
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyvinyl alcohol as solid proton donor to modify g-C3N4 via hydrogen bonding enabling efficient photocatalytic H2O2 production from H2O and O2†
Chen Chen‡
a,
Fengtiao Liao‡b,
Xiangcheng Zhangc,
Silian Chengc,
Yu Denga,
Chao Chen *b and
Mingce Longc
*b and
Mingce Longc
aCCCC Shanghai Dredging Co., Ltd, No. 850, Pudong Avenue, Pudong New Area, Shanghai, 200120, China
bShanghai Engineering Research Center of Biotransformation of Organic Solid Waste, Shanghai Key Lab for Urban Ecological Processes and Eco-Restoration, School of Ecological and Environmental Sciences, East China Normal University, Shanghai, 200241, China. E-mail: cchen@des.ecnu.edu.cn
cSchool of Environmental Science and Engineering, Key Laboratory of Thin Film and Microfabrication Technology (Ministry of Education), Shanghai Jiao Tong University, Shanghai, 200240, China
First published on 17th April 2024
Abstract
Polyvinyl alcohol (PVA) was used as a solid proton donor to improve the photocatalytic performance of graphitic carbon nitride (CN) for hydrogen peroxide (H2O2) production. The modified CN (CN/PVA) was prepared by mixing CN and PVA at room temperature. The H2O2 production efficiency of CN/PVA was 5.65 times higher than that of CN in pure water. Photocurrent measurement, electrochemical impedance spectroscopy (EIS), and photoluminescence (PL) analysis proved that PVA increased charge separation of CN. X-ray photoelectron spectroscopy (XPS), and Fourier-transform infrared (FTIR) analyses further suggested that PVA acted as the proton donor during H2O2 production by interacting with CN via hydrogen bonds. The combination of the charge separation enhancer and proton donor from PVA promoted the sequential two-step single-electron reduction of O2 for H2O2 production. This study paves the way for the modification of g-C3N4 with hydroxyl-containing materials as solid proton donors for photocatalytic H2O2 production.
Introduction
Hydrogen peroxide (H2O2) is a mild and environmentally friendly oxidant, and widely used in organic synthesis, wastewater treatment and disinfection.1–6 Currently, over 95% of the total H2O2 is industrially produced by the anthraquinone (AQ) oxidation method, which is complicated and suffers from high energy consumption and pollution emissions.7–10 Alternatively, photocatalytic H2O2 production has gained extensive attention due to its inherent advantages of low energy consumption and zero pollution discharge.11–13 Graphitic carbon nitride (g-C3N4) stands out as a common yet promising polymer catalyst for the photocatalytic production of H2O2.14–21 However, due to the significant recombination of photogenerated charges, proton donors such as propanol alcohols are required to promote oxygen reduction and H2O2 production.22 It is remains challenge to modify g-C3N4 for the efficiently promoted separation of photogenerated charge carriers and photocatalytic H2O2 production.23–27Modifying g-C3N4 with a solid proton donor through non-covalent bonds under ambient conditions proves to be an effective strategy for enhancing the separation of photogenerated charge carriers, thus realizing H2O2 production efficiently in the absence of organic sacrificial reagents. The modification of g-C3N4 using a hydroxyl-containing polymer, hydroxyethyl cellulose (HEC), as the solid proton donor were previously developed.28 The HEC modified g-C3N4 via hydrogen bonds between hydroxyl groups of HEC and g-C3N4 facilitated the separation of charge carriers by scavenging photogenerated holes, consequently boosting the photocatalytic H2O2 production.
PVA is a semi-crystalline polymer with a molecular formula of [CH2CH(OH)]n, and the molar ratio of hydroxyl groups to carbon atoms in it is 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, much higher than that in HEC. The abundant hydroxyl groups make PVA promising in modifying g-C3N4 via hydrogen bonds to boost H2O2 generation. In addition, PVA possesses several advantages like biodegradability, non-toxicity, adhesiveness and non-carcinogenicity.29 However, in the field of photocatalysis, PVA was mainly used to facilitate the formation of the photocatalyst films by combining with powder photocatalysts like TiO2 (ref. 30,31) and g-C3N4 (ref. 32) ZnS,33 and to promote the dispersion of nanoparticles in a polymer matrix34 for antibacterial applications.35 PVA was also investigated as the electron donor to promote the superoxide anion radical generation during TiO2 photocatalysis.36,37 To the best of our knowledge, there remains no report on the solid proton donor function of PVA in boosting photocatalytic performance in H2O2 production.
:1, much higher than that in HEC. The abundant hydroxyl groups make PVA promising in modifying g-C3N4 via hydrogen bonds to boost H2O2 generation. In addition, PVA possesses several advantages like biodegradability, non-toxicity, adhesiveness and non-carcinogenicity.29 However, in the field of photocatalysis, PVA was mainly used to facilitate the formation of the photocatalyst films by combining with powder photocatalysts like TiO2 (ref. 30,31) and g-C3N4 (ref. 32) ZnS,33 and to promote the dispersion of nanoparticles in a polymer matrix34 for antibacterial applications.35 PVA was also investigated as the electron donor to promote the superoxide anion radical generation during TiO2 photocatalysis.36,37 To the best of our knowledge, there remains no report on the solid proton donor function of PVA in boosting photocatalytic performance in H2O2 production.
Herein, we proposed to modify g-C3N4 with PVA(CN/PVA) under mild conditions via hydrogen bonds. The enhancing effect of PVA modification on H2O2 production of g-C3N4 has been experimentally demonstrated. The mechanism on solid proton donor and charge separation enhancer functions of PVA have been clarified.
Experimental
Materials
Urea and Polyethylene glycol (PEG) were purchased from Adamas (China). Polyvinyl alcohol (PVA) was purchased from Macklin (China). Hydroxyethyl Cellulose was purchased from Sigma-Aldrich (China). All the materials were utilized without further purification. Deionization water was used in all experiments.Synthetic procedures
Characterizations
Scanning electron microscopy (SEM) images were obtained by a Hitachi SU8020 electron microscope. Transmission electron microscopy (TEM) images were acquired on a FEI Talos F200X electron microscope operated at 200 kV. The crystalline structure of the samples was investigated by powder X-ray diffraction (SmartlabSE) using Cu Kα (λ = 0.15406 nm) radiation with a Nickel filter operating at 40 kV and 40 mA in the 2θ range of 5–80°. Fourier transform infrared (FTIR) spectra were obtained on a Thermo Scientific Nicolet IS5 spectrometer with the attenuated total reflectance (ATR) method and were recorded over the range 4000–400 cm−1 at 4 cm−1 resolutions and averaged over 16 scans per sample. X-ray photoelectron spectroscopy (XPS) measurements were carried out on an AXIS SUPRA XPS spectrometer. Thermal gravimetric analysis (TGA) was carried out by a SDTA851e. The samples were heated from 30 to 800 °C at the rate of 10 °C min−1 under flowing air. The steady-state and transient-state photoluminescence spectra were obtained on an Edinburgh FLS1000 instrument with an excitation wavelength at 365 nm.Photocatalytic H2O2 production
A cylindrical container (50 mL) equipped with a water circulating cooling system was used as reactor. In a typical photoreaction, 30 mg of catalyst was suspended in 30 mL ultrapure water, then purged with O2 for 10 min under dark condition (maintain oxygen flow and stirring during the reaction process). Then, a PLS-SXE300D xenon lamp with a UV-cutoff filter (λ ≥ 420 nm) was used as the light source. The light intensity on the surface of 30 mL liquid is 1.333 W. During the photoreaction, 2 mL of the suspension was withdrawn and filtrated with 0.45 μm Millipore filter at time intervals of every 10 min. Finally, the concentration of H2O2 was determined by N,N-diethylp-phenylenediamine–horseradish peroxidase (DPD–POD) colorimetry method.38 The mixture was detected by a Q/PG PXT0011-2012 UV-vis spectrophotometer at the peak of 551 nm.Apparent quantum yield (AQY)
The apparent quantum yield (AQY) is estimated by measuring the photocatalytic H2O2 production of CN/PVA0.05 under different monochromatic light irradiations. The incident monochromatic light intensities at 400, 420, 550, and 650 nm are 24, 44, 85, and 74 W m−2, respectively. The irradiation area was calculated to be 7.6 cm2. The AQY (%) is calculated by the equation as follows:39
 | (1) |
The number of incident photons (M) is calculated by the formula of Eλ/hc. Wherein, h and c are the average intensity of irradiation, the wavelength of the irradiation, Planck constant and the speed of light, respectively.
Photoelectrochemical measurements
The photoelectrochemical measurements were conducted using an electrochemical workstation (ZAHNER CIMPS) in a standard three-electrode system. A Pt wire served as the counter electrode, while Ag/AgCl (saturated KCl) was used as the reference electrode. The effective area of the working electrode with photocatalysts was 1.0 × 1.0 cm2. For transient photocurrent measurements, a 300 W xenon lamp (PLS-SXE300/300UV, Perfect Light) with a 420 nm cut-off filter was used as the light source, and the voltage was set at 1.0 V. Electrochemical impedance spectroscopy (EIS) was conducted over a frequency range of 102–105 Hz with a bias voltage of −0.4 V. The electrolyte used was a 0.05 M Na2SO4 aqueous solution. The equivalent circuit diagram simulation of is provided in Fig. S6.†Rotating ring-disk electrode (RRDE) measurements were performed on a Chenhua CHI 760E electrochemical workstation. The working electrode was prepared by coating 10 μL catalyst ink on glassy carbon electrodes and dried at room temperature. The catalyst ink was prepared by dispersing 5 mg photocatalyst in 1 mL ethanol and 50 μL Nafion (5%) under ultrasonic treatment. The linear sweep voltammetry (LSV) on the RRDE was conducted in O2 saturated Na2SO4 electrolyte (0.1 M, pH = 7) with a scan rate of 0.01 V s−1 at rotating speeds from 0 to 1600 rpm. The selectivity of H2O2 and electron transfer number (n) are calculated based on the following equations:40.
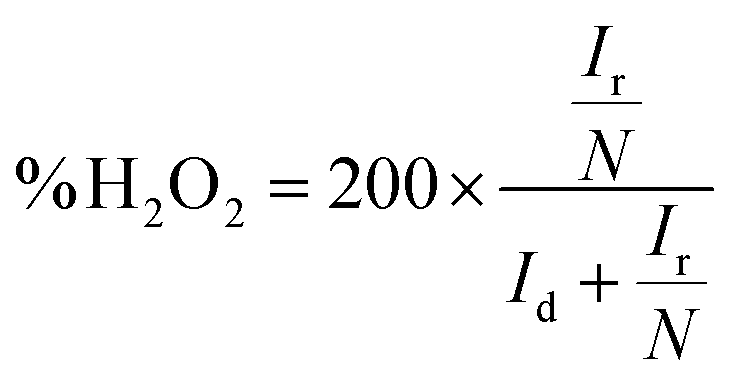 | (2) |
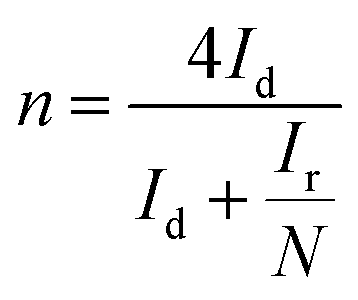 | (3) |
Results and discussion
Characterizations of CN/PVA
XRD patterns of PVA and CN were collected (Fig. 1a). The XRD pattern for PVA shows three peaks of 40.71°, 19.48° and 22.72°, corresponding to the (2,2,0), (1,0,1), and (1,0,−1) planes of semi-crystalline PVA, respectively.41 The XRD pattern for CN shows the peaks at 27.64° and 13.19°, ascribed to the stacked conjugated aromatic system and the tri-s-triazine structure of CN, respectively. These XRD peaks for CN exhibit a slight shift towards lower values when incorporating with PVA, suggests that there is a slight increase in the interplanar stacking distance between the aromatic units within the composite material.28 To further prove that the PVA was successfully combined with CN in CN/PVA, thermogravimetric analysis (TGA) was carried out. TGA results (Fig. 1b) suggest that PVA undergoes weight loss of around 94% in the range of 200–480 °C. For CN, a noticeable weight loss starts at approximately 470 °C. However, for CN/PVA, the weight loss begins in a temperature range of around 200 to 480 °C. This discrepancy in the weight loss patterns strongly suggests that PVA is indeed present in the CN/PVA composite. Derivative thermogravimetry (DTG) (inset in Fig. 1b) curve indicates that as the mass ratio of PVA in CN/PVA increases, there is an enhanced peak intensity at around 330 °C, further proving the presence of PVA in the CN/PVA. SEM images of CN and CN/PVA0.05 reveal a striking similarity in their nanosheet morphology (Fig. 1c and d), suggesting that the introduction of PVA into CN does not significantly alter the overall nanosheet structure, and PVA is homogenously dispersed over CN surface. Furthermore, TEM images provide additional insights into the structural changes (Fig. 1e and f). These images indicate the slightly expanded interplanar stacking distance of the aromatic units in CN/PVA0.05 in comparison to CN, as the interplanar spacing increases from 0.313 nm in CN to 0.318 nm in CN/PVA0.05, which is consistent with the results of XRD analysis.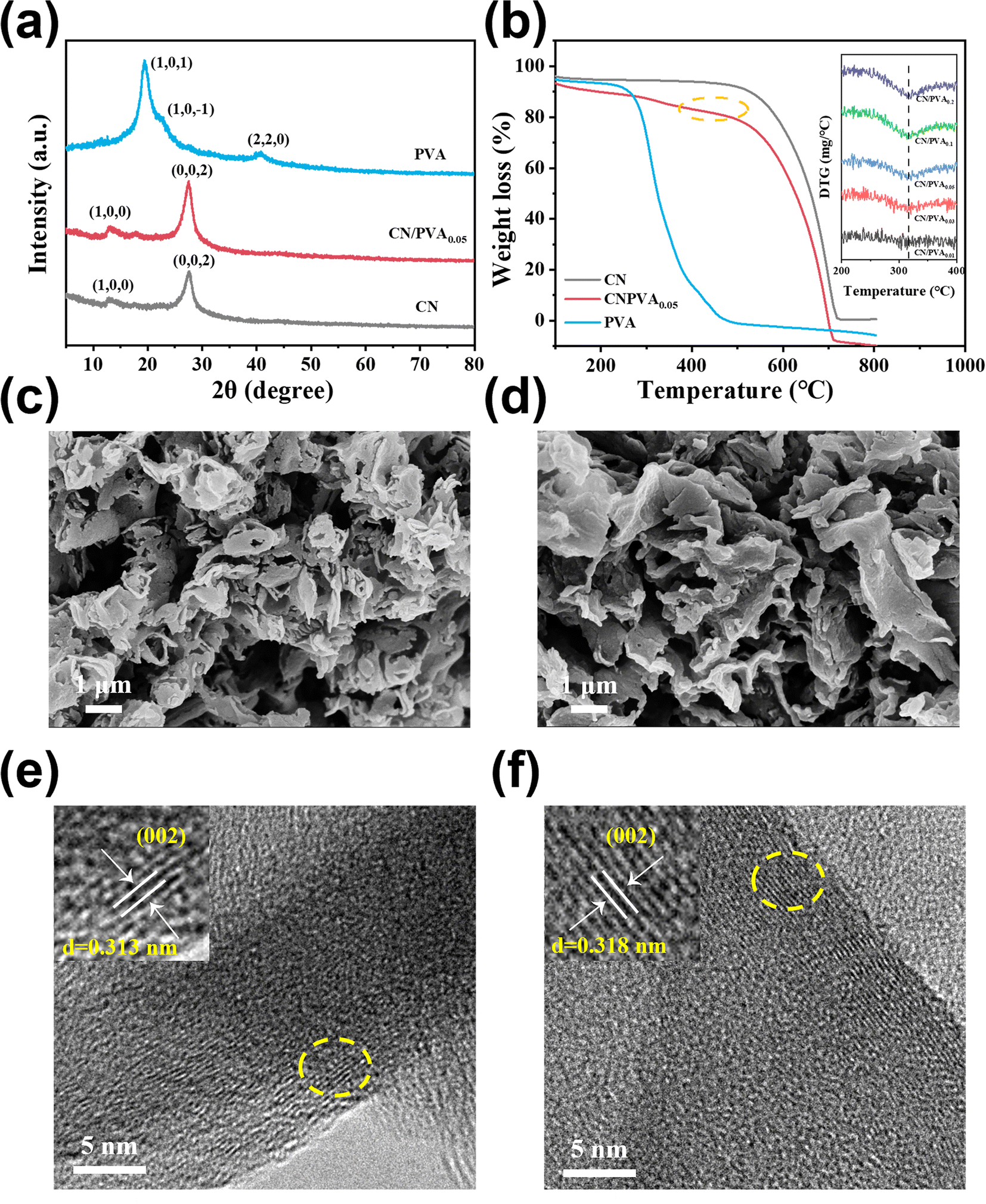 | ||
| Fig. 1 (a and b) XRD patterns and TGA curves of PVA and CN/PVA0.05, the inset of (b) shows the amplified DTG curves of CN/PVA0.01, CN/PVA0.03, CN/PVA0.05, CN/PVA0.1, and CN/PVA0.2 in the temperature range of 100–400 °C; (c and e) SEM and TEM images of CN; (d and f) SEM and TEM images of CN/PVA0.05; the insets of (e) and (f) are the HRTEM of CN and CN/PVA0.05. | ||
FTIR spectra were used to investigate the surface functional groups variation of CN after PVA modification (Fig. 2a). For PVA, a broad band observed in the range of 3030–3570 cm−1 is ascribed to the stretching vibration of ν(O–H), which is attributed to the intermolecular and intramolecular hydrogen bonding of PVA. The bands in the range of 2840–2970 cm−1 are related to the C–H stretching vibration of the alkyl group, while the bands at the fingerprint region (500–900 cm−1) and 1425 cm−1 are ascribed to C–H bending vibrations.42 The bands in the range of 1660–1750 cm−1 correspond to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O.43 The band at 1140 cm−1 is ascribed to the stretching vibration of C–O, and the band at 1087 cm−1 refers to the stretching vibration of C–O–C.44 For CN, bands in the range of 1200–1650 cm−1 is ascribed to aromatic C–N heterocycles, and the band at 809 cm−1 corresponds to triazine ring mode.45 The broad band at the range of 3000–3400 cm−1 corresponds to stretching vibration of terminal N–H groups.46 Differently, the FTIR spectrum of CN/PVA0.05 shows all the characteristic bands of CN without new band formation and band disappearance, which might be due to the low content of PVA in CN/PVA0.05. The absence of new band indicates that the interaction between CN and PVA should not be via covalent bond,28,47 but rather through the weak interactions like hydrogen bonds.
O.43 The band at 1140 cm−1 is ascribed to the stretching vibration of C–O, and the band at 1087 cm−1 refers to the stretching vibration of C–O–C.44 For CN, bands in the range of 1200–1650 cm−1 is ascribed to aromatic C–N heterocycles, and the band at 809 cm−1 corresponds to triazine ring mode.45 The broad band at the range of 3000–3400 cm−1 corresponds to stretching vibration of terminal N–H groups.46 Differently, the FTIR spectrum of CN/PVA0.05 shows all the characteristic bands of CN without new band formation and band disappearance, which might be due to the low content of PVA in CN/PVA0.05. The absence of new band indicates that the interaction between CN and PVA should not be via covalent bond,28,47 but rather through the weak interactions like hydrogen bonds.
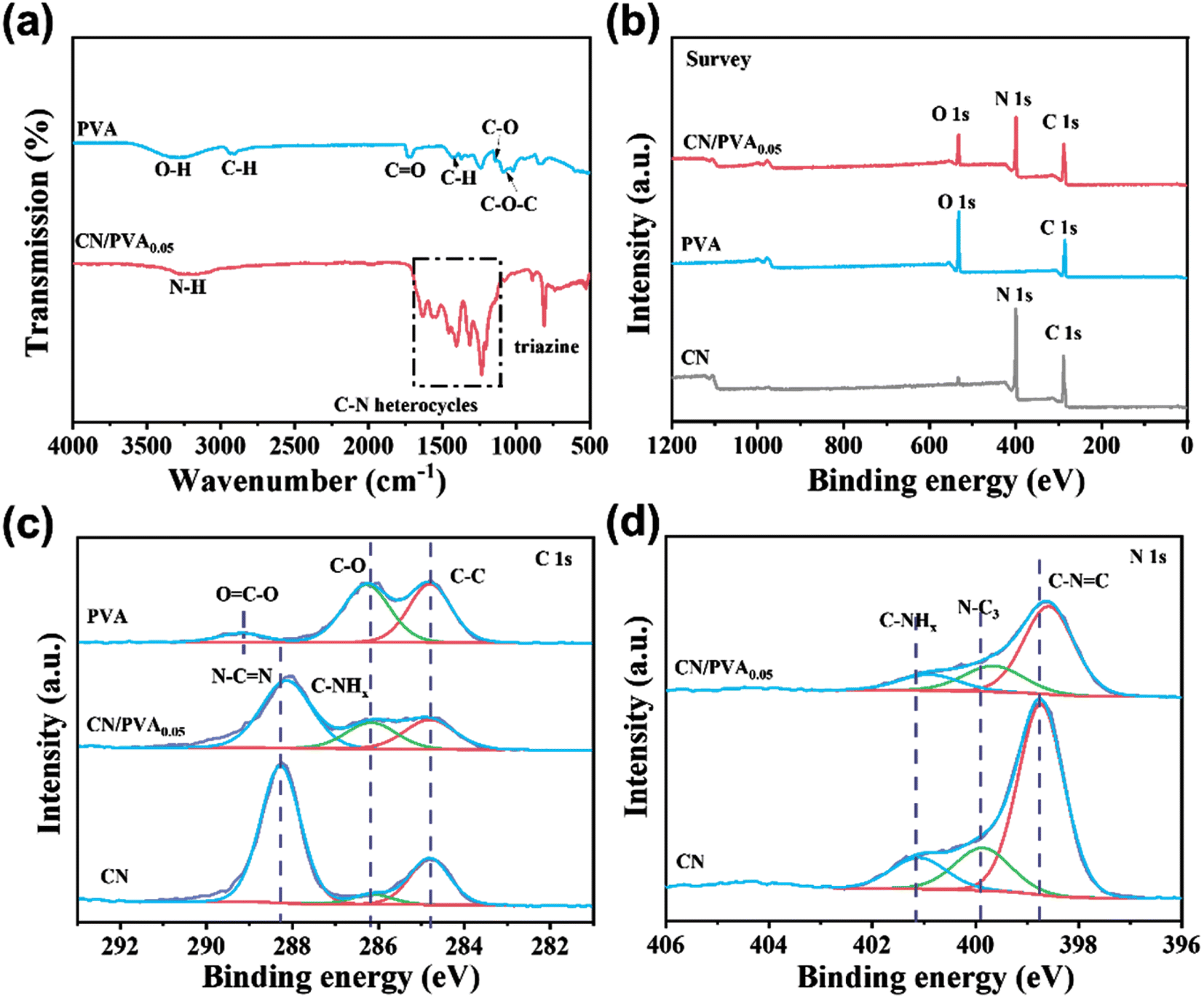 | ||
| Fig. 2 (a) FTIR spectra; (b) XPS survey spectra; (c and d) high-resolution C 1s and N 1s XPS spectra of PVA, CN and CN/PVA0.05. | ||
The effect of PVA modification on the chemical state of CN was further analyzed by XPS spectra. The survey spectrum of CN/PVA0.05 revealed the presence of C, N, and O elements. In contrast, PVA showed an absence of N, and CN exhibited minimal O content. This suggests that PVA has been successfully modified onto the CN surface (Fig. 2b). High-resolution C 1s XPS spectrum of CN shows three peaks at the binding energies of 284.8, 286.2 and 288.1 eV (Fig. 2c), which are ascribed to the C–C, C–NHx and NC–N groups, respectively.28,48 For PVA, the C 1s XPS spectrum also shows three peaks at binding energies of 284.8 eV, 286.2 eV, and 289.1 eV, corresponding to –CH2–CH2–, –C–O–, and OC–O groups, respectively.49 As modified CN with PVA, the C 1s peak for NC–N shifts around 0.2 eV to lower binding energy, and the N 1s peaks for C–NC, N–C3, and C–NHx groups shift in the range of 0.2–0.3 eV to lower binding energy. Moreover, three peaks at binding energies of 398.7 eV (C–NC), 400.0 eV (N–C3), and 401.1 eV (C–NHx) also can be observed in the high resolution N 1s XPS spectrum (Fig. 2d).28,50 Compared with CN, the binding energies of the three peaks both have obvious shifts toward lower binding energy after PVA modification. These results suggest that the PVA modification increases the electron density of C and N elements in g-C3N4, suggesting that non-covalent bond interaction between g-C3N4 and PVA. Thus, all above analyses demonstrates that PVA reacts with g-C3N4 via hydrogen bonds.28
Photocatalytic H2O2 production
The photocatalytic production of H2O2 over CN/PVAx, CN, and PVA was assessed. In Fig. 3a, PVA alone resulted in negligible H2O2 production after 60 min reaction. Similarly, when CN was used as the catalyst, the H2O2 production (16.87 μmol L−1) was also comparatively lower. Interestingly, the photocatalytic activity is significantly enhanced after PVA modification. As the mass ratio of PVA to CN increased from 0.01 to 0.03, the concentration of produced H2O2 under visible light increased significantly, reaching 87.19 μmol L−1, which was about 5.17 times that of CN under the same conditions. It exhibits promising performance compared to other g-C3N4-based catalysts (Fig. S3†). When the mass ratio further increased from 0.03 to 0.2, H2O2 production did not increase obviously. The possible reason for this phenomenon is that PVA and CN are connected by hydrogen bonds. When the ratio reaches 0.03, the hydrogen bonds are saturated, leading to the accumulation of PVA on the surface and preventing further formation of hydrogen bonds with CN.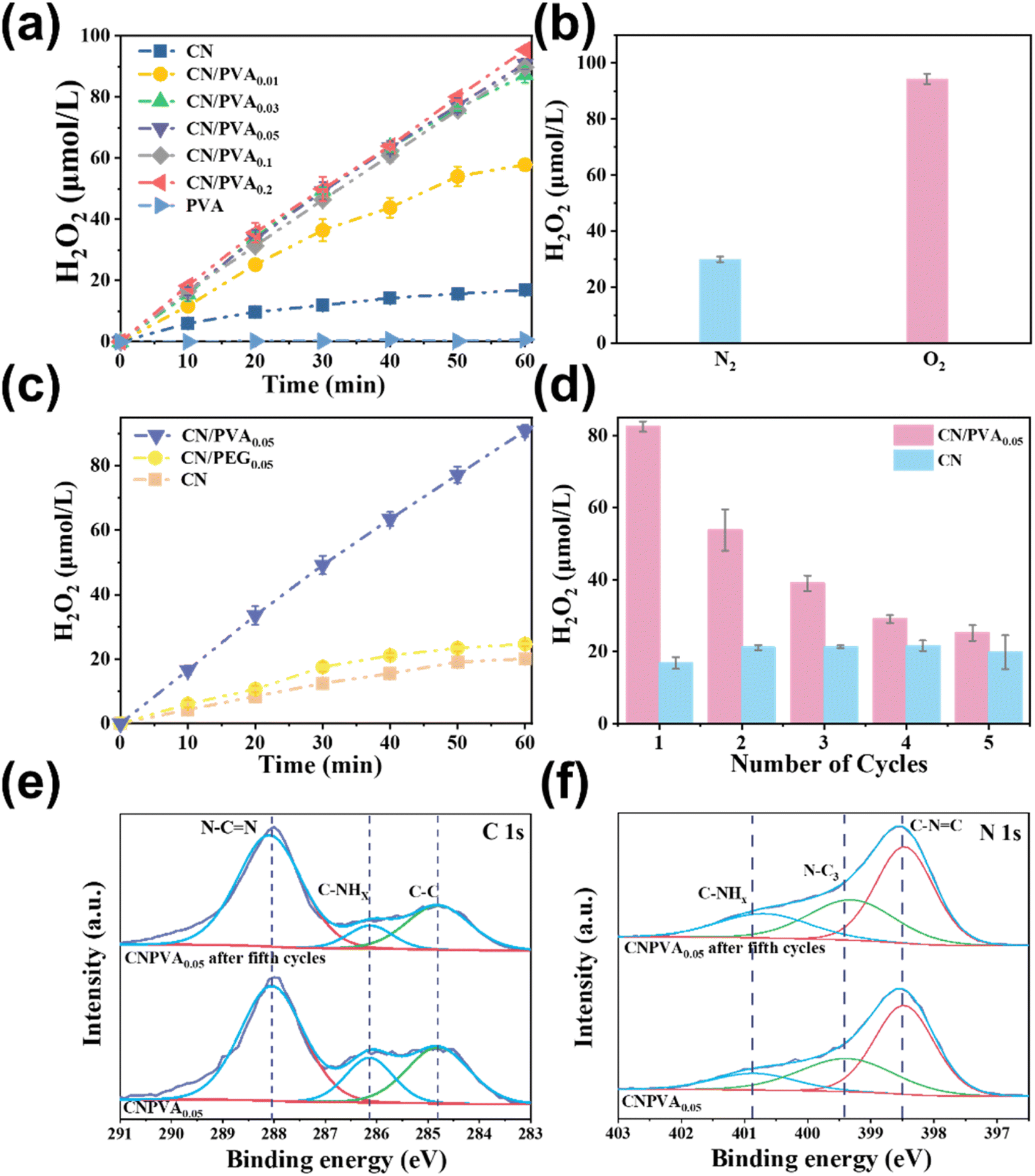 | ||
| Fig. 3 (a) Photocatalytic H2O2 production of CN/PVAx, CN and PVA in pure water and O2 under visible light irradiation (λ ≥ 420 nm); (b) photocatalytic H2O2 production over CN/PVA0.05 under different atmosphere; (c) photocatalytic H2O2 production over CN, CN/PVA0.05, and CN/PEG0.05, respectively; (d) recyclability study of CN and CN/PVA0.05 for H2O2 production from an oxygen/water solution; (e and f) high-resolution C 1s and N 1s XPS spectra of CN/PVA0.05 and CN/PVA0.05 after 5 cycles. | ||
The selectivity of H2O2 in O2-saturated electrolytes was studied using a rotating ring-disk electrode (RRDE). As shown in Fig. S4b,† the RRDE polarization curves of the CN/PVA catalyst at different rotation speeds were investigated. It can be clearly observed that the disk current (Id) becomes stronger with the increase in rotation speed. At the same time, due to the rapid diffusion of H2O2 generated on the disk to the ring part, the ring current (Ir) exhibits enhancement. By calculating the reduction current of O2 and the oxidation current of H2O2 at a rotation speed of 1600 rpm, the selectivity of the CN/PVA catalyst for producing H2O2 can reach 94.79%, which is much higher than the selectivity of CN (55.37%).
We then investigate the source of oxygen for H2O2 production by various control experiments. In comparison to oxygen atmosphere, the N2 purging system resulted in significantly inhibited H2O2 production (29.93 μmol L−1) (Fig. 3b), indicating the dominated contribution of ORR to H2O2 production. To verify if solid sacrificial reagents with abundant hydroxyl groups are favorable for promoting photocatalytic H2O2 production, another solid sacrificial reagent, PEG, with lower abundant hydroxyl groups, in which the ratio of hydroxyl to polymer chains is 2:1, are used as the references (Fig. 3c). CN/PEG0.05 is prepared as the same as CN/PVA0.05 except that PVA is replaced with PEG. The H2O2 production was in the following order: CN/PVA0.05 > CN/PEG0.05, which is in consistent with the abundance of hydroxyl groups in PVA and PEG. Notably, in comparison to pristine CN, the increase in H2O2 production is negligible for CN/PEG0.05 (24.69 μmol L−1). However, CN/PVA0.05 exhibits a significant increase in H2O2 production with a concentration approximately 4.01 times higher than CN/PEG0.05. These findings suggest that a higher hydroxyl content in the solid sacrificial regent strengthens the hydrogen bonding interaction with CN, thereby enhancing the photocatalytic H2O2 production.
To investigate the stability of the composite photocatalyst, cyclic experiments were conducted on CN and CN/PVA0.05 (Fig. 3d). After five cycles of testing, the performance of photocatalytic H2O2 production over CN remained constant, while the performance of CN/PVA0.05 gradually decreased. This phenomenon suggests that the modified sacrificial reagent of PVA is consumed during the photocatalytic H2O2 production. High-resolution of XPS spectra were further used to explore the PVA consumption in the H2O2 production. In the C 1s XPS spectra, after the fifth cycle, the N–CN peak of CN/PVA0.05 has shifted towards a higher binding energy by 0.02 eV (Fig. 3e). Similarly, the C–NHx, N–C3, and C–NC peaks in the N 1s spectra also have shifted towards higher binding energy by approximately 0.1 eV (Fig. 3f). These results suggest that the electron density of C and N in CN/PVA0.05 decreased, which may be due to the cleavage of hydrogen bonds between CN and PVA, resulting in PVA consumption during the photocatalytic H2O2 production. However, the performance of used CN/PVA can be easily restored by one more PVA modification.
After a long-term irradiation of 5 h, the H2O2 production on CN/PVA steadily increased (Fig. S4†), indicating that the catalyst has relatively stable performance. We then tested the total organic carbon (TOC) in the system before and after the reaction. Through TOC testing, we found that the TOC in the system after the reaction significantly increased (16.30 mg L−1), much higher than before the reaction (594.9 μg L−1). This may be due to the oxidation and transformation of PVA into small molecules, which promotes the consumption of holes and accelerates the separation of photogenerated charges.51 The AQY is measured to evaluate the conversion efficiency of photons during the generation of H2O2 on CN/PVA under monochromatic light irradiation (Fig. S5†). The AQY value is almost correlated with the light absorption of the catalyst. At 400 nm, the value reaches 2.06%.
Mechanism on the enhanced photocatalytic H2O2 production
Photocurrent and electrochemical impedance spectroscopy (EIS) were conducted to explore the photogenerated charge separation behaviors of CN and CN/PVA0.05. As shown in Fig. 4a, the photocurrent of CN/PVA0.05 is higher than that of CN in both N2 and O2 atmospheres. The photocurrent density in O2 atmosphere is significantly higher than that in N2 atmosphere, indicating that PVA modification of CN promotes the charge separation of the photocatalyst, and such promotion is more pronounced under O2 atmosphere. Furthermore, the EIS curves show that the arc radius of CN and CN/PVA0.05 is similar under N2 atmosphere, both larger than that under O2 atmosphere (Fig. 4b). Significantly, the smaller arc radius of CN/PVA0.05 under O2 atmosphere indicates the O2 enhances the charge carrier separation of CN, and the modification with PVA further enhances this effect. The fluorescence spectroscopy (PL) was further used to investigate the behavior of charge separation and transfer. Compared with CN, the weaker steady-state fluorescence intensity of CN/PVA0.05 indicates the high-efficient separation of photogenerated charge carrier (Fig. 4c). In addition, time-resolved fluorescence analysis shows shorter relaxation time of the electrons of CN/PVA0.05 (3.49 ns) than that of CN (3.97 ns) (Fig. 4d), suggesting the ORR is facilitated on the CN/PVA0.05. The above analysis demonstrated that the PVA modification enhances the charge separation efficiency of CN.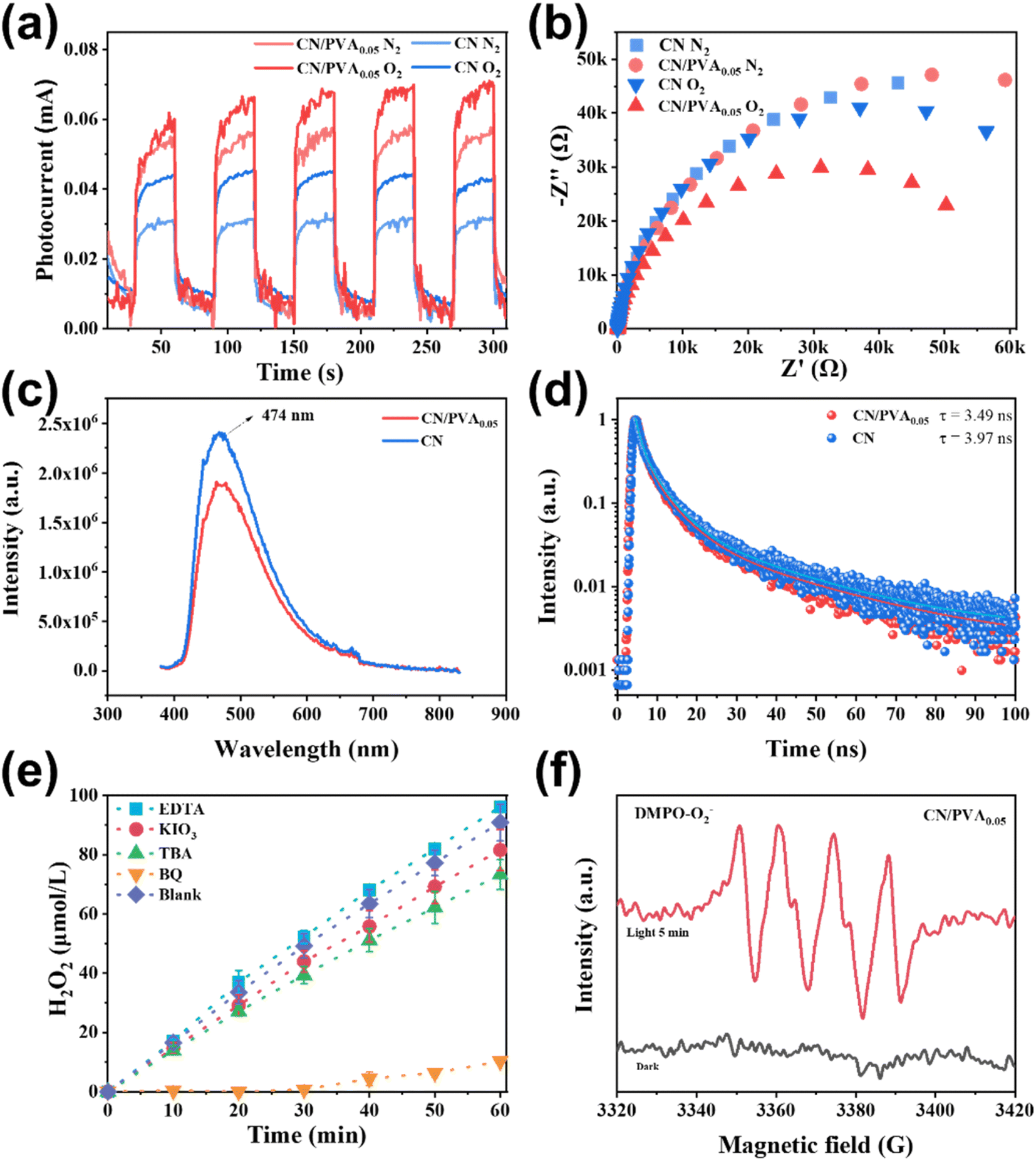 | ||
| Fig. 4 (a) The transient photocurrents and (b) electrochemical impedance spectroscopy Nyquist plots of CN and CN/PVA0.05 under visible light irradiation in the atmosphere of N2 and O2; (c) steady-state PL emission spectra (excitation at 365 nm) and (d) time-resolved PL spectra of CN and CNPVA0.05; (e) effects of different scavengers on the photocatalytic activity of CN/PVA0.05; (f) EPR spectra in CN/PVA0.05 suspension under dark and light irradiation. | ||
The contribution of active species on H2O2 production was analyzed by using active species trapping experiments. As shown in Fig. 4e, compared with the pure water system, the H2O2 production over CN/PVA0.05 in the EDTA solution slightly increased, and slightly decreased in the KIO3 and TBA solutions, indicating that e− and ˙OH partially participate in the photocatalytic H2O2 production. Notably, the H2O2 production in BQ solution was significantly decreased, demonstrating that ·O2− is the main active species involved in the photocatalytic H2O2 production. The EPR spectrum also shows strong characteristic signals of DMPO-˙O2− in CN/PVA0.05 after 5 minutes of visible light irradiation (Fig. 4f). The electron transfer number (n) for the oxygen reduction reaction (ORR) was measured for CN and CN/PVA using RRDE. The electron transfer numbers (n) for CN and CN/PVA are approximately 2.40 and 2.07, respectively (Fig. S8†). These results indicate that the H2O2 generation pathway for both CN and CN/PVA is a two-electron reaction. The lower electron transfer value for CN/PVA suggests that its electron transfer characteristics are more inclined towards the two-electron transfer pathway (n = 2), which promotes the efficient generation of H2O2. As shown in Fig. 5, under visible light irradiation, photogenerated e− in CN/PVA reacts with O2 forming ˙O2− radicals, while PVA acts as H+ donor, facilitating the proton coupled electron transfer reaction involved in the two-step single-electron oxygen reduction process.
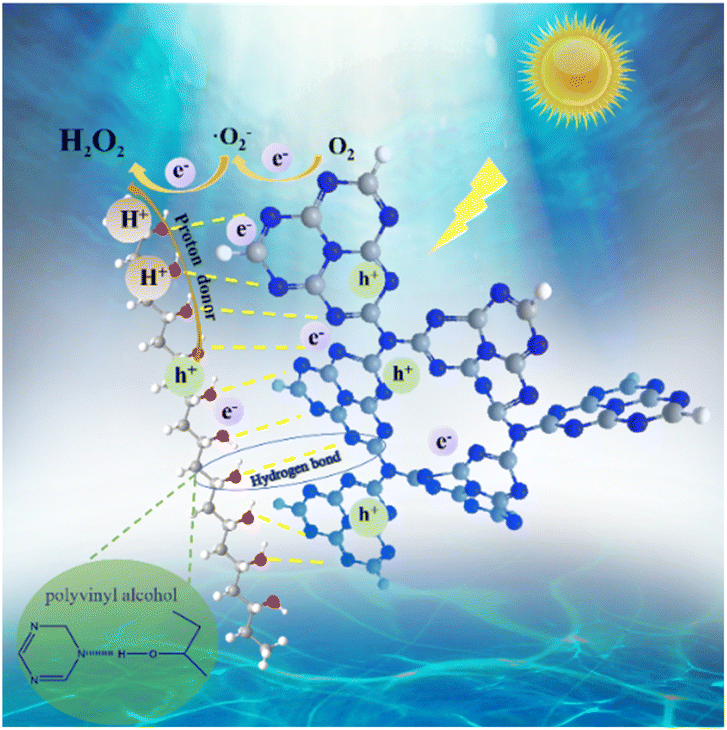 | ||
| Fig. 5 The mechanism of photocatalytic H2O2 production over CN/PVA. | ||
Conclusions
PVA with abundant hydroxyl groups was utilized to modify g-C3N4 to form an efficient photocatalyst composite CN/PVA for H2O2 production from only H2O and O2. Structural analyses revealed that PVA interacts with CN via hydrogen bonds. This interaction leads to the transfer of charges from PVA to CN enhancing charge density of the C and N elements in CN. Under visible light irradiation, the optimum catalyst, CN/PVA0.05, exhibited 5.65 times higher H2O2 production than CN. The mechanism underlying H2O2 production involves a two-step single-electron ORR process with ·O2− as the primary active species. In the H2O2 production process, PVA acts as both charge separation enhancer and solid proton donor. This study paves the way on enhancing H2O2 production performance of CN-based photocatalysts via modification with hydroxyl group-rich materials as solid proton donors.Author contributions
Chen Chen: conceptualization, methodology, investigation, writing – original draft. Fengtiao Liao: conceptualization, methodology, investigation, writing – original draft. Xiangcheng Zhang: writing – original draft, methodology, validation. Silian Cheng: formal analysis, methodology. Yu Deng: conceptualization, methodology, investigation. Chao Chen: supervision, funding acquisition, conceptualization, writing – review & editing. Mingce Long: supervision, funding acquisition, resources, writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work is supported by National Natural Science Foundation of China (No. 22105073, 52070128 and 22206125).Notes and references
- H. Hou, X. Zeng and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS PubMed.
- K. Mase, M. Yoneda, Y. Yamada and S. Fukuzumi, Nat. Commun., 2016, 7, 11470 CrossRef CAS PubMed.
- Y. Zhang, C. Pan, G. Bian, J. Xu, Y. Dong, Y. Zhang, Y. Lou, W. Liu and Y. Zhu, Nat. Energy, 2023, 8, 361–371 CrossRef CAS.
- Y. Luo, B. P. Zhang, C. C. Liu, D. H. Xia, X. W. Ou, Y. P. Cai, Y. Zhou, J. Jiang and B. Han, Angew. Chem., Int. Ed., 2023, 62, e202305355 CrossRef CAS PubMed.
- H. Li, B. Zhu, B. Cheng, G. Luo, J. Xu and S. Cao, J. Mater. Sci. Technol., 2023, 161, 192–200 CrossRef CAS.
- Y. Wu, Y. Yang, M. Gu, C. Bie, H. Tan, B. Cheng and J. Xu, Chin. J. Catal., 2023, 53, 123–133 CrossRef CAS.
- L. Xu, L. Li, Z. Hu and J. C. Yu, Appl. Catal., B, 2023, 328, 122490 CrossRef CAS.
- Q. Liao, Q. Sun, H. Xu, Y. Wang, Y. Xu, Z. Li, J. Hu, D. Wang, H. Li and K. Xi, Angew. Chem., Int. Ed., 2023, 62, e202310556 CrossRef CAS PubMed.
- J. N. Chang, J. W. Shi, Q. Li, S. Li, Y. R. Wang, Y. F. Chen, F. Yu, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2023, 62, e202303606 CrossRef PubMed.
- Z. Liao, J. Du, L. Wang, M. Yang, L. Zhao, X. Zhan, H. Yang, W. Yang and H. Hou, J. Alloys Compd., 2024, 976, 173322 CrossRef CAS.
- G. Liao, Y. Gong, L. Zhang, H. Gao, G.-J. Yang and B. Fang, Energy Environ. Sci., 2019, 12, 2080–2147 RSC.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- W. Yu, C. Hu, L. Bai, N. Tian, Y. Zhang and H. Huang, Nano Energy, 2022, 104, 107906 CrossRef CAS.
- L. L. Liu, F. Chen, J. H. Wu, J. J. Chen and H. Q. Yu, Proc. Natl. Acad. Sci. U.S.A., 2023, 120, e2215305120 CrossRef CAS PubMed.
- M. Gu, D. Lee, J. Mun, D. Kim, H. I. Cho, B. Kim, W. Kim, G. Lee, B. S. Kim and H. I. Kim, Appl. Catal., B, 2022, 312, 121379 CrossRef CAS.
- S. Yuan, M. Yang, X. Lan and J. Shi, Appl. Catal., A, 2022, 643, 118782 CrossRef CAS.
- Z. Wei, M. Liu, Z. Zhang, W. Yao, H. Tan and Y. Zhu, Energy Environ. Sci., 2018, 11, 2581–2589 RSC.
- Y. J. Lee, Y. J. Jeong, I. S. Cho, S. J. Park, C. G. Lee and P. J. J. Alvarez, J. Hazard. Mater., 2023, 449, 131046 CrossRef CAS PubMed.
- T. Wu, Q. He, Z. Liu, B. Shao, Q. Liang, Y. Pan, J. Huang, Z. Peng, Y. Liu, C. Zhao, X. Yuan, L. Tang and S. Gong, J. Hazard. Mater., 2022, 424, 127177 CrossRef CAS PubMed.
- X. Zhan, Z. Fang, B. Li, H. Zhang, L. Xu, H. Hou and W. Yang, J. Mater. Chem. A, 2021, 9, 27084–27094 RSC.
- S. Wan, J. Xu, S. Cao and J. Yu, Interdiscip. Mater., 2022, 1, 294–308 CrossRef CAS.
- S. Yan, Y. Li, X. Yang, X. Jia, J. Xu and H. Song, Adv. Mater., 2023, 36, 2307967 CrossRef PubMed.
- S. Li, G. Dong, R. Hailili, L. Yang, Y. Li, F. Wang, Y. Zeng and C. Wang, Appl. Catal., B, 2016, 190, 26–35 CrossRef CAS.
- S. Wu, H. Yu, S. Chen and X. Quan, ACS Catal., 2020, 10, 14380–14389 CrossRef CAS.
- X. Zhang, P. Ma, C. Wang, L. Gan, X. Chen, P. Zhang, Y. Wang, H. Li, L. Wang, X. Zhou and K. Zheng, Energy Environ. Sci., 2022, 15, 830–842 RSC.
- H. Chen, Y. Xing, S. Liu, Y. Liang, J. Fu, L. Wang and W. Wang, Chem. Eng. J., 2023, 462, 142038 CrossRef CAS.
- H. Hou, C. R. Bowen, D. Yang and W. Yang, Chem, 2024, 10, 800–831 CAS.
- P. Zhang, J. Zhang, D. Wang, F. Zhang, Y. Zhao, M. Yan, C. Zheng, Q. Wang, M. Long and C. Chen, Appl. Catal., B, 2022, 318, 121749 CrossRef CAS.
- T. Gaaz, A. Sulong, M. Akhtar, A. Kadhum, A. Mohamad and A. Al-Amiery, Molecules, 2015, 20, 22833–22847 CrossRef CAS PubMed.
- W. Yan, Q. Chen, X. Meng and B. Wang, Sci. China Mater., 2017, 60, 449–460 CrossRef CAS.
- E. Filippo, C. Carlucci, A. L. Capodilupo, P. Perulli, F. Conciauro, G. A. Corrente, G. Gigli and G. Ciccarella, Appl. Surf. Sci., 2015, 331, 292–298 CrossRef CAS.
- A. M. Ismail, M. I. Mohammed and I. S. Yahia, Opt. Laser Technol., 2021, 134, 106600 CrossRef CAS.
- M. M. Rahman Khan, S. Pal, M. M. Hoque, M. R. Alam, M. Younus and H. Kobayashi, ACS Omega, 2019, 4, 6144–6153 CrossRef CAS PubMed.
- Z. Guo, D. Zhang, S. Wei, Z. Wang, A. B. Karki, Y. Li, P. Bernazzani, D. P. Young, J. A. Gomes, D. L. Cocke and T. C. Ho, J. Nanopart. Res., 2009, 12, 2415–2426 CrossRef.
- J. H. Thurston, A. J. Clifford, B. S. Henderson, T. R. Smith, D. Quintana, K. F. Cudworth, T. J. Lujan and K. A. Cornell, ACS Appl. Bio Mater., 2020, 3, 1681–1689 CrossRef CAS PubMed.
- W. Yan, Q. Chen, M. Du, K. M. Yang, X. Cai, X. Meng and L. Wang, J. Nanosci. Nanotechnol., 2018, 18, 5660–5667 CrossRef CAS PubMed.
- I. A. Shkrob and M. C. Saucer Jr, J. Phys. Chem. B, 2004, 0405035 Search PubMed.
- Y. Wei, J. Zhang, Q. Zheng, J. Miao, P. J. Alvarez and M. Long, Chemosphere, 2021, 279, 130556 CrossRef CAS PubMed.
- X. Zhang, J. Zhang, J. Miao, X. Wen, C. Chen, B. Zhou and M. Long, Chem. Eng. J., 2023, 466, 143085 CrossRef CAS.
- G. Zhang, Q. Wei, X. Yang, A. C. Tavares and S. Sun, Appl. Catal., B, 2017, 206, 115–126 CrossRef CAS.
- A. Singhal, M. Kaur, K. A. Dubey, Y. K. Bhardwaj, D. Jain, C. G. S. Pillai and A. K. Tyagi, RSC Adv., 2012, 2, 7180 RSC.
- A. A. F. Sabirneeza and S. Subhashini, Int. J. Ind. Chem., 2014, 5, 111–120 CrossRef.
- M. Taspika, F. A. Permatasari, B. W. Nuryadin, T. R. Mayangsari, A. H. Aimon and F. Iskandar, RSC Adv., 2019, 9, 7375–7381 RSC.
- H. S. Mansur, C. M. Sadahira, A. N. Souza and A. A. P. Mansur, Mater. Sci. Eng. Carbon, 2008, 28, 539–548 CrossRef CAS.
- J. Hu, P. Zhang, W. An, L. Liu, Y. Liang and W. Cui, Appl. Catal., B, 2019, 245, 130–142 CrossRef CAS.
- P. Kalisamy, M. Lallimathi, M. Suryamathi, B. Palanivel and M. Venkatachalam, RSC Adv., 2020, 10, 28365–28375 RSC.
- X. Zeng, Y. Liu, Y. Kang, Q. Li, Y. Xia, Y. Zhu, H. Hou, M. H. Uddin, T. R. Gengenbach, D. Xia, C. Sun, D. T. McCarthy, A. Deletic, J. Yu and X. Zhang, ACS Catal., 2020, 10, 3697–3706 CrossRef CAS.
- Y. Zhang, Z. Chen, J. Li, Z. Lu and X. Wang, J. Energy Chem., 2021, 54, 36–44 CrossRef.
- T. Liu, C. Jiao, X. Peng, Y.-N. Chen, Y. Chen, C. He, R. Liu and H. Wang, J. Mater. Chem. B, 2018, 6, 8105–8114 RSC.
- S. Zhang, G. Li, L. Duan, H. Wang, Y. Zhao and Y. Zhang, RSC Adv., 2020, 10, 19669–19685 RSC.
- P. Miao, J. Zhao, P. Wang, R. Shi and T. Zhang, ACS Mater. Lett., 2024, 6, 590–597 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01746c |
| ‡ These authors have contributed equally to the paper and are considered co-first authors. |
| This journal is © The Royal Society of Chemistry 2024 |