
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA multi-channel microfluidic platform based on human flavin-containing monooxygenase 3 for personalised medicine†
Melissa De Angelisa,
Silvia Schobesberger b,
Florian Selingerb,
Viktor Laurin Sedlmayrb,
Martin Frauenlobb,
Orsola Corcionea,
Shiman Donga,
Gianfranco Gilardia,
Peter Ertl*b and
Sheila J. Sadeghi*a
b,
Florian Selingerb,
Viktor Laurin Sedlmayrb,
Martin Frauenlobb,
Orsola Corcionea,
Shiman Donga,
Gianfranco Gilardia,
Peter Ertl*b and
Sheila J. Sadeghi*a
aDepartment of Life Sciences and Systems Biology, University of Torino, via Accademia Albertina 13, 10123 Torino, Italy. E-mail: sheila.sadeghi@unito.it
bTU Wien, Faculty of Technical Chemistry, Getreidemarkt 9, 1060 Vienna, Austria
First published on 23rd April 2024
Abstract
Human flavin-containing monooxygenase 3 (FMO3) is a drug-metabolizing enzyme (DME) which is known to be highly polymorphic. Some of its polymorphic variants are associated with inter-individual differences that contribute to drug response. In order to measure these differences, the implementation of a quick and efficient in vitro assay is highly desirable. To this end, in this work a microfluidic immobilized enzyme reactor (μ-IMER) was developed with four separate serpentines where FMO3 and its two common polymorphic variants (V257M and E158K) were covalently immobilized via glutaraldehyde cross-linking in the presence of a polylysine coating. Computational fluid dynamics simulations were performed to calculate the selected substrate retention time in serpentines with different surface areas at various flow rates. The oxidation of tamoxifen, an anti-breast cancer drug, was used as a model reaction to characterize the new device in terms of available surface area for immobilization, channel coating, and applied flow rate. The highest amount of product was obtained when applying a 10 μL min−1 flow rate on polylysine-coated serpentines with a surface area of 90 mm2 each. Moreover, these conditions were used to test the device as a multi-enzymatic platform by simultaneously assessing the conversion of tamoxifen by FMO3 and its two polymorphic variants immobilized on different serpentines of the same chip. The results obtained demonstrate that the differences observed in the conversion of tamoxifen within the chip are similar to those already published (E158K > WT > V257M). Therefore, this microfluidic platform provides a feasible option for fabricating devices for personalised medicine.
1. Introduction
Recent advances in combinatorial chemistry and molecular biology have led to the identification of an increased number of proteins or pathways that can be targeted to give a therapeutic effect in a disease state. Drug-like molecules acting on these targets must be developed considering that they must be safe and not toxic for the human body. This is the main reason why the drug development process is expensive, time-consuming, and highly inefficient, with a probability of success lower than 10% from phase I clinical trials.1 Metabolism represents the major clearance route of 75% of drugs and other xenobiotics. It involves complex biotransformation processes where lipophilic molecules are converted to more water-soluble metabolites by various drug-metabolizing enzymes (DMEs) present in the human liver, such as cytochromes P450 (CYPs) and flavin-containing monooxygenases (FMOs).2,3 DMEs can generate pharmacologically active metabolites which might cause toxicity. Consequently, studies on drug metabolism are key processes to minimize potential safety liabilities.4 The implementation of quick and efficient in vitro assays that can predict whether new drug molecules are converted by major DMEs can help in reducing both time and costs in the drug development pipeline. To do so, many in vitro tools are available, including primary hepatocytes, liver cell lines, microsomes, and recombinant hepatic enzymes.5,6 The latter are easier to handle and therefore commonly used for high-throughput screening assays.7,8In the last few years, the application of microfluidic devices in analytical biochemistry has been growing. These platforms offer several advantages such as low reagent consumption, efficient reactions due to the high surface-to-volume ratio, and reduced analysis time, along with their inherent miniaturization, integration, portability, and automation.9,10 Therefore, the combination of enzymes and microfluidics has the potential to produce assays for a broad range of analytes and applications, such as drug screening and evaluation.11 Microfluidic immobilized enzyme reactors (μ-IMERs) have become more and more widespread due to the numerous advantages, including reusability, easy handling, prolonged lifetime, and substrate specificity.12 Moreover, enzyme immobilization has been shown not only to improve enzyme activity, stability and selectivity, but also to facilitate the recovery and reusability of the enzyme.13,14 Much effort has been directed towards studying various immobilization strategies, including physical adsorption, affinity binding, covalent binding and encapsulation.15–17
In this work, a new μ-IMER was designed to assess whether new drug-like molecules are converted by flavin-containing monooxygenase 3 (FMO3), a human liver enzyme that has a prominent role in the metabolism of drugs.2,18 Human FMOs are nicotinamide adenine dinucleotide phosphate (NADPH)-dependent enzymes that use molecular oxygen to catalyse the oxygenation of a large number of structurally different xenobiotics, including many pharmaceuticals.19,20 Human FMOs are the second most important class of monooxygenases and, unlike cytochromes P450, produce few toxic metabolites; for this reason, it is considered advantageous to design drugs that are metabolised by this family of enzymes.21 In particular, FMO3 is the most important isoform present in the adult liver, and it is known to exhibit single nucleotide polymorphisms (SNPs).22–24 SNPs can have a major impact on the pharmacokinetics and pharmacodynamics of a particular drug, and therefore contribute to variations in susceptibility and toxicity in individuals carrying them. For this reason, a better and more personalised treatment with tailored dosages could avoid not only treatment failure but also adverse reactions leading to toxicity.25
Initially, a bacterial enzyme from the same class of flavoprotein monooxygenases as human FMOs was exploited to identify the best immobilization strategy on a glass surface. The enzyme, a cytoplasmic Baeyer–Villiger monooxygenase (BVMO), is easier to express and purify with high yields in a heterologous system compared to the membrane-bound human FMO3.26,27 In addition, its capability to catalyse the formation of indigo, a blue-coloured dye, could be employed to develop a colorimetric assay to evaluate the activity of the enzyme after immobilization.28 Three strategies were investigated: ionic binding, affinity binding and cross-linking. In each case, the activity of the immobilized enzyme was assessed by looking at the formation of insoluble indigo crystals. The best strategy was found to be a glutaraldehyde-mediated cross-linking to a collagen coating. Therefore, the same strategy was applied to immobilize human FMO3 and its polymorphic variants on the new device.
In parallel, a multi-channel microfluidic chip was designed with the aim of evaluating the enzymatic conversion of a drug by FMO3 and its two common polymorphic variants (V257M and E158K) at the same time. Tamoxifen, an antiestrogen medication widely used in breast cancer therapy and chemoprevention, was chosen as a model drug, since it is a known substrate of FMO3.23,24,29,30
The chip was designed with a single inlet (connected to a syringe pump) and four separate channels so that the solution containing the drug is equally split to reach each immobilised enzyme at the same time. The channels were drawn with a serpentine shape to increase the available surface area for enzyme immobilization. Computational fluid dynamics simulations were performed to calculate the substrate retention time in serpentines with different surface areas at various flow rates.
For proof-of-concept purposes, the new platform was characterized in terms of channel coating (collagen versus polylysine, both workable for glutaraldehyde-mediated cross-linking), available surface area for immobilization, and applied flow rate. In the initial experiments, only wild type FMO3 was immobilized in the channels, and the product (tamoxifen N-oxide) obtained under each condition was separated and detected via high-performance liquid chromatography (HPLC). Once optimal conditions for maximal product formation were identified, the microfluidic device was tested as a multi-enzymatic platform by simultaneously assessing the conversion of tamoxifen by FMO3 and its two polymorphic variants immobilized on different serpentines of the same chip.
This microfluidic chip has a great potential not only for testing newly discovered drug-like molecules in a high-throughput manner, but also to measure differences in their metabolic conversion by polymorphic variants of DMEs. Therefore, directly relevant for applications to personalised medicine.
2. Results & discussion
2.1 Screening of enzyme immobilization strategies
Considering the structural diversity, complexity and variability of enzymes, the selection of an appropriate immobilization technique is one of the key points in obtaining human liver enzymes stable enough to perform their catalytic activity towards drug molecules on a surface. The exploration of simple, efficient, and widely adaptable enzyme immobilization methods is therefore needed to obtain a high-performing microfluidic device.31 Three commonly used immobilization strategies were investigated: ionic binding, affinity binding and cross-linking (Fig. 1). | ||
| Fig. 1 Schematic representation of the investigated enzyme immobilization methods. | ||
Given the higher stability of bacterial enzymes compared to their human counterparts, the initial investigation of the possible immobilization techniques was carried out with the bacterial BVMO. For ionic binding, the natural, biocompatible and positively charged chitosan was initially used to coat the surface followed by electrostatic interactions with the negatively charged amino acid residues present on the enzyme's surface. For affinity binding, the already existing His-tag at the C-terminus of the recombinant enzyme was exploited to obtain a site-specific and oriented immobilization on a nickel–nitrilotriacetic acid (Ni2+–NTA) functionalized surface. Finally, in the cross-linking procedure, the surface was coated with collagen, and glutaraldehyde was used as a chemical cross-linker to form covalent bonds between the amino groups of lysine residues present on both collagen fibrils and the BVMO surface.
To minimize the reaction volumes and mimic the final microfluidic device, a small channel structure with a central well was designed to carry out all the experiments (Fig. 2).
 | ||
| Fig. 2 Indigo formation and detection. Ar-BVMO R292A is immobilized on the bottom glass surface of a small well placed in the middle of a channel structure. The immobilized enzyme catalyses the hydroxylation of indole in presence of molecular oxygen and NADPH. The final product, indigo, is a coloured compound that can be detected spectrophotometrically (absorbance peak at 620 nm) or microscopically (formation of small blue particles). | ||
Only 5 μL were required to cover the surface of the well, considerably reducing the quantity of reagents necessary for the functionalization and the amount of enzyme needed for immobilization. The non-bound BVMO was removed by three consecutive rounds of filling and emptying the channel with 35 μL of 50 mM Tris–HCl buffer, pH 8.0. The catalytic activity of the enzyme was investigated by looking at the formation of indigo particles in the well. The produced indigo, which is not soluble in buffer, forms blue crystals that were observed using an inverted microscope. At the same time, the produced indigo was also solubilized in DMSO and detected spectrophotometrically by following the absorbance peak at 620 nm for its quantification (Fig. 2).
Three different concentrations of BVMO (10, 20, 40 μM) were used for the immobilization on chitosan-coated, Ni2+–NTA functionalized and glutaraldehyde-crosslinked collagen-coated glass surfaces. After washing out the excess enzyme, a solution containing 5 mM indole and 1 mM NADPH in 50 mM Tris–HCl buffer, pH 8.0 was injected into the channel and left for 1 h at room temperature to allow the reaction to proceed. The activity of the immobilized enzyme was subsequently measured by the formation of indigo, as mentioned above. Each condition (i.e. immobilization technique, BVMO concentration) was tested in four different well/channel structures at the same time.
Fig. 3A shows the correlation between (i) immobilization strategy, (ii) initial enzyme concentration used for the immobilization, and (iii) number of produced indigo particles. As can be seen in Fig. 3A, under all immobilization conditions, the number of produced indigo particles increases with increasing enzyme concentration. For example, for the cross-linking method, 207 ± 26 particles are produced using 10 μM BVMO; this number increases 9-folds (1821 ± 264) and 13-folds (2610 ± 500) when enzyme concentrations of 20 μM and 40 μM are used, respectively. This increasing trend is also observed for the other two immobilization strategies.
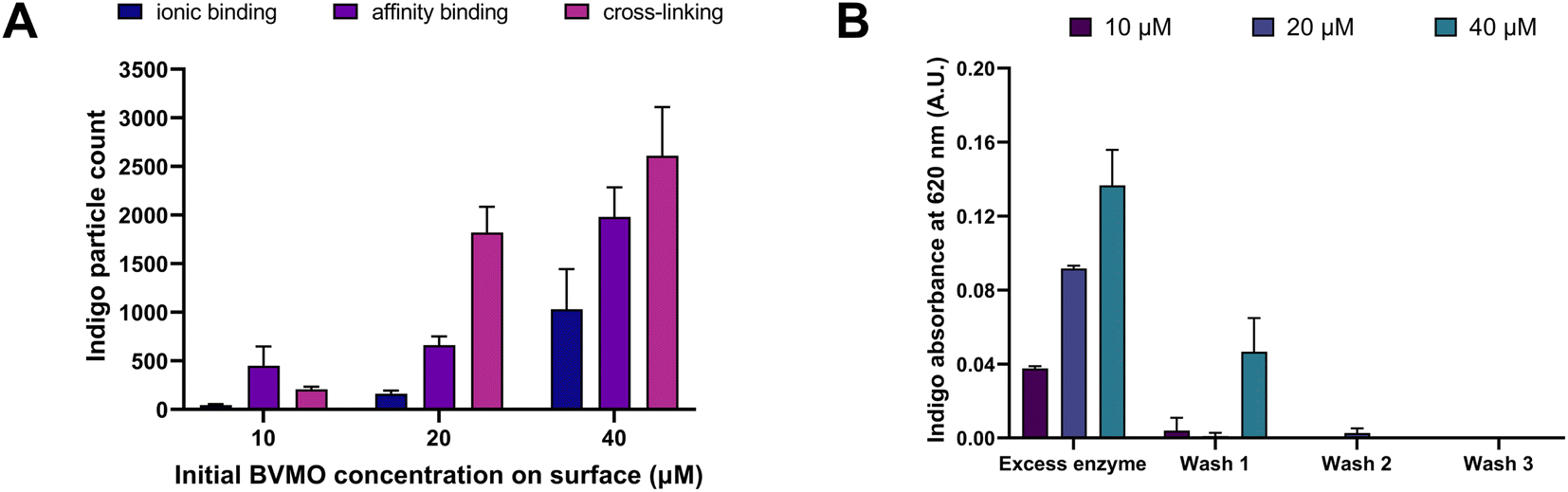 | ||
| Fig. 3 (A) Number of indigo dye particles produced by BVMO counted for each immobilization strategy at different initial enzyme concentrations. (B) Absorbance values at 620 nm (i.e. indigo absorbance peak) after indole conversion performed by the free enzyme washed out from the collagen-coated surface. | ||
When comparing the three different immobilization strategies, the highest number of indigo particles were observed with the cross-linking method (2610 ± 500). This number is 1.3 times higher than the one obtained with affinity binding (1981 ± 304) and 2.5 times higher than the one obtained with ionic binding (1031 ± 413). These results suggest that more catalytically active BVMO is immobilized when the enzyme is covalently bound – through glutaraldehyde – to a surface coating that provides lysine residues (e.g. collagen, polylysine).
In order to confirm that the product derived solely from the activity of the immobilized enzyme, the wash solutions obtained from this cross-linking methodology were transferred into a 96-well plate and the presence of free enzyme was assessed by adding 5 mM indole and 1 mM NADPH into each well. As shown in Fig. 3B, indigo was produced both by the excess enzyme initially removed from the surface with a micropipette, and by the non-bound enzyme progressively washed out with buffer injections in the channel. No indigo was detected in the third wash, indicating that all the non-bound enzyme was removed from the surface in the first two washes.
All together, given the ease in surface functionalization and the high enzyme retention and activity, the lysine-based cross-linking method was identified as the best strategy to immobilize human FMO3 on the multi-channel microfluidic platform.
2.2 Multi-channel microfluidic platform design and assembly
As mentioned in the introduction, the use of a multi-channel microfluidic platform could give us the opportunity to simultaneously measure the catalytic activity of wild-type FMO3 and its two polymorphic variants towards a specific drug. To this end, the chip was designed with four separate serpentines as described in the Experimental section. The chip consisted of four layers as shown in Fig. 4A: two glass microscope slides into which the single inlet (top) and the four outlet (bottom) exits were drilled; a bi-adhesive middle layer where the serpentines were introduced; and a PDMS bottom layer with outlets to assist the formation of the drops exiting from the chip. All the utilised materials are biocompatible and commonly used for chip fabrication, making this structure cheap, easy to fabricate and adaptable to different enzymatic systems. Different areas for enzyme immobilization were obtained by changing the length of the serpentines. Fig. 4B lists the characteristics of the channels (i.e. area, width and volume) of three serpentine types: small (S), medium (M) and large (L). Putting four type L serpentines next to each other made the best possible use of the chip's dimensions, allowing for the maximum enzyme loading. | ||
| Fig. 4 (A) Multi-enzymatic platform design. The chip consists of four layers and presents a single inlet and four outlets. (B) Characteristic area, width, and volume of three different serpentine-shaped channels: small (S), medium (M), and large (L). | ||
Once the chip was assembled, the surface of the serpentines was functionalized with a lysine-based glutaraldehyde-crosslinked coating. Subsequently, FMO3 was covalently bound to the functionalized surface and the excess enzyme was washed out at a 100 μL min−1 flow rate.
Since the activity of the enzyme could be to some degree dependent on the applied flow rate, computational simulations were carried out to determine the optimal flow rate for the activity of the immobilized enzyme in the presence of the specific substrate, tamoxifen.
2.3 Computational flow rate simulations
Computational fluid dynamics simulations were performed to predict the retention time of the substrate tamoxifen at various flow rates in the different serpentines. The aim was to identify the flow rate at which the on-chip retention time of the liquid in the channels was comparable to the usual 5–15 min incubation time of the batch enzymatic reactions.23 Fig. 5A shows the simulations based on type L serpentines (90 mm2, largest surface area), where tamoxifen retention times decreased with increasing flow rate, as expected. In particular, the amount of time required by a 100 μM tamoxifen solution in aqueous buffer to fill the channels at a 10 μL min−1 flow rate was found to be 7.5 min. This value falls within the previously mentioned 5–15 min incubation time. This flow rate (10 μL min−1) was selected to carry out more simulations on the other serpentine types (S and M, 45 and 70 mm2, respectively). Results obtained demonstrate that when the surface area of the channel was reduced, the substrate retention time decreased accordingly (4.5 < 6.0 < 7.5 min) (Fig. 5B). Therefore, the optimal conditions resulting from these simulations were found to be a flow rate of 10 μL min−1 in type L serpentines.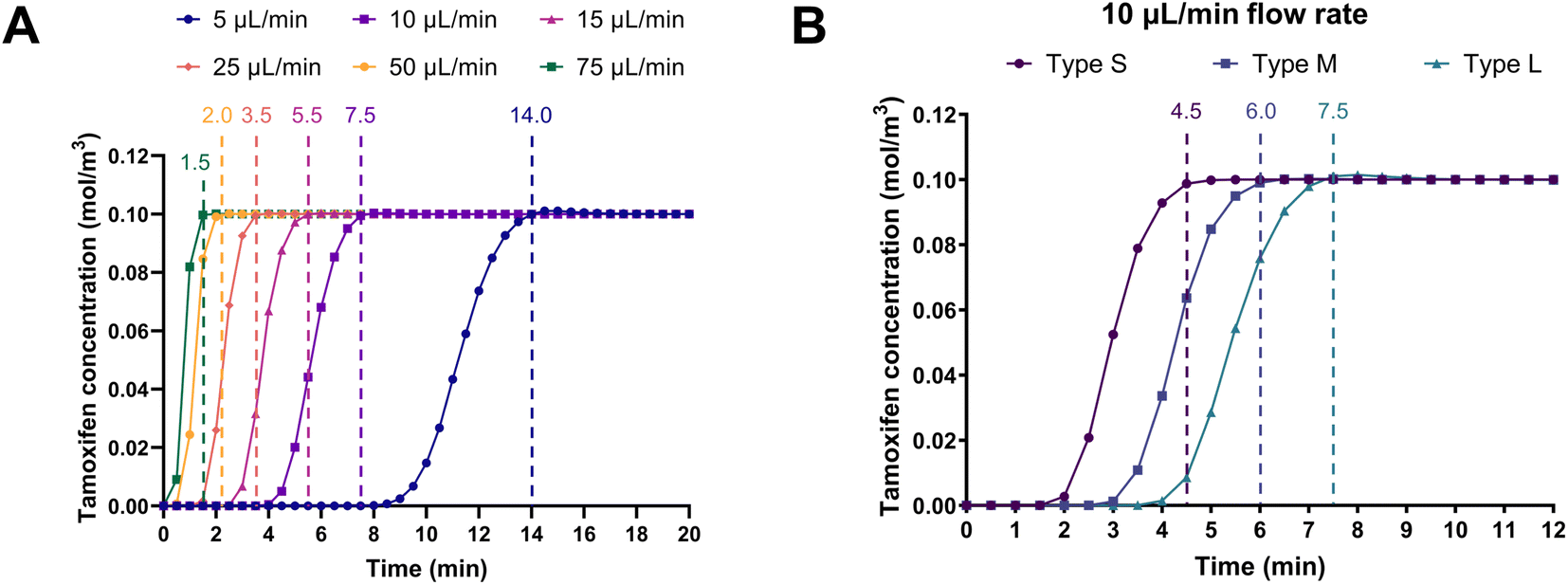 | ||
| Fig. 5 Computational flow rate simulations. (A) Tamoxifen retention times at different flow rates for a chip with type L serpentines. (B) Tamoxifen retention times for three different serpentine surface areas at a fixed flow rate (10 μL min−1). | ||
2.4 Multi-channel microfluidic platform characterization
The experiments for the characterization of the fabricated four-serpentine chip were carried out at room temperature based on the flow rate simulations.Initially, two coatings for lysine-based cross-linking via glutaraldehyde immobilization were investigated: collagen and polylysine. Both coatings were tested with all three serpentine types (S, M, and L) at a flow rate of 10 μL min−1. The catalytic activity of the enzyme was assessed in the presence of its substrate, tamoxifen. To determine which coating worked best, the product, tamoxifen N-oxide, was detected and quantified via HPLC. A solution containing 100 μM tamoxifen and 1 mM NADPH was constantly pumped in the chip using a syringe pump, and the flow through was collected in glass vials before injection into HPLC. Higher amounts of product were obtained with polylysine-coated chips for all three types of serpentines (Fig. 6A). Furthermore, the quantity of product was expected to rise with increasing surface area. This was true when using the polylysine coating: the amount of tamoxifen-N oxide increased from 42.8 ± 10.3 nM produced in type S serpentines (45 mm2) up to 66.5 ± 12.2 nM produced in type L serpentines (90 mm2). On the contrary, collagen showed the reverse trend, where less product was formed in the largest serpentine. One reason could be the higher non-specific binding of the product to the collagen-functionalized surface.32 These results highlighted the possibility to detect very small amounts of product deriving from the substrate conversion in this miniaturized system.
 | ||
| Fig. 6 Microfluidic platform characterization. (A) Tamoxifen N-oxide produced by FMO3 WT immobilized on chips with different serpentine surface areas (S, M, L) and lysine-based coatings. (B) Tamoxifen N-oxide produced by immobilized FMO3 WT at different flow velocities (10, 25, 50, 75 μL min−1). | ||
The impact of flow rate on the production of tamoxifen N-oxide was also investigated. Fig. 6B shows the correlation between velocity (flow rate divided by cross-sectional area of the channel) and product peak area from HPLC analysis. As expected, the quantity of product decreased with increasing velocity, indicating that more substrate was converted at a slower flow rate due to the longer on-chip incubation time with the immobilized enzyme. At 1.19 mm s−1 (= 10 μL min−1 flow rate) and 2.98 mm s−1 (= 25 μL min−1 flow rate), 69.2 ± 13.4 nM and 62.3 ± 6.5 nM tamoxifen N-oxide was produced, respectively. With increasing velocities (corresponding to 50 μL min−1 and 75 μL min−1 flow rates), the amount of product was halved.
Each condition (i.e. coating type, surface area available for immobilization, flow rate) was tested in parallel in four different serpentines on the same device to assess the repeatability and reliability of the microfluidic platform. In addition, device reusability was evaluated by quantifying the tamoxifen N-oxide produced at different flow rates applied to the same microfluidic platform in succession. The results (Fig. S1†) were compared to the data points obtained from single use chips (Fig. 6B) and confirmed that FMO3 remains not only covalently bound to the surface but also active for multiple usages in a reproducible manner.
The data obtained demonstrated that the optimal conditions for an FMO3-based microfluidic chip resulted to be: (i) high surface area for enzyme immobilization (90 mm2), (ii) polylysine coating, and (iii) low flow rate for drug conversion.
2.5 Simultaneous tamoxifen conversion by FMO3 and its polymorphic variants
The chip was further tested as a multi-enzymatic microfluidic platform by immobilizing wild-type FMO3 and its two common polymorphic variants (V257M and E158K). The enzymes were immobilized on three out of four available serpentines, where the fourth serpentine was used as control (glutaraldehyde-crosslinked polylysine coating without any enzyme). Given the previous results obtained from the platform characterization, a solution containing 100 μM tamoxifen and 1 mM NADPH in 50 mM potassium phosphate buffer, pH 7.4 was pumped through the serpentines first at 100 μL min−1 for 4 min to remove all the non-bound enzyme, and then at 10 μL min−1 for 7.5 min to make sure that the reaction occurred at the set flow rate for the required incubation time. This flow was kept constant until enough solution for each sample was collected for HPLC analysis (around 20 min). As shown in Fig. 7A the N-oxide product, with a retention time of 4.8 min, was detected for all serpentines except for the control, demonstrating that all the immobilized enzymes were catalytically active. | ||
| Fig. 7 (A) HPLC chromatograms of samples collected from the multi-enzymatic microfluidic platform with immobilized FMO3 WT, V257M and E158K. The substrate and product have retention times of 4.0 min and 4.8 min, respectively. (B) Comparison of tamoxifen N-oxide produced on-chip and off-chip by each enzyme. | ||
The amount of tamoxifen N-oxide produced by each variant on-chip was quantified via HPLC analysis and compared to the amount of tamoxifen N-oxide produced by the free enzymes in batch reactions. The latter were carried out at room temperature for 10 min in 50 mM potassium phosphate buffer, pH 7.4 using 1 μM enzyme, 100 μM tamoxifen and 1 mM NADPH, as previously reported.23 As shown in Fig. 7B, differences in the conversion of tamoxifen by each variant were detectable for both on-chip and batch reactions. The same trend can be observed in both cases: the highest amount of tamoxifen N-oxide was produced by the E158K variant (69.3 ± 6.50 nM on-chip and 4.85 ± 0.08 μM off-chip), while V257M converted even less substrate than the wild-type enzyme (46.8 ± 7.50 < 49.8 ± 9.55 nM on-chip, 1.79 ± 0.05 < 2.88 ± 0.04 μM off-chip). These results are in line with already published data suggesting that the differences in the catalytic performance of these enzymes could be reproduced in the microfluidic device.23
Moreover, this multi-enzymatic microfluidic platform resulted to be highly reproducible and reliable: data on tamoxifen conversion were in fact collected from separate chips, whose channels' surface was coated and functionalized with the three enzymes independently.
This set-up could be adapted to investigate not only whether newly discovered drug molecules are converted by different drug metabolizing enzymes simultaneously, but also to measure differences in the drug clearance by different polymorphic variants. In addition, coupling the system with liquid chromatography-mass spectrometry (LC-MS) or with gas chromatography-mass spectrometry (GC-MS) would allow to also identify the chemical structure of the enzymatic products.
3. Experimental
3.1 Materials
Chitosan, (3-aminopropyl)triethoxysilane (APTES), N-hydroxysuccinimide (NHS), nitrilotriacetic acid (NTA), nickel(II) chloride hexahydrate, type I collagen (from rat tail), poly-L-lysine, glutaraldehyde (50 wt% in H2O), indole, tamoxifen, Tris, 2-(N-morpholino)ethanesulfonic acid (MES), Dulbecco's phosphate buffered saline (PBS), potassium phosphate dibasic, potassium phosphate monobasic and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) was purchased from M&B Stricker Laborfachhandel GbR. Tamoxifen N-oxide was purchased from Szabo-Scandic (manufacturer: Cayman Chemical). Nicotinamide adenine dinucleotide phosphate, reduced form (NADPH) was purchased from VWR. Hellma™ Hellmanex III concentrated liquid cleaning solution was purchased from Fisher Scientific GmbH. Double-sided medical grade pressure-sensitive tape (ARcare® 90106NB) were purchased from Adhesives Research. Polydimethylsiloxane (PDMS) was purchased from MVQ Silicones.3.2 Enzyme expression and purification
The active site mutant R292A of the bacterial Baeyer–Villiger monooxygenase from Acinetobacter radioresistens (Ar-BVMO R292A) and human FMO3 wild-type (WT), V257M and E158K were expressed in E. coli and purified via affinity chromatography according to the previously published methodologies.28,33 Yields were 9 mg L−1 for BVMO, 37 mg L−1 for FMO3 WT, 14 mg L−1 for FMO3 E158K and 70 mg L−1 for FMO3 V257M. Before immobilization, the stock solutions were diluted to the desired concentrations in 50 mM Tris–HCl buffer (pH 8.0) for BVMO, and in 50 mM potassium phosphate buffer (pH 7.4) for FMO enzymes.3.3 Glass surface modification and enzyme immobilization
Prior to any surface modification, microscope glass slides were cleaned by immersion in a 2% Hellmanex III solution followed by pure isopropanol, and finally in double distilled water. Each cleaning step was performed for 5 min in an ultrasonic cleaner (VWR). The slides were then dried with pressurized air.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with a 10% glutaraldehyde solution, which was prepared by diluting the commercial stock in PBS. In this way, a 1% collagen + 5% glutaraldehyde solution or a 0.1 mg per mL polylysine + 5% glutaraldehyde solution was obtained. The glass surface was incubated with either of these two solutions for 1 h at 37 °C. Glutaraldehyde concentration was selected after coating thickness evaluation using the inverted microscope, as described in ESI (Fig. S2†).
:1 with a 10% glutaraldehyde solution, which was prepared by diluting the commercial stock in PBS. In this way, a 1% collagen + 5% glutaraldehyde solution or a 0.1 mg per mL polylysine + 5% glutaraldehyde solution was obtained. The glass surface was incubated with either of these two solutions for 1 h at 37 °C. Glutaraldehyde concentration was selected after coating thickness evaluation using the inverted microscope, as described in ESI (Fig. S2†).After rinsing with PBS, the coated surface was incubated for 2 h at 4 °C with BVMO (10, 20, 40 μM) in 50 mM Tris–HCl buffer, pH 8.0 for immobilization screening purposes or with a 50 μM FMO3 solution in 50 mM potassium phosphate buffer, pH 7.4 for on-chip tamoxifen conversion.
3.4 Colorimetric assay for BVMO activity
3.5 Multi-channel chip design and fabrication
The 2D design was drawn with AutoCAD based on microscope slides dimensions (76 × 26 cm). The chip consisted of four separate channels (corresponding to four outlets) connected to a single inlet. The channels had a serpentine shape to increase the available surface for enzyme immobilization. All four serpentines had the same surface area, in order to achieve the same flow in each. The serpentines were designed to have a surface area of 45 (S), 70 (M), or 90 (L) mm2 each.All the designs were cut on ARcare® 90106NB medical grade pressure-sensitive adhesive using a CAMM-1 GS-24 cutter (Roland). Inlet (top) and outlet (bottom) holes were drilled into microscope glass slides; outlet holes were also cut into a PDMS sheet (500 μm thickness). Both the PDMS layer and the bottom glass slide were activated using a Plasma Cleaner (Harrik Plasma), and the two surfaces were aligned and bonded overnight at 80 °C. The previously cut adhesive was then used to glue the two microscope slides together.
3.6 FMO3 immobilization on the fabricated microfluidic platform
The surface of the serpentines was coated with a lysine-based glutaraldehyde-crosslinked coating, either using collagen or polylysine. The coating stability at different flow rates was evaluated as described in ESI (Fig. S3†). Then, 50 μM FMO3 was injected into the serpentines from the outlets to cover only the channels surface. The injected volume depended on the serpentine type (6.4 μL for S, 9.9 μL for M, and 12.7 μL for L). After 2 h incubation at 4 °C, the excess enzyme was removed from the outlets with a micropipette. Afterwards, a syringe pump (KD Scientific, KDS-250-CE) was connected to the inlet and 50 mM potassium phosphate buffer, pH 7.4 was pumped at 100 μL min−1 for 4 min to remove the remaining unbound enzyme. The washing time was decided based on the amount of tamoxifen produced by unbound FMO3 washed-out from the polylysine-coated serpentines after immobilization (Fig. S4†).3.7 Computational fluid dynamics simulation of the fabricated chip
A computational fluid dynamics simulation was performed in COMSOL Multiphysics 6.1 to identify the flow rate at which the on-chip retention time of the liquid in the channels is comparable to the incubation time usually used for off-chip enzymatic reactions (5–15 min).23 In a 2D geometry model, the velocities from 5 to 75 μL min−1 and the concentration profiles (Cmax: 0.1 mol L−1) were studied considering a physical model based on transport of diluted species in laminar flow over the course of maximum 20 min. The simulation assumed fluid properties of water, no slip conditions at the wall and an extra fine element size.3.8 Activity of the immobilized enzyme(s) in the fabricated multi-channel platform
Initially, all four serpentines contained the wild-type enzyme. Subsequently, FMO3 WT and its two polymorphic variants (V257M and E158K) were immobilized in three different serpentines of the same chip. The fourth serpentine was used as a control. A solution containing 100 μM tamoxifen and 1 mM NADPH in 50 mM potassium phosphate buffer was constantly fed to the chip at the desired flow rate (10, 25, 50 or 75 μL min−1). The solution was equally split in the four channels, so that the final flow rate in each serpentine was ¼ of the set flow rate. The solution coming out from each outlet was collected into glass vials until at least 50 μL were obtained for HPLC injection. The microfluidic setup (i.e. connection to the syringe pump, sample collection) is shown in Fig. S5.†3.9 Product detection by HPLC
HPLC analysis of tamoxifen and tamoxifen N-oxide was performed on a Vanquish Flex HPLC system (Thermo Fisher Scientific) using an Accucore C18 reversed-phase column (150 × 3 mm, 2.6 μm; Thermo Fisher Scientific). Tamoxifen and tamoxifen N-oxide were separated by isocratic elution of 55% Milli-Q water (+0.1% trifluoroacetic acid) and 45% acetonitrile (+0.1% trifluoroacetic acid) using a column temperature of 68 °C and a flow rate of 1.1 mL min−1. The detection wavelength was set to 276 nm. Retention times of the commercially available standards of tamoxifen and tamoxifen N-oxide were 4.0 and 4.8 min, respectively.374. Conclusions
In this study, a four-channel microreactor with immobilized recombinant enzymes was designed and characterized as a proof-of-concept for the development of a multi-enzymatic microfluidic platform for in vitro screening of newly discovered drugs. The enzyme of interest was FMO3, the second most important monooxygenase present in the human liver, and its two common polymorphic variants: V257M and E158K. These genetic variants have been demonstrated to be associated with inter-individual differences that contribute to drug response.A microfluidic system containing four separate channels connected to a single inlet was designed and fabricated. The channels were drawn as serpentines to increase the surface area available for immobilization and hence enzyme retention by the surface. The enzymes were covalently bound to the chip surface using a glutaraldehyde-mediated cross-linking methodology, which resulted in the highest catalytic activity after immobilization.
The best possible substrate retention time and flow rate (according to the surface area of the serpentines) were determined via computational fluid dynamics simulations.
Initially, only the wild-type enzyme was immobilized in the microfluidic platform, where it was able to successfully convert one of its drug substrates, tamoxifen, into its N-oxide product. The amount of detected N-oxide product varied when changing different parameters in the microfluidic set-up, such as surface coating, surface area available for enzyme immobilization and flow rate. The highest amount of product was obtained when using a 10 μL min−1 flow rate on polylysine-coated serpentines with the largest surface area (90 mm2).
Subsequently, this simple configuration was upgraded to perform simultaneous measurements of tamoxifen conversion by not only the wild-type FMO3, but also its two polymorphic variants. The obtained results demonstrated that the already known differences in the catalytic performance of these enzymes could be reproduced in the microfluidic device.
Moreover, the so-developed microfluidic platform could be exploited to test whether newly discovered drug-like molecules are substrates of specific drug-metabolizing enzymes in a highly efficient and high-throughput format. In fact, four enzymes can be immobilized and work separately on the same surface, making it possible to quickly evaluate if and how a new molecule is converted by all of them. Further developments of this proof-of-concept (e.g. increasing the number of serpentines, adding more inlets, direct connection of the outlets to HPLC-MS and/or GC-MS) could make this platform a feasible option for testing many different compounds with several drug-metabolizing enzymes (and their polymorphic variants) present in the human liver. In addition, the set-up is highly adaptable: enzymes to be immobilized, drugs to be tested, catalysis conditions (i.e. flow rate, temperature and incubation time), and detection method can be easily adjusted as required. Finally, this device could be applied to detect differences in the catalytic performance of other DMEs and their polymorphic variants, making this microfluidic platform directly relevant to personalised medicine.
Author contributions
Conceptualization: PE and SJS; data curation: MDA; formal analysis: MDA, MF; investigation: MDA, SS and FS; methodology: MDA, SS, FS, MF, VLS, OC and SD; resources: PE, GG and SJS; supervision: PE and SJS; visualization: MDA; writing (original draft): MDA and SJS; writing (review & editing): MDA, SS, FS, MF, GG, PE and SJS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Melissa De Angelis is the recipient of a three-year PhD scholarship (PON D.M.1061), University of Torino.References
- H. Dowden and J. Munro, Nat. Rev. Drug Discovery, 2019, 18, 495–496 CrossRef CAS PubMed.
- G. Catucci, G. Gilardi and S. J. Sadeghi, Comprehensive Pharmacology, ed. T. P. Kenakin, Elsevier, Amsterdam, 1st edn, 2022, pp. 509–562 Search PubMed.
- P. R. Ortiz de Montellano, Cytochrome P450: Structure, Mechanism, and Biochemistry, Springer, Berlin, 2005 Search PubMed.
- L. Di, Expert Opin. Drug Metab. Toxicol., 2014, 10, 379–393 CrossRef CAS PubMed.
- P. Fasinu, P. J. Bouic and B. Rosenkranz, Curr. Drug Metab., 2012, 13, 215–224 CrossRef CAS PubMed.
- E. F. Brandon, C. D. Raap, I. Meijerman, J. H. Beijnen and J. H. Schellens, Toxicol. Appl. Pharmacol., 2003, 189, 233–246 CrossRef CAS PubMed.
- M. Ooka, C. Lynch and M. Xia, Int. J. Mol. Sci., 2020, 21, 8182 CrossRef CAS PubMed.
- F. Esteves, D. Campelo, P. Urban, S. Bozonnet, T. Lautier, J. Rueff, G. Truan and M. Kranendonk, Biochem. Pharmacol., 2018, 158, 134–140 CrossRef CAS PubMed.
- Y. Liu and X. Jiang, Lab Chip, 2017, 17, 3960–3978 RSC.
- S. F. Berlanda, M. Breitfeld, C. L. Dietsche and P. S. Dittrich, Anal. Chem., 2021, 93, 311–331 CrossRef CAS PubMed.
- Y. Liu, L. Sun, H. Zhang, L. Shang and Y. Zhao, Chem. Rev., 2021, 121, 7468–7529 CrossRef CAS PubMed.
- A. Kecskemeti and A. Gaspar, Talanta, 2017, 166, 275–283 CrossRef CAS PubMed.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS.
- R. C. Rodrigues, Á. Berenguer-Murcia, D. Carballares, R. Morellon-Sterling and R. Fernandez-Lafuente, Biotechnol. Adv., 2021, 52, 107821 CrossRef CAS PubMed.
- O. Barbosa, C. Ortiz, A. Berenguer-Murcia, R. Torres and R. Fernandez-Lafuente, RCS Adv., 2014, 4, 1583–1600 CAS.
- A. Sassolas, L. J. Blum and B. D. Leca-Bouvier, Biotechnol. Adv., 2012, 30, 489–511 CrossRef CAS PubMed.
- S. A. Ansari and Q. Husain, Biotechnol. Adv., 2012, 30, 512–523 CrossRef CAS PubMed.
- J. R. Cashman and J. Zhang, Annu. Rev. Pharmacol. Toxicol., 2006, 46, 65–100 CrossRef CAS PubMed.
- I. R. Phillips and E. A. Shephard, Trends Pharmacol. Sci., 2008, 29, 294–301 CrossRef CAS PubMed.
- D. M. Ziegler, Drug Metab. Rev., 2002, 34, 503–511 CrossRef CAS PubMed.
- I. R. Phillips and E. A. Shephard, Xenobiotica, 2019, 50, 19–33 CrossRef PubMed.
- S. K. Krueger and D. E. Williams, Pharmacol. Ther., 2005, 106, 357–387 CrossRef CAS PubMed.
- G. Catucci, S. Bortolussi, G. Rampolla, D. Cusumano, G. Gilardi and S. J. Sadeghi, Basic Clin. Pharmacol. Toxicol., 2018, 123, 687–691 CrossRef CAS PubMed.
- S. Bortolussi, G. Catucci, G. Gilardi and S. J. Sadeghi, Arch. Biochem. Biophys., 2021, 697, 108663 CrossRef CAS PubMed.
- M. Oscarson, CCLM, 2003, 41, 573–580 CAS.
- D. Minerdi, I. Zgrablic, S. J. Sadeghi and G. Gilardi, Microb. Biotechnol., 2012, 5, 700–716 CrossRef PubMed.
- G. Catucci, I. Zgrablic, F. Lanciani, F. Valetti, D. Minerdi, D. P. Ballou, G. Gilardi and S. J. Sadeghi, Biochim. Biophys. Acta, 2016, 1864, 1177–1187 CrossRef CAS PubMed.
- G. Catucci, S. Turella, H. Cheropkina, M. De Angelis, G. Gilardi and S. J. Sadeghi, Biocatal. Agric. Biotechnol., 2022, 44, 102458 CrossRef CAS.
- E. Hodgson, R. L. Rose, Y. Cao, S. S. Dehal and D. Kupfer, J. Biochem. Mol. Toxicol., 2000, 14, 118–120 CrossRef CAS PubMed.
- S. K. Krueger, J. E. VanDyke, D. E. Williams and R. N. Hines, Drug Metab. Rev., 2006, 38, 139–147 CrossRef CAS PubMed.
- Y. Zhu, Q. Chen, L. Shao, Y. Jia and X. Zhang, React. Chem. Eng., 2020, 5, 9–32 RSC.
- Y. Zhang, V. Lukacova, K. Reindl and S. Balaz, J. Biochem. Biophys. Methods, 2006, 67, 107–122 CrossRef CAS PubMed.
- G. Catucci, G. Gilardi, L. Jeuken and S. J. Sadeghi, Biochem. Pharmacol., 2012, 83, 551–558 CrossRef CAS PubMed.
- H. Bao, Q. Chen, L. Zhang and G. Chen, Analyst, 2011, 136, 5190 RSC.
- C. Cherkouk, L. Rebohle, J. Lenk, A. Keller, X. Ou, M. Laube, C. Neuber, C. Haase-Kohn, W. Skorupa and J. Pietzsch, Clin. Hemorheol. Microcirc., 2015, 61, 523–539 CAS.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- S. Castrignano, G. Gilardi and S. J. Sadeghi, Anal. Chem., 2015, 87, 2974–2980 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01516a |
| This journal is © The Royal Society of Chemistry 2024 |