
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSyntheses, structural, photophysical and theoretical studies of heteroleptic cycloplatinated guanidinate(1−) complexes bearing acetylacetonate and picolinate ancillary ligands†
Vasudha Thakur a,
Jisha Mary Thomasb,
Mohammad Adnanc,
Chinnappan Sivasankarb,
G. Vijaya Prakashc and
Natesan Thirupathi*a
a,
Jisha Mary Thomasb,
Mohammad Adnanc,
Chinnappan Sivasankarb,
G. Vijaya Prakashc and
Natesan Thirupathi*a
aDepartment of Chemistry, University of Delhi, Delhi 110 007, India. E-mail: tnat@chemistry.du.ac.in; thirupathi_n@yahoo.com
bDepartment of Chemistry, Catalysis and Energy Laboratory, Pondicherry University, Puducherry 605 014, India. E-mail: siva.che@pondiuni.edu.in
cDepartment of Physics, Nanophotonics Laboratory, Indian Institute of Technology-Delhi, New Delhi 110 016, India. E-mail: prakash@physics.iitd.ac.in
First published on 23rd April 2024
Abstract
Cycloplatination of symmetrical N,N′,N′′-triarylguanidines, (ArNH)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NAr with cis-[Pt(TFA)2(S(O)Me2)2] in toluene afforded cis-[Pt(TAG)(TFA)(S(O)Me2)] (TAG = triarylguanidinate(1−)-κC,κN; TFA = OC(O)CF3; 6–9) in 75–82% yields. The reactions of 6–9 and the previously known cis-[Pt(TAG)X(S(O)Me2)] (X = Cl (1) and TFA (2–5)) with acetylacetone (acacH) or 2-picolinic acid (picH) in the presence of a base afforded [Pt(TAG)(acac)] (acac = acetylacetonate-κ2O,O′; 10–18) and [Pt(TAG)(pic)] (pic = 2-picolinate-κN,κO; 19) in high yields. The new complexes were characterised by analytical, IR and multinuclear NMR spectroscopies. Further, molecular structures of 11, 12, 13·0.5 toluene and 14–19 were determined by single crystal X-ray diffraction. Absorption spectra of 10–19 in solution and their emission spectra in crystalline form were measured. Platinacycles 10–19 are bluish green light emitter in the crystalline form, and emit in the λPL = 488–529 nm range (11 and 13–19) while 12 emits at λPL = 570 nm. Unlike other platinacycles, the emission band of 12 is broad, red shifted, and this pattern is ascribed to the presence of an intermolecular N–H⋯Pt interaction involving the endocyclic amino unit of the six-membered [Pt(TAG)] ring and the Pt(II) atom in the adjacent molecule in an asymmetric unit of the crystal lattice. Lifetime measurements were carried out for all platinacycles in crystalline form, which revealed lifetime in the order of nanoseconds. The origin of absorption and emission properties of 11, 15, 18 and 19 were studied by TD-DFT calculations.
NAr with cis-[Pt(TFA)2(S(O)Me2)2] in toluene afforded cis-[Pt(TAG)(TFA)(S(O)Me2)] (TAG = triarylguanidinate(1−)-κC,κN; TFA = OC(O)CF3; 6–9) in 75–82% yields. The reactions of 6–9 and the previously known cis-[Pt(TAG)X(S(O)Me2)] (X = Cl (1) and TFA (2–5)) with acetylacetone (acacH) or 2-picolinic acid (picH) in the presence of a base afforded [Pt(TAG)(acac)] (acac = acetylacetonate-κ2O,O′; 10–18) and [Pt(TAG)(pic)] (pic = 2-picolinate-κN,κO; 19) in high yields. The new complexes were characterised by analytical, IR and multinuclear NMR spectroscopies. Further, molecular structures of 11, 12, 13·0.5 toluene and 14–19 were determined by single crystal X-ray diffraction. Absorption spectra of 10–19 in solution and their emission spectra in crystalline form were measured. Platinacycles 10–19 are bluish green light emitter in the crystalline form, and emit in the λPL = 488–529 nm range (11 and 13–19) while 12 emits at λPL = 570 nm. Unlike other platinacycles, the emission band of 12 is broad, red shifted, and this pattern is ascribed to the presence of an intermolecular N–H⋯Pt interaction involving the endocyclic amino unit of the six-membered [Pt(TAG)] ring and the Pt(II) atom in the adjacent molecule in an asymmetric unit of the crystal lattice. Lifetime measurements were carried out for all platinacycles in crystalline form, which revealed lifetime in the order of nanoseconds. The origin of absorption and emission properties of 11, 15, 18 and 19 were studied by TD-DFT calculations.
Introduction
Cycloplatinated nitrogen donor complexes that contain one or two Pt–C(aryl) bonds are known for their intriguing structural, bonding, photophysical properties and biological applications.1 Heteroleptic complexes [Pt(ND)(acac)] (ND = Monoanionic nitrogen donor ligands-κC,κN; acac = acetylacetonate-κ2O,O′ (A)) and [Pt(ND)(pic)] (pic = 2-picolinate-κN,κO (B)) are a well-studied class of cycloplatinated nitrogen donor complexes due to their interesting structural, bonding and emission properties (see Fig. 1). The availability of a range of 2-arylpyridyl type nitrogen donor ligands for cycloplatination reaction, tunability of substituents on both the chelate rings in A and B makes this class of complexes as suitable substrates for structural and photophysical studies.2–14 The photophysical properties such as color purity, quantum yield, ΦPL and life time of the excited state, τ of complexes of the types A and B were changed by tuning the substituent on either of the chelate rings. Further, this type of platinacycles were shown to act as metallomesogens15 and as O2 sensors.16–19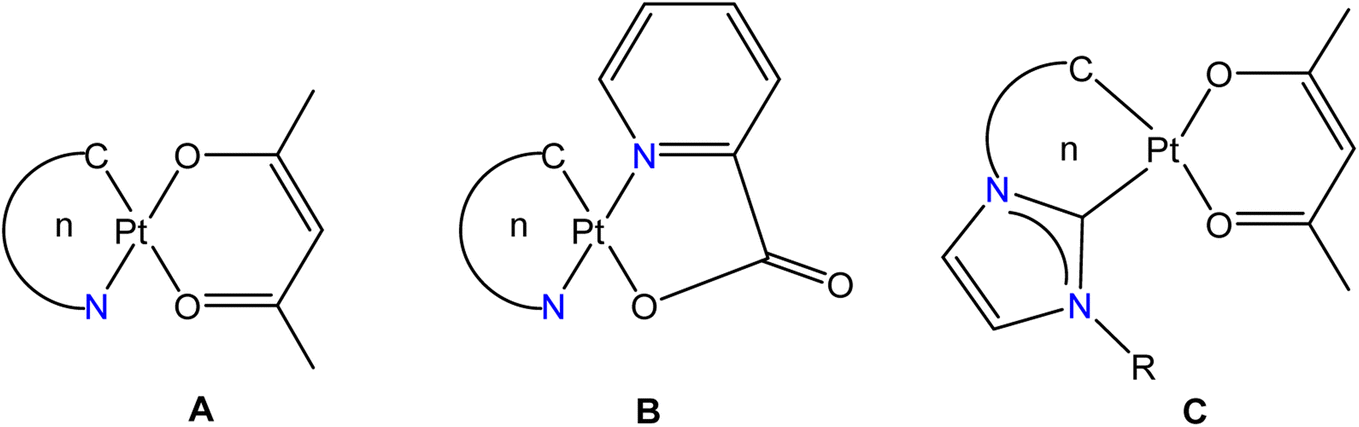 | ||
| Fig. 1 Types of heteroleptic cycloplatinated nitrogen- and carbon-donor complexes known in the literature. The letter n refers to the ring size of the [Pt(ND)] (ND = monoanionic nitrogen donor ligand-κC,κN; (A and B)) and [Pt(CD)] (CD = monoanionic carbon donor ligand-κ2C,C′; (C)) units. | ||
The strongly σ-donating aryl carbon and π-accepting nitrogen atom of the C,N chelate ring in A and B were shown to widen the d–d energy gap so that the non-radiative decay of excited state is minimized. Other structural features such as geometry of the platinum, degree of steric encumbrance around the Pt(II) atom, substitution pattern and conjugation in the chelate rings and degree of non-covalent interactions in the crystal lattice were shown to influence the photophysical properties of these complexes significantly. It has been suggested that platinacycles with rigid scaffolds are desirable for developing luminescent materials with high quantum yield, ΦPL.1f The square planar geometry of the Pt(II) atom in sterically unencumbered A permits aggregation in the crystal lattice through Pt⋯Pt and π–π interactions thereby leading to the formation of excimer and thus influences the shape and wavelength of emission bands.13b,20,21 A detailed theoretical calculations have been carried out on type A complexes to understand the origin of emission, band shapes and structure-emission property relationship.22
Cycloplatinated carbon donor complexes derived from imidazoles such as C are studied due to the presence of strongly σ-donating carbene carbon, which rises the energy of non-radiative d–d states on the metal center thereby increasing the energy spacing with emissive excited states and thus improving the quantum yield, ΦPL. The presence of a stable M–Ccarbene bond in C could increase the lifetime of these materials in organic electronic devices.23,24 Five-membered platinacycles of the types A and B are well-known (n = 5) while to the best of our knowledge only two six-membered platinacycles A (n = 6) and none of the type B (n = 6) are known in the literature.25,26
The C,N chelate rings in A and B are usually derived from a rigid nitrogen donor ligands such as 2-arylpyridyls. We wanted to utilize a range of triarylguanidinate(1−) ligand as monoanionic C,N chelate ring in A and B to investigate whether the resulting heteroleptic complexes are emissive or non-emissive. Further, we wanted to tune the substituents in the aryl rings in the C,N chelate of A and B to investigate whether this endeavor has any influence on the structural and photophysical properties of these complexes.
The photophysical properties of cycloplatinated imino(1−) complexes are sparsely studied in the literature.27–29 Considering high basicity and tunability of N-aryl guanidines and their propensity to undergo cycloplatination with cis-[PtX2(S(O)Me2)2] (X = Cl and TFA) under mild reaction condition, we began a systematic investigation aimed at understanding the structural and photophysical properties of heteroleptic platinacycles, [Pt(TAG)(acac)] (TAG = triarylguanidinate(1−)-κC,κN; 10–18) and [Pt(TAG)(pic)] (19). The new complexes were fully characterized. DFT and TD-DFT calculations were carried out on 11, 15, 18 and 19 to understand the origin of electronic absorption and emission bands.
Results and discussion
Syntheses
Cycloplatinated guanidinate(1−) complexes 1–5 were prepared following the literature procedures (see Fig. 2).30–32 Cycloplatination reactions of sym N,N′,N′′-triarylguanidines, (ArNH)2CNAr (sym = symmetrical; Ar = 2-XC6H4 (X = F, Cl and Br) and 4-FC6H4) with cis-[Pt(TFA)2(S(O)Me2)2] (TFA = OC(O)CF3) in toluene under reflux condition for 8 h following the procedure established for 1–5 in our laboratories afforded 6–9 in 75–82% yields as outlined in Scheme 1. Reactions of 1–9 with one equiv. of acetylacetone (acacH) in the presence of one equiv. of K2CO3 in MeCN at 75 °C for 36 h afforded 10, 11, 15 and 16 as green crystals and 12–14, 17 and 18 as yellow crystals in 81–94% yields after work up and crystallization events (see Scheme 2).
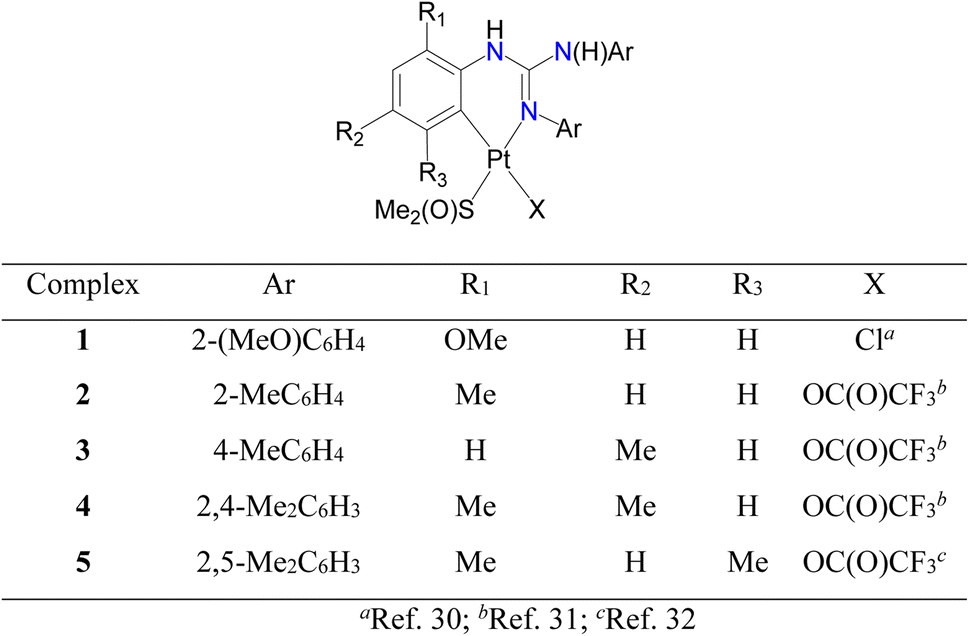 | ||
| Fig. 2 Known cycloplatinated guanidinate(1−) complexes, 1–5. | ||
 | ||
| Scheme 1 Syntheses of cycloplatinated guanidinate(1−) complexes, 6–9. | ||
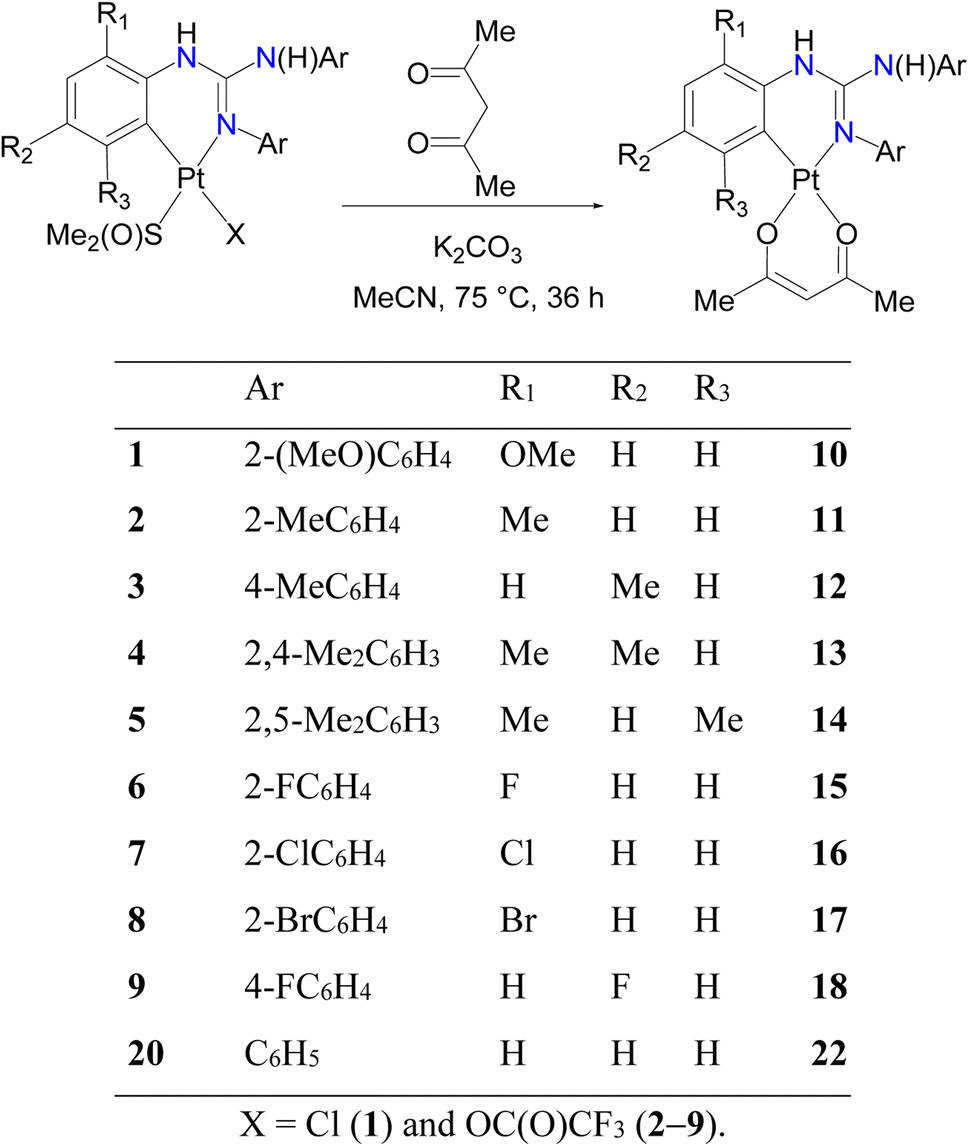 | ||
| Scheme 2 Syntheses of heteroleptic cycloplatinated guanidinate(1−) complexes, 10–18 and 22. | ||
Cycloplatination of nitrogen donor ligands can be carried out by thermal method with various Pt(II) sources such as K2PtCl4, (Bu4N)2PtCl4 and cis-[PtCl2(S(O)Me2)2]. However, this method is plagued with issues such as decomposition of the Pt(II) source to Pt(0) and requires a longer reaction period up to a week thereby producing platinacycles in low to moderate yields. Further, platinacycles of the type A have been isolated using more expensive and air-sensitive [PtMe2(μ-SMe2)]2 as the Pt(II) precursor.33 Recently, Gonzalez-Herrero and co-workers reported photochemical pathway for [Bu4N][Pt(ND)Cl2] (ND = monoanionic 2-aryl pyridines-κC,κN and related C,N ligands) from [Bu4N]2[Pt2Cl6] and the corresponding nitrogen donor ligands. The reaction of [Bu4N][Pt(ND)Cl2] even with Na(acac) afforded platinacycles of the type A only in 31–65% yields.34 Thus, synthetic route outlined in Scheme 2 is operationally simple but yet this route gave the products in good to very good yields.
We have also employed picolinic acid (picH) in place of acacH in the reaction with 1 in order to understand the ring size effect and higher ligand field strength of the picolinate(1−) ligand1h on the photophysical properties of the resulting platinacycle, 19. Thus, 1 was treated with picH under the condition identical to that described in Scheme 2, which enabled us to isolate 19 as green crystals in 89% yield as illustrated in Scheme 3. The reaction involving 20 and acacH in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in the presence of K2CO3 under the optimised conditions always gave a known dinuclear platinacycle 2135 instead of the anticipated mononuclear platinacycle, 22 (see Schemes 2 and S1 in the, ESI†).
:1 molar ratio in the presence of K2CO3 under the optimised conditions always gave a known dinuclear platinacycle 2135 instead of the anticipated mononuclear platinacycle, 22 (see Schemes 2 and S1 in the, ESI†).
 | ||
| Scheme 3 Synthesis of cycloplatinated guanidinate(1−) complex, 19. | ||
Solution studies
195Pt NMR spectroscopy is an important tool to shed light on the coordination environment around the Pt atom in organoplatinum complexes.30–32,35–39 Platinacycles of the type A have been sparsely characterized by 195Pt NMR spectroscopy with a few exceptions,6,40,41 possibly due to their poor solubility in commonly used NMR solvent, CDCl3. Platinacycle, 6 was not sufficiently soluble in CDCl3 for 195Pt and 13C{1H} NMR NMR measurements. 195Pt NMR spectra of precursors 7–9 revealed a singlet at δPt −3606, −3591 and −3622, respectively. The aforementioned δPt values are somewhat comparable with those shifts reported for 1–5 (δPt −3737 (1),30 −3650 (2), −3674 (3), −3655 (4)31 and −3717 (5)32).Platinacycles 10–18 revealed a single peak in the interval of −2385 to −2527 ppm as listed in Table 1. These δPt values of 10–18 are deshielded relative to their precursors 1–9 as reflected from the positive coordination chemical shift, Δδ. This trend either resembles40 or opposes41 the trend reported for five-membered cycloplatinated nitrogen-donor complexes, [Pt(ND)(acac)] and their precursors depending upon the steric/electronic properties of the C,N chelate rings. The downfield δPt values of 10–18 is ascribed to the Pt(II) → acac(1−) charge transfer process as reported for the related complexes known in the literature.6,39a,40,41 Platinacycle 19 revealed a singlet at δPt −2906 with a smaller Δδ value of 831 ppm than that of 10 (Δδ = 1312 ppm), which is likely ascribed to the difference in the ring size and the difference in the donor atoms associated with the ancillary ligands.36,42
The δPt value observed for platinacycles are influenced by both steric and electronic factors.30,31,43 The δPt value of platinacycles which are ligated with more basic guanidinate(1−) ligand is more shielded than those platinacycles which are ligated with a less basic guanidinate(1−) ligand as can be seen from the shifts of the 11/15 and 12/18 pairs. The more shielded δPt value of 12 than that of 11, as is also observed between 2 and 3, can be ascribed to the difference in the electronic/steric factors associated with the C,N chelates. This trend is in line with the trend reported for the known Pt(II) complexes.30,31,39b Complex 14 revealed a smaller Δδ = 1190 than 13 (Δδ = 1217) which is likely due to greater rigidity of the six-membered [Pt(TAG)] ring in the former complex due to the presence of Me group on the carbon which, is present adjacent to the Pt–C bond. The δPt shifts toward downfield upon going from 15 → 16 → 17 and this trend could be ascribed to the difference in both the steric and electronic factors associated with the C,N chelate rings.
The δPt values reported for 10–18 compare favourably well with that reported for the six-membered cycloplatinated 1,2-diarylimidazolate(1−) complex, [Pt(ND)(acac)] (−2522 ppm).25 The δPt value reported for five-membered platinacycles, [Pt(ND)(acac)] varied from −2700 to −2800 ppm,6,40,41 more shielded than those shifts reported for 10–18 listed in Table 1. This shift difference could arise due to a combination of electronic, steric factors associated with the C,N chelate rings and the difference in the ring size of the [Pt(ND)] units in these two types of platinacycles.42
Platinacycles 10–18 contain two types CH3 protons in the acac(1−) ligand and three types of OCH3 (10)/CH3 protons (11 and 12) or six types of CH3 protons (13 and 14) in the guanidinate(1−) ligands. 1H NMR spectroscopy was used to estimate the ratio of CH3 protons of the acac(1−) ligand to the OCH3/CH3 protons present in the guanidinate(1−) ligands. The estimated ratios of about 6:9 (10–12) or 6:18 (13 and 14) matched with the anticipated ratios thereby confirming retention of the solid state structures of these platinacycles in solution as well. The ratio of CH3 protons of the acac(1−) ligand to the aryl protons of the guanidinate(1−) ligands was estimated and the ratios in 10–12 and 13–14 were found to be about 6:11 and 6:8 ratios, respectively as anticipated for the solid state structures. The presence of a chelating acac(1−) ligand in 10–18 was also inferred from a signature signal assignable to the CH proton at about 5.20 ppm. Platinacycle 18 revealed the presence of two species in solution in about 1.00:0.07 ratio as revealed by 1H NMR spectroscopy and the presence of two solution species was independently confirmed by 19F NMR spectroscopy. We believe that these two species arise due to the restricted rotation of the exocyclic (N2)C–N(H)Ar single bond in the six-membered [Pt(TAG)] ring.32,37
The 1H NMR spectrum of 19 revealed three singlets at δH 3.80, 3.82 and 4.02 assignable to OCH3 protons of the guanidinate(1−) ligand and the estimated ratio of these protons to the sum of the aryl protons of the guanidinate(1−) and pic(1−) ligands was found to be 9:15 thereby confirming retention of the solid structure in solution as well. The 13C{1H} NMR spectral pattern of 10–19 complements their respective 1H NMR spectral pattern and as anticipated, conforming to their respective solid-state structures.
Molecular and crystal structures
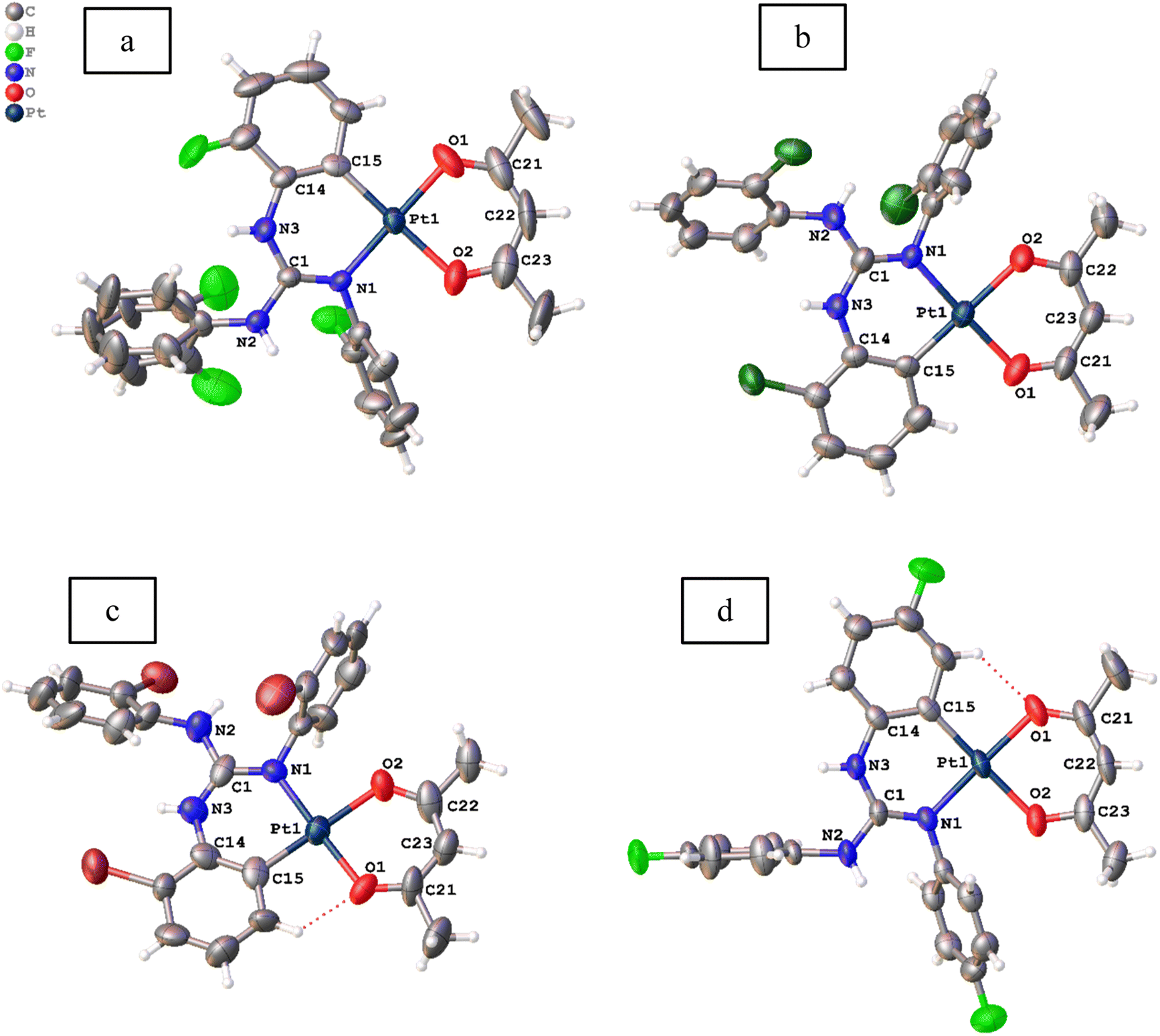
Molecular structures of 11, 12, 13·0.5 toluene and 14–19 were determined by single crystal X-ray diffraction (SCXRD) and are illustrated in Fig. 3–5. The bond parameters around the Pt(II) atom in the above-mentioned complexes are listed in Tables S3–S8 in the ESI.† The Pt(II) atom in all platiancycles except 19 is simultaneously bonded to the guanidinate(1−) and acac(1−) ligands in a chelating fashion and simultaneously becomes part of two six-membered [Pt(TAG)] and [Pt(acac)] rings. The key structural parameters of the aforementioned platinacycles are listed in Table 2. The six-membered [Pt(TAG)] ring revealed a shallow boat (11, 12 and 13·0.5 toluene), sofa (14) or deep boat (19) conformations. The distinct conformation of the six-membered [Pt(TAG)] ring in 14 arises due to the presence of Me substituent on the aryl carbon which present adjacent to the Pt–C bond while that in 19 arises due to the fact that the Pt(II) atom is also part of the five-membered [Pt(pic)] ring. | ||
| Fig. 3 Molecular structures of 11 (a), 12 ((b) Z′ = 2), 13·0.5 toluene ((c) Z′ = 2) and 14 (d) at the 50% probability level. Solvent molecule is omitted from 13·0.5 toluene for clarity. | ||
 | ||
| Fig. 4 Molecular structures of 15 (a), 16 (b), 17 (c) and 18 (d) at the 50% probability level. | ||
 | ||
| Fig. 5 Molecular structure of 19 at the 50% probability level. | ||
|
|||||
|---|---|---|---|---|---|
| θ1a (deg) | θ2b (deg) | θ3c (deg) | θ4d (deg) | θ5e (deg) | |
| a θ1 is the angle between the mean plane defined by the N1Pt1C15 and C1N1C14C15 planes.b θ2 is the angle between the mean plane defined by the C1N3C14 and C1N1C14C15 planes.c θ3 the angle between the mean plane defined by the N1Pt1C15 and O1Pt1O2 planes.d θ4 is the angle between the mean plane defined by the O1Pt1O2 and O1C21O2C23 planes.e θ5 the angle between the mean plane defined by the C21C22C23 and O1C21O2C23 planes. | |||||
| 11 | 13.2(7) | 19.9(3) | 5.7(3) | 2.7(6) | 1.9(2) |
| 12 | |||||
| Molecule 1 | 15.3(1) | 19.3(1) | 1.4(1) | 1.2(1) | 2.9(3) |
| Molecule 2 | 16.0(1) | 25.0(1) | 2.5(1) | 1.1(1) | 1.7(3) |
| 13·0.5 toluene | |||||
| Molecule 1 | 5.55(6) | 12.59(9) | 3.10(3) | 10.99(9) | 2.78(9) |
| Molecule 2 | 10.68(4) | 9.60(7) | 0.66(3) | 2.38(4) | 2.58(9) |
| 14 | 34.8(2) | 28.4(7) | 4.6(3) | 2.6(5) | 0.4(1.1) |
| 15 | 12.1(7) | 10.5(1) | 0.7(7) | 3.3(9) | 1.9(3) |
| 16 | 5.3(5) | 9.1(8) | 0.4(4) | 0.7(5) | 1.6(3) |
| 17 | 11.7(2) | 4.6(9) | 1.8(8) | 1.6(1) | 2.4(3) |
| 18 | 4.1(3) | 1.1(7) | 2.5(3) | 12.8(4) | 4.3(1) |

The degree of puckering of the six-membered [Pt(TAG)] and [Pt(acac)] rings and the degree of deviation of the geometry of the Pt(II) atom from the square plane are quantified with θ1, θ2, θ3, θ4 and θ5 parameters. The values of θ1 = 15.3(1)° (molecule 1) and 16.0(1)° (molecule 2) in 12 are higher than the corresponding values observed for 11 (13.2(7)°), which possibly arises due to the difference in the packing pattern observed in the solid state. The value of θ1 = 34.8(2)° in 14 is significantly greater than the corresponding value observed for 13·0.5 toluene (5.55(6)°(molecule 1) and 10.68(4)° (molecule 2)). The greater value of θ1 observed for 14 is likely caused by the greater steric hindrance imparted by the Me group on the aryl carbon, which is present adjacent to the Pt–C bond.
The differences in θ2 value between 11 and 12 (molecule 2) on one hand and 13 and 14 on the other arise due to the difference in the packing pattern of the first pair and variable positions of two Me substituents in the platinated aryl ring in the second pair. The Pt(II) atom deviates from the square plane either slightly (13·0.5 toluene (molecule 2), 15 and 16), moderately (12 (molecules 1 and 2), 13·0.5 toluene (molecule 1), 17 and 18) or significantly (11 and 14) as reflected from value of θ3. The extent of folding of Pt1 atom from the basal plane constituted by O1, C21, O2 and C23 atoms in the [Pt(acac)] ring is minimal in 11–18 except in 13·0.5 toluene (molecule 1) and 18 as reflected from the value of θ4. The greater folding of acac(1−) ligand along the O1⋯O2 vector in 13·0.5 toluene (molecule 1) and 18 could arise due to the difference in the intermolecular interactions (see the ESI†). The folding of methine C22 of the acac(1−) ligand from the basal plane constituted by O1, C21, O2, and C23 in all complexes is minimal as reflected from the value of θ5. The values of θ1 = 29.6(2)°, θ2 = 23.6(4)° and θ3 = 8.2(1)° observed for 19 are greater than the corresponding values observed for 11 as the Pt(II) is part of the five-membered [Pt(pic)] ring in the former complex.
Crystal structures of platinacycles were analysed to understand the nature and types of intermolecular interactions and their possible influence on the shape of the emission bands. Various types intermolecular interactions present in the crystal lattice of 12 are illustrated in Fig. 6 while these interactions in the remaining platinacycles are illustrated in the Fig. S1–S8 in the ESI.† Two molecules were found in an asymmetric unit of 12, and these two molecules are linked through intermolecular N–H⋯π, C–H⋯π and N–H⋯Pt interactions as illustrated in Fig. 6.
 | ||
| Fig. 6 Packing diagram of 12 (Z′ = 2) illustrating intermolecular interactions. The intermolecular distances (Å) and angles (deg) are: (i) N3B⋯Pt1A = 3.531, H3B⋯Pt1A = 2.767 and N3B–H3B⋯Pt1A = 148.79. (ii) C3A⋯C12B = 3.714, H3A1⋯C12B = 2.851 and C3A–H3A1⋯C12B = 154.84. (iii) N2B⋯C16A = 3.086, H2B⋯C16A = 2.823 and N2B–H2B⋯C16A = 119.91. | ||
The binding of d8 metals to the H–X (X = C, N and O) bond through X–H⋯M interaction is being actively studied due to their relevance in the fields of crystal engineering, metal mediated and catalysed X–H bond activation and optical properties.44 The bonding in the X–H⋯M unit can be classified as agostic, anagostic and weak hydrogen bonding interactions. An agostic interaction occurs when the X–H⋯M unit is stabilised by 3c–2e bonding with the electron deficient metals, anagostic interaction occurs when the X–H⋯M unit is stabilised by 3c–4e bonding with the electron rich metals. The N–H⋯Pt interaction found in 12 is considered as a weak hydrogen bonding interaction as it fulfils four essential criteria set out by Brammer and co-workers for the N–H⋯Pt hydrogen bonding.45 Moreover, the H3B⋯Pt1A = 2.767 Å distance and N3B–H3B⋯Pt1A = 148.79° angle found in 12 clearly matched with the corresponding values of 2.1–2.8 Å and 140−170° respectively anticipated for the N–H⋯Pt hydrogen bonding interaction reported in the in the literature.45–48 This hydrogen bond is believed to influence both the shape and position of the band in the photoluminescence spectrum of 12 (see later).
Photophysical properties
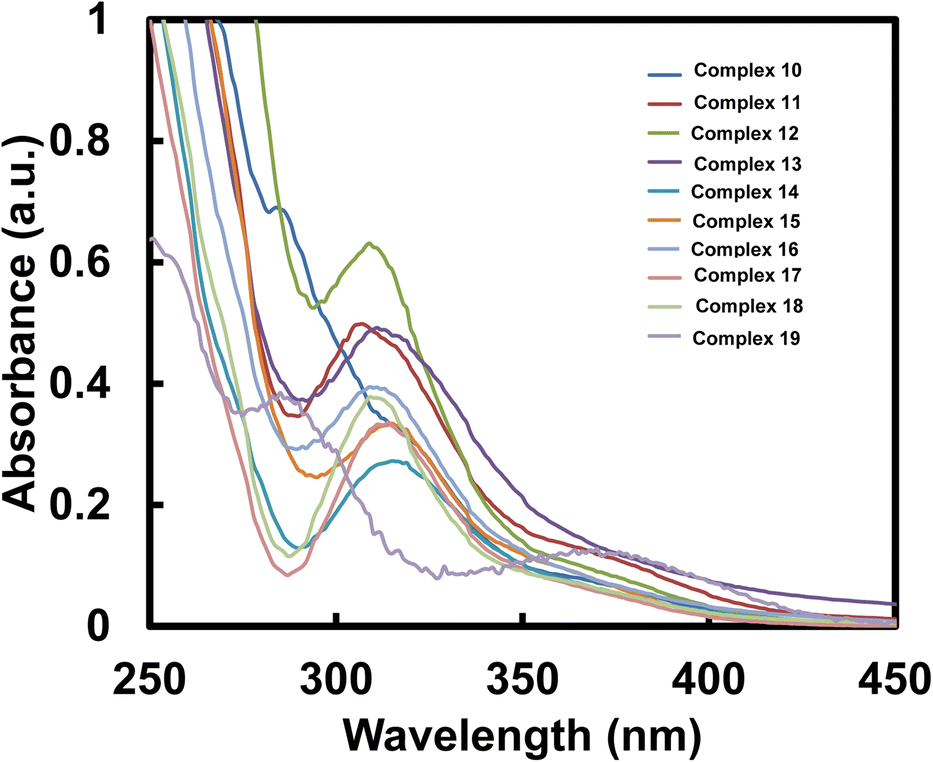 | ||
| Fig. 7 UV-Visible absorption spectra of 10–19 (10−5 M, CH2Cl2) measured at RT. | ||
| Complex | λabs (nm) | ε (×104 M−1 cm−1) | Complex | λabs (nm) | ε (×104 M−1 cm−1) |
|---|---|---|---|---|---|
| 10 | 284, 318 | 6.9, 3.234 | 15 | 315 | 3.346 |
| 11 | 307 | 4.985 | 16 | 309 | 3.948 |
| 12 | 309 | 6.315 | 17 | 310 | 3.273 |
| 13 | 311 | 4.923 | 18 | 311 | 3.766 |
| 14 | 315 | 2.727 | 19 | 285, 370 | 3.85, 1.28 |
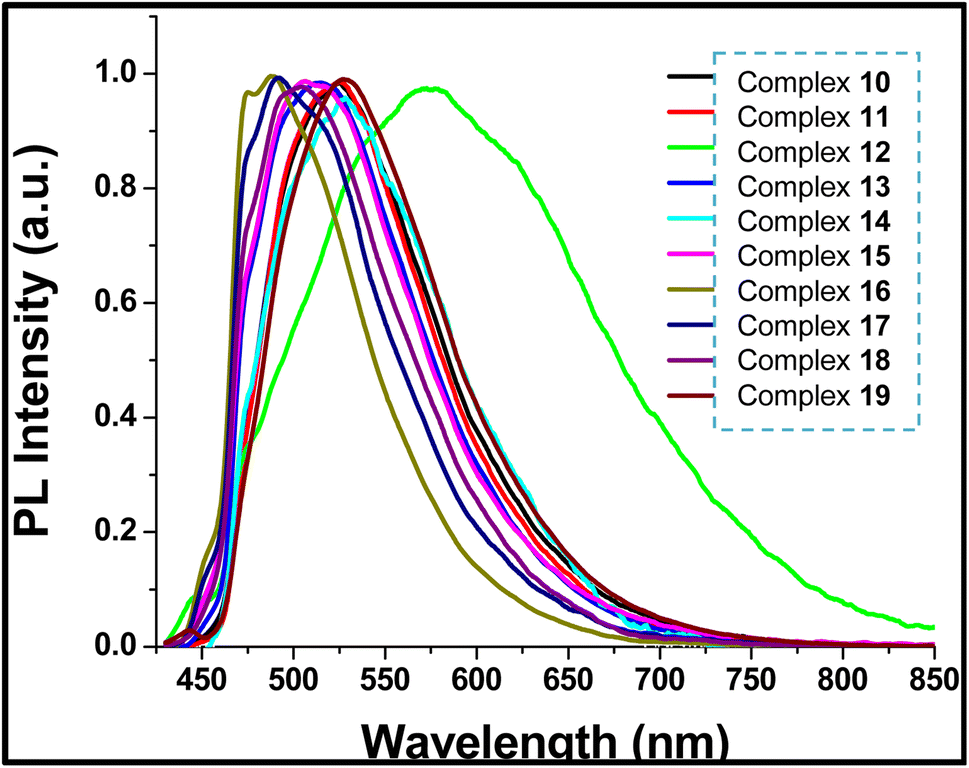 | ||
| Fig. 8 Normalized room temperature photoluminescence spectra of crystalline samples of 10–19 (excited by 405 nm CW diode laser). | ||
 | ||
| Fig. 9 Photoluminescence microscope images of 10–19 in crystalline form excited by 405 nm CW diode laser. | ||
The photoluminescence spectrum of 12 is broader and red shifted than the rest of the platinacycles (λPL = 570 nm (12) and 488–529 nm (10, 11 and 13–19)). In a family of N-heterocyclic carbene derived heteroleptic platinacycles of the type C, one complex used to show a broad emission band, which exhibited red shift from the rest of the complexes.49,50 However, the reason(s) for the above-mentioned spectral behaviour was not addressed clearly. The broad and red shifted emission band of 12 are ascribed to the presence of an intermolecular N–H⋯Pt, N–H⋯π and C–H⋯π interactions between two crystallographically distinct molecules in an asymmetric unit (Z′ = 2) found in the crystal lattice. The red shift observed for 12 is likely ascribed to destabilisation of the HOMO level, decreasing the luminescence energy.51 The broadening of photoluminescence spectrum of 12 is likely ascribed to the distortion in the excited state.52
The presence of fluoro substituent in the aryl rings of the guanidinate(1−) ligand in 15 and 18 causes a blue shift of 21 nm and 67 nm, respectively when compared with analogous complexes 11 and 12. Complexes 16 and 17 are also blue shifted by 39 nm and 35 nm respectively when compared to 11. It is to be noted that λPL observed for 19 closely matches with that observed for 10 suggesting the absence of any significant effect of ancillary ligands pic(1−) and acac(1−) respectively in these complexes, upon the λPL.
| I(t) ∝ exp(t/τexp) |
 | ||
| Fig. 10 Time correlated single photon counting (TCSPC) decay curves of 10–19 in crystalline form, monitored at respective emission peak maxima (excited by Ti-sapphire femtosecond laser: 400 nm, 120 fs, 80 MHz). | ||
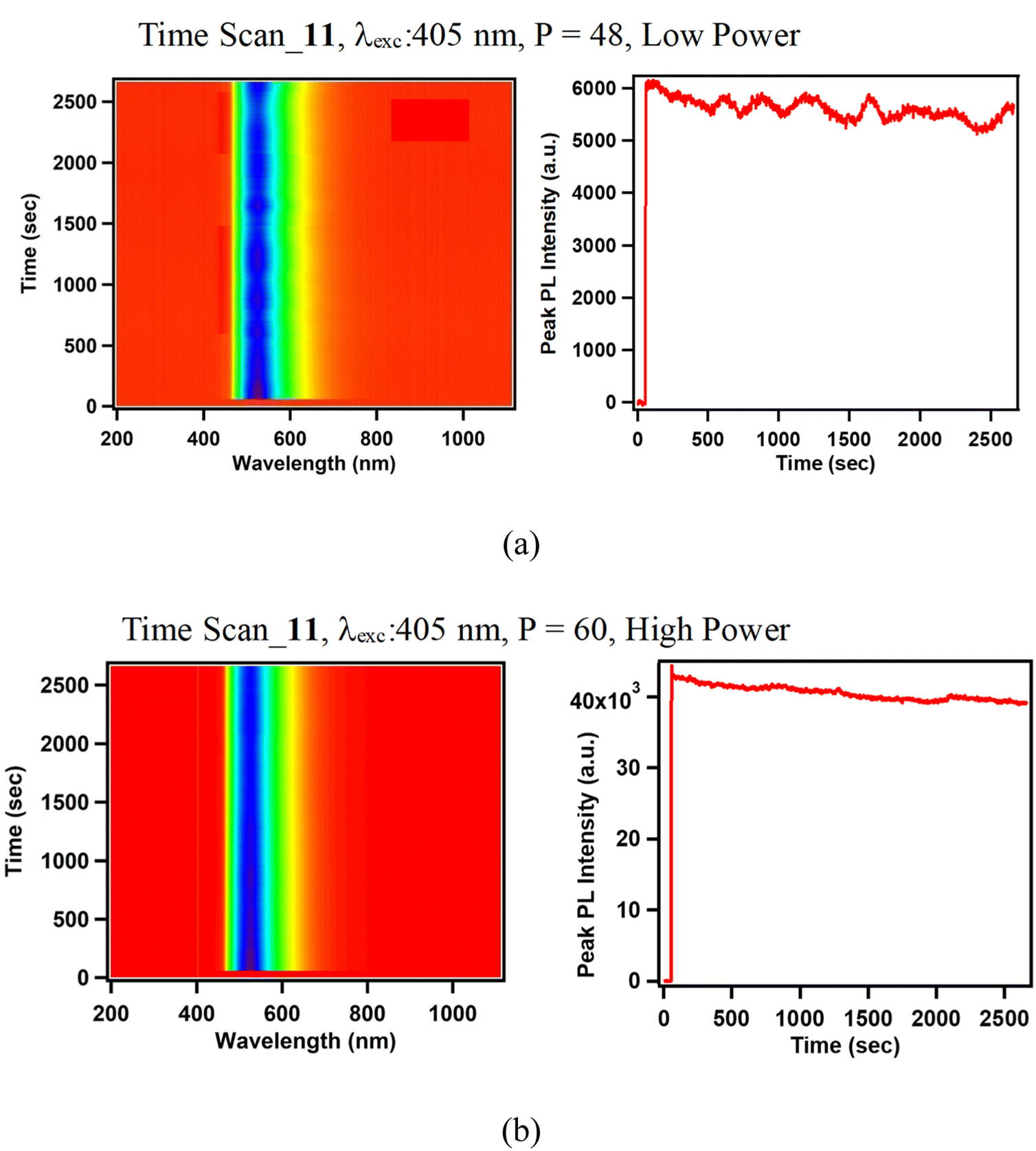 | ||
| Fig. 11 Photoluminescence stability (time-spectral intensity maps and PL peak maxima time variation) of platinacycle 11 at (a) low power and (b) high power, excited by 405 nm CW diode laser. | ||
DFT and TD-DFT studies
The TD-DFT calculations were carried out for 12 excited states and the vertical excitation energies (nm) are computed along with their oscillator strength, f. The TD-DFT results for the absorption study in CH2Cl2 agree well with that of the experimental data. Maximum absorption is centred around 300 nm for complexes 11, 15 and 18 and for 19, additional bands ranging from 350–400 nm are obtained. The frontier molecular orbitals (FMOs) and their calculated energy levels for complexes 11, 15, 18 and 19 are illustrated in Fig. 12. For 11, 15 and 18, the highest probable transition centered on 300 nm arises due to the singlet transition from HOMO−1 → LUMO level (f > 0.1). The HOMO−1 of these complexes is located on the Pt-primary ligand (containing the C,N donor atoms), whereas the LUMO is located mainly on the acac(1−) ligand. Thus, HOMO−1 → LUMO transition can be interpreted as a mixture of MLCT (Pt to the π orbital of primary ligand) and LLCT from the primary ligand to the acac(1−) ligand. For complex 19, the most probable transition is centred on 395 nm and it arises due to the HOMO → LUMO transition (f = 0.0721). The simulated UV-Visible profiles of complexes 11, 15 and 18 and 19 are compiled in the ESI† while their corresponding transitions are listed in Table S11.† Similarly, the corresponding LUMO → HOMO−1 transition from the singlet excited state to the ground state occurs in 11, 15 and 18 while complex 19 shows emission from LUMO → HOMO level (see Table S12†).
 | ||
| Fig. 12 Molecular orbital diagrams and their calculated energy levels (in eV) for complexes 11, 15, 18 and 19. | ||
On comparing the experimental and theoretical emission spectra, only the computed spectra of 19 showed most probable emission (LUMO → HOMO, 96%) at 525 nm which is closer to the experimental value (λPL = 529 nm). However, the computed emission spectra of 11, 15 and 18 showed the most probable emission around 400 nm. These values are different from those experimentally observed λPL (527 nm (11), 506 nm (15) and 503 nm (18)) for single crystals. The theoretical λPL values obtained at the B3LYP/6-31G*/LANL2DZ level of theory deviate considerably from those of the experimental results. However, in the emission spectra of 11, 15 and 18 computed theoretically, a low intensity transition is observed around 500 nm, which occurs due to LUMO → HOMO transition of the complexes involved. This deviation may be rectified perhaps by the use of higher basis set, but due to computational limitations we present the results obtained by the use of the above mentioned level of theory.
Conclusions
In conclusion, we have isolated ten heteroleptic cycloplatinated guanidinate(1−) complexes in good to very good yields. The new complexes were characterized by analytical and IR and multinuclear NMR spectroscopic techniques. Molecular and crystal structures of nine complexes were determined by SCXRD. 195Pt{1H} NMR spectroscopy of new complexes prepared in this investigation enabled us to unravel the factors such as steric and electronic factors, ring size and nature of donor atom on the observed δPt shifts. The variation of steric property of the aryl substituents in the guanidinate(1−) ligands influenced the extent of puckering of the six-membered [Pt(TAG)] ring in the structurally characterized complexes more than the geometry of the Pt(II) atom.A detailed crystal structure analyses carried out on 12 suggest the significance of intermolecular N–H⋯π, C–H⋯π and N–H⋯Pt hydrogen bonding interactions on the anomalous emission spectrum of this complex in the crystalline form. The new complexes were shown to be a green to blue light emitting materials in the solid state. The tunability of aryl substituents in the six-membered [Pt(TAG)] ring affected the emission properties of certain complexes to some extent while the ancillary ligand affected the emission to a lesser extent (10 versus 19). The FMOs involved in the absorption and emission of 11, 15 and 18 were shown to be HOMO−1 and LUMO while the FMOs of 19 were shown to be HOMO and LUMO. By a judicious combination of the primary and ancillary ligands in the new platinacycles, better materials for the purpose of OLED fabrication can be further developed.
Experimental section
Platinacycle 6
Cis-[Pt(TFA)2(S(O)Me2)2] (50.0 mg, 0.09 mmol) and the guanidine (ArNH)2CNAr (Ar = 2-FC6H4; 34.2 mg, 0.09 mmol) were taken in a 25 mL RB flask and dispersed in toluene (10 mL). The RB flask was fitted with a double surface condenser capped with a freshly prepared anhydrous CaCl2 guard tube. The reaction mixture was refluxed for 8 h, cooled and filtered. The volume of the filtrate was reduced to about 3 mL and stored at RT for two days to afford 6 as colourless crystals. Yield = 75% (47.4 mg, 0.065 mmol). Mp: 240.2 °C. ATR-IR (cm−1): ν(NH) 3306 (m); νa(OCO) 1688 (s); ν(CN) 1622 (m); νs(OCO) 1344 (m); ν(SO) 1190 (s). Anal. calcd for C23H19N3O3F6SPt (Mw = 726.56 g mol−1): C, 38.02; H, 2.64; N, 5.78; S, 4.41. Found: C, 37.88; H, 2.63; N, 5.83; S, 4.61. ESI mass (HRMS) m/z [ion]: calcd 613.0849 [M − TFA]+. Found 613.0851. 1H NMR (CDCl3, 400 MHz): δH 3.19 (br, 6H, (CH3)2S(O)), 6.40 (s, 1H, NH), 6.83–6.88, 6.92–6.99 (each m, 2 × 1H, ArH), 7.16–7.23 (m, 4H, ArH), 7.25 (br, 1H, NH), 7.27–7.38 (m, 4H, ArH), 7.72 (d, JHH = 8.4 Hz; 1H, ArH). 19F{1H} NMR (CDCl3, 376.31 MHz): δF −74.4, −119.5, −122.7, −133.5 (JF–Pt = 41.4 Hz).
Platinacycle 7
Platinacycle 7 was prepared from cis-[Pt(TFA)2(S(O)Me2)2] (50.00 mg, 0.087 mmol) and the guanidine (ArNH)2CNAr (Ar = 2-ClC6H4; 34.2 mg, 0.087 mmol) in toluene (10 mL) by following the procedure previously mentioned for 6. Platinacycle 7 was obtained as colourless crystals in 82% (55.3 mg, 0.071 mmol) yield. Mp: 209.8 °C. ATR-IR (cm−1): ν(NH) 3364 (m); νa(OCO) 1680 (s); ν(CN) 1607 (m), 1568; νs(OCO) 1356 (m); ν(SO) 1186 (s). Anal. calcd for C23H19N3O3F3Cl3SPt (Mw = 775.91): C, 35.60; H, 2.47; N, 5.42; S, 4.13. Found: C, 35.66; H, 2.74; N, 5.68; S, 4.43. ESI mass (HRMS) m/z [ion]: calcd 662.0040 [M − TFA + H]+. Found 661.9937. 1H NMR (CDCl3, 400 MHz): δH 3.17 (br, 6H, (CH3)2S(O)), 6.39 (s, 1H, NH), 6.88 (t, JHH = 8.0 Hz, 1H, ArH), 7.09 (dd, JHH = 8.0 Hz; 1.2 Hz, 1H, ArH), 7.24–7.28, 7.30–7.33 (each m, 2 × 1H, ArH), 7.34 (s, 1H, NH), 7.37–7.40, 7.48–7.51 (each m, 2 × 3H, ArH), 7.86 (dd, JHH = 8.0 Hz; 1.6 Hz, 1H, ArH). 13C{1H} NMR (CDCl3, 100.5 MHz): δC 45.1, 45.9, 110.5, 117.0 (q, JC–F = 290.9 Hz), 119.9, 124.2 (JPt–C = 34.7 Hz), 125.6, 128.0, 128.3, 128.7, 128.9, 129.1, 129.6, 131.1, 131.4, 131.7, 132.2, 132.6, 137.4, 139.6, 148.2, 161.7 (q, JC–F = 36.3 Hz). 19F{1H} NMR (CDCl3, 376.31 MHz): δF −74.4. 195Pt{1H} NMR (CDCl3, 85.78 MHz): δPt −3606.
Platinacycle 8
Platinacycle 8 was prepared from cis-[Pt(TFA)2(S(O)Me2)2] (50.00 mg, 0.087 mmol) and the guanidine (ArNH)2CNAr (Ar = 2-BrC6H4; 46.1 mg, 0.087 mmol) in toluene (10 mL) by following the procedure previously mentioned for 6. Platinacycle 8 was obtained as colourless crystals in 76% (60.0 mg, 0.066 mmol) yield. Mp: 198.1 °C. ATR-IR (cm−1): ν(NH) 3354 (m); νa(OCO) 1686 (s); ν(CN) 1613 (m); νs(OCO) 1360 (m); ν(SO) 1130 (s). Anal. calcd for C23H19N3O3F3Br3SPt (Mw = 905.83): C, 30.38; H, 2.11; N, 4.62; S, 3.53. Found: C, 30.04; H, 2.09; N, 4.56; S, 3.91. ESI mass (HRMS) m/z [ion]: calcd 795.8504 [M − TFA + H]+. Found 795.8481. 1H NMR (CDCl3, 400 MHz): δH 3.16, 3.20 (each s, 2 × 3H, (CH3)2S(O)), 6.32 (s, 1H, NH), 6.79 (t, JHH = 7.8 Hz, 1H, ArH), 7.17–7.24 (m, 4H, ArH), 7.37–7.44 (m, 4H, ArH (3H), NH (1H)), 7.68, 7.70 (each d, JHH = 3.2 Hz, 2 × 1H, ArH), 7.88 (d, JHH = 8.4 Hz, 1H, ArH). 13C{1H} NMR (CDCl3, 100.5 MHz): δC 44.6, 46.1, 110.1, 110.8, 116.9 (q, JC–F = 292.0 Hz), 122.3, 122.5, 124.6, 128.5, 128.8, 128.9, 129.0, 129.2, 129.5, 130.2, 133.3, 133.5, 134.2, 134.3, 138.0, 141.0, 148.5, 161.6 (q, JC–F = 36.3 Hz). 19F{1H} NMR (CDCl3, 376.31 MHz): δF −74.4. 195Pt{1H} NMR (CDCl3, 85.78 MHz): δPt −3591.
Platinacycle 9
Platinacycle 9 was prepared from cis-[Pt(TFA)2(S(O)Me2)2] (50.0 mg, 0.087 mmol) and the guanidine (ArNH)2CNAr (Ar = 4-FC6H4; 30.0 mg, 0.087 mmol) in toluene (10 mL) by following the procedure previously mentioned for 6. Platinacycle 9 was obtained as colourless crystals in 78% (49.4 mg, 0.068 mmol) yield. Mp: 227.7 °C. ATR-IR (cm−1): ν(NH) 3314 (m); νa(OCO) 1686 (s); ν(CN) 1618 (m); νs(OCO) 1343 (m); ν(SO) 1113 (s). Anal. calcd for C23H19N3O3F6SPt (Mw = 726.56): C, 38.02; H, 2.64; N, 5.78; S, 4.41. Found: C, 38.41; H, 3.01; N, 6.17; S, 4.78. ESI mass (HRMS) m/z [ion]: calcd: 613.0849 [M − TFA]+. Found: 613.0913. 1H NMR (CDCl3, 400 MHz): δH 3.16 (br, 6H, (CH3)2S(O)), 6.39–6.42 (m, 1H, ArH), 6.46 (s, 1H, NH), 6.73 (dt, JHH = 8.0 Hz; 2.8 Hz, 1H, ArH), 7.07–7.22 (m, 9H, ArH (8H), NH (1H)), 7.69 (dd, JHH = 10.6 Hz; 2.6 Hz, 1H, ArH). 13C{1H} NMR (CDCl3, 100.5 MHz): δC 45.42, 111.01 (d, JC–F = 6.8 Hz), 112.11 (d, JC–F = 23.1 Hz), 115.70 (d, JC–F = 8.6 Hz), 116.90 (q, JC–F = 290.5 Hz), 116.91 (d, JC–F = 23.1 Hz), 117.66 (d, JC–F = 23.1 Hz), 124.43 (d, JC–F = 21.2 Hz), 128.45 (d, JC–F = 8.7 Hz), 128.70 (d, JC–F = 7.6 Hz), 131.11, 133.53, 138.35, 149.17, 158.52 (d, JC–F = 244.6 Hz), 160.54 (d, JC–F = 62.6 Hz), 161.75 (q, JC–F = 36.6 Hz), 163.02 (d, JC–F = 65.5 Hz). 19F{1H} NMR (CDCl3, 376.31 MHz): δF −74.6, −111.6, −113.8, −118.3. 195Pt{1H} NMR (CDCl3, 85.78 MHz): δPt −3622.
Platinacycle 10
Platinacycle 10 (50.0 mg, 0.073 mmol) and acetylacetone (8.7 mg, 0.0087 mmol) were dispersed in a freshly distilled acetonitrile (15 mL) in a 25 mL RB-flask and the flask was fitted with an air condenser. K2CO3 (12.1 mg, 0.087 mmol) was added to the reaction mixture, the contents in the flask were simultaneously stirred and heated to 75 °C for 36 h. Subsequently, the reaction mixture was cooled and the volatiles were completely removed under vacuum to afford yellow solid. To the solid, CH2Cl2 (10 mL) was added and filtered using a Whatman filter paper. The filtrate was concentrated under vacuum to about 2 mL, layered with toluene (2 mL) and stored at RT for 10 days to afford 10 as green crystals suitable for SCXRD. Yield = 92% (44.9 mg, 0.067 mmol). Mp: 206.0 °C. Anal. calcd for C27H29O5N3Pt (MW = 670.63): C, 48.36; H, 4.36; N, 6.27. Found: C, 48.53; H, 3.95; N, 6.25. ATR-IR (cm−1): ν(N–H) 3382 (m), ν(CN) 1624 (m), ν(C–O) (acac(1−)) 1572 (s), ν(C–O) (acac(1−)) 1546 (s). ESI mass (HRMS) m/z [ion]: calcd: 671.1833 [M + H]+. Found: 671.1804. 1H NMR (CDCl3, 400 MHz): δH 1.36, 1.84 (each s, 2 × 3H, CH3, acac(1−)), 3.74, 3.81, 3.95 (each s, 3 × 3H, OCH3), 5.20 (s, 1H, CH, acac(1−)), 6.59 (dd, JHH = 8.0 Hz; 1.2 Hz, 1H, ArH), 6.84 (s, 1H, NH), 6.88–6.96 (m, 5H, ArH), 7.09 (dt, JHH = 8.0 Hz; 1.3 Hz, 1H, ArH), 7.18 (dt, JHH = 7.8 Hz; 1.6 Hz, 1H, ArH), 7.27 (dd, JHH = 8.2 Hz; 1.8 Hz, 1H, ArH), 7.33 (dt, JHH = 6.4 Hz; 1.6 Hz, 2H, ArH), 8.05 (s, 1H, NH). 13C{1H} NMR (CDCl3, 100.5 MHz): δC 27.0, 27.1, 55.5, 55.9, 56.2, 101.3, 105.1, 111.1, 111.5, 111.7, 121.0, 121.7, 125.1, 126.3, 127.2, 127.3, 130.2, 133.8, 143.8, 145.0, 150.8, 155.0, 183.2, 184.6.
Platinacycle 11
Platinacycle 11 was prepared from 2 (50.0 mg, 0.070 mmol), acetylacetone (8.4 mg, 0.084 mmol) and K2CO3 (11.6 mg, 0.084 mmol) in acetonitrile (15 mL) and purified as described previously for platinacycle 10. The solid obtained after removal of the volatiles from the reaction mixture was dissolved in CH2Cl2 (2 mL), layered with toluene (2 mL) and stored at RT for 7 days to afford 11 as bright yellow crystals suitable for SCXRD. Yield = 94% (41.1 mg, 0.066 mmol). Mp: 236.2 °C. Anal. calcd for C27H29O2N3Pt (MW = 622.19): C, 52.08; H, 4.69; N, 6.75. Found: C, 52.09; H, 4.69; N, 6.75. ATR-IR (cm−1): ν(N–H) 3404 (m), ν(CN) 1624 (m), ν(C–O) (acac(1−)) 1574 (s), ν(C–O) (acac(1−)) 1541 (s). ESI mass (HRMS) m/z [ion]: calcd: 623.1986 [M + H]+. Found: 626.1814. 1H NMR (CDCl3, 400 MHz): δH 1.35, 1.70 (each s, 2 × 3H, CH3, acac(1−)), 1.85, 2.19, 2.42 (each s, 3 × 3H, CH3), 5.22 (s, 1H, CH, acac(1−)), 5.69, 6.54 (each s, 2 × 1H, NH), 6.74 (d, JHH = 7.2 Hz, 1H, ArH), 6.81 (t, JHH = 7.6 Hz, 1H, ArH), 7.15–7.24 (m, 4H, ArH), 7.27–7.32 (m, 4H, ArH), 7.65 (d, JHH = 7.6 Hz, 1H, ArH). 13C{1H} NMR (CDCl3, 100.5 MHz): δC 17.1, 17.9, 18.2, 26.8, 27.1, 101.3, 108.5, 119.3, 120.8, 125.6, 126.7, 126.8, 127.9, 128.2, 128.4, 128.8, 130.6, 131.9, 132.4, 134.5, 135.6, 136.1, 136.3, 142.2, 144.1, 183.3, 184.9.
Platinacycle 12
Platinacycle 12 was prepared from 3 (50.0 mg, 0.070 mmol), acetylacetone (8.4 mg, 0.084 mmol) and K2CO3 (11.6 mg, 0.084 mmol) in acetonitrile (15 mL) and purified as described previously for platinacycle 10. The solid obtained after removal of the volatiles from the reaction mixture was dissolved in CH2Cl2 (2 mL), layered with toluene (2 mL) and stored at RT for 7 days to afford 12 as yellow crystals suitable for SCXRD. Yield = 89% (38.6 mg, 0.062 mmol). Mp: 166.9 °C. Anal. calcd for C27H29O2N3Pt (MW = 622.63): C, 52.08; H, 4.69; N, 6.75. Found: C, 52.35; H, 4.89; N, 6.98. ATR-IR (cm−1): ν(N–H) 3399 (m), ν(CN) 1624 (m), ν(C–O) (acac(1−)) 1574 (s), ν(C–O) (acac(1−)) 1510 (s). ESI mass (HRMS) m/z [ion]: calcd: 623.1986 [M + H]+. Found: 623.1956. 1H NMR (CDCl3, 400 MHz): δH 1.45, 1.87 (each s, 2 × 3H, CH3, acac(1−)), 2.29 (s, 3H, CH3), 2.35 (s, 2 × 3H, CH3), 5.24 (s, 1H, CH, acac(1−)), 5.94 (s, 1H, NH), 6.26 (d, JHH = 8.0 Hz, 1H, ArH), 6.50 (s, 1H, NH), 6.73 (d, JHH = 8.0 Hz, 1H, ArH), 6.96 (d, JHH = 8.0 Hz, 2H, ArH), 7.15–7.20 (m, 6H, ArH), 7.51 (s, 1H, ArH). 13C{1H} NMR (CDCl3, 100.5 MHz): δC 21.0, 21.1, 21.3, 27.1, 27.2, 101.4, 109.6, 112.9, 124.4, 125.3, 128.0, 129.5, 130.6, 130.8, 133.9, 134.0, 134.8, 135.7, 135.9, 137.0, 141.1, 145.4, 183.1, 184.8.
Platinacycle 13
Platinacycle 13 was prepared from 4 (50.0 mg, 0.066 mmol), acetylacetone (7.9 mg, 0.079 mmol) and K2CO3 (11.2 mg, 0.079 mmol) in acetonitrile (15 mL) and purified as described previously for platinacycle 10. The solid obtained after removal of the volatiles from the reaction mixture was dissolved in CH2Cl2 (2 mL), layered with toluene (2 mL) and stored at RT for 5 days to afford 13·0.5 toluene as yellow crystals suitable for SCXRD. Yield = 83% (36.9 mg, 0.055 mmol). Mp: 152.4 °C. Anal. calcd for C30H35O2N3Pt (MW = 664.71): C, 54.21; H, 5.31; N, 6.32. Found: C, 54.45; H, 5.66; N, 6.50. ATR-IR (cm−1): ν(N–H) 3406 (m), ν(CN) 1620 (m), ν(C–O) (acac(1−)) 1574 (s), ν(C–O) (acac(1−)) 1512 (s). ESI mass (HRMS) m/z [ion]: calcd: 665.2455 [M + H]+. Found: 665.2456. 1H NMR (CDCl3, 400 MHz): δH 1.40, 1.69 (each s, 2 × 3H, CH3, acac(1−)), 1.86, 2.14, 2.24, 2.32, 2.33, 2.36 (each s, 6 × 3H, CH3), 5.22 (s, 1H, CH, acac(1−)), 5.65, 6.45 (each s, 2 × 1H, NH), 6.55 (s, 1H, ArH), 7.02–7.07 (m, 4H, ArH), 7.10 (s, 1H, ArH), 7.14 (d, JHH = 7.6 Hz, 1H, ArH), 7.42 (s, 1H, ArH). 13C{1H} NMR (CDCl3, 100.5 MHz): δC 17.1, 17.9, 18.2, 21.1, 21.2, 27.0, 27.2, 101.2, 108.6, 119.0, 126.6, 127.1, 128.1, 128.3, 128.4, 129.6, 131.2, 131.8, 132.5, 132.8, 133.6, 135.7, 136.0, 136.1, 138.7, 139.6, 144.6, 183.1, 184.8.
Platinacycle 14
Platinacycle 14 was prepared from 5 (50.0 mg, 0.066 mmol), acetylacetone (7.9 mg, 0.079 mmol) and K2CO3 (11.2 mg, 0.079 mmol) in acetonitrile (15 mL) and purified as described previously for platinacycle 10. The solid obtained after removal of the volatiles from the reaction mixture was dissolved in CH2Cl2 (2 mL), layered with toluene (2 mL) and stored at RT for 9 days to afford 14 as yellow crystals suitable for SCXRD. Yield = 86% (37.8 mg, 0.057 mmol). Mp: 190.4 °C. Anal. calcd for C30H35O2N3Pt (MW = 664.71): C, 54.21; H, 5.31; N, 6.32. Found: C, 54.57; H, 5.62; N, 6.27. ATR-IR (cm−1): ν(N–H) 3417 (m), ν(CN) 1631 (m), ν(C–O) (acac(1−)) 1613 (s), ν(C–O) (acac(1−)) 1582 (s). ESI mass (HRMS) m/z [ion]: calcd: 665.2455 [M + H]+. Found: 665.2467. 1H NMR (CDCl3, 400 MHz): δH 1.60, 1.70 (each s, 2 × 3H, CH3, acac(1−)), 1.71, 2.09, 2.25, 2.29, 2.34, 2.64 (each s, 6 × 3H, CH3), 5.23 (s, 1H, CH, acac(1−)), 5.81, 6.35 (each s, 2 × 1H, NH), 6.58, 6.68 (each d, JHH = 7.2 Hz, 2 × 1H, ArH), 6.91 (s, 1H, ArH), 6.97 (d, JHH = 6.8 Hz, 1H, ArH), 7.04 (d, JHH = 8.0 Hz, 1H, ArH), 7.14 (t, JHH = 7.0 Hz, 2H, ArH), 7.20 (s, 1H, ArH). 13C{1H} NMR (CDCl3, 100.5 MHz): δC 16.6, 17.5, 17.9, 20.8, 21.1, 23.7, 26.2, 27.7, 101.3, 113.1, 117.2, 124.3, 125.5, 127.4, 128.2, 128.9, 129.1, 130.9, 131.6, 132.2, 134.5, 136.0, 137.7, 138.4, 141.6, 144.3, 148.3, 182.0, 184.4.
Platinacycle 15
Platinacycle 15 was prepared from 6 (50.0 mg, 0.069 mmol), acetylacetone (8.3 mg, 0.083 mmol) and K2CO3 (11.5 mg, 0.083 mmol) in acetonitrile (15 mL) and purified as described previously for platinacycle 10. The solid obtained after removal of the volatiles from the reaction mixture was dissolved in CH2Cl2 (2 mL), layered with toluene (2 mL) and stored at RT for 5 days to afford 15 as green crystals suitable for SCXRD. Yield = 81% (35.5 mg, 0.056 mmol). Mp: 201.8 °C. Anal. calcd for C24H20F3O2N3Pt (MW = 634.52): C, 45.43; H, 3.18; N, 6.62. Found: C, 45.69; H, 3.09; N, 6.25. ATR-IR (cm−1): ν(N–H) 3406 (m), ν(CN) 1630 (m), ν(C–O) (acac(1−)) 1581 (s), ν(C–O) (acac(1−)) 1544 (s). ESI mass (HRMS) m/z [ion]: calcd: 635.1234 [M + H]+. Found: 635.1238. 1H NMR (CDCl3, 400 MHz): δH 1.42, 1.88 (each s, 2 × 3H, CH3, acac(1−)), 5.26 (s, 1H, CH, acac(1−)), 5.93 (s, 1H, NH), 6.72–6.77, 6.89–6.93 (each m, 2 × 1H, ArH), 7.14–7.24 (m, 5H, ArH (4H), NH (1H)), 7.27–7.39 (m, 4H, ArH), 7.51 (d, JHH = 8.0 Hz, 1H, ArH). 13C{1H} NMR (CDCl3, 100.5 MHz): δC 26.8, 27.0, 101.6, 109.1 (d, JC–F = 18.3 Hz), 112.6, 116.1 (d, JC–F = 20.2 Hz), 117.2 (d, JC–F = 19.3 Hz), 121.6 (d, JC–F = 6.7 Hz), 124.2 (d, JC–F = 12.5 Hz), 124.5 (d, JC–F = 2.9 Hz), 125.2 (d, JC–F = 5.7 Hz), 125.6 (d, JC–F = 3.8 Hz), 126.2, 128.5 (d, JC–F = 7.7 Hz), 128.7 (d, JC–F = 7.7 Hz), 129.0 (d, JC–F = 1.9 Hz), 130.3, 130.8 (d, JC–F = 13.6 Hz), 143.7, 149.4 (d, JC–F = 244.7 Hz), 155.9 (d, JC–F = 185.8 Hz), 158.4 (d, JC–F = 184.9 Hz), 183.6, 185.2. 19F{1H} NMR (CDCl3, 376.31 MHz): δF −137.4 (JPt–F = 40.3 Hz), −123.6, −120.6.
Platinacycle 16
Platinacycle 16 was prepared from 7 (50.0 mg, 0.064 mmol), acetylacetone (7.7 mg, 0.077 mmol) and K2CO3 (10.6 mg, 0.077 mmol) in acetonitrile (15 mL) and purified as described previously for platinacycle 10. The solid obtained after removal of the volatiles from the reaction mixture was dissolved in CH2Cl2 (2 mL), layered with toluene (2 mL) and stored at RT for 5 days to afford 16 as green crystals suitable for SCXRD. Yield = 94% (40.9 mg, 0.060 mmol). Mp: 228.0 °C. Anal. calcd for C24H20Cl3O2N3Pt (MW = 683.88): C, 42.15; H, 2.95; N, 6.14. Found: C, 42.45; H, 2.98; N, 6.21. ATR-IR (cm−1): ν(N–H) 3368 (m); ν(CN) 1628 (m); ν(C–O) (acac(1−)) 1576 (s); ν(C–O) (acac(1−)) 1516 (s). ESI mass (HRMS) m/z [ion] calcd: 683.0347 [M + H]+. Found: 683.0416. 1H NMR (CDCl3, 400 MHz): δH1.41, 1.88 (each s, 2 × 3H, CH3, acac(1−)), 5.26 (s, 1H, CH, acac(1−)), 6.01 (s, 1H, NH), 6.87 (t, JHH = 7.8 Hz, 1H, ArH), 7.03 (dd, JHH = 7.8 Hz; 1.4 Hz, 1H, ArH), 7.23–7.25 (m, 2H, ArH (1H), NH (1 H)), 7.34 (dq, JHH = 8.0 Hz; 1.5 Hz, 2H, ArH), 7.40–7.49 (m, 4H, ArH), 7.69–7.72 (m, 2H, ArH). 13C{1H} NMR (CDCl3, 100.5 MHz): δC 26.7, 27.0, 101.6, 111.6, 117.9, 122.1, 124.0, 126.3, 127.5, 128.1, 128.5, 129.5, 129.8, 129.9, 130.9, 132.5, 132.6, 132.8, 133.2, 140.6, 143.6, 183.5, 185.1.
Platinacycle 17
Platinacycle 17 was prepared from 8 (50.0 mg, 0.055 mmol), acetylacetone (6.6 mg, 0.066 mmol) and K2CO3 (9.1 mg, 0.066 mmol) in acetonitrile (15 mL) and purified as described previously for platinacycle 10. The solid obtained after removal of the volatiles from the reaction mixture was dissolved in CH2Cl2 (2 mL), layered with toluene (2 mL) and stored at RT for 4 days to afford 17 as yellow crystals suitable for SCXRD. Yield = 84% (37.4 mg, 0.046 mmol). Mp: 244.9 °C. Anal. calcd for C24H20Br3N3O2Pt (MW = 813.88): C, 35.27; H, 2.47; N, 5.14. Found: C, 35.48; H, 2.81; N, 5.47. ATR-IR (cm−1): ν(N–H) 3350 (m); ν(CN) 1626 (m); ν(C–O) (acac(1−)) 1574 (s); ν(C–O) (acac(1−)) 1520 (s). 1H NMR (CDCl3, 400 MHz): δH 1.41, 1.88 (each s, 2 × 3H, CH3, acac(1−)), 5.26 (s, 1H, CH, acac(1−)), 5.95 (s, 1H, NH), 6.79 (t, JHH = 7.6 Hz, 1H, ArH), 7.14–7.20 (m, 3H, ArH), 7.36–7.48 (m, 4H, ArH (3H), NH (1H)), 7.65 (dt, JHH = 8.6 Hz; 1.6 Hz, 3H, ArH), 7.75 (dd, JHH = 7.6 Hz; 1.2 Hz, 1H, ArH). 13C{1H} NMR (CDCl3, 100.5 MHz): δC 26.7, 27.1, 101.6, 108.6, 111.7, 120.5, 122.6, 123.3, 127.2, 127.3, 128.1, 128.3, 128.6, 129.3, 129.9, 133.0, 133.3, 133.5, 134.1, 134.4, 142.0, 143.9, 183.5, 185.1.
Platinacycle 18
Platinacycle 18 was prepared from 9 (50.0 mg, 0.069 mmol), acetylacetone (8.2 mg, 0.082 mmol) and K2CO3 (11.3 mg, 0.082 mmol) in acetonitrile (15 mL) and purified as described previously for platinacycle 10. The solid obtained after removal of the volatiles from the reaction mixture was dissolved in CH2Cl2 (2 mL), layered with toluene (2 mL) and stored at RT for 3 days to afford 18 as yellow crystals suitable for SCXRD. Yield = 86% (37.4 mg, 0.059 mmol). Mp: 260.1 °C. Anal. calcd for C24H20F3O2N3Pt (MW = 634.52): C, 45.43; H, 3.18; N, 6.62. Found: C, 45.63; H, 3.33; N, 7.01. ATR-IR (cm−1): ν(N–H) 3404.36 (m); ν(CN) 1624 (m); ν(C–O) (acac(1−)) 1578 (s); ν(C–O) (acac(1−)) 1503 (s). ESI mass (HRMS) m/z [ion] calcd: 635.1234 [M + H]+. Found: 635.1298. Platinacycle 18 exists in two isomeric forms with their approximate ratios being 1:0.07 as estimated from integrals of methyl protons of the acac(1−) ligand. 1H NMR (CDCl3, 400 MHz): δH 1.46 (s, 2 × 3H, CH3, acac(1−), isomers 1 & 2), 1.87 (s, 3H, CH3, acac(1−), isomer 2), 1.89 (s, 3H, CH3, acac(1−), isomer 1), 5.26 (s, 1H, CH, acac(1−), isomer 2), 5.27 (s, 1H, CH, acac(1−), isomer 1), 5.84 (s, 2 × 1H, NH, isomers 1 & 2), 6.30 (dd, JHH = 8.6 Hz; 5.0 Hz, 2 × 1H, ArH, isomers 1 & 2), 6.43 (s, 2 × 1H, NH, isomers 1 & 2), 6.63 (dt, JHH = 7.6 Hz; 2.9 Hz, 2 × 1H, ArH, isomers 1 & 2), 7.06–7.14 (m, 2 × 6H, ArH, isomers 1 & 2), 7.25–7.28 (m, 2 × 2H, ArH, isomers 1 & 2), 7.45 (dd, JHH = 10.8 Hz; 3.0 Hz, 2 × 1H, ArH, isomers 1 & 2). 13C{1H} NMR (CDCl3, 100.5 MHz): δC 27.1, 101.7, 110.0 (d, JC–F = 24.1 Hz), 112.2 (d, JC–F = 5.7 Hz), 114.2 (d, JC–F = 7.7 Hz), 115.8 (d, JC–F = 22.1 Hz), 117.4 (d, JC–F = 22.2 Hz), 119.6 (d, JC–F = 19.3 Hz), 127.9 (d, JC–F = 8.6 Hz), 129.7 (d, JC–F = 8.6 Hz), 132.0, 133.8, 139.2, 145.1, 157.3 (d, JC–F = 241.7 Hz), 160.2 (d, JC–F = 42.4 Hz), 162.6 (d, JC–F = 45.3 Hz), 183.6, 185.2. 19F{1H} NMR (CDCl3, 376.31 MHz): δF −121.4 (isomers 1 & 2), −115.8 (isomer 2), −115.6 (isomer 1), −113.1 (isomer 2), −112.9 (isomer 1).
Platinacycle 19
Platinacycle 19 was prepared from 1 (50.0 mg, 0.073 mmol), 2-picolinic acid (10.8 mg, 0.088 mmol) and K2CO3 (12.1 mg, 0.088 mmol) in acetonitrile (15 mL) and purified as described previously for platinacycle 10. The solid obtained after removal of the volatiles from the reaction mixture was dissolved in CH2Cl2 (2 mL), layered with toluene (2 mL) and stored at RT for 2 days to afford 19 as green crystals suitable for SCXRD. Yield = 89% (45.1 mg, 0.065 mmol). Mp: 248.3 °C. Anal. calcd for C28H26O5N4Pt (MW = 693.62): C, 48.49; H, 3.78; N, 8.08. Found: C, 48.44; H, 3.68; N, 7.96. ATR-IR (cm−1): ν(N–H) 3385 (m), ν(>CO) 1657 (s), ν(CN) 1611 (m). ESI mass (HRMS) m/z [ion]: calcd: 694.1629 [M + H]+, Found: 694.1670; calcd: 1387.3237 {[Pt(TAG)(μ2-N,O-pic)]2 + H}, Found: 1387.3265. 1H NMR (CDCl3, 400 MHz): δH 3.80, 3.82, 4.02 (each s, 3 × 3H, OCH3), 6.68 (d, JHH = 8.4 Hz, 1H, ArH), 6.90–7.06 (m, 6H, ArH), 7.11–7.23 (m, 4H, ArH), 7.27–7.33 (m, 2H, ArH), 7.87 (s, 1H, NH), 7.89–7.96 (m, 2H, ArH (1H), NH (1H)), 8.72 (d, JHH = 5.6 Hz, 1H, ArH). 13C{1H} NMR (CDCl3, 100.5 MHz): δC 55.4, 55.9, 56.6, 105.5, 111.6, 112.7, 117.3, 120.9, 121.5, 122.4, 123.5, 126.0, 126.5, 126.9, 127.4, 128.0, 128.2, 129.4, 129.6, 133.6, 137.5, 145.8, 147.8, 148.9, 151.3, 154.0, 154.5, 172.3.
Author contributions
Project conceptualization, secured funding and supervision (NT), syntheses, characterization including SCXRD (VT), emission measurements (MA), emission data analyses (GVP), DFT and TD-DFT calculations (JMT), DFT and TD-DFT analyses (CS). The manuscript was written with contributions from all authors.Conflicts of interest
There is no conflicts to declare.Acknowledgements
We acknowledge SERB for research grant (EMR/2014/000698) and one of us (V. T.) acknowledges UGC for a fellowship. University Science Instrumentation Center, University of Delhi is acknowledged for infrastructural facilities. We thank Dr Nitish Kumar Sinha for some experimental assistance related to complexes 6 and 13⋅0.5 toluene.References
- (a) J. A. Gareth Williams, S. Develay, D. L. Rochester and L. Murphy, Coord. Chem. Rev., 2008, 252, 2596–2611 CrossRef CAS; (b) J. Kalinowski, V. Fattori, M. Cocchi and J. A. Gareth Williams, Coord. Chem. Rev., 2011, 255, 2401–2425 CrossRef CAS; (c) X. Yang, C. Yao and G. Zhou, Platinum Met. Rev., 2013, 57, 2–16 CrossRef CAS; (d) T. Fleetham and J. Li, J. Photonics Energy, 2014, 4, 040991 CrossRef CAS; (e) S. Huo, J. Carroll and D. A. K. Vezzu, Asian J. Org. Chem., 2015, 4, 1210–1245 CrossRef CAS; (f) K. Li, G. S. Ming Tong, Q. Wan, G. Cheng, W.-Y. Tong, W.-H. Ang, W.-L. Kwong and C.-M. Che, Chem. Sci., 2016, 7, 1653–1673 RSC; (g) C. Cebrián and M. Mauro, Beilstein J. Org. Chem., 2018, 14, 1459–1481 CrossRef PubMed; (h) X. Wang and S. Wang, Chem. Rec., 2019, 19, 1693–1709 CrossRef CAS PubMed; (i) J. Herberger and R. F. Winter, Coord. Chem. Rev., 2019, 400, 213048 CrossRef; (j) A. Haque, H. El Moll, K. M. Alenezi, M. S. Khan and W.-Y. Wong, Materials, 2021, 14, 4236 CrossRef CAS PubMed.
- J. Brooks, Y. Babayan, S. Lamansky, P. I. Djurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055–3066 CrossRef CAS PubMed.
- J. C.-H. Chan, W. H. Lam, H.-L. Wong, N. Zhu, W.-T. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2011, 133, 12690–12705 CrossRef CAS PubMed.
- D. N. Kozhevnikov, V. N. Kozhevnikov, M. M. Ustinova, A. Santoro, D. W. Bruce, B. Koenig, R. Czerwieniec, T. Fischer, M. Zabel and H. Yersin, Inorg. Chem., 2009, 48, 4179–4189 CrossRef CAS PubMed.
- A. Bossi, A. F. Rausch, M. J. Leitl, R. Czerwieniec, M. T. Whited, P. I. Djurovich, H. Yersin and M. E. Thompson, Inorg. Chem., 2013, 52, 12403–12415 CrossRef CAS PubMed.
- O. J. Stacey, B. D. Ward, S. J. Coles, P. N. Horton and S. J. A. Pope, Dalton Trans., 2016, 45, 10297–10307 RSC.
- M. Z. Shafikov, D. N. Kozhevnikov, M. Bodensteiner, F. Brandl and R. Czerwieniec, Inorg. Chem., 2016, 55, 7457–7466 CrossRef CAS PubMed.
- Y.-J. Cho, S.-Y. Kim, H.-J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2017, 19, 5486–5494 RSC.
- N. Okamura, T. Maeda, H. Fujiwara, A. Soman, K. N. Narayanan Unni, A. Ajayaghosh and S. Yagi, Phys. Chem. Chem. Phys., 2018, 20, 542–552 RSC.
- Y. Zhou, J. Jia, L. Cai and Y. Huang, Dalton Trans., 2018, 47, 693–699 RSC.
- P.-H. Lanoë, A. Moreno-Betancourt, L. Wilson, C. Philouze, C. Monnereau, H. Jamet, D. Jouvenot and F. Loiseau, Dyes Pigm., 2019, 162, 967–977 CrossRef.
- A. F. Henwood, J. Webster, D. Cordes, A. M. Z. Slawin, D. Jacquemin and E. Zysman-Colman, RSC Adv., 2017, 7, 25566–25574 RSC.
- (a) X. Mou, Y. Wu, S. Liu, M. Shi, X. Liu, C. Wang, S. Sun, Q. Zhao, X. Zhou and W. Huang, J. Mater. Chem., 2011, 21, 13951–13962 RSC; (b) C.-H. Chen, F.-I. Wu, Y.-Y. Tsai and C.-H. Cheng, Adv. Funct. Mater., 2011, 21, 3150–3158 CrossRef CAS.
- W. Zeng, M.-J. Sun, Z.-L. Gong, J.-Y. Shao, Y.-W. Zhong and J. Yao, Inorg. Chem., 2020, 59, 11316–11328 CrossRef CAS PubMed.
- (a) K. Venkatesan, P. H. J. Kouwer, S. Yagi, P. Müller and T. M. Swager, J. Mater. Chem., 2008, 18, 400–407 RSC; (b) A. Santoro, A. C. Whitwood, J. A. Gareth Williams, V. N. Kozhevnikov and D. W. Bruce, Chem. Mater., 2009, 21, 3871–3882 CrossRef CAS; (c) T. Sato, H. Awano, O. Haba, H. Katagiri, Y.-J. Pu, T. Takahashi and K. Yonetake, Dalton Trans., 2012, 41, 8379–8389 RSC; (d) M. Spencer, A. Santoro, G. R. Freeman, Á. Díez, P. R. Murray, J. Torroba, A. C. Whitwood, L. J. Yellowlees, J. A. Gareth Williams and D. W. Bruce, Dalton Trans., 2012, 41, 14244–14256 RSC.
- W. Wu, C. Cheng, W. Wu, H. Guo, S. Ji, P. Song, K. Han, J. Zhao, X. Zhang, Y. Wu and G. Du, Eur. J. Inorg. Chem., 2010, 2010, 4683–4696 CrossRef.
- C. Liu, X. Song, Z. Wang and J. Qiu, ChemPlusChem, 2014, 79, 1472–1481 CrossRef CAS.
- C. Liu, X. Song, X. Rao, Y. Xing, Z. Wang, J. Zhao and J. Qiu, Dyes Pigm., 2014, 101, 85–92 CrossRef CAS.
- (a) Y. Xing, C. Liu, J.-H. Xiu and J.-Y. Li, Inorg. Chem., 2015, 54, 7783–7790 CrossRef CAS PubMed; (b) Y. Xing, C. Liu, X. Song and J. Li, J. Mater. Chem. C, 2015, 3, 2166–2174 RSC.
- A. S. Ionkin, W. J. Marshall and Y. Wang, Organometallics, 2005, 24, 619–627 CrossRef CAS.
- H. Li, W. Yuan, X. Wang, H. Zhan, Z. Xie and Y. Cheng, J. Mater. Chem. C, 2015, 3, 2744–2750 RSC.
- A. Heil and C. M. Marian, Inorg. Chem., 2019, 58, 6123–6136 CrossRef CAS PubMed.
- Z. M. Hudson, C. Sun, M. G. Helander, Y.-L. Chang, Z.-H. Lu and S. Wang, J. Am. Chem. Soc., 2012, 134, 13930–13933 CrossRef CAS PubMed.
- T. Strassner, Acc. Chem. Res., 2016, 49, 2680–2689 CrossRef CAS PubMed.
- M. Micksch, M. Tenne and T. Strassner, Organometallics, 2014, 33, 3464–3473 CrossRef CAS.
- W. Wu, H. Guo, W. Wu, S. Ji and J. Zhao, Inorg. Chem., 2011, 50, 11446–11460 CrossRef CAS PubMed.
- A. Caubet, C. Lopez, X. Solans and M. Font-Bardia, J. Organomet. Chem., 2003, 669, 164–171 CrossRef CAS.
- Y. Yu. Scaffidi-Domianello, A. A. Nazarov, M. Haukka, M. Galanski, B. K. Keppler, J. Schneider, P. Du, R. Eisenberg and V. Yu. Kukushkin, Inorg. Chem., 2007, 46, 4469–4482 CrossRef CAS PubMed.
- S. U. Pandya, K. C. Moss, M. R. Bryce, A. S. Batsanov, M. A. Fox, V. Jankus, H. A. Al Attar and A. P. Monkman, Eur. J. Inorg. Chem., 2010, 2010, 1963–1972 CrossRef.
- P. Elumalai, N. Thirupathi and M. Nethaji, Inorg. Chem., 2013, 52, 1883–1894 CrossRef CAS PubMed.
- V. Mishra and N. Thirupathi, ACS Omega, 2018, 3, 6075–6090 CrossRef CAS PubMed.
- N. K. Sinha, V. Mishra and N. Thirupathi, Inorg. Chem., 2023, 62, 7644–7661 CrossRef CAS PubMed.
- Z. M. Hudson, B. A. Blight and S. Wang, Org. Lett., 2012, 14, 1700–1703 CrossRef CAS PubMed.
- D. Poveda, A. Vivancos, D. Bautista and P. Gonzalez-Herrero, Inorg. Chem., 2023, 62, 6207–6213 CrossRef CAS PubMed.
- V. Thakur and N. Thirupathi, J. Organomet. Chem., 2020, 911, 121138 CrossRef CAS.
- R. Ujjval, M. Deepa, J. M. Thomas, C. Sivasankar and N. Thirupathi, Organometallics, 2020, 39, 3663–3678 CrossRef CAS.
- V. Mishra, N. K. Sinha and N. Thirupathi, Inorg. Chem., 2021, 60, 3879–3892 CrossRef CAS PubMed.
- V. Thakur and N. Thirupathi, J. Organomet. Chem., 2022, 959, 122200 CrossRef CAS.
- (a) J. R. L. Priqueler, I. S. Butler and F. D. Rochon, Appl. Spectrosc. Rev., 2006, 41, 185–226 CrossRef CAS; (b) B. M. Still, P. G. A. Kumar, J. R. Aldrich-Wright and W. S. Price, Chem. Soc. Rev., 2007, 36, 665–686 RSC.
- O. J. Stacey, J. A. Platts, S. J. Coles, P. N. Horton and S. J. A. Pope, Inorg. Chem., 2015, 54, 6528–6536 CrossRef CAS PubMed.
- J. A. Lowe, O. J. Stacey, P. N. Horton, S. J. Coles and S. J. A. Pope, J. Organomet. Chem., 2016, 805, 87–93 CrossRef CAS.
- E. Lindner, R. Fawzi, H. A. Mayer, K. Eichele and W. Hiller, Organometallics, 1992, 11, 1033–1043 CrossRef CAS.
- S. Fuertes, A. J. Chueca and V. Sicilia, Inorg. Chem., 2015, 54, 9885–9895 CrossRef CAS PubMed.
- (a) M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U.S.A., 2007, 104, 6908–6914 CrossRef CAS PubMed; (b) K. A. Siddiqui and E. R. T. Tiekink, Chem. Commun., 2013, 49, 8501–8503 RSC; (c) M. Baya, U. Belio and A. Martin, Inorg. Chem., 2014, 53, 189–200 CrossRef CAS PubMed; (d) J. Kozelka, in Noncovalent Forces, ed. S. Scheiner, Springer International Publishing, Cham, 2015, vol. 19, pp. 129–158 Search PubMed; (e) A. Pérez-Bitrián, M. Baya, J. M. Casas, A. Martín and B. Menjón, Dalton Trans., 2021, 50, 5465–5472 RSC.
- L. Brammer, M. C. McCann, R. M. Bullock, R. K. McMullan and P. Sherwood, Organometallics, 1992, 11, 2339–2341 CrossRef CAS.
- L. Brammer, J. M. Charnock, P. L. Goggin, R. J. Goodfellow, T. F. Koetzle and A. G. Orpen, J. Chem. Soc., Chem. Commun., 1987, 443–445 RSC.
- W. Yao, O. Eisenstein and R. H. Crabtree, Inorg. Chim. Acta, 1997, 254, 105–111 CrossRef CAS.
- Y. Zhang, J. C. Lewis, R. G. Bergman, J. A. Ellman and E. Oldfield, Organometallics, 2006, 25, 3515–3519 CrossRef CAS.
- Y. Unger, D. Meyer, O. Molt, C. Schildknecht, I. Munster, G. Wagenblast and T. Strassner, Angew. Chem., Int. Ed., 2010, 49, 10214–10216 CrossRef CAS PubMed.
- S. Stipurin, F. Wurl and T. Strassner, Organometallics, 2022, 41, 313–320 CrossRef CAS.
- S. Poirier, F. Rahmani and C. Reber, Dalton Trans., 2017, 46, 5279–5287 RSC.
- S. Poirier, H. Lynn, C. Reber, E. Tailleur, M. Marchivie, P. Guionneau and M. R. Probert, Inorg. Chem., 2018, 57, 7713–7723 CrossRef CAS PubMed.
- L. F. Gildea and J. A. Gareth Williams, Iridium and platinum complexes for OLEDs, in Organic Light-Emitting Diodes: Materials, Devices and Applications, ed. A. Buckley, Woodhead, Cambridge, 2013, pp. 77–113 Search PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- T. H. Dunning Jr and P. J. Hay, in Modern Theoretical Chemistry, ed. H. F. Schaefer III, Plenum, New York, 1976 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 284–298 CrossRef.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- M. T. Cancès, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef.
- M. Cossi, V. Barone, B. Mennucci and J. Tomasi, Chem. Phys. Lett., 1998, 286, 253–260 CrossRef CAS.
- B. Mennucci and J. Tomasi, Chem. Phys., 1997, 106, 5151–5158 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 09, Rev. A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: XYZ files containing the coordinates of the DFT optimised species and TD-DFT related data. CCDC 1887641–1887649. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra00828f |
| This journal is © The Royal Society of Chemistry 2024 |