
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of novel bioactive pyrido[2,3-d]pyrimidine derivatives with potent cytotoxicity through apoptosis as PIM-1 kinase inhibitors†
Eman S. Tantawya,
Mohamed S. Nafie *bc,
Hesham A. Morsyd,
Hassan A. El-Sayeda,
Ahmed H. Moustafaa and
Samar M. Mohammeda
*bc,
Hesham A. Morsyd,
Hassan A. El-Sayeda,
Ahmed H. Moustafaa and
Samar M. Mohammeda
aDepartment of Chemistry, Faculty of Science, Zagazig University, Zagazig 44519, Egypt
bDepartment of Chemistry, College of Sciences, University of Sharjah, P. O. 27272, Sharjah, United Arab Emirates. E-mail: mohamed.elsayed@sharjah.ac.ae
cChemistry Department, Faculty of Science, Suez Canal University, P. O. 41522, Ismailia, Egypt. E-mail: mohamed_nafie@science.suez.edu.eg
dHigher Institution of Engineering & Modern Technology, Elmarg, Cairo, 13774, Egypt
First published on 5th April 2024
Abstract
Direct synthesis and cytotoxicity activity of new series of pyrido[2,3-d]pyrimidine was described. Nicotinamide 2 was synthesized via cyclization of N-cyclohexyl derivative with cyanoacetamide. The o-aminonicotinonitrile 2 was subjected to acylation or thio acylation process followed by intramolecular heterocyclization to afford the desired pyrido[2,3-d]pyrimidine (3–10) and pyrido triazine 11. Compounds 4 and 11 exhibited remarkable cytotoxicity against MCF-7 cells with IC50 values of 0.57 μM and 1.31 μM and IC50 values of 1.13 μM and 0.99 μM against HepG2 cells. Interestingly, compounds 4 and 10 had potent PIM-1 kinase inhibition with IC50 values of 11.4 and 17.2 nM, respectively, with inhibition of 97.8% and 94.6% compared to staurosporine (IC50 = 16.7 nM, with 95.6% inhibition). Moreover, compound 4 significantly activated apoptosis in MCF-7 cells, increasing the cell apoptosis by 58.29-fold by having 36.14% total apoptosis in treated cells compared to 0.62% for control. Moreover, it arrested the cell cycle at the G1 phase. PIM-1 kinase inhibition was virtually elucidated by the molecular docking study, highlighting binding interactions of the lead compound 4 towards the PIM-1 protein. Accordingly, compound 4 was validated as a promising PIM-1 targeted chemotherapeutic agent to treat breast cancer.
1 Introduction
Cancer is among the worst diseases that people face today. It ranks as the world's second-leading killer.1,2 Cancer ranks high among the leading causes of mortality on a global scale, according to global statistics.3,4 Modern medicine increasingly relies on medicinal chemists to identify targeted chemotherapy drugs.5,6 By binding to specific molecular targets, these medications cause severe damage to cancer cells, therefore inhibiting their proliferation.7,8 Because of their many biological uses, pyridine nuclei are significant nitrogen-containing heterocyclic systems. Important biological processes include anticancer,9 anti-Alzheimer's disease,10 antibacterial,11 antitubercular,12 antifungal,13 anti-inflammatory14 activities, and the management of several cardiovascular disorders, including hypertension and angina.15 In addition, pyridine derivatives were shown to halt the proliferation of human MCF-7 and HepG-2 cancer cells by increasing JNK levels and causing G2/M phase arrest via a p53–p21-driven pathway. Among the many well-known pharmacophores in medicinal chemistry are pyridopyrimidines and other bicyclic nitrogen-containing heterocyclic compounds. A great deal of research has focused on pyrido[2,3-d]pyrimidines. There have been reports of their anticonvulsant, antibacterial, anticancer, antipyretic, and analgesic properties. PIM-1 is highly expressed in many different types of cancer and has an impact on carcinogenesis, cell cycle progression, cell proliferation, cell death, and cell migration, making it a promising target for the development of new cancer drugs. The phosphorylation and regulation of several proteins involved in cell survival, proliferation, and apoptosis are crucial functions of PIM-1 kinase, which is why it is associated with numerous cancer forms.16 It has been shown to be excessively expressed in numerous forms of solid tumors such as prostate as well as hematological malignancy such as leukemia, multiple myeloma, and diffuse large B cell lymphomas (DLBCL).17 Fig. 1 shows a number of pyridine and pyridopyrimidine (I–IV) compounds that have potent anticancer effects, and it also includes some of our earlier publications on cyanopyridine-based compounds that have been demonstrated to be effective PIM-1 kinase inhibitors that induce cell death.18–20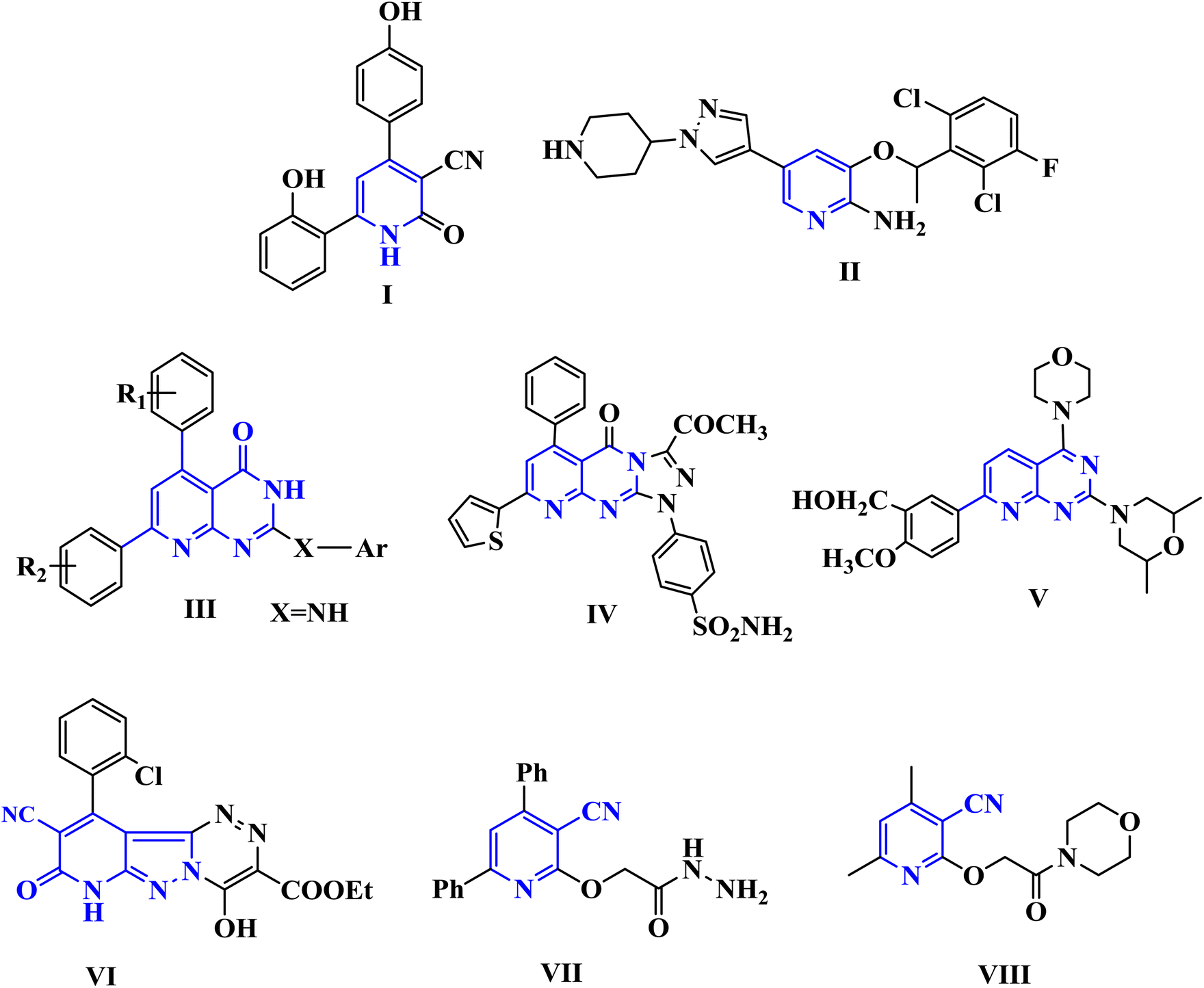 | ||
| Fig. 1 Biologically active pyridine and pyridopyrimidines containing anti-cancer agents (I–V) and cyanopyridine-based compounds (VI–VIII) as PIM-1 kinase inhibitors. | ||
Accordingly, new series of pyridines and pyridopyrimidines were rationally designed and synthesized, as highlighted Fig. 2, where the three pharmacophoric regions of lipophilic, hydrophilic moieties with the main scaffold of pyridopyrimidine were highlighted. It was our intention to synthesize new derivatives with the same scaffold and different incorporated derivatives and to assess the anticancer activity of new substituted pyridine and pyridopyrimidines derivatives for more effective and target-oriented chemotherapeutics, as well as to study their molecular target, cytotoxicity, and cell death mechanism in both in situ and in vitro studies.
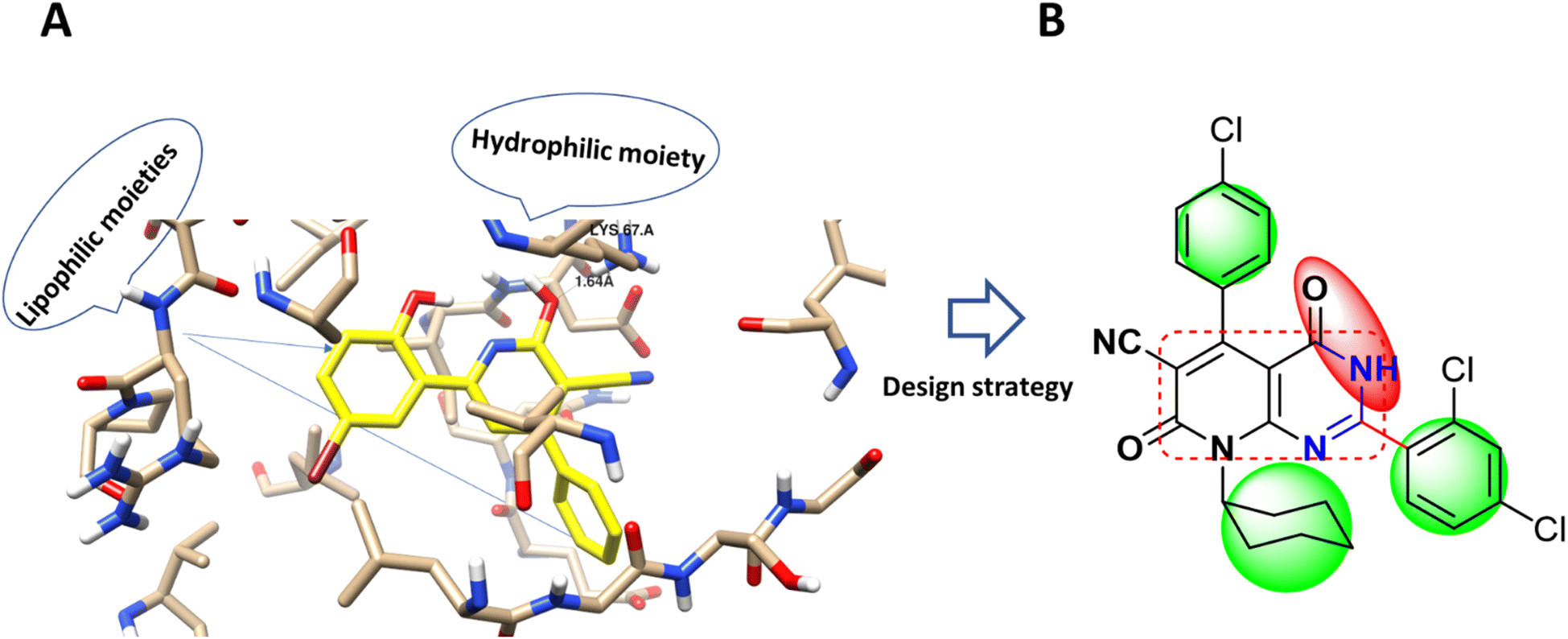 | ||
| Fig. 2 (A) Molecular modeling of PIM-1 binding site (PDB 2OBJ) and the interaction pose with the co-crystallized ligand as a cyano-pyridine derivative (yellow-colored). (B) Design strategy of the target compound with the anchored pharmacophoric groups; lipophilic moiety (green) and hydrophilic (red) with the main scaffold (square circled). | ||
It was reported that incorporating pyridine derivatives exhibited potent anticancer activity against cancer cell lines such as HepG2 and MCF-7.10
2 Material and methods
2.1. Chemistry
All melting points were uncorrected and were measured using an Electrothermal IA 9100 apparatus. The IR spectra (KBr discs) were recorded on a Pye Unicam Sp-3-300 or a Shimadzu FTIR 8101 PC infrared spectrophotometer “Cairo University, Cairo, Egypt”. The operation frequency was 400 MHz for 1H NMR and 100 MHz for 13C NMR using BRUKER 400 MHz spectrometer at Zagazig University, Faculty of Science, Zagazig, Egypt “Nucleic Acid Center Research”. Elemental analyses were determined using PerkinElmer 240 “Cairo University, Cairo, Egypt”. The coupling constants (J) are given in hertz. The chemical shifts are expressed on the δ (ppm) scale using TMS as the standard reference.3-(4-Chlorophenyl)-2-cyano-N-cyclohexylacrylamide (1) was synthesized as reported method.21 Mp = 178–180 °C. yield 95% [reported mp = 169–170 °C].
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) and 1665, 1650, (2C
N) and 1665, 1650, (2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, amide). 1H NMR (DMSO-d6), δ (ppm): 0.72–1.46 (m, 11H, Hcyclohexyl), 6.96 (s, 2H, NH2, exchange with D2O), 7.29 (d, 2H, J = 8.80 Hz, Ar–H), 7.51 (d, 2H, J = 8.80 Hz, Ar–H), 11.45 (s, 2H, NH2, exchange with D2O). 13C NMR (DMSO-d6), δ (ppm): 23.72, 25.04, 25.35, 32.10, 81.61, 116.3 (CN), 118.6, 128.4, 128.7, 129.9, 130.4, 132.8, 133.1, 160.9 and 166.0. Anal. calcd for C19H19ClN4O2 (370.83): C, 61.54; H, 5.16; N, 15.11. Found: C, 61.53; H, 5.14; N, 15.10.N) and 1664 (2CO, amide). 1H NMR (DMSO-d6), δ(ppm): 1.08–1.66 (m, 11H, Hcyclohexyl), 7.25 (d, 2H, J = 8.40 Hz, Ar–H), 7.32 (d, 2H, J = 8.48 Hz, Ar–H), 7.38 (m, 2H, Ar–H), 7.50 (t, 1H, J = 7.69 Hz, Ar–H), 8.17 (d, 2H, J = 8.40 Hz, Ar–H), 11.48 (s, 1H, NH, exchange with D2O). Anal. calcd for C26H21ClN4O2 (456.92): C, 68.34; H, 4.63; N, 12.26. Found: C, 68.32; H, 4.64; N, 12.27.N) and 1698, 1676 (2CO, amide). 1H NMR (DMSO-d6) δ (ppm): 1.08–1.71 (m, 11H, Hcyclohexyl), 7.08–7.95 (m, 7H, Ar–H), 11.60 (s, 1H, NH, exchange with D2O). 13C NMR (DMSO-d6), δ (ppm): 23.72, 24.14, 25.38, 34.29, 85.10, 100.3, 104.7, 117.5 (CN), 127.9, 128.0, 128.7, 129.9, 133.4, 133.8, 135.2, 153.2, 155.3, 157.2, 159.7, 160.0, 162.3, 163.2. Anal. calcd for C26H19Cl3N4O2 (525.81): C, 59.39; H, 3.64; N, 10.66. Found: C, 59.28; H, 3.60; N, 10.71.N) and 1649 (br, 2CO amide). 1H NMR (DMSO-d6) δ (ppm): 1.05–1.47 (m, 11H, Hcyclohexyl), 6.98 (s, 1H, Hfuryl), 7.29–7.52 (m, 6H, Ar–H), 11.49 (s, 1H, NH, exchange with D2O). 13C NMR (DMSO-d6), δ (ppm): 22.39, 24.13, 25.04, 31.88, 67.40, 117.6 (CN), 128.0, 128.3, 128.4, 128.6, 128.7, 129.9, 130.9, 131.5, 133.7, 135.3, 155.6, 160.8, 163.3, 166.9. Anal. Calcd for C24H19ClN4O3 (446.89): C, 64.50; H, 4.29; N, 12.54. Found: C, 64.59; H, 4.30; N, 12.49.N) and 1665 (br, 2CO, amide). 1H NMR (DMSO-d6) δ (ppm): 0.70–1.84 (m, 11H, Hcyclohexyl), 4.73 (s, 2H, CH2), 7.49 (d, 2H, J = 8.28 Hz, Ar–H), 8.14 (d, 2H, J = 8.28 Hz, Ar–H), 11.88 (s, 1H, NH exchange with D2O). Anal. calcd for C21H18Cl2N4O2 (429.30): C, 58.75; H, 4.23; N, 13.05. Found: C, 58.64; H, 4.21; N, 13.10.N) and 1718, 1659, (3CO ester and amide). 1H NMR (DMSO-d6), δ (ppm): 1.18 (t, 3H, 3J = 5.72 Hz, CH2C
O, amide). 1H NMR (DMSO-d6), δ (ppm): 0.72–1.46 (m, 11H, Hcyclohexyl), 6.96 (s, 2H, NH2, exchange with D2O), 7.29 (d, 2H, J = 8.80 Hz, Ar–H), 7.51 (d, 2H, J = 8.80 Hz, Ar–H), 11.45 (s, 2H, NH2, exchange with D2O). 13C NMR (DMSO-d6), δ (ppm): 23.72, 25.04, 25.35, 32.10, 81.61, 116.3 (CN), 118.6, 128.4, 128.7, 129.9, 130.4, 132.8, 133.1, 160.9 and 166.0. Anal. calcd for C19H19ClN4O2 (370.83): C, 61.54; H, 5.16; N, 15.11. Found: C, 61.53; H, 5.14; N, 15.10.N) and 1664 (2CO, amide). 1H NMR (DMSO-d6), δ(ppm): 1.08–1.66 (m, 11H, Hcyclohexyl), 7.25 (d, 2H, J = 8.40 Hz, Ar–H), 7.32 (d, 2H, J = 8.48 Hz, Ar–H), 7.38 (m, 2H, Ar–H), 7.50 (t, 1H, J = 7.69 Hz, Ar–H), 8.17 (d, 2H, J = 8.40 Hz, Ar–H), 11.48 (s, 1H, NH, exchange with D2O). Anal. calcd for C26H21ClN4O2 (456.92): C, 68.34; H, 4.63; N, 12.26. Found: C, 68.32; H, 4.64; N, 12.27.N) and 1698, 1676 (2CO, amide). 1H NMR (DMSO-d6) δ (ppm): 1.08–1.71 (m, 11H, Hcyclohexyl), 7.08–7.95 (m, 7H, Ar–H), 11.60 (s, 1H, NH, exchange with D2O). 13C NMR (DMSO-d6), δ (ppm): 23.72, 24.14, 25.38, 34.29, 85.10, 100.3, 104.7, 117.5 (CN), 127.9, 128.0, 128.7, 129.9, 133.4, 133.8, 135.2, 153.2, 155.3, 157.2, 159.7, 160.0, 162.3, 163.2. Anal. calcd for C26H19Cl3N4O2 (525.81): C, 59.39; H, 3.64; N, 10.66. Found: C, 59.28; H, 3.60; N, 10.71.N) and 1649 (br, 2CO amide). 1H NMR (DMSO-d6) δ (ppm): 1.05–1.47 (m, 11H, Hcyclohexyl), 6.98 (s, 1H, Hfuryl), 7.29–7.52 (m, 6H, Ar–H), 11.49 (s, 1H, NH, exchange with D2O). 13C NMR (DMSO-d6), δ (ppm): 22.39, 24.13, 25.04, 31.88, 67.40, 117.6 (CN), 128.0, 128.3, 128.4, 128.6, 128.7, 129.9, 130.9, 131.5, 133.7, 135.3, 155.6, 160.8, 163.3, 166.9. Anal. Calcd for C24H19ClN4O3 (446.89): C, 64.50; H, 4.29; N, 12.54. Found: C, 64.59; H, 4.30; N, 12.49.N) and 1665 (br, 2CO, amide). 1H NMR (DMSO-d6) δ (ppm): 0.70–1.84 (m, 11H, Hcyclohexyl), 4.73 (s, 2H, CH2), 7.49 (d, 2H, J = 8.28 Hz, Ar–H), 8.14 (d, 2H, J = 8.28 Hz, Ar–H), 11.88 (s, 1H, NH exchange with D2O). Anal. calcd for C21H18Cl2N4O2 (429.30): C, 58.75; H, 4.23; N, 13.05. Found: C, 58.64; H, 4.21; N, 13.10.N) and 1718, 1659, (3CO ester and amide). 1H NMR (DMSO-d6), δ (ppm): 1.18 (t, 3H, 3J = 5.72 Hz, CH2C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3), 1.29–1.79 (m, 11H, Hcyclohexyl), 4.37 (q, 2H, 3J = 5.72 Hz, C2CH3), 7.63 (d, 2H, J = 8.48 Hz, Ar–H), 7.93 (d, 2H, J = 8.48 Hz, Ar–H), 8.10 (s, 1H, NH, exchange with D2O). Anal. calcd for C23H21ClN4O4 (452.89): C, 61.00; H, 4.67; N, 12.37. Found: C, 61.02; H, 4.70; N, 12.35.N) and 1647, 1668 (2CO, amide). 1H NMR (DMSO-d6), δ (ppm): 1.14 (t, 3H, 3J = 12.68 Hz, CH2C3), 1.25–1.80 (m, 11H, Hcyclohexyl), 4.31 (q, 2H, 3J = 11.84 Hz, C2CH3), 7.33 (d, 2H, J = 8.24 Hz, Ar–H), 7.51 (d, 2H, J = 8.24 Hz, Ar–H), 13.22 (s, 1H, NH exchange with D2O). 13C NMR (DMSO-d6), δ (ppm): 25.00, 25.82, 31.68, 54.10 (O–CH2), 101.5, 105.2, 115.4 (CN), 128.4, 128.7, 129.5, 133.8, 135.7, 153.7, 155.8, 156.9, 160.1 and 160.5. Anal. calcd for C22H21ClN4O3 (424.88): C, 62.19; H, 4.98; N, 13.19. Found: C, 62.22; H, 4.97; N, 13.18.N) and 1681 (br, 2CO, amide). 1H NMR (DMSO-d6), δ (ppm): 1.23–1.87 (m, 11H, Hcyclohexyl), 7.35 (m, 4H, Ar–H), 7.95 (s, 1H, Ar–H), 13.22 (s, 1H, NH exchange with D2O). Anal. calcd for C20H17ClN4O2 (380.83): C, 63.08; H, 4.50; N, 14.71. Found: C, 63.04; H, 4.51; N, 14.73.N) and 1670, 1643 (2CO, amide) and 1323 (CS). 1H NMR (DMSO-d6) δ (ppm): 1.08–1.66 (m, 11H, Hcyclohexyl), 7.25 (d, 2H, J = 8.32 Hz, Ar–H), 7.38 (d, 2H, J = 8.20 Hz, Ar–H), 11.20, 12.60 (s, 2H, 2NH, exchange with D2O). 13C NMR (DMSO-d6), δ (ppm): 23.48, 24.14, 25.06, 32.10, 107.7, 116.7 (CN), 128.2, 129.3, 130.3, 130.7, 131.5, 132.7, 133.1, 134.8, 148.5, 161.5. Anal. calcd for C20H17ClN4O2S (412.89): C, 58.18; H, 4.15; N, 13.57. Found: C, 58.09; H, 4.17; N, 13.52.N), 1681 (2CO, amide) and 1549 (NN). 1H NMR (DMSO-d6), δ (ppm): 1.04–1.58 (m, 11H, Hcyclohexyl) 7.18 (d, 2H, J = 8.32 Hz, Ar–H), 7.29 (d, 2H, J = 8.32 Hz, Ar–H), 11.81 (s, 1H, NH exchange with D2O). Anal. calcd for C19H16ClN5O2 (381.82): C, 59.77; H, 4.22; N, 18.34. Found: C, 59.78; H, 4.25; N, 18.35.
3), 1.29–1.79 (m, 11H, Hcyclohexyl), 4.37 (q, 2H, 3J = 5.72 Hz, C2CH3), 7.63 (d, 2H, J = 8.48 Hz, Ar–H), 7.93 (d, 2H, J = 8.48 Hz, Ar–H), 8.10 (s, 1H, NH, exchange with D2O). Anal. calcd for C23H21ClN4O4 (452.89): C, 61.00; H, 4.67; N, 12.37. Found: C, 61.02; H, 4.70; N, 12.35.N) and 1647, 1668 (2CO, amide). 1H NMR (DMSO-d6), δ (ppm): 1.14 (t, 3H, 3J = 12.68 Hz, CH2C3), 1.25–1.80 (m, 11H, Hcyclohexyl), 4.31 (q, 2H, 3J = 11.84 Hz, C2CH3), 7.33 (d, 2H, J = 8.24 Hz, Ar–H), 7.51 (d, 2H, J = 8.24 Hz, Ar–H), 13.22 (s, 1H, NH exchange with D2O). 13C NMR (DMSO-d6), δ (ppm): 25.00, 25.82, 31.68, 54.10 (O–CH2), 101.5, 105.2, 115.4 (CN), 128.4, 128.7, 129.5, 133.8, 135.7, 153.7, 155.8, 156.9, 160.1 and 160.5. Anal. calcd for C22H21ClN4O3 (424.88): C, 62.19; H, 4.98; N, 13.19. Found: C, 62.22; H, 4.97; N, 13.18.N) and 1681 (br, 2CO, amide). 1H NMR (DMSO-d6), δ (ppm): 1.23–1.87 (m, 11H, Hcyclohexyl), 7.35 (m, 4H, Ar–H), 7.95 (s, 1H, Ar–H), 13.22 (s, 1H, NH exchange with D2O). Anal. calcd for C20H17ClN4O2 (380.83): C, 63.08; H, 4.50; N, 14.71. Found: C, 63.04; H, 4.51; N, 14.73.N) and 1670, 1643 (2CO, amide) and 1323 (CS). 1H NMR (DMSO-d6) δ (ppm): 1.08–1.66 (m, 11H, Hcyclohexyl), 7.25 (d, 2H, J = 8.32 Hz, Ar–H), 7.38 (d, 2H, J = 8.20 Hz, Ar–H), 11.20, 12.60 (s, 2H, 2NH, exchange with D2O). 13C NMR (DMSO-d6), δ (ppm): 23.48, 24.14, 25.06, 32.10, 107.7, 116.7 (CN), 128.2, 129.3, 130.3, 130.7, 131.5, 132.7, 133.1, 134.8, 148.5, 161.5. Anal. calcd for C20H17ClN4O2S (412.89): C, 58.18; H, 4.15; N, 13.57. Found: C, 58.09; H, 4.17; N, 13.52.N), 1681 (2CO, amide) and 1549 (NN). 1H NMR (DMSO-d6), δ (ppm): 1.04–1.58 (m, 11H, Hcyclohexyl) 7.18 (d, 2H, J = 8.32 Hz, Ar–H), 7.29 (d, 2H, J = 8.32 Hz, Ar–H), 11.81 (s, 1H, NH exchange with D2O). Anal. calcd for C19H16ClN5O2 (381.82): C, 59.77; H, 4.22; N, 18.34. Found: C, 59.78; H, 4.25; N, 18.35.2.2. Biology
2.2.3.1. Annexin V/PI staining and cell cycle analysis. MCF-7 cells were seeded into 6-well culture plates (3–5 × 105 cells per well) and incubated overnight. Cells were treated with compound 4 at their IC50 values for 48 h. Next, media supernatants and cells were collected and rinsed with ice-cold PBS. Then, cells were suspended the cells in 100 μL of annexin binding buffer solution “25 mM CaCl2, 1.4 M NaCl, and 0.1 M Hepes/NaOH, pH 7.4” and incubated with “annexin V-FITC solution (1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100) and propidium iodide (PI)” at a concentration equals 10 μg mL−1 in the dark for 30 min. Stained cells were then acquired by BD FACSCalibur™ Flow Cytometer.26–28
:100) and propidium iodide (PI)” at a concentration equals 10 μg mL−1 in the dark for 30 min. Stained cells were then acquired by BD FACSCalibur™ Flow Cytometer.26–28
2.2.3.2. Real time-polymerase chain reaction for the selected genes. Gene expression of Bcl-2, the anti-apoptotic gene, and the pro-apoptotic genes P53, Bax, and caspases-3,8,9 were examined to delve deeper into the apoptotic pathway. MCF-7 cells were treated with compound 4 at their IC50 values for 48 h. After treatment, the RT-PCR reaction was carried out following routine work. Then, the Ct values were collected to calculate the relative genes' expression in all samples by normalization to the β-actin housekeeping gene.26,29
3 Results and discussion
3.1. Chemistry
The synthesis of the target o-aminonicotinoamide 2 was outlined in (Scheme 1) via Michael type addition of cyanoacetamide to 2-cyano-N-cyclohexyl acrylamide (1) followed by intramolecular cyclization in the presence of piperidine as a mild catalytic base. The reaction smoothly proceeded in ethanol, and the product was obtained in high yield (91%).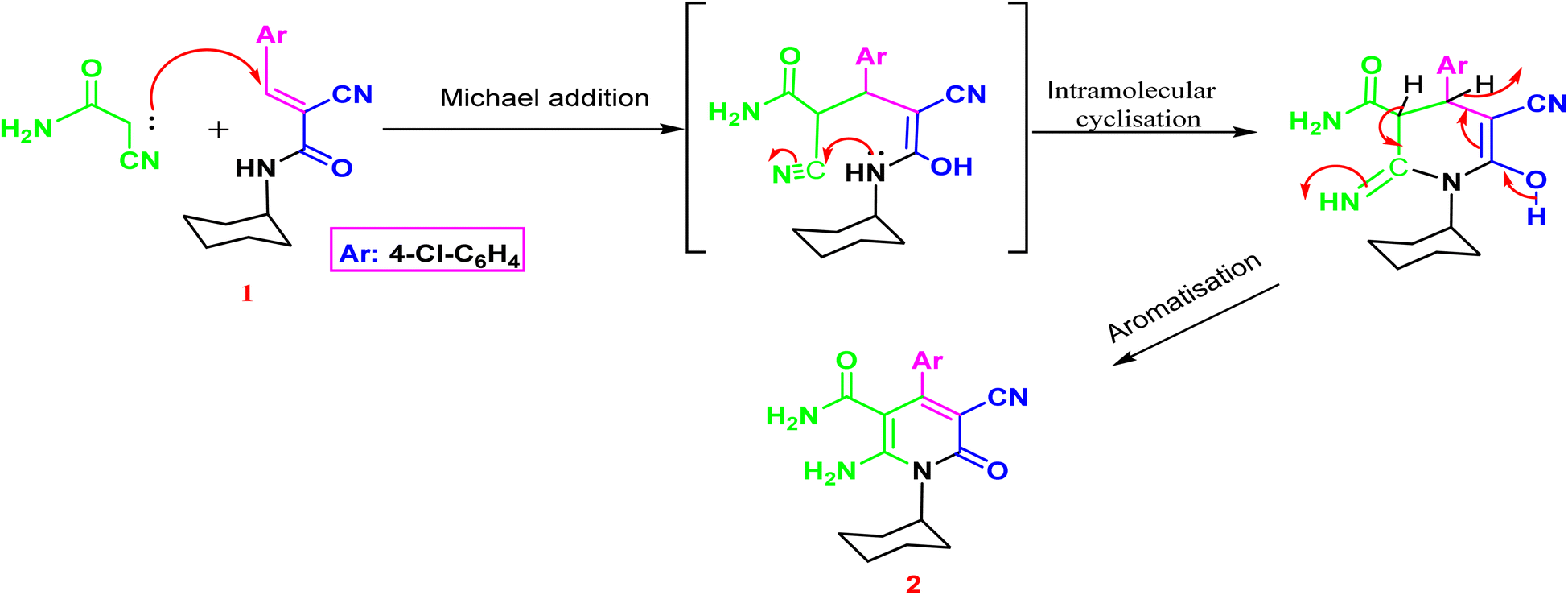 | ||
| Scheme 1 The plausible mechanism for the formation of 1,6-dihydropyridine-3-carboxamide 2. | ||
The characteristic IR bands of 2 show the presence of stretching bands at 3325, 3300, 2210, 1665 and 1650 cm−1 for 2NH2, CN, and 2CO, which evidence the formation of the structure. The 1H NMR signals confirm its formation via the presence of cyclohexyl protons as multiplet at 0.72–1.46 ppm, and the two amino groups appeared as two main singlets at δH 6.96 and 11.45 ppm, which exchange with D2O. Furthermore, the aromatic protons split into two doublets at 7.29 and 7.51 ppm with a coupling constant (J = 8.80 Hz). The nicotinamide 2 contains o-amino-amide functions that enable such compounds to act as trapping for an electron-deficient carbon atom, resulting in cyclization and formation of a new annulated pyrimidine nucleus Fig. 3.
 | ||
| Fig. 3 The plausible cyclization of pyrimidine nucleus. | ||
The acylation of nicotinamide 2 with acid chloride, namely, (benzoyl chloride, 2,4-dichloro benzoyl chloride, furoyl chloride, chloroacetyl chloride) and ester (e.g. diethyl oxalate), triethylorthoformate and formamide or thio acylation with carbon disulfide afforded the corresponding pyrido[2,3-d]pyrimidine 3–10 according to (Schemes 2 and 3). The structure of the desired compound is agreed with their spectral analysis. For example, the IR confirms the disappearance of the primary NH2 groups and the presence of secondary NH between 3441 and 3271. The 1H NMR spectra are another evidence for the formation of the target pyrimidines.
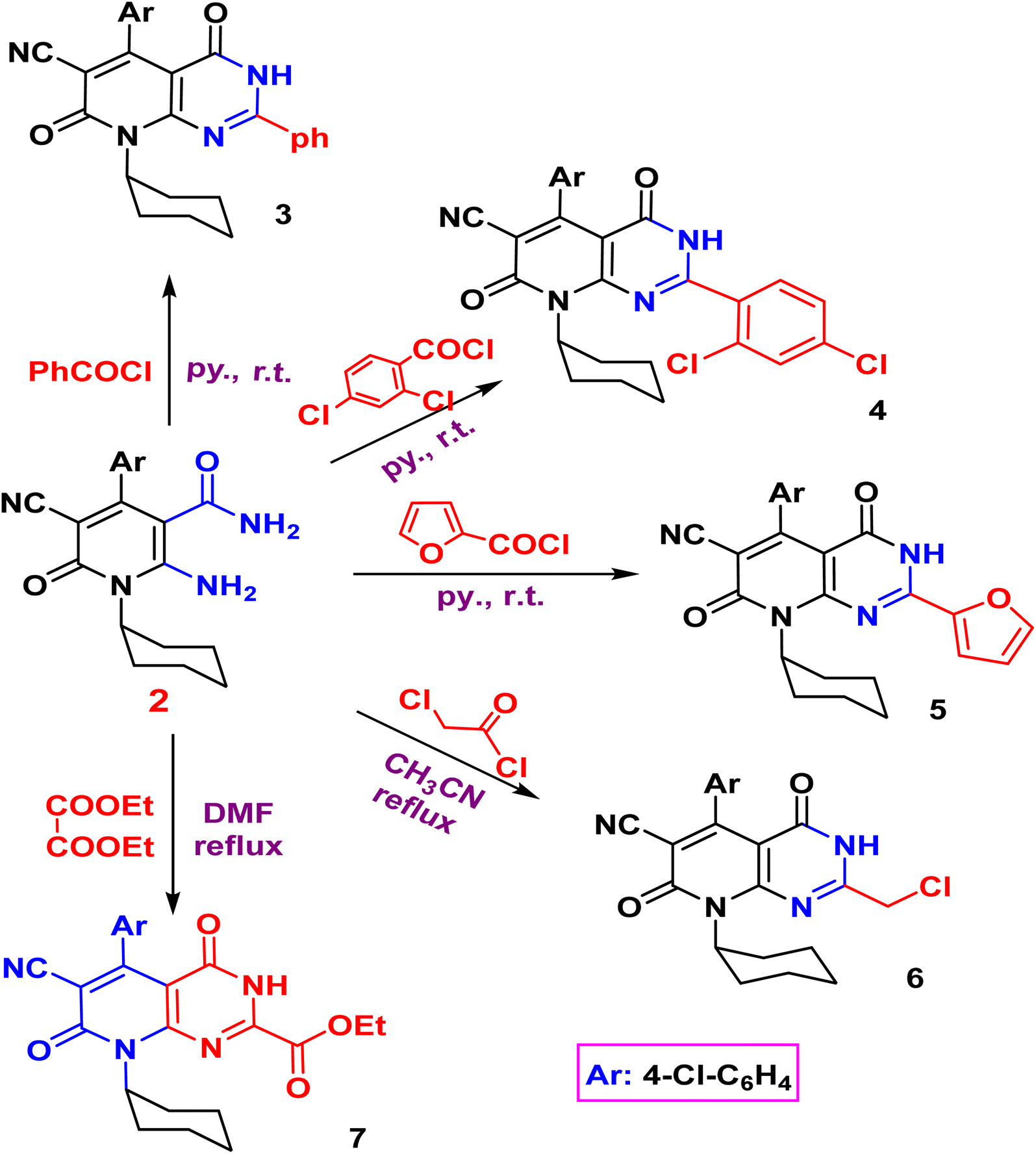 | ||
| Scheme 2 Synthesis of 2-arylpyrido[2,3-d]pyrimidines 3–7. | ||
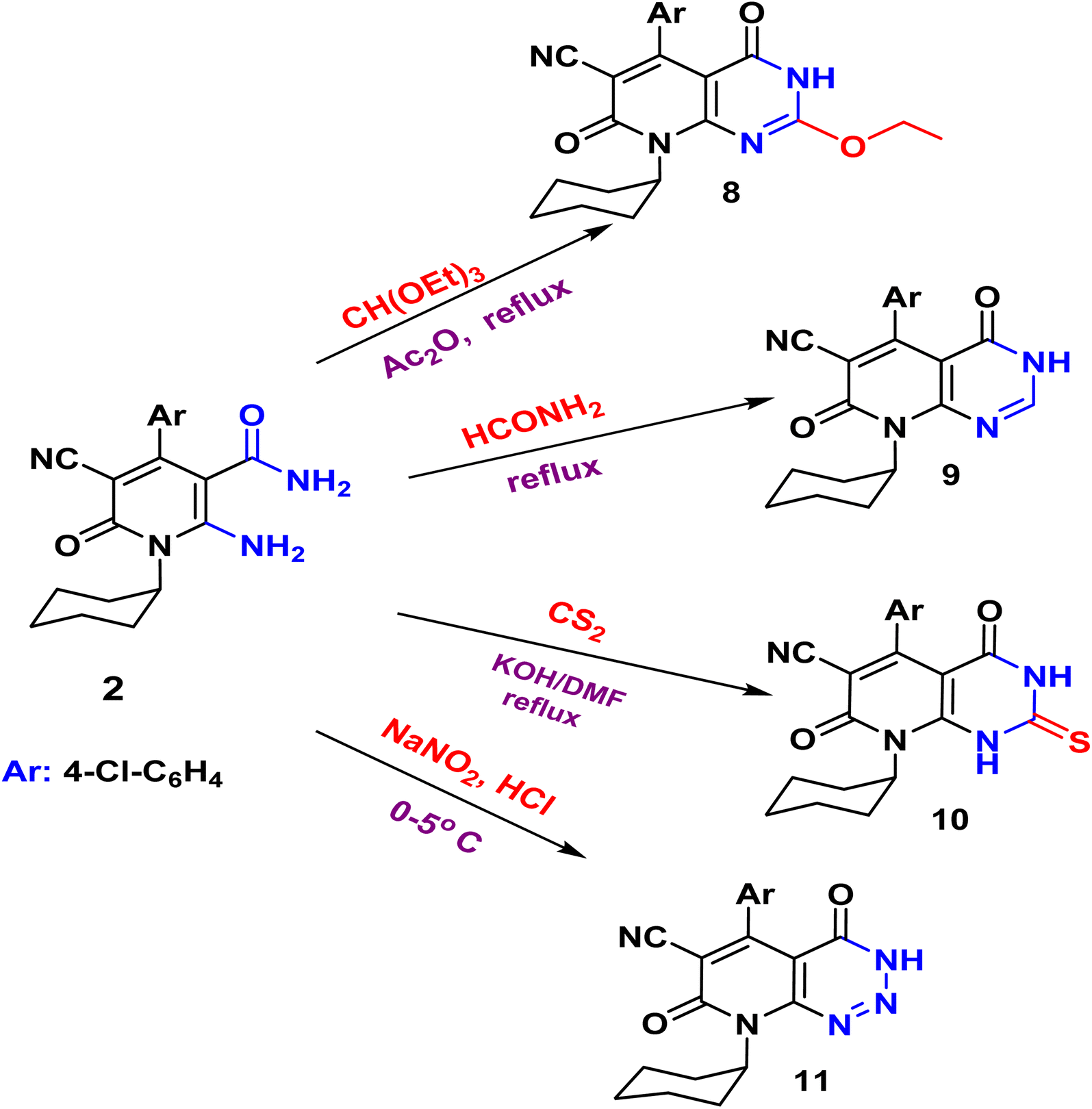 | ||
| Scheme 3 Synthesis of pyridopyrimidines and triazine (8–10 and 11). | ||
Compound 3 shows the presence of phenyl group in its 1H NMR spectrum as doublet, multiplet, and triplet at 7.32, 7.38, and 7.50 ppm, respectively. Cyclization of 2-amino-1,6-dihydropyridine-3-carboxamide 2 with chloroacetyl chloride yielded pyridopyrimidinone 6. The IR spectrum revealed the absorption band of NH at 3429 cm−1 and CN function at 2215 cm−1. While the two carbonyl amide groups appeared as broad band at 1665 cm−1. The 1H NMR spectrum showed a singlet signal at 4.73 ppm assigned for the two protons of CH2 and a singlet signal at δ = 11.88 ppm for NH proton. On the other hand, compound 2 underwent refluxing with diethyl oxalate in DMF to afford the pyridopyrimidine ester derivative 7. Compound 7 shows the presence of ethyl ester group via the presence of CO ester in its IR at 1718 cm−1 and ethyl group of ester as triplet and quartet at 1.18 and 4.37 ppm in its 1H NMR, in addition to NH proton at 8.10 ppm. Also, the structures of 8, 9, and 10 were confirmed by the usual spectroscopic techniques and elemental analysis. The IR spectra of compound 8 displayed a lack of the significant two amino group bands and the presence of a strong stretching NH group at 3441 cm−1. Additionally, the 1H NMR spectra for the same compound 8 assigned specific triplet and quartet signals for ethoxy protons at δ 1.14 and 4.31 ppm, respectively, as well as, NH proton at δ 13.22 as a singlet. The IR spectrum of compound 9 showed characteristic absorption bands at 3390 cm−1 for (NH). Another evidence for pyrimidine ring formation is the significant singlet signal corresponding to (CH) at 7.95 ppm.
Finally, the pyrido[2,3-d]triazine 11 was obtained in high yield (97%) via diazotization of nicotinamide 2 with nitrous acid. The expected structure was elucidated through the IR spectrum, in which a significant band for (NH), (2CO), and (NN, triazine) stretched at ν 3233, 1681, and 1549 cm−1, respectively. Moreover, the 1H NMR spectrum displayed significant signals at δ 1.04–1.58 ppm for cyclohexyl protons, and the aromatic protons appeared as doublets at 7.18 and 7.29 ppm, in addition to the characteristic singlet at 11.81 ppm for NH group.
3.2. Biology
| Compounds no. | IC50 ± SD [μM] | ||
|---|---|---|---|
| MCF-7 | HepG2 | MCF-10A | |
| a Values are expressed as mean ± SD of three independent triplets (n = 3). NT: non-tested. Staurosporine is a potent anticancer agent as a potent PIM-1 kinase inhibitor.31 | |||
| 2 | 16.9 ± 0.45 | 15.6 ± 0.69 | NT |
| 3 | ≥50 | 1.17 ± 0.24 | NT |
| 4 | 0.57 ± 0.14 | 1.13 ± 0.19 | 47.6 ± 3.1 |
| 5 | 11.2 ± 0.54 | 13.4 ± 0.27 | NT |
| 6 | 1.29 ± 0.23 | 2.1 ± 0.24 | 43.5 ± 2.9 |
| 7 | 29.6 ± 0.8 | ≥50 | NT |
| 8 | 9.6 ± 0.24 | 8.1 ± 0.24 | NT |
| 9 | 3.15 ± 0.14 | 4.6 ± 0.34 | NT |
| 10 | 1.4 ± 0.24 | 1.79 ± 0.14 | ≥50 |
| 11 | 1.31 ± 0.28 | 0.99 ± 0.17 | ≥50 |
| Staurosporine | 6.76 ± 0.51 | 5.07 ± 0.36 | NT |
Against HepG2 cells, compounds 4, 6, 9, 10, and 11 exhibited potent cytotoxicity against HepG2 with IC50 values range (0.99–4.16 μM) compared to staurosporine (IC50 = 5.07 μM). Additionally, compounds 2, 5, and 8 exhibited promising cytotoxicity with IC50 values range (8.1–15.6 μM). At the same time, other compounds were non-toxic with higher IC50 values.
As shown in Fig. 4 for the dose–response curve of cell viability versus the tested concentration of compound 4 against MCF-7, HepG2 and MCF-10A cells. It caused the highest percentage of cell viability of cancer cells at the highest concentration, while it caused the lowest viability of normal cells; this highlighted the selectivity profile of compound 4. Furthermore, compounds 4, 6, 10, and 11 were worthy of investigation for their cytotoxicity against normal MCF-10A cells, and they were non-toxic with much higher IC50 values.
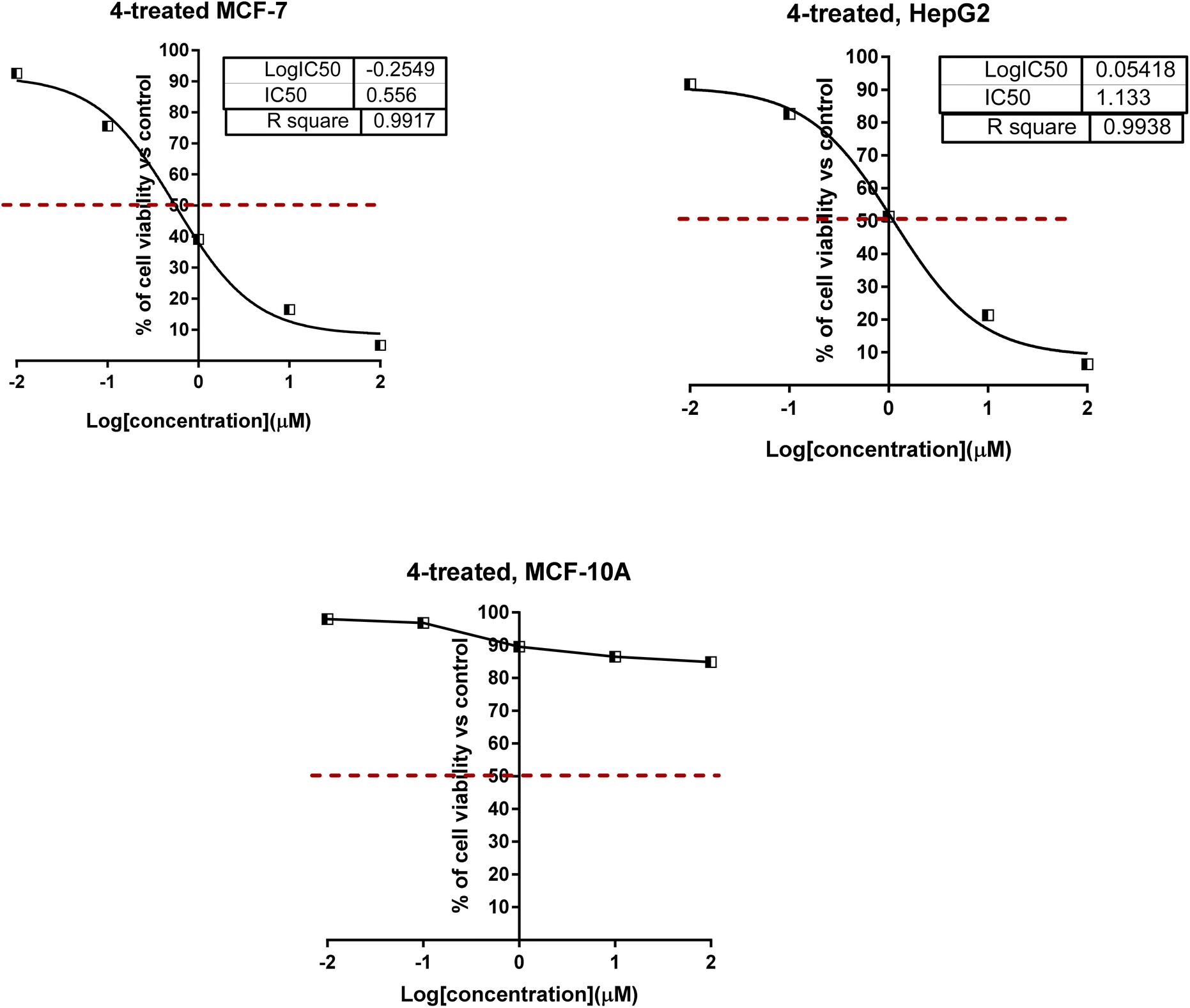 | ||
| Fig. 4 Dose–response nonlinear regression curve fitting the percentage of cell viability vs. log[con. μM], R square ≈ 1 using the GraphPad prism. | ||
Comparing the structure and the cytotoxicity of tested compounds, a valid SAR model can be generated as summarized in Fig. 5. Incorporating hydrophilic, aromatic, hydrogen bond donors and acceptors caused an increase in cytotoxicity, particularly in compounds 4, 6, 10 and 11.
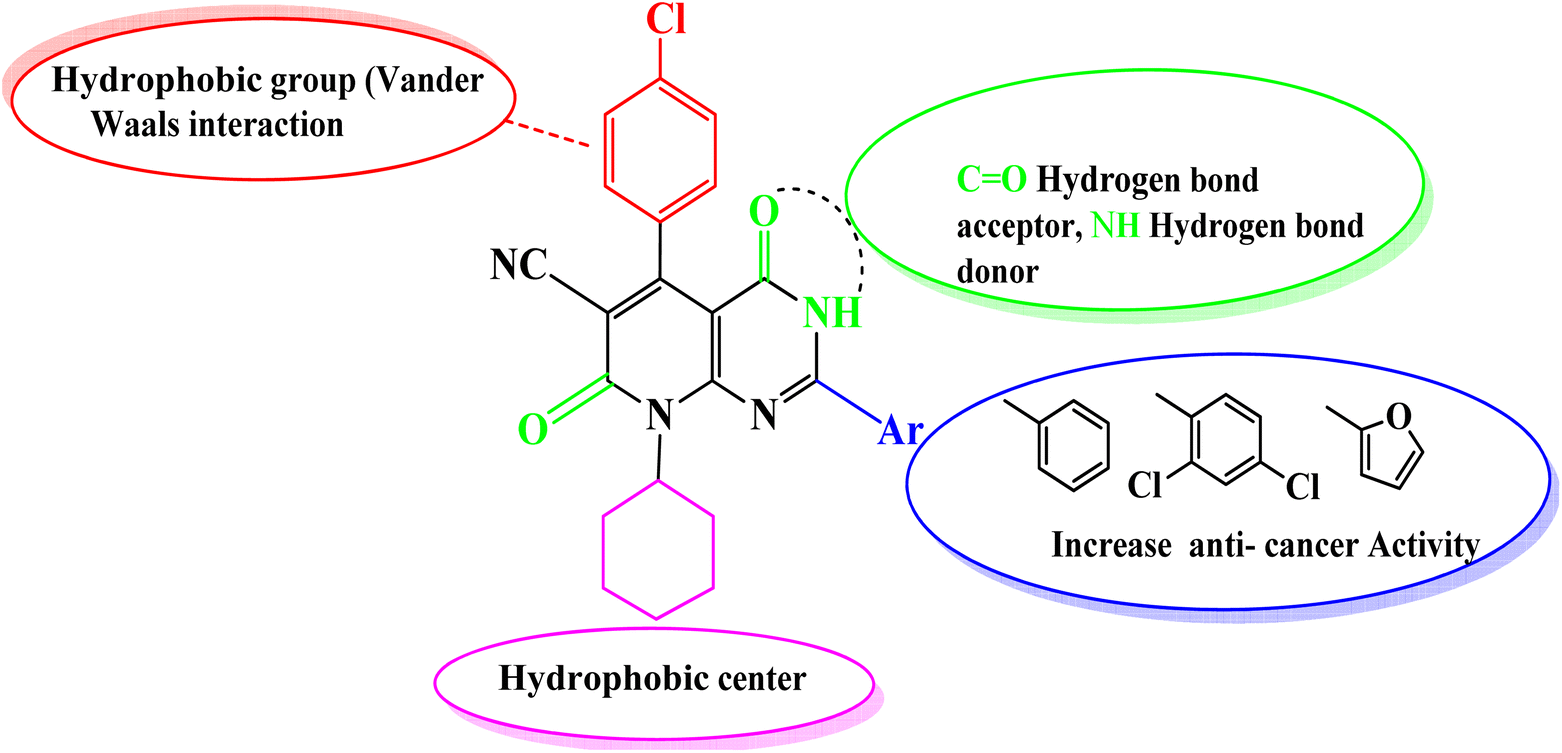 | ||
| Fig. 5 Structure–activity relationship (SAR) model for the activity of the designed derivatives. Substituents anchored on the main scaffold with cytotoxic activities. | ||
| Compound | % of PIM-1 inhibition | IC50 ± SDa (nM) |
|---|---|---|
| a Values are expressed as an average of three independent replicates. “IC50 values were calculated using sigmoidal non-linear regression curve fit of percentage inhibition against five concentrations of each compound”. | ||
| 4 | 97.8 ± 1.8 | 11.4 ± 0.13 |
| 6 | 89.6 ± 2.1 | 34.6 ± 0.18 |
| 10 | 94.6 ± 2.1 | 17.2 ± 0.20 |
| 11 | 92.1 ± 1.9 | 21.4 ± 0.24 |
| Staurosporine | 95.6 ± 2.4 | 16.7 ± 0.32 |
| Pyrido[4,3-d]pyrimidine-derivative inhibitor (SKI-O-068)32 | — | 123 ± 14 |
3.2.3.1. Annexin V/PI staining with cell cycle analysis. The apoptotic activity of compound 4 (IC50 = 0.57 μM, 48 h) was determined by using flow cytometric analysis of annexin V/PI staining to investigate apoptotic cell death in both untreated and treated MCF-7 cells. Fig. 6 (top panel) showed compound 4 dramatically induced apoptotic cell death in MCF-7 cells, increasing the death rate by 58.3-fold; it induced total apoptosis by 36.14% (14.03% for late apoptosis, 22.11% for early apoptosis) compared to the untreated control group (0.62%).
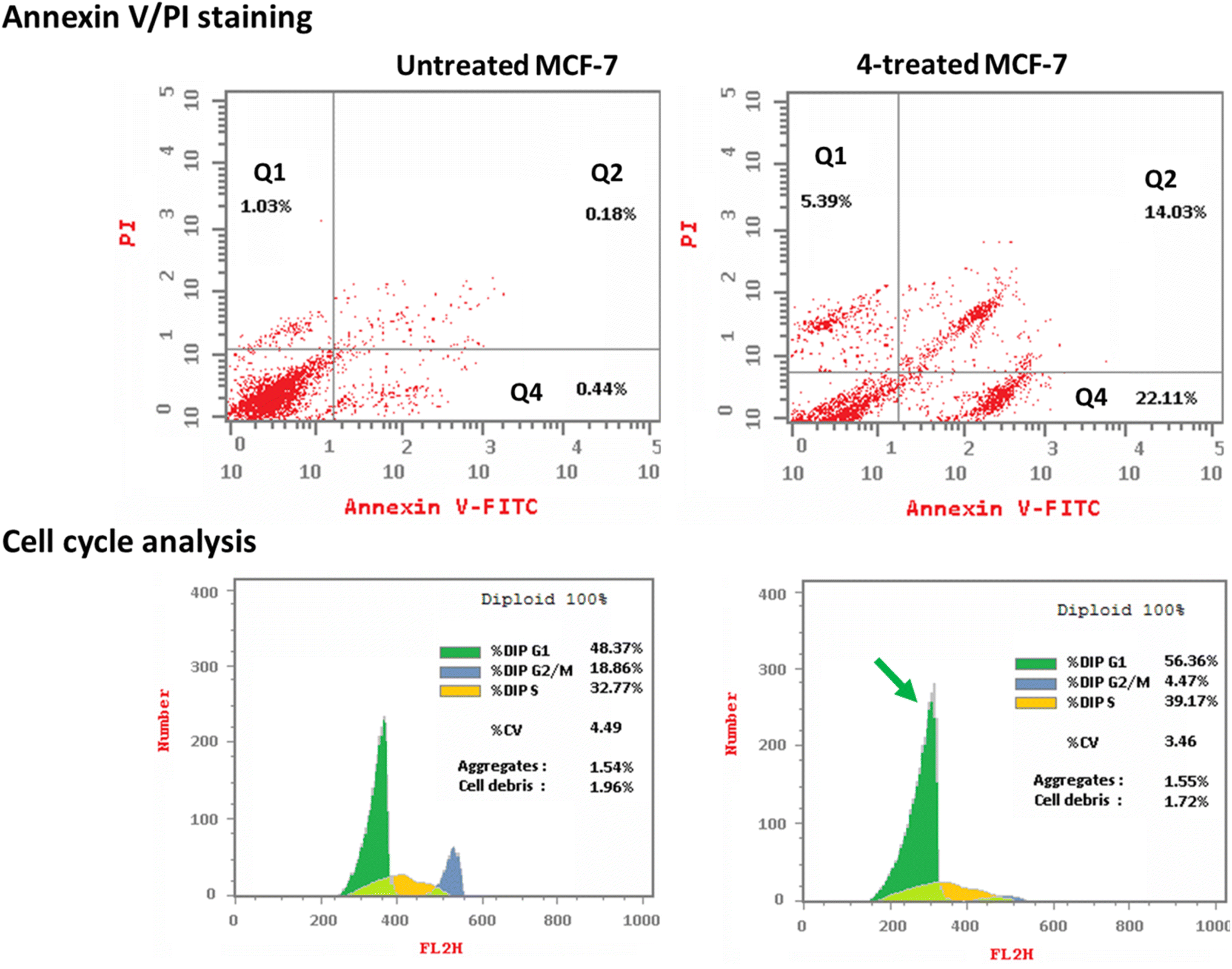 | ||
| Fig. 6 Cryptographs of annexin-V/propidium iodide staining of untreated and 4-treated MCF-7 cells with (IC50 = 0.57 μM, 48 h). Percentage of cell population at each cell cycle “G1, S, and G2/M” using DNA content-flow cytometry aided cell cycle analysis. | ||
The cell population in each cell phase after cytotoxic agent treatment was then determined by DNA flow cytometry. As seen in Fig. 6 (bottom panel), compound 4 treatment significantly increased the cell population at the G1-phase by 56.36% compared to control 48.37%, while cells in the G2/M-phase were decreased (4.47% compared to 18.86%). Compound 4 caused cell death in MCF-7 cells, stopping their growth in the G1 phase.
3.2.3.2. RT-PCR. By comparing the RT-PCR levels of the apoptosis-mediated genes P53, Bax, caspase-3,8,9, and Bcl-2 in both the untreated and treated MCF-7 cells, we could confirm that compound 4 caused cell death in these cells. As seen in Fig. 7, compound 4 upregulated caspase-3,8,9 levels by 11.2, 1.23, and 8.9-fold, Bax by 6.7-fold, and P53 by 7.9-fold, while it downregulated the Bcl-2 expression by 0.11-fold and inhibited the PIM-1 expression by 0.31-fold. Consequently, our results demonstrated that compound 4 treatment caused cell death by apoptosis via an intrinsic mechanism through inhibition of PIM-1 kinase.
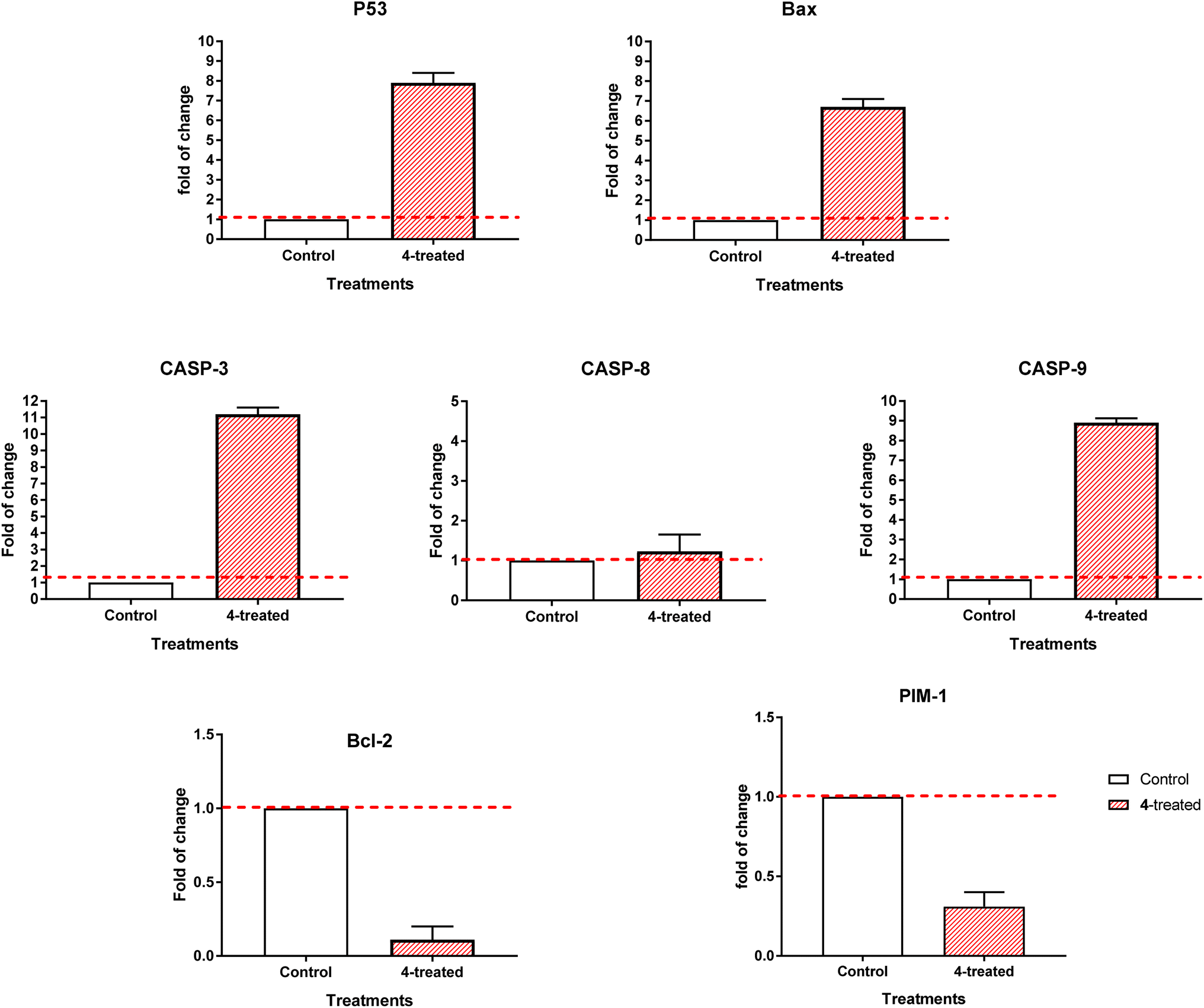 | ||
| Fig. 7 Quantitative RT-PCR results analysis of the apoptosis-related genes; P53, Bax, caspases-3,8,9, Bcl-2 and PIM-1 kinase, respectively in MCF-7 cells treated with compound 4 with (IC50 = 0.57 μM, 48 h). The data illustrated is the average of 3 independent experimental runs (mean ± SD). The red dashed line represents the fold change of control = 1. | ||
The ubiquitous PIM kinases regulate various biological events, including cell proliferation, cell differentiation, and cell death. When expressed in ways that worsen cancer, these genes, which function as weak oncogenes, cause a variety of human cancers. Thus, PIM kinase inhibition may be a valuable therapeutic target for cancer.33 Among heterocycles, pyridine derivatives showed cell cycle stop and apoptosis induction capabilities and could be considered a potential PIM-1 kinase inhibitor. These compounds could be explored as potential treatments for breast cancer.34 Compound 4 as concluded from this study; it may solve some problems of pyridine and pyridopyrimidine containing anticancer molecules by having more potent and target-oriented chemotherapeutics (apoptosis-induction) with selective profile against normal cells.
3.3. In silico studies
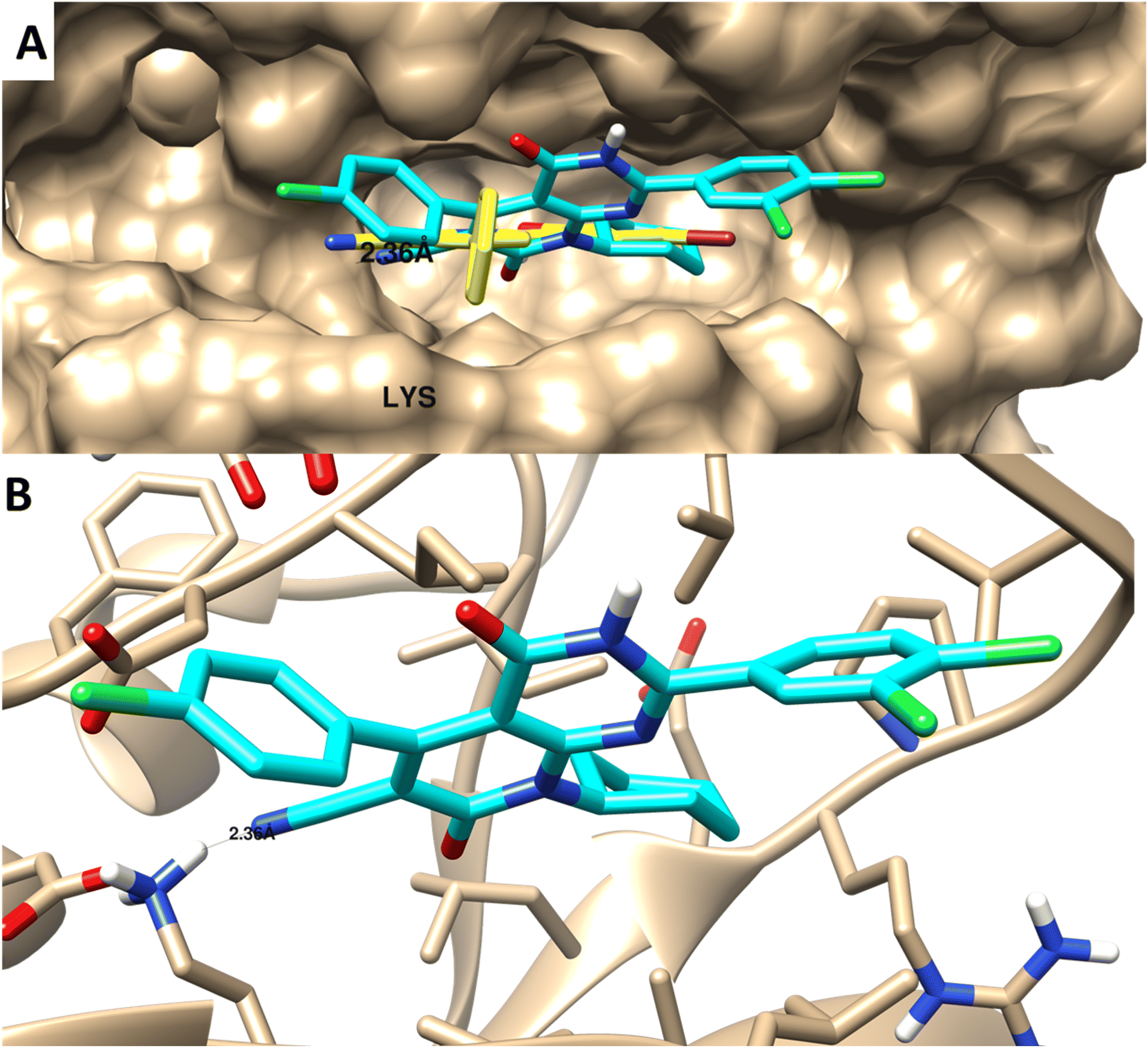 | ||
| Fig. 8 Molecular docking of 4 towards PIM-1 protein (PDB 2OBJ). (A) Surface disposition of the co-crystallized ligand (yellow-colored) and the docked compound (cyan-colored) and (B) interactive view of the compound with the highlighted key amino acids. 3D images were generated by Chimera-UCSF software. | ||
| # | Molsoft | Molinspiration 2018.10 | SwissADME | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HBA | HBD | Solubility (mg L−1) | Drug score | MWt (D) | MV (Å3) | PSA (Å2) | Logp |
BBB score | nviolations | Drug likeness (Lipinski Pfizer filter) | |
| a “Mwt: molecular weight, MV: molecular volume, PAS: polar surface area, logp: logp: octanol–water partition coefficient, nrotb: number of rotatable bonds, nviolations: number of violations, HBA: hydrogen bond acceptor, HBD: hydrogen bond donor, drug-likeness score, compounds having negative or zero value should not be considered as drug-like”. Drug likeness (Lipinski Pfizer filter)/“yes, drug-like” MW ≤ 500, logp ≤ 4.25, HBA ≤ 10 and HBD ≤ 5”. |
|||||||||||
| 4 | 4 | 1 | 1.25 | 0.84 | 490 | 507.4 | 65.99 | 5.0 | 3.84 | 0 | Yes |
| 6 | 4 | 1 | 134.68 | 0.12 | 394.12 | 433.08 | 66.27 | 3.23 | 3.71 | 0 | Yes |
| 10 | 4 | 2 | 91.36 | 0.0 | 378.12 | 430.63 | 67.20 | 3.12 | 2.74 | 0 | Yes |
| 11 | 5 | 1 | 230.53 | −0.22 | 347.14 | 393.24 | 81.13 | 2.76 | 3.12 | 0 | Yes |
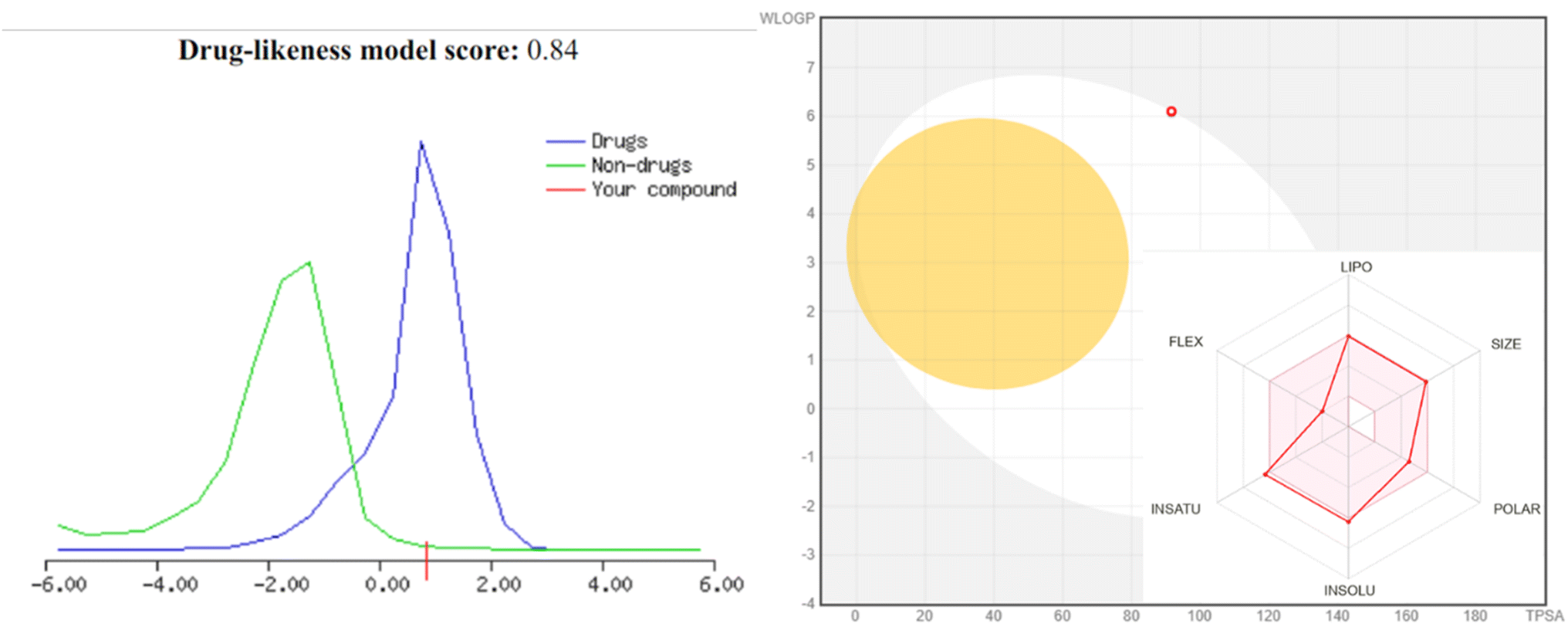 | ||
| Fig. 9 Drug likeness score of compound 4 using MolSoft “the green color means non-drug-like behavior, and those that fall under blue color area are considered as drug-like. Those compounds having negative or zero value should not be considered as a drug like”. BOILED-Egg model for compound 4 using SwissADME molecules projected to passively cross the blood–brain barrier (BBB) is positioned in the yolk of the BOILED-Egg, while those predicted to be passively absorbed by the gastrointestinal tract (GI) are in the white of the egg. | ||
4 Conclusion and recommendations
New series of pyrido[2,3-d]pyrimidine was described. Nicotinamide 2 was synthesized via cyclization of N-cyclohexyl derivative with cyanoacetamide. The o-aminonicotinonitrile 2 was subjected to acylation or thioacylation process followed by intramolecular heterocyclization to afford the desired pyrido[2,3-d]pyrimidine (3–11). Among these derivatives, compound 4 exhibited remarkable cytotoxic activities against MCF-7 and HepG2 cells with IC50 values of 0.57 μM and 1.13 μM, respectively. Interestingly, compound 4 had potent PIM-1 kinase inhibition with IC50 values of 11.4 nM, respectively, with inhibition of 97.8% compared to staurosporine (IC50 = 16.7 nM, with 95.6% inhibition). Moreover, compound 4 significantly induced apoptosis in MCF-7 cells by 58.29-fold by having 36.14% total apoptosis in treated cells compared to 0.62% for control. Moreover, arresting the cell cycle at the G1-phase. Accordingly, compound 4 was validated as a promising PIM-1 targeted chemotherapeutic agent to treat breast cancer. Accordingly, the anticancer activity of compound 4 will be further validated in the future using in vivo study as a real animal model.Author contributions
E. S. Tantawy, S. M. Mohammed, H. A. Morsy, H. A. El-Sayed, and A. H. Moustafa synthesized the entire series of derivatives with the characterization of structure elucidation, while M. S. Nafie initiated the idea and design of the biology part by carrying out in vitro cytotoxic screening, flow cytometry, RT-PCR analysis, and in silico studies with the linguistic revision and manuscript finalizing. All authors contributed to data analysis and manuscript writing original draft in their corresponding parts. All authors validated it in the final submitted form.Conflicts of interest
The authors declare no conflict of interest.References
- T. Al-Warhi, A. A. Al-Karmalawy, A. A. Elmaaty, M. A. Alshubramy, M. Abdel-Motaal, T. A. Majrashi, M. Asem, A. Nabil, W. M. Eldehna and M. Sharaky, Biological evaluation, docking studies, and in silico ADME prediction of some pyrimidine and pyridine derivatives as potential EGFR(WT) and EGFR(T790M) inhibitors, J. Enzyme Inhib. Med. Chem., 2023, 38, 176–191, DOI:10.1080/14756366.2022.2135512.
- W. Cao, H.-D. Chen, Y.-W. Yu, N. Li and W.-Q. Chen, Changing profiles of cancer burden worldwide and in China: a secondary analysis of the global cancer statistics 2020, China Med. J., 2021, 134, 783–791, DOI:10.1097/CM9.0000000000001474.
- E. A. Madbouly, E.-S. M. Lashine, A. A. Al-Karmalawy, M. M. Sebaiy, H. Pratsinis, D. Kletsas and K. Metwally, Design and synthesis of novel quinazolinone–chalcone hybrids as potential apoptotic candidates targeting caspase-3 and PARP-1: in vitro, molecular docking, and SAR studies, New J. Chem., 2022, 46, 22013–22029, 10.1039/d2nj04053k.
- W. M. Eldehna, R. M. Maklad, H. Almahli, T. Al-Warhi, E. B. Elkaeed, M. A. S. Abourehab, H. A. Abdel-Aziz and A. M. El Kerdawy, Identification of 3-(piperazinylmethyl)benzofuran derivatives as novel type II CDK2 inhibitors: design, synthesis, biological evaluation, and in silico insights, J. Enzyme Inhib. Med. Chem., 2022, 37, 1227–1240, DOI:10.1080/14756366.2022.2062337.
- T. Al-Warhi, M. M. Elbadawi, A. Bonardi, A. Nocentini, A. A. Al-Karmalawy, N. Aljaeed, O. J. Alotaibi, H. A. Abdel-Aziz, C. T. Supuran and W. M. Eldehna, Design and synthesis of benzothiazole-based SLC-0111 analogues as new inhibitors for the cancer-associated carbonic anhydrase isoforms IX and XII, J. Enzyme Inhib. Med. Chem., 2022, 37, 2635–2643, DOI:10.1080/14756366.2022.2124409.
- A. Sabt, W. M. Eldehna, T. Al-Warhi, O. J. Alotaibi, M. M. Elaasser, H. Suliman and H. A. Abdel-Aziz, Discovery of 3,6-disubstituted pyridazines as a novel class of anticancer agents targeting cyclin-dependent kinase 2: synthesis, biological evaluation and in silico insights, J. Enzyme Inhib. Med. Chem., 2020, 35, 1616–1630, DOI:10.1080/14756366.2020.1806259.
- L. Akl, A. A. Abd El-Hafeez, T. M. Ibrahim, R. Salem, H. M. M. Marzouk, R. A. El-Domany, P. Ghosh, W. M. Eldehna and S. M. Abou-Seri, Identification of novel piperazine-tethered phthalazines as selective CDK1 inhibitors endowed with in vitro anticancer activity toward the pancreatic cancer, Eur. J. Med. Chem., 2022, 243, 114704, DOI:10.1016/j.ejmech.2022.114704.
- L. K. Gediya and V. C. Njar, Promise and challenges in drug discovery and development of hybrid anticancer drugs, Expert Opin. Drug Discovery, 2009, 4, 1099–1111, DOI:10.1517/17460440903341705.
- B. Mansour, Y. A. Salem, K. M. Attallah, O. A. El-Kawy, I. T. Ibrahim and N. I. Abdel-Aziz, Cyanopyridinone- and Cyanopyridine-Based Cancer Cell Pim-1 Inhibitors: Design, Synthesis, Radiolabeling, Biodistribution, and Molecular Modeling Simulation, ACS Omega, 2023, 8, 19351–19366, DOI:10.1021/acsomega.2c08304.
- M. A. Waly, I. I. Elhawary and T. M. Elgogary, Utilization of nicotinonitrile-2-thiol in the synthesis of new thiepino[2,3-b]pyridine derivative as an in vitro novel antitumor potent, Med. Chem. Res., 2012, 22, 1674–1678, DOI:10.1007/s00044-012-0161-4.
- A. Krauze, S. Grinberga, L. Krasnova, I. Adlere, E. Sokolova, I. Domracheva, I. Shestakova, Z. Andzans and G. Duburs, Thieno[2,3-b]pyridines—a new class of multidrug resistance (MDR) modulators, Bioorg. Med. Chem., 2014, 22, 5860–5870, DOI:10.1016/j.bmc.2014.09.023.
- M. M. Ghorab, F. A. Ragab, H. I. Heiba, M. G. El-Gazzar and S. S. Zahran, Synthesis, anticancer and radiosensitizing evaluation of some novel sulfonamide derivatives, Eur. J. Med. Chem., 2015, 92, 682–692, DOI:10.1016/j.ejmech.2015.01.036.
- A. Samadi, M. de la Fuente Revenga, C. Pérez, I. Iriepa, I. Moraleda, M. I. Rodríguez-Franco and J. Marco-Contelles, Synthesis, pharmacological assessment, and molecular modeling of 6-chloro-pyridonepezils: new dual AChE inhibitors as potential drugs for the treatment of Alzheimer's disease, Eur. J. Med. Chem., 2013, 67, 64–74, DOI:10.1016/j.ejmech.2013.06.021.
- A. Samadi, M. Estrada, C. Pérez, M. I. Rodríguez-Franco, I. Iriepa, I. Moraleda, M. Chioua and J. Marco-Contelles, Pyridonepezils, new dual AChE inhibitors as potential drugs for the treatment of Alzheimer's disease: synthesis, biological assessment, and molecular modeling, Eur. J. Med. Chem., 2012, 57, 296–301, DOI:10.1016/j.ejmech.2012.09.030.
- N. M. Khalifa, M. A. Al-Omar, H. M. Alkahtani and A. H. Bakheit, Kinase Inhibitors of Novel Pyridopyrimidinone Candidates: Synthesis and In Vitro Anticancer Properties, J. Chem., 2019, 2019, 1–10, DOI:10.1155/2019/2635219.
- S. M. Mohammed, A. A. Masry, A. H. Moustafa, H. A. El-Sayed, E. M. Gad and H. A. Morsy, K2CO3-nanoparticles catalyzed the synthesis of 3-arylidine imidazo[1,2-c]pyrimidine candidates: Cytotoxic activity and docking study, Synth. Commun., 2023, 53, 1319–1332, DOI:10.1080/00397911.2023.2220442.
- W. M. Eldehna, M. A. El Hassab, M. F. Abo-Ashour, T. Al-Warhi, M. M. Elaasser, N. A. Safwat, H. Suliman, M. F. Ahmed, S. T. Al-Rashood, H. A. Abdel-Aziz and R. El-Haggar, Development of isatin-thiazolo[3,2-a]benzimidazole hybrids as novel CDK2 inhibitors with potent in vitro apoptotic anti-proliferative activity: synthesis, biological and molecular dynamics investigations, Bioorg. Chem., 2021, 110, 104748, DOI:10.1016/j.bioorg.2021.104748.
- A. T. A. Boraei, E. H. Eltamany, I. A. I. Ali, S. M. Gebriel and M. S. Nafie, Synthesis of new substituted pyridine derivatives as potent anti-liver cancer agents through apoptosis induction: in vitro, in vivo, and in silico integrated approaches, Bioorg. Chem., 2021, 111, 104877, DOI:10.1016/j.bioorg.2021.104877.
- M. S. Nafie, A. M. Amer, A. K. Mohamed and E. S. Tantawy, Discovery of novel pyrazolo[3,4-b]pyridine scaffold-based derivatives as potential PIM-1 kinase inhibitors in breast cancer MCF-7 cells, Bioorg. Med. Chem., 2020, 28, 115828, DOI:10.1016/j.bmc.2020.115828.
- S. M. Shaban, E. H. Eltamany, A. T. A. Boraei, M. S. Nafie and E. M. Gad, Design and Synthesis of Novel Pyridine-Based Compounds as Potential PIM-1 Kinase Inhibitors, Apoptosis, and Autophagy Inducers Targeting MCF-7 Cell Lines: In Vitro and In Vivo Studies, ACS Omega, 2023, 8, 46922–46933, DOI:10.1021/acsomega.3c06700.
- A. Fahim, A. Farag, M. Shaaban and E. Ragab, Microwave Assisted Synthesis of Pyrazolo[1,5-a]pyrimidine, Triazolo[1,5-a]pyrimidine, Pyrimido[1,2-a]benzimdazole, Triazolo[5,1-c][1,2,4]triazine and Imidazo[2,1-c][1,2,4]triazine, Current Microwave, Chemistry, 2019, 5, 111–119, DOI:10.2174/2213335605666180425144009.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65, 55–63, DOI:10.1016/0022-1759(83)90303-4.
- E. S. Tantawy, A. M. Amer, E. K. Mohamed, M. M. Abd Alla and M. S. Nafie, Synthesis, characterization of some pyrazine derivatives as anti-cancer agents: in vitro and in silico approaches, J. Mol. Struct., 2020, 1210, 128013, DOI:10.1016/j.molstruc.2020.128013.
- M. E. Abdelaziz, M. M. M. El-Miligy, S. M. Fahmy, M. A. Mahran and A. A. Hazzaa, Design, synthesis and docking study of pyridine and thieno[2,3-b]pyridine derivatives as anticancer PIM-1 kinase inhibitors, Bioorg. Chem., 2018, 80, 674–692, DOI:10.1016/j.bioorg.2018.07.024.
- CST – HTScan® Pim-1 Kinase Assay Kit, https://www.cellsignal.com/products/cellular-assay-kits/htscan-pim-1-kinase-assay-kit/7573, accessed July 17, 2020.
- M. S. Nafie, K. Arafa, N. K. Sedky, A. A. Alakhdar and R. K. Arafa, Triaryl dicationic DNA minor-groove binders with antioxidant activity display cytotoxicity and induce apoptosis in breast cancer, Chem.-Biol. Interact., 2020, 324, 109087, DOI:10.1016/j.cbi.2020.109087.
- M. S. Nafie, A. M. Amer, A. K. Mohamed and E. S. Tantawy, Discovery of novel pyrazolo[3,4-b]pyridine scaffold-based derivatives as potential PIM-1 kinase inhibitors in breast cancer MCF-7 cells, Bioorg. Med. Chem., 2020, 28, 115828, DOI:10.1016/j.bmc.2020.115828.
- E. M. Gad, M. S. Nafie, E. H. Eltamany, M. S. A. G. Hammad, A. Barakat and A. T. A. Boraei, Discovery of New Apoptosis-Inducing Agents for Breast Cancer Based on Ethyl 2-Amino-4,5,6,7-Tetra Hydrobenzo[b]Thiophene-3-Carboxylate: Synthesis, In Vitro, and In Vivo Activity Evaluation, Molecules, 2020, 25, 2523, DOI:10.3390/molecules25112523.
- M. S. Nafie, S. Mahgoub and A. M. Amer, Antimicrobial and antiproliferative activities of novel synthesized 6-(quinolin-2-ylthio)pyridine derivatives with molecular docking study as multi-targeted JAK2/STAT3 inhibitors, Chem. Biol. Drug Des., 2021, 97, 553–564, DOI:10.1111/cbdd.13791.
- M. S. Nafie, M. A. Tantawy and G. A. Elmgeed, Screening of different drug design tools to predict the mode of action of steroidal derivatives as anti-cancer agents, Steroids, 2019, 152, 108485, DOI:10.1016/j.steroids.2019.108485.
- M. D. Jacobs, J. Black, O. Futer, L. Swenson, B. Hare, M. Fleming and K. Saxena, Pim-1 Ligand-bound Structures Reveal the Mechanism of Serine/Threonine Kinase Inhibition by LY294002, J. Biol. Chem., 2005, 280, 13728–13734, DOI:10.1074/jbc.M413155200.
- S. J. Lee, B.-G. Han, J.-W. Cho, J.-S. Choi, J. Lee, H.-J. Song, J. S. Koh and B. I. Lee, Crystal Structure of Pim1 Kinase in Complex with a Pyrido[4,3-D]Pyrimidine Derivative Suggests a Unique Binding Mode, PLoS One, 2013, 8, e70358, DOI:10.1371/journal.pone.0070358.
- V. Asati, D. K. Mahapatra and S. K. Bharti, PIM kinase inhibitors: structural and pharmacological perspectives, Eur. J. Med. Chem., 2019, 172, 95–108, DOI:10.1016/j.ejmech.2019.03.050.
- K. Li, Y. Li, D. Zhou, Y. Fan, H. Guo, T. Ma, J. Wen, D. Liu and L. Zhao, Synthesis and biological evaluation of quinoline derivatives as potential anti-prostate cancer agents and Pim-1 kinase inhibitors, Bioorg. Med. Chem., 2016, 24, 1889–1897, DOI:10.1016/j.bmc.2016.03.016.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic characterizations of the synthesized compounds. See DOI: https://doi.org/10.1039/d4ra00902a |
| This journal is © The Royal Society of Chemistry 2024 |