
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSystematic surface bowing in 2D III-nitride monolayers†
Imdad Hussainab,
Adnan Ali Khan ac,
Iftikhar Ahmad*ab,
Rashid Ahmadac and
Saif Ullahab
ac,
Iftikhar Ahmad*ab,
Rashid Ahmadac and
Saif Ullahab
aDepartment of Physics, University of Malakand, Chakdara, Pakistan. E-mail: ahma5532@gmail.com
bCenter for Computational Materials Science, University of Malakand, Chakdara, Pakistan
cDepartment of Chemistry, University of Malakand, Chakdara, Pakistan
First published on 18th March 2024
Abstract
In this article we report novel composite materials of bucky ball (C60 fullerene) and III-nitrides (BN, AlN, GaN, InN). The experimental feasibility of the novel composite materials is confirmed through negative binding energies and molecular dynamics simulations performed at 500 K. The structural properties of the novel composites are explored through density functional theory. An unusual phenomenon of surface bowing is observed in the 2D structure of the III-nitride monolayers due to the C60 sticking. This surface bowing systematically increases as one proceeds from BN → AlN → GaN → InN. The electron density difference and Hirshfeld charge density analysis show smaller charge transfer during the complexation, which is probably due to weak van der Waal's forces. The presence of van der Waal's forces is also confirmed by the Atom in Molecule analysis, Reduced Density Gradient Technique and Non-covalent Interaction analysis. This work provides a foundation for further theoretical and experimental studies of the novel phenomenon of systematic bowing in the 2D structure of III-nitride monolayers.
1 Introduction
The physical properties of materials are generally not dependent on size. However, if the dimension of a material is reduced to ultra-fine size within the nano range, then quantum confinement effects are induced in it due to reduced dimensionality. This reduction in size is possible by either reducing the size of a material in one direction (2D materials), or two directions (nanotubes etc.) or three directions (nanodots). Quantum confinement effects significantly modify the electronic properties of materials from their bulk counterparts. The critical size for these effects varies from material to material. These unusual quantum effects in materials due to their low dimensions have introduced a new class of emerging nanomaterials with exotic properties. These nanomaterials have attracted enormous attention in science and technology for their extraordinary physical properties and potential applications in high-tech devices.This reduction in size is possible by either reducing the size of a material in one direction (2D materials), or two directions (nanotubes etc.) or three directions (nanodots). The reduced dimensions of materials, with enhanced properties caused by quantum confinement effects, have introduced a new class of nanomaterials. Nanomaterials have attracted much attention in both science and technology for their potential high-tech applications in modern technologies.
The wide range of direct band gap nature of III nitrides (BN, AlN, GaN and InN) make them extensively applicable in the optoelectronic industry. The band gap of III nitrides can also be engineered for a specific device application using various techniques like doping, pressure, strain etc. Rare-earth doped III nitrides are commonly used as phosphor materials in optical displays.1,2
Since the discovery of graphene in 2005,3 two dimensional (2D) monolayers have attracted much attention for their unusual properties, due to the quantum confinement effect caused by the reduced dimensionality. The search for suitable non-toxic, environment-friendly, cheaper and long-lasting novel materials for various technological applications is the growing need of the day. Two dimensional (2D) monolayers, 2D heterostructures4–6 and 2D Janus materials are recently at the forefront of this search for their potential applications.5–10 Among 2D materials, 2D III-nitrides (BN, AlN, GaN, InN) are wide band gap, sp2 hybridized semiconductors. The 2D III-nitrides were synthesized by different experimental techniques like Chemical-Vapor Deposition (CVD), Physical-Vapor Transport (PVT), Metal–Organic Vapor Phase Epitaxy (MOVPE), Molecular-Beam Epitaxy (MBE) and Metal–Organic Chemical Vapor Deposition (MOCVD) methods.11–13 The 2D III-nitrides semiconductors have numerous applications like field emitter, biosensor, ideal layer for fabricating flexible devices, as a substrate in graphene electronic devices, sensors, anode material for light emitting diode, field effect transistors, piezoelectric materials, solar cells and high-speed electronics.
Fullerene (C60) is a carbon allotrope having closed cage like structure, discovered in 1985 by Kroto et al. has opened a new era of nanoscience.14 It consists of sp2 hybridized carbon atoms and is the only soluble material among all elemental carbon allotropes.15 The C60 is also called Buckminster fullerene is the mostly studied fullerene due to its unique 0D cage like structure, charge acceptor character (holding up to six electrons), high temperature, nanometer dimension and large polarizability.16 The properties of fullerene can be tuned by interaction with metals and semiconductors. The unique cage like closed 0D structure of fullerenes enables it to be used as important building blocks for the construction of supramolecular assemblies and micro/nano hybrid materials, having numerous technological applications like fast phototransistor, hydrogen storage, photocatalyst, thermoelectric devices and UV-visible photodetectors.17–21 Fullerene can also be regarded as an electron deficient-polyalkene, so, it is chemically reactive.22 Hence, to extend the functionalities of Fullerene, derivation is feasible. Many efforts have been made to adsorb fullerene on graphene and its derivative for to modify its properties.23–25 The adsorption of C20, C34, C42 and C60 fullerenes on graphene was studied and a sizeable band gap opening was observed.26 The adsorption of C20 and C60 fullerenes on the nitrogenated-holy graphene (NHG) sheet for investigating band gap engineering was performed by DFT method. A decrease in the band gap energy of NHG sheet by fullerene adsorption was observed. Furthermore, the calculations revealed that C20 adsorption on NHG sheet to p-type semiconductor and C60 fullerene adsorption changes NHG sheet to n-type semiconductor.
In this paper we report novel composite materials of C60 fullerene and 2D-III nitrides (h-BN, h-AlN, h-GaN, h-InN monolayer). The experimental feasibility of the novel materials is confirmed by using the binding energies and molecular dynamics simulations at 500 K. Density functional theory calculations are used to explore the structural properties of these compounds. The fascinating, aspect of these studies was the bowing effect in the III nitride monolayers. The bowing effect was increased as we proceeded from BN to InN. The Atom in Molecule, Electron Density Difference, Reduced Density Gradient and Non-covalent Interaction analysis are performed to understand the bonding nature (forces) among the complexes and stability of the complexes. This study is a baseline for further experimental and theoretical studies on the surface bowing effect and physical properties for the potential applications of the novel composite materials.
2 Computational method
We used density functional theory (DFT) to simulate our materials. We cleave 5 × 5 × 1 hexagonal supercell of X–N (where X = BN, AlN, GaN, InN) monolayer of 50 atoms for our DFT simulations with vacuum gap of 20 Å to avoid the interaction of different layers. The geometry optimizations were performed by Dmol3 code27 in material studio software using GGA-PBE functional28 with double numerical plus polarization (DNP) basis set.29 The k-point grid of 7 × 7 was generated for 2D monolayers and 4 × 4 for composites from the Monkhorst pack-scheme. The double numerical plus polarization (DNP) basis was used for the geometries optimization and electronic properties calculations, where the orbital cut-off and energy cut-off were set at 4.6 Å and 450 eV to improve computational efficiency. For van der Waal's corrections in intermolecular interactions the DFT-D2 Grimme method30 was chosen. The 2D structures were fully relaxed until the force on each atom was less than 0.002 Ha Å−1. For SCF density convergence 1.0 × 10−5 hartree was set for energy. All composites structures were fully relaxed until the force on each atom was less than 0.002 Ha Å−1 to improve computational efficiency. The calculated lattice constants of these 2D optimized structures were 2.55 Å for BN, 3.13 Å for AlN, 3.19 Å for GaN and 3.55 Å for InN and the calculated bond lengths for these materials were 1.48 Å, 1.81 Å, 1.84 Å and 2.05 Å, which match well with the experimental lattice constants given in Table 1, so our calculations are logical. We used ab initio molecular dynamic (AIMD) simulations with GGA-PBE functional by DMol3 code to examine the thermodynamic stability of the studied systems. The AIMD simulations were performed in the NVT ensemble with Nose–Hoover thermostat with a time step of 1 fs at 500 K temperature. The atom in molecule (AIM) analysis was used to study the nature of bonding in these 2D materials using Multiwfn code.31 The binding energy (Eb) was calculated by the following formula| Eb = (EC60–XN) − (EC60 + EX–N) | (1) |
3 Results and discussion
3.1 Structural and energetic analysis of C60@XN composite system
The optimized geometries of 2D-III nitrides and C60-fullerene nanostructure is represented in Fig. S1 and S2.† The overall geometrical parameters like bond distances, lattice parameters are equivalent (as shown in Table 1) to already reported results.32–36 We investigated the composites of C60-fullerene and 2D-III nitrides.The optimized geometries of the composites (C60@h-BN, C60@h-AlN, C60@h-GaN and C60@h-InN) material is illustrated in Fig. 1, while the optimized structural parameters like lattice parameters, bond lengths are collected in Table 2. The composite materials are designed as the C60 fullerene is placed over the top of the III-nitrides monolayers and fully geometry relaxation is performed. Initially the fullerene molecule is kept onto the top of monolayers by keeping it at about 2.00 Å distance. After geometry relaxation it is found that the fullerene system is physiosorbed over the surface of nitrides monolayers by interacted through van der Waal's forces having an average bond distance up to 3.00 Å between the C atoms of C60 fullerene and X–N (X = B, Al, Ga, In) atoms of monolayers. The bond lengths X–N increases from 0.03 to 0.07 Å shown by Δ in Table 2 due to the fullerene adsorptions on these monolayers. Moreover, the geometry relaxation shows some interesting results about the planar structure of III-nitrides. As we moved from C60@h-BN composite to C60@h-BN, C60@GaN and AlN@C60 composite the structure distortion enhanced. The planar geometry of monolayers (except h-BN monolayer) changes from planar to bowl shape. This variation is more pronounced in the order C60@h-InN > C60@h-GaN > C60@h-AlN > C60@h-BN (Fig. 1). This result shows that the C60@h-BN composite is more stable compared to other studied composites. This distortion may be due to the enhanced repulsive forces between the pi-conjugated electrons of fullerene system and lone-pair electrons of N atoms of III-nitrides monolayer. This can be explained as there is opposite forces operational between these system like the positive B atoms of III-nitrides and pi-electron of fullerene atoms and the electropositive C atoms of fullerene and lone-pair of N atoms of III-nitrides. Thus, there is a competition between the attractive and repulsive forces between the two materials. Finally, the repulsive forces dominant over the attractive forces and both systems repels each other and they are stabilized at certain distance and formed stable composite material. The sequence of distortion is in the order C60@h-InN > C60@h-GaN > C60@h-AlN > C60@h-BN because lattice parameter i.e. X–N (X = B, Al, Ga, In) distance is larger as we moved down the group. This resulted dominant vibrations of X–N bond moving from B–N to In–N system. Thus, the rigidity in h-BN system is more compared to other III-nitrides which has the ability to these vibrations without any structural distortion and evince the most stable composite structure.
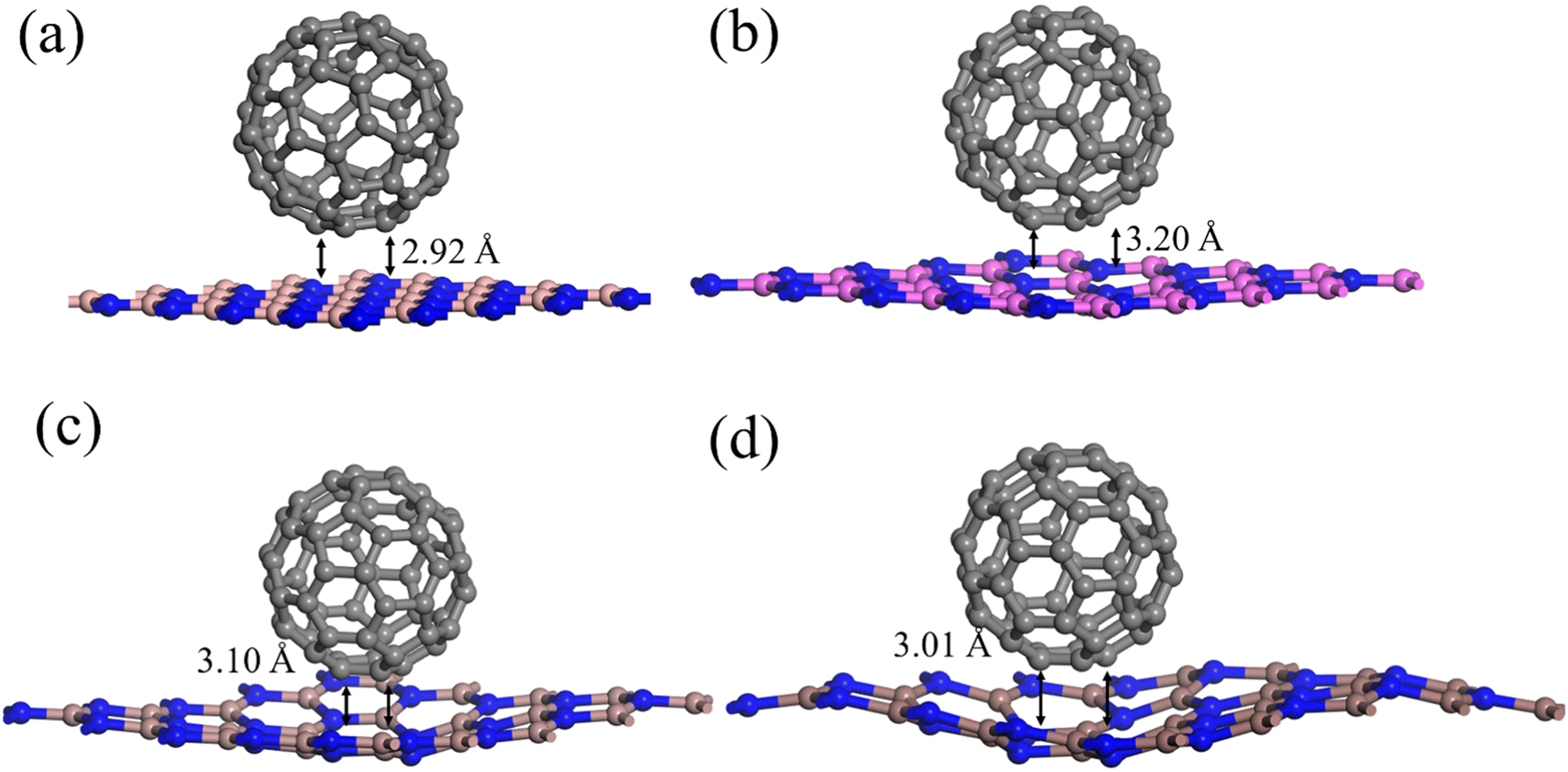 | ||
| Fig. 1 The optimized geometry of C60@h-BN (a), C60@h-AlN (b), C60@h-GaN (c) and C60@h-InN (d) composite. | ||
| Composite | X–C | X–N | C–N | a | Δ | Eb | V |
|---|---|---|---|---|---|---|---|
| C60@BN | 2.9 | 1.48 | 3.07 | 12.78 | 0.03 | −0.94 | 2903.19 |
| C60@AlN | 3.17 | 1.82 | 3.18 | 15.64 | 0.02 | −1.8 | 4241.9 |
| C60@GaN | 3.24 | 1.87 | 3.29 | 15.94 | 0.07 | −2.17 | 4445.8 |
| C60@InN | 3.0 | 2.11 | 2.92 | 17.75 | 0.06 | −2.20 | 5713.49 |
The binding energy for all these composites (calculated by eqn (1)) are negative (−0.94 to −2.20 eV), confirm the thermodynamic stability and feasibility of experimental synthesis. The binding energy increases from −0.45 eV which represent the chemisorption state but actually the composites are in physiosorbed state. This increasement in the binding energies is due to the dominant distortion in these systems. This order of binding energy is C60@h-InN (−2.20 eV) > C60@h-GaN (−2.17 eV) > C60@h-AlN (−1.80 eV) > C60@h-BN (−0.94 eV) as shown in Table 2.
The molecular dynamic (MD) simulations of these composites were performed to check the thermal stability of a material at 500 K for 2.0 ps equilibrium at a time period of 1 fs. The potential energy plots and structures variation after MD simulation are displayed in Fig. 2. Due to out of planes vibrations modes at 500 K temperature, there are dangling bonds in these structures and a small variations in potential energies specially for C60–GaN and C60–InN which are consistent with the reported result.37 It is clear from the Fig. 2 the potential energy varies nearby a specific magnitude and no breakdown of any bond in any material is observed so all these materials are thermodynamically stable at 500 K, which are consistent with the energetic analysis.
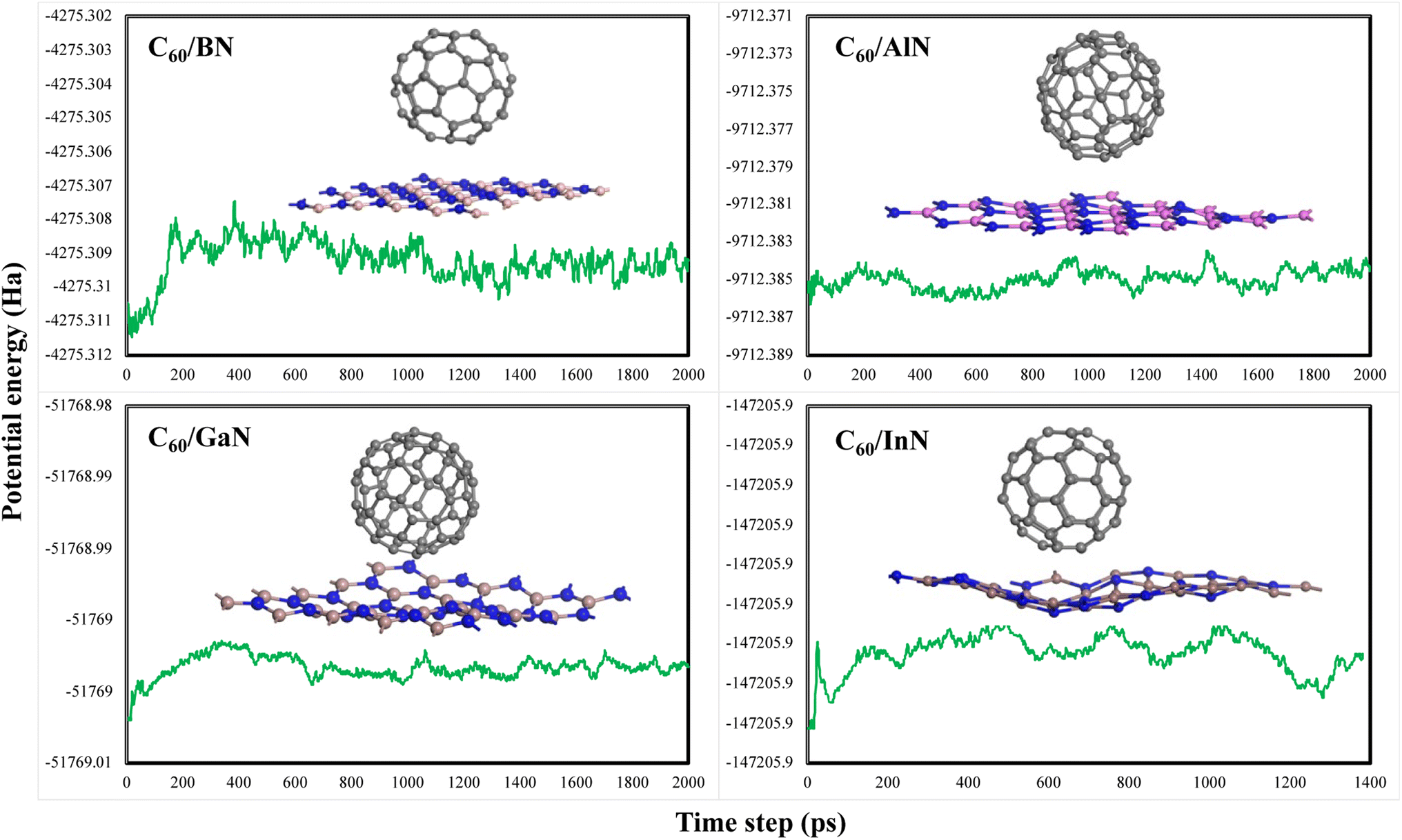 | ||
| Fig. 2 Geometries of C60–XN (X = B, Al, Ga, In) composites after MD simulations and corresponding potential energy fluctuation plots. | ||
The charge transfer was determined by Hirshfeld charge transfer analysis. Here it is clear from Table 3 in case of C60–BN the charge transfer take place from fullerene to BN, same is the case of C60–AlN. In these composites the fullerene is a charge donor molecule and behaves as n-type dopant. While in case of C60–GaN and C60–InN charge is transferred from monolayers sheets to fullerene, so fullerene is charge acceptor molecule and behave as p-type dopants. The charge transfer between the 2D III-nitrides monolayers and fullerene molecule is small. The large bond distances and small charge transfer identify the weak interactions between 2D III-nitrides sheets and fullerene molecule. Hence fullerene is weakly physiosorbed on these monolayers.
| Materials | ρ | ∇2ρ | H | QC | QX | QN | Gravimetric density W (%) |
|---|---|---|---|---|---|---|---|
| a QC, QX, QN represent charge on carbon atom, X atom and N atom. | |||||||
| C60–BN | 0.001 | 0.029 | 0.0011 | 0.0088 | 0.1901 | −0.2000 | 54 |
| C60–AlN | 0.01 | 0.027 | 0.008 | 0.01 | 0.4243 | −0.4346 | 41 |
| C60–GaN | 0.012 | 0.026 | 0.005 | −0.001 | 0.3950 | −0.3940 | 26 |
| C60–lnN | 0.012 | 0.025 | 0.004 | −0.016 | 0.4500 | −0.4364 | 18 |
3.2 Electronic properties analysis of C60@XN composite system
The interaction among fullerene and 2D III-nitrides monolayers is also confirmed by electron density difference (EDD) plots shown in Fig. 3. The figure clearly shows that there is no over lapping exists between the red lobes (high electron density) of fullerene and III-nitrides while there is small overlapping occurred between the yellow regions (less electron dense region) of fullerene and III-nitrides. This gives evidence of small charge shifting between the two-system due to weak interactions. The results are in agreement with the Hirshfeld charge shift analysis.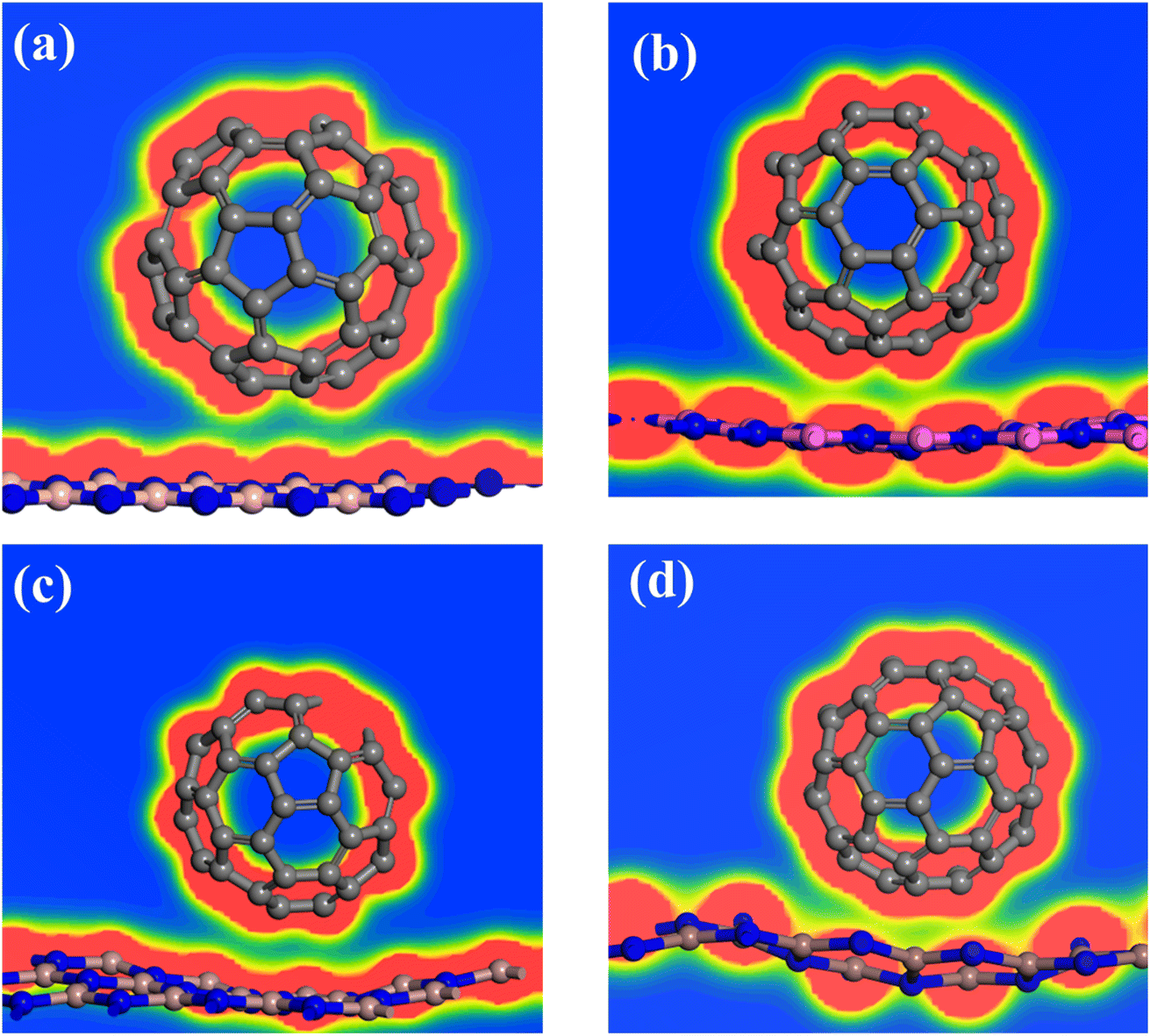 | ||
| Fig. 3 Electron density difference plots of (a) BN-C60, (b) AlN-C60, (c) GaN-C60, and (d) InN-C60 composites. Red lobes represent rich electron density regions and yellow represents poor electron density region. | ||
We used Bader quantum atom in molecule theory of atoms in molecules38 to understand the bond strength and nature formed between the fullerene and III-nitrides in composite. For studying the nature of bonding in molecules we need to exploit charge density or electron density (ρ). The AIM analysis of the fullerene binding onto 2D III-nitrides is shown in Fig. 4. In characteristics of bonding, the Laplacian (∇2ρ) plays an important role. In case if ∇2ρ < 0 clarify charge concentrations towards interaction line. This results in lowering potential energy creating attractive forces. On the other hand, ∇2ρ > 0 indicates the repulsive forces act on the nuclei. The electron energy density (H) is another parameter also helpful in identifying the nature of bonding. According to Bader and Essen the interactions are covalent if values of ∇2ρ and H are negative and electron density at BCP > 10−1 a.u while values of electron density less than 10−1 a.u and positive ∇2ρ and H attribute to Vander Waal interactions. The calculated values of electron density (ρ), Laplacian (∇2ρ) and total electron energy density (H) are shown in Table 3. The calculated values of electron density (ρ) for the C60–XN (X = B, Al, Ga and In) are 0.001 a.u, 0.01 a.u, 0.012 a.u and 0.012 a.u are less than 10−1 a.u and the values of Laplacian and total electron energy density for these materials are all positive. The calculated values of electron density for these composites are less than 10−1 a.u and positive ∇2ρ and H attribute to van der Waal's interactions. The calculated bond lengths X–N of these materials shown in Table 1 are in the range of 1.00 to 2.00 Å. The bond distance i.e. X–C and C–N are nearly in the range of 3.00 Å. It is also clear from our calculations of bond length and AIM analysis that the bond strength decreases as we move from C60–BN to C60–InN. The same situations appear in AIMD calculations performed at 500 K, where the wiggles in the structure increase from h-BN to h-InN composites.
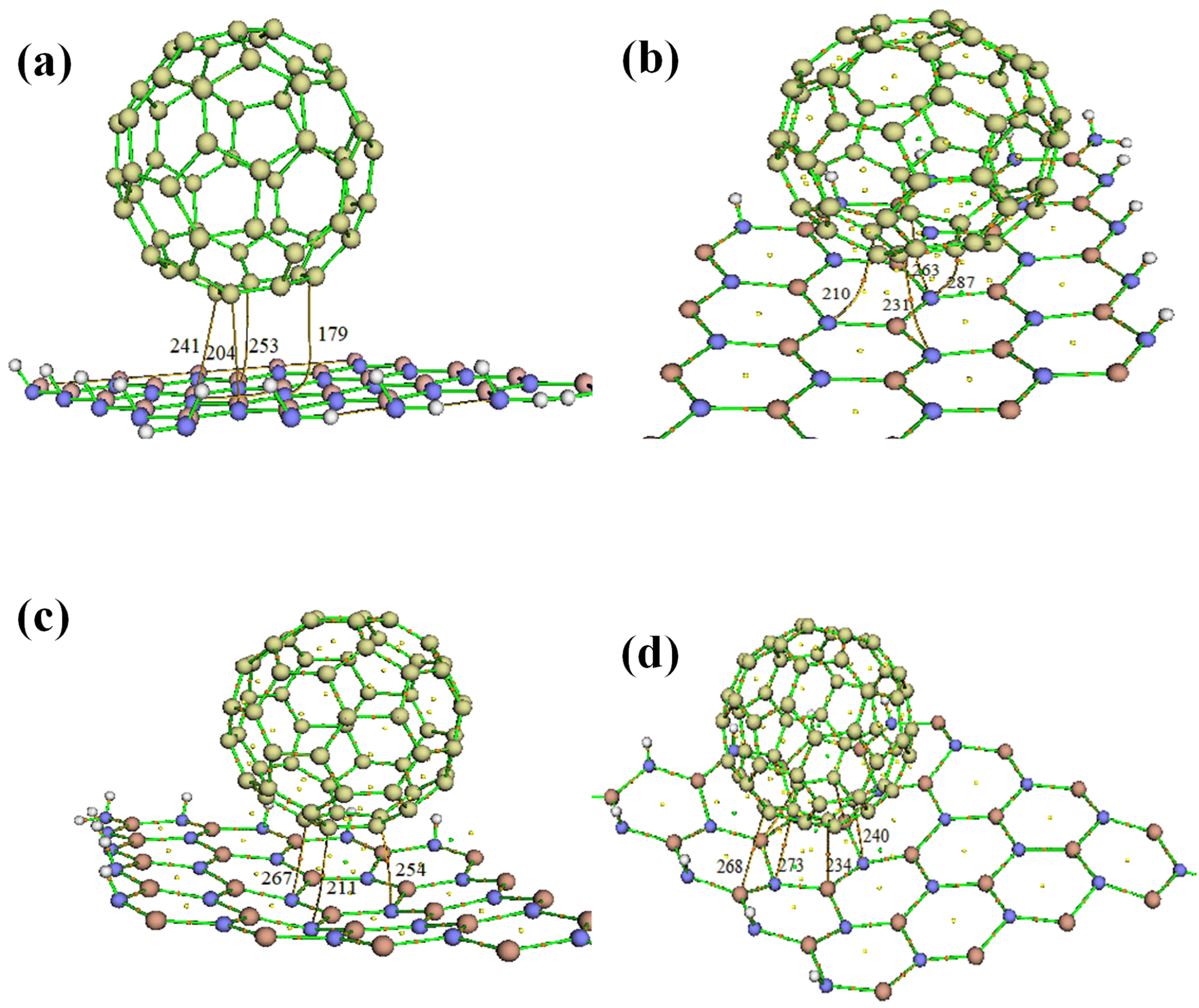 | ||
| Fig. 4 Atom in molecule analysis plots of (a) BN-C60, (b) AlN-C60, (c) GaN-C60, and (d) InN-C60 composites. The number represents the bond critical points. | ||
The maximum storage capacity of fullerene over the 2D III-nitrides is estimated from gravimetric density39 which was calculated from the following equation
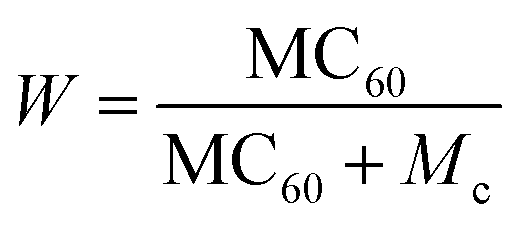 | (2) |
According to Table 3 the storage capacity of fullerenes are 54, 41, 26 and 18 for BN, AlN, GaN and InN. Next, the reduced density gradient (RDG) and non-covalent interaction (NCI) analysis are investigated to further understand the nature of interactions and structural distortion in the studied composite. The 2D-RDG and 3D-NCI plots are illustrated in Fig. 5, the red, green and dark blue spikes in the RDG plot represent the repulsive, van der Waal's and strong electrostatic (covalent or ionic) forces in the complex system. The RDG clearly shows that there are green spikes appear at x-axis at the range from −0.005 au to −0.02 au, confirm the presence of van der Waal's forces between the C60 fullerene and III-nitrides materials. This is justified by NCI analysis, the appearance of 3D green spot between the C60 fullerene and III-nitrides evidenced the presence of van der Waal's forces. While the appearance of red spots over the 3D green spot in the composite materials indicates the existence of repulsive forces. The red-spots over green spot is more dominant by moving from C60@h-BN composite to C60@h-InN. While this red spot teardown in case of C60@h-GaN and C60@h-InN composite which justify the more prominent repulsive forces between these systems, which caused larger distortion and instability. These results are in good agreement with the geometrical and AIM analysis.
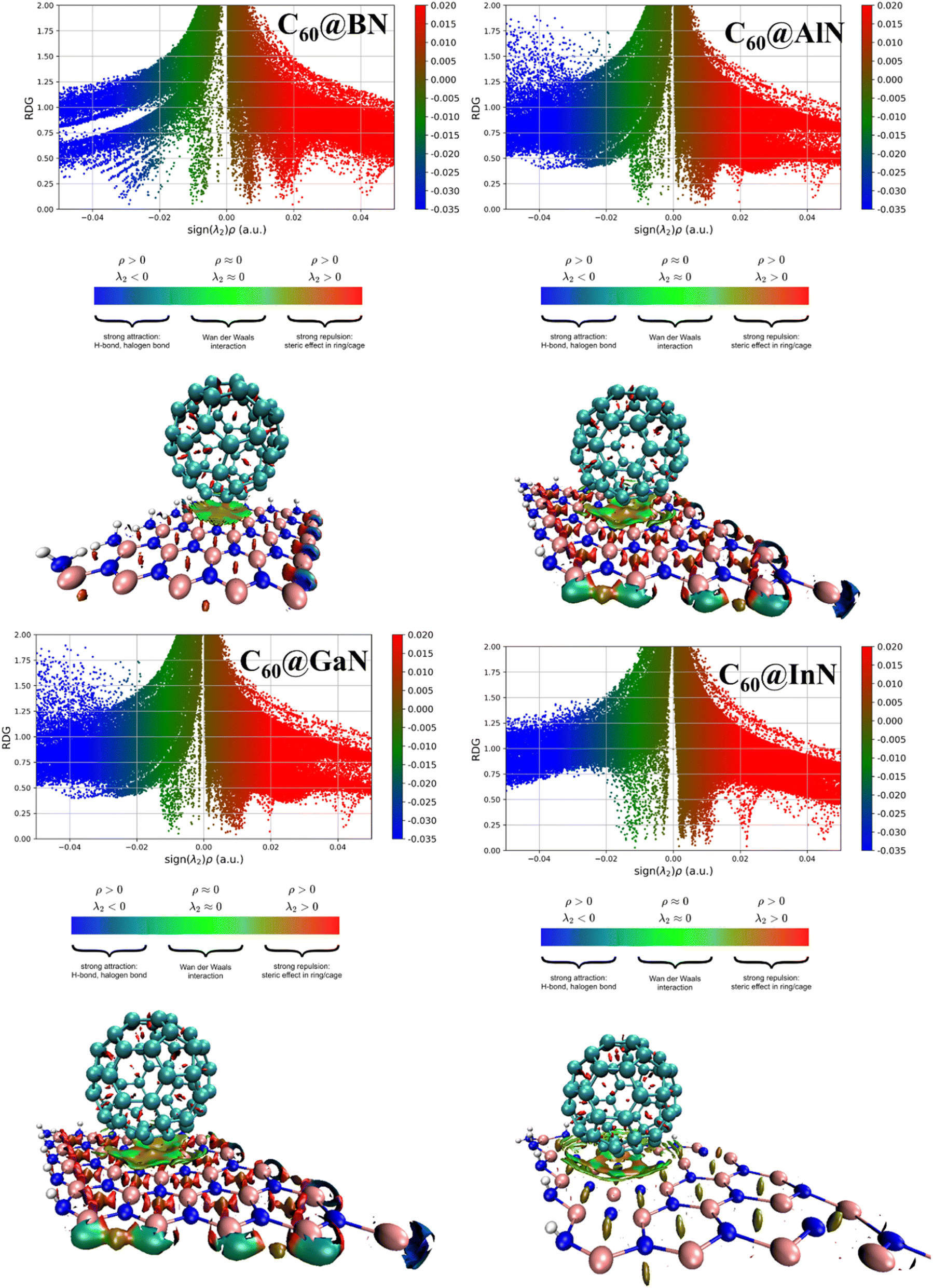 | ||
| Fig. 5 Reduced density gradient and non-covalent interaction plots for the studied composite materials. | ||
4 Conclusions
In this article we report novel composite materials of C60 fullerene and III Nitrides (BN, AlN, GaN, InN) using MD simulations and DFT calculations. The negative binding energies of the compounds and MD simulations at 500 K confirm that these materials are thermodynamically stable and experimentally feasible. The geometrical studies show that the C60 fullerene is bonded over the top of III nitride monolayers and formed composites, C60@h-BN, C60@h-AlN, C60@h-GaN and C60@h-InN. The striking feature of this study is the unusual systematic bowing induced in the planer monolayers of nitrides by the C60 fullerene except for BN. The induced surface bowing in the monolayers increases as we proceed from BN to InN. The Atom in Molecule, Hirshfeld charge density, Electron Density Difference, Reduced Density Gradient and Non-covalent Interactions analysis reveal that van der Waal's forces are operational in these novel composite materials. This study would provide a baseline for the further investigation of the interesting systematically induced bowing effect in the III nitride monolayers and other related fascinated properties.Conflicts of interest
There are no conflicts to declare.References
- M. Maqbool, I. Ahmad, H. H. Richardson and M. E. Kordesch, Direct Ultraviolet Excitation of an Amorphous AlN:Praseodymium Phosphor by Codoped Gd3+ Cathodoluminescence, Appl. Phys. Lett., 2007, 91, 3–6 CrossRef.
- M. Maqbool, G. Ali, S. O. Cho, I. Ahmad, M. Mehmood, M. E. Kordesch, M. Maqbool, G. Ali, S. O. Cho, I. Ahmad and M. Mehmood, Nanocrystals Formation and Intense Green Emission in Thermally Annealed AlN : Ho Films for Microlaser Cavities and Photonic Applications, J. Appl. Phys., 2010, 108, 043528 CrossRef.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Two-Dimensional Gas of Massless Dirac Fermions in Graphene, Nature, 2005, 438, 197–200 CrossRef CAS PubMed.
- A. Gupta, T. Sakthivel and S. Seal, Progress in Materials Science Recent Development in 2D Materials beyond Graphene, Prog. Mater. Sci., 2015, 73, 44–126 CrossRef CAS.
- H. U. Din, M. Idrees, A. Albar, M. Shafiq, I. Ahmad, C. V. Nguyen and B. Amin, Rashba Spin Splitting and Photocatalytic Properties of GeC-MSSe (M=Mo, W) van Der Waals Heterostructures, Phys. Rev. B, 2019, 100, 1–9 CrossRef.
- G. Rehman, S. A. Khan, B. Amin, I. Ahmad, L. Y. Gan and M. Maqbool, Intriguing Electronic Structures and Optical Properties of Two-Dimensional van Der Waals Heterostructures of Zr2CT2 (T = O, F) with MoSe2 and WSe2, J. Mater. Chem. C, 2018, 6, 2830–2839 RSC.
- W. Zhang, C. Chuu, J. Huang, C. Chen, M. Tsai, Y. Chang, C. Liang, Y. Chen, Y. Chueh, J. He, M. Chou and L. Li, Ultrahigh-Gain Photodetectors Based on Atomically Thin Graphene-MoS2,Heterostructure, Sci. Rep., 2014, 4(1–8), 3826 CrossRef PubMed.
- J. D. Mehew, S. Unal, E. T. Alonso, G. F. Jones, S. F. Ramadhan, M. F. Craciun and S. Russo, Fast and Highly Sensitive Ionic-Polymer-Gated WS 2 – Graphene Photodetectors, Sci. Rep., 2017, 29, 1700222 Search PubMed.
- A. Cho and J. Kwon, Hexagonal Boron Nitride for Surface Passivation of Two- Dimensional van Der Waals Heterojunction Solar Cells, ACS Appl. Mater. Interfaces, 2019, 11, 39765–39771 CrossRef CAS.
- T. Roy, M. Tosun, J. S. Kang, A. B. Sachid, S. B. Desai, M. Hettick, C. C. Hu and A. Javey, Terms of Use Field-E Ff Ect Transistors Built from All Two-Dimensional Material Components, ACS Nano, 2014, 8, 6259–6264 CrossRef CAS PubMed.
- M. Corso, M. Corso, W. Auwa, M. Muntwiler and A. Tamai, Boron Nitride Nanomesh, Science, 2004, 303, 217–220 CrossRef CAS PubMed.
- X. Zhang, Z. Liu and S. Hark, Synthesis and Optical Characterization of Single-Crystalline AlN Nanosheets, Solid State Commun., 2007, 143, 317–320 CrossRef CAS.
- Z. Wang, G. Wang, X. Liu, S. Wang, T. Wang, S. Zhang, J. Yu, G. Zhao and L. Zhang, Two-Dimensional Wide Band-Gap Nitride Semiconductor GaN and AlN Materials: Properties, Fabrication and Applications, J. Mater. Chem. C, 2021, 9, 17201–17232 RSC.
- H. W. Kroto, J. R. Heath, S. C. O’Brien, R. F. Curl and R. E. Smalley, C60: Buckminsterfullerene, Nature, 1985, 318, 162–163 CrossRef CAS.
- A. A. Popov, S. Yang and L. Dunsch, Endohedral Fullerenes, Chem. Rev., 2013, 113, 5989–6113 CrossRef CAS PubMed.
- J. Coro, M. Suárez, L. S. R. Silva, K. I. B. Eguiluz and G. R. Salazar-Banda, Fullerene Applications in Fuel Cells: A Review, Int. J. Hydrogen Energy, 2016, 41, 17944–17959 CrossRef CAS.
- R. Pan, Y. Cai, F. Zhang, S. Wang, L. Chen and X. Feng, High Performance Graphene – C 60 – Bismuth Telluride – C 60 – Graphene Nanometer Thin Film Phototransistor with Adjustable Positive and Negative Responses, Adv. Sci., 2023, 1–8 Search PubMed.
- S. Qin, H. Jiang, Q. Du, Z. Nie, X. Wang, W. Wang, X. Wang, Y. Xu, Y. Shi, R. Zhang and F. Wang, Planar Graphene-C 60 -Graphene Heterostructure for Sensitive UV-Visible Photodetection, Carbon, 2019, 146, 486–490 CrossRef CAS.
- P. Zong, J. Liang, P. Zhang, C. Wan, Y. Wang and K. Koumoto, Graphene-Based Thermoelectrics, ACS Appl. Energy Mater., 2022, 03, 2224–2239 CrossRef.
- A. Kuc, L. Zhechkov, S. Patchkovskii and G. Seifert, Hydrogen Sieving and Storage in Fullerene Intercalated Graphite, Nano Lett., 2007, 7, 1–5 CrossRef CAS.
- M. Q. Yang, N. Zhang and Y. J. Xu, Synthesis of Fullerene-, Carbon Nanotube-, and Graphene-TiO2 Nanocomposite Photocatalysts for Selective Oxidation: A Comparative Study, ACS Appl. Mater. Interfaces, 2013, 5, 1156–1164 CrossRef CAS PubMed.
- A. Hirsch, and M. Brettreich, Fullerenes Chemistry and Reactions, Wiley-VCH, Weinheim, 2005 Search PubMed.
- R. Majidi, Electronic Properties of Graphyne Nanotubes Filled with Small Fullerenes : A Density Functional Theory Study, J. Comput. Electron., 2016, 15, 1263–1268 CrossRef CAS.
- R. Majidi and M. Ghorbani, Density Functional Theory Study of Fullerenes Adsorption on Nitrogenated Holey Graphene Sheet, Fullerenes, Nanotubes Carbon Nanostruct., 2019, 27, 601–606 CrossRef CAS.
- S. S. J. Wonsang Koh, J. H. Lee and S. Geol Lee, Li Adsorption on a Graphene- Fullerene Nanobud System : Density Functional Theory Approach, RSC Adv., 2015, 5, 32819–32825 RSC.
- X. Liu, Y. Wen, Z. Chen, H. Lin, R. Chen, K. Cho, B. Shan, X. Liu, Y. Wen, Z. Chen, H. Lin and R. Chen, Modulation of Dirac Points and Band-Gaps in Graphene via Periodic Fullerene Adsorption Modulation of Dirac Points and Band-Gaps in Graphene via Periodic Fullerene Adsorption, AIP Adv., 2013, 3, 052126 CrossRef.
- B. Delley, From Molecules to Solids with the DMol3 Approach, J. Phys. Chem., 2000, 113, 7756–7764 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. Liu and J. A. Rodriguez, Catalysts for Hydrogen Evolution from the [NiFe] Hydrogenase to the Ni2P (001) Surface: The Importance of Ensemble Effect, J. Am. Chem. Soc., 2005, 127, 14871–14878 CrossRef CAS.
- L. Goerigk and S. Grimme, Benchmark of Density Functional Methods for General Main Group Thermochemistry, Kinetics, and Noncovalent Interactions, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC.
- T. Lu and F. Chen, Multiwfn : A Multifunctional Wavefunction Analyzer, Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- M. Topsakal, E. Aktürk and S. Ciraci, First-Principles Study of Two- and One-Dimensional Honeycomb Structures of Boron Nitride, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 1–11 Search PubMed.
- J. Ben, X. Liu, C. Wang, Y. Zhang, Z. Shi, Y. Jia, S. Zhang, H. Zhang, W. Yu, D. Li and X. Sun, 2D III-Nitride Materials : Properties , Growth , and Applications, Adv. Mater., 2021, 1–32 Search PubMed.
- B. L. Yin, Y. Bando and Y. Zhu, Single-Crystalline AlN Nanotubes with Carbon-Layer Coatings on the Outer and Inner Surfaces via a Multiwalled-Carbon-Nanotube- Template-Induced Route, Adv. Mater., 2005, 213–217 CrossRef.
- I. Vurgaftman, J. R. Meyer, I. Vurgaftman and J. R. Meyer, Band Parameters for Nitrogen-Containing Semiconductors, J. Appl. Phys., 2012, 94, 3675–3696 CrossRef.
- A. Costales, M. A. Blanco and Ä. Martı, Chemical Bonding in Group III Nitrides, J. Am. Chem. Soc., 2002, 124, 4116–4123 CrossRef CAS PubMed.
- Q. Peng, X. Sun, H. Wang, Y. Yang, X. Wen, C. Huang and S. Liu, Theoretical Prediction of a Graphene-like Structure of Indium Nitride : A Promising Excellent Material for Optoelectronics, Appl. Mater. Today, 2017, 7, 169–178 CrossRef.
- R. F. W. Bader, Atoms in Molecules, Acc. Chem. Res., 1985, 9–15 CrossRef CAS.
- R. Akilan, M. Malarkodi, S. Vijayakumar, S. Gopalakrishnan and R. Shankar, Modeling of 2-D Hydrogen-Edge Capped Defected & Boron-Doped Defected Graphene Sheets for the Adsorption of CO2, SO2 towards Energy Harvesting Applications, Appl. Surf. Sci., 2019, 463, 596–609 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01310g |
| This journal is © The Royal Society of Chemistry 2024 |