
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spectroscopic characterization of europium binding to a calmodulin-EF4 hand peptide–polymer conjugate†
Dini Marlina a,
Yannic Müllersbc,
Ulrich Glebebc and
Michael U. Kumke*a
a,
Yannic Müllersbc,
Ulrich Glebebc and
Michael U. Kumke*a
aUniversity of Potsdam, Institute of Chemistry, Optical Sensing and Spectroscopy, Karl-Liebknecht-Str. 24–25, 14476 Potsdam-Golm, Germany. E-mail: kumke@uni-potsdam.de
bUniversity of Potsdam, Institute of Chemistry, Polymer Materials and Polymer Technologies, Karl-Liebknecht-Str. 24–25, 14476 Potsdam-Golm, Germany
cFraunhofer Institute for Applied Polymer Research IAP, Geiselbergstr. 69, 14476 Potsdam-Golm, Germany
First published on 29th April 2024
Abstract
The emergence of biological ligand as an alternative to chemical ligands enables a sustainable lanthanide extraction route. In this study, a peptide originating from the loop of domain 4 calmodulin (EF4) was synthesized and the interaction with europium ions was monitored using time resolved laser fluorescence spectroscopy (TRLFS). Despite being retracted from its full protein structure, the twelve amino acids of calmodulin-EF4 showed binding to europium. Europium-peptide complex formation was evident by an increase in decay time from 110 to 187 μs. The spectra of europium bound to peptide can be easily distinguished from the free europium ion as the 5D0 → 7F2 peak intensifies. When europium bound to the peptide–polymer conjugate, the decay time was further increased to 259 μs. This suggests that lanthanide binding can be enhanced by immobilizing the short peptide into a polymer matrix. The europium-peptide/conjugate bond was reversible, triggered by pH, promoting peptide reusability. Due to the fact that the study was conducted exclusively in water, it suggests minimal use of chemicals is possible while maintaining peptide affinity. This makes the calmodulin-EF4 peptide an ideal candidate as biological ligand. This study lays the groundwork for developing a peptide-based filter material for lanthanide separation.
Introduction
Lanthanides are enabling elements in contemporary technologies. As a consequence, the demand for lanthanides has increased rapidly along with technological developments, e.g., related to renewable energy. The lanthanides are 4f elements starting from La (Z = 57) to Lu (Z = 71). The primary source of lanthanides are minerals, e.g., monazite and bastnaesite. Lanthanides occur simultaneously in minerals, and the extraction as well as the separation from minerals are energy- and chemical–intensive processes.1,2 The challenges in lanthanides separation arise from the similarity of their physicochemical properties. In the minerals the +3 oxidation state prevails and the difference in ionic radii is extremely small, ranging from 1.2 Å for La3+ to ∼1.0 Å for Lu3+.3 This situation makes the tailoring of complexing agents specifically designed to recognize a particular lanthanide ion very challenging.4 As a result, the separation process needs to be repeated many times which releases large amounts of chemical waste that may cause water, soil, and air pollution.5,6 Moreover, mineral deposits also contain radioactive materials (Th, U), that if left unmitigated, can pose a severe risk to human health, for example, through inhaling contaminated dusts.1According to a review conducted by McNulty et al.,1 the recoveries of lanthanides from their primary source are limited by current technology and plant practice to 50–80%.1 The increasing demand has caused the need to extract lanthanides from secondary sources such as acid mine drainage (AMD), coal and coal by-product, iron ore and apatite.7 Common complexing agents for lanthanides are, citrate, α-hydroxy-butyrate, or ethylenediaminetetraacetic acid (EDTA)8 In case of EDTA, it lacks in selectivity as it can bind divalent ions (Mg2+, Cu2+, Fe2+, Mn2+, Ni2+, and Zn2+).9 An alternative approach to overcome the lack of selectivity is to utilize specialized biological ligands.10,11 This way, the separation process can be trimmed reducing chemical as well as energy consumption being less harmful to the environment.
Biological ligands can be derived from proteins found in nature.12–14 Particular amino acid sequences located in calmodulin (CaM), a calcium ion (Ca2+) sensor protein, were found to form strong complexes with lanthanides. CaM is composed of 148 amino acids and binds Ca2+ in a putative helix-loop-helix domain known as the EF-Hand.15 The interactions of lanthanide ions with CaM have been known for decades. Due to the similar ionic radii, Ln3+ can replace Ca2+ and due to their unique optical properties (e.g., Eu3+) can serve as a spectroscopic probe in CaM structure determination studies.12,15–19 Using the full protein as a potential ligand in a technical process might be difficult for various reasons. On the other side using a shorter amino acid sequence, which is directly involved in the metal binding is more favourable. CaM has four EF-Hands, labelled as EF1, EF2, EF3, and EF4, each consisting of 12 amino acids. The amino acid sequences of EF1–EF4 are almost identical, only differing in certain positions. The amino acid sequences of the EF-Hand motif are intriguing as a starting point in the course of finding efficient and selective novel ligands for lanthanide complexation.
Many metal binding affinity evaluations were carried out using the full CaM structure.12,15–19 CaM has a cooperative effect increasing the affinity for further Ln3+ ions after binding the first Ln3+ ion. Until now, most of the experiments have been carried out in a buffer solution, which is unlikely to be encountered in technical applications, e.g., extraction of lanthanides from AMD. Some buffers (MOPS, MES, HEPES, and PIPES) at high concentration (>10 mM) were also found to interact with Eu3+ changing the speciation and because of the very similar chemical properties this can also be expected for the lanthanides in general.20 Consequently, buffers should be used with care to provide unbiased results that represent purely peptide–lanthanide interactions, which would be relevant in a remediation process for lanthanides from secondary sources.
When envisioning an effective and sustainable lanthanide separation system, the peptide should be functionalized or immobilized onto a solid support to enable an easy recovering and reuse of the peptide. Polymers are often used as solid support for the immobilization of biomolecules. In the search for a biomimetic peptide–polymer conjugate, a first but important step is to manage the coupling between both without eliminating the binding capabilities, but instead improving them due to a synergistic effect. This synergy is highly wanted since it is known from previous studies that short peptides lack the advantage of a pre-structuring due to the backbone in the full protein and subsequently show a low(er) binding affinity.
In this study as a first step to a biomimetic peptide-polymer-toolbox, we synthesized EF4 CaM peptide and its conjugate with the polymer poly(dimethyl acrylamide) (pDMA). Solid-phase peptide synthesis (SPPS) was exploited, being automatized, and enabling high purity and large scale especially for peptides with less than 50 amino acids.21 We investigated a model peptide for lanthanide binding based on the EF4 CaM, having the sequence DIDGDGQVNYEE. While still being immobilized on the resin for SPPS, a chain-transfer agent (CTA) for reversible addition–fragmentation chain-transfer (RAFT) polymerization was conjugated to the N-terminus of the peptide. After cleavage from the resin, pDMA was grown in a grafting-from approach.
The complexation of Eu3+ with peptide or peptide-polymer conjugate was investigated under mild acidic conditions (pH 5), without any buffer solutions, to simulate real-life wastewater conditions frequently found for mining drainage. It is advantageous to use pH 5 to avoid the formation of europium carbonates or hydroxides complexes since these complexes start to form at pH 7 and higher.22 Moreover, aspartic and glutamic acid, which are potential binding sites of peptides, are already deprotonated at this pH (with pKa values of 3.9 and 4.2, respectively) and are subsequently fully available for cation binding.23 Complexation was monitored using time resolved laser fluorescence spectroscopy (TRLFS) and the luminescence spectra were deconvoluted using parallel factor analysis (PARAFAC) to identify the chemical species. The reversibility of binding between an Eu3+ ion and peptide was tested by lowering the solution pH and was monitored qualitatively using luminescence spectroscopy.
Experimental
Materials and reagents
Tert-butanoic acid butyl trithiocarbonate (BTMP) was synthesized according to a published procedure.24 DMA was purified over alumina to remove the inhibitors before polymerization. Millipore pure water with a resistivity of 18.2 MΩ cm was used. Europium was purchased as chloride salt (EuCl3·6H2O) from Sigma Aldrich and was used as received. It dissolved as a stock solution in deionized water and further diluted to desired concentration. A commercial EF4 Hand peptide (purity 95.9%) as a reference peptide was purchased from Peptide&Elephants.Peptide synthesis and characterization
Binding studies of peptide-Eu3+
Before luminescence measurement, the solution was mixed using a shaker (Vortex Genie 2, Scientific Industries) for at least 10 s. The measurements were carried out at room temperature. Eu3+ was excited at λex = 394 nm with a pulsed Nd:YAG laser (Quanta Ray, Spectra Physics, with repetition rate 20 Hz or 10 Hz). The luminescence was monitored at 570 nm < λem < 720 nm. The detector used was an iCCD camera coupled to a spectrograph (iStar DH734-18H-13, Andor technology coupled to Shamrock 303i-A, SR0275, Andor technology for the 20 Hz laser or iStar sCMOS, Andor technology coupled to Kymera328i, Andor technology for the 10 Hz laser). In both detection systems a grating with 300 lines per mm with λblaze = 760 nm (20 Hz laser) or 500 nm (10 Hz laser) was used.
The boxcar technique was used to measure time resolved emission spectra and subsequently to obtain the emission kinetics. The slit size, gate width, initial delay time, and gate delay step were adjusted according to the laser used (Table S1†). The luminescence spectra were corrected for the spectral sensitivity of the grating and of the iCCD camera, respectively.
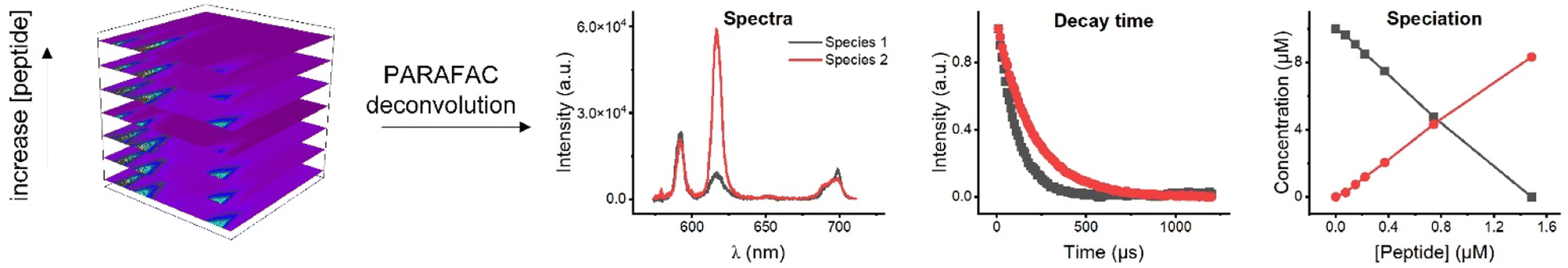 | ||
| Fig. 1 Four-dimensional data cube in PARAFAC analysis consists of emission wavelength, intensity, and time as x-, y-, z- axis, respectively, with different peptide concentration as the fourth axis. PARAFAC deconvolution yielded information regarding the species. | ||
In the luminescence measurements the data were collected for the spectral range between 570 nm < λem < 720 nm, covering the transitions up to 5D0 → 7F4. Using the species-selective emission spectra obtained from the PARAFAC analysis the luminescence quantum efficiencies of the different species were calculated based on the Judd–Ofelt theory using an in-lab written routine for MATLAB 2022b. The quantum efficiencies (ϕ) of the different Eu3+ species in the solutions were used to determine the concentrations and subsequently the dissociation constant (KD) values. The relative concentration of each species (Ci) was calculated from its luminescence intensity (Ii) and the quantum yield (ϕi) according to the eqn (1):25
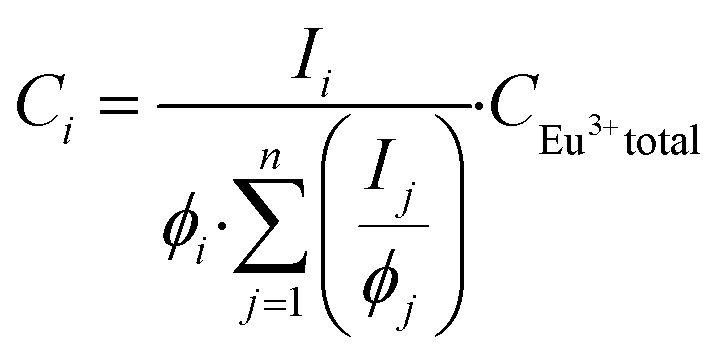 | (1) |
To connect the photophysical data in a more intuitive way to the Eu3+-complexes, the fluorescence decay time was used. The estimation of the number of water molecules (n) in the first coordination sphere of Eu3+ was calculated based on eqn (2):15
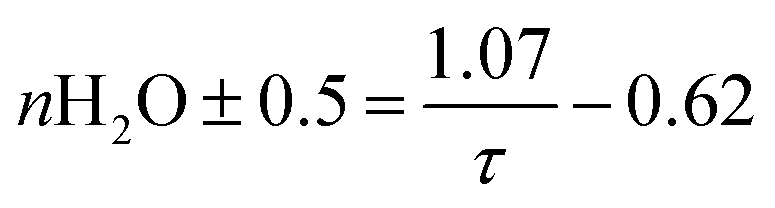 | (2) |
Results and discussion
Peptide and peptide-polymer characteristic
The EF4 peptide was synthesized in a standard SPPS approach on an automated peptide synthesizer.21 A resin with 2-chlorotrityl (2-CT) linker and preloaded with glycine was exploited for SPPS, resulting in DIDGDGQVNYEEG (EF4G) peptide (Scheme S1†). Subsequently, while still being immobilized on the resin, the peptide was functionalized with a CTA for RAFT polymerization. As SPPS takes place from the C- to the N-terminus, the CTA tert-butanoic acid butyl trithiocarbonate (BTMP) with carboxylic acid functionality was chosen to be linked to the peptide's N-terminus via the same amide bond forming chemistry as the amino acids before. In addition, the trithiocarbonate structure of the CTA is more stable towards amines than other CTAs.26 The aim of our study was to investigate the influence of polymers that are covalently connected to the peptide on the Ln binding properties. To synthesize peptide–polymer conjugates, we chose RAFT polymerization which is a frequently used controlled radical polymerization. For RAFT, a CTA is required to mediate the polymerization process. In this reaction, the peptide species (R-group) functions as a radical, while the long alkyl chain in the CTA (Z-group) mediates the stability of the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond and the adduct radical. Theoretically, it would be possible to connect peptide and CTA via the Z-group, however this would be synthetically more complicated with unknown influence on polymerization.
C bond and the adduct radical. Theoretically, it would be possible to connect peptide and CTA via the Z-group, however this would be synthetically more complicated with unknown influence on polymerization.
After cleavage from the resin and precipitation in cold diethylether, ESI mass spectrometry confirmed the expected masses (Fig. S1†). HPLC analysis demonstrated a sufficient purity of the peptide (Fig. S2†). As shown in Fig. S3,† the conjugation of BTMP to the peptide resulted in a mixture of unmodified peptide and CTA-peptide. This was probably caused by a non-quantitative coupling reaction due to high steric hindrance of the carboxylic acid group of BTMP through both alpha-methyl groups. The purity of the conjugate was determined to be 52% and was calculated by the ratio of absorbance at 220 nm of modified and unmodified peptide. Since EF4G does not interfere in the polymerization reaction, the CTA-peptide was used for polymerization without further purification. Such a grafting-from approach is advantageous regarding ease of purification and ensures that nearly all polymers carry a peptide end group.27 DMA was polymerized with a targeted degree of polymerization of 160. Monomer conversion was 96% as determined by NMR spectroscopy of the crude polymer mixture. GPC analysis of pDMA-EF4G (Fig. S4†) showed a well-controlled reaction with a product dispersity Ð of 1.3. The number weighted molar mass Mn was determined to 17.6 kDa which fits well with the theoretical mass of 17.2 kDa calculated through monomer conversion. 1H-NMR spectroscopy of both peptide and peptide-polymer conjugate (Fig. S5†) show the characteristic proton signals of the amide backbone as well as asparagine and glutamine sidechains in the low field of the spectrum. This indicates the successful implementation of EF4G into the polymer.
Luminescence spectroscopy studies
The Eu3+ luminescence originates from the 5D0 excited state to the 7FJ (J = 0 to 6) ground states and the spectral intensity distribution of the Eu3+ luminescence is very sensitive to the actual ligand field. Therefore, Eu3+ was used as the luminescence probe of choice to establish a procedure for the investigation of novel polymer–peptide conjugates as binders for the extraction of lanthanides. However, since the 5D0 → 7FJ (J = 5,6) transitions are in the NIR spectral range and usually low in intensity, in this study the luminescence spectra were monitored only in the wavelength range of 570 nm < λem < 720 nm, covering the transitions from 5D0 → 7FJ (J = 0 to 4), which is sufficient to apply Judd–Ofelt theory for the further calculation of specific photophysical parameters such as the luminescence quantum yield (vide infra).In Fig. 2 the alterations of the Eu3+ luminescence spectrum upon addition of EF4G or pDMA-EF4G are shown. In the absence of EF4G (or pDMA-EF4G), at pH 5 Eu3+ exists as an aquo ion characterized by the dominance of the 5D0 → 7F1 transition peak. Because in the aquo ion the coordination sphere of the Eu3+ ion with 8 to 9 water molecules is highly symmetric, the 5D0 → 7F0 transition is strictly forbidden and not seen in the emission spectrum at RT.28 Meanwhile, for the Eu3+ peptide complexes (EF4G as well as pDMA-EF4G), the spectral intensity distribution of the Eu3+ emission is distinctly changed. In contrast to the aquo ion now the luminescence spectra are dominated by the 5D0 → 7F2 transition peak (λem = 616 nm). This transition is often indicated as “hypersensitive”, which means that its intensity is strongly influenced by the local symmetry of the Eu3+ ion and the nature of the ligand. In combination with the 5D0 → 7F1 transition (magnetic transition, which is not affected by the ligand field and therefore useful as internal standard), the intensity ratio of (5D0 → 7F2/5D0 → 7F1) peaks (asymmetry ratio) is often used as an indicator of alterations in the Eu3+ coordination site.29 It is worth mentioning that asymmetry ratios can already be extracted from the raw data without advanced data treatments to visualize the progress of the complexation upon addition of peptide (inset Fig. 2a and b). The asymmetry ratio of the respective complex can be extracted from the species-selective spectra obtained from PARAFAC analysis (vide infra, Table 1).
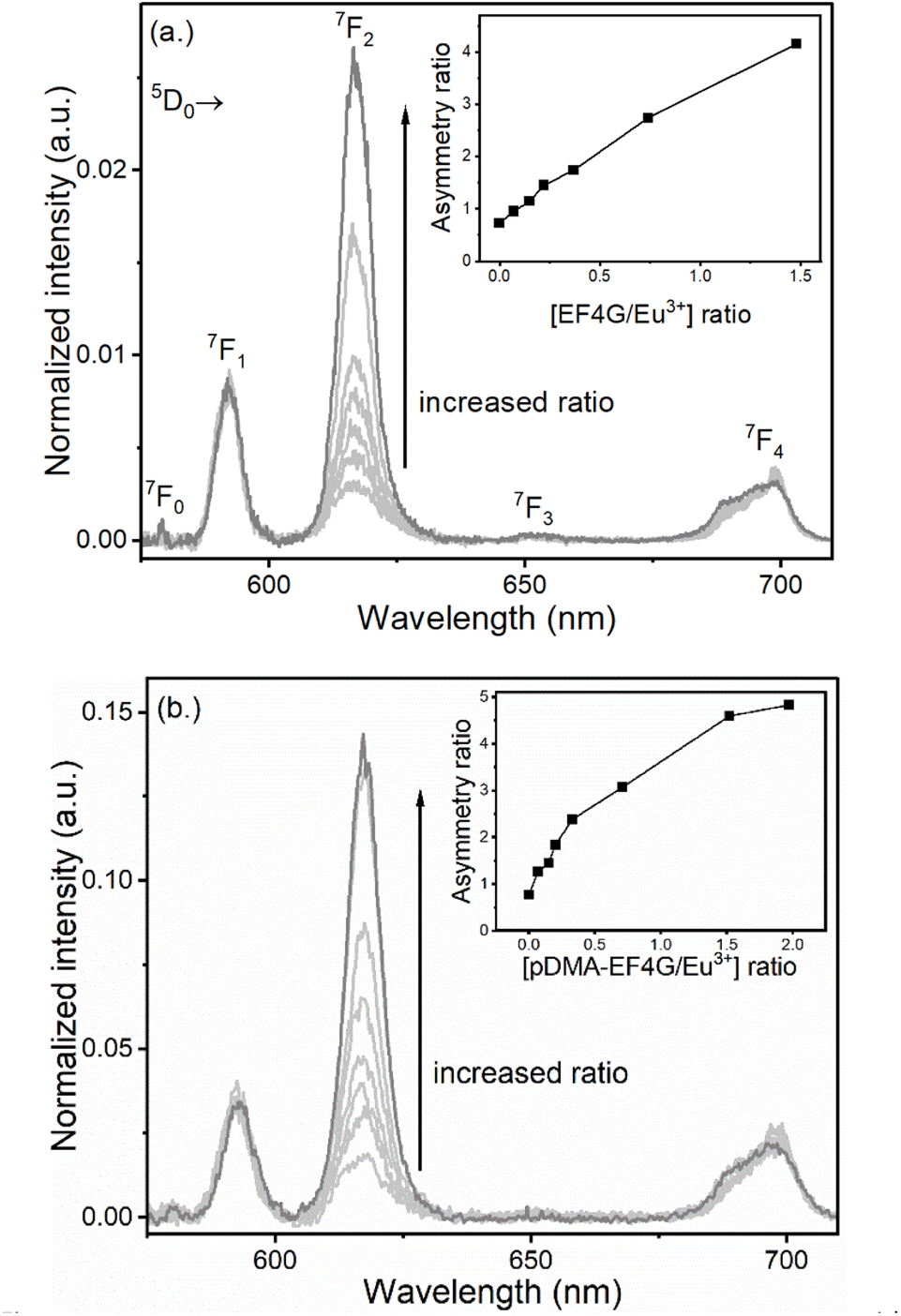 | ||
| Fig. 2 Luminescence spectra of Eu3+ at pH 5 taken at gate delay 10 μs with increasing concentration of (a) EF4G and (b) pDMA-EF4G. The complexation of Eu3+ with the peptide indicated by the increase of the 5D0 → 7F2, “the hypersensitive” peak intensity; inset: asymmetry ratio. | ||
| Eu3+ aquo ion | Eu3+-EF4 (ref.) | Eu3+-EF4G | Eu3+-pDMA-EF4G | |
|---|---|---|---|---|
| a Data averaged from three experiments.b Standard deviation of Kd was estimated to be 50% from the stated value. | ||||
| 5D0 → 7F0 peak position (nm) | — | 579.37 | 579.15 | 579.9 |
| Asymmetry ratio | 0.8 | 3.5 | 3.7 | 4.9 |
| Decay time (τ) in μs | 110 ± 10a | 163 ± 1 | 187 ± 1 | 259 ± 2 |
| Number of water molecules (n) | 9 ± 0.5 | 6 ± 0.5 | 5 ± 0.5 | 3.5 ± 0.5 |
| Quantum yield (φ) in % | 1 ± 0.2 | 3.2 ± 0.2 | 3.8 ± 0.2 | 7.1 ± 0.2 |
| JO parameter | ||||
| (Ω2) (10−20 cm2) | 1.3 | 6.2 | 6.7 | 9.0 |
| (Ω4) (10−20 cm2) | 2.7 | 2.8 | 2.6 | 4.4 |
| Dissociation constant (KD) in μMb | — | 8.9 | 8.0 | 0.4 |
Another direct indication for the complexation of Eu3+ by the peptides is the appearance of the 5D0 → 7F0 peak (see Table 1 for λem). For the Eu3+ complex with EF4G the 5D0 → 7F0 peak was more prominent meanwhile for the pDMA-EF4G conjugate the 5D0 → 7F0 peak was weaker and monitoring the complexation based on this particular parameter would be challenging.
In addition, also the 5D0 → 7F4 spectral distribution and relative intensity was slightly changing upon complexation with EF4G or pDMA-EF4G (see Fig. 2). In general, the intensity of this band is weaker than the other luminescence peaks in the Eu3+ luminescence spectra.29
Based on the raw luminescence data presented in Fig. 2, it can already be concluded that EF4G as well as pDMA-EF4G are complexing Eu3+ in an aqueous solution at pH 5, which is lower than the physiological pH under which peptides or the full calmodulin normally would be tested.12,16 Moreover, based solely on the alteration of the spectral intensity distribution (asymmetry ratio, intensity of luminescence peaks), which was found to be similar but not identical for both ligands (EF4G vs. its polymer conjugate). From the distinct differences an influence of the polymer on the overall binding is indicated.
As a comparison, we also tested the affinity of a commercially obtained EF4 (“only” 12 amino acids) and compared to our EF4G (13 amino acids) we found identical binding characteristics. This shows that the additional glycine added to the EF4 sequence does not influence the peptide binding (see Table 1 and Fig. S6†). Additionally, the potential contribution to Eu3+ binding from the polymer itself was checked by adding free polymer (pDMA) only. Here, no alteration in the spectral intensity distribution of the Eu3+ luminescence was found indicating that, in absence of peptide, the polymer does not complex the Eu3+ ion (see Fig. S7†).
Although the increased binding by EF4G (or pDMA-EF4G) can be qualitatively shown by the dependence of the asymmetry ratio and partly, by the intensity increase of the 5D0 → 7F0 transition, the determination of the dissociation constant (KD) values to quantify the binding based on the raw data is not straightforward since either the luminescence signal is a sum of contributions from the aquo ion and the Eu3+ bound to the peptide (e.g., affects the absolute value of the asymmetry ratio) or the overall intensity of the 5D0 → 7F0 transition is small and therefore not very precise. In order to determine the KD, the concentrations of aquo ion as well as of the peptide complex needs to be known at each step of the titration. Therefore, the raw data have to be deconvoluted into the species related fractions. This was carried out applying a PARAFAC analysis in combination with Judd–Ofelt theory to the experimental TRLFS data (vide infra), because for a quantitative analysis of the luminescence data the luminescence quantum yields of the species is needed.
TRLFS data analysis
TRLFS titration data of Eu3+ with EF4G and pDMA-EF4G were deconvoluted using parallel factor analysis (PARAFAC). PARAFAC is a routine analysis used to deconvolute multidimensional emission spectra.25,30–36 In the PARAFAC analysis, fitting the data with a two-species-model yielded for both ligands the best result.For the investigated molar ratios between Eu3+ and the ligands no indication of a further species was found and therefore the two species were attributed to the Eu3+ aquo ion and the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Eu3+-EF4G complex (or the Eu3+-pDMA-EF4G complex). The obtained species-selective luminescence spectra, the respective luminescence decay kinetics (and luminescence decay times) as well as the relative fractions of the species at different molar ratios of Eu3+ and ligand are shown in Fig. 3.
:1 Eu3+-EF4G complex (or the Eu3+-pDMA-EF4G complex). The obtained species-selective luminescence spectra, the respective luminescence decay kinetics (and luminescence decay times) as well as the relative fractions of the species at different molar ratios of Eu3+ and ligand are shown in Fig. 3.
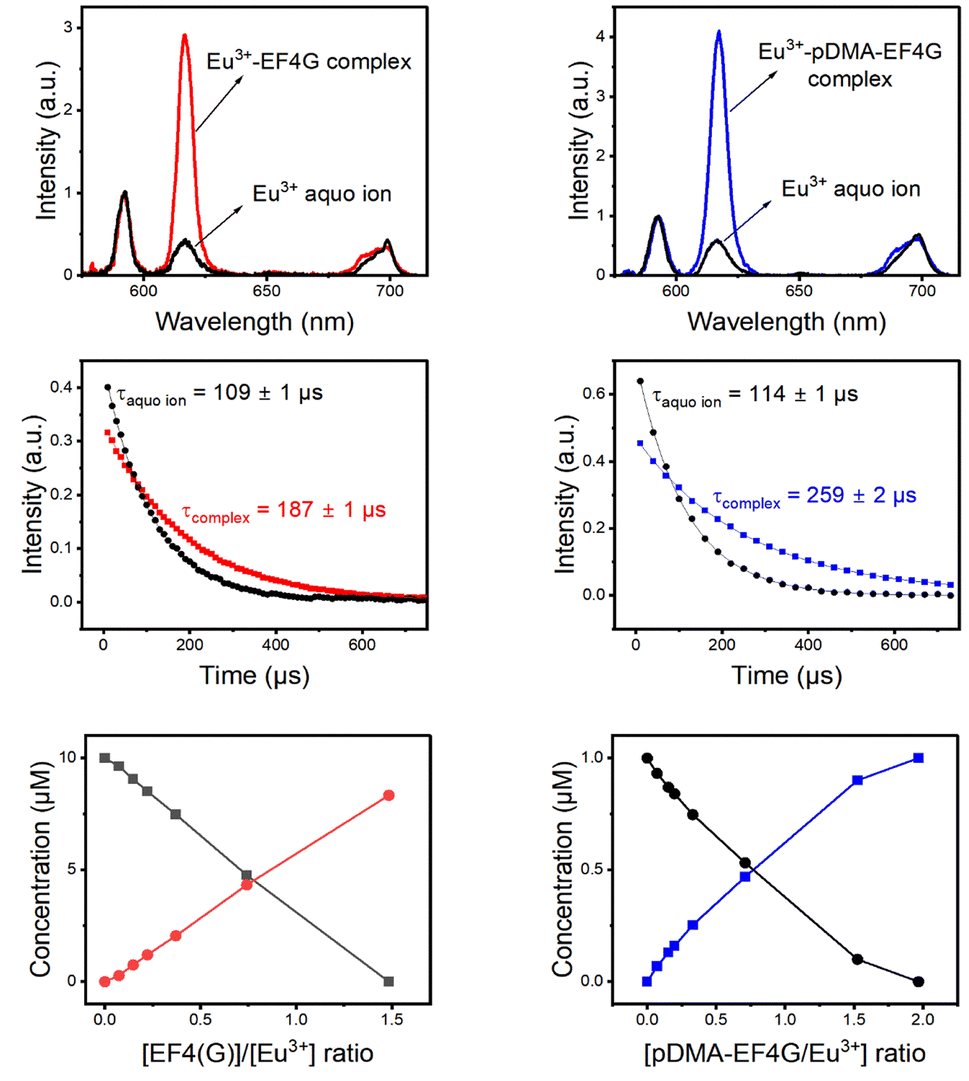 | ||
| Fig. 3 PARAFAC deconvolution of TRLFS of Eu3+ complexation with EF4G and pDMA-EF4G. From top to bottom: spectrum of each species, decay time, and species distribution. | ||
The decay time (τ) of Eu3+ aquo ion is found to match with literature data.15,22 A τ of 110 ± 10 μs was determined for the aquo ion for all ligand systems (including controls) and based on eqn (2) this corresponds to 8–9 water molecules in the first coordination sphere. The decay time of Eu3+ bound to EF4G is determined to τ = 187 ± 1 μs, which corresponds to an average of 5 ± 0.5 water molecules. This decay time is distinctly shorter in comparison with EF4 in an intact calmodulin (τ = 405 ± 2.0 μs) which indicates that the binding situation using a short peptide is very different due to the missing pre-structuring and additional binding by the protein's backbone.15 In a short water-soluble peptide, the interactions between hydrophobic side chain and hydrophilic backbone segments are interspersed, precluding the formation of specific intramolecular hydrophobic clusters. In consequence, the short linear peptides would adopt random structures in water solution.37 This behaviour is confirmed by a report on isolated EF3 calmodulin that was found to be in a random configuration.19 As EF3 and EF4 calmodulin sequences differ only slightly, we assume that this random structure applies to isolated EF4 calmodulin as well.
Considering the larger average number of water molecules in the first coordination sphere (5 for EF4G vs. 2 for the full protein, calculated from the luminescence decay time using eqn (2)), a multi-dentate binding can be ruled out (indication of only two water molecules being removed from first coordination sphere of aquo ion), which means the peptide seems to bind only via bidentate motif.
The conjugation with the polymer pDMA increased the decay time of the Eu3+-complex to τ = 259 μs corresponding to an average of 3–4 water molecules in the first coordination. This implicates that more binding sites from the pDMA-EF4G are attached to the Eu3+ ion. The attachment of the peptide to polymer enhances the rearrangement of the binding sites, improving the folding structure of the peptide. A similar effect was observed in the complexation of europium with 33-mer peptides that bind to DNA. There is a decrease in the number of water molecules in the first coordination sphere of europium, which results in a longer decay time.38
The complexes Eu3+-EF4G and Eu3+-pDMA-EF4G have asymmetry ratios of 3.7 and 4.9, respectively. According to literature data, this finding is consistent with the ratio of Eu3+ bound to sites 2–4 in calmodulin, which has a ratio of 4.15 From the fact that the observed asymmetry ratios are all very similar for the different complexes, it is shown that this parameter is more sensitive to the overall symmetry instead of the nature of the ligands. Here, at least in an aqueous milieu the luminescence decay time is the stronger indicator.
Influence of pH on the complex formation
The reversibility of the Eu3+ binding to the peptide material is of high importance for further potential technical applications. We have investigated the pH as a potential trigger to release the Eu3+ from the polymer-peptide complexes. A variation of the pH in the range of 3 < pH < 6 was tested, which on the one hand side can be expected for natural water (e.g., from mining drainage) and on the other hand should not cleave the peptide. The Eu3+-peptide-polymer (at a molar ratio 1:1) binding was qualitatively monitored by observing the changes in the Eu3+ luminescence spectrum (Fig. 4). At the initial pH, the spectrum is characteristic for the Eu3+: EF4G/pDMA-EF4G complex. As the pH is decreased to around 3.8, the spectrum now becomes characteristic for the aquo Eu3+ ion indicating a release of the Eu3+ from the peptide complex. Re-binding of Eu3+ is achieved when the pH is adjusted back to 5–6. The asymmetry ratio (Fig. 5) points to different Eu3+ species according to the solution pH. At pH 5–6 the asymmetry ratio is ≥3 indicating Eu3+ binding to the peptide and at pH ∼3, the asymmetry ratio is <1, characteristic for Eu3+ aquo ion. As isolated EF Hand has no particular conformational constraints in aqueous solution, the binding process is governed by the functional group present in the side chains. According to the literature, the coordination positions of EF Hand in intact protein are located at position 1, 3, 5, 7, 9 and 12.39 In the case of isolated EF4 CaM, not all amino acids at those positions bind to Eu3+. Since the binding reversibility is highly affected by the pH, it is likely that the acidic side chain of aspartic and glutamic acid play role in the coordination with Eu3+. The binding reversibility of lanthanide ions with bioinspired ligands has also been observed with Lanmodulin.40,41
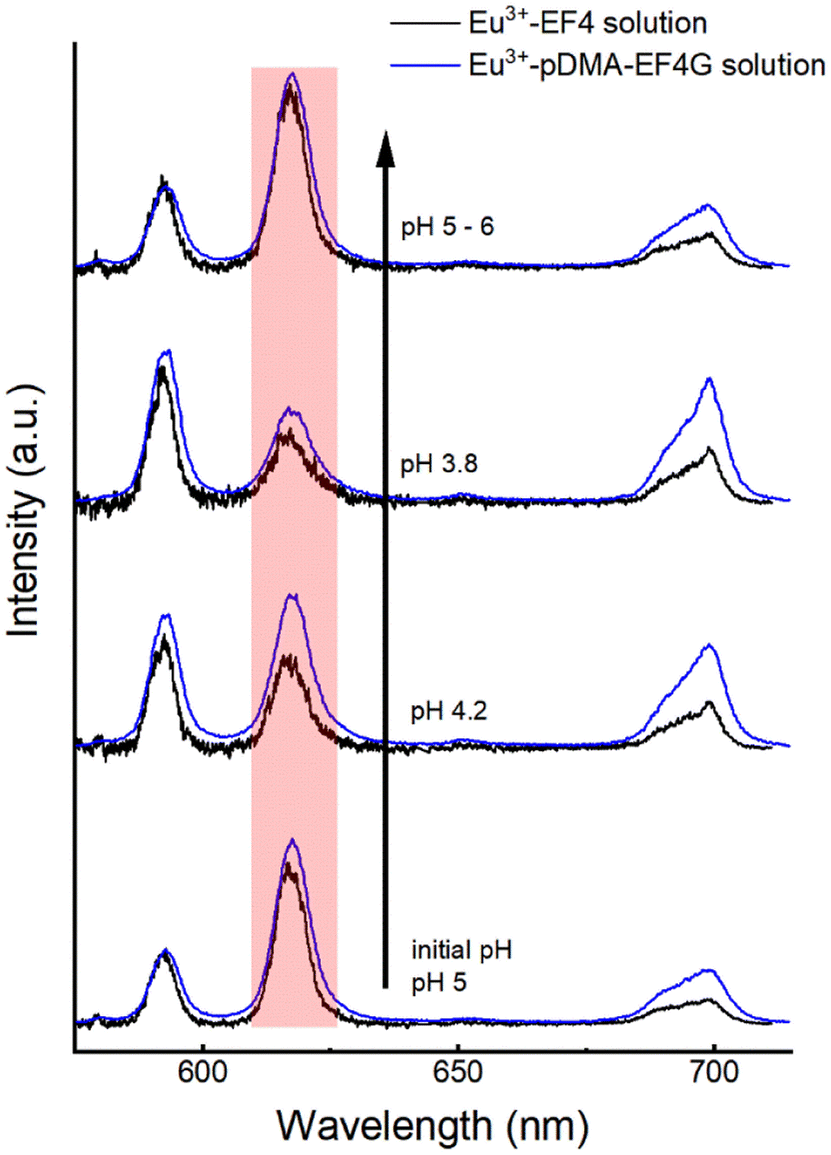 | ||
| Fig. 4 Spectral change of Eu3+-EF4/pDMA-EF4G (1:1 molar ratio) luminescence upon alteration of the pH. The change in intensity of 5D0 → 7F2 (hypersensitive) peak indicates the binding reversibility: at pH 4.2 the intensity started to decrease, at pH 3.8 the luminescence spectrum resembles largely the Eu3+ aquo ion upon increasing the pH back above 5 the luminescence of the peptide complex is observed again. | ||
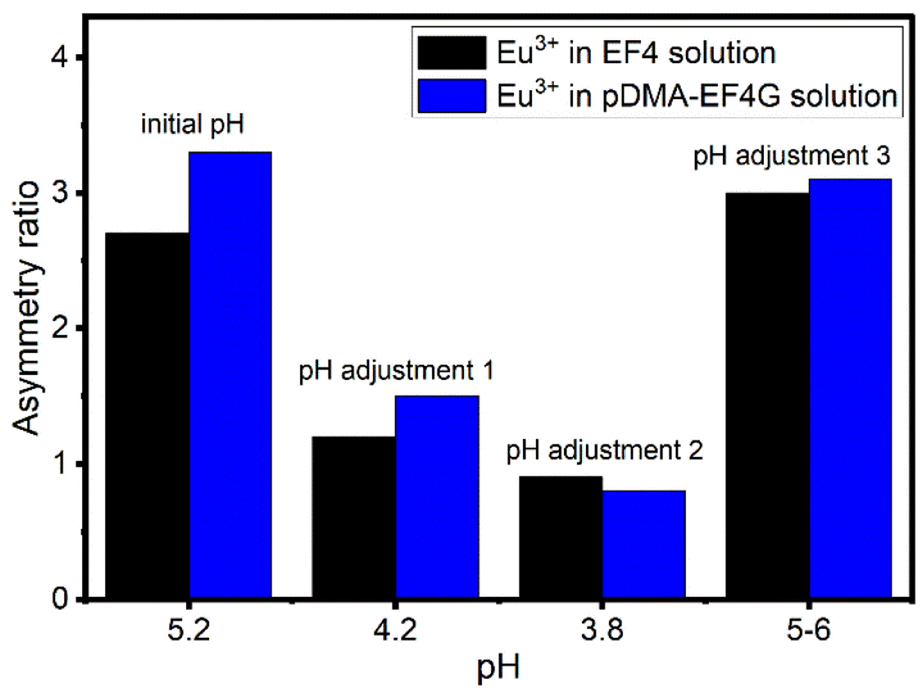 | ||
| Fig. 5 The change in asymmetry ratio of Eu3+ in EF4/pDMA-EF4G solution at different pH. Black: Eu3+ in EF4 solution, blue: Eu3+ in pDMA-EF4G solution. Asymmetry ratio < 1 indicates the free Eu3+ aquo ion. | ||
Conclusions
The utilization of peptides as biological ligands for lanthanide complexation was demonstrated for a small peptide fragment of the metal-binding site of calmodulin and Eu3+ as a representative of lanthanide elements. First, the peptide EF4G was synthesized using the SPPS method and showed in control experiments binding of Eu3+. In the next step a chain-transfer agent (CTA) for RAFT polymerization was conjugated to the N-terminus of the peptide, which was still immobilized on the SPPS resin. With this precursor material an important step for the further synthesis of peptide–polymer conjugates was made. In a first prove of concept, pDMA was grown in a grafting-from approach to obtain the pDMA-EF4G conjugate which was further characterized for its Eu3+ binding properties.Conjugation of the peptide to the polymer pDMA has a synergistic effect on the Eu3+ binding as can be evidenced from TRLFS-PARAFAC analysis. The increased decay time (compared to EF4G) of the Eu3+-conjugate complex is caused by the exclusion of more water from the Eu3+ first coordination sphere. It is attractive to assume that the polymer improves the binding of Eu3+ by pre-structuring the peptide sequence (like the contribution of the backbone in the full protein) making a multidentate binding more favourable even for such a small molecule. In order to be used in the recycling process of lanthanide ions, in addition to high affinity and high selectivity, the reversibility of the binding process to the peptide material is of high importance. Hence, a key requirement for the envisioned biomimetic lanthanide binding polymer–peptide conjugates has to be the reversibility of Ln binding to such ligands. For the pDMA-EF4G conjugate the release of Eu3+ was easily achieved by changing the pH of the solution. This feature is very promising for the future development of bio-based resins for lanthanide extraction.
Future research will focus on varying amino acid sequence of the peptide, the polymer type and length to optimize the binding with respect to strength but also for selectivity towards different lanthanides ions. Especially stimuli-responsive polymers seem to be very attractive to tailor the properties for ion binding and release. Through the use of thermoresponsive polymers such as poly(N-isopropylacrylamide), a thermal release of ions could be envisioned.
With the successful synthesis of the pDMA-EF4G conjugate a blueprint for a novel class of peptide-based filter materials for lanthanide separation is on hand. In our future work we will extend this approach towards a toolbox with different polymers available for conjugation. Work is in progress to tackle the other equally important tasks which are the investigation of the selectivity towards different lanthanides and the competition with other metal ions (e.g., alkali metals, d-metals) most likely present in real-world samples.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by The Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung (BMBF)), Germany. [grant number 031B1122]. D. M. thank Dr Sascha Eidner and Sophie Dettmann, MSc for their guidance on PARAFAC analysis.Notes and references
- T. McNulty, N. Hazen and S. Park, Processing the ores of rare-earth elements, MRS Bull., 2022, 47(March), 1–9, DOI:10.1557/s43577-022-00288-4.
- K. Binnemans, P. T. Jones, T. Müller and L. Yurramendi, Rare Earths and the Balance Problem: How to Deal with Changing Markets?, J. Sustain. Metall., 2018, 4, 126–146 CrossRef.
- J. A. Mattocks and J. A. Cotruvo, Biological, biomolecular, and bio-inspired strategies for detection, extraction, and separations of lanthanides and actinides, Chem. Soc. Rev., 2020, 49, 8315–8334 RSC.
- J. C. G. Bunzli, N. Andre, M. Elhabiri, G. Muller and C. Piguet, Trivalent lanthanide ions: versatile coordination centers with unique spectroscopic and magnetic properties, J. Alloys Compd., 2000, 303–304, 66–74 CrossRef CAS . Available from: https://www.elsevier.com/locate/jallcom.
- R. Pell, L. Tijsseling, K. Goodenough, F. Wall, Q. Dehaine and A. Grant, et al., Towards sustainable extraction of technology materials through integrated approaches, Nat. Rev. Earth Environ., 2021, 2, 665–679 CrossRef.
- S. Tang, C. Zheng, M. Chen, W. Du and X. Xu, Geobiochemistry characteristics of rare earth elements in soil and ground water: a case study in Baotou, China, Sci. Rep., 2020, 10(1), 11740 CrossRef CAS PubMed.
- M. Peiravi, F. Dehghani, L. Ackah, A. Baharlouei, J. Godbold and J. Liu, et al., A Review of Rare-Earth Elements Extraction with Emphasis on Non-conventional Sources: Coal and Coal Byproducts, Iron Ore Tailings, Apatite, and Phosphate Byproducts, Min. Metall. Explor., 2021, 38, 1–26, DOI:10.1007/s42461-020-00307-5.
- S. Cotton, Two Centuries of the Rare Earths, 2020, available from, https://upload.wikimedia.org/wikipedia/commons/0/09/Elemental_abundances.svg Search PubMed.
- A. Lopata, B. Jójárt, É. V. Surányi, E. Takács, L. Bezúr and I. Leveles, et al., Beyond chelation: EDTA tightly binds taq DNA polymerase, MutT and dUTPase and directly inhibits dNTPase activity, Biomolecules, 2019, 9(10), 621 CrossRef CAS PubMed.
- L. J. Daumann, Essenziell und weitverbreitet: Lanthanoid-Metalloproteine, Angew. Chem., 2019, 131(37), 12926–12933 CrossRef.
- J. A. Cotruvo, The Chemistry of Lanthanides in Biology: Recent Discoveries, Emerging Principles, and Technological Applications, ACS Cent. Sci., 2019, 5(9), 1496–1506 CrossRef CAS PubMed.
- P. Mulqueen, J. M. Tingey and W. D. W. Horrocks, Characterization of Lanthanide(III) Ion Binding to Calmodulin Using Luminescence Spectroscopy, Biochemistry, 1985, 24(23), 6639–6645 CrossRef CAS PubMed.
- J. A. Cotruvo, E. R. Featherston, J. A. Mattocks, J. V. Ho and T. N. Laremore, Lanmodulin: A Highly Selective Lanthanide-Binding Protein from a Lanthanide-Utilizing Bacterium, J. Am. Chem. Soc., 2018, 140(44), 15056–15061 CrossRef CAS PubMed.
- M. P. Bemquerer, C. Bloch, H. F. Brito, E. E. S. Teotonio, M. T. M. Miranda and S. M. Gutenthaler, et al., Steady-state luminescence investigation of the binding of Eu(III) and Tb(III) ions with synthetic peptides derived from plant thionins, J. Inorg. Biochem., 2002, 91(2), 363–370 CrossRef CAS PubMed . Available from: https://chemrxiv.org/engage/chemrxiv/article-details/61c43bd09efae74c7b240f0e.
- B. Drobot, M. Schmidt, Y. Mochizuki, T. Abe, K. Okuwaki and F. Brulfert, et al., Cm3+/Eu3+ induced structural, mechanistic and functional implications for calmodulin, Phys. Chem. Chem. Phys., 2019, 21(38), 21213–21222 RSC.
- M. C. Kilhoffer, J. G. Demaille and D. Gerard, Terbium as luminescent probe of calmodulin calcium-binding sites domains I and II contain the high-affinity sites, FEBS Lett., 1980, 116, 269–272 CrossRef CAS PubMed.
- R. W. Wallace, E. A. Tallant, M. E. Dockter and W. Y. Cheung, Calcium binding domains of calmodulin. Sequence of fill as determined with terbium luminescence, J. Biol. Chem., 1982, 257(4), 1845–1854 CrossRef CAS PubMed.
- J. A. Bruno, W. D. W. Horrocks and R. J. Zauhar, Europium(III) Luminescence and Tyrosine to Terbium(III) Energy-Transfer Studies of Invertebrate (Octopus) Calmodulin, Biochemistry, 1992, 31(31), 7016–7026 CrossRef CAS PubMed . Available from: https://pubs.acs.org/sharingguidelines.
- G. Borin, P. Ruzza, M. Rossi, A. Calderan, F. Marchiori and E. Peggion, Conformation and ion binding properties of peptides related to calcium binding domain III of bovine brain calmodulin, Biopolymers, 1989, 28(1), 353–369 CrossRef CAS PubMed.
- P. Mandal, J. Kretzschmar and B. Drobot, Supporting Informations Not just a background: pH buffers do interact with lanthanide ions - a Europium(III) case study, J. Biol. Inorg. Chem., 2022, 27, 249–260 CrossRef CAS PubMed.
- G. B. Fields, Introduction to Peptide Synthesis, Curr. Protoc. Protein Sci., 2001, 26(1), 18.1 Search PubMed.
- G. Plancque, V. Moulin, P. Toulhoat and C. Moulin, Europium speciation by time-resolved laser-induced fluorescence, Anal. Chim. Acta, 2003, 478(1), 11–22 CrossRef CAS.
- W. R. Forsyth, J. M. Antosiewicz and A. D. Robertson, Empirical relationships between protein structure and carboxyl pKa values in proteins, Proteins: Struct., Funct., Genet., 2002, 48(2), 388–403 CrossRef CAS PubMed.
- C. Bray, R. Peltier, H. Kim, A. Mastrangelo and S. Perrier, Anionic multiblock core cross-linked star copolymers: Via RAFT polymerization, Polym. Chem., 2017, 8(36), 5513–5524 RSC.
- S. Dettmann, N. M. Huittinen, N. Jahn, J. Kretzschmar, M. U. Kumke and T. Kutyma, et al., Influence of gluconate on the retention of Eu(III), Am(III), Th(IV), Pu(IV), and U(VI) by C-S-H (C/S = 0.8), Front. Nucl. Eng., 2023, 2, 1124856 CrossRef.
- H. G. Börner, Bioconjugates of polymers and sequence-defined peptides by reversible addition fragmentation chain transfer radical polymerization, in ACS Symposium Series, 2009 Search PubMed.
- L. Wu, U. Glebe and A. Böker, Surface-initiated controlled radical polymerizations from silica nanoparticles, gold nanocrystals, and bionanoparticles, Polym. Chem., 2015, 6, 5143–5184 RSC.
- B. Marmodée, K. Jahn, F. Ariese, C. Gooijer and M. U. Kumke, Direct spectroscopic evidence of 8- and 9-fold coordinated europium(III) species in H2O and D2O, J. Phys. Chem. A, 2010, 114(50), 13050–13054 CrossRef PubMed.
- K. Binnemans, Interpretation of europium(III) spectra, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- T. Haubitz, B. Drobot, S. Tsushima, R. Steudtner, T. Stumpf and M. U. Kumke, Quenching Mechanism of Uranyl(VI) by Chloride and Bromide in Aqueous and Non-Aqueous Solutions, J. Phys. Chem. A, 2021, 125, 4380–4389 CrossRef CAS PubMed.
- S. Chemura, T. Haubitz, P. A. Primus, M. Underberg, T. Hülser and M. U. Kumke, Europium-Doped Ceria-Gadolinium Mixed Oxides: PARAFAC Analysis and High-Resolution Emission Spectroscopy under Cryogenic Conditions for Structural Analysis, J. Phys. Chem. A, 2020, 124(24), 4972–4983 CrossRef CAS PubMed.
- T. Haubitz, L. John, D. Freyse, P. Wessig and M. U. Kumke, Investigating the Sulfur “Twist” on the Photophysics of DBD Dyes, J. Phys. Chem. A, 2020, 124(22), 4345–4353 CrossRef CAS PubMed.
- T. Haubitz, L. John, P. Wessig and M. U. Kumke, Photophysics of Acyl- and Ester-DBD Dyes: Quadrupole-Induced Solvent Relaxation Investigated by Transient Absorption Spectroscopy, J. Phys. Chem. A, 2019, 123(22), 4717–4726 CrossRef CAS PubMed.
- B. Drobot, R. Steudtner, J. Raff, G. Geipel, V. Brendler and S. Tsushima, Combining luminescence spectroscopy, parallel factor analysis and quantum chemistry to reveal metal speciation - A case study of uranyl(VI) hydrolysis, Chem. Sci., 2015, 6(2), 964–972 RSC.
- T. Saito, H. Sao, K. Ishida, N. Aoyagi, T. Kimura and S. Nagasaki, et al., Application of parallel factor analysis for time-resolved laser fluorescence spectroscopy: implication for metal speciation study, Environ. Sci. Technol., 2010, 44(13), 5055–5060 CrossRef CAS PubMed.
- S. Friedrich, C. Sieber, B. Drobot, S. Tsushima, A. Barkleit and K. Schmeide, et al., Eu(III) and Cm(III) Complexation by the Aminocarboxylates NTA, EDTA, and EGTA Studied with NMR, TRLFS, and ITC—An Improved Approach to More Robust Thermodynamics, Molecules, 2023, 28(12), 4881 CrossRef CAS PubMed.
- H. J. Dyson and P. E. Wright, Defining solution conformations of small linear peptides, Annu. Rev. Biophys. Biophys. Chem., 1991, 20, 519–538 CrossRef CAS PubMed.
- S. Jain, J. T. Welch, W. D. Horrocks and S. J. Franklin, Europium Luminescence of EF-Hand Helix-Turn-Helix Chimeras: Impact of pH and DNA-Binding on Europium Coordination, Inorg. Chem., 2003, 42(24), 8098–8104 CrossRef CAS PubMed.
- R. H. Kretsinger, V. N. Uversky and E. A. Permyakov, Encyclopedia of Metalloproteins, Springer, New York, 2013 Search PubMed.
- G. J. P. Deblonde, J. A. Mattocks, D. M. Park, D. W. Reed, J. A. Cotruvo and Y. Jiao, Selective and Efficient Biomacromolecular Extraction of Rare-Earth Elements using Lanmodulin, Inorg. Chem., 2020, 59(17), 11855–11867 CrossRef CAS PubMed.
- Z. Hussain, S. Kim, J. Cho, G. Sim, Y. Park and I. Kwon, Repeated Recovery of Rare Earth Elements Using a Highly Selective and Thermo-Responsive Genetically Encoded Polypeptide, Adv. Funct. Mater., 2022, 32(13), 2109158 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01505c |
| This journal is © The Royal Society of Chemistry 2024 |