
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePregnenolone derivatives for the treatment of Alzheimer's disease: synthesis, and in vitro inhibition of amyloid β1–42 peptide aggregation, acetylcholinesterase and carbonic anhydrase-II†
Ayesha Tahir‡
a,
Bushra Mobeen‡a,
Fahad Hussaina,
Abdul Sadiqb and
Umer Rashid *a
*a
aDepartment of Chemistry, COMSATS University Islamabad, Abbottabad Campus, Abbottabad, 22060, Pakistan. E-mail: umerrashid@cuiatd.edu.pk
bDepartment of Pharmacy, University of Malakand, Chakdara, KP 18000, Pakistan
First published on 7th May 2024
Abstract
The amyloid state, which is a specific conformation of proteins, offers valuable information about both functional protein structures and the pathological assemblies associated with various diseases. One of the major hallmarks of Alzheimer's disease includes primarily the extracellular build-up of a peptide known as amyloid-β, which has a sequence consisting of 39 to 42 amino acid residues, and the formation of intracellular neurofibrillary tangles mostly consisting of hyperphosphorylated tau protein. Drugs that are expected to reduce Aβ production, prevent Aβ aggregation, and promote Aβ clearance are promising approaches for treating AD. Current work is focused on identifying the compounds that have balanced even mild biological activities against multiple targets instead of finding one-target compound with high potency. We synthesized pregnenolone derivatives and evaluated their potential against inhibition of eeAChE/eqBChE, hCA-II and self-mediated Aβ1–42 peptide aggregation. Our synthesized derivatives 23, and 25–27 exhibited concomitant inhibition of all the tested macromolecular targets. All the active compounds were found to be BBB penetrants in the PAMPA assay. Furthermore, these selected compounds were found to be non-neurotoxic in the MTT assay on neuroblastoma SH-SY5Y cells. Docking studies support dual binding site (PAS and CAS) inhibition of AChE which showed Aβ1–42 aggregation and AChE inhibition. Moreover, docking studies carried out on the 3D crystallographic structure of Aβ1–42 peptide (PDB ID = 1IYT) showed significant interactions with amino acid residues Asp 23 and Lys 28, and hydrophobic interactions with the Phe19, Phe20, and Ala 30 effectively impeding the formation of β-sheet structures.
Introduction
The amyloid state, which is a specific conformation of proteins, offers valuable information about both functional protein structures and the pathological assemblies associated with various diseases.1 Amyloids are characterized by their insoluble, fibrillary deposits consisting of β-sheet secondary structures.2 These structures are observed in numerous diseases and tend to accumulate as plaques and inclusions with advancing age. Notable examples of key proteins that form amyloid fibrils in specific diseases include Aβ in Alzheimer's disease (AD), hunting-tin in Huntington's disease, and α-synuclein in Parkinson's disease.3 AD, a neurodegenerative condition, is characterized by the accumulation of a peptide called Amyloid-β1–42 peptide.4,5Compared to other fragments formed when amyloid precursor protein (APP) is sliced, Aβ is chemically “stickier”. Gradually, it builds up into tiny amyloid plaques, which are recognized as a hallmark of an Alzheimer's-affected brain.6 First, the fragments form tiny clusters known as oligomers, followed by chains of clusters known as fibrils, and finally, “mats” of fibrils known as beta-sheets. Plaques, the last stage, are composed of various substances and beta-sheet clumps. These phases of beta-amyloid accumulation consequently disrupt neuronal function, interfere with cell-to-cell communication and trigger immunological responses, as per the amyloid hypothesis. The brain cells are ultimately destroyed when inflammation is brought on by these immune cells. Thus, the two main pathogenic AD mechanisms are plaque deposition and massive amyloid beta (Aβ) aggregates.7–9
The complex mechanisms responsible for Aβ aggregation involve genetic, environmental, and metabolic factors. As amyloid plaques are a hallmark of AD because the aggregate formation of Aβ sets off a series of actions that eventually cause brain cell degradation and cognitive loss as well as memory decline.10
Scientists are devoting an enormous amount of time and energy to finding out the potential therapeutic approaches that stop beta-amyloid aggregation as a possible therapy for the condition.11,12 Amyloid aggregation inhibitors, including polyoxometalates (POM), metallohelices, nanomaterials, and peptides, have also been rationally designed. However, there are still hazards associated with BBB permeability alterations, even with the effectiveness of therapeutic medicines in brain uptake.13,14 Additionally, they are searching for strategies to stop the first contact between Aβ and nerve cells that cause toxicity since some data indicates that the harmful effects of beta-amyloid happen before plaques and oligomers develop.15–17 Alvarez in 1997 and Inestrosa in 1996 suggested that the “peripheral anionic site” (PAS) of AChE is involved in the aggregation and formation of toxic Aβ oligomers. The 42-mer peptide, Aβ1–42, is the more toxic among these two (i.e., Aβ1–42 and Aβ1–40).18–20 Therefore, blocking the PAS could serve to reduce Aβ aggregation in order to increase the chance to prevent harmful effects. Moreover, a number of authors studied the effect of AChE PAS inhibitors (propidium and fasciculin) and active site inhibitor (edrophonium) on Aβ aggregation process experimentally.21–25 Therefore, small molecules that can inhibit the PAS of AChE may also prevent Aβ from aggregating.26 Moreover, the Aβ-dependent toxicity is elevated by the AChE-Aβ and it depends on the bulk of complexes that are formed. The amyloidogenic pathway is one of the pathways for the breakdown of amyloid precursor protein or APP.27,28 Beta secretase which is also known as BACE-1 and gamma secretases are responsible for this commonly named amyloidogenic pathway. So, by inhibiting the BACE-1 and gamma secretases, Aβ levels can be degraded.29,30
Zinc-containing metallo-enzyme carbonic anhydrase (CA) was initially discovered in cow RBC in 1933.31,32 A number of investigations provided fresh insights into the potential of carbonic anhydrases (CAs) as therapeutic targets for Alzheimer's disease (AD). In prokaryotes and eukaryotes, carbonic anhydrases (CAS) are zinc-bearing metalloenzymes that initiate the reversible reaction between carbon dioxide and bicarbonate ions.33 The zinc ion at the CA active site is surrounded by hydroxide ions (OH) and possesses the same catalytic activity.34,35 Three histidine residues (His 94, His 96, and His 119) as well as side-chain residues that make coordination bonds with the zinc ion are found in the CA enzyme.36 When developing CA inhibitors, the design of the inhibitors that target hCA-II is crucial. The main sulfonamide group (RSO2NH2)-containing compounds make up the foundation of all CA inhibitors (hCA-II). Sulfonamide hCA-II inhibitors, including acetazolamide, methazolamide, ethoxzolamide, dichlorphenamide, and others, have demonstrated significant inhibition against the majority of human hCA isoforms at generally low nanomolar doses. So far, a number of compounds have been created to inhibit different isoforms of carbonic anhydrase.37 Clinically available carbonic anhydrase inhibitors, such as acetazolamide (AZA), dorzolamide, methazolamide, brinzolamide, ethoxazolamide, diclofenamide, indisulam, zonisamide, etc., are used to treat a variety of conditions, including ocular hypertension, glaucoma, and epilepsy.38–41
Steroids include various structurally related compounds primarily originating from plant and animal kingdoms.42 Perhydro-cyclopentanophenanthrene is the common structural nucleus present in this class of compounds.43 Steroids either natural or semi-synthetic have been involved in the handling of inflammatory, immunology, infectious and metabolic diseases.44 In 2004, Abbate et al., reported hCA-I, II and IX inhibitions by Estrone-3-O-sulfamate (EMATE, 1).45 In 2014, the inhibition mechanism of human carbonic anhydrase-II by bile acid (2) was first explored (see Fig. 1).
In 2018 Nocentini et al. reported the various carbonic anhydrase isoforms inhibition of 22 steroids having pendants and functional groups of sulfonates, phenols and carboxylates. Among these 22 bile acids/steroids, hyocholic acid (3, Ki = 38.9 μM), α-estradiol (4, Ki = 40.4 μM), deoxycholic acid (5, Ki = 51.0 μM), and oestrone (Ki = 50.8 μM) showed good CA-II inhibition.46 A. Khalid et al. reported that four recognized bases sarcosine, sarcodine, sarcosine, and alkaloid-C as well as a novel steroidal alkaloid, is isosarcodine 6, were extracted from extract (MeOH) of Sarcococca saligna.47 After AChE and butyrylcholinesterase (BuChE) inhibition experiments were directed on these compounds, isosarcodine was discovered as a non-competitive inhibitor of AChE (having Ki = 21.8 μM). As, carbonic anhydrases' catalytic activity is inhibited by sulfonamides. Compound 7 was reported as a potent CA IX inhibitor by Cecchi, Alessandro et al.48 In 2022, compound 8 was reported by Peschiulli et al. It is an oral bioavailable and highly effective (hAβ1–42 cell IC50 = 1.3 nM) molecule that produced a persistent decrease in Aβ in mouse and dog animal models (see Fig. 1).49
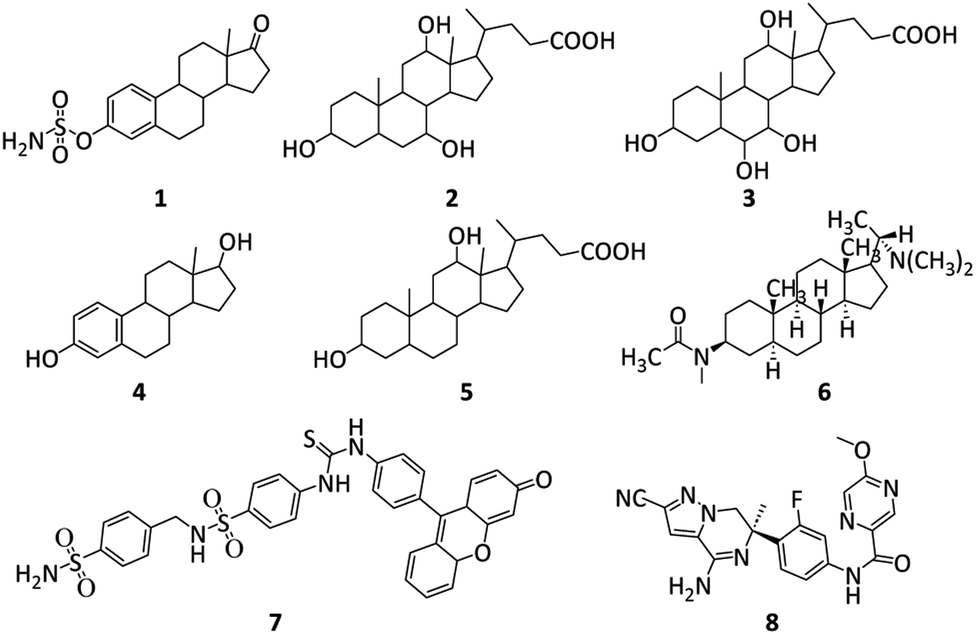 | ||
| Fig. 1 Structures of reported AChE, Aβ and CA inhibitors.45–49 | ||
Pregnenolone (PREG) is a neurosteroid synthesized from cholesterol in both mammals and invertebrates.50 Neurosteroids are known for their rapid activity, as they are synthesized within the brain and for the brain. Within the brain, pregnenolone and its various metabolic derivatives (pregnenolone sulfate, allopregnanolone, and dehydroepiandrosterone), have demonstrated the ability to improve learning and memory, alleviate depression, and regulate cognitive functions.51 Moreover, these compounds play a neuroprotective role in addressing neuroinflammatory diseases like AD and multiple sclerosis (MS), as well as in managing neuropsychiatric disorders such as schizophrenia, depression, and autism.29,52
Our research group is involved in the structural modification of steroidal nuclei for various disease treatments. Recently, we reported structural modification of pregnenolone at C-3 and C-17 positions.53 Excellent in vitro inhibition results were exhibited by Pyrimidine derivatives of pregnenolone for human dihydrofolate reductase (hDHFR). In another report, we have also synthesized C-3 pregnenolone dihydropyrimidine derivatives for the breast cancer treatment.54 In the current research, we planned to modify the pregnenolone core at C-3 and C-17 positions. 4-(Sulfamoylphenyl)thiourea moiety was incorporated at position C-3. While α-bromination of the acetyl group present at position C-17 resulted in the synthesis of various amine derivatives.
Materials and methods
General
The purified reagents/solvents were acquired from viable vendors (standard) and were employed. From Sigma Aldrich and Alfa-Aesar, we have purchased the starting materials. Tetramethyl silane (TMS, internal reference) was employed for recording 1H and 13C-NMR spectra by utilizing deuterated solvents at 400 MHz and 100 MHz on a Bruker spectrometer. Chemical shifts are specified in δ scale (ppm). TLC was utilized to investigate the progress of all the reactions that is 2.0 × 5.0 cm aluminium sheets (having silica gel 60F254 on it and 0.25 mm thickness). The purity of the synthesized compounds was analysed by using Shimadzu HPLC system with C18 RP column. The mobile phase was composed of acetonitrile/water (30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70 v/v). The system was run in an isocratic mode with at a 1.0 mL min−1 flow rate. The compounds with a purity greater than 95% were used for bioevaluation studies. The CHN elemental analysis was carried out by utilizing the LECO-932 CHN analyzer (LECO Corporation, USA).
:70 v/v). The system was run in an isocratic mode with at a 1.0 mL min−1 flow rate. The compounds with a purity greater than 95% were used for bioevaluation studies. The CHN elemental analysis was carried out by utilizing the LECO-932 CHN analyzer (LECO Corporation, USA).
General method for synthesis of 1-((8S,9S,10R,13S,14S)-3-amino-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)ethan-1-one (12)
In a round bottom flask, 25 mmol of pregnenolone and 25.5 mmol of p-toluene sulfonyl chloride in 30 mL dichloromethane (CH2Cl2), trimethylamine (Et3N, 30 mmol) were added and heat under reflux condition. TLC was utilized to monitor the completion of the reaction. After the completion of the reaction, ice water was poured into it and stirred. The resulting aqueous solution was extracted from ethyl acetate (3 × 20 mL). Finally, saturated NaHCO3 was used for washing the combined organic layer and anhydrous NaSO4 was utilized for drying. The organic layer was removed and the resulting tosylate 11 was dried and used for the next step without any further purification.The tosylate 11 (20 mmol) was suspended in acetone. Then ammonium acetate (40 mmol) was added and at room temperature for 3–4 hours mixture was stirred. The resulting precipitates were filtered off, diethyl ether was used for washing and drying to afford compound 12.
Yield = 70%; 1H NMR (400 MHz, CDCl3) δ 5.25 (brs, 1H), 4.54 (d, J = 6.0 Hz, 2H), 3.74 (t, J = 5.6 Hz, 1H), 2.55 (t, 1H), 2.45–2.37 (m, 3H), 2.32–2.27 (m, 1H), 2.21–2.18 (m, 1H), 2.14 (s, 3H), 2.10–2.02 (m, 2H), 1.90–1.86 (m, 1H), 1.77–1.67 (m, 4H), 1.66–1.63 (m, 1H), 1.59–1.52 (m, 1H), 1.50–1.42 (m, 2H), 1.35–1.28 (m, 1H), 1.25 (s, 3H), 1.18–1.14 (m, 2H), 0.68 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 208.5, 139.2, 120.9, 64.5, 53.8, 51.8, 48.2, 44.7, 41.2, 37.9 (2C), 34.4, 32.5 (2C), 30.5, 29.3, 26.6, 23.8, 21.9, 19.3, 15.9. Analysis calculated for C21H33NO: C, 79.95; H, 10.54; N, 4.44; O, 5.07. Observed: C, 80.04; H, 10.52; N, 4.46.
4-(3-((8S,9S,10R,13S,14S)-17-Acetyl-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3yl)thioureido)benzenesulfonamide (15)
To a stirred solution of 12 (20 mmol) in 30 mL acetone added 20 mmol o-(p-tolyl)chlorothionoformate 13. For 2h, the mixture was kept at continuous stirring at RT. The TLC technique was utilized to check the completion of the reaction. The mixture was filtered to obtain intermediate 14. In the second step, crude intermediate 14 in 5 mL 1,4-dioxane was added 8 mmol of sulfanilamide followed by 16 mmol triethylamine. It was then stirred at 60 °C for 3 h. The resulting precipitates, after that diethyl ether was employed for washing, and dried to afford compound 15.Light yellow powder. Yield = 61%; Rf = 0.38 (n-hexane/EtOAc 8:1); HPLC purity = 98.9%, Rt = 5.78 min, 1H NMR (400 MHz, CDCl3) δ 8.25 (s, 1H, –NH), 7.90 (d, 2H, J = 8.4 Hz, ArH), 7.74 (d, 2H, J = 8.4 Hz, ArH), 6.76 (s, 2H, NH2), 6.51 (s, 1H, NH), 5.27–5.21 (m, 1H), 3.73 (t, 1H, J = 5.6 Hz), 2.59–2.54 (m, 1H), 2.41–2.36 (m, 1H), 2.28–2.26 (m, 1H), 2.24–2.21 (m, 1H), 2.15 (s, 3H), 2.10–2.02 (m, 2H), 1.82–1.76 (m, 1H), 1.74–1.66 (m, 4H), 1.53–1.46 (m, 6H), 1.29–1.26 (m, 1H), 1.22 (s, 3H), 1.09–1.04 (m, 2H), 0.69 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 208.5, 180.6, 141.1, 140.4, 136.1, 125.2 (2C), 122.1 (2C), 63.2, 53.2, 50.2, 48.2, 43.8, 38.5, 35.9, 33.3 (1C), 32.1, 31.5 (2C), 28.1, 26.4, 24.3, 20.5, 18.3 (2C), 15.6 (2C). Analysis calculated for C28H39N3O3S2: C, 63.48; H, 7.42; N, 7.93; O, 9.06; S, 12.10. Observed: C, 63.36; H, 7.44; N, 8.78.
The general method for the synthesis of α-brominated derivative 16
A solution of 3-amino pregnenolone derivative (15, 10 mmol) in 25 mL of chloroform was stirred. To this solution, bromine (10 mmol) in CHCl3 was added drop-wise by using a dropping funnel. After that, it was stirred for 15–20 minutes at 50 °C and cooled to RT. The final crude product was recrystallized from acetic acid.Light brown powder. Yield = 79%; Rf = 0.36 (n-hexane/EtOAc 8:1); 1H NMR (400 MHz, CDCl3) δ 8.24 (s, 1H, –NH), 7.92 (d, 2H, J = 8.48 Hz, ArH), 7.74 (d, 2H, J = 8.48 Hz, ArH), 6.77 (s, 2H, NH2), 6.51 (s, 1H, NH), 5.26–5.20 (m, 1H), 3.71 (t, 1H, J = 5.6 Hz), 2.57–2.53 (m, 1H), 2.41–2.36 (m, 1H), 2.28–2.20 (m, 2H), 2.19 (s, 3H), 2.12–2.06 (m, 2H), 1.82–1.76 (m, 1H), 1.72–1.68 (m, 2H), 1.55–1.48 (m, 4H), 1.46–1.43 (m, 2H), 1.27–1.24 (m, 1H), 1.20 (s, 3H), 1.12–1.06 (m, 2H), 0.71 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 204.3, 181.8, 141.5, 140.7, 135.2, 129.4 (2C), 122.5 (2C), 61.3, 54.5, 52.6, 49.3, 45.6, 39.0, 37.2, 36.5, 36.1, 35.4, 33.6, 29.7, 27.1, 25.3, 23.8, 20.4, 18.2 (2C), 13.3. Analysis calculated for C28H38BrN3O3S2: C, 55.25; H, 6.29; Br, 13.13; N, 6.90; O, 7.89; S, 10.53. Observed: C, 55.36; H, 6.24; N, 6.88.
The general method for the synthesis of derivatives 23–28
To a solution of α-brominated 16 (5 mmol) in DMF, 5.25 mmol of trimethylamine (Et3N) and 7.5 mmol of amines (17–22) were added. The mixture was refluxed overnight. Upon completion of the reaction, the pregnenolone derivatives were purified by column chromatography using petroleum ether/ethyl acetate 20:1 for compound 23 and 10:1 for 24–28.
4-(3-((8S,9S,10R,13S,14S)-10,13-Dimethyl-17-((4-sulfa-moylphenethyl)glycyl)-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl)-thio-ureido)benzenesulfonamide (23)
Light yellow powder. Yield = 61%; Rf = 0.40 (n-hexane/EtOAc 20:1); m.p. = 195–197 °C; HPLC purity = 99%, Rt = 6.72 min, 1H NMR (400 MHz, CDCl3); δ 8.23 (s, 1H, –NH), 7.94 (d, 2H, J = 8.4 Hz, ArH), 7.77 (d, 2H, J = 8.4 Hz, ArH), 7.41 (d, 2H, J = 8.0 Hz, ArH), 7.24 (d, 2H, J = 8.0 Hz, ArH), 6.98 (s, 4H, 2 × NH2), 6.54 (s, 1H, NH), 5.35–5.29 (m, 1H), 4.25–4.20 (m, 1H), 3.42 (d, 2H, J = 5.6 Hz, CH2), 3.33–3.29 (m, 1H), 2.99–2.94 (m, 2H), 2.78 (t, 2H, J = 5.6 Hz, CH2), 2.73–2.69 (m, 1H, NH), 2.56–2.48 (m, 2H, CH2), 1.91–1.88 (m, 1H), 1.75–1.69 (m, 1H), 1.58–1.47 (m, 6H), 1.45 (m, 4H), 1.23 (m, 4H), 1.00 (s, 3H), 0.93 (m, 1H), 0.79 (s, 3H).13C NMR (100 MHz, CDCl3) δ 204.3, 180.5, 149.3, 141.5, 140.1, 138.0, 137.1, 136.3, 128.4 (2C), 127.8 (2C), 126.8 (2C), 126.0, 124.5, 121.4, 113.4, 77.0, 76.3, 62.3, 58.2, 55.5, 49.7, 48.1, 40.2, 39.8, 37.9, 37.0, 34.4, 28.8, 24.4, 23.4, 21.5, 17.2, 15.8. Analysis calculated for C36H49N5O5S3: C, 59.39; H, 6.78; N, 9.62; O, 10.99; S, 13.21. Observed: C, 59.45; H, 6.76; N, 9.59.
4-(3-((8S,9S,10R,13S,14S)-10,13-Dimethyl-17-(phenethylglycyl)-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradeca-hydro-1H-cyclopenta[a]phenanthren-3-yl)thioureido)benzenesulfonamide (24)
Off-white powder. Yield = 66%; Rf = 0.46 (n-hexane/EtOAc 8:1); m.p. = 152–154 °C; HPLC purity = 98%, Rt = 7.5 min, 1H NMR (400 MHz, CDCl3) δ 8.23 (s, 1H, –NH), 7.93 (d, 2H, J = 8.4 Hz, ArH), 7.75 (d, 2H, J = 8.4 Hz, ArH), 7.24–7.19 (m, 5H, ArH), 6.75 (s, 2H, NH2), 6.51 (s, 1H, NH), 5.29–5.24 (m, 1H), 4.13 (d, 1H, J = 7 Hz), 3.97–3.95 (m, 2H), 3.89–3.86 (m, 2H), 3.30 (d, 2H, J = 5.2 Hz, CH2), 2.50–2.35 (m, 5H), 2.22–2.17 (m, 4H), 1.95–1.85 (m, 6H), 1.76–1.69 (m, 2H), 1.62–1.54 (m, 2H), 1.24–1.19 (m, 2H), 0.96 (s, 3H), 0.77 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 204.8, 180.6, 149.3, 141.3, 138.0, 136.5, 128.3 (2C), 126.6 (2C), 124.5, 122.0, 121.4, 113.4, 76.3, 77.0, 76.3, 62.3, 58.2, 55.5, 50.5, 48.7, 49.7, 43.4, 40.2, 39.8, 37.6, 37.3, 34.4, 32.2, 28.8, 24.4, 23.2, 21.5, 17.6, 15.5. Analysis calculated for C36H48N4O3S2: C, 66.63; H, 7.46; N, 8.63; O, 7.40; S, 9.88. Observed: C, 66.70; H, 7.47; N, 8.61.
4-(3-((8S,9S,10R,13S,14S)-17-(Benzylglycyl)-10,13-di-methyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradeca-hydro-1H-cyclopenta[a]phenanthren-3-yl)thioureido)benzenesulfonamide (25)
Off white powder. Yield = 68%; Rf = 0.48 (n-hexane/EtOAc 8:1); m.p. = 163–165 °C; HPLC purity = 97%, Rt = 8.8 min, 1H NMR (400 MHz, CDCl3) δ 8.27 (s, 1H, –NH), 7.96 (d, 2H, J = 8.4 Hz, ArH), 7.75 (d, 2H, J = 8.4 Hz, ArH), 7.53–7.45 (m, 5H, ArH), 6.73 (s, 2H, NH2), 6.53 (s, 1H, NH), 5.37–5.31 (m, 1H), 4.88 (d, 2H, J = 5.2 Hz, CH2), 4.12–4.05 (m, 1H), 3.51–3.47 (m, 2H), 2.98 (t, 1H, J = 5.4 Hz), 2.92–2.86 (m, 1H), 2.53–2.54 (m, 4H), 2.27–2.09 (m, 4H), 1.94–1.77 (m, 2H), 1.76–1.61 (m, 2H), 1.59–1.50 (m, 1H), 1.37–1.30 (m, 2H), 1.25–1.21 (m, 1H), 0.95 (s, 3H), 0.89–0.81 (m, 1H), 0.77 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 204.9, 180.5, 149.3, 141.6, 139.7, 136.8, 129.4 (2C), 127.6 (2C), 123.4, 122.5, 121.0, 113.6, 77.5, 77.1, 76.6, 62.4 (2C), 58.2, 55.5, 50.5, 49.7, 43.4, 39.2, 37.6, 37.1, 34.4, 32.2, 28.8, 24.4, 23.4, 21.5, 17.8, 15.6. Analysis calculated for C35H46N4O3S2: C, 66.21; H, 7.30; N, 8.82; O, 7.56; S, 10.10. Observed: C, 66.26; H, 7.31; N, 8.81.
4-(3-((8S,9S,10R,13S,14S)-10,13-Dimethyl-17-(phenylglycyl)-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl)thioureido)benzenesulfonamide (26)
Off white powder. Yield = 59%; Rf = 0.44 (n-hexane/EtOAc 8:1); m.p. = 183–185 °C; HPLC purity = 99%, Rt = 8.3 min, 1H NMR (400 MHz, CDCl3) δ 8.24 (s, 1H, –NH), 7.90 (d, 2H, J = 8.0 Hz, ArH), 7.77 (d, 2H, J = 8.0 Hz, ArH), 7.25–7.22 (m, 2H, ArH), 7.18–7.14 (m, 2H, ArH), 7.11–7.08 (m, 1H, ArH), 6.75 (s, 2H, NH2), 6.55 (s, 1H, NH), 5.26–5.23 (m, 1H), 4.99 (t, 1H, J = 4.8 Hz, NH), 3.99 (d, 2H, J = 4.8 Hz, CH2), 3.76–3.72 (m, 1H), 2.56–2.47 (m, 2H), 2.37–2.28 (m, 2H), 2.22–2.16 (m, 2H), 1.99–1.88 (m, 1H), 1.77–1.63 (m, 6H), 1.57–1.43 (m, 4H), 1.29–1.15 (m, 5H), 1.10–1.02 (m, 1H), 0.94 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 204.7, 180.6, 149.0, 141.6, 138.5, 136.8, 129.3 (2), 127.9 (2), 121.2 (2), 113.7 (2C), 77.4, 77.1, 76.8, 57.3, 55.6, 52.2, 50.5, 49.9, 43.4, 39.3, 37.6, 37.0, 34.6, 32.2, 28.8, 24.4, 23.4, 21.0, 17.6, 15.9. Analysis calculated for C34H44N4O3S2: C, 65.77; H, 7.14; N, 9.02; O, 7.73; S, 10.33. Observed: C, 65.84; H, 7.16; N, 9.00.
4-(3-((8S,9S,10R,13S,14S)-17-(2-(10H-Phenothiazin-10-yl)acetyl)-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl)thioureido)benzenesulfonamide (27)
Yellow powder. Yield = 69%; Rf = 0.49 (n-hexane/EtOAc 8:1); m.p. = 170–172 °C; HPLC purity = 99%, Rt = 9.43 min, 1H NMR (400 MHz, CDCl3) δ 8.24 (s, 1H, –NH), 7.91 (d, 2H, J = 8.4 Hz, ArH), 7.77 (d, 2H, J = 8.4 Hz, ArH), 6.91–6.85 (m, 8H), 6.78 (s, 2H, NH2), 6.55 (s, 1H, NH), 5.27–5.25 (m, 1H), 3.96 (s, 2H, CH2), 3.77–3.73 (m, 1H), 3.33 (m, 2H), 2.74–2.63 (m, 2H), 2.42–2.32 (m, 1H), 2.15–1.88 (m, 9H), 1.75–1.71 (m, 2H), 1.60–1.51 (m, 3H), 1.36–1.24 (m, 4H), 0.96 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 207.2, 181.1, 144.5, 140.1, 139.0, 136.2, 128.7, 128.7, 127.8, 127.8, 127.3, 127.3, 126.9, 126.9, 122.5, 122.4, 122.2, 120.8, 120.8, 116.7, 116.4, 57.8, 56.4, 55.6, 50.4, 49.6, 43.2, 39.2, 37.7, 37.1, 34.6, 32.2, 32.1, 27.7, 24.4, 24.4, 23.5, 21.1, 19.0, 16.1. Analysis calculated for C40H46N4O3S3: C, 66.08; H, 6.38; N, 7.71; O, 6.60; S, 13.23. Observed: C, 66.18; H, 6.40; N, 7.69.
4-(3-((8S,9S,10R,13S,14S)-10,13-Dimethyl-17-((1,2,3,4-tetrahydroanthracen-9-yl)glycyl)-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl)thioureido)benzenesulfonamide (28)
Yellow powder. Yield = 64%; Rf = 0.55 (n-hexane/EtOAc 8:1); m.p. = 214–216 °C; HPLC purity = 99%, Rt = 11.20 min, 1H NMR (400 MHz, CDCl3) δ 8.25 (s, 1H, –NH), 7.91 (d, 2H, J = 8.4 Hz, ArH), 7.78 (d, 2H, J = 8.4 Hz, ArH), 7.63–7.58 (m, 2H), 7.17–7.13 (m, 2H), 6.97 (s, 2H, NH2), 6.52 (s, 1H, NH), 5.32–5.28 (m, 1H), 3.43 (d, 2H, CH2), 3.38–3.36 (m, 2H), 3.34–3.26 (m, 2H), 2.58–2.46 (m, 3H), 2.34–2.28 (m, 1H), 2.15–1.88 (m, 9H), 1.94–1.78 (m, 9H), 1.71–1.68 (m, 2H), 1.58–1.55 (m, 1H), 1.53–1.49 (m, 2H), 1.42–1.36 (m, 2H), 1.23–1.18 (m, 1H), 0.96 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 209.5, 181.1, 140.5, 140.1, 139.0, 136.2, 134.2, 128.1, 127.9, 127.8(2), 127.2, 126.1, 125.3(2), 125.1, 123.3, 122.2, 120.8(2), 58.1, 55.8, 54.1, 50.4, 49.6, 43.3, 39.2, 37.7, 37.1, 34.6, 32.6, 32.1, 31.0, 29.5, 27.7, 24.9, 24.4, 24.0, 23.5, 21.1, 19.0, 16.1. Analysis calculated for C42H52N4O3S2: C, 69.58; H, 7.23; N, 7.73; O, 6.62; S, 8.84. Observed: C, 69.65; H, 7.24; N, 7.71.
In vitro CA-II inhibition assay
CA inhibition studies were performed using human CA-II (Sigma, Catalogue No. C6165) reported assay procedures with little modification.55–57In vitro acetyl/butyrylcholinesterase inhibition assays
Ellman method is utilized to evaluate the inhibition potential (IC50 values) of synthesized compounds and their derivatives against electric eel acetylcholinesterase (eeAChE).57,58 Enzymes AChE is used for the hydrolysis of acetylcholine/butyrylcholine iodide which is used as a basis for the principle of this inhibition assay. An anion 5-thio-2-nitrobenzoate is yielded as a result of this hydrolysis which afterwards form a yellow colour complex with Ellman's reagent; 5,5-dithio-bis-2-nitrobenzoic acid (DTNB) solution and that can be determined by utilizing spectrophotometer.For the preparation of solution, the compound is dissolved separately in a 0.1 M phosphate buffer and different concentrations are formed that can range from 250 to 1000 micrograms per millilitre. To prepare the solution of phosphate buffer with a certain pH, few micrograms per litre of monopotassium phosphate and dipotassium phosphate are mixed in a ratio and for adjusting the pH, potassium hydroxide is used. Then solid AChE/BChE is diluted in a freshly formed phosphate buffer as far as required final concentration is achieved. Subsequently, the solutions of acetylcholine/butyrylcholine and DTNB are prepared in distilled water and these solutions are placed in the refrigerator after enclosing them in an Eppendorf Cap. Compound and positive control are dissolved in methanol and then further dilutions of compound are prepared.
Self-mediated Aβ1–42 aggregation assay
The impact of synthesized compounds on self-mediated Aβ aggregation was evaluated by using a thioflavin T-based fluorometric test.10,59–61 Initially pretreated Aβ1–42 peptide was dissolved in DMSO to prepare 100 μM stock solution. The Aβ1–42 peptide stock solution was diluted to a concentration of 10 μM in 150 mM phosphate buffer (pH 7.4) containing 150 mM NaCl prior to the incubations. Next, 10 μL of the test compounds and 20 μL of Aβ1–42 was combined, and the mixture was applied to the appropriate wells on 96-well plates with black walls. After that, the mixture was quickly diluted with 70 μL of thioflavin T solution (ThT) to get a final volume of 100 μL (2 μM final Aβ1–42 concentration). Every sample was run in triplicate. The sealed 96-well plate was placed in a plate reader (Synergy H4) and heated to 37 °C to start the aggregation process. Every 90 seconds, the ThT fluorescence was monitored through the plate's bottom at an excitation wavelength of 440 nm and an emission wavelength of 490 nm. The media was shaken constantly in between measurements to quantify the development of amyloid fibrils. After 1 hour, the Aβ1–42's ThT emission started to grow, reached a plateau after 8 hours, and then stayed essentially unaltered for an additional 16 hours of incubation. The average fluorescence intensities at the plateau were calculated by summing the fluorescence intensities at the corresponding wells at t = 0 h, both in the presence and absence of the various doses of test substances (1, 5, 10, 15, 20, 25 μM). The amount of inhibition of Aβ1–42 self-induced aggregation was measured using the formula:| (1 − Fi/F0) × 100%, |
Cell viability assay test
To evaluate the Aβ1–42 toxicity in the presence or absence of designed compounds, we performed the cell viability assays. Promega cat. CellTiter 96 aqueous non-radioactive cell proliferation assay kit was used to perform this assay. To analyze the cytotoxicity of Aβ under various conditions, SH-SY5Y cell lines were used. RPMI 1640 medium (ATCC formulated) was utilized to culture the SH-SY5Y cells supplemented with 10% Fetal bovine serum (FBS). SH-SY5Y cells were plated in 96-well plates at 10000 cells per well and then it was cultured in 5% carbon dioxide for 20 hours at 37 °C. After that, pre-disaggregated and purified samples of Aβ were solvated in phosphate-buffered buffered saline (PBS) up to an ultimate concentration of 5 micromolars for preparing the solutions of peptide inhibitors and Aβ mixture. Then at various concentrations, different peptide inhibitors were added. A 0.22 micromolar filter was utilized to filter the mixture solution and then without shaking incubate it for 16 hours at 37 °C. In each well that contains a 90 microlitre medium, a pre-incubated mixture of 10 microliters was added to begin the cell viability assay and then for 24 hours it was incubated in 5% carbon dioxide at 37 °C. Then, in each well a dye solution of 15 microliters was added. After incubating it at 37 °C for four hours, 100 microliters stop mix or solubilization solution was put into a solution. It was followed by further incubation for 12 hours at room temperature and then at 570 nm reading of absorbance was taken along with the background reading at 700 nm. Quadruplicate readings for each sample were collected in parallel. Then, the rate for cell survival was normalized by utilizing the cells treated with PBS at 100% and 0.02 per cent cells treated with SDS at 0% viability.
Docking studies
Molecular operating environment (MOE, 2016.0802) was employed for docking simulations on human carbonic anhydrase II complexed with acetazolamide (PDB accession: 3HS4), AChE complexed with bis-tacrine (PDB accession: 2CKM) and Aβ1–42 (PDB accession: 1IYT) and β-sheet pentamer or Aβ42 protofibril oligomer (PDB ID = 2BEG).The downloaded enzyme was subjected to 3D protonation and energy minimization for stable conformation. Protonation of synthesized compound was done and after that energy minimization was employed by utilizing parameter at default operation (Gradient 0.00001, Amber 10EHT Force field). After the preparation of enzyme and ligand structures, the reliability of the docking procedure to reproduce the correct binding orientation was determined by the re-docking of the extracted ligand from the crystal structure. The protocol with an RMSD value less than 2 Å was used for docking runs. For the current study, triangle matcher (placement), affinity dG (scoring), GBVI/WSA (re-scoring) protocol was used. Ten conformations for each ligand were generated and top-ranked conformations were selected for analysis. Three-dimensional/two-dimensional ligand interactions were visualized by using MOE interaction plots and discovery studio visualizer.
Results and discussion
Design rational
Structural modification of the steroidal nucleus has got considerable attention in recent years. This is because steroid-based therapeutics have several advantages and possess interesting structural features for the treatment of various diseases.Multitarget-directed ligands (MTDLs) have emerged as a successful strategy for the design of therapeutics for multifactorial diseases like Alzheimer's disease. The combination of two or more pharmacophoric moieties into a large molecular weight single multipotent entity has been reported to have increased therapeutic advantages and chances of success. Initially, several predictive docking analyses were conducted on target enzyme X-ray crystal structures to anticipate optimum moieties on the left and right sides of pregnenolone. Steroidal cores are complex lipophilic molecules and are responsible for the improvement of BBB permeability. At the C-3 position, 4-(sulfamoylphenyl)thiourea moiety was chosen due to its ability to form hydrogen bond interactions with mid-gorge amino acid residues of AChE and carbonic anhydrase-II. Moreover, the sulfonamide group, also known as the zinc binding group (ZBG), is considered highly important for designing CA inhibitors. We then pursued exploring the influence of different amines with diverse sizes (tricyclic/monocyclic systems) and linker lengths (17–22) in the C-17 acetyl group (Fig. 2). This iterative rational approach allows for designing a pregnenolone-based framework that enhances cholinesterase, carbonic anhydrase and Aβ aggregation inhibition.
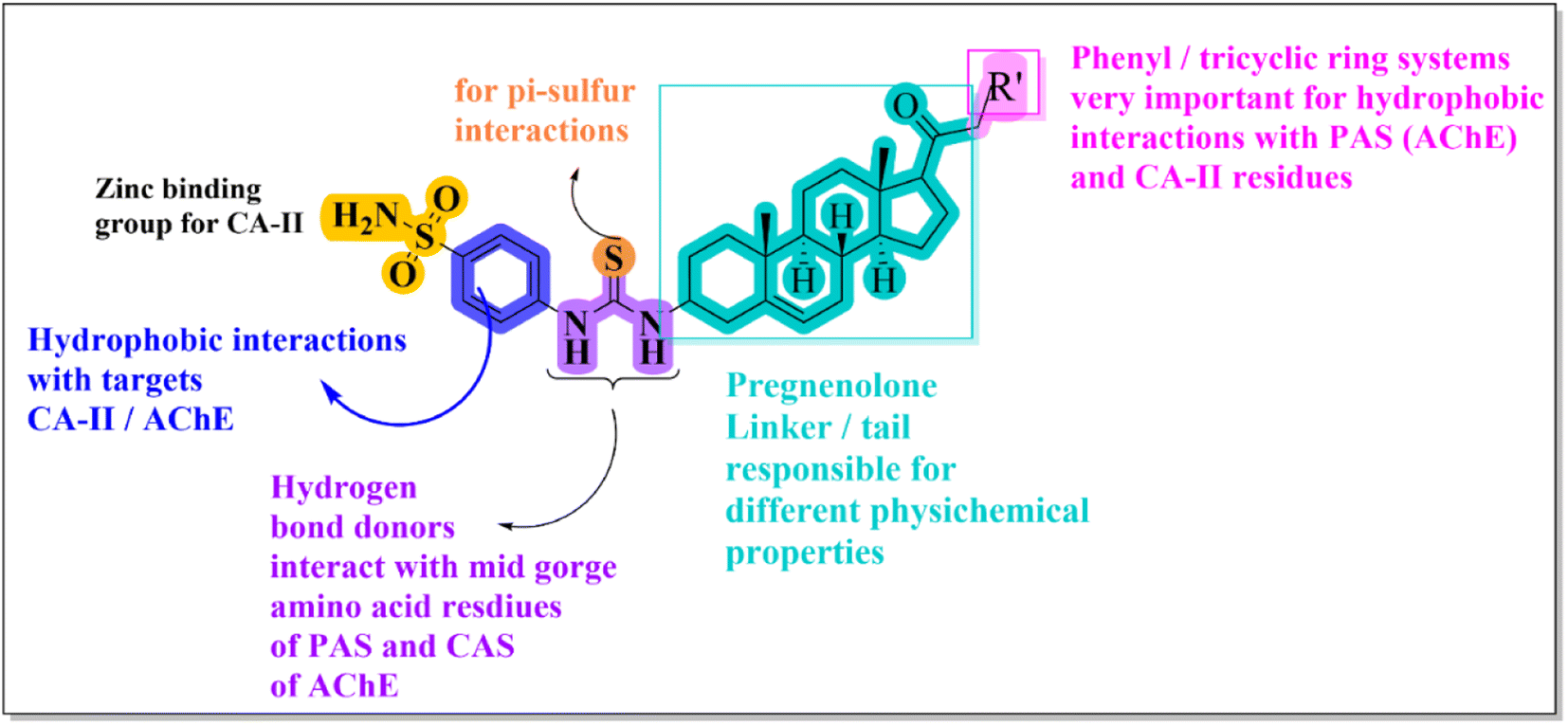 | ||
| Fig. 2 Design rationale for current research. | ||
Chemistry
Synthesis of the 3-amino derivative of pregnenolone 12 is outlined in Scheme 1. Pregnenolone 9 was reacted with p-toluene sulfonyl chloride 10 to give tosylate 11. For the synthesis of C-3 amine derivative of pregnenolone, ammonium acetate was used. Matloobi et al. used ammonium acetate in DMSO for the introduction of amine. In the current study, we used acetone instead of DMSO.62,63 Tosylate 11 was further reacted with ammonium acetate in acetone to give the target compound 12.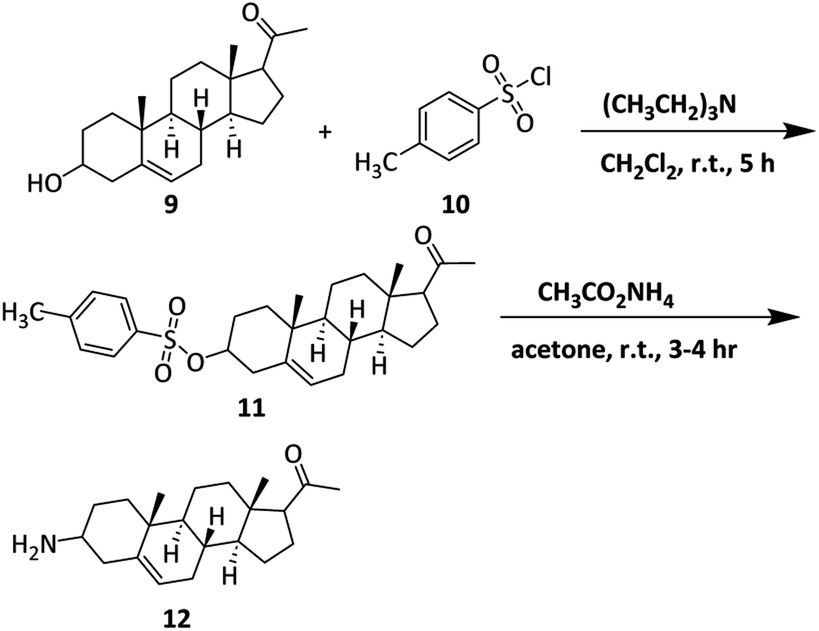 | ||
| Scheme 1 Synthesis of 3-amino derivative of pregnenolone. | ||
The synthesis of sulfonamide derivative 15 is outlined in Scheme 2. Here, we used o-(p-tolyl)chlorothionoformate 13 for the synthesis of sulfonamide-thiourea adduct 15. Reaction of 3-amino derivative of pregnenolone 12 with o-(p-tolyl) chlorothionoformate 13 in acetone at room temperature stirring gave intermediate 14 in good yield. The synthesized intermediate finally reacted with sulfanilamide in 1,4-dioxane solvent under reflux conditions to give product 15.
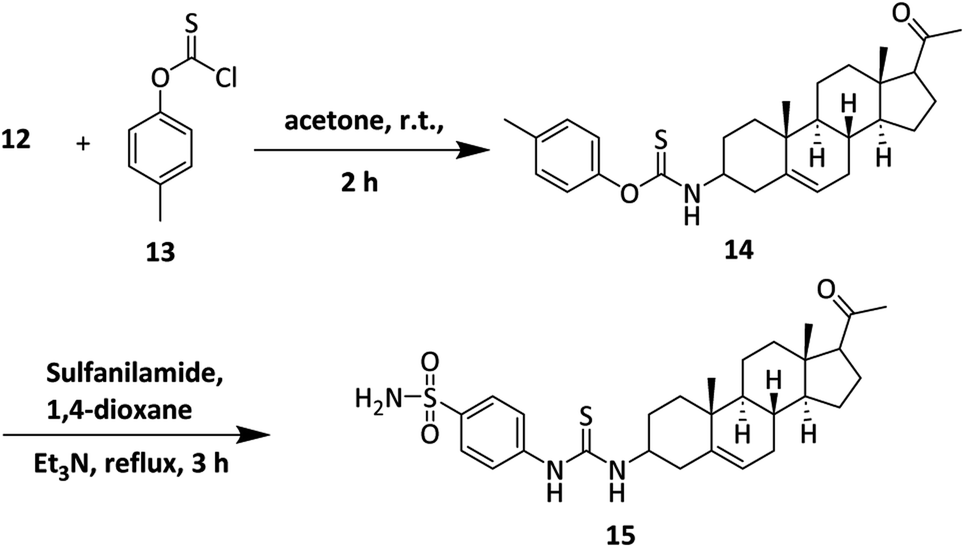 | ||
| Scheme 2 Synthesis of sulfonamide-thiourea adduct 15. | ||
Finally, the synthesis of target sulfonamide derivatives 23–28 is outlined in Scheme 3. The acetyl group was brominated by using bromine in chloroform.64 The bromo-compound was used for the next step without any further purification. Compound 16 was reacted with various amine derivatives 17–22 in DMF to obtain target compounds 23–28.
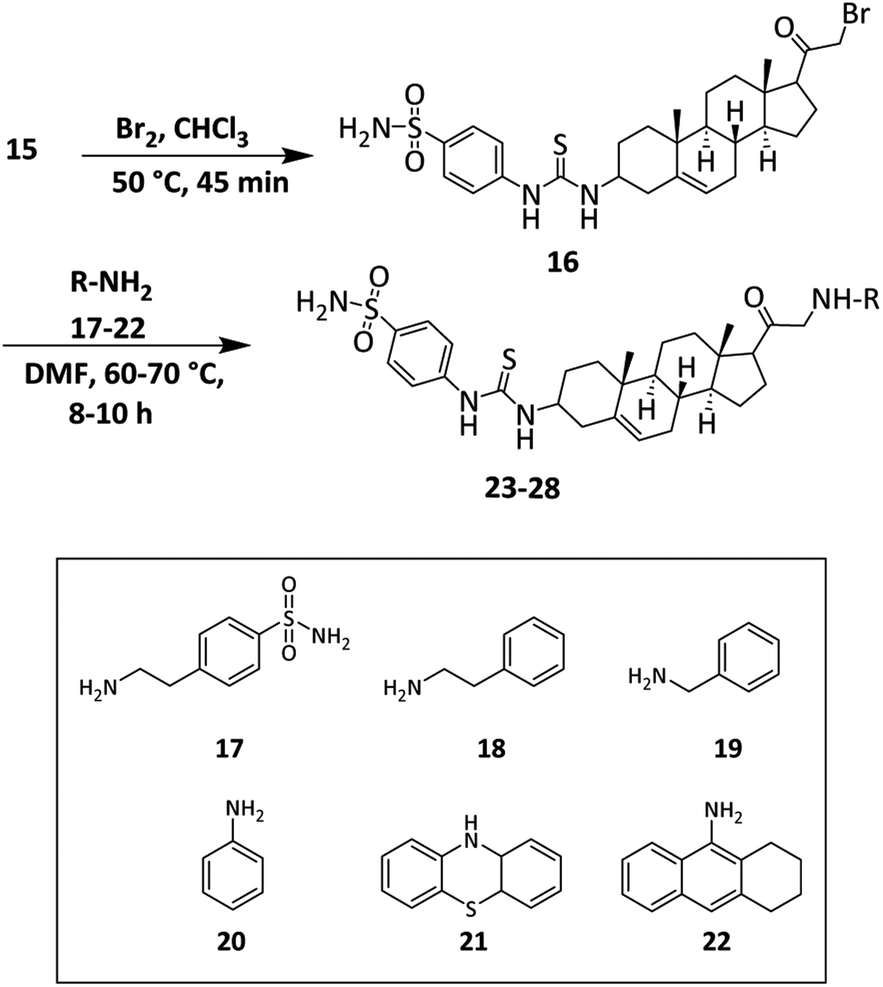 | ||
| Scheme 3 Synthesis of target compounds (23–28) of pregnenolone. | ||
In vitro pharmacology
| Compd no. | Structures | IC50a (μM) ± SEM | ||||
|---|---|---|---|---|---|---|
| hCA-II | eeAChE | eqBChE | SIb | Amyloid β1–42 peptide aggregation inhibition c | ||
| a Values represent mean ± SEM; n = 3.b Selectivity index = IC50 of BChE/IC50 of AChE.c The Aβ1–42 was incubated in the absence or presence of different concentrations of the inhibitors (pregnenolone derivatives).d NA = no activity found in tested concentrations; yellow highlighted boxes represent the compounds that have balanced (moderate to excellent) biological activities against all tested target. | ||||||
| 9 | 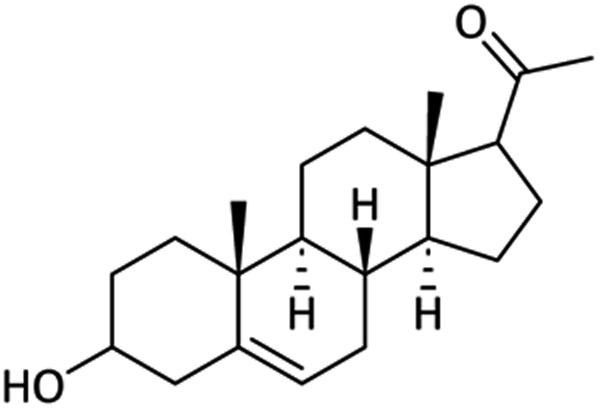 |
89.38 ± 1.21 | 73.18 ± 0.83 | <100 | — | NAd |
| 12 | 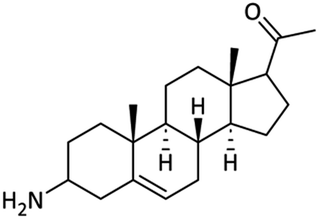 |
73.19 ± 1.14 | 67.67 ± 1.09 | <100 | — | NA |
| 15 | 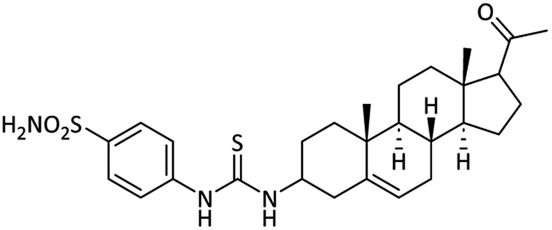 |
48.91 ± 1.37 | 12.98 ± 0.18 | 63.02 ± 1.23 | 4.8 | 9.25 ± 0.17 |
| 23 | 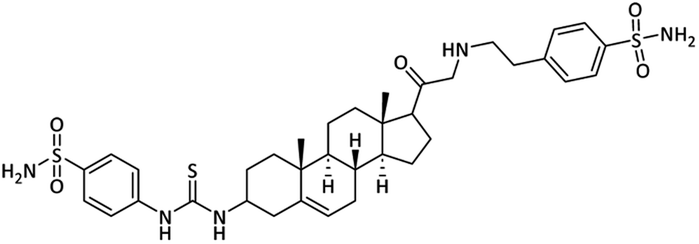 |
0.67 ± 0.04 | 0.043 ± 0.001 | 0.58 ± 0.03 | 14.5 | 13.08 ± 0.16 |
| 24 | 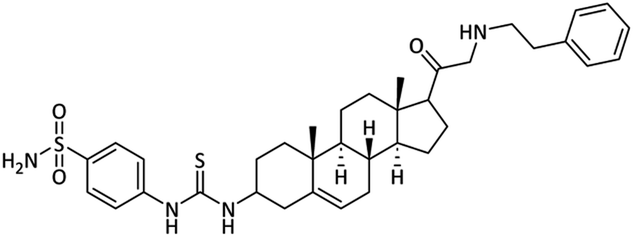 |
5.44 ± 0.14 | 1.61 ± 0.10 | 12.84 ± 0.45 | 8.0 | 29.64 ± 1.21 |
| 25 | 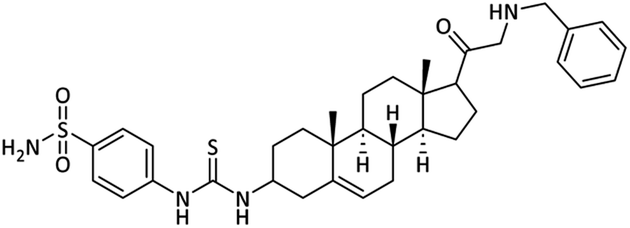 |
1.02 ± 0.01 | 0.98 ± 0.10 | 11.27 ± 0.11 | 11.5 | 12.84 ± 0.15 |
| 26 | 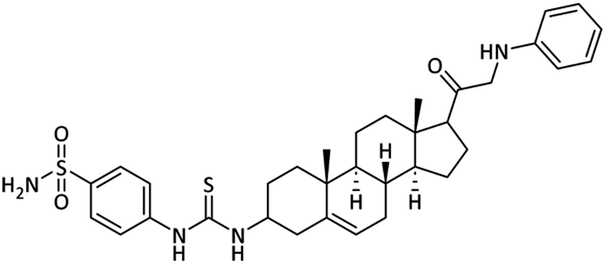 |
1.56 ± 0.02 | 0.12 ± 0.01 | 2.84 ± 0.04 | 23.6 | 1.04 ± 0.01 |
| 27 | 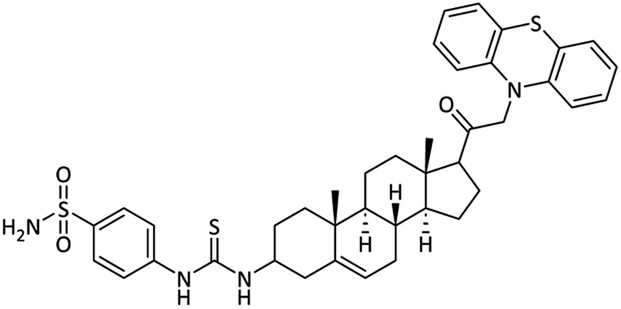 |
5.18 ± 0.12 | 0.094 ± 0.002 | 15.01 ± 0.03 | 166.7 | 5.78 ± 0.07 |
| 28 | 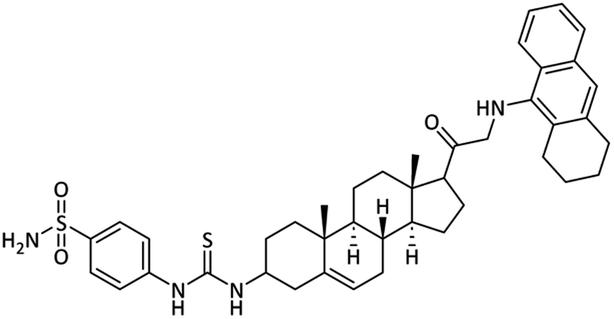 |
31.83 ± 1.08 | 0.32 ± 0.01 | 13.50 ± 0.09 | 42.2 | 26.37 ± 0.25 |
| Acetazolamide | 0.028 ± 0.001 | — | — | — | — | |
| Donepezil | — | 0.054 ± 0.001 | — | — | — | |
| Curcumin | — | — | — | — | 7.29 ± 0.03 | |
Self-mediated Aβ1–42 aggregation inhibition
The selected synthesized compounds were used in the Thioflavin-T (ThT) fluorescence assay test to evaluate the effect on Aβ aggregation.10 For that purpose, each compound with one and half equivalent concentrations (1:0.5 and 1:1) was employed with Aβ1–42 peptide. Using ThT, a fluorescent dye that attaches specifically to fibrous structures, we were able to track the development of Aβ aggregation over time in the presence of certain compounds (15, 23, 26 and 27). Control experiment Aβ1–42 alone in the ThT research demonstrated an increase in fluorescence intensity, but reduced fluorescence intensity was observed when Aβ1–42 was incubated with synthesized derivatives and curcumin (standard drug/positive control). All of the synthesized derivatives have the potential to interact with the Aβ peptide and prevent self-aggregation, based on the findings of the experiments. Initially, the fluorescence intensity of Aβ1–42 was evaluated independently. The percentage of aggregates for the samples treated with synthesized compounds was evaluated by taking the Aβ1–42 aggregation alone as 100%. The samples treated with curcumin, compound 15, compound 23, compound 26 and compound 27 fabricated minor amyloid fibrils. Furthermore, the sample treated with compound 27 exhibited less aggregation than compound 26 to some extent. After 24 hours of equimolar treatment, curcumin and compound 15 showed the inhibition of approximately 60% while compound 27 and compound 26 showed 85% and >80% Aβ inhibition of fibrils respectively (Fig. 3).
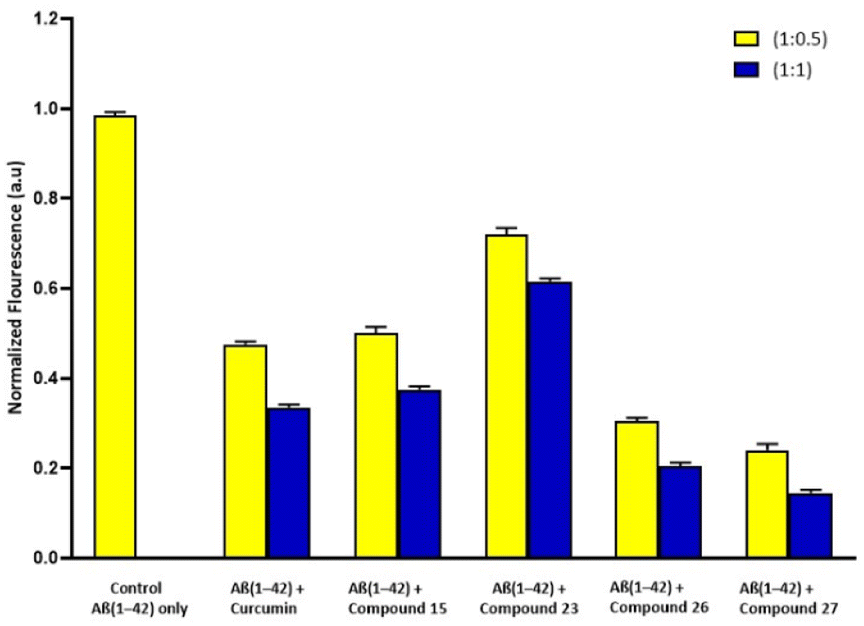 | ||
| Fig. 3 Bar diagram illustration of the effect of curcumin and compounds (15, 23, 26 and 27) on ThT fluorescence and aggregation of Aβ1–42 peptide after incubated for 24 hours. | ||
Concentration-dependent self-mediated Aβ1–42 aggregation inhibition
Following that, we explored the Aβ1–42 aggregation (self-mediated) inhibition, with a focus on concentration dependence. Firstly Aβ1–42 was incubated in both conditions (absence or presence) of different concentrations of the inhibitors (pregnenolone derivatives). The calculated IC50 values in μM of the compounds are presented in Table 1. Pregnenolone derivatives 15, 26 and 27 showed good inhibition potential towards self-mediated Aβ1–42 aggregation having IC50 values of 9.25 μM, 1.04 μM and 5.78 μM, respectively (Table 1). Furthermore, curcumin (standard drug) exhibited an IC50 value of 7.29 μM. The dose–response curve is shown in Fig. 4. This curve displays the inhibitory compound's (15, 23–28 and curcumin) concentration on the x-axis and the percentage inhibition on the y-axis. Results have shown that the amount of inhibition rises along with the concentration of the inhibitory compound.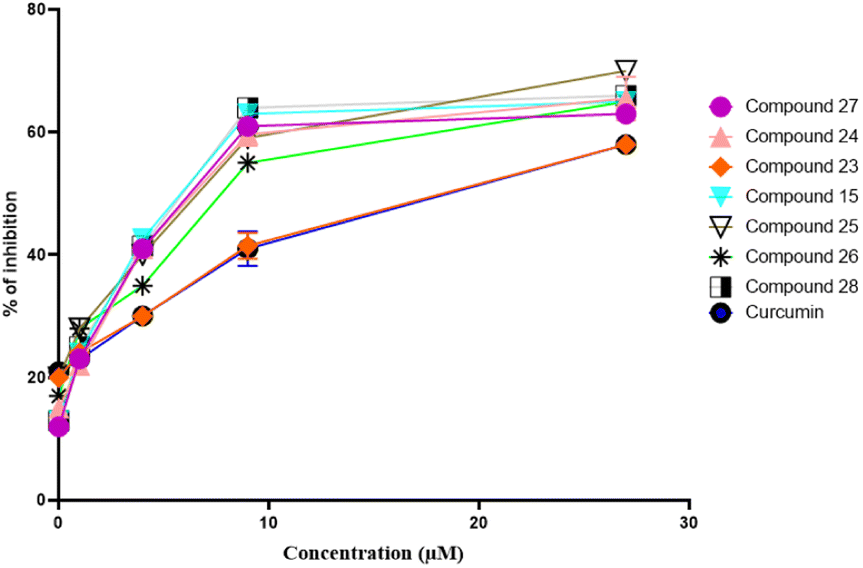 | ||
| Fig. 4 Dose–response curve of compounds 15, 23–28, and curcumin against self-induced Aβ1–42 aggregation. | ||
Cell viability/neurotoxicity
The cell lines of human neuroblastoma SH-SY5Y were employed to assess the profile cytotoxicity of Aβ1–42 aggregates with the selected synthesized compounds.65,66 Experiments for cell viability were conducted to figure out whether these compounds can abandon the neuronal cells from the toxicity of Aβ-species. MTT assay was accomplished to compare induced Aβ1–42 death of neurons in the incidence and absence of distinct concentrations. Cell viability was assessed at 1, 5, and 10 μM doses along with the standard drug donepezil. Most potent compounds 15, 26 and 27 were selected for this MTT assay. Results are summarized in Fig. 5. The results showed that the viability of cells treated with Aβ1–42 was reduced in contrast to control cells which were without any compound or peptide. Compound 27 at 5 μM was completely non-cytotoxic. Significant toxicity was observed on incubating 5 μM Aβ1–42 with cells of SH-SY5Y, as the death of cells was about 65% enhanced than in the control. On incubation of cells with Aβ1–42 having the selected compounds, an obvious dose–response neuroprotection was perceived. Compound 27, cells of human neuronal SH-SY5Y were effectively protected from the toxicity of the Aβ peptide. Donepezil (standard drug), compound 15 and compound 26 along with Aβ1–42 peptide increased cell viability up to approximately 60%, 75% and 90% respectively at the equimolar concentrations.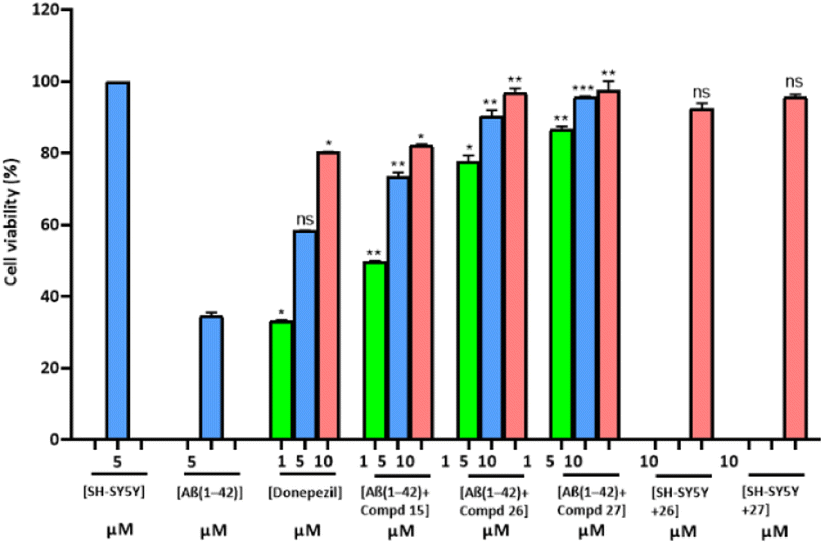 | ||
| Fig. 5 Viability of the SH-SY5Y cells with Aβ1–42 in the presence and absence of designated concentrations of donepezil and compounds (15, 26, and 27) after 48 h, as determined using the MTS assay. The control group was observed as 100% cell viability, and the assays were performed in triplicate. *P < 0.05, **P < 0.01, ***P < 0.001 and ns = non-significant. | ||
Hence, Aβ1–42 peptide in the presence of these compounds, especially 26 and 27 exhibited low toxicity at high concentrations.
The SH-SY5Y cells were also contemplated with compound 26 and compound 27 at 10 μM concentrations to analyze the cytotoxicity of compounds. Compound 27 showed a more noticeable effect than compound 26 on comparing the average viability. So, the results have shown that both compounds 26 and 27 had a reduced level of Aβ aggregates and also had reduced cellular cytotoxicity (Fig. 5).
PAMPA BBB assay
In AD therapies the blood–brain barrier (BBB) penetration is a necessary concern. Parallel artificial membrane permeation assay (PAMPA) was employed by utilizing a previously reported procedure.59,61,67 Pregnenolone 9 and its 3-amino derivative 12 showed high BBB penetration. Selected all three compounds i.e. 23, 26 and 27 also found to be BBB penetrants in PAMPA assay (Table 2). The presence of a hydrophobic steroidal nucleus could be the reason for this permeation.| Compounds number | (PAMPA-BBB)a Pe(tested) (10−6 cm s−1) | Prediction of CNS penetrationb,c |
|---|---|---|
| a Data represent are the assay mean for the marketed drugs (n = 3).b ‘CNS+’ (prediction of high BBB permeation); Pe (10−6 cm s−1) > 4.39.c ‘CNS−’ (prediction of low BBB permeation); Pe (10−6 cm s−1) < 1.78. | ||
| Pe(tested) evaluation (10−6 cm s−1) for the standard and compounds | ||
| 23 | 16.4 | High |
| 26 | 13.8 | High |
| 27 | 18.1 | High |
| Pregnenolone 9 | 20.4 | High |
| C-3 amino-pregnenolone (12) | 19.6 | High |
| Donepezil | 16.7 | High |
|
||
| Validation of the model by four commercial drugs | ||
| Diazepam | 15.30 | High |
| Atenolol | 0.75 | No |
| Alprazolam | 5.60 | High |
| Lomefloxacin | 1.12 | No |
Docking studies
Docking simulations were employed via molecular operating environment software (MOE, 2016.0208) on human carbonic anhydrase II complexed with acetazolamide (PDB accession: 3HS4). The reliability of the docking procedure to reproduce the correct binding orientation was determined by the re-docking of the extracted ligand from the crystal structure. The protocol with an RMSD value less than 2 Å was used for docking runs.Docking studies on hCA-II
Molecular docking runs showed favourable binding modes and hydrophilic/hydrophobic binding affinities with the important residues (amino acid) present in the binding site. In all cases, the sulfonamide nucleus was found to coordinate with the Zn centre. A two-dimensional (2D) binding affinity plot of potent compound 23 is illustrated in Fig. 6. 4-(Sulfamoylphenyl)thiourea moiety was found in contact with the Zn centre via coordination bond with one of the nitrogen atoms of sulfonamide at a distance of 2.07 Å. The computed binding energy for compound 23 is −9.087 kcal mol−1. The binding pose showed that the 4-(sulfamoylphenyl)ethylamino group oriented towards the zinc centre and penetrated deep into the catalytic site. While all the other compounds contacted the zinc centre via (4-sulfamoylphenyl)thiourea moiety (Fig. 6a).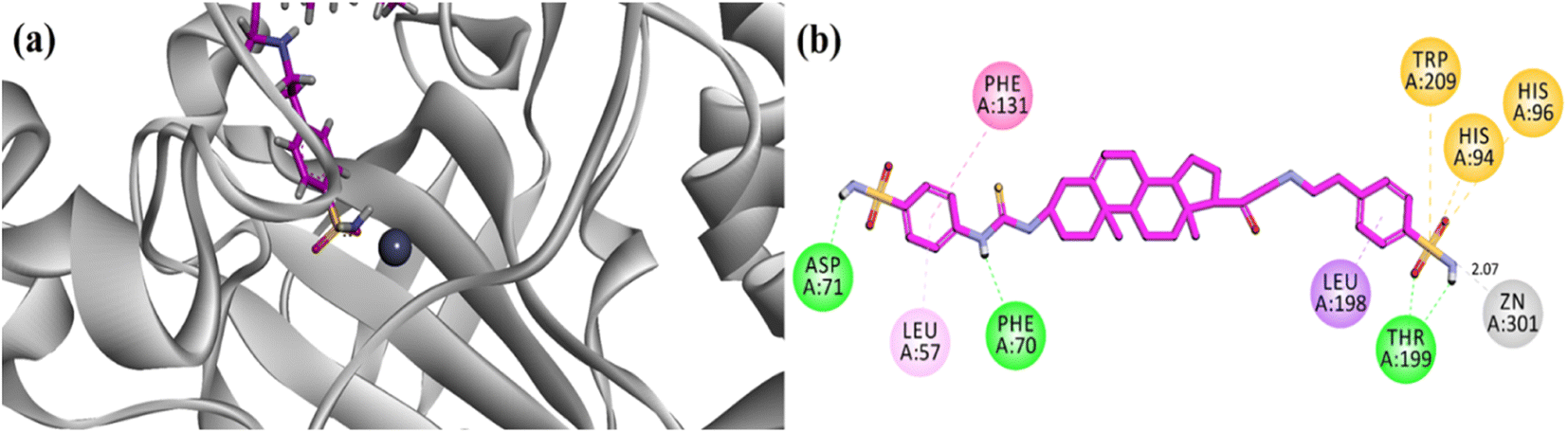 | ||
| Fig. 6 (a) Three-dimensional ribbon diagram of most potent compound 23 into the binding region of hCA-II. (b) Two-dimensional (2-D) interaction plots of potent compound 23 into the binding site of hCA-II (PDB accession: 3HS4). | ||
Overall, the steroidal derivative 23 established three hydrogen bond binding affinity and five hydrophobic interactions (π–π stacking, π-sulfur and π-alkyl). Phe70, Asp71 and Thr200 established hydrophilic (hydrogen bond) binding affinities, while Phe131 forms π–π stacking interactions and amino acid residues His94, His96 and Trp209 are implied in the π-sulfur type of hydrophobic interactions (Fig. 6b).
The binding orientation of native acetazolamide is shown in (Fig. 7a). It showed a strong contact of acetazolamide with zinc ion at a distance of 1.83 Å. Pregnenolone (9), C-3 amine derivative 12 and 4-(sulfamoylphenyl)thiourea analogue 15 experimentally showed poor CA-II inhibition having IC50 values of 89.38 μM, 73.19 μM, and 48.91 μM respectively. The binding orientation in the binding site of hCA-II showed that these compounds bind far from the Zn centre at a distance of 4.64 Å, 4.90 Å and 4.83 Å respectively (Fig. 7a–d).
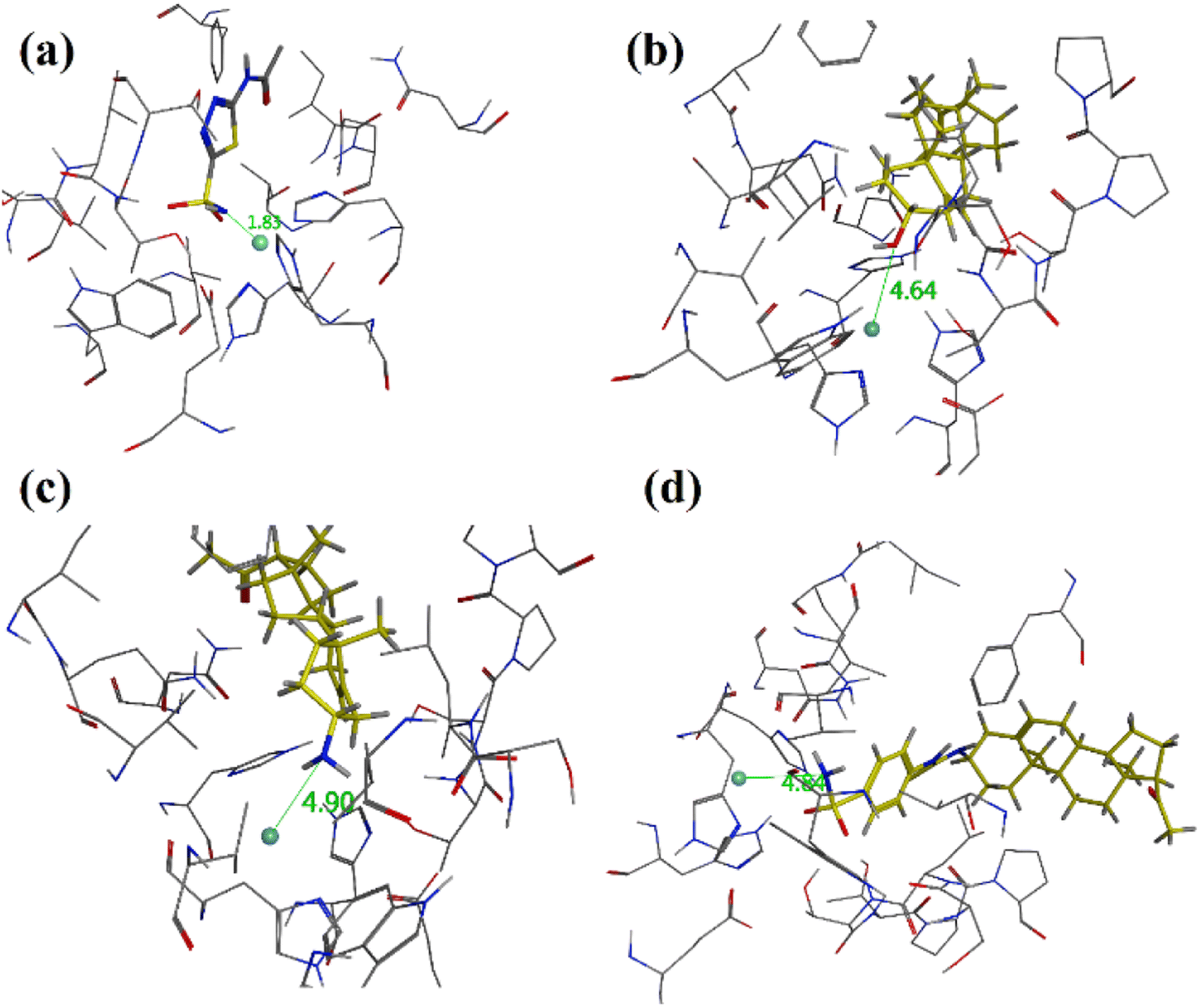 | ||
| Fig. 7 Three dimensional diagrams of (a) native acetazolamide, (b) pregnenolone (9), (c) 3-amino-pregnenolone derivative 12 and (d) derivative 15 into the binding site of hCA-II. The distance from zinc centre is shown as dotted green lines and distance is in Å (PDB accession: 3HS4). | ||
Docking studies on AChE
Docking studies on AChE was carried out on Torpedo californica acetylcholinesterase (TcAChE) complexed with alkylene-linked bis-tacrine (PDB ID = 2CKM). After validation of the docking protocol, the docking simulation studies were employed for synthesized pregnenolone derivatives into the binding site of 2CKM. Docking studies support dual binding site (PAS and CAS) inhibition of AChE which ultimately results in Aβ1–42 aggregation and AChE inhibition. All the active compounds showed interactions with amino acid residues present in PAS (Tyr334, Trp279, Tyr121 and Tyr70) and CAS (Phe331, Phe330 and Trp84). Two-dimensional (2-D) binding affinity plots of active compounds 23, 26 and 27 into the binding site of TcAChE are shown in Fig. 8a–c. It is important to note from 2D interaction plots that all three compounds under study interact with His440 (catalytic triad residue) via hydrogen (compounds 23 and 27) or π–π stacking interactions (compound 26). The high inhibition potential of these compounds towards AChE may be due to these interactions.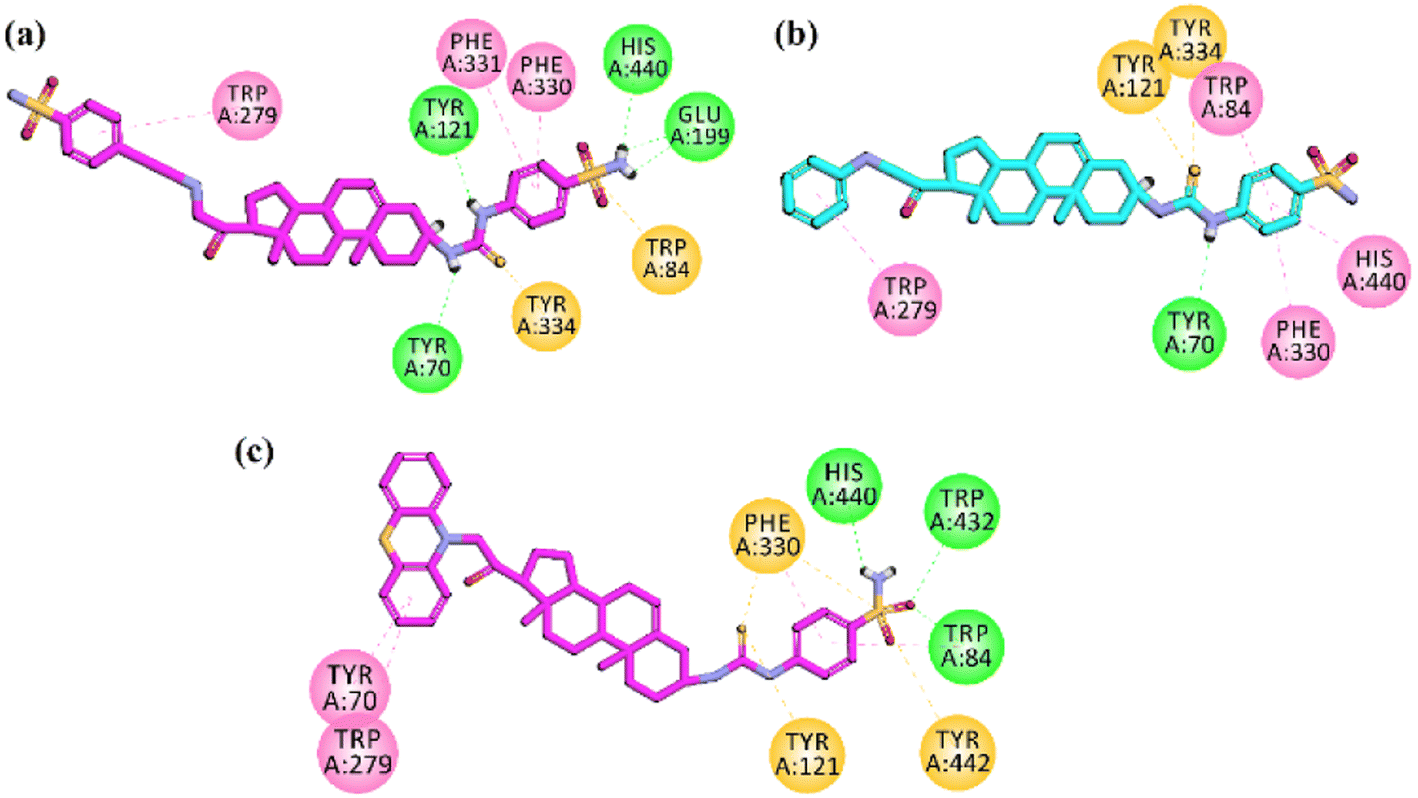 | ||
| Fig. 8 Two-dimensional (2-D) interaction plots of potent compound (a) 23 (b) 26 and (c) 27 into the binding region of AChE (PDB accession: 2CKM). | ||
Docking studies on amyloid-β42 peptide
We applied molecular modelling studies to explore how the synthesized compounds interact with amyloid-β. The analysis was performed on two different forms of amyloid-β. We consider helical monomer (PDB ID = 1IYT) and β-sheet pentamer or Aβ42 protofibril oligomer (PDB ID = 2BEG). Both the structures have been determined by NMR spectroscopy. The β-sheet pentamer or Aβ42 protofibril oligmer (PDB ID = 2BEG) has been used as a suitable model in several recent computational studies. The pentameric model, 2BEG, has been used as a suitable model in several recent computational studies.Docking studies on helical monomer
To prevent the Aβ (self-aggregation), the small molecule is needed to impede the formation of structures of β-sheet.68,69 The stability of these β-sheet structures relies on the creation of salt bridges involving Asp23 and Glu22, as well as interactions with hydrophobic residues such as Phe20, Glu22, Asp23, Gly29, and Ala30. We conducted molecular docking experiments with newly synthesized compounds, targeting the binding region of Aβ1–42, utilizing the protein data bank code 1IYT. There were significant hydrophobic interactions observed with the amino acids Asp23, Glu22, Phe20, and Phe19, effectively impeding the formation of β-sheet structures. Additionally, when analysing the docking results for compound 15, hydrogen bond interactions with residues Asp23, and Glu22 were found.Moreover, Asn27, Lys28 and Gly25 also established hydrogen bond interactions. A π-sulphur interaction with important residue Asn27 was also observed. Docking results of compound 26 exhibited binding affinities with the binding region of Aβ through hydrophilic (hydrogen bond) formation through the oxygens of –SO2OH with Leu34 and Lys28, A π–π interaction from benzene ring with Phe19 was observed. Moreover, –NH also formed hydrogen bond interactions with Asp23, Glu22, and π-anion interaction with residue Phe20. This array of binding affinities might contribute to α-helical stabilization and hindrance of the formation of β-sheet. The binding affinities in docking results of the compound 27 explained its extraordinary affinity as it shows the important interactions with the amino acid residues Lys28, Asp23, Glu22, Phe20 and Phe19, and through π-sulphur, π-anion and conventional hydrogen bonding. NH2 group showed binding through hydrogen bonding through the residues Asp23, and Glu22, while sulphur showed π-sulphur binding affinity with residue Phe20 (Fig. 9). The binding modes of compounds 26 and 27 are shown in Fig. 10.
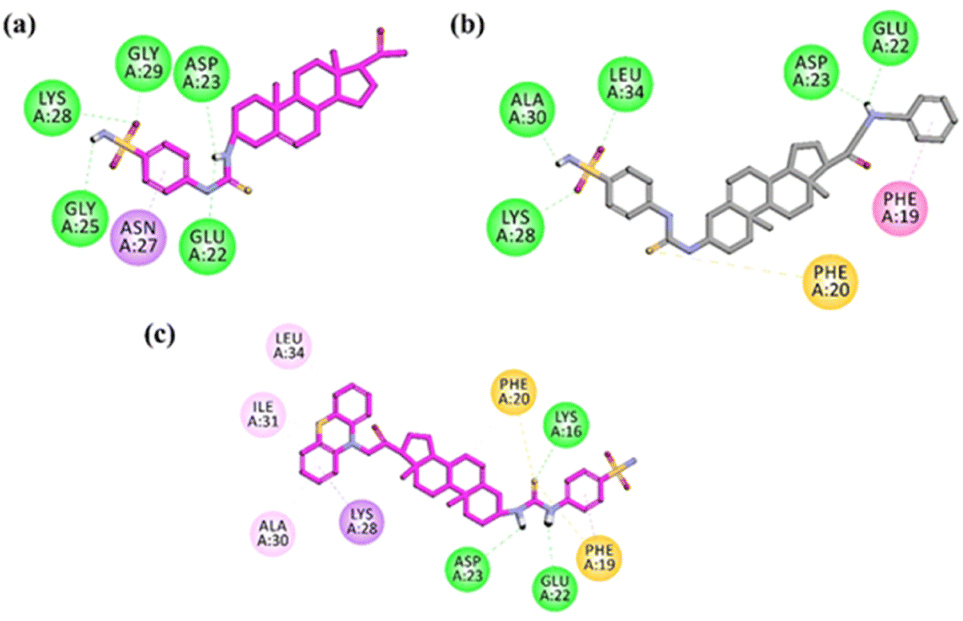 | ||
| Fig. 9 Two-dimensional (2-D) interaction plots of active compound (a) 15, (b) 26 and (c) 27 into the 3D crystallographic structure of amyloid-β42 peptide (PDB ID = 1IYT). | ||
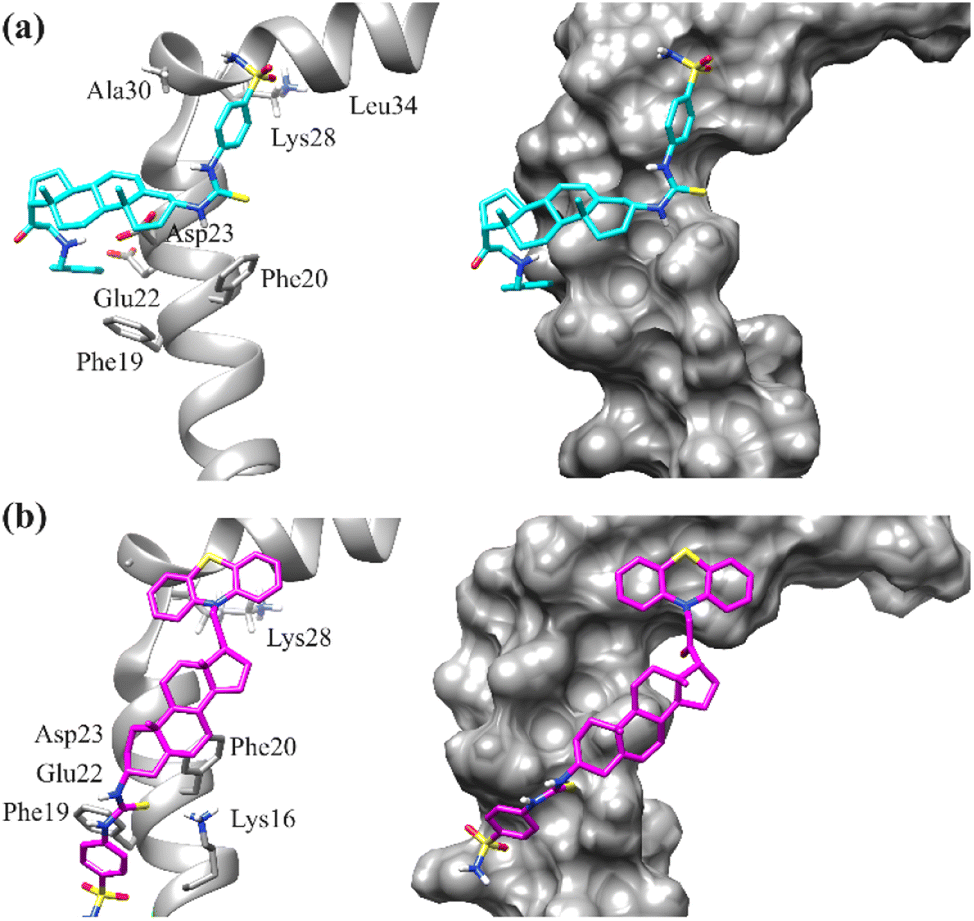 | ||
| Fig. 10 Binding mode of compound (a) 26 (b) 27 into the binding site of amyloid-β42 peptide (PDB ID = 1IYT). Left: ribbon diagrams; right: surface diagrams. | ||
Docking studies on β-sheet pentamer (oligomer)
Next, we docked selected compounds 15, 23, 26 and 27 with β-sheet pentamer or Aβ42 protofibril (PDB ID = 2BEG) to determine the preferred binding region. Two possible binding modes of selected compounds were observed (Fig. 11). Compounds 15 and 23 were docked at the C-terminal of the pentamer.70 The 2-D interaction plots are shown in Fig. S1 (ESI†). While compounds 26 and 27 were docked outside the amyloid fibrils.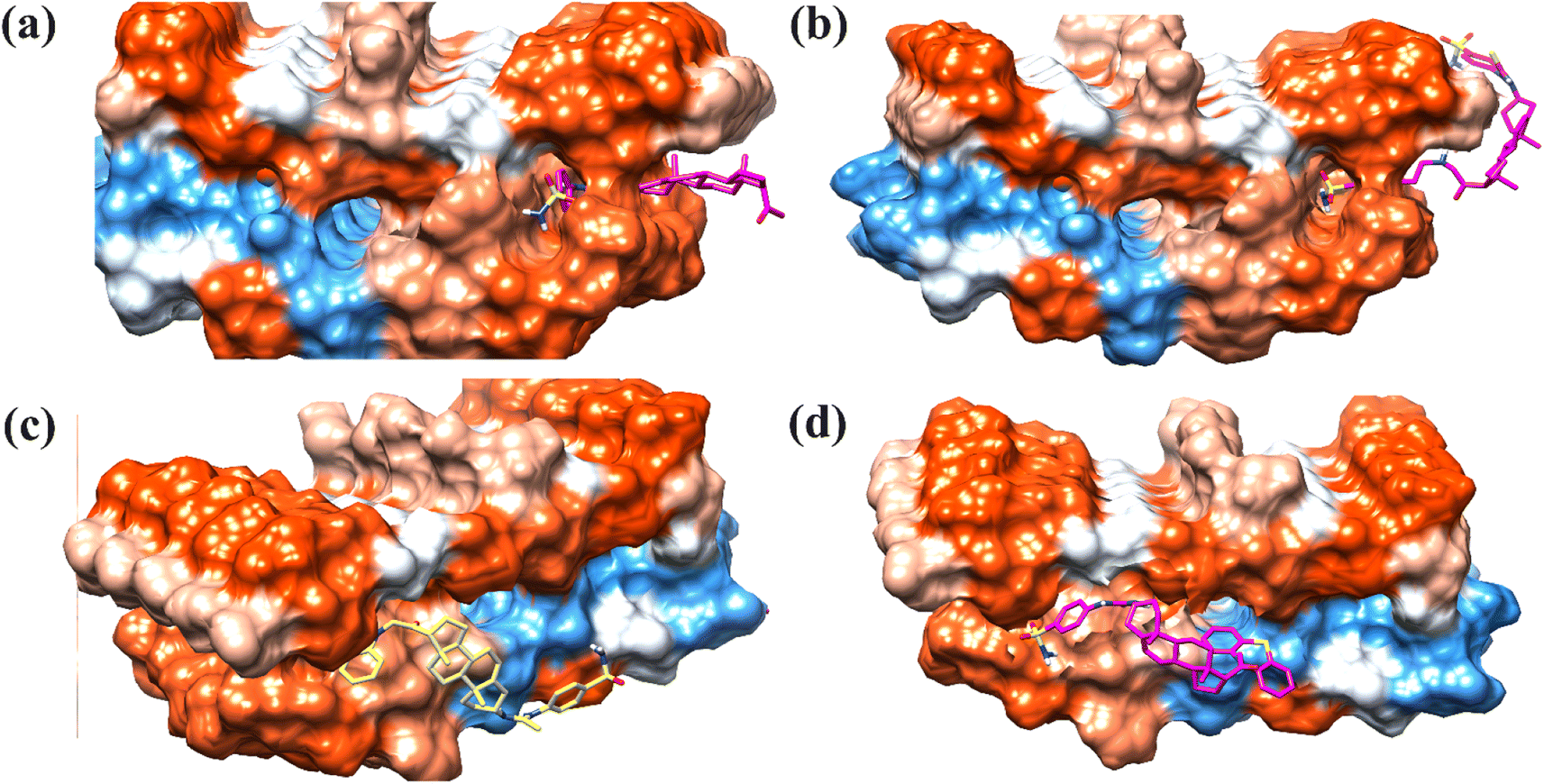 | ||
| Fig. 11 T3-D docked pose of compounds (a) 15, (b), 23, (c) 26 and (d) 27 into the Aβ protofibrils (PDB ID: 2BEG). | ||
The 2-D interaction plot of 26 showed three hydrogen bond interactions with Glu22, Asp23 and Gly25. While weak hydrophobic interactions were also observed with Val24 and Val36 (Fig. 12).26,70
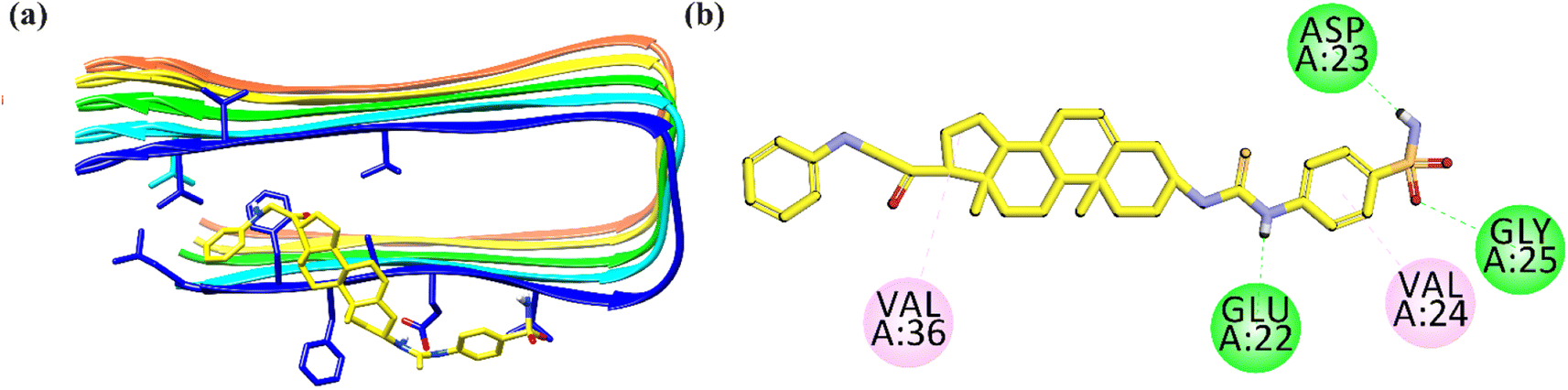 | ||
| Fig. 12 Binding mode of most active Aβ-aggregation inhibitor 26 with Aβ protofibrils (a) ribbon diagram; (b) 2-D interaction plot. | ||
Conclusions
Current work is focused on identifying the compounds that have balanced even mild biological activities against multiple targets instead of finding one-target compound with high potency. Structural modification of the steroidal nucleus has got considerable attention in recent years. This is because steroid-based therapeutics have several advantages and possess interesting structural features for the treatment of various diseases. Alzheimer's disease, a neurodegenerative condition, is characterized by the accumulation of a peptide called amyloid-β1–42 peptide. Drugs that are expected to reduce Aβ1–42 production, prevent Aβ1–42 aggregation, and promote Aβ1–42 clearance are promising approaches for treating AD. Moreover, small molecules having the ability to bind with the peripheral anionic site (PAS) will inhibit Aβ1–42 aggregation and AChE. Additionally, studies have revealed that some carbonic anhydrase inhibitors can diminish the neuroinflammation and deposition of Aβ1–42 aggregates at the walls of vessels of the brain in vivo. Keeping these points in mind, our current study involved the structural modification of pregnenolone. Compound 23 with two sulfonamide moieties present on two different phenyl rings emerged as potent hCA-II inhibitors with an IC50 value of 0.67 ± 0.09 μM. Self-mediated Aβ1–42 aggregation inhibition assay showed that compounds 26 and 27 have shown >80% and 85% inhibition of Aβ fibrils respectively. In concentration-dependent self-mediated Aβ1–42 aggregation inhibition assay compounds 15, 26 and 27 showed good inhibition potential with IC50 values of 9.25 μM, 1.04 μM and 5.78 μM respectively. In cholinesterase inhibition assay 4-(sulfamoylphenyl)thiourea derivative 23 emerged as a nanomolar inhibitor of AChE and BChE with IC50 = 0.04 μM and 0.58 μM respectively with SI = 14.5.Analysis of docked complexes showed that all the compounds confined in the active site coordinated with zinc centre and formed hydrogen bond interactions with key amino acid residues. Docking studies support dual binding site (PAS and CAS) inhibition of AChE which showed Aβ1–42 aggregation and AChE inhibition. Moreover, docking studies carried out on 3D crystallographic structure of Aβ1–42 peptide (PDB ID = 1IYT) showed significant hydrophobic interactions with the amino acids Phe19, Phe20, Glu22, and Asp23, effectively impeding the formation of β-sheet structures.
In conclusion, our current study is the first-ever report on the structural modification of pregnenolone at C-3 (–OH) and C-17 (acetyl group). Our synthesized derivatives 23, 25–27 exhibited concomitant inhibition of all the tested macromolecular targets and hence emerged as multitarget hybrid compounds among all synthesized pregnenolone derivatives.
Author contributions
UR conceived, designed, and supervised this study. He was involved in all the phases (from designing to synthesis/pharmacological evaluation and manuscript writing/editing) that led to the completion of the manuscript. AT and BM synthesized the compounds. In vitro experiments were performed by FH and AS. Docking studies were performed and written by AT, BM and UR. The manuscript was drafted by AT, BM, and FH. UR reviewed and edited the drafts.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The research is financially supported by a Project Grant from the Higher Education Commission Pakistan to Umer Rashid (PI) under the National Research Program for Universities (NRPU) (20-14513/NRPU/R&D/HEC/2021 2021).References
- U. Rashid and F. L. Ansari, in Drug Design and Discovery in Alzheimer's Disease, Elsevier, 2014, pp. 40–141 Search PubMed.
- E. Cerf, R. Sarroukh, S. Tamamizu-Kato, L. Breydo, S. Derclaye, Y. F. Dufrêne, V. Narayanaswami, E. Goormaghtigh, J.-M. Ruysschaert and V. Raussens, Biochem. J., 2009, 421, 415–423 CrossRef CAS PubMed.
- B. S. Gadad, G. B. Britton and K. Rao, J. Alzheimer's Dis., 2011, 24, 223–232 CAS.
- D. A. Butterfield, A. M. Swomley and R. Sultana, Antioxid. Redox Signaling, 2013, 19, 823–835 CrossRef CAS PubMed.
- H. Hampel, F. Caraci, A. C. Cuello, G. Caruso, R. Nisticò, M. Corbo, F. Baldacci, N. Toschi, F. Garaci and P. A. Chiesa, Front. Immunol., 2020, 11, 456 CrossRef CAS PubMed.
- V. Tiwari and S. Shukla, Front. Genet., 2023, 14, 1057068 CrossRef CAS PubMed.
- G. Dixit and A. Prabhu, J. Mol. Struct., 2023, 1294, 136498 CrossRef CAS.
- H. Hampel, J. Hardy, K. Blennow, C. Chen, G. Perry, S. H. Kim, V. L. Villemagne, P. Aisen, M. Vendruscolo and T. Iwatsubo, Mol. Psychiatry, 2021, 26, 5481–5503 CrossRef CAS PubMed.
- S. S. Hirschbeck, E. T. Lindberg, J. H. Jang, M. R. Jacob, K. L. Lazar Cantrell and T. D. Do, ACS Chem. Neurosci., 2024, 15(7), 1523–1532 CrossRef CAS PubMed.
- F. Hussain, A. Tahir, M. S. Jan, N. Fatima, A. Sadiq and U. Rashid, RSC Adv., 2024, 14, 10304–10321 RSC.
- V. Kumar and K. Roy, Computational Modeling of Drugs against Alzheimer's Disease, 2023, pp. 3–47 Search PubMed.
- J. Bieschke, Neurotherapeutics, 2013, 10, 429–439 CrossRef CAS PubMed.
- Z. Du, M. Li, J. Ren and X. Qu, Acc. Chem. Res., 2021, 54, 2172–2184 CrossRef CAS PubMed.
- X. Han and G. He, ACS Chem. Neurosci., 2018, 9, 198–210 CrossRef CAS PubMed.
- S. Sharari, N. N. Vaikath, M. Tsakou, S. S. Ghanem and K. Vekrellis, Int. J. Mol. Sci., 2023, 24, 11326 CrossRef CAS PubMed.
- G. E. Vitek, B. Decourt and M. N. Sabbagh, Expert Opin. Invest. Drugs, 2023, 32, 89–94 CrossRef CAS PubMed.
- A. V. Atoki, P. M. Aja, E. N. Ondari and T. S. Shinkafi, Int. J. Food Prop., 2023, 26, 2091–2127 CrossRef CAS.
- N. Stern, A. Gacs, E. Tátrai, B. Flachner, I. Hajdú, K. Dobi, I. Bágyi, G. Dormán, Z. Lőrincz and S. Cseh, Int. J. Mol. Sci., 2022, 23, 13098 CrossRef CAS PubMed.
- A. Alvarez, C. Opazo, R. Alarcón, J. Garrido and N. C. Inestrosa, J. Mol. Biol., 1997, 272, 348–361 CrossRef CAS PubMed.
- N. C. Inestrosa, A. Alvarez, C. A. Perez, R. D. Moreno, M. Vicente, C. Linker, O. I. Casanueva, C. Soto and J. Garrido, Neuron, 1996, 16, 881–891 CrossRef CAS PubMed.
- A. Alvarez, R. Alarcon, C. Opazo, E. O. Campos, F. J. Munoz, F. H. Calderon, F. Dajas, M. K. Gentry, B. P. Doctor and F. G. De Mello, J. Neurosci., 1998, 18, 3213–3223 CrossRef CAS PubMed.
- F. J. Carvajal and N. C. Inestrosa, Front. Mol. Neurosci., 2011, 4, 19 CAS.
- J. L. Sussman, M. Harel, F. Frolow, C. Oefner, A. Goldman, L. Toker and I. Silman, Science, 1991, 253, 872–879 CrossRef CAS PubMed.
- M. Bartolini, C. Bertucci, V. Cavrini and V. Andrisano, Biochem. Pharmacol., 2003, 65, 407–416 CrossRef CAS PubMed.
- N. C. Inestrosa, M. C. Dinamarca and A. Alvarez, FEBS J., 2008, 275, 625–632 CrossRef CAS PubMed.
- O. M. Waly, K. M. Saad, H. I. El-Subbagh, S. M. Bayomi and M. A. Ghaly, Eur. J. Med. Chem., 2022, 231, 114152 CrossRef CAS PubMed.
- P. Agostinho, A. Pliassova, C. R. Oliveira and R. A. Cunha, J. Alzheimer's Dis., 2015, 45, 329–347 CAS.
- H. M. Al-Kuraishy, M. S. Jabir, A. I. Al-Gareeb, A. K. Albuhadily, S. Albukhaty, G. M. Sulaiman and G. E.-S. Batiha, Ageing Res. Rev., 2023, 102119 CrossRef CAS PubMed.
- M. Vallée, J. Steroid Biochem. Mol. Biol., 2016, 160, 78–87 CrossRef PubMed.
- Y. Xia, Z.-H. Wang, Z. Zhang, X. Liu, S. P. Yu, J.-Z. Wang, X.-C. Wang and K. Ye, Prog. Neurobiol., 2021, 204, 102113 CrossRef CAS PubMed.
- A. D. Corbett, Pseudo-Dynamic Combinatorial Libraries, 2003 Search PubMed.
- T. K. Hurst, D. Wang, R. B. Thompson and C. A. Fierke, Biochim. Biophys. Acta, 2010, 1804, 393–403 CrossRef CAS PubMed.
- C. T. Supuran, Expert Opin. Ther. Targets, 2023, 27, 897–910 CrossRef CAS PubMed.
- C. T. Supuran and A. Scozzafava, Expert Opin. Ther. Pat., 2000, 10, 575–600 CrossRef CAS.
- A. Nocentini, A. Angeli, F. Carta, J.-Y. Winum, R. Zalubovskis, S. Carradori, C. Capasso, W. A. Donald and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2021, 36, 561–580 CrossRef CAS PubMed.
- S. Kumar, S. Rulhania, S. Jaswal and V. Monga, Eur. J. Med. Chem., 2021, 209, 112923 CrossRef CAS PubMed.
- C. T. Supuran, Curr. Pharm. Des., 2010, 16, 3233–3245 CrossRef CAS PubMed.
- S. Ghorai, S. Pulya, K. Ghosh, P. Panda, B. Ghosh and S. Gayen, Bioorg. Chem., 2020, 95, 103557 CrossRef CAS PubMed.
- A. Angeli, A.-M. Alaa, A. Nocentini, A. S. El-Azab, P. Gratteri and C. T. Supuran, Bioorg. Med. Chem., 2017, 25, 5373–5379 CrossRef CAS PubMed.
- M. A. Mohamed, A.-M. Alaa, H. M. Sakr, A. S. El-Azab, S. Bua and C. T. Supuran, Bioorg. Med. Chem., 2017, 25, 2524–2529 CrossRef CAS PubMed.
- C. T. Supuran, Clin. Sci., 2021, 135, 1233–1249 CrossRef CAS PubMed.
- D. Tarkowská, Molecules, 2019, 24, 2585 CrossRef PubMed.
- A. Gupta, B. S. Kumar and A. S. Negi, J. Steroid Biochem. Mol. Biol., 2013, 137, 242–270 CrossRef CAS PubMed.
- N. H. Mustafa, M. Sekar, S. Fuloria, M. Y. Begum, S. H. Gan, N. N. I. M. Rani, S. Ravi, K. Chidambaram, V. Subramaniyan and K. V. Sathasivam, Molecules, 2022, 27, 2032 CrossRef CAS PubMed.
- J. Y. Winum, A. Scozzafava, J. L. Montero and C. T. Supuran, Med. Res. Rev., 2005, 25, 186–228 CrossRef CAS PubMed.
- A. Nocentini, A. Bonardi, P. Gratteri, B. Cerra, A. Gioiello and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2018, 33, 1453–1459 CrossRef CAS PubMed.
- A. Khalid, M. N. Ghayur, F. Feroz, A. Gilani and M. I. Choudhary, J. Steroid Biochem. Mol. Biol., 2004, 92, 477–484 CrossRef CAS PubMed.
- A. Cecchi, A. Hulikova, J. Pastorek, S. Pastoreková, A. Scozzafava, J.-Y. Winum, J.-L. Montero and C. T. Supuran, J. Med. Chem., 2005, 48, 4834–4841 CrossRef CAS PubMed.
- A. Peschiulli, D. Oehlrich, M. Van Gool, N. Austin, S. Van Brandt, M. Surkyn, M. De Cleyn, A. Vos, G. Tresadern and F. J. Rombouts, ACS Med. Chem. Lett., 2021, 13, 76–83 CrossRef PubMed.
- Z. Y. Wang, M. R. Pergande, C. W. Ragsdale and S. M. Cologna, Curr. Biol., 2022, 32, 2572–2579 CrossRef CAS PubMed.
- D. S. Reddy, Prog. Brain Res., 2010, 186, 113–137 CAS.
- V. Kolas, J. S. A. Bandonil, N. Wali, K.-C. Hsia, J.-J. Shie and B.-c. Chung, Cell Biosci., 2022, 12, 190 CrossRef CAS PubMed.
- M. B. Tufail, M. A. Javed, M. Ikram, M. H. Mahnashi, B. A. Alyami, Y. S. Alqahtani, A. Sadiq and U. Rashid, Steroids, 2021, 168, 108801 CrossRef PubMed.
- B. A. Alyami, I. Ejaz, M. H. Mahnashi, Y. S. Alqahtani, A. O. Alqarni, M. S. Jan, A. Sadiq and U. Rashid, Steroids, 2022, 185, 109059 CrossRef CAS PubMed.
- Y. Pocker and J. Meany, Biochemistry, 1967, 6, 239–246 CrossRef CAS PubMed.
- N. Ur Rehman, S. A. Halim, M. Khan, H. Hussain, H. Yar Khan, A. Khan, G. Abbas, K. Rafiq and A. Al-Harrasi, Pharmaceuticals, 2020, 13, 94 CrossRef CAS PubMed.
- A. Khan, M. Khan, S. A. Halim, Z. A. Khan, Z. Shafiq and A. Al-Harrasi, Front. Chem., 2020, 8, 598095 CrossRef CAS PubMed.
- M. A. Javed, S. Bibi, M. S. Jan, M. Ikram, A. Zaidi, U. Farooq, A. Sadiq and U. Rashid, RSC Adv., 2022, 12, 22503–22517 RSC.
- S. K. Bowroju, N. Mainali, S. Ayyadevara, N. R. Penthala, S. Krishnamachari, S. Kakraba, R. J. S. Reis and P. A. Crooks, Molecules, 2020, 25, 3610 CrossRef CAS PubMed.
- C. B. Mishra, S. Shalini, S. Gusain, A. Prakash, J. Kumari, S. Kumari, A. K. Yadav, A. M. Lynn and M. Tiwari, RSC Adv., 2020, 10, 17602–17619 RSC.
- B. Brus, U. Kosak, S. Turk, A. Pislar, N. Coquelle, J. Kos, J. Stojan, J.-P. Colletier and S. Gobec, J. Med. Chem., 2014, 57, 8167–8179 CrossRef CAS PubMed.
- M. Matloobi and C. O. Kappe, J. Comb. Chem., 2007, 9, 275–284 CrossRef CAS PubMed.
- F. Iftikhar, F. Yaqoob, N. Tabassum, M. S. Jan, A. Sadiq, S. Tahir, T. Batool, B. Niaz, F. L. Ansari and M. I. Choudhary, Bioorg. Chem., 2018, 80, 99–111 CrossRef CAS PubMed.
- F. Sonmez, B. Zengin Kurt, I. Gazioglu, L. Basile, A. Dag, V. Cappello, T. Ginex, M. Kucukislamoglu and S. Guccione, J. Enzyme Inhib. Med. Chem., 2017, 32, 285–297 CrossRef CAS PubMed.
- R. Saini, G. R. Navale, S. Singh, H. K. Singh, R. Chauhan, S. Agrawal, D. Sarkar, M. Sarma and K. Ghosh, Int. J. Biol. Macromol., 2023, 248, 125847 CrossRef CAS PubMed.
- M. A. Javed, N. Ashraf, M. Saeed Jan, M. H. Mahnashi, Y. S. Alqahtani, B. A. Alyami, A. O. Alqarni, Y. I. Asiri, M. Ikram and A. Sadiq, ACS Chem. Neurosci., 2021, 12, 4123–4143 CrossRef CAS PubMed.
- S. Manzoor, M. T. Gabr, B. Rasool, K. Pal and N. Hoda, Bioorg. Chem., 2021, 116, 105354 CrossRef CAS PubMed.
- T. Umar, S. Shalini, M. K. Raza, S. Gusain, J. Kumar, P. Seth, M. Tiwari and N. Hoda, Eur. J. Med. Chem., 2019, 175, 2–19 CrossRef CAS PubMed.
- A. Pasieka, D. Panek, N. Szałaj, A. Espargaró, A. Więckowska, B. Malawska, R. Sabaté and M. Bajda, ACS Chem. Neurosci., 2021, 12, 2057–2068 CrossRef CAS PubMed.
- A. A. Mohammed, S. S. Barale, S. A. Kamble, S. B. Paymal and K. D. Sonawane, Int. J. Biol. Macromol., 2023, 242, 124880 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01536c |
| ‡ These authors have equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |