar, (d) 300 °C, and 10 bar. | ||
The re-adsorption of olefin is significantly influenced by the relative strength of the Fe–CO2 bond. The saturation of the chain can be attributed to the re-adsorption of olefins, leading to a reduction in the O/P ratio and subsequent formation of paraffins, as depicted in Fig. 4(b). It is imperative to acknowledge that once paraffin is produced, it does not undergo re-adsorption.65 In contrast, the ramifications of strengthened Fe–CO2 bonds include the prevention of olefin re-adsorption, the impeding of paraffin synthesis through hydrogen insertion, and ultimately leading to increased olefin selectivity as depicted in Fig. 4(b). To assess the stability of the catalysts under examination, both were subjected to continuous TOS for a duration of 70 hours at a constant temperature of 340 °C and pressure of 20 bar. Fig. 4 (c) demonstrates remarkable stability since there was no evidence of deactivation noticed for both the two catalysts. The XPS results (Fig. S2†) for the catalyst before and after the reaction showed that the catalyst composition and oxidation state did not affected remarkably during the reaction.
Fig. 5 presents the hydrocarbon distribution produced at different operation conditions using the ST- and RT-based catalytic systems. The olefin distribution was observed clearly when using the ST-based catalyst at different temperature and pressure values which refer to stronger Fe–CO2 bond leading to reduced probability for olefin re-adsorption. This observation might refer to enhanced basic properties of the ST-based catalyst, meanwhile having lower affinity to the olefins. On contrary, the olefin distribution found to be very limited when using RT-based catalyst referring to stronger affinity toward re-adsorption of olefins followed by further reduction.
3.3. H2 and CO2 temperature programed desorption
During the adsorption process, it has been shown that carbon dioxide could take electrons from iron, whereas hydrogen exhibits a propensity to donate electrons to iron.64 This rationale led us to suggest that the disparities in the affinities of CO2 and H2 for the catalysts are accountable for the contrasting selectivity of hydrocarbons. To make it clear, the hydrogen chemisorption shown in Fig. 6(a) and Table S2† demonstrates that the H2 desorption curve of the RT-based catalyst illustrates a larger quantity of desorbed H2 than the ST-based catalyst. | ||
| Fig. 6 TPD of (a) H2, and (b) CO2 of carbide samples of MIL-100(Fe)-RT, and MIL-100(Fe)-ST derived catalysts. | ||
This observation offers a rationale for the previously documented low O/P ratio. It was found that the catalyst exhibited high affinity to the electron donating H2 and olefin which induces the olefin re-adsorption and further reduction to paraffin. In contrast, as illustrated in Fig. 6(b) it can be observed that the strength of the Fe–CO2 bond caused a notable increase in the CO2 adsorption on the MIL-100(Fe)-ST catalyst, particularly when the particle size is bigger. Consequently, this phenomenon facilitates the adsorption of carbon dioxide (CO2) and enhance the selectivity of olefins.66
It is noteworthy to mention that hydrocarbons characterized by a lower molecular weight range of C2–C4, consisting of olefins and paraffins, exhibit a higher proportion under conditions of elevated pressure and temperature, at 340 °C and 20 bar. Conversely, hydrocarbons with a molecular weight exceeding C5+ demonstrate an increased percentage under conditions of lower pressure and temperature, at 300 °C and 10 bar. Therefore, based on the current work, it is conceivable that by manipulating the iron particle size in the catalytic system, we could regulate the effectiveness of CO2 conversion and the features of the hydrocarbons produced during the process. The synthesis of iron particles with an average size of 3 nm resulted in the production of a hydrocarbon product that exhibited minimal presence of olefins in both its light components (C2–C4) and heavier fractions (C5+). In contrast, we achieved a notable enhancement in the proportion of olefins to paraffin (O/P), elevating it from 0.01 to 0.15, through the utilization of iron particles with an average size of 10 nm.
4. Conclusion
In conclusion, this study has provided valuable insights into the influence of iron particle size on the process of CO2 hydrogenation and the resulting products distribution. By systematically examining the effects of particle size on chemisorption behaviors and selectivity, we have uncovered critical nuances in the catalytic performance of MIL-100(Fe)-derived catalytic systems. The study demonstrates that the smaller active phase particles exhibit higher chemisorption of hydrogen, leading to increased selectivity for paraffins. Conversely, larger particles favor CO2 chemisorption, resulting in higher selectivity towards olefins. In addition, the observed trends highlight the importance of tailoring particle size to optimize surface area and, consequently, catalytic activity. These findings contribute to the ongoing efforts in designing more efficient and selective catalysts for CO2 conversion and sustainable hydrocarbon production, crucial for addressing global challenges related to carbon emissions and fuel sustainability.Data availability
Data will be made available on request.Author contributions
Hany E. Ahmed: writing original draft, investigation, and formal analysis. Mohamed K. Albolkany: writing, review & editing, visualization, conceptualization, and methodology. Mohamed E. El-Khouly: supervision. Ahmed Abd El-Moneim: writing, review & editing, methodology, visualization, project administration, funding acquisition, resources, supervision.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors express their gratitude to the Mission Sector of the Ministry of Higher Education for providing support for the first author's Ph.D. degree. The study was facilitated by the provision of laboratories, analysis facilities, and materials by the Graphene Center of Excellence at EJUST. This research was conducted under the funding of the Academy of Scientific Research and Technology (ASRT) for the project titled “Integrated Green Solar Fuel Production System: Two-Step and Direct FT Synthesis Pathways” (ID: 7825) and the project “Graphene Center for Energy and Electronic Applications GCEE” (ID: 31306) supported by Science, Technology and Innovation Funding (STDF). We would like to express our gratitude to Dr Ahmed El-Sayed Rashed for his thorough feedback on the initial version of the manuscript.References
- Carbon Dioxide, Vital Signs – Climate Change: Vital Signs of the Planet (nasa.gov), 2024, https://climate.nasa.gov/vital-signs/carbon-dioxide/ Search PubMed.
- IEA, CO2 Emissions in 2022 report, 2023, https://www.iea.org/news/global-CarbonDioxide-rebounded-to-their-highest-level-in-history-in-2021 Search PubMed.
- A. E. Rashed, M. F. Elkady, Y. Matsushita, A. Nasser and A. A. El-moneim, Chem. Eng. J., 2023, 473, 145125 CrossRef CAS.
- R. Zhao, X. Meng, W. Dai, D. Jin, B. Xu, F. Xu, D. Yang and Z. Xin, Fuel, 2023, 351, 128926 CrossRef CAS.
- Y. Zheng, M. Ma and H. Shao, Carbon Neutrality, 2023, 2, 23 CrossRef.
- Q. Zhang, L. Pastor-Pérez, Q. Wang and T. Ramirez Reina, J. Energy Chem., 2022, 66, 635–646 CrossRef CAS.
- A. Dokania, A. Ramirez, G. Shterk, J. L. Cerrillo and J. Gascon, ChemPlusChem, 2022, 87, 1–6 CrossRef PubMed.
- A. Ramirez, S. M. Sarathy and J. Gascon, Trends Chem., 2020, 2, 785–795 CrossRef CAS.
- M. Ezzat, A. E. Rashed, S. A. Sabra, M. Haroun and A. A. El-Moneim, Mater. Today Commun., 2023, 37, 107549 CrossRef CAS.
- Q. Yang, H. Lund, S. Bartling, F. Krumeich, A. S. Skrypnik and E. V. Kondratenko, J. Catal., 2023, 426, 126–139 CrossRef CAS.
- F. Ding, A. Zhang, M. Liu, Y. Zuo, K. Li, X. Guo and C. Song, Ind. Eng. Chem. Res., 2014, 53, 17563–17569 CrossRef CAS.
- H. E. Ahmed, M. Hassan, M. Nour, A. B. Shehata and M. Helmy, Mapan - J. Metrol. Soc. India, 2017, 32, 215–222 Search PubMed.
- D. S. Alqarni, M. Marshall, T. R. Gengenbach, R. Lippi and A. L. Chaffee, Microporous Mesoporous Mater., 2022, 336, 111855 CrossRef CAS.
- A. E. Rashed, K. Essam, M. F. El Kady, M. Yoshihisa, A. A. El-Moneim and A.-H. M. Nasser, Key Eng. Mater., 2021, 891, 56–61 Search PubMed.
- J. Xu, X. Su, H. Duan, B. Hou, Q. Lin, X. Liu, X. Pan, G. Pei, H. Geng, Y. Huang and T. Zhang, J. Catal., 2016, 333, 227–237 CrossRef CAS.
- S. Ma, X. Li, Z. Yang and H. Li, J. Catal., 2023, 417, 368–378 CrossRef CAS.
- A. Lotfollahzade Moghaddam and M. J. Hazlett, J. Environ. Chem. Eng., 2023, 11, 110307 CrossRef CAS.
- A. H. Nasser, L. Guo, H. Elnaggar, Y. Wang, X. Guo, A. Abdelmoneim and N. Tsubaki, RSC Adv., 2018, 8, 14854–14863 RSC.
- A. L. H. Nasser, H. El-Naggar, H. El-Bery, I. Basha and A. Abdelmoneim, RSC Adv., 2019, 9, 10937 RSC.
- H. E. Ahmed, I. F. Tahoun and S. Zakel, Egypt. J. Pet., 2021, 30, 7–10 CrossRef.
- Y. Xu, M. Wang, Z. Xie, D. Tian, G. Sheng, X. Tang, H. Li, Y. Wu, C. Song, X. Gao, S. Yao, D. Ma and L. Lin, Chem. Eng. J., 2023, 470, 144006 CrossRef CAS.
- A. E. Rashed, A. Nasser, M. F. Elkady, Y. Matsushita and A. A. El-Moneim, ACS Omega, 2022, 7, 8403–8419 CrossRef CAS PubMed.
- L. Wang, E. Guan, Y. Wang, L. Wang, Z. Gong, Y. Cui, X. Meng, B. C. Gates and F. S. Xiao, Nat. Commun., 2020, 11, 1–9 CrossRef PubMed.
- C. Wang, J. L. Zhang, X. H. Gao and T. S. Zhao, J. Fuel Chem. Technol., 2023, 51, 67–84 CrossRef CAS.
- M. H. Eldesouki, A. E. Rashed and A. A. El-Moneim, Clean Technol. Environ. Policy, 2023, 25, 3131–3148 CrossRef CAS.
- A. E. Rashed, A. Nasser, M. F. Elkady, Y. Matsushita and A. A. El-moneim, Case Stud. Chem. Environ. Eng., 2023, 7, 100300 CrossRef CAS.
- H. Yang, Y. Dang, X. Cui, X. Bu, J. Li, S. Li, Y. Sun and P. Gao, Appl. Catal., B, 2023, 321, 122050 CrossRef CAS.
- M. Gamil, O. Tabata, K. Nakamura, A. M. R. F. El-Bab and A. A. El-Moneim, Key Eng. Mater., 2014, 605, 207–210 Search PubMed.
- A. Shaker, A. H. Hassanin, N. M. Shaalan, M. A. Hassan and A. A. El-Moneim, Smart Mater. Struct., 2019, 28, 75029 CrossRef CAS.
- Y. Jiang, K. Wang, Y. Wang, X. Gao, J. Zhang, T. S. Zhao and M. Yao, Mol. Catal., 2024, 556, 113950 CrossRef CAS.
- A. Nasser, L. Guo, H. El Naggar, Y. Wang, X. Guo and A. A. El-Moneim, RSC Adv., 2018, 8(27), 14854–14863 RSC.
- C. R. Kwawu and A. Aniagyei, J. Mol. Model., 2021, 27, 200 CrossRef CAS PubMed.
- L. Dai, Y. Chen, R. Liu, X. Li, N. Ullah and Z. Li, Appl. Organomet. Chem., 2021, 35, 1–13 Search PubMed.
- A. Hamed, A. Hessein and A. A. El-moneim, Appl. Surf. Sci., 2021, 551, 149457 CrossRef CAS.
- N. Luo, W. Yang, L. Feng, S. Huang, P. Huang and M. Wei, Mol. Catal., 2024, 553, 113683 CrossRef CAS.
- H. E. Ahmed, A. E. Rashed, M. E. El-Khouly, M. K. Albolkany and A. A. El-Moneim, J. Environ. Chem. Eng., 2023, 11, 111071 CrossRef CAS.
- Y. Chen, X. Li, J. Zhang, N. Zhao, L. Dai, X. Jiang, C. Liu, S. Lyu and Z. Li, Appl. Catal., B, 2021, 289, 120027 CrossRef CAS.
- A. E. Rashed and A. A. El-Moneim, Mater. Today Energy, 2017, 3, 24–31 CrossRef.
- M. A. Belal, R. Yousry, G. Taulo, A. A. AbdelHamid, A. E. Rashed and A. A. El-Moneim, ACS Appl. Mater. Interfaces, 2023, 15, 53632–53643 CrossRef CAS PubMed.
- S. Guo, Z. Ma, Q. Wang, J. Wang, H. Guo, C. Chen, B. Hou, L. Jia and D. Li, Mol. Catal., 2024, 557, 113962 CrossRef CAS.
- R. Zhang, X. Wang, K. Wang, H. Wang, S. Song and H. Zhang, Mater. Chem. Front., 2023, 7, 6411–6426 RSC.
- K. Liu, A. Ramirez, X. Zhang, M. Çağlayan, X. Gong, J. Gascon and A. D. Chowdhury, ChemSusChem, 2023, 16, 202300608 CrossRef PubMed.
- L. Zhao, L. Zhang, Z. Wu, C. Huang, K. Chen, H. Wang and F. Yang, Catalysts, 2023, 13, 1190 CrossRef CAS.
- L. Hao, Q. Xia, Q. Zhang, J. Masa and Z. Sun, Chin. J. Catal., 2021, 42, 1903–1920 CrossRef CAS.
- N. Mohammad, J. Abdi, M. Oveisi and M. Alinia, Mater. Res. Bull., 2018, 100, 357–366 CrossRef.
- J. M. Crawford, B. E. Petel, M. J. Rasmussen, T. Ludwig, E. M. Miller, S. Halingstad, S. A. Akhade, S. H. Pang and M. M. Yung, Appl. Catal., A, 2023, 663, 119292 CrossRef CAS.
- N. L. Visser, J. C. Verschoor, L. C. J. Smulders, F. Mattarozzi, D. J. Morgan, J. D. Meeldijk, J. E. S. van der Hoeven, J. A. Stewart, B. D. Vandegehuchte and P. E. de Jongh, Catal. Today, 2023, 418, 114071 CrossRef CAS.
- T. Xie, J. Wang, F. Ding, A. Zhang, W. Li, X. Guo and C. Song, J. CO2 Util., 2017, 19, 202–208 CrossRef CAS.
- Y. Liu, Z. Li, P. Luo, N. Cui, K. Wang and W. Huang, Appl. Catal., B, 2023, 336, 122949 CrossRef CAS.
- L. Wu, R. Zou, C. Shen and C. J. Liu, Energy Fuels, 2023, 37, 18120–18127 CrossRef CAS.
- J. Liu, K. Li, Y. Song, C. Song and X. Guo, Energy Fuels, 2021, 35, 10703–10709 CrossRef CAS.
- P. Horcajada, S. Surblé, C. Serre, D. Y. Hong, Y. K. Seo, J. S. Chang, J. M. Grenèche, I. Margiolaki and G. Férey, Chem. Commun., 2007, 100, 2820–2822 RSC.
- K. M. El-Khatib, M. O. Abou Helal, A. A. El-Moneim and H. Tawfik, Anti-Corros. Methods Mater., 2004, 51, 136–142 CrossRef CAS.
- A. Gebert, A. A. El-Moneim, O. Gutfleisch and L. Schultz, IEEE Trans. Magn., 2002, 38(5), 2979–2981 CrossRef CAS.
- S. Hassan, M. Suzuki and A. A. El-Moneim, Am. J. Mater. Sci. Eng., 2012, 2(2), 11–14 CrossRef.
- M. S. El-Asfoury, M. N. A. Nasr, K. Nakamura and A. A. El-Moneim, J. Alloys Compd., 2018, 745, 331–340 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- P. M. Maitlis, R. Quyoum, H. C. Long and M. L. Turner, Appl. Catal., A, 1999, 186, 363–374 CrossRef CAS.
- K. Jeske, A. C. Kizilkaya, I. López-Luque, N. Pfänder, M. Bartsch, P. Concepción and G. Prieto, ACS Catal., 2021, 11, 4784–4798 CrossRef CAS PubMed.
- G. P. Van Der Laan and A. A. C. M. Beenackers, Catal. Rev. - Sci. Eng., 1999, 41, 255–318 CrossRef CAS.
- H. Schulz and M. Claeys, Appl. Catal., A, 1999, 186, 71–90 CrossRef CAS.
- B. Yao, T. Xiao, O. A. Makgae, X. Jie, S. Gonzalez-Cortes, S. Guan, A. I. Kirkland, J. R. Dilworth, H. A. Al-Megren, S. M. Alshihri, P. J. Dobson, G. P. Owen, J. M. Thomas and P. P. Edwards, Nat. Commun., 2020, 11, 6395 CrossRef CAS PubMed.
- B. Yuan, X. Wang, X. Zhou, J. Xiao and Z. Li, Chem. Eng. J., 2019, 355, 679–686 CrossRef CAS.
- A. Ramirez, L. Gevers, A. Bavykina, S. Ould-Chikh and J. Gascon, ACS Catal., 2018, 8, 9174–9182 CrossRef CAS.
- M. E. Dry, T. Shingles, L. J. Boshoff and G. J. Oosthuizen, J. Catal., 1969, 15, 190–199 CrossRef CAS.
- H. Gao, Z. Miao, Z. Qin, J. Yang, T. Wang, C. Gao, H. Dong and W. Hu, Adv. Mater., 2022, 34, 2108795 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01772b |
| This journal is © The Royal Society of Chemistry 2024 |