DOI:
10.1039/D4RA01201A
(Paper)
RSC Adv., 2024,
14, 16170-16193
Ion Trap LC/MS reveals the generation of reactive intermediates in acalabrutinib metabolism: phase I metabolic profiling and bioactivation pathways elucidation†
Received
16th February 2024
, Accepted 14th May 2024
First published on 20th May 2024
Abstract
Acalabrutinib (CALQUENCE; ACB) is a Bruton tyrosine kinase inhibitor (BTKI) used to treat mantle cell lymphoma, small lymphocytic lymphoma (SLL), and chronic lymphocytic leukemia (CLL). On 21 November 2019, ACB was approved by the U.S. FDA for the use as a single therapy for the treatment of CLL/SLL. In silico studies were first done to propose vulnerable sites of metabolism and reactivity pathways by StarDrop software and Xenosite online software; respectively. ACB metabolites and stable adducts were characterized in vitro from rat liver microsomes (RLMs) using Ion Trap LC/MS. Generation of reactive intermediates (RIs) in the in vitro metabolism of ACB was investigated using glutathione, potassium cyanide, and methoxylamine as trapping nucleophiles for the RIs including iminopyridinone, iminium, and aldehyde, respectively, to form stable adducts that can be identified and characterized by Ion Trap LC/MS. Five phase I metabolites, seven 6-iminopyridin-3(6H)-one and five aldehyde RIs of ACB were identified. Based on literature reviews, the generation of RIs of ACB, and the subsequent drug-induced organ toxicity (DIOT) reactions may provide an explanation of ACB ADRs. Additional drug discovery investigations can be performed to facilitate the creation of novel medications with improved safety characteristics.
1. Introduction
Cancer is the second leading cause of mortality worldwide. The number of cancer-related fatalities in 2020 exceeded 10 million.1 Nevertheless, the administration of anticancer drugs is linked to detrimental consequences in the treatment of cancer. Despite their effectiveness, these treatments are generally abandoned due to the toxicity they induce.2 B-cell lymphomas are the most common types of blood cancers. The treatment of B-cell lymphomas with pharmacological therapy has undergone substantial changes since the introduction of targeted drugs such as Bruton tyrosine kinase inhibitors (BTKI).3 Acalabrutinib (ACB; Fig. 1) is a second-generation inhibitor of BTK that received approval in 2017 under the fast approval pathway of the United States Food and Drug Administration (U.S. FDA). Additionally, it was granted Orphan Drug status, which provides support and incentives for the development of drugs targeting uncommon disorders. It is employed for the treatment of many types of B-cell malignancies, such as mantle cell lymphoma, small lymphocytic lymphoma (SLL), and chronic lymphocytic leukemia (CLL). ACB (CALQUENCE), developed by AstraZeneca company, was granted approval by the U.S. FDA on November 21, 2019, for use as a monotherapy in the treatment of CLL/SLL.4 The development of ACB aimed to enhance the effectiveness and specificity of first BTK inhibitors, such as ibrutinib.5 ACB has a more limited range of kinase inhibition compared to ibrutinib. The molecule creates a covalent link with the cysteine 481 residue located on the binding site of BTK.6,7 Calquence is a pharmaceutical product that was created by AstraZeneca. The medication is taken orally twice a day as part of the treatment plan.8
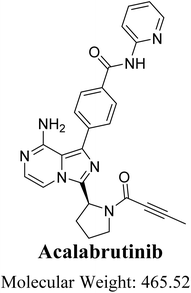 |
| | Fig. 1 Chemical structure of acalabrutinib (4-{8-amino-3-[(2S)-1-(2-butynoyl)-2-pyrrolidinyl]imidazo[1,5-a]pyrazin-1-yl}-N-(2-pyridinyl)benzamide). | |
While ACB is generally regarded as a safer medication in comparison to other anticancer drugs, it is nonetheless linked to numerous adverse effects. The predominant side effects observed in over 20% of patients receiving ACB included thrombocytopenia, anemia, headache, neutropenia, tiredness, diarrhea, myalgia, and bruising. 1.6% of patients experienced dose reductions, while 6.5% of patients had to discontinue treatment owing to adverse effects. The primary deleterious consequences induced by ACB include neutropenia (23%), significant bleeding (3%), thrombocytopenia (7%), severe infections (19%), cardiac arrhythmias (4%), anemia (8%), and lymphopenia (7%).6 In order to identify substances that may have harmful effects, particular focus is placed on structural alerts. These alerts refer to molecular intermediates or fragments that exhibit high chemical reactivity and have the potential to be transformed into highly reactive intermediates (RIs) through metabolic bioactivation. The presentation of the perception aims to minimize the likelihood of anticipated adverse toxic consequences in new pharmacological candidates. The process of metabolically activating small-molecule medicines into electrophilic RIs is commonly recognized as a sign of idiosyncratic adverse drug responses (IADRs).9–11 Enzymes create toxic RIs of medicines. These substances are electrophilic and highly reactive, capable of forming covalent connections with large molecules in the body, leading to harmful consequences. They induce cellular damage and provoke immune-mediated toxicities due to their capacity to attach to proteins and DNA.
Characterizing and identifying RIs is a crucial step in the development of novel drugs with improved toxicological properties.12,13 In vivo, the identification of RIs is not possible due to their exceptionally reactive and unstable characteristics. In vitro, trapping agents are employed to aid in the capture and identification of these intermediates.14 Ion Trap LC/MS is widely regarded as the most effective analytical technique for determining the structure and properties of RIs.15–17 The inherent instability of RIs renders them unlikely to be detected using direct means. Instead, a capturing agent was employed to ensnare the RIs, resulting in the formation of stable adducts that can be analyzed by LC-ITMS.18 The production of RIs was investigated by employing potassium cyanide, glutathione, and methoxylamine as nucleophiles to capture the transient and RIs including iminium, aldehyde, and iminopyridinone species, respectively. This process results in the formation of stable cyanide, oximer, and GSH adducts, which can be analyzed and identified using Ion Trap LC/MS. The chemical structure of ACB contains one chiral center at the pyrrolidine as indicated by the red label on the chemical structure (Fig. 1). Compounds that contain a cyclic tertiary amine ring, such as pyrrolidine, have the ability to create RIs (iminium species) when metabolized. These intermediates can react with nucleophilic macromolecules. The iminium intermediates are classified as hard RIs.19,20 GSH or its derivatives are ineffective in trapping Hard RIs, while potassium cyanide (KCN) is highly successful in trapping them.20 The absence of cyano adducts in the current study can be attributed to the acidic properties of tertiary nitrogen (amidic group), which has been observed in other medications studied previously.21–23 The ACB metabolites were subjected to in silico toxicity evaluations using the DEREK software. The bioactivation theory was validated using the StarDrop software and the XenoSite.
2. Chemicals and methods
2.1. Chemicals
The production of RLMs entailed the utilization of Sprague Dawley rats.24 The substances including formic acid, acetonitrile, methoxylamine, and glutathione were obtained from the Sigma-Aldrich company based in St. Louis, MO, USA. The target analyte; ACB (ACP-196; 99.88%) was obtained from MedChemExpress, a company based in Princeton, NJ, USA. The water utilized was of HPLC grade and sourced from the Milli-Q plus purification system situated at the Billerica, MA, USA facility. The solvents used are of HPLC grade, whereas the chemicals are of analytical grade. The Animal Ethics Committee at King Saud University has authorized the animal research (No. KSU-SE-22-83) after obtaining ethical approval. The animal studies were conducted following the experimental Animal Use and Care Guidelines of the National Institutes of Health and under the supervision of the Animal Studies Committee. The Sprague-Dawley rats were acquired from the experimental animal care facility at King Saud University in Riyadh, Saudi Arabia.
2.2. Chromatographic conditions
The analysis of samples was conducted using a 6320 Ion Trap LC/MS instrument manufactured by Agilent Technologies, located in Palo Alto, CA, USA. The electrospray ionization was conducted under ambient conditions using positive ion mode. The temperature in the dry environment was 350 °C, the pressure of the nebulizer was 60 psi, the temperature of the capillary was 325 °C, and the flow rate of the dry gas was 10 L per minute. The column employed was an Eclipse plus C18 with dimensions of 4.6 mm internal diameter, 150 mm length, and 3.5 μm particle size, manufactured by Agilent Technologies in Palo Alto, CA, USA. The LC separation was performed using a mobile phase consisting of solvent A, which was composed of 0.1% formic acid in water, and solvent B, which was acetonitrile. The run commenced with a mobile phase A concentration of 95%. Subsequently, the proportion of mobile phase B was elevated from 5% to 60% over a duration of 20 minutes. At the 25 minute mark, it was further increased to 65% and maintained at this level until the 30 minute mark. The process of gradient chromatography was carried out using a mobile phase consisting of a mixture of water and acetonitrile. The flow rate of the mobile phase was set at 0.4 mL per minute, and the entire process took a total of 45 minutes. The samples were injected with a volume of 5 μl, which was extracted from vials and loaded into the autosampler. Table S1† summarizes the chromatographic parameters recommended for studying ACB metabolites (ESI file†).
2.3. In silico prediction of ACB metabolites and structural alerts using StarDrop software (P450 and DEREK modules)
The StarDrop software was utilized to forecast the crucial locations where metabolism occurs and to anticipate the level of instability at these sites, as indicated by the composite site lability (CSL). The software includes the P450 module, which enables the prediction of regioselectivity of metabolism by various isoforms. In addition, it generates a pie chart illustrating the predominant CYP450 isoform responsible for ACB metabolism. The DEREK module was utilized to detect structural alarms in ACB that may induce specific toxicities.
2.4. In silico prediction of ACB RIs using XenoSite reactivity model
The XenoSite web predictor offers a variety of modules that aid in the prediction of small molecule biochemistry. It is accessible for free at: https://swami.wustl.edu/xenosite. The XenoSite reactivity module was employed to forecast potential locations of RIs. The software possesses the benefit of a brief execution duration. The SMILES format of ACB was submitted to the XenoSite reactivity module on the online website.
2.5. Preparation of RLMs
Three male Sprague-Dawley rats were obtained from the experimental animal care unit at the College of Pharmacy, King Saud University in Riyadh, Saudi Arabia. The animal experimental protocol received approval from the University's Ethics Review Committee. Initially, cervical dislocation of the rats was performed, followed by making an incision in the peritoneal cavity.25 Subsequently, the livers of the rats were extracted and transferred to a sterile beaker for weighing. A chilled solution of KCl/sucrose buffer was administered to the rat liver at a ratio of 1 part buffer to 4 parts liver by weight/volume. The buffer consisted of 0.25 M sucrose, 0.04 M KH2PO4/NaH2PO4, 0.15 M KCl, and had a pH of 7.4. The liver was dissected into minute fragments and then pulverized using an OMNI homogenizer. The liver homogenate underwent two rounds of centrifugation. The initial centrifugation step was performed at a force of 9000 times the acceleration due to gravity (9000g) for a duration of 25 minutes at a temperature of 4 °C. This process resulted in the separation of the supernatant, known as S9. The second step of centrifugation involved subjecting the supernatant to a centrifugal force of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for a duration of 65 minutes. Next, the liquid portion (supernatant) was taken out while the solid portion (pellets or RLMs) was mixed with a solution containing KCl and sucrose. The RLMs suspension was stored in a deep freezer at a temperature of −76 °C.26
000g for a duration of 65 minutes. Next, the liquid portion (supernatant) was taken out while the solid portion (pellets or RLMs) was mixed with a solution containing KCl and sucrose. The RLMs suspension was stored in a deep freezer at a temperature of −76 °C.26
2.6. RLMs incubations
The protein concentration of the produced RLMs was assessed using the Lowery technique.19 Given the increased solubility of ACB in dimethyl sulfoxide (DMSO; 93 mg mL−1), ACB was dissolved in DMSO. The compound ACB, at a concentration of 5 μM, was mixed with microsomes derived from rat liver, at a concentration of 1 mg mL−1, in a phosphate buffer containing 50 mM of sodium and potassium ions, as well as 3.3 mM of magnesium chloride, at a pH of 7.4. A concentration of 1 mM of NADPH was introduced to commence the metabolic process. The in vitro tests utilized trapping agents, namely KCN (1.0 mM), GSH (1.0 mM), and methoxyl amine (2.5 mM), to capture iminium, iminopyridinone, and aldehyde RId, respectively. The reactions were conducted in a temperature-controlled water bath with shaking at 37 °C for 60 minutes. Two mL of ice-cold acetonitrile were added to halt the metabolic reaction by the denaturation of protein enzymes. Proteins were precipitated by subjecting the sample to centrifugation at 9000g for 12 minutes at a temperature of 4 °C. The transparent liquid portion was subjected to evaporation using a flow of nitrogen gas, and subsequently restored by mixing with an equal proportion of the mobile phase (50:50). Control samples were created without the presence of RLMs or NADPH in order to validate the metabolic production of ACB RIs and relative adducts.20,27–29
2.7. Characterization of ACB RIs
The ACB metabolites and RIs were characterized using product ion chromatograms (PIC) obtained from various incubation mixtures. Product ions (PIs) were employed to reconstitute the chemical composition of in vitro ACB metabolites and RIs.
3. Results and discussion
3.1. Results of in silico ACB metabolites prediction
The Metabolic Landscape for ACB, as predicted by StarDrop software, reveals the susceptibility of each site to be metabolized by various isoforms of CYP450 enzymes. This suggests that the C1 carbon atom in the butynoyl group is susceptible to being metabolized by CYP450 enzymes, particularly CYP3A4 (99%) and CYP2D6 (100%). The CSL is displayed in the upper-right portion of the metabolic landscape (Fig. 2), with a value of 0.9980. This number indicates that ACB is very prone to being metabolized by CYP450 isoforms. The pie chart displayed in Fig. 2 presents the outcomes of the WhichP450™ module, which predicts the CYP450 isoforms that are most likely to play a significant role in ACB metabolism. Based on these findings, the CYP3A4 isoform plays a predominant role in the metabolism of ACB. The butynoyl moiety is anticipated to have a significant role in the pharmacological metabolism of ACB.
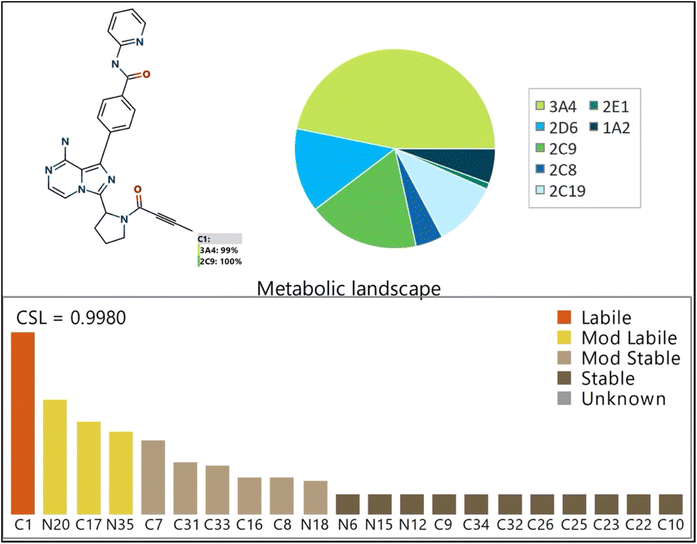 |
| | Fig. 2 Proposed metabolic sites for ACB by StarDrop WhichP450™ module. | |
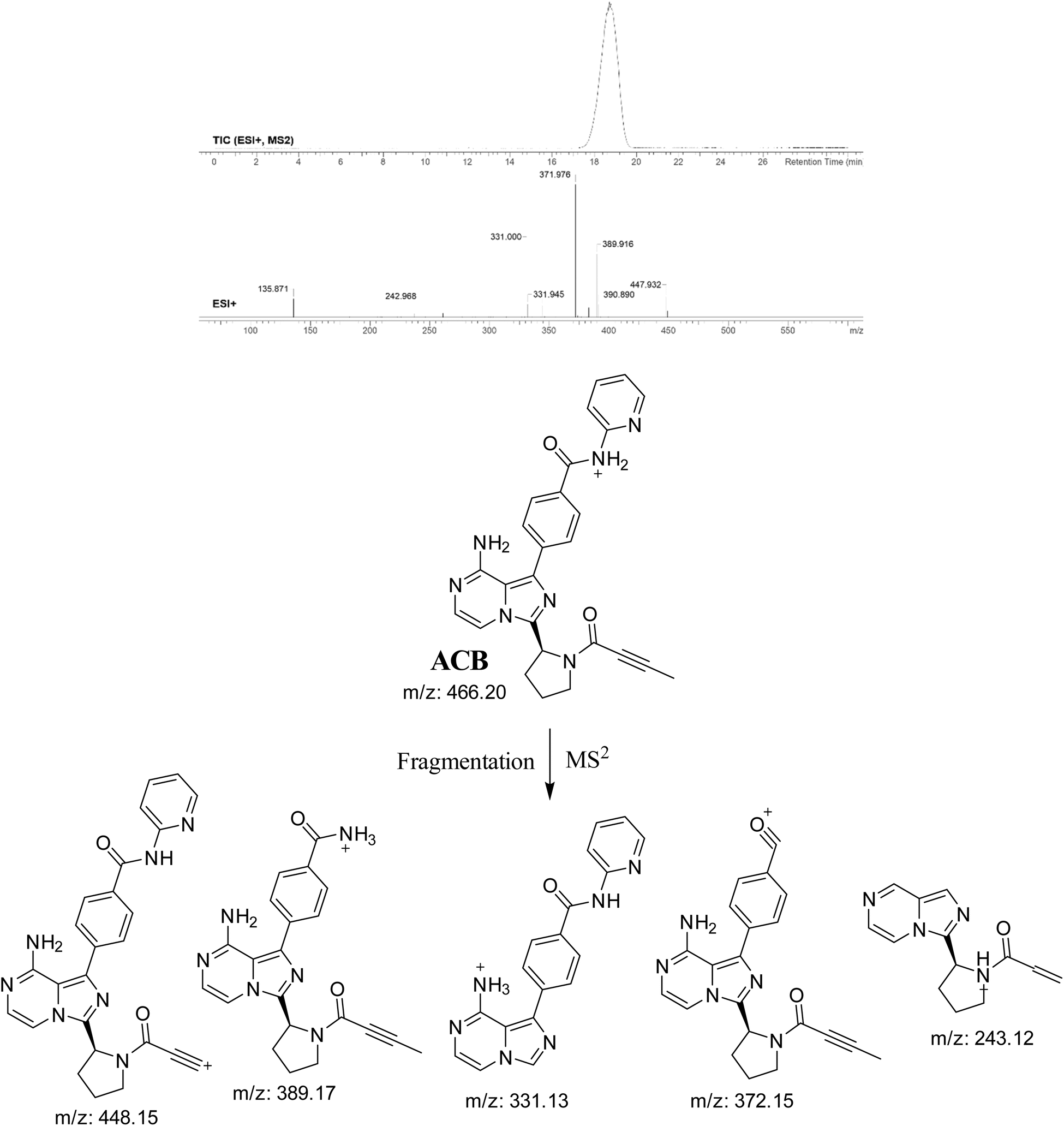
3.2. PI study of ACB
The ACB chromatographic peak is eluted at a retention time of 19.1 minutes in the PIC. The ACB ion dissociates at mass-to-charge ratio (m/z) 466 in the collision cell, resulting in the formation of five distinct and recognizable PIs at m/z 448, m/z 389, m/z 372, m/z 331, and m/z 243 (Fig. 3). The PIs serve as a reference for the analysis and estimation of the chemical composition of ACB metabolites and stable adducts.
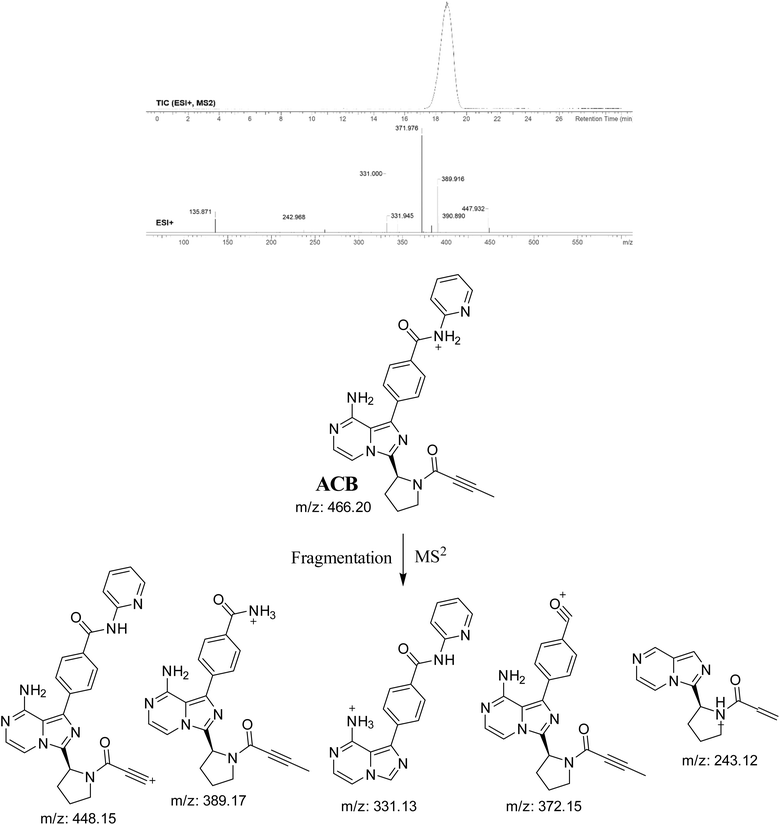 |
| | Fig. 3 PIC of ACB, PI mass spectrum of ACB and the predicted interpretation of the fragmentation pattern of ACB. | |
3.3. Results of in silico ACB bioactivity and toxicity prediction
The ACB reactivity was determined using the reactivity module on the Xenosite web page, as depicted in Fig. 4. The probable locations for bioactivation in the ACB chemical structure are indicated in Fig. 4. The DEREK software was utilized to assess the potential toxicities of ACB by analyzing its chemical structure (Fig. 5). ACB exhibits the potential to cause cutaneous sensitization (equivocal) as a result of the group highlighted in red in Fig. 5.
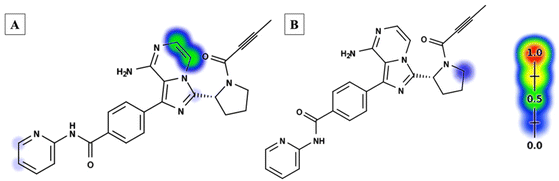 |
| | Fig. 4 Xenosite web predictor results of acalabrutinib (A) predicted sensitive sites for GSH bioactivation. (B) Predicted site of cyanide binding. The presence of red and green colors signifies a significant likelihood of bioactivation occurring at these places, while the light blue hue indicates a smaller likelihood of RIs being formed. | |
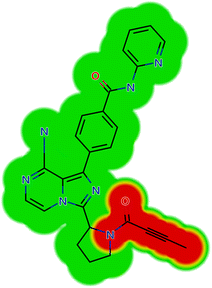 |
| | Fig. 5 Structural alerts of acalabrutinib by DEREK module (skin sensitization; equivocal). | |
3.4. Identification of ACB phase metabolites proposed by StarDrop software
The incubation of ACB in RLM resulted in the identification of five metabolites through in vitro analysis. The mechanisms involved in the formation of these metabolites are believed to be oxidation, N-dealkylation, oxidative dealkylation, and hydroxylation, as shown in Table 1. The metabolites were produced in all metabolic incubations when conducted three times, indicating the effectiveness of the applied method. The additional file contains the data for ACB metabolites, specifically M2, M3, M4 and M5 (Fig. S1–S4†).
Table 1 Proposed phase 1 metabolites of ACB
| |
m/z |
Fragments |
Retention time |
Proposed metabolic reaction |
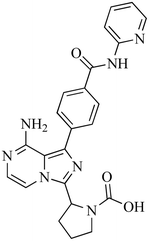
Proposed chemical structure |
| M1 |
482 |
392, 366, 356, 348, 338 |
16 |
Hydroxylation |
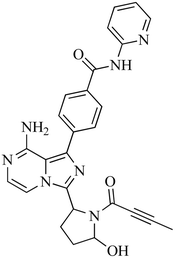 |
| M2 |
480 |
408, 390, 384, 372, 338 |
19.8 |
Oxidation |
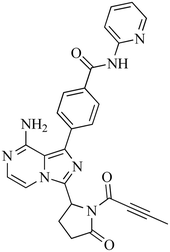 |
| M3 |
400 |
408, 394, 384, 366, 372 |
18.46 |
N-Dealkylation |
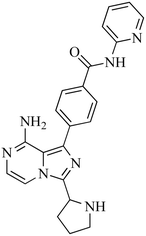 |
| M4 |
416 |
422, 390, 382, 386, 340, 278 |
23.05 |
N-Dealkylation and hydroxylation |
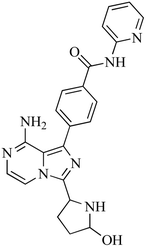 |
| M5 |
444 |
422, 386, 380, 382, 398 |
19.79 |
Oxidative dealkylation |
 |
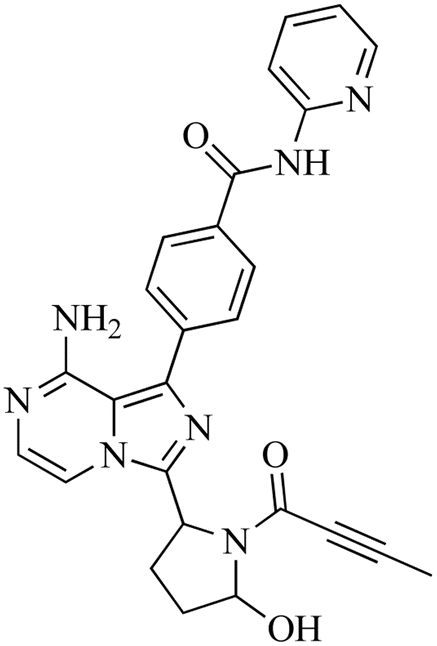
3.4.1. Identification of M1. M1, with a m/z of 482, is expected to be produced through the process of hydroxylation of ACB. The peak of this metabolite is detected at 16.2 minutes in the PIC. The M1 ion undergoes fragmentation within the collision cell, resulting in the production of four PIs with m/z of 464, 388, 381, 348, and 331 (Fig. 6). The DEREK module's predictions of M1's toxicities indicated that hydroxylation did not alter the toxicity alert for skin sensitivity.
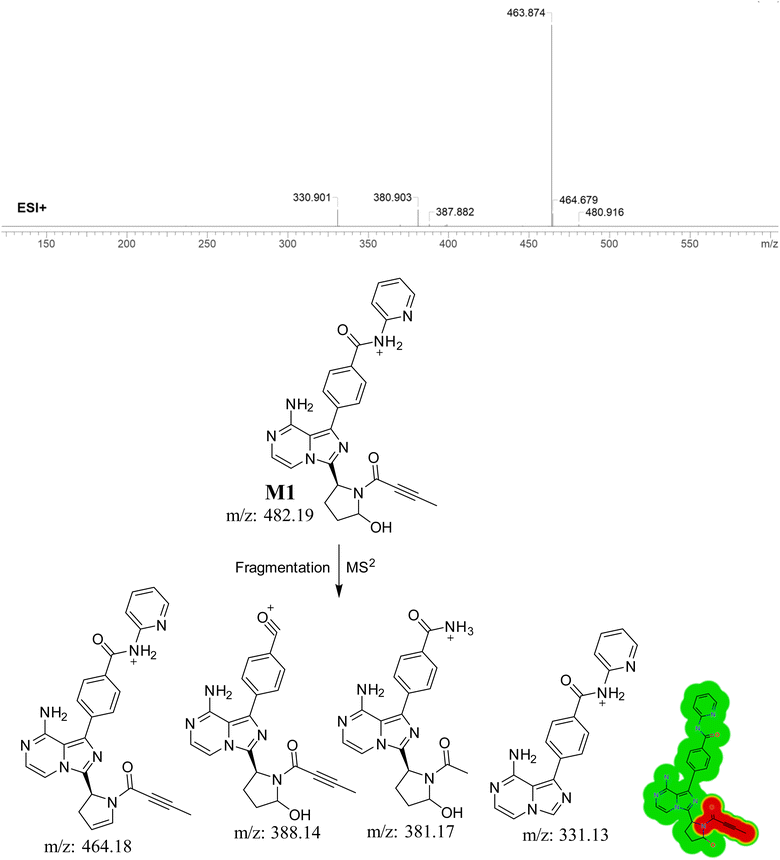 |
| | Fig. 6 PI mass spectrum of M1, the proposed interpretation of fragmentation pattern of M1 and the predicted new toxicities of M1 by DEREK module. | |
3.5. Identification of 6-iminopyridin-3(6H)-one RIs using GSH as a trapping agent
3.5.1. Identification of ACB787 (GSH adduct). The formation of ACB787 is anticipated by the reaction of GSH with the 6-iminopyridin-3(6H)-one RIs of ACB. The ACB787 peak, with a m/z of 787, was seen during a retention time of 16.8 minutes in the PIC. The dissociation of the ACB787 ion within the collision cell produces six PIs with m/z of 769, 668, 658, 531, 488, and 398 (Fig. 7).
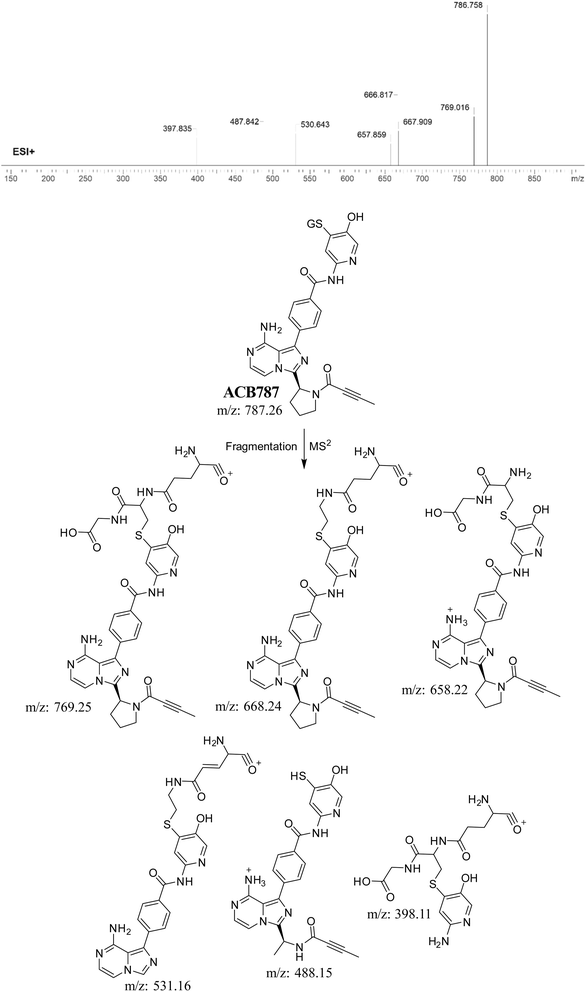 |
| | Fig. 7 PI mass spectrum of ACB787 GSH adduct and the predicted fragmentation pattern of ACB787. | |
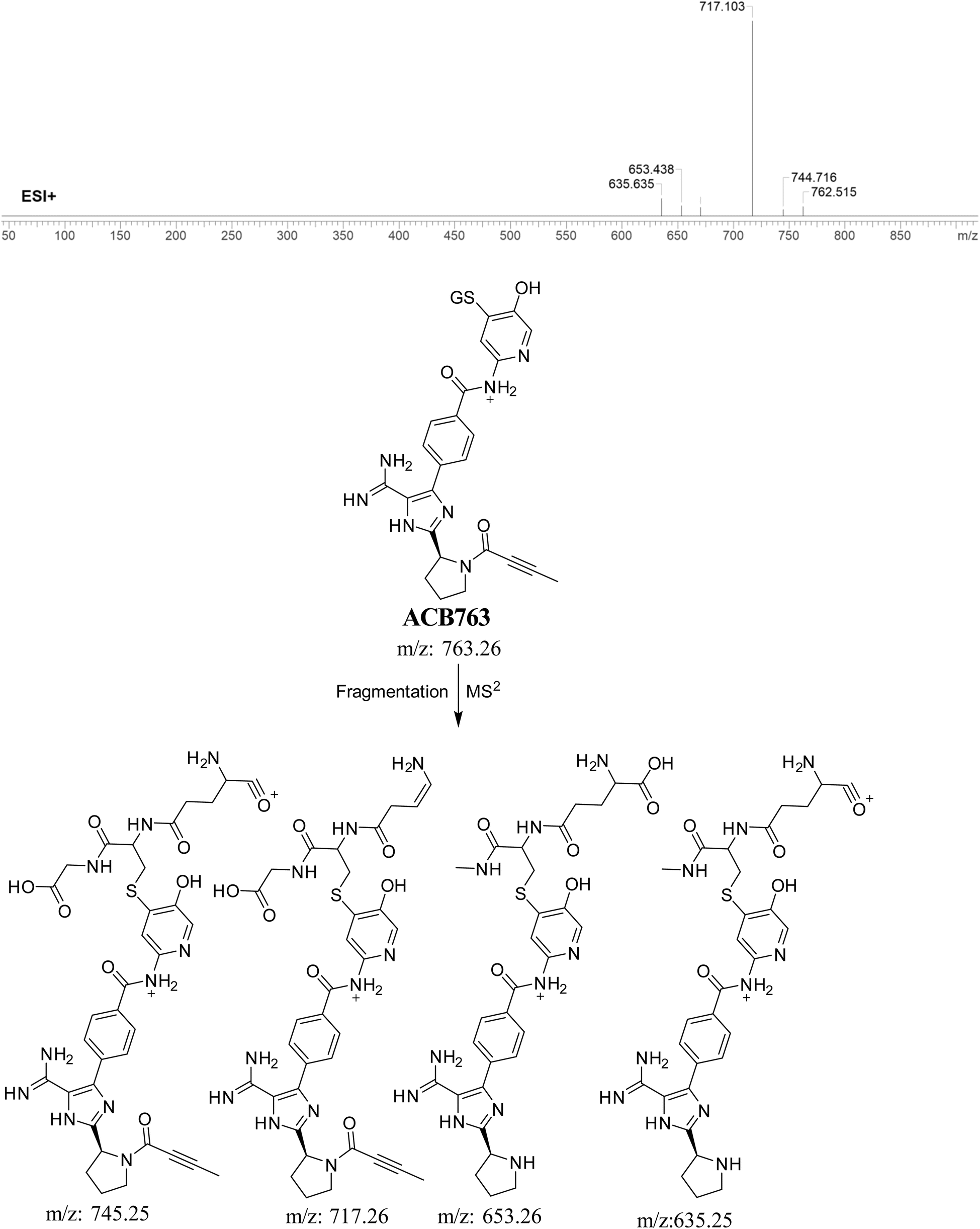
3.5.2. Identification of ACB763 (GSH adduct). ACB763 is suggested to be formed through the conjugation of GSH with the RIs, namely the 6-iminopyridin-3(6H)-one after metabolic hydroxylation reaction. The ACB763 peak, with a m/z of 763, was seen during a retention time of 24.9 minutes in the PIC. The ACB763 ion undergoes dissociation within the collision cell, resulting in the formation of five PIs with m/z of 762, 745, 717, 653, and 635 (Fig. 8).
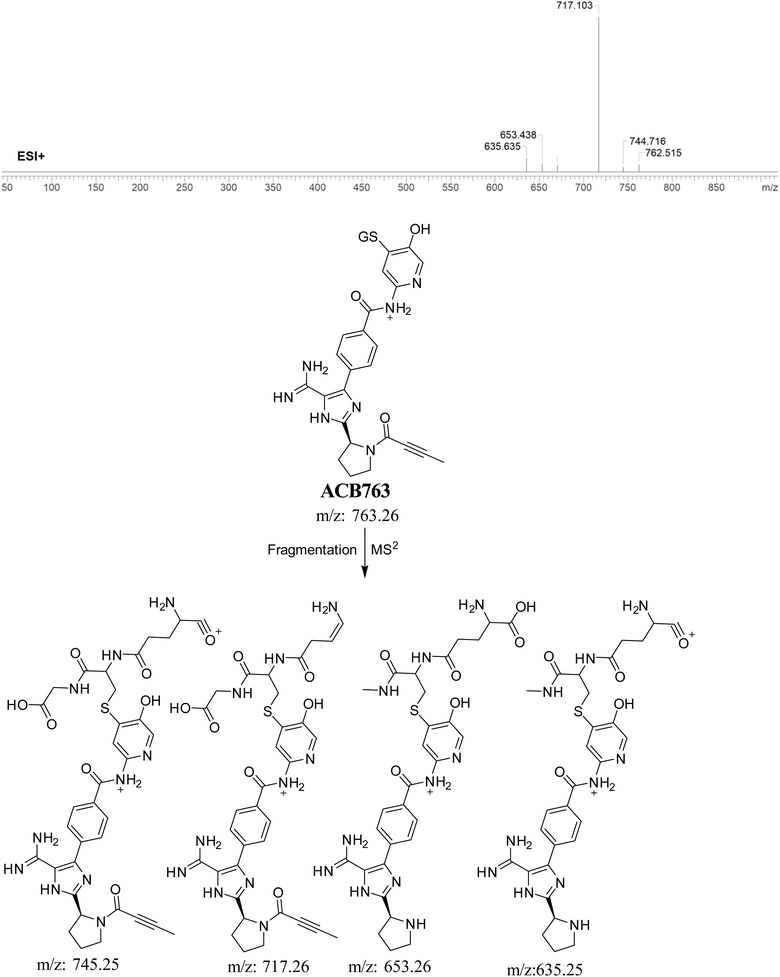 |
| | Fig. 8 PI mass spectrum of ACB763 GSH adduct and the proposed interpretation of fragmentation of ACB763. | |
3.5.3. Identification of GSH adduct ACB803a. The formation of ACB803a is suggested to occur through the conjugation of GSH with the RIs, specifically the 6-iminopyridin-3(6H)-one after double metabolic hydroxylation. The ACB803a peak, with a m/z of 803, was observed during a retention time of 23.5 minutes in the PIC. The dissociation of the ACB803a ion within the collision cell results in the formation of four PIs with a m/z of 745, 701, 661, and 608 (Fig. 9).
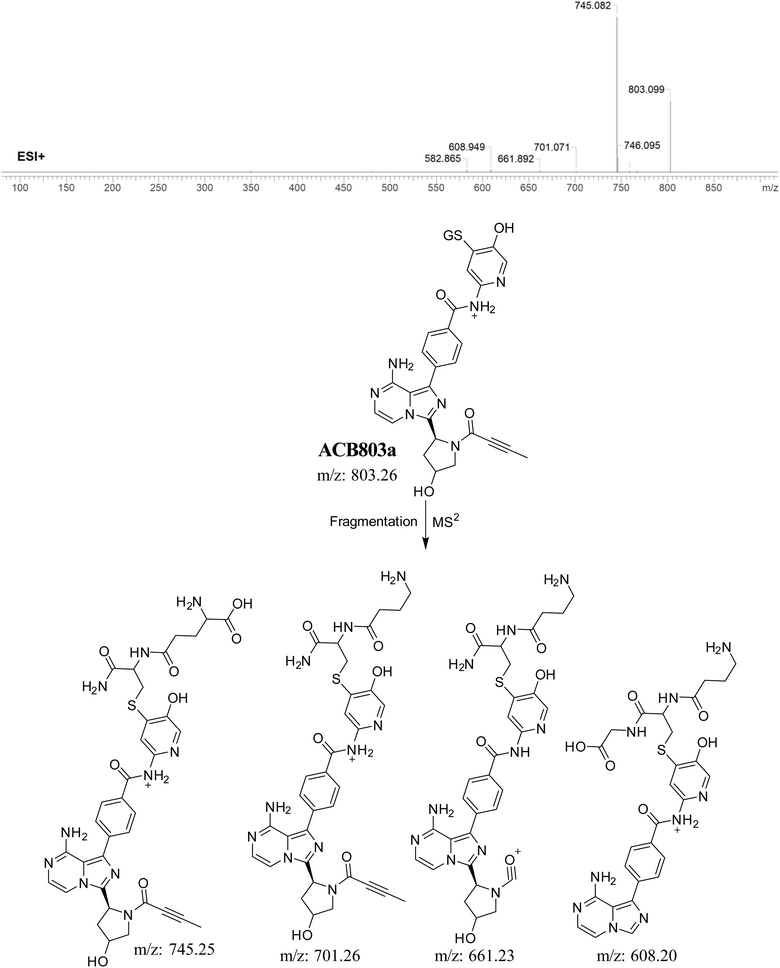 |
| | Fig. 9 PI mass spectrum of ACB803a GSH adduct and the proposed interpretation of fragmentation of ACB803a. | |
3.5.4. Identification of GSH adduct ACB803b. The formation of ACB803b is hypothesized to occur through the addition of GSH to the RIs, namely the 6-iminopyridin-3(6H)-one after pyrrolidine ring breakdown. The ACB803b peak with a m/z of 803 was observed during a retention time of 20.9 minutes in the PIC. The dissociation of the ACB803b ion within the collision cell results in the formation of three PIs with m/z of 714, 684, and 639 (Fig. 10).
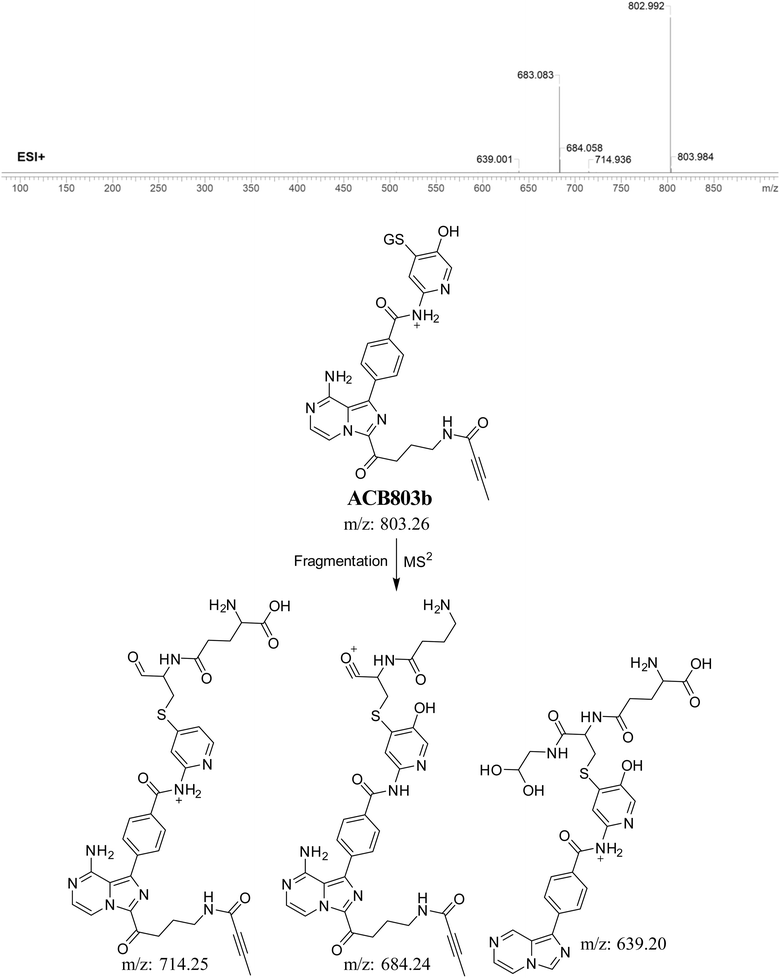 |
| | Fig. 10 PI mass spectrum of ACB803b GSH adduct and the proposed interpretation of fragmentation of ACB803b. | |
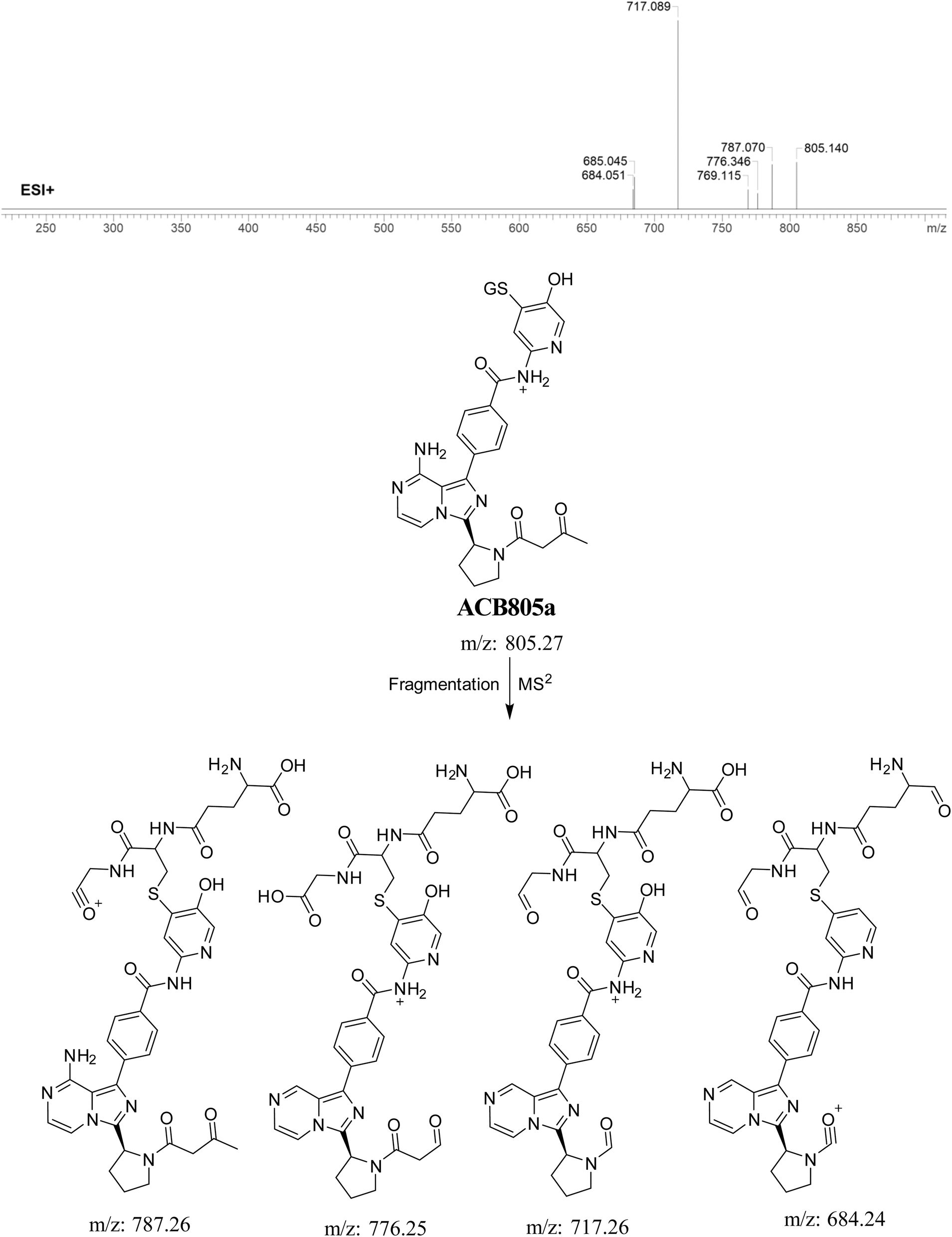
3.5.5. Identification of GSH adduct ACB805a. ACB805a is suggested to be produced through the reaction of GSH with the 6-iminopyridin-3(6H)-one RIs after butynoyl group oxidation. The ACB805a peak with a m/z of 803 was observed at 24.7 minutes in the PIC. The dissociation of the ACB805a ion within the collision cell results in the formation of four PIs with m/z of 787, 776, 717, and 684 (Fig. 11).
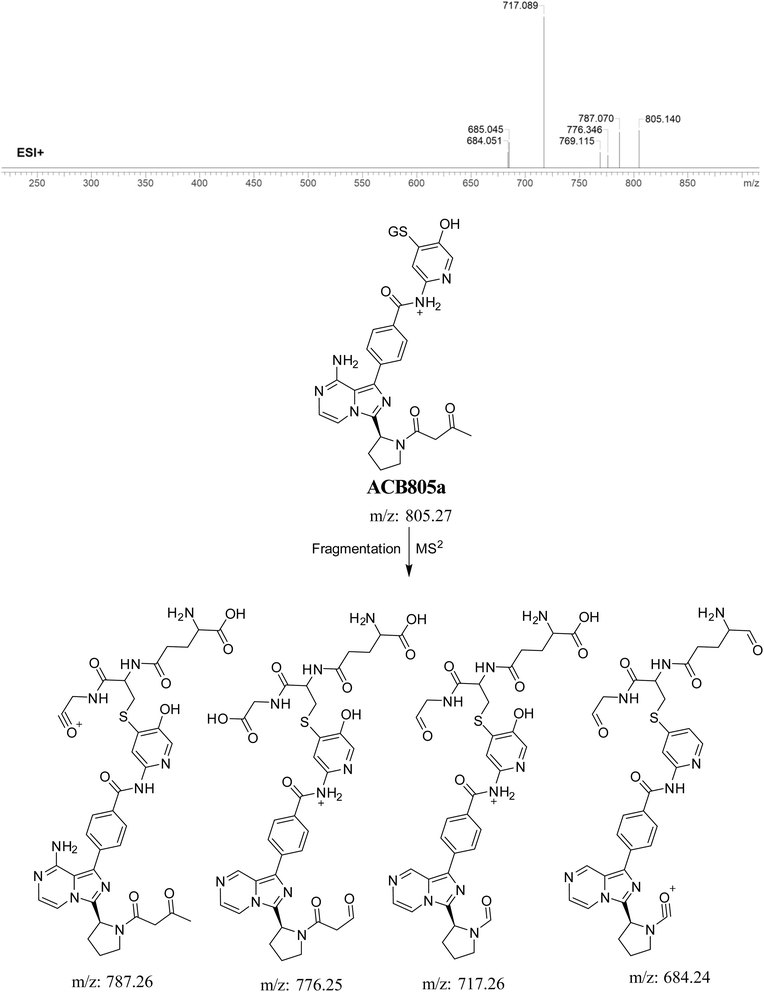 |
| | Fig. 11 PI mass spectrum of ACB805a GSH adduct and the proposed interpretation of fragmentation of ACB805a. | |
3.5.6. Identification of GSH adduct ACB805b. The formation of ACB805b is suggested to occur by the conjugation of GSH with the RIs, namely the 6-iminopyridin-3(6H)-one after butyne group dealkylation and pyridine ring hydroxylation. The ACB805b peak, with a m/z of 803, was observed during a retention time of 20.7 minutes in the PIC. The dissociation of the ACB805b ion within the collision cell results in the formation of four PIs with m/z of 761, 685, 669, and 642 (Fig. 12).
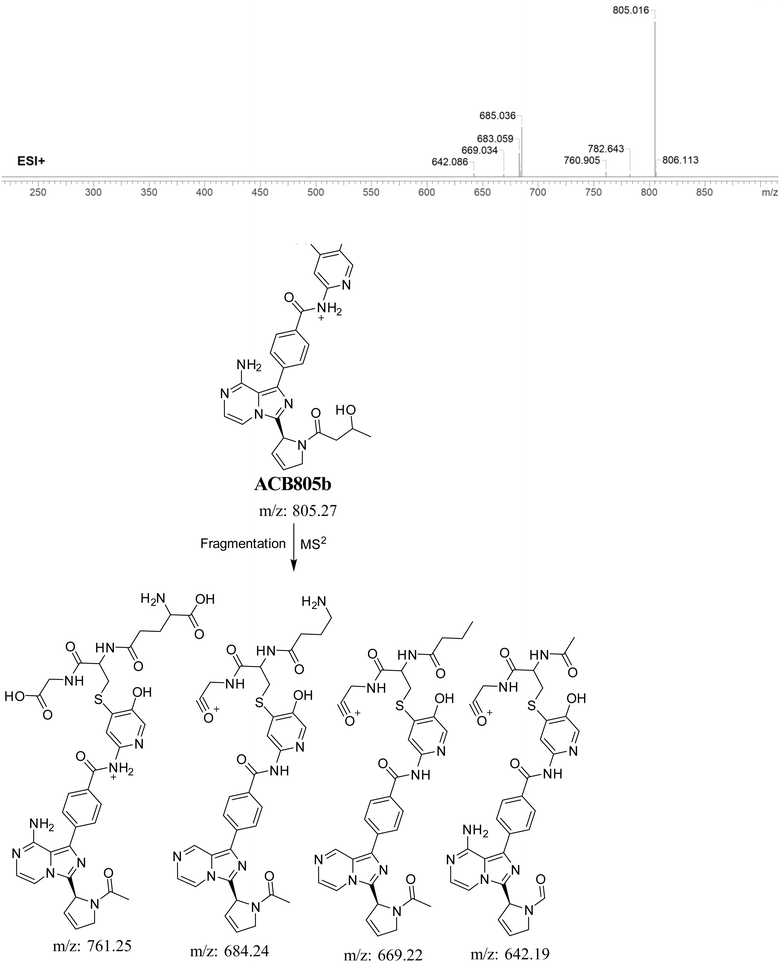 |
| | Fig. 12 PI mass spectrum of ACB805b GSH adduct and the proposed interpretation of fragmentation of ACB805b. | |
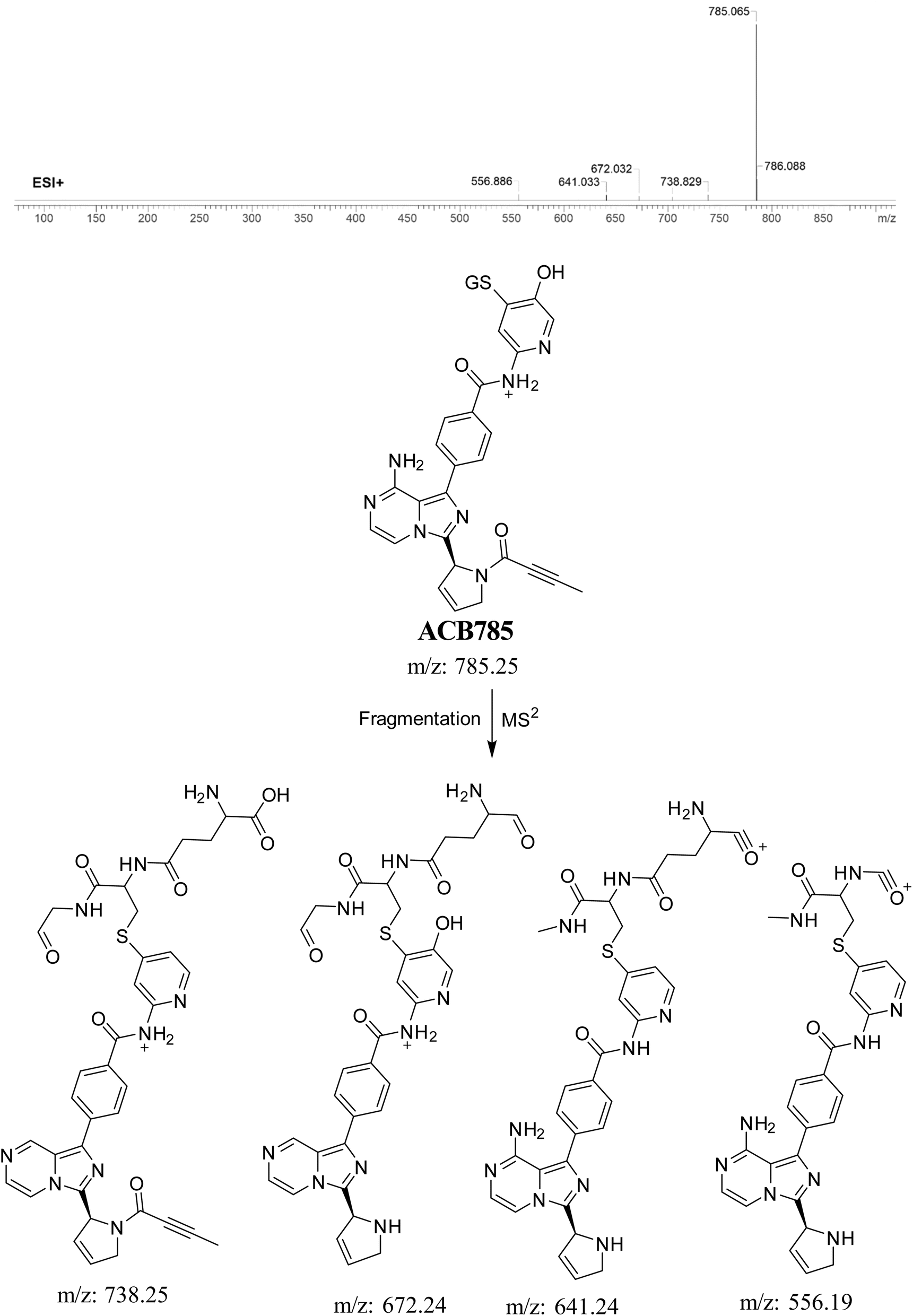
3.5.7. Identification of ACB785 (GSH adduct). ACB785 is formed through the addition of GSH to the RIs, specifically the 6-iminopyridin-3(6H)-one after butynoyl group dealkylation and pyridine ring hydroxylation. The ACB785 peak, with a m/z of 785, was observed at 22.3 minutes in the PIC. The dissociation of the ACB785 ion within the collision cell results in the formation of four PIs with m/z of 738, 672, 641, and 556 (Fig. 13).
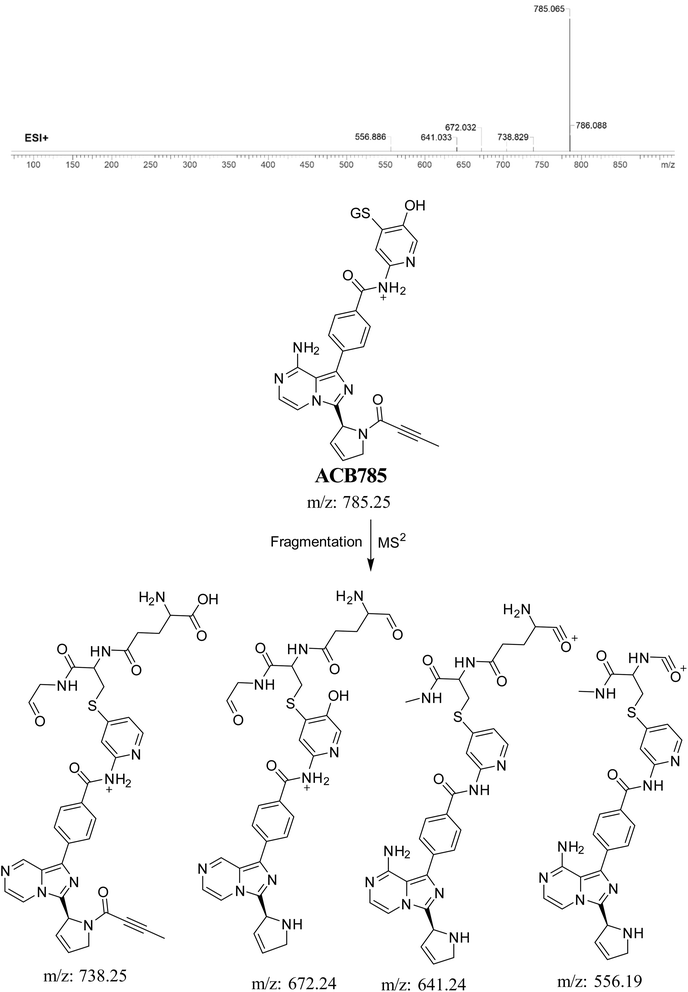 |
| | Fig. 13 PI mass spectrum of ACB785 GSH adduct and the proposed interpretation of fragmentation of ACB785. | |
3.6. Proposed bioactivation mechanism of ACB to 6-iminopyridin-3(6H)-one RIs
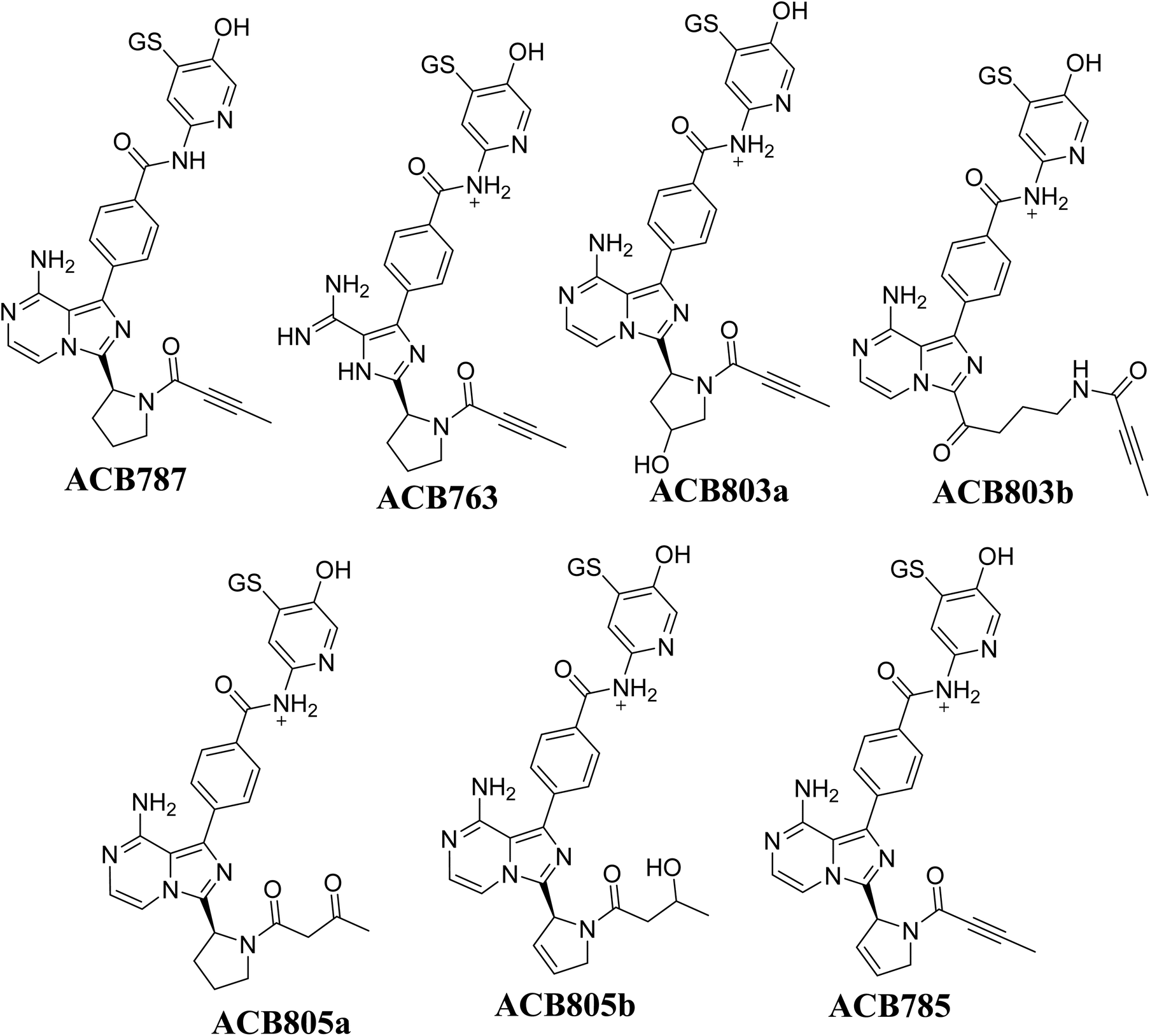
Fig. 14 illustrates the process by which ACB is converted into 6-iminopyridin-3(6H)-one intermediates by bioactivation. The presence of several GSH adducts suggests the production of unstable intermediates of ACB, specifically 6-iminopyridin-3(6H)-one. The pyridine ring undergoes hydroxylation catalyzed by CYP450 enzymes, which is subsequently followed by oxidation, leading to the generation of highly RIs (6-iminopyridin-3(6H)-one). The GSH captured and identified these RIs by LC/MS analysis. The Fig. 15 and Table 2 includes the information for ACB763, ACB803a, ACB803b, ACB805a, ACB805b, and ACB785.
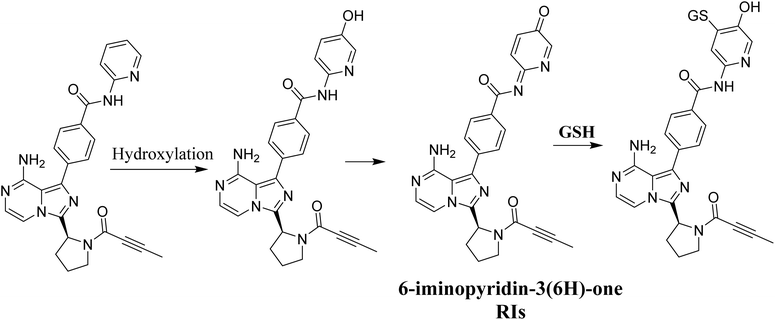 |
| | Fig. 14 Proposed bioactivation pathway of ACB to 6-iminopyridin-3(6H)-one RIs captured by GSH. | |
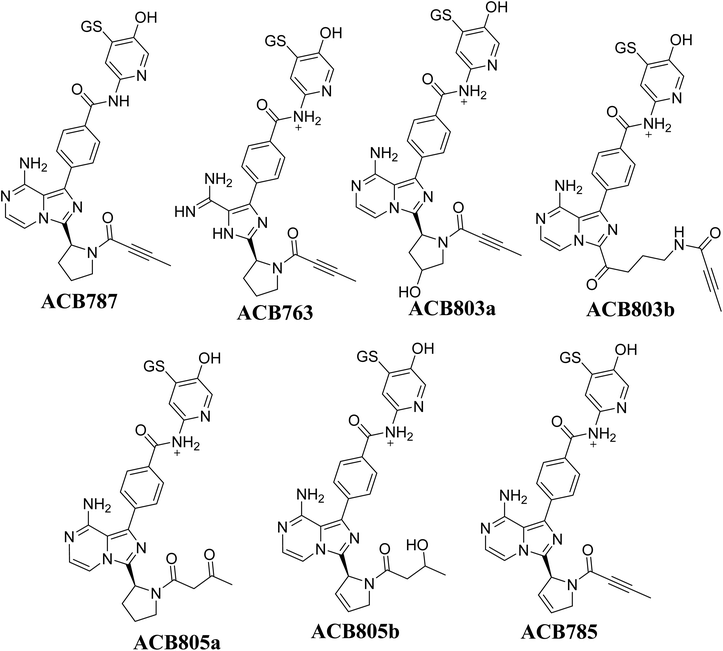 |
| | Fig. 15 Summary of proposed GSH adducts of ACB. | |
Table 2 Summary of proposed GSH adducts of ACB
| |
m/z |
Fragments |
Retention time (minutes) |
| ACB787 |
787 |
769, 668, 658, 531, 488, 398 |
16.8 |
| ACB763 |
763 |
745, 717, 670, 653, 635 |
24.9 |
| ACB803a |
803 |
759, 745, 701, 661, 608 |
23.5 |
| ACB803b |
803 |
714, 684, 639 |
20.9 |
| ACB805a |
805 |
787, 776, 769, 717, 684 |
24.7 |
| ACB805b |
805 |
782, 761, 684, 642, 669 |
20.7 |
| ACB785 |
785 |
738, 672, 641, 556 |
22.3 |
3.7. Identification of ACB oximes (aldehyde intermediates and methoxylamine)
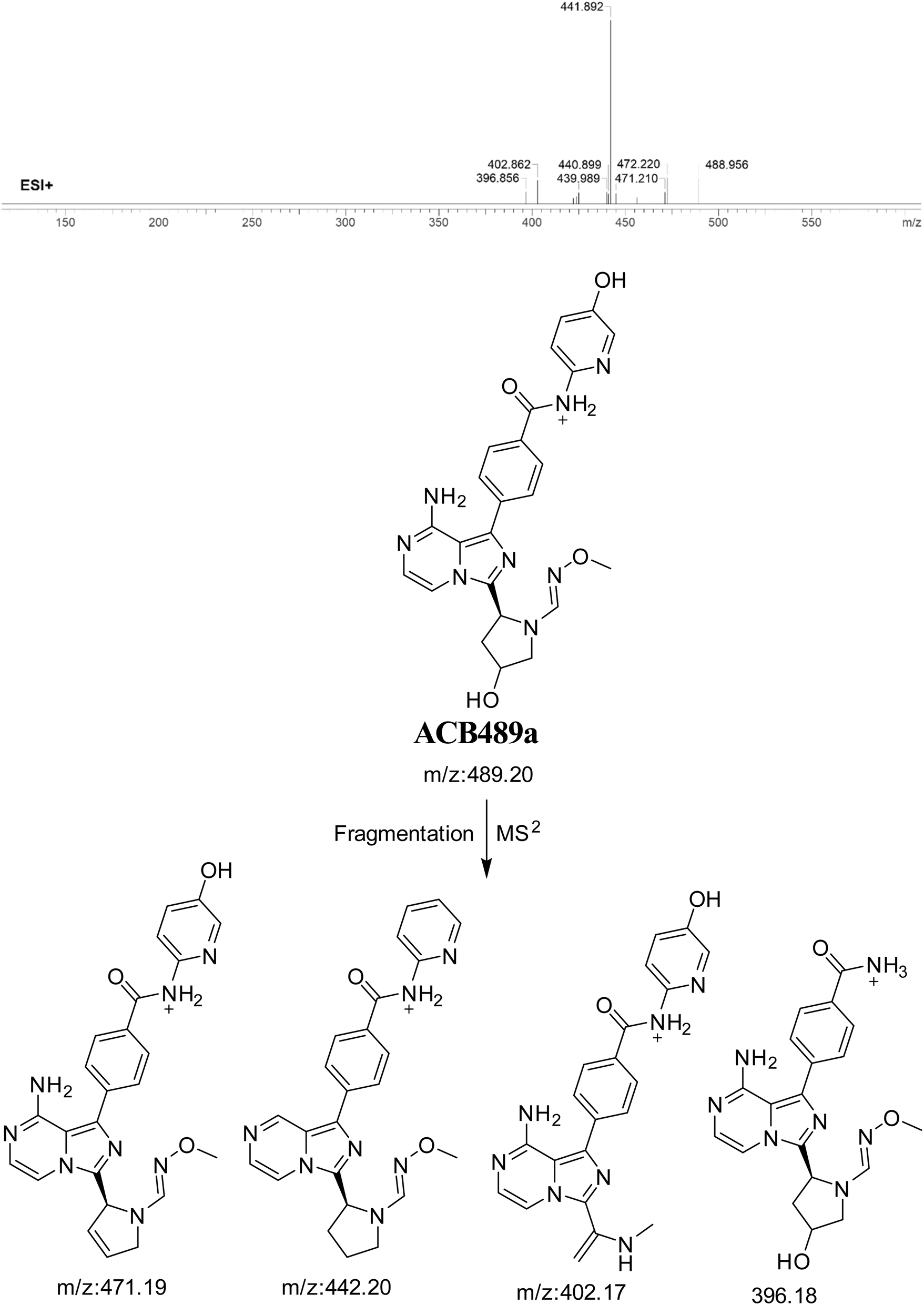
3.7.1. Identification of ACB489a oxime. The formation of ACB489a is anticipated through the addition of methoxylamine to the aldehyde RIs after double metabolic hydroxylation reactions. The ACB489a peak, with a m/z of 489, was observed during a retention time of 20.2 minutes in the PIC. The dissociation of the ACB489a ion within the collision cell produces four PIs with m/z of 471, 442, 402, and 396 (Fig. 16).
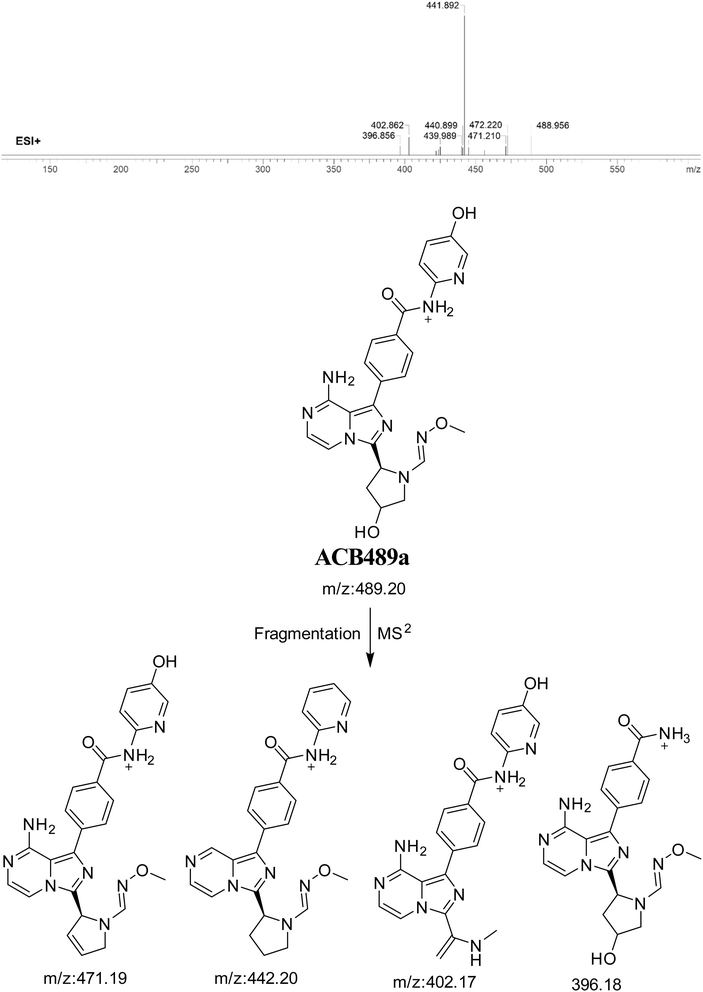 |
| | Fig. 16 PI mass spectrum of ACB489a oxime and the proposed interpretation of fragmentation of ACB489a. | |
3.7.2. Identification of ACB489b oxime. The formation of ACB489b is hypothesized to occur by the addition of methoxylamine to the aldehyde RIs after double metabolic hydroxylation reaction at pyrrolidine ring and oxidative dealkylation of butynoyl group. The ACB489a peak, with a m/z of 489, was seen during a retention time of 24.5 minutes in the PIC. The ACB489b ion dissociates within the collision cell, resulting in the formation of three PIs with m/z of 471, 412, and 396 (Fig. 17).
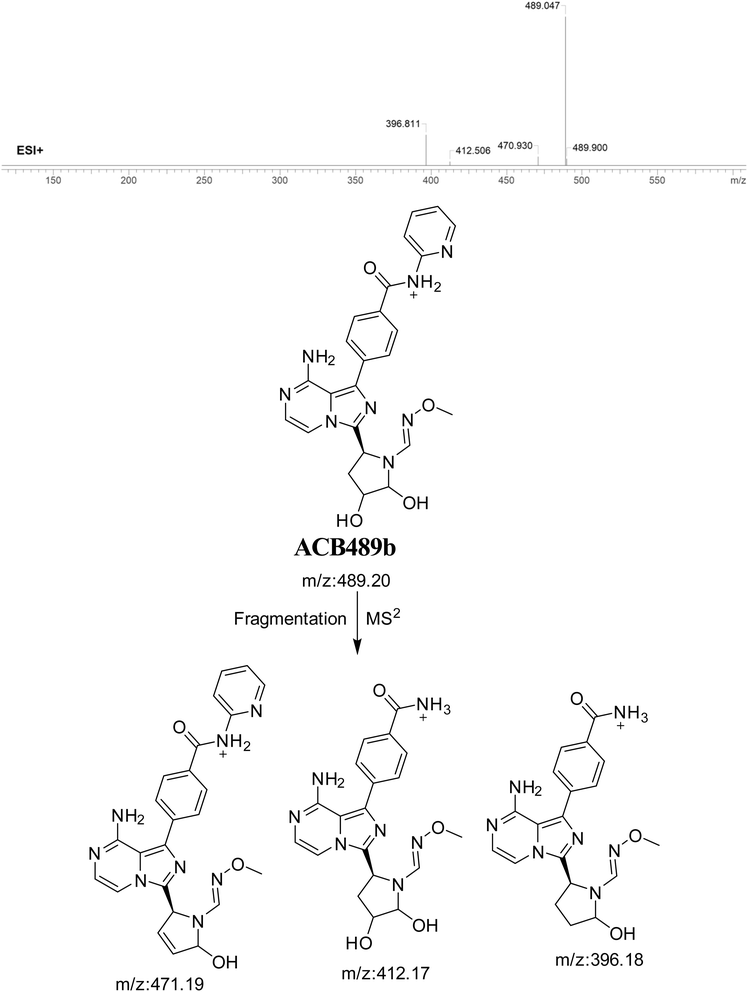 |
| | Fig. 17 PI mass spectrum of ACB489b oxime and the proposed interpretation of fragmentation of ACB489b. | |
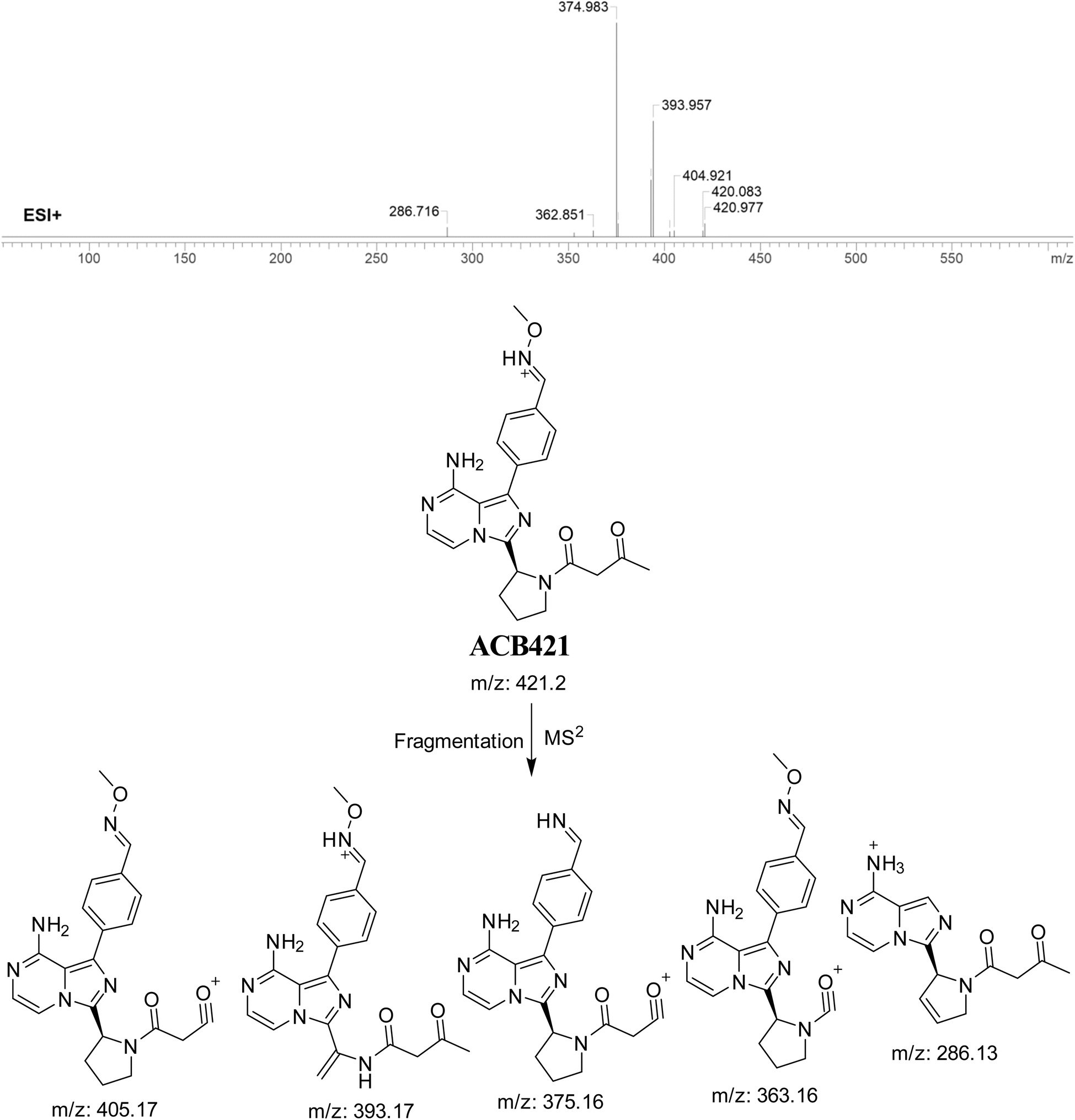
3.7.3. Identification of ACB421 oxime. The formation of ACB421 is suggested to occur by the addition of methoxylamine to the aldehyde RIs after oxidation metabolic reaction at butynoyl ring and oxidative dealkylation of pyridine group. The ACB421 peak, with a m/z of 421, was seen during a retention time of 18.5 minutes in the PIC. The ACB421 ion dissociates within the collision cell, resulting in the formation of five PIs with m/z of 405, 393, 375, 363, and 286 (Fig. 18).
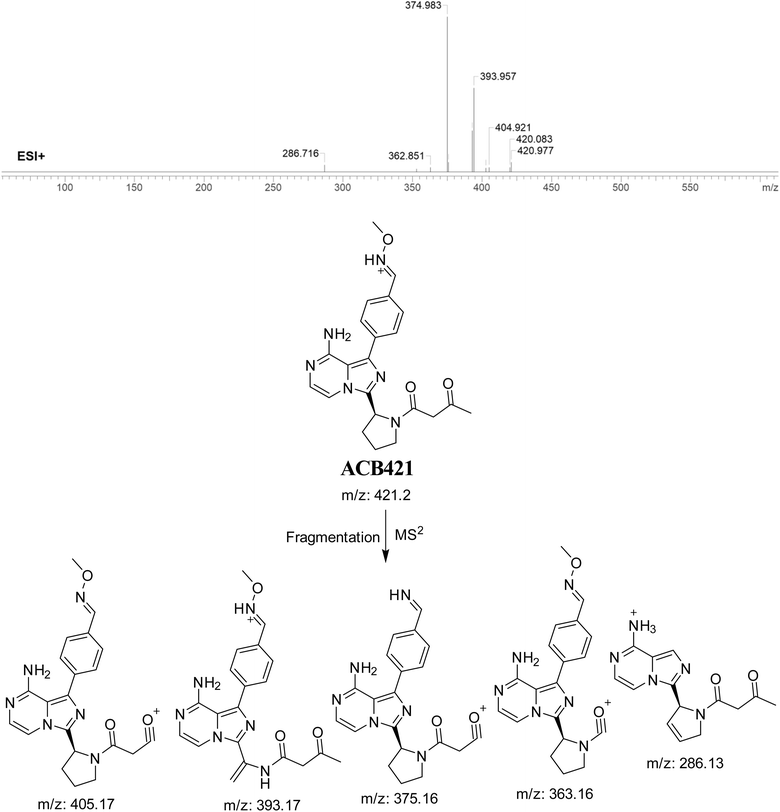 |
| | Fig. 18 PI mass spectrum of ACB421 oxime and the proposed interpretation of fragmentation of ACB421. | |
3.7.4. Identification of ACB419 oxime. ACB419 is suggested to be formed through the addition of methoxylamine to the aldehyde RIs after hydroxylation reactions at pyrrolidine ring and oxidative dealkylation of pyridine group. The ACB419 peak, with a m/z of 419, was seen during a retention time of 25.8 minutes in the PIC. The ACB419 ion undergoes dissociation within the collision cell, resulting in the formation of five PIs with m/z of 401, 395, 363, 347, and 326 (Fig. 19).
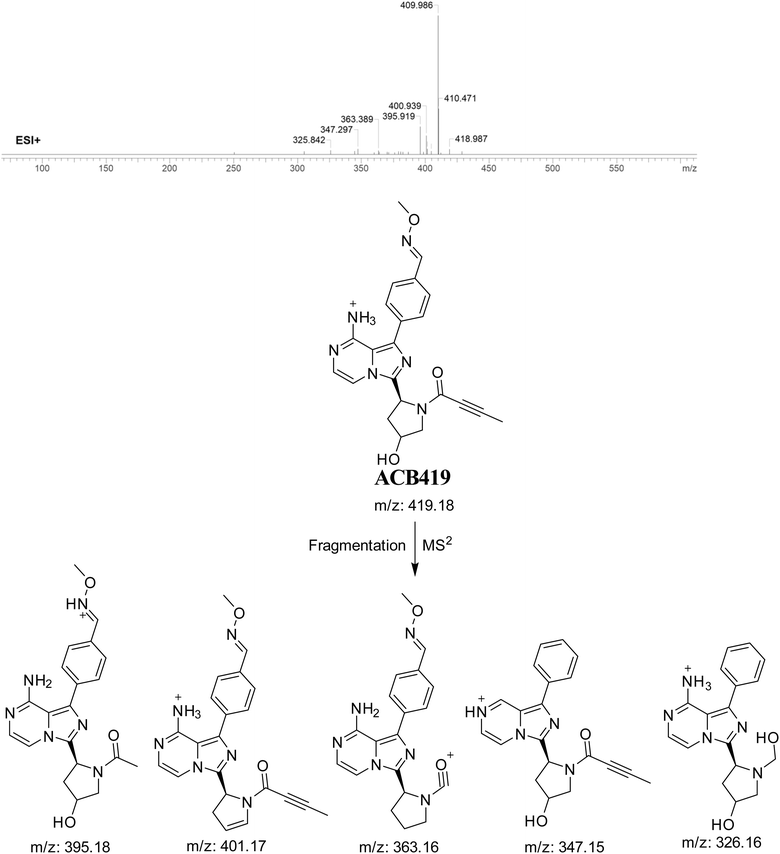 |
| | Fig. 19 PI mass spectrum of ACB419 oxime and the proposed interpretation of fragmentation of ACB419. | |
3.7.5. Identification of ACB435 oxime. The formation of ACB435 is suggested to occur by the addition of methoxylamine to the aldehyde RIs after double metabolic hydroxylation reaction at pyrrolidine ring and oxidative dealkylation of pyridine group. The ACB435 peak, with a m/z of 435, was seen during a retention time of 25.2 minutes in the PIC. The dissociation of the ACB435 ion within the collision cell results in the formation of five PIs with m/z of 425, 417, 372, 336, and 308 (Fig. 20).
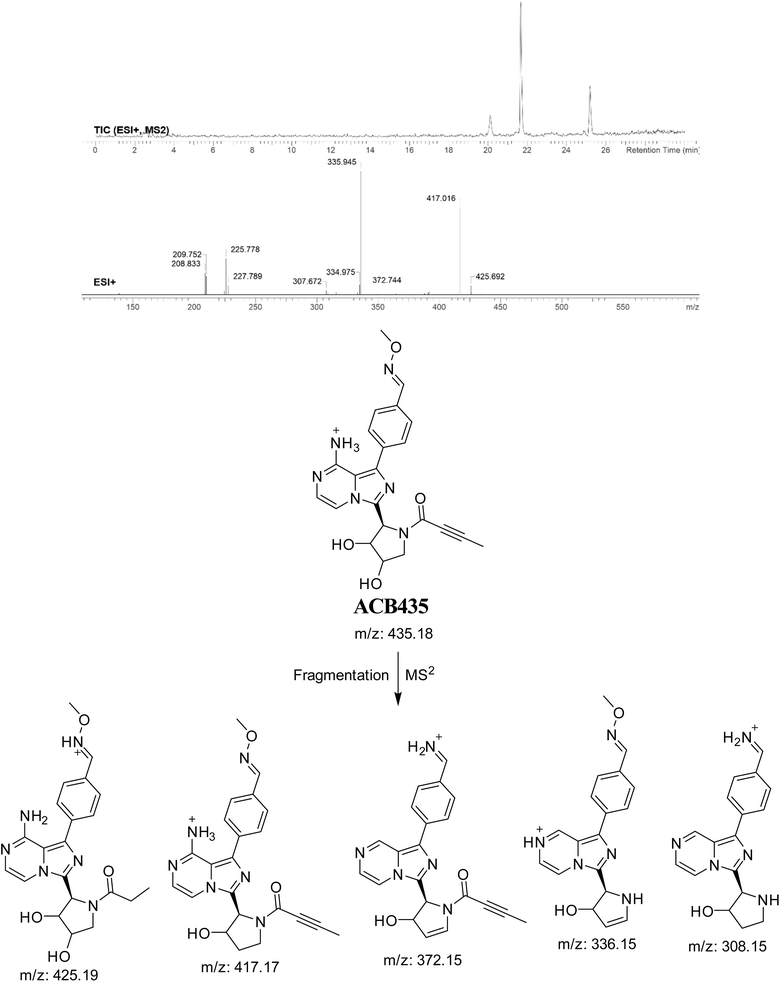 |
| | Fig. 20 PI mass spectrum of ACB435 oxime and the proposed interpretation of fragmentation of ACB435. | |
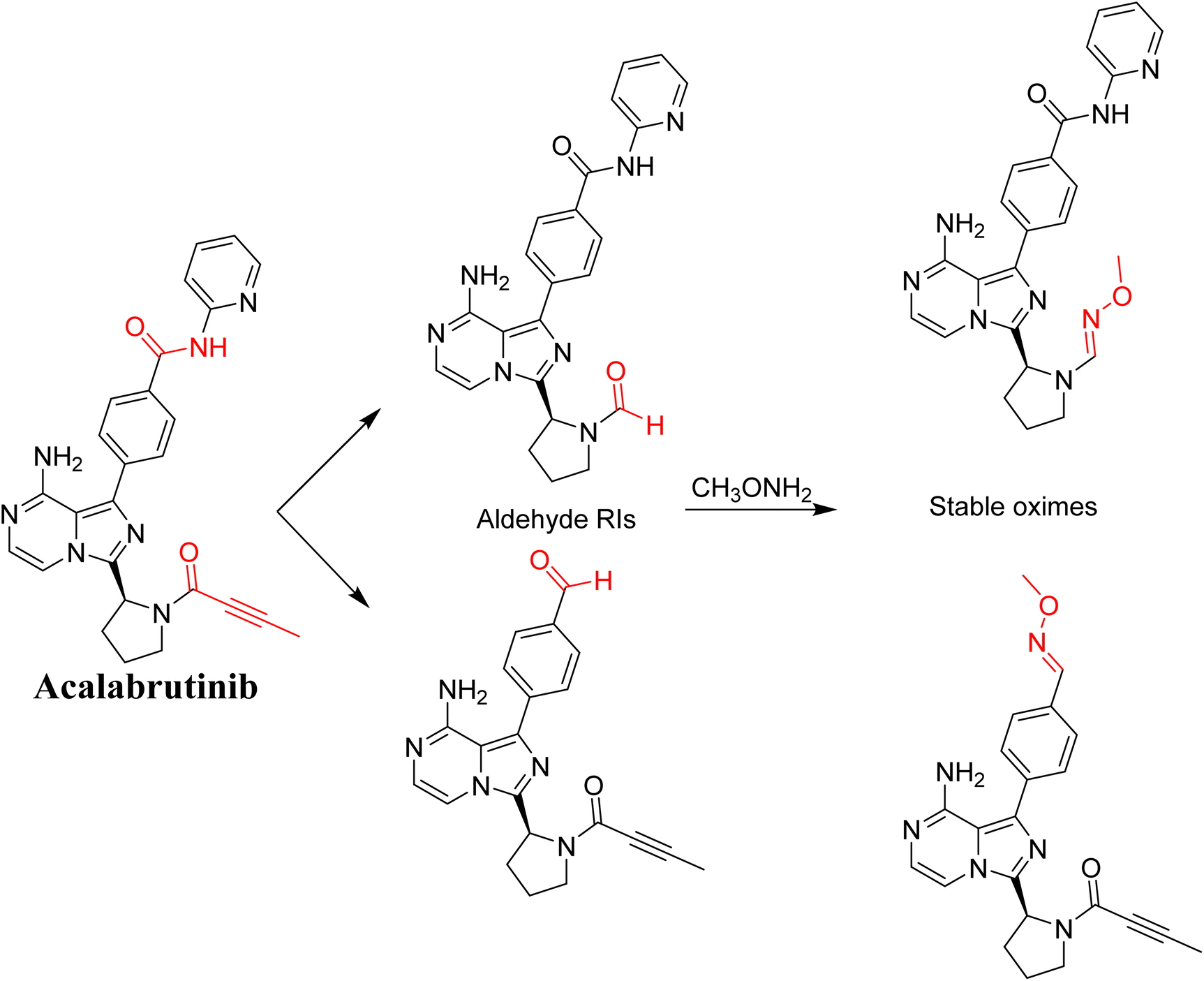
3.8. Proposed bioactivation mechanism of ACB to aldehyde RIs
Fig. 21 illustrates the bioactivation pathways of ACB leading to the formation of aldehyde intermediates. The presence of methoxylamine adducts (oximes) confirmed the production of aldehyde RIs at two locations in ACB during in vitro metabolism following incubation with RLMs. The pyridine amide and pyrrolidine ring undergo metabolism by CYP450 enzymes, resulting in the creation of aldehyde RIs at these locations. Methoxylamine can be used to capture the unstable intermediates, resulting in the formation of stable oximes. These oximes can then be characterized by LC-ITMS. The ESI file† (Fig. 22; Table 3) now includes the information for ACB489b, ACB421, ACB419, and ACB435.
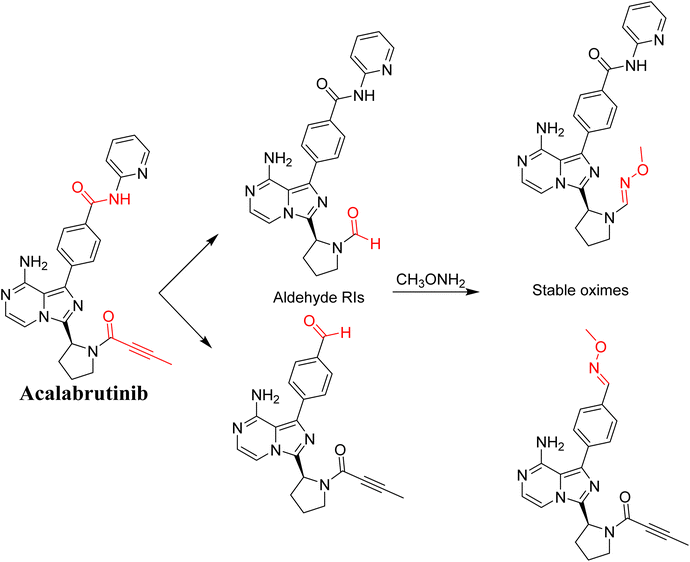 |
| | Fig. 21 Predicted bioactivation pathways of ACB to aldehyde RIs captured by methoxylamine. | |
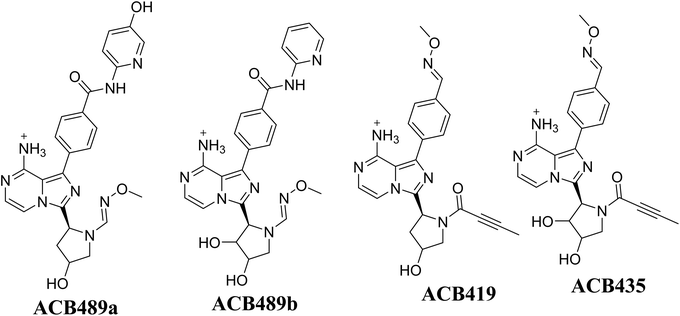 |
| | Fig. 22 Summary of proposed methoxylamine oximes of ACB. | |
Table 3 Summary of proposed methoxylamine adducts of ACB
| |
m/z |
Fragments |
Retention time (minutes) |
| ACB489a |
489 |
472, 441, 402, 396, 369 |
20.2 |
| ACB489b |
489 |
471, 412, 396 |
24.5 |
| ACB421 |
421 |
405, 393, 375, 363, 286 |
18.5 |
| ACB419 |
419 |
401, 395, 363, 347, 326 |
25.8 |
| ACB435 |
435 |
425, 417, 372, 336, 308, 277 |
25.2 |
4. Conclusions
This study involved in vitro (RLMs) bioactivation identification of ACB using LC-MS/MS. Trapping agents: glutathione, potassium cyanide and methoxylamine were used to help capturing unstable intermediates that may cause adverse effects in patients by binding to DNA and proteins in the body. Five phase I metabolites of ACB were identified. Seven 6-iminopyridin-3(6H)-one RIs of ACB were characterized. These intermediates were formed through the hydroxylation of the pyridine ring in ACB and its metabolites. Five aldehyde RIs of ACB were identified that are formed by further metabolism of ACB phase I metabolites by CYP450 enzymes. Aldehyde intermediates are formed at two different sites of ACB: the pyridine amide and the pyrrolidine ring. The generation of RIs of ACB, and the subsequent DIOT reactions may provide an explanation of ACB numerous ADR's. The alpha beta-unsaturated amide group butenoyl group, present in ACB, is considered a structural alert for toxicity as proposed by the DEREK software in silico prediction of toxicity and is therefore the likely chemical moiety involved in the formation of some of the observed RIs. Further drug discovery studies can be conducted allowing the development of new drugs with enhanced safety profile.
Ethical approval
The male Sprague-Dawley rats were housed in compliance with the Animal Care Center rules of King Saud University, which were approved by the Local Animal Care and Use Committee of KSU. The animal experimental approach utilized in our current research has received approval and authorization from the Ethics Review Committee of King Saud University (Number: KSU-SE-22-83).
Author contributions
Contributions to the study design were made by A. M. A., M. W. A., A. A. K., H. I. A., and A. S. A. The data analysis was performed by A. M. A. and M. W. A., who also did experimental work and provided assistance in manuscript writing. A. A. K., A. S. A., H. I. A., and M. W. A. supervised the laboratory experimental work. M. W. A. is the author responsible for correspondence regarding this paper. Each author contributed to the revision of the final draft manuscript. The submission to the journal was endorsed by all authors.
Conflicts of interest
“The authors declare no conflict interests.”
Acknowledgements
“This study was funded by the Researcher Supporting Project Number (RSPD2024R750), King Saud University, Riyadh, Saudi Arabia”.
References
- J. Ferlay, M. Colombet, I. Soerjomataram, D. M. Parkin, M. Piñeros, A. Znaor and F. Bray, Int. J. Cancer, 2021, 149, 778–789 CrossRef CAS PubMed.
- K. Nurgali, R. T. Jagoe and R. Abalo, Front. Pharmacol, 2018, 9, 245 CrossRef PubMed.
- H. Wang, H. Guo, J. Yang, Y. Liu, X. Liu, Q. Zhang and K. Zhou, Exp. Hematol. Oncol., 2022, 11, 60 CrossRef CAS PubMed.
- S. Gaballa and J. Pinilla-Ibarz, Curr. Hematol. Malig. Rep., 2021, 1–11 Search PubMed.
- C. Owen, N. L. Berinstein, A. Christofides and L. H. Sehn, Curr. Oncol., 2019, 26, 233–240 CrossRef PubMed.
- H. A. Abbas and W. G. Wierda, Front. Oncol., 2021, 11, 668162 CrossRef CAS PubMed.
- J. A. Burger, Cancer J., 2019, 25, 386–393 CrossRef CAS PubMed.
- D. S. Wishart, Y. D. Feunang, A. C. Guo, E. J. Lo, A. Marcu, J. R. Grant, T. Sajed, D. Johnson, C. Li, Z. Sayeeda, N. Assempour, I. Iynkkaran, Y. Liu, A. Maciejewski, N. Gale, A. Wilson, L. Chin, R. Cummings, D. Le, A. Pon, C. Knox and M. Wilson, Nucleic Acids Res., 2018, 46, D1074–d1082 CrossRef CAS PubMed.
- S. M. Attia, Oxid. Med. Cell. Longevity, 2010, 3, 238–253 CrossRef PubMed.
- T. A. Baillie and A. E. Rettie, Drug Metab. Pharmacokinet., 2011, 26, 15–29 CrossRef CAS PubMed.
- A. Alhoshani, F. E. Alanazi, M. R. Alotaibi, M. W. Attwa, A. A. Kadi, A. Aldhfyan, S. Akhtar, S. Hourani, A. Agouni, A. Zeidan and H. M. Korashy, Chem. Res. Toxicol., 2020, 33, 1719–1728 Search PubMed.
- A. F. Stepan, D. P. Walker, J. Bauman, D. A. Price, T. A. Baillie, A. S. Kalgutkar and M. D. Aleo, Chem. Res. Toxicol., 2011, 24, 1345–1410 Search PubMed.
- M. W. Attwa, A. A. Kadi, A. S. Abdelhameed and H. A. Alhazmi, Drug Des., Dev. Ther., 2020, 14, 783–793 Search PubMed.
- T. A. Baillie and K. Kassahun, Adv. Exp. Med. Biol., 2001, 500, 45–51 CrossRef CAS PubMed.
- S. Ma and R. Subramanian, J. Mass Spectrom., 2006, 41, 1121–1139 CrossRef CAS PubMed.
- A. Tolonen, M. Turpeinen and O. Pelkonen, Drug Discovery Today, 2009, 14, 120–133 CrossRef CAS PubMed.
- M. W. Attwa, A. A. Kadi and A. S. Abdelhameed, RSC Adv., 2018, 8, 38733–38744 RSC.
- S. Ma and M. Zhu, Chem.–Biol. Interact., 2009, 179, 25–37 CrossRef CAS PubMed.
- L. P. Mašič, Curr. Drug Metab., 2011, 12, 35–50 CrossRef PubMed.
- A. A. Kadi, S. M. Amer, H. W. Darwish and M. W. Attwa, RSC Adv., 2017, 7, 36279–36287 RSC.
- A. S. Abdelhameed, M. W. Attwa and A. A. Kadi, R. Soc. Open Sci., 2019, 6, 190852 CrossRef CAS PubMed.
- A. S. Abdelhameed, M. W. Attwa and A. A. Kadi, Drug Des., Dev. Ther., 2020, 14, 5259–5273 CrossRef CAS PubMed.
- M. W. Attwa, A. A. Kadi and A. S. Abdelhameed, RSC Adv., 2020, 10, 5412–5427 RSC.
- H. Yamada, H. Hayashi and Y. Natori, J. Biochem., 1984, 95, 1155–1160 CrossRef CAS PubMed.
- J. M. Clarkson, J. E. Martin and D. E. F. McKeegan, Lab. Anim., 2022, 56, 419–436 CrossRef CAS PubMed.
- R. von Jagow, H. Kampffmeyer and M. Kinese, Naunyn-Schmiedebergs Arch. Exp. Pathol. Pharmakol., 1965, 251, 73–87 CrossRef CAS PubMed.
- A. A. Kadi, H. W. Darwish, M. W. Attwa and S. M. Amer, RSC Adv., 2016, 6, 72575–72585 RSC.
- M. W. Attwa, A. A. Kadi, H. W. Darwish, S. M. Amer and N. S. Al-shakliah, Chem. Cent. J., 2018, 12, 99–115 CrossRef CAS PubMed.
- S. M. Amer, A. A. Kadi, H. W. Darwish and M. W. Attwa, RSC Adv., 2017, 7, 4479–4491 RSC.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Mohamed W. Attwa
,
Mohamed W. Attwa