
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA high-performance liquid chromatography method validation and a Box–Behnken experimental design for the extraction optimization of quercitrin from Nectandra reticulata
Juanita Pulido Teutaa,
Carlos-Eduardo Narváez-Cuenca *b and
Mónica Ávila Murilloa
*b and
Mónica Ávila Murilloa
aUniversidad Nacional de Colombia, Sede Bogotá, Facultad de Ciencias, Departamento de Química, Quipronab, Carrera 30 No. 45-03, Bogotá, Colombia
bUniversidad Nacional de Colombia, Sede Bogotá, Facultad de Ciencias, Departamento de Química, Food Chemistry Research Group, Carrera 30 No. 45-03, Bogotá, Colombia. E-mail: cenarvaezc@unal.edu.co; Tel: +57-3165000, ext. 14458
First published on 9th July 2024
Abstract
The ethanolic extract of Nectandra reticulata contains a high amount of quercetin-3-O-rhamnoside (quercitrin) that has exhibited a significant activity toward Alzheimer's disease, specifically with LXR receptors. In this work, a methodology was validated following the specifications of the International Conference of Harmonization in terms of linearity, limit of detection (LOD), limit of quantification (LOQ), accuracy (recovery), repeatability (intra-assay), intermediate precision (intra-laboratory), reproducibility (inter-laboratory), robustness, and specificity. The effect of location (Oiba, Granada, and Chiquinquira) and the extraction method (percolation, maceration, and ultrasound-assisted extraction) towards the chromatographic profile and quercitrin recovery was studied. Furthermore, a Box–Behnken design was conducted to optimize quercitrin extraction and extraction yield by ultrasonic-assisted extraction. The chromatographic method was validated, with a linear range from 5 to 180 mg quercitrin per L, LOD 0.26 mg L−1, and LOQ 0.86 mg L−1. Accuracy [recovery of 93.8% (w/w)], repeatability (relative standard deviation, RSD, 3.3%), intermediate precision (RSD 5.4%), and reproducibility (RSD 1.4%) were within the acceptable values. The method was robust and specific, except for the variation in the formic acid concentration. The location had a greater influence than the extraction method towards both the chromatographic profile and quercitrin recovery. Quercitrin extraction was maximized at 60% (v/v) ethanol and 50 °C, independent of the solvent![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :material ratio used. The highest yield values were achieved at 60% (v/v) ethanol and 50 °C, with a solvent:material ratio of 40 mL g−1.
:material ratio used. The highest yield values were achieved at 60% (v/v) ethanol and 50 °C, with a solvent:material ratio of 40 mL g−1.
Introduction
Nectandra reticulata is a species of the Lauracae family that has shown significant activity against LXRs.1 The ethanolic extract of N. reticulata is composed of a large amount of flavonoids, with quercitrin (quercetin-3-O-rhamnoside) as the main representative.2 Quercitrin has anti-inflammatory, anti-leishmanial, osteogenic, anti-nociceptive, and neuroprotective properties.3 Research has shown that quercitrin inhibits GSK3 and enhances the signaling pathway Wnt/catenin.3 Moreover, quercitrin has antioxidant and free radical scavenger activity,4 and it inhibits acetylcholinesterase.5 Recent studies show that quercitrin inhibits activation and proliferation of microglia and secretion of inflammatory cytokines and chemokines and reduces the accumulation of amyloid-β plaques in Alzheimer's disease (AD) model mice.6 Each of the reported quercitrin bioactive activities is of importance in the progression of AD. It is known that AD is a neurodegenerative disease of unknown etiology, characterized by a marked deposition of amyloid-β (Aβ) plaques and progressive cognitive impairment,6 therefore, the reduction of protein aggregates and the improvement of memory may be a treatment option.7 The effects shown by quercitrin producing improvement in memory by enhancing the signaling pathway Wnt/catenin and reducing reactive oxygen species (ROS) and its cholinergic inhibition activity are related to a decrease in the formation of amyloid plaque.8 In our previous research, we have shown that quercitrin exhibits agonistic activity as evidenced through molecular docking with LXR-β and LXR-α. This flavonoid was also shown to increase the expression of mRNA in both APOE and ABCA1 target genes of LXRs as seen by in vitro experiments.2 This effect was related to the modulation of APP processing, Aβ production and clearance, and reversal of memory deficits by LXR ligands, the potential of LXR agonists to diminish AD pathogenesis to improve cognitive performance, increase the clearance of Aβ, and diminish senile plaque levels.9Due to the importance of quercitrin bioactivity including its beneficial activity in AD, and its presence in N. reticulata leaves it is relevant to have a suitable methodology not only for its quantification but also for its extraction. Performance parameters in a quantification methodology can be validated by following, for example, the specifications described by the International Conference on Harmonization (ICH).10 Within the validation of a chromatographic method, aspects such as linearity, the limit of detection (LOD), limit of quantification (LOQ), accuracy (determined as recovery), precision [in terms of repeatability (intra-assay), intermediate precision (intra-laboratory), and reproducibility (inter-laboratory)], robustness, and specificity are studied. Validation of reversed-phase (RP)-high-performance liquid chromatography (HPLC) with diode array detection (DAD) methods, following the ICH requirements, have been previously reported for the quantification of quecitrin extracted from leaves of e.g. Cosmos caudatus,11 Houttuynia cordata Thunb,12 Buchanania lanzan Spreng, and Buchanania siamensis Miq.13
Once a proper validation method is developed, there are other aspects that are important to evaluate as they affect the concentration of a metabolite in an extract. It is known that the metabolites present in a plant can vary depending on the ecophysiological and geographical conditions.14,15 Furthermore, the extraction conditions such as the extraction method, the ratio between plant material and volume of extraction solvent, the temperature, and the extraction time might affect the extraction of a particular metabolite.15 Such extraction conditions can be optimized by diverse methodologies that allow for minimizing costs and maximizing the extraction of a particular compound or a group of compounds.16 Within these methodologies there is the surface design strategy with which few experiments allow studying simultaneously the effect of different factors towards a response variable.16 Within the surface design strategies, the Box–Behnken design (BBD) has different advantages. Reports in the literature show that the BBD is slightly more efficient than other experimental designs such as the central composite design.17 Moreover, BBD does not present simultaneous combinations of all their highest or lowest levels. This means that experiments with extreme conditions that might generate degradation of the compounds or other unsatisfactory results will never occur.17,18 Additionally, this experimental design allows using the response surface methodology (RSM) as a statistical and mathematical tool for the construction of an empirical model using the most relevant variables and their responses.19
On the one hand, optimal extraction conditions depend on the compound of interest to be extracted. In this regard, when rutin (quercetin-3-O-rutinoside) and quercetin were extracted from forestry waste of Cyclobalanopsis leaves using alcohol–acetic acid–water as the extraction solvent, different optimal conditions were obtained.20 On the other hand, optimal conditions for a specific compound extraction seem to depend on the vegetal material as seen when quercetin was extracted by ultrasound-assisted extraction from onion skin,21 leaves and stems of Dendrobium officinale,22 or from the stalks of Euonymus alatus (Thunb.) Sieb.23 The extract of N. reticulata leaves has a promising activity due to the presence of quercitrin. To the best of our knowledge, no attempts to validate a chromatographic method focused on the analysis of quercitrin in N. reticula leaves have been made. Furthermore, no reports are available for the best extraction conditions of quercitrin from N. reticulata. The aim of this study was, therefore, (i) to validate a chromatographic method for the quantification of quercitrin and (ii) to optimize its extraction from N. reticulate by means of a BBD.
Experimental
Plant material and chemicals
N. reticulata leaves were collected in three different locations in Colombia: Granada (Department of Cundinamarca), Chiquinquirá (Department of Boyacá), and Oiba (Department of Santander). A specimen of the species was deposited and identified by the Herbario Nacional de Colombia at Universidad Nacional de Colombia whose code is COL547368. Leaves from the three locations were dried separately at room temperature and ground to a particle size lower than 2 mm.The quercitrin primary reference standard was obtained from the HWI group (Frankfurt, Germany). Methanol and acetonitrile, both of HPLC grade, were purchased from Honeywell (Muskegon, Michigan, USA). Analytical grade formic acid was purchased from Merck (Darmstadt, Germany). Ethanol (96%, v/v) was purchased from El Alquimista (Bogotá, Colombia). This solvent was redistilled by simple distillation in a tower of 1 m. Milli-Q water was processed in the Direct-Pure Adept equipment of the Rephile brand (Miami, FL, USA).
Selection of chromatographic conditions
To find a suitable chromatographic method for the analysis of quercitrin, an extract obtained by maceration from N. reticulata collected in Granada was analysed on a Dionex Ultimate 3000 UHPLC system (Thermo Scientific, San Jose, California, USA) with a photodiode array detector (DAD), using a Hypersyl Gold RP column (Thermo Scientific; 150 mm × 2.1 mm id; 1.9 μm particle size) operated at 40 °C. The extract (5 μL) was analyzed by reversed-phase high-performance liquid chromatography (RP-HPLC). The mobile phase consisted of water/formic acid (99.9/0.1, v/v) (eluent A) and pure acetonitrile (eluent B). The flow rate was 400 μL min−1. The detection wavelength was set to 350 nm. Before injection, all samples were passed through a 0.22 μm filter. Initially, an elution program based on the literature was tested (method 1).24 In total, four elution programs were tested (method 1–method 4). Method 1: 0–30 min, linear gradient from 5 to 70% B; 30–33 min, linear gradient from 70 to 95% B; 33–36 min, isocratic at 95% B; 36–41 min, linear gradient from 95 to 5% B; 41–46 min, isocratic at 5% B. Method 2: 0–4 min, linear gradient from 5 to 20% B; 4–30 min, linear gradient from 20 to 70% B; 30–33 min, linear gradient from 70 to 95% B; 33–36 min, isocratic at 95% B; 36–41 min, linear gradient from 95 to 5% B; 41–46 min, isocratic at 5% B. Method 3: 0–4 min, linear gradient from 5 to 25% B; 4–6 min, linear gradient from 25 to 28% B; 6–10 min, isocratic at 28% B; 10–20 min, linear gradient from 28 to 50% B; 20–24 min, isocratic at 50% B; 24–30 min, linear gradient from 50 to 70% B; 30–33 min, linear gradient from 70 to 95% B; 33–36 min, isocratic at 95% B; 36–41 min, linear gradient from 95 to 5% B; 41–46 min, isocratic at 5% B. Method 4: 0–4 min, linear gradient from 5 to 20% B; 4–10 min, isocratic at 20% B; 10–12 min, linear gradient from 20 to 25% B; 12–16 min, isocratic at 25% B; 16–22 min, linear gradient from 25 to 52% B; 22–26 min, isocratic at 52% B; 26–30 min, linear gradient from 52 to 70% B; 30–33 min, linear gradient from 70 to 100% B; 33–36 min, isocratic at 100% B; 36–41 min, linear gradient from 100 to 5% B; 41–46 min, isocratic at 5% B. The presence of quercitrin in each chromatographic method was verified by matching the retention time and UV-vis spectrum with an authentic standard. Selection of the method was based on the response variables: number of peaks with chromatographic both area greater than 0.15 mAU min and minimum height of 2.5 mAU, peak width range, and resolution of the critical pair of chromatographic peaks.Once, the chromatographic method was selected, extracts were analysed. Quercitrin quantification was performed by the external standard method. This was done by taking the area under the curve at 350 nm for the chromatographic peak of quercitrin and interpolating it in calibration curves of authentic quercitrin. The concentration of quercitrin was expressed in mg L−1 extract and in mg g−1 plant material.
Calibration curve preparation
On three different days, 5 mg of authentic quercitrin was weighed and dissolved in 5 mL of 100% (v/v) methanol, to obtain a stock solution of 1000 mg mL−1. With the stock solution, 19 solutions with concentrations ranging from 0.2 to 240 mg mL−1 were prepared. Then, 5 μL of each solution was injected into the HPLC system.Validation of the RP-HPLC-DAD method
The validation of the selected chromatographic method (method 4) was performed according to the parameters described by the ICH guidelines. The calibration curve was prepared with the authentic quercitrin standard both in a high concentration range between 10 to 240 mg mL−1 and in a low concentration range between 0.2 to 10 mg mL−1. The limit of detection (LOD) and limit of quantification (LOQ) were determined with the area of quercitrin of the standard solutions in the low concentration range, according to the following equations: LOD = 3.3σ/S and LOQ = 10σ/S, where σ is the residual standard deviation of the regression curves and S is the slope. Before analyzing the linearity of the calibration curve, the Shapiro–Wilk normality test was performed. Then, the linear range was estimated by evaluating the adjusted R-squared (near 1), the studentized residuals [no more than (NMT) 2], the analysis of variance (ANOVA) (NMT 0.05), and the Durbin–Watson statistic [more than (MT) 0.05]. When a linear range w to be determined, it is convenient to avoid values with studentized residuals greater than 2, since they would represent atypical measurements of the linear model.25 The studentized residuals measure how many standard deviations each observed value of the quercitrin area deviates from the fitted model. Statistical analyses were performed in R-studio (2022.02.3+492).Accuracy was determined using the recovery method by spiking. Extract samples were spiked with a known analytical concentration of quercitrin (40 mg L−1) and the percent recovery was determined.
Precision was determined at three levels: repeatability (intra-assay), intermediate precision (intra-laboratory), and reproducibility (inter-laboratory), with authentic quercitrin standard solutions prepared at 80 mg L−1. Repeatability was evaluated using six replicates (individual replications) of the authentic quercitrin on the same day. Intermediate precision was studied by analysing such standard solutions on different days (n = 9). Reproducibility was evaluated by changing the laboratory, both placed at Universidad Nacional de Colombia (n = 3). In all studied cases, repeatability, intermediate precision, and reproducibility, the percentage of the relative standard deviation (RSD) and the standard error of the mean (SEM) were calculated.
The robustness was evaluated by performing small variations in the parameters of the chromatographic method as follows: concentration of formic acid in eluent A (±0.1% v/v, this is either 0.0 or 0.2% v/v), flow rate (±10 μL min−1, this is either 390 or 410 μL min−1), column temperature (±2 °C, this is either 38 or 42 °C), and wavelength at which the chromatographic area is recorded (±2 nm, this is either 348 or 352 nm), performing 6 runs on each parameter with both the authentic quercitrin standard (80 mg L−1) and the extract. In each case, the retention time was measured as a response variable. A t-test was performed to determine whether the retention time was significantly affected by the variation in the chromatographic method. For doing so, the results of the original method were compared with those obtained with the variations.
The specificity of the chromatographic method was evaluated by comparing the chromatographic profiles of the authentic quercitrin standard (240 mg L−1) and the extract (10 mg mL−1) with an electro-light scattering detector (ELSD), with respect to the peak eluting before quercitrin, which is the one that elutes at a closer time. In this case, an ELSD was used instead of a DAD since ELSD is a universal detector and DAD only responds to active compounds in UV-vis. By ELSD, therefore, it was possible to verify if there were other compounds with similar retention times that could affect the quercitrin response due to cooperative or antagonistic effects.26,27
Effect of the extraction method and agroclimatic conditions towards the chromatographic profile and quercitrin recovery
Leaves of N. reticulata collected from three locations in Colombia (Oiba, Granada, and Chiquinquirá) were extracted by three different extraction methods (percolation, maceration, and ultrasound-assisted extraction) following the protocols developed by the QUIPRONAB research group.28 For each extraction, 96% (v/v) ethanol was used. Once each extraction was completed, the extract was roto-evaporated (40 °C) until dryness and 10 mg of the dried extract was dissolved in 1 mL of 100% (v/v) methanol and filtered using a 0.22 μm membrane filter. Each dissolved extract was analyzed by RP-HPLC-DAD as described in the section Selection of chromatographic conditions, method 4. The chromatographic profile was evaluated in terms of the efficacy of the HPLC method to separate the compounds as recorded at 350 nm. Quercitrin recovery was accessed in terms of both quercitrin yield (mg quercitrin/g extract) and relative abundance of quercitrin as compared to other chromatographic peaks at 350 nm.Ultrasound-assisted extraction optimization of quercitrin
For each point of the experimental design, approximately 1.5 g of dry plant material of N. reticulata collected in Granada was taken. Ethanol (at different ratios water:ethanol) was added in different volumes to this plant material, and it was sonicated for 15 min. The supernatant was filtered, and the extraction procedure was repeated once more. The two supernatants were combined and roto-evaporated at 40 °C. The dry extract was then solubilized in 100% (v/v) methanol, filtered using a 0.22 μm membrane filter, and analyzed by RP-HPLC-DAD as described in the section Selection of chromatographic conditions, method 4.
To define the factors that affected the most and the domain, before performing the BBD, different screening assays were carried out. Five factors (and their domains) were evaluated: solvent:plant material ratio (20–40 mL g−1), number of extractions (1–3), percentage of aqueous ethanol (40–96% v/v), extraction time (5–40 min), and extraction temperature (30–50 °C). Taking into account the risk assessment recommendation mentioned in the ICH, chromatograms of the obtained extracts were checked for the formation of new chromatographic peaks, which might be related to the artifact formation. After such verification, new chromatographic peaks were not observed.
When evaluating the effect of the solvent:plant material ratio it was found that the higher the such ratio the higher the quercitrin extraction. Moreover, the more extractions that were performed the higher the extraction of quercitrin. In relative values, the second and third extraction represented 1.2 and 1.6 times what was extracted in the first extraction. Despite the number of extractions being among the factors that affected the response variable, it was decided to extract twice taking into account the costs and time the third extraction represents. The percentage of ethanol in the extraction solvent had an important effect towards the response variable, with the greatest values being obtained at 60–70% (v/v) aqueous ethanol and the lowest at either 40 or 96% (v/v) aqueous ethanol, showing a parabolic behavior. Between 10 and 30 min of extraction the quercitrin content was similar, at shorter times (5 min) and longer times (40 min) lower quercitrin values were found; this allowed selecting an extraction time of 15 min. Furthermore, when increasing the temperature there was an increase in the extraction of quercitrin, without the formation of new compounds. After conducting these preliminary tests, the factors with the greatest effect towards quercitrin extraction were selected. This is the solvent:plant material ratio, percentage of aqueous ethanol, and the extraction temperature.
Once the factors and the domain that affected the most were defined, a BBD was conducted. In the experimental design (Table 1), three levels of each factor were used, and four replicates of the central point were carried out. The three factors (and the domain) were as follows: percentage of ethanol (60–96% v/v), material:solvent ratio (20–40 mL g−1), and temperature (30–50 °C). The response variables evaluated were (i) quercitrin extraction (mg of quercitrin/g of plant material) and (ii) extraction yield (g dry extract/g dry leaves × 100). A total of 16 experiments were performed in a randomized order. Once the experiments were completed, the normality of the data was verified by the Shapiro–Wilk normality test. Then, for each response variable, the effect of the factors was evaluated using a complete quadratic equation. To verify the efficiency of the mathematical equation to describe each response variable, the adjusted R-squared, the p-value from the ANOVA, the Durbin–Watson statistic, and the absolute average deviation were evaluated. To eliminate the terms of the equation that were statistically not significant, a stepwise backward elimination procedure was performed. With the equation that satisfied the statistical parameters, each of the two responses was represented by response surfaces and the optimal values of the model were determined. The model processing was performed using R-studio (2022.02.3+492).
| Independent variables | Symbol | Levels | ||
|---|---|---|---|---|
| −1 | 0 | +1 | ||
| Percentage of aqueous ethanol (%, v/v) | A | 60 | 78 | 96 |
| Solvent:vegetable material ratio (mL g−1) |
B | 20 | 30 | 40 |
| Temperature (°C) | C | 30 | 40 | 50 |
Results and discussion
Selection of chromatographic conditions
Among the four chromatographic methods tested, method 4 was selected. The four tested methods yielded a well-resolved symmetric chromatographic peak corresponding to quercitrin (Fig. 1). Nevertheless, because the chromatographic method was developed not only to analyse quercitrin but also to identify other compounds,2 further chromatographic features were taken into account (Table 2). More peaks were obtained with method 2 (10 peaks) and method 4 (10 peaks) than with methods 1 and 3 (6 peaks in each one) (Table 2). Both methods 2 and 4 provided chromatographic peaks with comparable peak width range at half high (0.036–0.131 and 0.035–0.131 min). Nevertheless, method 4 was more convenient than method 2 when evaluating the resolution of the critical pair of peaks to find a suitable one. Finally, method 4 was selected for the analysis of extracts.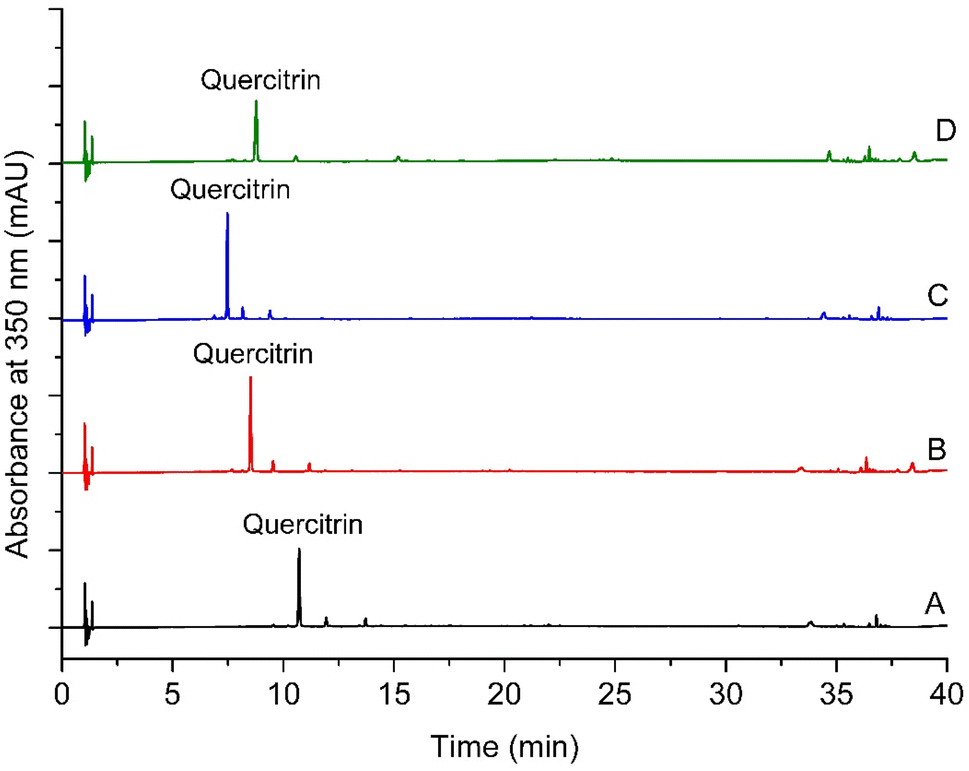 | ||
| Fig. 1 Chromatographic profiles of an extract of Nectandra reticulata prepared by maceration. (A) Method 1. (B) Method 2. (C) Method 3. (D) Method 4. Each method is described in the subheading Selection of chromatographic conditions in the Experimental section. | ||
| Feature | Method 1 | Method 2 | Method 3 | Method 4 |
|---|---|---|---|---|
| a W50%: peak width at half high. | ||||
| Number of peaks | 6 | 8 | 6 | 8 |
| W50% range | 0.044–0.074 | 0.036–0.131 | 0.042–0.064 | 0.035–0.131 |
| Resolution of the critical pair of peaks | 0.23 | 0.27 | 0.24 | 0.52 |
Validation of the RP-HPLC-DAD method
The quantification method of quercitrin in N. reticulata by RP-HPLC-DAD was validated in terms of linearity, LOD, LOQ, accuracy, intra-assay, intra-laboratory, and inter-laboratory precision, robustness, and specificity according to the ICH. Area data in the full evaluated concentration range (0.2–240 mg L−1) followed a normal distribution as evaluated by the Shapiro–Wilk normality test. The linear range concentration was found between 5–180 mg L−1 (Fig. 2). When the fully tested concentration interval was evaluated, the adjusted R-squared decreased, the studentized residues increased, and the p-value of the ANOVA remained lower than 0.05 as compared to the values obtained in the selected linear range (5–180 mg L−1.) At the selected linear range, the R-squared and the studentized errors were 0.9933 and ±2.5, respectively, while at 0.2–240 mg L−1 they became 0.9897 and ±4.5, respectively. Additionally, linearity between 5–180 mg L−1 was evaluated by analyzing the presence of autocorrelation using the Durbin–Watson statistic. As the p-value (2.501) was greater than 0.05, Ho was not rejected. Then, it could be concluded that the residuals of the model were not autocorrelated. The linear range found in the current research was narrower than those found elsewhere for the same analyte (Table 3).29–31 The intercept and slope of the linear equation were determined for the selected range (5–180 mg L−1) by examining the relationship between quercitrin concentration and the chromatographic area for such a peak. The intercept and slope were, respectively, −0.527 mAU min and 0.328 mAU min L mg−1 (Table 3). Furthermore, LOD was found to be 0.26 mg L−1, while the LOQ was 0.86 mg L−1 (Table 3). Both LOD and LOQ obtained in the current research for quercitrin are within those reported in previous works (Table 3).29–31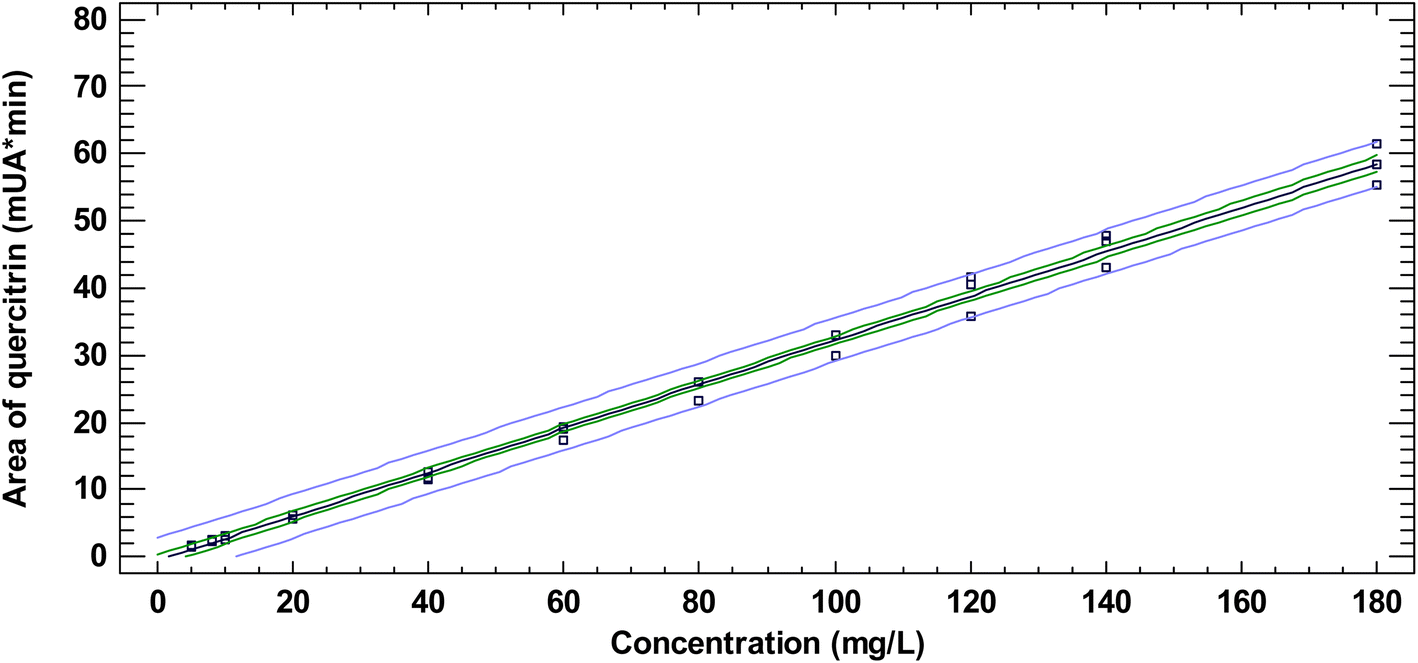 | ||
| Fig. 2 Linear range determined for the certified quercitrin standard at 350 nm. For each concentration point, a triplicate of the measurement was performed. The blue lines (the outer interval) are the prediction limits for the new observations at 95% confidence. The green lines (internal interval) are the confidence limits (95%) of the mean. | ||
| Parameter | Present work | Sharifuldin et al.11 | Nguyen et al.12 | Prompanya et al.13 |
|---|---|---|---|---|
| a LOD: limit of detection. LOQ: limit of quantification. RSD: relative standard deviation. SEM: standard error of the mean. | ||||
| Linearity range (mg L−1) | 5–180 | 0.5–500 | 48–1000 | 31–500 |
| Adjusted R-squared | 0.9933 | 1.000 | 0.9999 | 0.9990 |
| Regression equation | Area350 nm (mUA min) = −0.527 + 0.328[quercitrin (mg L−1)] | Area254 nm = 0.035 + 0.220[quercitrin] (no units reported) | Area355 nm = −2.8326 + 0.0001772[quercitrin] (no units reported) | Area254 nm = −11111 + 466.06[quercitrin] (no units reported) |
| ANOVA (p-value) | 2.2 × 10−16 | — | — | — |
| Shapiro–Wilk normality test (p-value) | 0.081 | — | — | — |
| Durbin–Watson statistic | 2.501 | — | — | — |
| LOD (mg L−1) | 0.26 | 0.08 | 9.13 | 7.81 |
| LOQ (mg L−1) | 0.86 | 0.24 | 33.44 | 23.67 |
| Recovery (%, w/w) | 93.8 | 80.4–93.3 | 101.0–101.1 | 108.1 |
| Repeatibility (intra-assay, n = 6) RSD//SEM | 3.27//0.35 | 0.25//— | 0.25//— | 1.66/— |
| Intermediate precision (intra-laboratory, n = 9) RSD//SEM | 5.45//0.47 | 0.31//— | 0.95//— | 2.69//— |
| Reproducibility (inter-laboratory, n = 3) RSD//SEM | 1.42//0.09 | —//— | —//— | —//— |
The recovery value, indicative of accuracy, was 93.8% (w/w), the value that is within the acceptability range (90–107%, w/w), this means that there is no matrix effect with the other components of the extract.32,33 The tested RP-HPLC-DAD method had a good repeatability (intra-assay), intermediate precision (intra-laboratory), and reproducibility (inter-laboratory) (Table 3), since for all the cases, the RSD values were below the accepted limit (11.3%).33 Good recovery, repeatability (intra-assay), and intermediate precision is in agreement with those from previous works (Table 3).29–31
When robustness was assessed, the results showed that for almost all the variations the p-values were above 0.05 (except when the retention time of the quercitrin standard was evaluated after formic acid variation in the mobile phase). Then, since the retention times were not significantly affected in most of the cases, it is concluded that the HPLC-DAD method is robust (Table 4). The precision and robustness analyses allow us to conclude that the developed method is not sensitive to changes within the laboratory and additionally, it is possible to transfer it to other laboratories without a significant change in the chromatographic behavior. Finally, the method was specific. As shown in Fig. 3, when the sample was analyzed, with the temperature of the evaporation tube of the ELSD set at either 70 or 75 °C, the resolution of the quercitrin peak with respect to the peak that eluted before was close to 1.5. In addition, the peak was observed to be symmetrical.
| Parameter | 80 mg L−1 quercitrin | Extract sample |
|---|---|---|
| t-Test (p-value) | t-Test (p-value) | |
| Flow rate ± 0.01 mL min−1 (n = 6) | 9.9313 × 10−1 | 4.6124 × 10−1 |
| [Formic acid] ± 1% v/v (n = 6) | 4.9005 × 10−2 | 2.7771 × 10−1 |
| Column temperature ± 2 °C (n = 6) | 9.3028 × 10−1 | 7.7549 × 10−1 |
| Wavelength ± 2 nm (n = 6) | 1.0000 | 1.0000 |
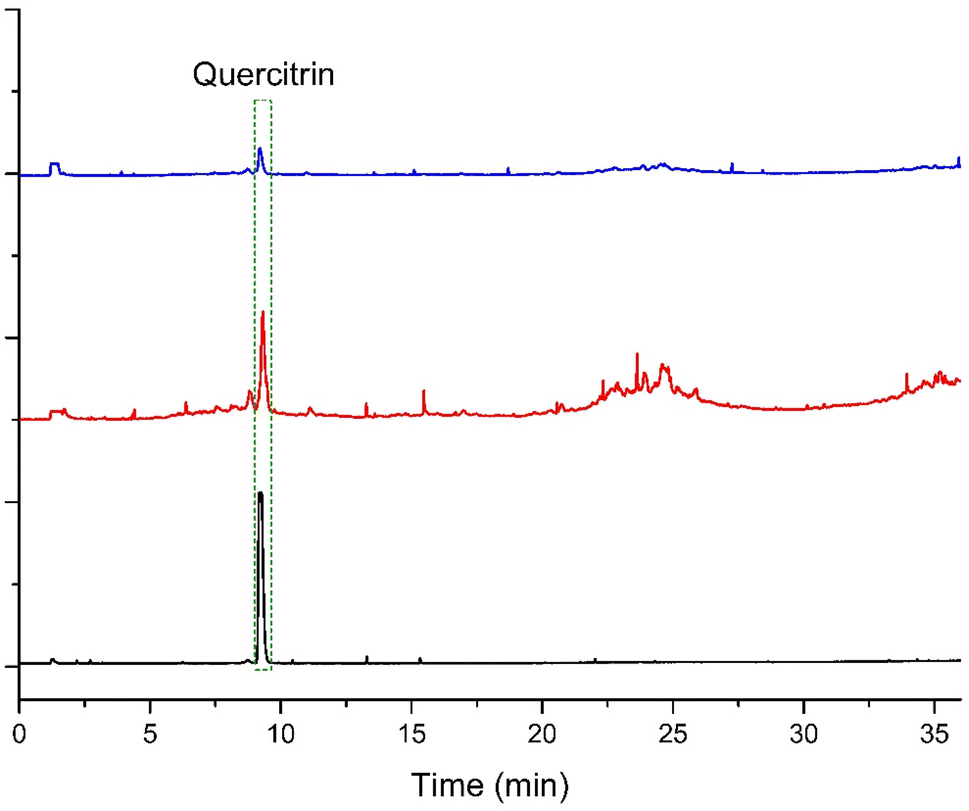 | ||
| Fig. 3 Comparison of the chromatographic profiles of Nectandra reticulata from Granada in the ELSD. The chromatogram in blue corresponds to the extract of N. reticulata measured at an evaporator tube temperature of 75 °C, the red one is a measurement of the same extract with the evaporator tube at 70 °C, and the chromatogram in black corresponds to the authentic standard of quercitrin at an evaporator tube temperature of 70 °C. | ||
Effect of the extraction method and agroclimatic conditions towards the chromatographic profile and quercitrin recovery
The location had a greater influence than the extraction method towards both the chromatographic profile (Fig. 4) and quercitrin recovery (Table 5). A simple chromatographic profile with few metabolites was observed when the samples were collected in Granada and Oiba. In both cases, the resolution of quercitrin was greater than 3.5. Nevertheless, for the extract of the plant material collected in Chiquinquirá, a decrease in the resolution of the peaks of compounds different from those found in the extracts from Granada and Oiba was observed. Despite this, the chromatographic peak corresponding to quercitrin could be distinguished without any problem. This means that the chromatographic method has the capacity to separate quercitrin in HPLC from other metabolites that the plant can produce.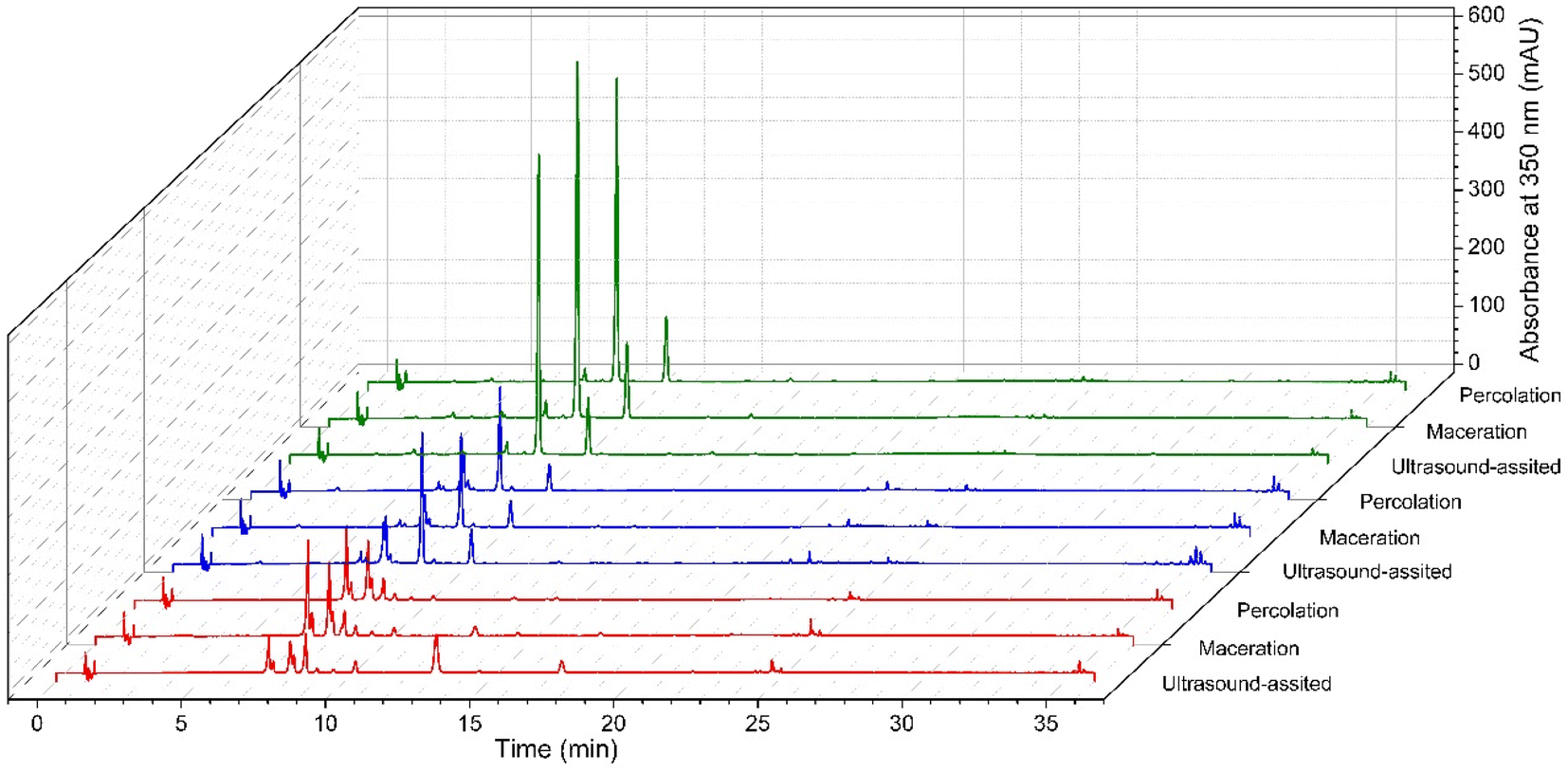 | ||
| Fig. 4 Comparison of the chromatographic profiles of Nectandra reticulata recorded at 350 nm by percolation, maceration, and ultrasound-assisted extraction of samples collected in Chiquinquira (red), Oiba (blue), and Granada (green). | ||
| Location | Extraction method | mg quercitrin/g extract | Area of quercitrin/total area × 100 |
|---|---|---|---|
| Chiquinquirá | Percolation | 0.490 | 10.2 |
| Maceration | 0.579 | 16.6 | |
| Ultrasonic-assisted | 0.272 | 9.1 | |
| Granada | Percolation | 3.399 | 73.8 |
| Maceration | 2.447 | 69.4 | |
| Ultrasonic-assisted | 2.380 | 73.2 | |
| Oiba | Percolation | 1.140 | 35.6 |
| Maceration | 1.144 | 37.9 | |
| Ultrasonic-assisted | 1.755 | 46.5 |
When comparing the effect of location on quercitrin yield, the greatest value was obtained for the Granada sample (2.380–3.399 mg of quercitrin/g of extract, depending on the extraction method). In decreasing order of quercitrin content, Oiba (1.140–1.755 mg quercitrin/g of extract) was found, followed by Chiquinquirá (0.272–0.579 mg quercitrin/g of extract). When looking at the relative abundance of quercitrin as compared to other chromatographic peaks (area of quercitrin divided by total area at 350 nm, in percentage), the greatest values were obtained for the sample from Granada (69.4–73.8%). Along with the analysis of the effect of location, the quercitrin recovery was studied according to the extraction method. When analysing the effect of the extraction method, a unique optimal method was not observed, since for each location both the greatest yield and its relative abundance were evidenced with a different method (Table 5). While for the Chiquinquirá sample, the best method in terms of both response variables was maceration, for Oiba it was ultrasonic-assisted extraction. When the sample was collected in Granada, percolation resulted in yielding the highest content of quercitrin, while the greatest relative abundance of quercitrin was achieved with either percolation or ultrasonic-assisted extraction. Despite no single optimal method was observed for all collections, ultrasound-assisted extraction was selected to evaluate the effect of different factors in the experimental design. Among the criteria for selecting this method are that this methodology has low extraction times and is friendly to the environment because of the reduction in solvent consumption as compared to other methods.34 In previous reports, ultrasonic-assisted extraction has been proven to be an efficient extraction method of quercitrin-like compounds as compared to other ones. For instance, this extraction method yielded higher quercetin extraction from leaves of Raphanus sativus L. as compared to other extraction methods including maceration, digestion, or Soxhlet.35 Furthermore, when quercetin was extracted from onion skin, the ultrasonic-assisted extraction method was more efficient than conventional solvent extraction.21 In agreement with the effect of location towards quercitrin extraction found in the current research, it was reported that the concentration of a set of flavonoids, including derivatives of quercetin, changed in a wild population of white birch (Betula pubescens EHRH) when collected from different locations in Finland.36 While we were interested in tracking a single compound and its relative abundance, previous research has demonstrated that location affects the entire chromatographic profile of phenolics, including for instance hydroxybenzoic acids, hydroxycinnamic acids, and flavonoids.37,38
It is known that specific metabolites, formerly called secondary metabolites, are synthesized only under certain circumstances. The content of such metabolites can vary with changes in different agroclimatic conditions, such as environmental and soil nutrients.39 For most plants, external factors such as light, temperature, soil water, soil fertility, and salinity, can significantly affect some processes associated with growth and development, even affecting their ability to synthesize secondary metabolites, which eventually leads to the change of general phytochemical profiles that play a strategic role in the production of bioactive substances.40 Although there are no studies that document how environmental factors affect quercitrin content, it is known that climatic conditions of low rainfall, arid soils, low nutrient deficit, and high salinity are stimuli that can increase the levels of phenolic compounds in response to oxidative stress generated by the formation of reactive oxygen species in hostile environments.41 Polyhydroxylated flavonoids such as quercitrin are good free radical scavengers. Due to the presence of OH in the 3′, 4′, and 7 positions, and the conjugated system, polyhydroxylated flavonoids can stabilize the radical formed by the canonical resonance structures typical of conjugated aromatic systems.42,43 It can be presumed, therefore, that the plant material collected in Granada was subjected to greater environmental stress as compared to the other materials characterized in this research.
Experimental design
The response variables measured in the BBD [quercitrin extraction (mg of quercitrin/g of plant material) and extraction yield (g dry extract/g dry leaves × 100), Table 6] followed a normal distribution as observed by the Shapiro–Wilk normality test (p-values of 0.253 and 0.302, respectively).| Percentage of ethanol (%, v/v) | Solvent:vegetable material ratio (mL:g) |
Temperature (°C) | mg of quercitrin/g of plant material | Extraction yield (% g:g) |
|---|---|---|---|---|
| 78(0) | 30(0) | 40(0) | 0.613 | 19.00 |
| 60(−1) | 20(−1) | 40(0) | 0.573 | 16.90 |
| 78(0) | 30(0) | 40(0) | 0.524 | 15.47 |
| 78(0) | 40(+1) | 50(+1) | 0.657 | 21.61 |
| 96(+1) | 20(−1) | 40(0) | 0.311 | 11.47 |
| 60(−1) | 30(0) | 50(+1) | 0.692 | 20.57 |
| 78(0) | 20(−1) | 30(−1) | 0.484 | 13.61 |
| 96(+1) | 40(+1) | 40(0) | 0.324 | 12.51 |
| 96(+1) | 30(0) | 30(−1) | 0.330 | 11.16 |
| 60(−1) | 40(+1) | 40(0) | 0.639 | 21.02 |
| 78(0) | 40(+1) | 30(−1) | 0.482 | 14.86 |
| 78(0) | 30(0) | 40(0) | 0.543 | 17.12 |
| 60(−1) | 30(0) | 30(−1) | 0.577 | 17.37 |
| 96(+1) | 30(0) | 50(+1) | 0.402 | 14.25 |
| 78(0) | 30(0) | 40(0) | 0.533 | 19.25 |
| 78(0) | 20(−1) | 50(+1) | 0.606 | 19.77 |
When the complete quadratic equation was obtained for the response variable mg of quercitrin/g of plant material (quercitrin extraction) the only significant coefficients, according to the Pareto diagram (Fig. 5, Left), were linear and quadratic components of the percentage of ethanol (A and A2, respectively). In the complete quadratic model, the values of the adjusted R-squared, p-value in the ANOVA, Durbin–Watson statistic, and absolute average deviation were 0.9232, 7.162 × 10−4, 0.416, and 3.0%, respectively. After performing the stepwise backward elimination to obtain only the coefficients that statistically contributed to the mathematical model (Fig. 5, Right) eqn (1) was obtained. For such an equation the R-squared (0.9228), p-value in the ANOVA (1.578 × 10−7), and absolute average deviation (5.1%) were within the acceptable values. Furthermore, the presence of autocorrelation was evaluated using the Durbin–Watson statistic, obtaining a p-value of 0.412; since this number is greater than 0.05 (Ho is not rejected), it can be stated that the residuals of the model do not follow autocorrelation, which means they are random. All evaluated statistical parameters indicate that the model equation fits the data correctly, so it can be predictive. In the process of stepwise backward elimination, the independent variables A (percentage of ethanol) and C (temperature) remained in the equation.
| y = 2.8039 × 10−2A + 6.0377 × 10−3C − 2.2938 × 10−4A2 − 0.47771 | (1) |
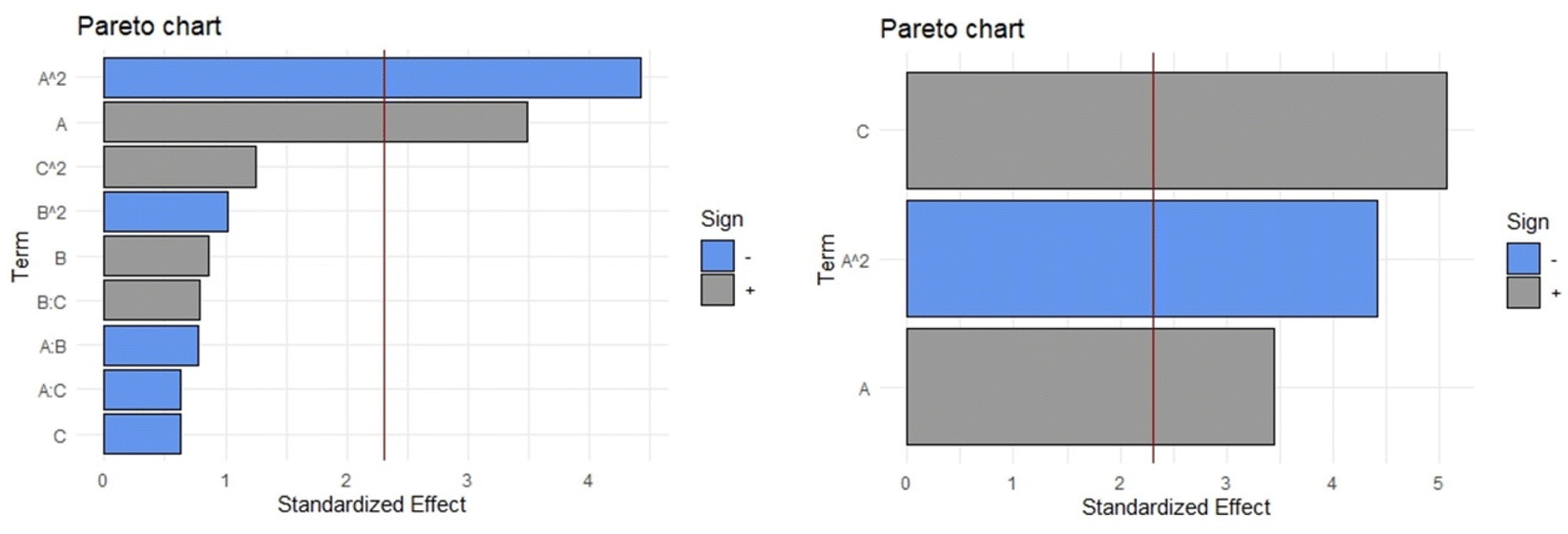 | ||
| Fig. 5 Pareto diagram for the response variable “mg of quercitrin/g of plant material”. Left: Complete model. Right: After stepwise backward elimination. Percentage of ethanol (A), material solvent ratio (B) and temperature (C). | ||
Optimization of the extraction parameters by means of eqn (1) indicated that, within the studied range, the extraction of quercitrin (mg of quercitrin/g of plant material) was maximized at 60% (v/v) ethanol at 50 °C, independent of the solvent:plant material ratio used, in agreement with what is shown in Fig. 6. If the response surface diagram (Fig. 6) is observed, the higher the percentage of ethanol and the higher the temperature, the higher the quercitrin yield. This is due to the fact that quercitrin, having a sugar moiety in its structure dissolves better in water than alcohol, with the last one being less polar than water.
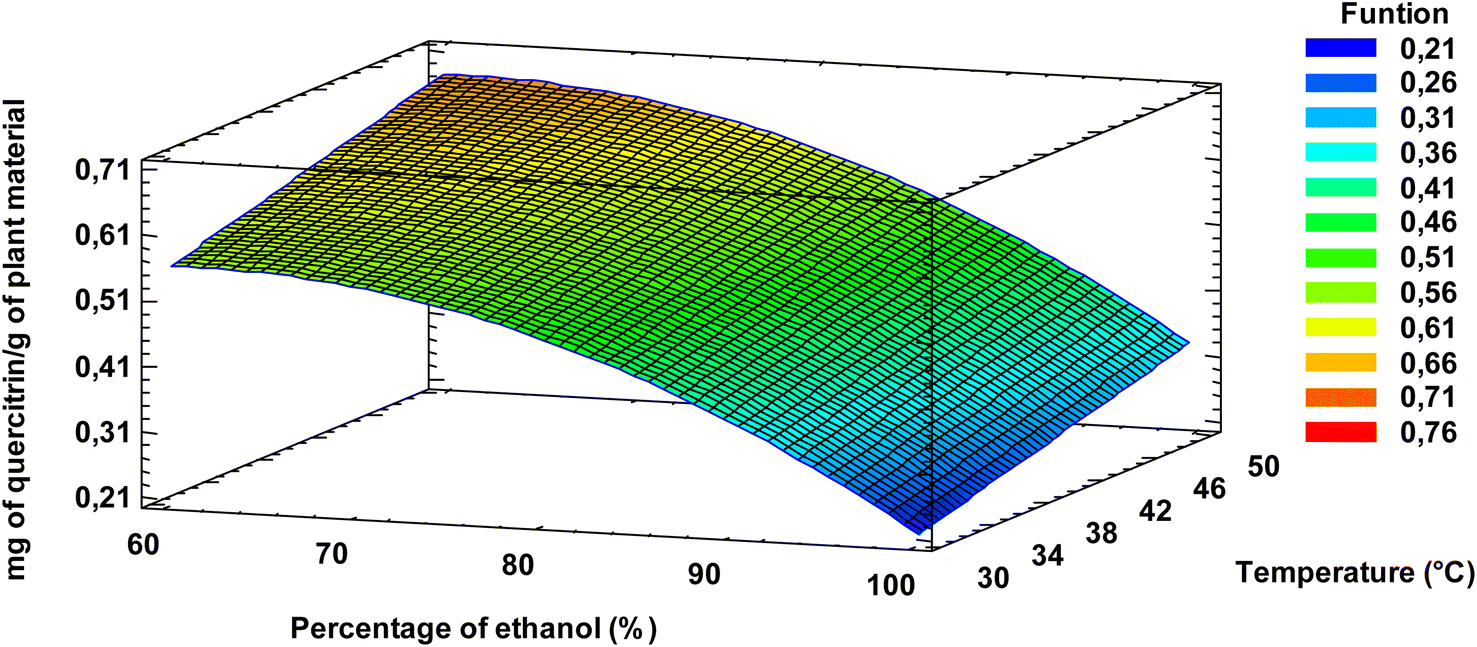 | ||
| Fig. 6 Response surface for the response variable mg of quercitrin/g of plant material. Figures related to the solvent:plant material factor (mL g−1) are not presented, because the equation obtained after stepwise backward elimination did not consider any parameter of that factor. | ||
Next, the analysis was performed for the second response variable, extraction yield (g dry extract/g dry leaves × 100). The Pareto diagram (Fig. 7, Left) shows that when the complete quadratic equation is used, the only significant coefficient corresponds to the one related to the quadratic component of the factor percentage of ethanol (A2). In this complete model, the values of adjusted R-squared, ANOVA (p-value), Durbin–Watson statistic, and absolute average deviation were 0.7765, 0.015, 0.946, and 5.1%, respectively. After eliminating the non-significant coefficients in the mathematical equation, by using the strategy of stepwise backward elimination, the coefficients A2, A, B, and C remained in the equation (Fig. 7, Right, eqn (2)). As a result, an outstanding improvement of the adjusted R-squared was found. The starting point was an adjusted R-squared of 0.7765 in the complete quadratic equation and after the stepwise backward elimination, the adjusted R-squared was increased to 0.8559. After this procedure, the p-value obtained in the ANOVA (2.533 × 10−5) and the absolute average deviation (5.1%) were within the acceptance limits.
| y = 0.74522A + 0.10309B + 0.24002C − 0.5955 × 10−2A2 − 17.00429 | (2) |
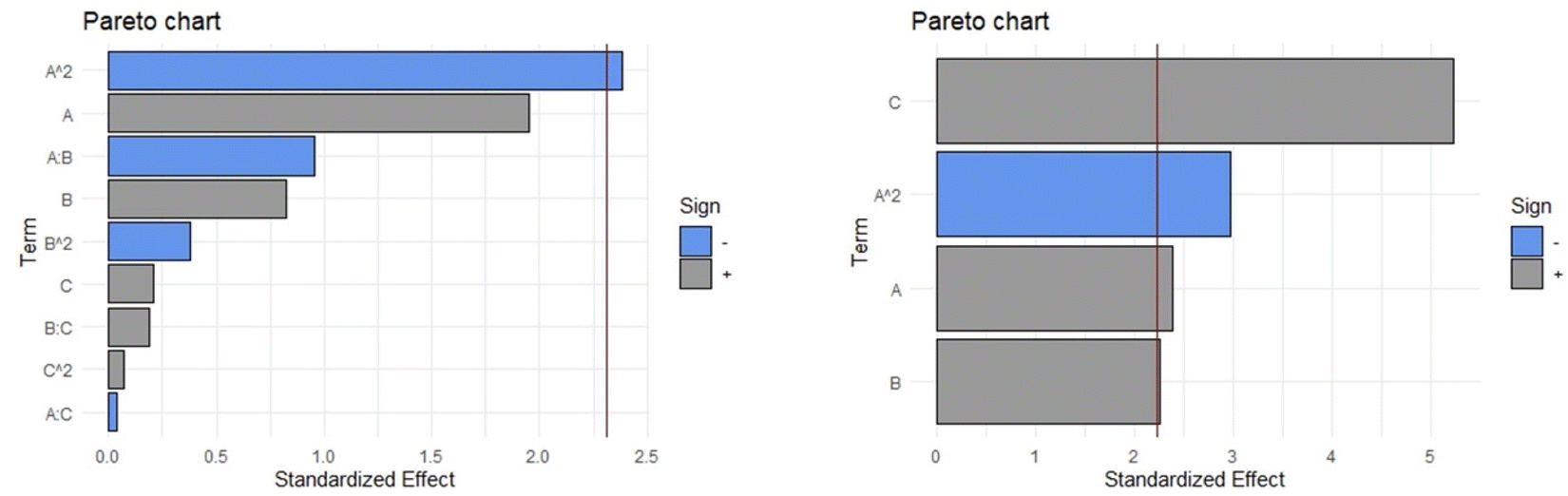 | ||
| Fig. 7 Pareto diagram for the response variable “extraction yield”. Left: Complete model. Right: After stepwise backward elimination. Percentage of ethanol (A), material solvent ratio (B) and temperature (C). | ||
The Durbin–Watson statistic (p-value 0.985) of this simplified equation indicates that the residuals of the model do not follow autocorrelation. Evaluated statistical parameters indicate that the model generated only with the significant coefficients, three of the linear components and one of the quadratic components (eqn (2)), allows adequate modelling of the obtained experimental data.
Optimum extraction conditions were calculated from eqn (2). Within the studied range of each factor, an optimal extraction yield is achieved at 60% (v/v) ethanol, with a solvent:material ratio of 40 mL g−1 at 50 °C. Such maxima can be visualised in the response surface diagram (Fig. 8).
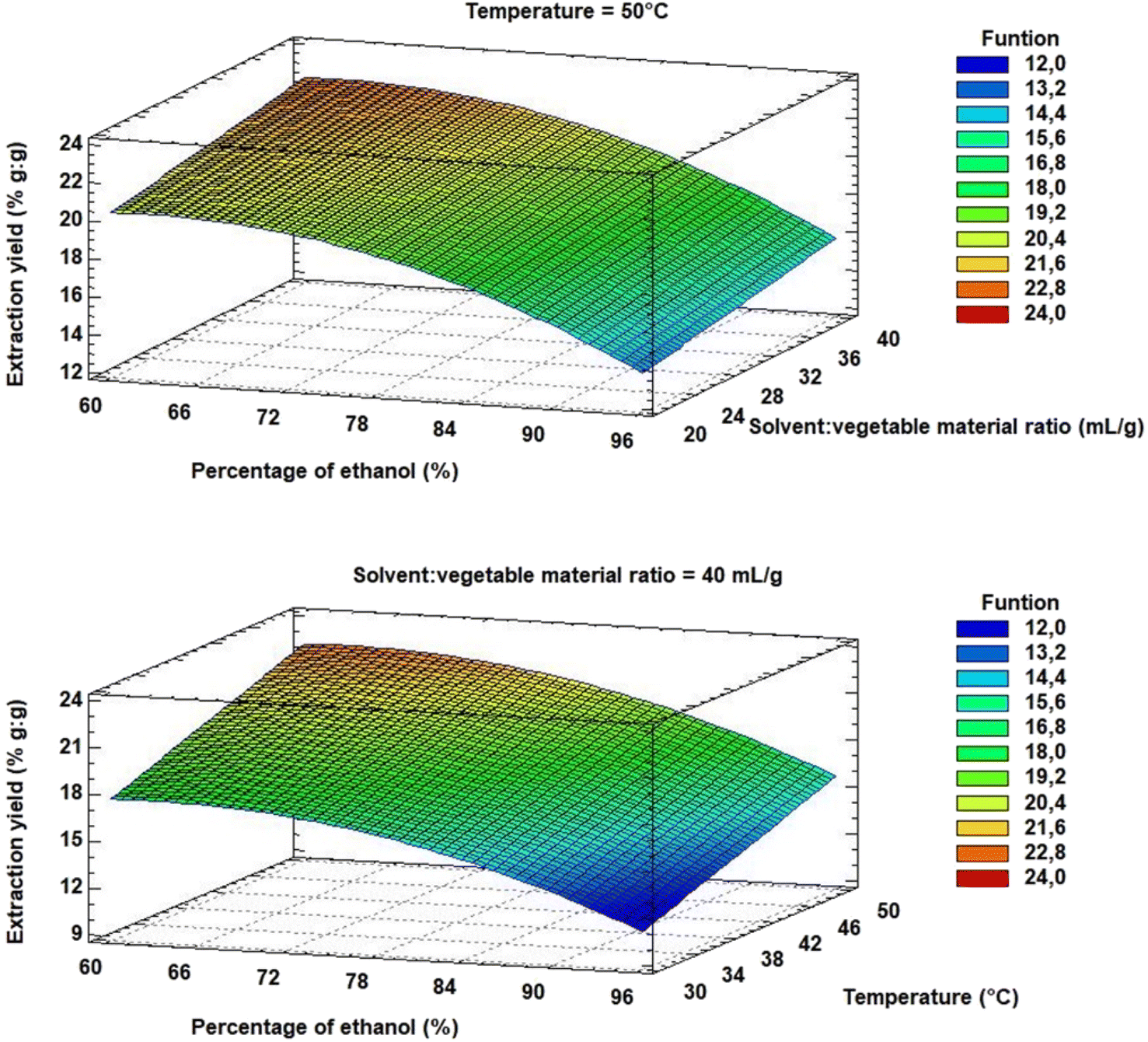 | ||
| Fig. 8 Response surface for the response variable extraction yield. | ||
Ultrasound-assisted extraction has been optimized previously for the extraction of quercetin from other sources. Optimum conditions for the maximum extraction of quercetin from onion skin were attained at 44% (v/v) ethanol, 22 min, and 606 W.21 Furthermore, extraction at 82% (v/v) ethanol, 41 mL solvent/g plant material ratio, 60 °C, 156 W, and 30 min gave maximum extraction of quercetin from leaves and stems of Dendrobium officinale.22 Moreover, the parameters obtained for maximum extraction of quercetin from the stalks of Euonymus alatus (Thunb.) Sieb were: 60% (v/v) ethanol, solvent volume:sample ratio 40 mL g−1, 200 W, 30 min, 30 °C, and ultrasound frequency 80 kHz.23 The differences in the optimal extraction conditions for the extraction of quercetin explain the differences found in the optimal extraction conditions found for quercitrin extraction in the current research [60% (v/v) ethanol at 50 °C, independent of the solvent:plant material ratio used] as compared with the previous report when extracted from Polygonum capitatum Buch.-Ham. ex D. Don [66% (v/v) aqueous ethanol, with an extractant:plant material ratio of 36 mL g−1].44
Conclusions
In this research, a chromatographic RP-HPLC-DAD method was validated for the quantification of quercitrin (quercetin-3-O-rhamnoside) from Nectandra reticulata leaves. Furthermore, the extraction of quercitrin was successfully optimized by means of a Box–Behnken design. The validation of the chromatographic method to analyze quercitrin allowed us to find that between the range of 5–180 mg quercitrin per L the response in absorbance at 350 nm presented a linear behavior. Recovery [93.8% (w/w)], and precision as measured by (i) repeatability (intra-assay, RSD 3.3% and SEM 0.35), (ii) intermediate precision (intra-laboratory, RSD 5.4% and SEM 0.47), and (iii) reproducibility (inter-laboratory, RSD 1.4% and SEM 0.09) were within the acceptable values as defined by ICH. Furthermore, the chromatographic method was proven to be robust, in terms of the stability of the retention time of quercitrin, when small variations in flow rate, column temperature, and wavelength of detection were done. In contrast, variations in formic acid concentration in the mobile phase statistically affected the retention time of quercitrin. Despite this aspect, the chromatographic method was specific. Quercitrin yield and chromatographic profile were found to depend on the location, with the sample collected in Granada yielding the greatest amount of quercitrin yield (2.380–3.399 mg of quercitrin/g of extract, depending on the extraction method), followed by those from Oiba (1.140–1.755 mg quercitrin/g of extract), and Chiquinquirá (0.272–0.579 mg quercitrin/g of extract). Quercitrin extraction (mg of quercitrin/g of plant material) by ultrasound-assisted extraction was highly affected by the composition of aqueous ethanol and temperature as evidenced by the Box–Behnken design. The highest quercitrin extraction was obtained at 60% (v/v) aqueous ethanol and 50 °C, independent of the solvent/plant material ratio. Furthermore, extraction yield (g extract/g plant material × 100) was affected by the three studied factors: the composition of aqueous ethanol, temperature, and solvent/plant material ratio, with the greatest values being obtained at 60% (v/v) aqueous ethanol, 50 °C, and 40 mL g−1 solvent/plant material ratio. The methodology is suitable not only for quercitrin quantification but also for its extraction from N. reticulata leaves developed in this research provides an important foundation for the formulation of a phytotherapeutic.Data availability
The data supporting this article have been included in the tables and figures provided in the manuscript.Author contributions
Juanita Pulido Teuta: investigation (lead); methodology (lead); data curation (equal). Carlos-Eduardo Narváez-Cuenca: supervision (equal); data curation (equal); writing-review; editing (equal). Mónica Ávila Murillo: conceptualization (equal); funding acquisition (equal); project administration (equal); supervision (equal); writing-review; editing (equal). Furthermore, all data were generated in-house, and no paper mill was used. All authors agree to be accountable for all aspects of work ensuring integrity and accuracy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Ministerio de Ciencia, Tecnología e Innovación, Contract number 727-2018.References
- A. M. Bustos Rangel, Búsqueda de agonistas LXR en plantas colombianas con potencial terapéutico para la enfermedad de Alzheimer, 2021, https://repositorio.unal.edu.co/handle/unal/80277, accessed March 12, 2022 Search PubMed.
- J. Pulido-Teuta, F. Lopez-Vallejo, A. Sandoval-Hernandez, C.-E. Narváez-Cuenca and M. Avila-Murillo, Bioactive glycosylated flavonoids with LXRs agonist activity in an ethanolic extract from Nectandra reticulate, submitted to.
- D. Predes, L. A. Maia, I. Matias, H. P. M. Araujo, C. Soares, F. G. Barros-Aragão and J. G. Abreu, The flavonol quercitrin hinders GSK3 activity and potentiates the Wnt/β-catenin signaling pathway, Int. J. Mol. Sci., 2022, 23, 12078, DOI:10.3390/ijms232012078.
- S. Rattanajarasroj and S. Unchern, Comparable attenuation of Aβ 25-35-induced neurotoxicity by quercitrin and 17β-estradiol in cultured rat hippocampal neurons, Neurochem. Res., 2010, 35, 1196–1205, DOI:10.1007/s11064-010-0175-6.
- G. Rajamanickam and S. L. Manju, Bio-guided isolation of anti-Alzheimer’s compounds from Phyllanthus niruri and role of niruriflavone in the reversal of aluminum chloride-induced neurobehavioral and biochemical changes in an animal model, Med. Chem. Res., 2022, 31, 1740–1753, DOI:10.1007/s00044-022-02944-5.
- L. Wang, J. Sun, Z. Miao, X. Jiang, Y. Zheng and G. Yang, Quercitrin improved cognitive impairment through inhibiting inflammation induced by microglia in Alzheimer's disease mice, Neuroreport, 2022, 33, 327–335, DOI:10.1097/WNR.0000000000001783.
- M. Kurkinen, M. Fułek, K. Fułek, J. A. Beszłej, D. Kurpasand and J. Leszek, The amyloid cascade hypothesis in Alzheimer's disease: Should we change our thinking?, Biomolecules, 2023, 13, 453, DOI:10.3390/biom13030453.
- A. Bustos-Rangel, J. Muñoz-Cabrera, L. Cuca, G. Arboleda, M. Ávila Murillo and A. G. Sandoval-Hernández, Neuroprotective and antioxidant activities of Colombian plants against paraquat and C2-ceramide exposure in SH-SY5Y cells, Frontiers in Natural Products, 2023, 2, 1169182, DOI:10.3389/fntpr.2023.1169182.
- N. F. Fitz, K. N. Nam, R. Koldamova and I. Lefterov, Therapeutic targeting of nuclear receptors, liver X and retinoid X receptors, for Alzheimer's disease, Br. J. Pharmacol., 2019, 176, 3599–3610, DOI:10.1111/bph.14668.
- S. K. Branch, Guidelines from the International Conference on Harmonisation (ICH), J. Pharm. Biomed. Anal., 2005, 38, 798–805, DOI:10.1016/j.jpba.2005.02.037.
- M. M. A. Sharifuldin, Z. Ismail, A. F. A. Aisha, E. K. Seow and H. K. Beh, Quantification of rutin, quercitrin and quercetin in Cosmos caudatus Kunth by reverse phase high performance liquid chromatography, Qual. Assur. Saf. Crops Foods, 2016, 8, 617–622, DOI:10.3920/QAS2015.0839.
- M. H. Nguyen, D. L. Ha, B. M. Do, N. T. N. Chau, T. H. Tran, N. T. H. Le and M. T. Le, RP-HPLC-based flavonoid profiling accompanied with multivariate analysis: an efficient approach for quality assessment of Houttuynia cordata Thunb leaves and their commercial products, Molecules, 2023, 28, 6378, DOI:10.3390/molecules28176378.
- C. Prompanya, A. Petchsomrit and B. Vongsak, Antioxidant evaluation and HPLC analysis of Buchanania lanzan and Buchanania siamensis leaf extracts, J. Res. Pharm., 2023, 27, 2480–2486, DOI:10.29228/jrp.535.
- V. Cambier, T. Hance and E. de Hoffmann, Variation of DIMBOA and related compounds content in relation to the age and plant organ in maize, Phytochemistry, 2000, 53, 223–229, DOI:10.1016/S0031-9422(99)00498-7.
- C. Xu, Y. Zhang, L. Zhu, Y. Huang and J. Lu, Influence of growing season on phenolic compounds and antioxidant properties of grape berries from vines grown in subtropical climate, J. Agric. Food Chem., 2011, 59, 1078–1086, DOI:10.1021/jf104157z.
- R. Leardi, Experimental design in chemistry: A tutorial, Anal. Chim. Acta, 2009, 652, 161–172, DOI:10.1016/j.aca.2009.06.015.
- S. L. C. Ferreira, R. E. Bruns, H. S. Ferreira, G. D. Matos, J. M. David, G. C. Brandão, E. G. P. da Silva, L. A. Portugal, P. S. dos Reis, A. S. Souza and W. N. L. dos Santos, Box-Behnken design: An alternative for the optimization of analytical methods, Anal. Chim. Acta, 2007, 597, 179–186, DOI:10.1016/j.aca.2007.07.011.
- V. Goud, A. Ramasamy, A. Das and D. Kalyanasundaram, Box-Behnken technique based multi-parametric optimization of electrostatic spray coating in the manufacturing of thermoplastic composites, Mater. Manuf. Processes, 2019, 34, 1638–1645, DOI:10.1080/10426914.2019.1666991.
- S. Raissi and R.-E. Farsani, Statistical process optimization through multi-response surface methodology, International Journal of Mathematical and Computational Science, 2009, 3, 197–201 Search PubMed.
- C. Guo, X. Gao, X. Zhao, B. Zhang, J. Chen, C. Chang and Z. Chen, Response surface optimization of extraction of rutin and quercetin from Cyclobalanopsis leaves by hydrothermal treatment catalyzed by ethanol-acetic acid, Biomass Convers. Biorefin., 2023, 13, 12291–12301, DOI:10.1007/s13399-021-02116-2.
- E. Y. Jin, S. Lim, S. oh Kim, Y.-S. Park, J. K. Jang, M.-S. Chung, H. Park, K.-S. Shim and Y. J. Choi, Optimization of various extraction methods for quercetin from onion skin using response surface methodology, Food Sci. Biotechnol., 2011, 20, 1727–1733, DOI:10.1007/s10068-011-0238-8.
- Y. Zhu, J. Yu, C. Jiao, J. Tong, L. Zhang, Y. Chang, W. Sun, Q. Jin and Y. Cai, Optimization of quercetin extraction method in Dendrobium officinale by response surface methodology, Heliyon, 2019, 5, e02374, DOI:10.1016/j.heliyon.2019.e02374.
- J. Liao, B. Qu and N. Zheng, Effects of process parameters on the extraction of quercetin and rutin from the stalks of Euonymus alatus (Thumb.) Sieb and predictive model based on least squares support vector machine optimized by an improved fruit fly optimization algorithm, Appl. Sci., 2016, 6, 340, DOI:10.3390/app6110340.
- Z. Lei, B. W. Sumner, A. Bhatia, S. J. Sarma and L. W. Sumner, UHPLC-MS analyses of plant flavonoids, Curr. Protoc. Plant Biol., 2019, 4, e20085, DOI:10.1002/cppb.20085.
- Á. A. Balaguer Beser and L. Á. Ruiz Fernández, Selección de un modelo de regresión lineal múltiple para el cálculo de la precipitación media en verano, 2021, https://riunet.upv.es/handle/10251/167659, accessed October 5, 2022 Search PubMed.
- I. García, M. C. Ortiz, L. Sarabia and J. M. Aldama, Validation of an analytical method to determine sulfamides in kidney by HPLC-DAD and PARAFAC2 with first-order derivative chromatograms, Anal. Chim. Acta, 2007, 587, 222–234, DOI:10.1016/j.aca.2007.01.054.
- S. Stipičević, S. Fingler, L. Zupančič-Kralj and V. Drevenkar, Comparison of gas and high performance liquid chromatography with selective detection for determination of triazine herbicides and their degradation products extracted ultrasonically from soil, J. Sep. Sci., 2003, 26, 1237–1246, DOI:10.1002/jssc.200301420.
- O. C. Ruge, S. L. E. Cuca and V. J. C. Martínez, Estudio químico y microbiológico del extracto etanólico de las hojas y corteza de Virola calophylla (Myristicaceae), Rev. Colomb. Cienc. Quim.-Farm., 1998, 27, 25–29 Search PubMed , https://revistas.unal.edu.co/index.php/rccquifa/article/view/56440/55406.
- M. M. A. Sharifuldin, Z. Ismail, A. F. A. Aisha, E. K. Seow and H. K. Beh, Quantification of rutin, quercitrin and quercetin in Cosmos caudatus Kunth by reverse phase high performance liquid chromatography, Qual. Assur. Saf. Crops Foods, 2016, 8, 617–622, DOI:10.3920/QAS2015.0839.
- M. H. Nguyen, D. L. Ha, B. M. Do, N. T. N. Chau, T. H. Tran, N. T. H. Le and M. T. Le, RP-HPLC-based flavonoid profiling accompanied with multivariate analysis: an efficient approach for quality assessment of Houttuynia cordata Thunb leaves and their commercial products, Molecules, 2023, 28, 6378, DOI:10.3390/molecules28176378.
- C. Prompanya, A. Petchsomrit and B. Vongsak, Antioxidant evaluation and HPLC analysis of Buchanania lanzan and Buchanania siamensis leaf extracts, J. Res. Pharm., 2023, 27, 2480–2486, DOI:10.29228/jrp.535.
- J. Agalloco, P. DeSantis, A. Grilli and A. Pavell, Handbook of Validation in Pharmaceutical Processes, CRC Press, 4th edn, 2021 Search PubMed.
- I. Taverniers, M. De Loose and E. Van Bockstaele, Trends in quality in the analytical laboratory. II. Analytical method validation and quality assurance, TrAC, Trends Anal. Chem., 2004, 23, 535–552, DOI:10.1016/j.trac.2004.04.001.
- C. Ramón and M. A. Gil-Garzón, Efecto de los parámetros de operación de la extracción asistida por ultrasonido en la obtención de polifenoles de uva: una revisión, Tecnlógicas, 2021, 24, e1822, DOI:10.22430/22565337.1822.
- N. Sharifi, S. Mahernia and M. Amanlou, Comparison of different methods in quercetin extraction from leaves of Raphanus sativus L, Pharmaceut. Sci., 2016, 23, 59–65, DOI:10.15171/PS.2017.09.
- S. Stark, R. Julkunen-Tiitto, E. Holappa, K. Mikkola and A. Nikula, Concentrations of foliar quercetin in natural populations of white birch (Betula pubescens) increase with latitude, J. Chem. Ecol., 2008, 34, 1382–1391, DOI:10.1007/s10886-008-9554-8.
- M. L. Pérez-Ochoa, A. M. Vera-Guzmán, D. M. Mondragón-Chaparro, S. Sandoval-Torres, J. C. Carrillo-Rodríguez and J. L. Chávez-Servia, Effects of growth conditions on phenolic composition and antioxidant activity in the medicinal plant Ageratina petiolaris (Asteraceae), Diversity, 2022, 14, 595, DOI:10.3390/d14080595.
- G. Vilkickyte and L. Raudone, Phenological and geographical effects on phenolic and triterpenoid content in Vaccinium vitis-idaea L. leaves, Plants, 2021, 10, 1986, DOI:10.3390/plants10101986.
- L. Yang, K. S. Wen, X. Ruan, Y. X. Zhao, F. Wei and Q. Wang, Response of plant secondary metabolites to environmental factors, Molecules, 2018, 23, 762, DOI:10.3390/molecules23040762.
- M. M. Qaderi, A. B. Martel and C. A. Strugnell, Environmental factors regulate plant secondary metabolites, Plants, 2023, 12, 447, DOI:10.3390/plants12030447.
- R. Ksouri, W. Megdiche, H. Falleh, N. Trabelsi, M. Boulaaba, A. Smaoui and C. Abdelly, Influence of biological, environmental and technical factors on phenolic content and antioxidant activities of Tunisian halophytes, C. R. Biol., 2008, 331, 865–873 CrossRef CAS PubMed.
- S. H. Hassanpour and A. Doroudi, Review of the antioxidant potential of flavonoids as a subgroup of polyphenols and partial substitute for synthetic antioxidants, Avicenna J. Phytomed., 2023, 13, 354, DOI:10.22038/AJP.2023.21774.
- N. V. Zagoskina, M. Y. Zubova, T. L. Nechaeva, V. V. Kazantseva, E. A. Goncharuk, V. M. Katanskaya and M. A. Aksenova, Polyphenols in plants: Structure, biosynthesis, abiotic stress regulation, and practical applications, Int. J. Mol. Sci., 2023, 24, 13874, DOI:10.3390/ijms241813874.
- F. Ma, Y. Zhao, X. Gong, Y. Xie and X. Zhou, Optimization of quercitrin and total flavonoids extraction from Herba Polygoni Capitati by response surface methodology, Pharmacogn. Mag., 2014, 10, S57–S64, DOI:10.4103/0973-1296.127343.
| This journal is © The Royal Society of Chemistry 2024 |