
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMono and di ortho-C–H acetoxylation of 2-aryloxyquinoline-3-carbaldehydes†
Manickam Bakthadoss *a,
Oluwafemi S. Ainaab,
Tadiparthi Thirupathi Reddya,
Josephat U. Izunobib and
Oluwole B. Familoni*b
*a,
Oluwafemi S. Ainaab,
Tadiparthi Thirupathi Reddya,
Josephat U. Izunobib and
Oluwole B. Familoni*b
aDepartment of Chemistry, Pondicherry University, Pondicherry 605 014, India. E-mail: bhakthadoss@yahoo.com
bDepartment of Chemistry, University of Lagos, Lagos, Nigeria
First published on 23rd April 2024
Abstract
2-Aryloxyquinolines are well known for various biological activities. In this report, we have developed a novel protocol for introducing an acetoxy functional group on the aryl sp2 carbon of 2-aryloxyquinoline-3-carbaldehyde using a palladium catalyst for the first time. Interestingly, this C–H acetoxylation reaction is highly chemo- and site-selective. By modifying the reaction conditions, mono or di ortho-C–H acetoxylation products have been synthesized selectively with good yields and with good functional group tolerance.
Quinoline-based compounds are an important class of N-heterocyclics with a diverse range of biological activities. In the class of quinolines, 2-aryloxyquinolines have gained a lot of interest as they contain important pharmacophores which are frequently useful in many biologically active compounds.1 Because of the significance of these molecules, many research groups have made efforts to develop more efficient methods for their synthesis.2 Moreover, these 2-aryloxyquinoline molecules have antiasthmatic, antiviral, antifungal, antiprotozoal, anthelmintic, cardiotonic, and local anesthetic properties3 (Fig. 1).
 | ||
| Fig. 1 Structures of bioactive aryloxyquinoline derivatives. | ||
Over the last two decades, tremendous progress has been made in transition-metal-catalyzed C–H activation reactions.4 In general, the construction of a C–C bond and C-hetero atom bond by activating the inert C–H bonds of arenes has gained a lot of interest in both academic research and industry. In that direction, the construction of a C–O bond via the C–H activation reaction is challenging due to low reactivity and regioselectivity. Using the directing group (DG) strategy, a number of C–O bond-forming reactions have been reported in recent years.5 Among the various C–O bond-forming reactions, the acetoxylation of aromatic compounds is important. Furthermore, acetoxylation of aromatic compounds is essential since they can be easily converted into phenolic derivatives which are an integral part of many natural products and biologically active molecules. Also, aromatic C–H acetoxylation and hydroxylation reactions are complementary reactions to each other for the incorporation of an acetoxy or hydroxyl group into the product. Towards this end, Sanford and her co-workers developed a regio- and chemoselective method for the acetoxylation of the 2-aryl pyridine C(sp2)–H bond using pyridine as the DG via a palladium catalyst for the first time. Later, various DGs were successfully utilized for selective C(sp2)–H acetoxylation by using various transition metal catalysts6 (Scheme 1).
 | ||
| Scheme 1 Metal-catalyzed C–H acetoxylation. | ||
Among nitrogen-directing-group-assisted C–H activation reactions, pyridine, oxime, and imine groups are very much utilized as chelating groups. However, the nitrogen of the quinoline-moiety-directed C–H activation has not been explored much in the literature and quinolines are most commonly found in various natural products, alkaloids, and pharmacologically active scaffolds. Due to the importance of quinolones, quinolone-containing molecules, such as 2-aryloxyquinoline can be easily transformed into a variety of new scaffolds and this will be helpful in finding new or active pharmaceutical ingredients (APIs) as well as in improving the biological activity of existing drug molecules. Moreover, from the point of view of the C–H activation reaction, functionalization of the C–H bond in the presence of another reactive functional group such as aldehyde is not known and is highly challenging due to the competitive reaction associated with the aldehyde functional group. Further, the aldehyde group will be very useful in late-stage modifications and for the creation of new pharmacophores through various functional group transformations. In continuation of our interest in C–H activation,7 herein we disclose a novel site-selective ortho-C–H activation reaction for the introduction of an acetoxyl group on aryl ether derivatives. To execute our idea, we chose 2-aryloxyquinolines (1a) as the model substrate. Interestingly, we optimized the reaction conditions for both the mono- and di-acetoxylation of 2-aryloxyquinolines with moderate to good yields.
In order to execute our idea, we prepared 2-aryloxyquinoline-3-carbaldehyde (1a) and treated it with PhI(OAc)2 in the presence of Pd(OAc)2 as a catalyst and AcOH as a solvent at 110 °C for 6 hours (see Table 1, entry 1). Interestingly, the reaction produced the desired mono-acetoxylated product 2a in 30% yield. In order to increase the yield of mono-acetoxylated product, we increased the reaction time from 6 h to 9 h, which yielded the mono-acetoxylated product in moderate yield (45%) (see Table 1, entry 2). To improve the yield further, we carried out the reaction with increased time i.e., from 9 h to 12 h. Interestingly, in this case, we observed the formation of mono-acetoxylated product 2a in 62% yield and di-acetoxylated product 3a in 13% yield (see Table 1, entry 3). However, the best result was obtained when we carried out the reaction with increased catalyst loading from 5 mol% to 10 mol%, which produced the expected mono-acetoxylated product 2a in 85% yield and di-acetoxylated product 3a in 10% yield (see Table 1, entry 4). We also observed that decreasing the load of the catalyst and even increasing the equivalents of PhI(OAc)2 did not provide 2a with better yield (see Table 1, eEntries 5 and 6). Hence the standardized conditions for the mono-acetoxylated product are Pd(OAc)2 10 mol%, PhI(OAc)2 (1.5 equiv.) at 110 °C for 12 hours.
|
|||||
|---|---|---|---|---|---|
| Entry | Pd(OAc)2 (mol%) | PhI(OAc)2 (equiv.) | Time (h) | Yieldb (2a %) | Yieldb (3a %) |
| a Reaction conditions: 1a (0.3 mmol), catalyst (5–10 mol%), oxidant (1.5–3.5 equiv.), AcOH (2 mL), 110 °C, 6–15 h.b Isolated yields. | |||||
| 1 | 5 | 1.5 | 6 | 30 | 0 |
| 2 | 5 | 1.5 | 9 | 45 | 0 |
| 3 | 5 | 1.5 | 12 | 62 | 13 |
| 4 | 10 | 1.5 | 12 | 85 | 10 |
| 5 | 5 | 3.0 | 6 | 40 | 32 |
| 6 | 5 | 3.0 | 9 | 50 | 45 |
| 7 | 5 | 3.0 | 12 | 18 | 62 |
| 8 | 10 | 3.0 | 9 | 40 | 50 |
| 9 | 10 | 3.0 | 12 | 25 | 70 |
| 10 | 5 | 3.5 | 12 | 21 | 66 |
| 11 | 10 | 3.5 | 12 | 15 | 78 |
| 12 | 10 | 3.5 | 15 | 14 | 76 |
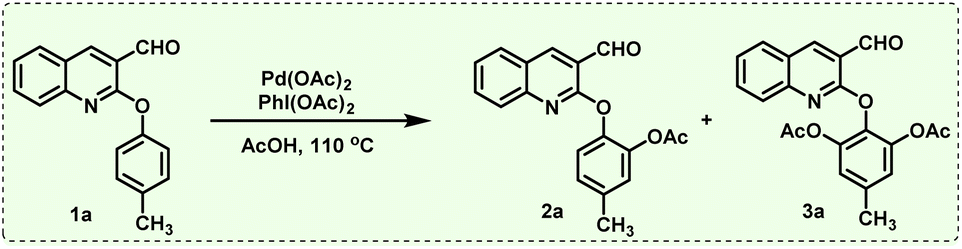
Then we focussed our attention on improving the yield of di-acetoxylation product. Accordingly, we treated 2a with 5 mol% of Pd(OAc)2 and 3 equiv. of PhI(OAc)2 at 110 °C for 12 h, which produced mono-acetoxylated product 2a in 18% yield and the di-acetoxylated product in 62% yield. Then we carried out the reaction by increasing Pd(OAc)2 loading from 5 mol% to 10 mol% and 3 equiv. of PhI(OAc)2 at 110 °C for 9 h, which produced 2a in 40% yield and the desired di-acetoxylated product 3a in 50% yield (see Table 1, entry 8). The same reaction was carried out with 10 mol% of Pd(OAc)2 and 3 equiv. of PhI(OAc)2 at 110 °C for 12 h, which produced mono-acetoxylated product 2a in 25% yield while the di-acetoxylated product 3a was formed in 70% yield (see Table 1, entry 9).
Decreasing the Pd(OAc)2 loading from 10 mol% to 5 mol% and increasing the equivalents of PhI(OAc)2 from 3 equiv. to 3.5 equiv. at 110 °C for 12 h also produced mono-acetoxylated product 2a in 21% yield and di-acetoxylated product 3a in 66% yield (see Table 1, entry 10). Finally, the best result was obtained when we carried out the reaction with 10 mol% of Pd(OAc)2 and 3.5 equiv. of PhI(OAc)2 at 110 °C for 12 h, which provided the desired di-acetoxylated product 3a in 78% yield and mono-acetoxylated product 2a in 15% yield (see Table 1, entry 11). Further increasing the reaction time of the same reaction from 12 h to 15 h did not improve the yield of 3a (see Table 1, entry 12).
With the standardized conditions for the formation of mono-acetoxylated product 2a in hand, we focussed on studying the scope of the ortho-C–H mono-acetoxylation reaction with a wide range of substrates. Initially, we tuned the quinoline ring with different electron-donating and electron-withdrawing groups. In all cases, we observed the formation of mono-acetoxylated products (2a–f) in good yields (60–85%). Later, we changed the substitution on the aryloxy ring, which provided the desired mono-acetoxylated products (2g–j) in good yields (51–65%) (Table 2). It is important to note that the C–H functionalization reaction provided the desired product without any interference from aldehyde functional groups.
| a Reaction conditions: 1a (0.3 mmol), Pd(OAc)2 (10 mol%), PhI(OAc)2 (1.5 equiv.), AcOH (2 mL), 110 °C, 12 h. |
|---|
 |
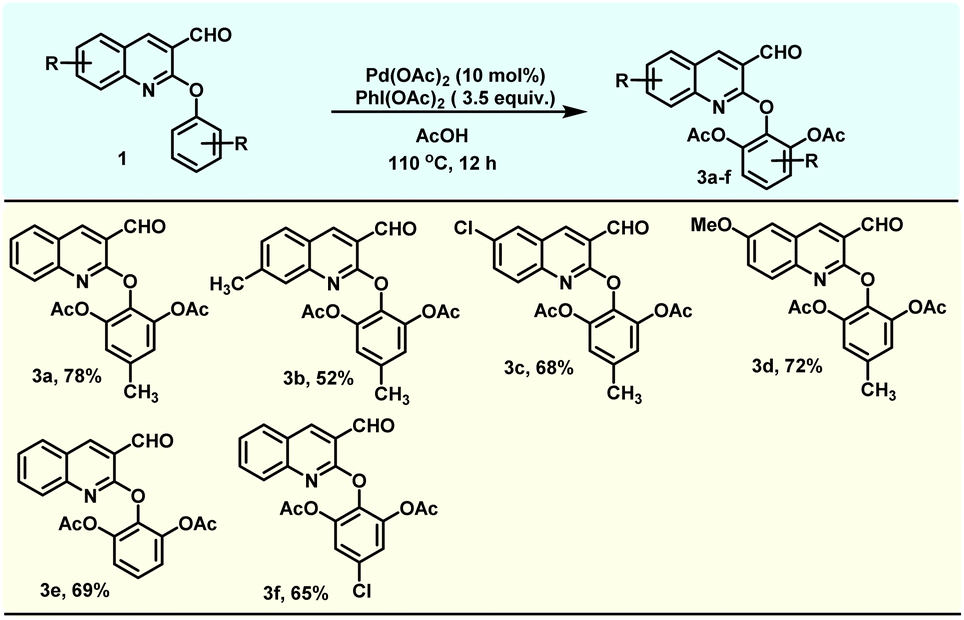
After studying the scope of the mono-acetoxylation reaction, we focussed our attention on the scope of the di-acetoxylated product 3. Accordingly, we investigated the scope of the di-acetoxylation reaction with electron-donating groups (EDG) and electron-withdrawing groups (EWG) on the quinoline part. Both EDG and EWG consisting of substrates (1) were compatible and provided the desired di-acetoxylated products in good yields (52–78%) (Table 3).
| a Reaction conditions: 1a (0.3 mmol), Pd(OAc)2 (10 mol%), PhI(OAc)2 (3.5 equiv.), AcOH (2 mL), 110 °C, 12 h. |
|---|
 |
Subsequently, we conducted a competitive experiment between p-methyl and p-chloro derivatives (1a and i), which produced the p-methyl ortho acetoxylated product (2a) in 65% yield and the p-chloro ortho acetoxylated product (2i) in 17% yield. This reaction clearly indicates that the electron-donating group on the aryloxy ring facilitated the reaction compared to the electron-withdrawing group (Scheme 2).
 | ||
| Scheme 2 Competition reaction. | ||
To understand further which functional group (the quinoline nitrogen or aldehyde group) acts as a directing group, we prepared substrate 4, where we kept the reactive site containing the aryloxy group at the 4th position of the quinoline moiety, which in turn increased the distance and changed the positions between the nitrogen atom and the reactive site containing the aryloxy moiety. Accordingly, we carried out a couple of control experiments with 4-(p-tolyloxy)quinoline-3-carbaldehyde (4) under the standard reaction conditions, which did not provide the expected product (5). This indicates that the close proximity of the quinoline nitrogen to the reactive site of the aryloxy moiety is essential for the reaction. Therefore, the quinoline nitrogen acts as a directing group, not the aldehyde group. Next, we prepared 3-formyl-2-(p-tolyloxy)quinoline 1-oxide (6), in this substrate we converted the quinoline nitrogen into N-oxide to understand the importance of iminium nitrogen. Accordingly, substrate 6 under the optimized conditions for the ortho-C–H acetoxylation reaction did not lead to the expected C–H activation product (7). This clearly indicates that the quinoline nitrogen acts as the DG and not the N-oxide group (Scheme 3).
 | ||
| Scheme 3 Control experiments. | ||
The proposed mechanism for the ortho-C–H acetoxylation reaction is depicted in Scheme 4. The catalytic reaction proceeds via preferential coordination of the N atom of compound 1 with Pd(OAc)2 to generate intermediate A. In the next step, intermediate A undergoes C–H activation and forms palladacycle B by the liberation of AcOH. Subsequently, palladacycle B transforms to complex C via oxidative addition using PhI(OAc)2. Finally, desired product 2a is obtained from complex C via reductive elimination. The catalytic cycle is completed by the regeneration of the Pd(II) species, as shown in Scheme 4.
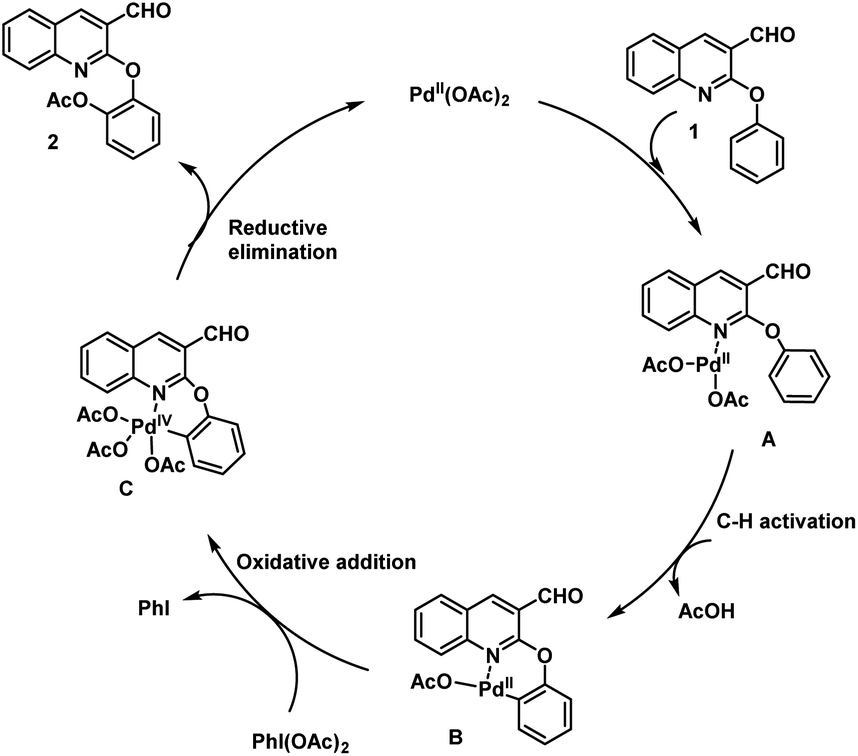 | ||
| Scheme 4 Plausible mechanism for the formation of acetoxylated 2-aryloxyquinoline-3-carbaldehyde (2). | ||
Conclusions
We have developed a novel and efficient protocol for the synthesis of mono- and di-acetoxylated 2-aryloxyquinoline-3-carbaldehydes using a palladium catalyst for the first time. A variety of mono- and di-acetoxylated 2-aryloxyquinoline-3-carbaldehydes were synthesized in very good yields with excellent site selectivity. Various functional groups on the aryl rings as well as the more reactive aldehyde functional group were well tolerated in this reaction. Keeping the aldehyde functional group intact in the reaction will help in further useful transformations. Additionally, the incorporation of acetoxy groups on 2-aryloxyquinoline-3-carbaldehydes is useful for making a variety of phenolic scaffolds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank DST-SERB for the Financial support (Ref. No. CRG/2023/007946). T. T. R. thanks CSIR, New Delhi, for a direct Senior Research Fellowship (D-SRF) and O. S. A. thanks University of Lagos Tetfund IBR (CRC/TETFUND/No.2018/016) for providing fellowship. We thank Pondicherry University for ESI-HRMS and NMR facilities.Notes and references
- (a) J. L. McCormick, T. C. McKee, J. H. Cardellina and M. R. Boyd, J. Nat. Prod., 1996, 59, 469–471 CrossRef CAS PubMed; (b) I. S. Chen, H. F. Chen, M. J. Cheng, Y. L. Chang, C. M. Teng, I. Tsutomu, J. J. Chen and I. L. Tsai, J. Nat. Prod., 2001, 64, 1143–1147 CrossRef CAS PubMed; (c) M. E. Wall, M. C. Wani, C. E. Cook, K. H. Palmer, A. T. McPhail and G. A. Sim, J. Am. Chem. Soc., 1966, 88, 3888–3890 CrossRef CAS; (d) A. Marella, O. P. Tanwar, R. Saha, M. R. Ali, S. Srivastava, M. Akhter, M. Shaquiquzzaman and M. M. Alam, Saudi Pharm. J., 2013, 21, 1–12 CrossRef PubMed.
- X. Yang, G. Zhou and W. Y. Wong, Chem. Soc. Rev., 2015, 44, 8484–8575 RSC.
- (a) M. P. LaMontagne, P. Blumbergs and D. C. Smith, J. Med. Chem., 1989, 32, 1728–1732 CrossRef CAS PubMed; (b) S. T. Hazeldine, L. Polin, J. Kushner, K. White, N. M. Bouregeois, B. Crantz, E. Palomino, T. H. Corbett and J. P. Horwitz, J. Med. Chem., 2002, 45, 3130–3137 CrossRef CAS PubMed; (c) S. T. Hazeldine, L. Polin, J. Kushner, K. White, T. H. Corbett and J. P. Horwitz, Bioorg. Med. Chem., 2006, 14, 2462–2467 CrossRef CAS PubMed; (d) R. S. Upadhayaya, G. M. Kulkarni, N. R. Vasireddy, J. K. Vandavasi, S. S. Dixit, V. Sharma and J. Chattopadhyaya, Bioorg. Med. Chem., 2009, 17, 4681–4692 CrossRef CAS PubMed; (e) U. J. Ries, H. W. M. Priepke, N. H. Hauel, E. E. J. Haaksma, J. M. Stassen, W. Wienen and H. Nar, Bioorg. Med. Chem. Lett., 2003, 13, 2291–2295 CrossRef CAS PubMed; (f) D. G. Batt, J. J. Petraitis, S. R. Sherk, R. A. Copeland, R. L. Dowling, T. L. Taylor, E. A. Jones, R. L. Magolda and B. D. Jaffee, Bioorg. Med. Chem. Lett., 1998, 8, 1745–1750 CrossRef CAS PubMed; (g) F. Caijo, P. Mosset, R. Grée, V. Audinot-Bouchez, J. Boutin, P. Renard, D. H. Caignard and C. Dacquet, Bioorg. Med. Chem. Lett., 2005, 15, 4421–4426 CrossRef CAS PubMed; (h) D. Mabire, S. Coupa, C. Adelinet, A. Poncelet, Y. Simonnet, M. Venet, R. Wouters, A. S. J. Lesage, L. V. Van Beijsterveldt and F. Bischoff, J. Med. Chem., 2005, 48, 2134–2153 CrossRef CAS PubMed.
- (a) R. Giri, B. F. Shi, K. M. Engle, N. Maugel and J. Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC; (b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed; (c) J. Le Bras and J. Muzart, Chem. Rev., 2011, 111, 1170–1214 CrossRef CAS PubMed; (d) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC; (e) H. M. L. Davies and D. Morton, ACS Cent. Sci., 2017, 3, 936–943 CrossRef CAS PubMed; (f) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS PubMed; (g) X. S. Xue, P. Ji, B. Zhou and J. P. Cheng, Chem. Rev., 2017, 117, 8622–8648 CrossRef CAS PubMed; (h) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199–222 CrossRef CAS; (i) S. K. Sinha, S. Guin, S. Maiti, J. P. Biswas, S. Porey and D. Maiti, Chem. Rev., 2022, 122, 5682–5841 CrossRef CAS PubMed; (j) A. Banerjee, A. Bera, S. K. Santra, S. Guin and B. K. Patel, RSC Adv., 2014, 4, 8558–8566 RSC; (k) A. Mandal, S. Dana, D. Chowdhury and M. Baidya, Chem.–Asian J., 2019, 14, 4074 CrossRef CAS PubMed; (l) S. B. Jadhav, S. Maurya, N. Navaneetha and R. Chegondi, Chem. Commun., 2021, 57, 13598–13601 RSC; (m) S. Basak, T. Paul, S. Mandal, P. Karjee, M. V. Nanjegowda and T. Punniyamurthy, Synthesis, 2023, 55, 3454–3469 CrossRef CAS; (n) S. M. Patel, P. Paradakshini, M. Bakthadoss and D. S. Sharada, Org. Lett., 2021, 23, 257–261 CrossRef CAS PubMed; (o) A. Murugan, K. R. Gorantla, B. S. Mallik and D. S. Sharada, Org. Biomol. Chem., 2018, 16, 5113–5118 RSC.
- (a) I. B. Krylov, V. A. Vil' and A. O. Terent'ev, Beilstein J. Org. Chem., 2015, 11, 92–146 CrossRef PubMed; (b) B. Liu and B. F. Shi, Tetrahedron Lett., 2015, 56, 15–22 CrossRef CAS; (c) S. Moghimi, M. Mahdavi, A. Shafiee and A. Foroumadi, Eur. J. Org Chem., 2016, 3282 CrossRef CAS; (d) Y. F. Liang and N. Jiao, Acc. Chem. Res., 2017, 50, 1640–1653 CrossRef CAS PubMed; (e) F. Yang, H. Zhang, X. Liu, B. Wang and L. Ackermann, Chin. J. Org. Chem., 2019, 39, 59 CrossRef CAS; (f) X. Wang, H. Wang, C. Zhou, L. Yang, L. Fu and G. Li, Chem. Commun., 2022, 58, 4993–4996 RSC.
- (a) A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 2300–2301 CrossRef CAS PubMed; (b) Z. Ren, J. E. Schulz and G. Dong, Org. Lett., 2015, 17, 2696–2699 CrossRef CAS PubMed; (c) T. Okada, K. Nobushige, T. Satoh and M. Miura, Org. Lett., 2016, 18, 1150–1153 CrossRef CAS PubMed; (d) Q. Zhang, X. Xie, J. Peng, F. Chen, J. Ma, C. Li, H. Liu, D. Wang and J. Wang, Org. Lett., 2021, 23, 4699–4704 CrossRef CAS PubMed; (e) L. V. Desai, K. J. Stowers and M. S. Sanford, J. Am. Chem. Soc., 2008, 130, 13285–13293 CrossRef CAS PubMed; (f) S. Gu, C. Chen and W. Chen, J. Org. Chem., 2009, 74, 7203–7206 CrossRef CAS PubMed; (g) L. Wang, L. Pan, Y. Huang, Q. Chen and M. He, Eur. J. Org Chem., 2016, 3113–3118 CrossRef CAS; (h) I. Urruzuno, P. A. Sampedro and A. Correa, Eur. J. Org Chem., 2023, e202201489 CrossRef CAS; (i) Z. Peng, Z. Yu, T. Li, N. Li, Y. Wang, L. Song and C. Jiang, Organometallics, 2017, 36, 2826–2831 CrossRef CAS.
- (a) M. Bakthadoss and P. V. Kumar, Adv. Synth. Catal., 2018, 36, 2650–2658 CrossRef; (b) M. Bakthadoss, T. T. Reddy, V. Agarwal and D. S. Sharada, Chem. Commun., 2022, 58, 1406–1409 RSC; (c) M. Bakthadoss, T. T. Reddy and D. S. Sharada, RSC Adv., 2020, 10, 31570–31574 RSC; (d) M. Bakthadoss, P. V. Kumar, R. Kumar, M. Surendar and D. S. Sharada, New J. Chem., 2019, 43, 14190–14195 RSC; (e) M. Bakthadoss, P. V. Kumar, R. Kumar and V. Agarwal, Org. Biomol. Chem., 2019, 17, 4465–4469 RSC; (f) M. Bakthadoss, T. T. Reddy and D. S. Sharada, RSC Adv., 2020, 10, 31570–31574 RSC; (g) M. Bakthadoss, P. V. Kumar and T. T. Reddy, Eur. J. Org Chem., 2017, 4439 CrossRef CAS; (h) M. Bakthadoss and T. T. Reddy, Chem. Sci., 2023, 14, 5880–5886 RSC; (i) M. Bakthadoss, M. A. Hussain and T. T. Reddy, Chem. Commun., 2023, 59, 5249–5252 RSC; (j) S. Arepally, V. N. Babu, M. Bakhadoss and D. S. Sharada, Org. Lett., 2017, 19, 5014–5017 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01289e |
| This journal is © The Royal Society of Chemistry 2024 |