
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceY-tube assisted coprecipitation synthesis of iron-based Prussian blue analogues cathode materials for sodium-ion batteries†
Ruizhong Zhang,
Yuao Liu,
Hongquan Liu,
Yanjun Zhong *,
Yuan Zhang,
Zhenguo Wu and
Xinlong Wang
*,
Yuan Zhang,
Zhenguo Wu and
Xinlong Wang
Engineering Research Center of Comprehensive Utilization and Clean Processing of Phosphorus Resources of Ministry of Education, School of Chemical Engineering, Sichuan University, Chengdu 610065, China. E-mail: yjzhong@scu.edu.cn; Fax: +86-28-85405235; Tel: +86-28-85405235
First published on 16th April 2024
Abstract
Prussian blue analogues possess numerous advantages as cathode materials for sodium-ion batteries, including high energy density, low cost, sustainability, and straightforward synthesis processes, making them highly promising for practical applications. However, during the synthesis, crystal defects such as vacancies and the incorporation of crystal water can lead to issues such as diminished capacity and suboptimal cycling stability. In the current study, a Y-tube assisted coprecipitation method was used to synthesize iron-based Prussian blue analogues, and the optimized feed flow rate during synthesis contributed to the successful preparation of the material with a formula of Na1.56Fe[Fe(CN)6]0.90□0.10·2.42H2O, representing a low-defect cathode material. This approach cleverly utilizes the Y-tube component to enhance the micro-mixing of materials in the co-precipitation reaction, featuring simplicity, low cost, user-friendly, and the ability to be used in continuous production. Electrochemical performance tests show that the sample retains 69.8% of its capacity after 200 cycles at a current density of 0.5C (1C = 140 mA g−1) and delivers a capacity of 71.9 mA h g−1 at a high rate of 10C. The findings of this research provide important insights for the development of high-performance Prussian blue analogues cathode materials for sodium-ion batteries.
1. Introduction
Lithium-ion batteries (LIBs) are heralded for their substantial energy density, prolonged cycle life, elevated working voltage, and absence of memory effect, making them a widely adopted choice in both everyday life and industrial electrochemical energy storage applications.1,2 Nonetheless, their high costs and the looming shadow of lithium resource scarcity curtail their broader implementation in large-scale energy storage systems. The ideal electrode materials for such storage solutions should be derived from abundant resources, cost-effective, enduring in life cycles, and intrinsically safe, with less emphasis on energy density compared to the higher benchmarks expected in portable devices.3,4 Consequently, battery technologies capitalizing on the wealth of more common elements hold a competitive advantage in the realm of large-scale storage. Sodium stands out in stark contrast to lithium with its greater abundance, and sodium-ion batteries (NIBs), sharing a kinship in electrochemical mechanisms with their lithium-ion counterparts, are poised for significant growth. They are regarded as one of the prime choices for integrating renewable energy sources like wind into smart grid storage infrastructures, with the potential to evolve into an efficient, eco-friendly, and sustainable energy solution.5–7 However, industrializing NIBs technology is fraught with challenges, among which the quest for suitable cathode materials looms large. Owing to the larger ionic radius of sodium compared to lithium, many cathode materials that are effective in LIBs fall short of NIBs.Prussian blue analogs (PBAs, NaxM1[M2(CN)6]y□1−y·nH2O, where M1 and M2 are transition metals, 0 < x ≤ 2, 0 < y ≤ 1, and □ denotes vacancies at the Fe(CN)6 sites), with their open-framework structure, provide an ample array of sites and pathways that facilitate the reversible insertion and extraction of sodium ions, thus earning widespread interest and attention within the research community.8–11 Iron-based PBAs, represented as NaxFe[Fe(CN)6]y□1−y·nH2O (abbreviated as NaFeHCF), are heralded for a high theoretical capacity of approximately 170 mA h g−1 and lower cost, positioning it as one of the most promising PBAs cathode materials.12–16 However, the synthesis of NaFeHCF tends to generate a significant number of vacancy defects, which can lead to several adverse effects on its electrochemical performance. To be specific, an increase in vacancy defects introduces more crystal water molecules, diminishing the number of available sodium-ion sites.17 These water molecules compete with sodium ions for space within the crystal lattice, obstructing sodium-ion ingress and thus reducing the capacity utilization of the PBAs framework.18,19 Water present in the lattice can migrate into the electrolyte and undergo electrochemical decomposition, causing degradation of the organic electrolyte and presenting potential safety hazards.20 The vacancy defects disrupt the Fe–CN–Fe bridging within the framework, creating distorted and unstable lattice structures prone to collapse during the sodium-ion insertion/extraction cycling.21 The presence of vacancy defects also interrupts electronic conduction along the Fe–CN–Fe pathways, resulting in localized blockage of the sodium-ion insertion reaction and exacerbating ohmic polarization within the material.22,23 Consequently, the realized capacity and cycle life of the material are compromised. Despite efforts to enhance electrochemical performance through various strategies, such as ion doping or optimizing the composition or framework structure of PBAs,24–29 nanoparticle sizing,9,14,30 designing porous structures or unique morphologies,31 post-synthesis dehydration processing,32–34 surface coating modifications,31,35,36 and optimizing electrolyte composition,37–39 these measures are yet insufficient to fundamentally overcome the issues posed by vacancy defects and associated crystal water. At the heart of the matter, crystal vacancy defects and crystal water primarily form during the synthesis process.
The synthesis of PBAs materials for SIBs research primarily involves two main methods: coprecipitation and hydrothermal. Additionally, methods like mechanical ball milling, electrodeposition, microemulsion, and microwave-assisted techniques are also studied.8,9,11 Among these methods, the coprecipitation technique is regarded as the most promising for industrial-scale production of PBAs. The precipitation reaction of PBAs is extremely rapid, resulting in nucleation, growth, and aggregation of crystals before achieving uniform supersaturation in the reactor. This uneven supersaturation field generates different driving forces, affecting the fundamental structural characteristics of the products and resulting in the formation of crystal vacancy defects and coordinated water.9,15 Consequently, micro-mixing has a significant impact on the quality of the PBAs product. To obtain high-quality PBAs, it is crucial to achieve a high degree of supersaturation in the solution swiftly, facilitating the instantaneous formation of nuclei. Subsequently, it is essential to provide excellent micro-mixing to create a uniform reaction environment for the growth of the nuclei. In recent years, many distinguished scholars have conducted extensive research on reducing vacancy defects and crystal water in PBAs by slowing down the rate of reaction precipitation. The main approaches can be summarized as follows: (1) control of reagent addition rate: By regulating the concentration and flow rate of reagents, the reagent ions are made to participate in the reaction slowly.40,41 (2) Single iron source method: Utilizing the Fe2+ released by the hydrolysis of Na4Fe(CN)6 under acidic conditions to combine with unreacted Fe(CN)64−, allowing the crystal to grow slowly.42–46 (3) Chemical inhibitor method: Introducing chelating agents such as citric acid or sodium citrate, causing the transition metal ions to coordinate with the chelating agents first, reducing the concentration of metal ions in the monomer solution.47–49 Lowering the nucleation rate can improve the microstructure of crystals to a certain extent, but this often comes at the expense of reaction efficiency and product yield, and it is challenging to ensure batch stability when scaling up for industrial production. Therefore, there is an urgent need to focus on enhancing micro-mixing to balance product quality and production efficiency.
By enhancing micro-mixing efficiency and reducing concentration gradients within the reactor, the saturation distribution becomes more uniform. During crystal nucleation and growth, reactants experience sufficient micro-mixing, resulting in uniform saturation fields and equivalent driving forces. Through the coupling of micro-mixing and reaction processes, the micro-mixing rate is synchronized with the reaction rate, ultimately yielding products with fewer vacancy defects and crystal water.50–52 In traditional stirred-tank reactors, the micro-mixing efficiency is low, leading to material concentration gradients within the vessel, resulting in uneven saturation distribution. Micro-mixing times are often prolonged, leading to varying micro-mixing conditions in different regions. As a result, PBAs crystals obtained often contain a higher concentration of Fe(CN)6 vacancies and crystal water, which adversely affect the product quality.16 Some scholars have adopted microreactors to control reaction conditions and achieve more uniform supersaturation and reaction environments. For example, Xi et al.50 developed a T-shaped membrane dispersed microreactor for synthesizing PBAs. The excellent micro-mixing in this approach facilitates explosive nucleation of PBAs within a homogeneous environment, achieving high nucleation rates. Consequently, the resulting in PBAs exhibit reduced vacancy defects and a narrower particle size distribution. Zhu et al.53 employed a stator–rotor reactor (RSSDR) combined with precipitation transformation to enhance the crystallization and precipitation process for continuous production of iron-based PBAs. By leveraging intense micromixing and aging, they successfully prepared PBAs with high sodium content and minimal defects. However, their complex and expensive production processes pose significant challenges to scaling up.
In this study, we have developed an innovative Y-tube assisted coprecipitation technique for the synthesis of NaFeHCF characterized by minimal vacancy defects and reduced content of crystal water. We also explored the influence of the flow rate of reactants on the structure and performance of NaFeHCF. The Y-tube design offers significant advantages due to its small size, such as a larger surface area to volume ratio, enhancing efficiency in mass and heat exchange. This design ensures rapid and uniform mixing of reactant fluids, thereby creating a consistent and stable environment conducive to crystal growth. Additionally, isolating the reaction environment reduces contact with air, thus minimizing oxidation. The Y-tube design is also flexible and easily integrated with other equipment. As a method conducive to continuous production, it not only ensures the quality and stability of batch products but also offers advantages in terms of low cost and simplicity in synthesis.54–56 The key to synthesizing high-performance PBAs lies in controlling the reaction process and minimizing oxidation as much as possible. Therefore, theoretically, the Y-tube is suitable for the production of PBAs. To our knowledge, there are no reports on the synthesis of PBAs assisted by Y-tube. The findings from this research will offer vital theoretical insights for the cost-effective and high-yield industrial fabrication of advanced PBAs cathode materials.
2. Experimental
2.1 Materials
Ten-hydrate sodium ferrocyanide (Na4Fe(CN)6·10H2O, 96%), dihydrate trisodium citrate (C6H5Na3O7·2H2O, 98%), and heptahydrate ferrous sulfate (FeSO4·7H2O, 98%) were procured from Shanghai Macklin Co., Ltd. Ascorbic acid (C6H8O6, analytical grade), anhydrous ethanol (C2H6O, analytical grade), conductive agent (Super-P, analytical grade), separator (Whatman GF/D glass fiber filter membrane, diameter 125 mm), aluminum foil (10 μm), and sodium chunks (analytical grade) were purchased from Chengdu Cologne Chemical Co., Ltd. The NaClO4 electrolyte (NC-004, 1 M NaClO4 in EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PC = 1:1 vol% with 5% FEC, battery-grade) was supplied by DuoDuo Chemical Co., Ltd. All chemicals were of reagent grade and used without additional purification. Deionized water with 18.25 MΩ cm resistivity was used throughout the experiments.
:PC = 1:1 vol% with 5% FEC, battery-grade) was supplied by DuoDuo Chemical Co., Ltd. All chemicals were of reagent grade and used without additional purification. Deionized water with 18.25 MΩ cm resistivity was used throughout the experiments.
2.2 Synthesis of NaFeHCF
The schematic diagram of NaFeHCF preparation process by Y-tube assisted coprecipitation method is shown in Fig. 1. 6 mmol of Na4Fe(CN)6·10H2O and 30 mmol of C6H5Na3O7·2H2O were dissolved in 100 mL of deionized water and stirred thoroughly. This resultant mixture is designated as solution A. For solution B, 9 mmol of FeSO4·7H2O, 100 mmol of C6H5Na3O7·2H2O, and 0.4 mmol of C6H8O6 were dissolved in 100 mL of deionized water and stirred thoroughly. Both solution A and solution B were simultaneously injected into a Y-tube (inner diameter: 1 mm) at a controlled flow rate using a peristaltic pump and then dripped into a three-neck flask containing 100 mL of deionized water. Nitrogen gas was introduced into the three-neck flask to provide an inert atmosphere. After the addition was complete, stirring was continued for 2 hours, followed by aging for 12 hours. The precipitate was thoroughly washed with deionized water through centrifugation and then dried under vacuum at 100 °C for 18 hours to obtain the iron-based Prussian blue material. The flow rates tested were 0.3 mL min−1, 15 mL min−1, 30 mL min−1, and 41 mL min−1. The samples obtained were correspondingly labeled as Y-PB-0.3, Y-PB-15, Y-PB-30, and Y-PB-41. For comparison, NaFeHCF materials synthesized without employing a Y-tube with a flow rate of 0.3 mL min−1 were named C-PB-0.3. | ||
| Fig. 1 Schematic diagram of NaFeHCF preparation process by Y-tube assisted coprecipitation method. | ||
2.3 Materials characterization
The phase composition of the samples was characterized using an X-ray diffraction (XRD) analyzer, specifically the X'Pert Pro Super model equipped with Cu Kα radiation (λ = 1.5406 Å). The diffraction patterns were collected over a 2θ range of 10° to 80° with a step size of 0.02626°. The microstructures of the samples were examined with a VEGA3 TESCAN scanning electron microscope (SEM), and a Tecnai G2 F20 S-TWIN transmission electron microscope (TEM). X-ray photoelectron spectroscopy (XPS) measurements were performed using a Thermo Scientific K-Alpha system to assess the valence states of transition metal elements within the samples. The spectra were calibrated using C1s at 284.8 eV as the reference. Thermogravimetric analysis (TG) was conducted on an EXSTAR6000 instrument from Seiko Instruments to quantify the crystal water content in the materials. Fourier-transform infrared spectroscopy (FTIR) analysis was carried out using a Nicolet-5700 from Spectrum to identify chemical bond types and functional groups present in the Prussian blue analogs. Elemental analysis (EA) for the determination of carbon and nitrogen contents in the materials was performed with an EL cube from Elementar Vario. Finally, the concentrations of sodium (Na) and iron (Fe) in the NaFeHCF were measured using an Agilent 5100 SVDV inductively coupled plasma atomic emission spectrometer (ICP-AES).2.4 Electrochemical measurements
The active material, Super-P conductive carbon, and polyvinylidene fluoride (PVDF) were combined in a mass ratio of 7:2:1, utilizing N-methyl-2-pyrrolidone (NMP) as the solvent. This slurry was uniformly coated onto an aluminum foil current collector, followed by vacuum drying at 110 °C for 20 hours to remove solvent residues. The dried film was then punched into electrode disks with a 14 mm diameter. In an argon-filled glove box with oxygen and water levels below 0.1 ppm, the electrodes were assembled into CR2025 coin cells along with a 125 mm diameter GF/D glass fiber filter separator. The electrolyte consisted of 1.0 M sodium perchlorate (NaClO4) in a 1:1 volumetric mixture of ethylene carbonate (EC) and propylene carbonate (PC), with an additional 5.0 wt% fluoroethylene carbonate (FEC) as an additive. After the cell assembly, the battery was allowed to equilibrate for 24 hours to ensure thorough wetting of the electrodes and separator before proceeding to electrochemical evaluation. Galvanostatic charge–discharge was employed within a voltage range of 2.0–4.0 V. The usual procedure involves activating the battery during the first cycle with a relatively small current of 0.1C (where 1C corresponds to 140 mA g−1) before evaluating cycling performance. Cyclic voltammetry (CV) testing is conducted with a voltage scan range of 2.0 to 4.0 V and a scan rate of 0.1 mV s−1. Electrochemical impedance spectroscopy (EIS) testing applies a voltage amplitude of 5 mV with a frequency range from 10−2 Hz to 105 Hz. All electrochemical tests were conducted at room temperature (25 °C).
3. Results and discussion
3.1 Phase and composition characteristics of materials
The XRD patterns of five NaFeHCF samples are presented in Fig. 2(a). Based on the characteristics of the peak shapes and positions, the XRD diffraction peaks of NaFeHCF samples match those of the standard Prussian blue (JCPDS, No. 52-1907), with no visible impurity phases. The C-PB-0.3 sample has an Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group and a cubic phase structure. After employing a Y-tube in the synthesis process, significant peak splitting at 24.2° and 38.7° is observed, indicating a structural transition from a sodium-deficient cubic phase to a sodium-rich monoclinic phase with the space group P21/n.57 The increased peak intensity and reduced full width at half maximum (FWHM) of Y-PB-0.3 suggest that the use of a Y-tube to enhance micro-mixing can improve the material's crystallinity. With the use of the Y-tube and increasing flow rates, the intensity of the characteristic peaks initially increases and then decreases. The highest peak intensity is observed for Y-PB-15, indicating the highest degree of crystallinity. Notably, the primary diffraction peaks corresponding to the (200), (220), and (400) planes for NaFeHCF samples are shifted to lower angles compared to the standard card. This shift is attributed to the incorporation of sodium ions into the crystal lattice, leading to an increase in the lattice parameters and distortion of the lattice, resulting in the peak shift. The lattice volume increases with the sodium content, which is beneficial for the insertion and extraction of Na+ ions.58
m space group and a cubic phase structure. After employing a Y-tube in the synthesis process, significant peak splitting at 24.2° and 38.7° is observed, indicating a structural transition from a sodium-deficient cubic phase to a sodium-rich monoclinic phase with the space group P21/n.57 The increased peak intensity and reduced full width at half maximum (FWHM) of Y-PB-0.3 suggest that the use of a Y-tube to enhance micro-mixing can improve the material's crystallinity. With the use of the Y-tube and increasing flow rates, the intensity of the characteristic peaks initially increases and then decreases. The highest peak intensity is observed for Y-PB-15, indicating the highest degree of crystallinity. Notably, the primary diffraction peaks corresponding to the (200), (220), and (400) planes for NaFeHCF samples are shifted to lower angles compared to the standard card. This shift is attributed to the incorporation of sodium ions into the crystal lattice, leading to an increase in the lattice parameters and distortion of the lattice, resulting in the peak shift. The lattice volume increases with the sodium content, which is beneficial for the insertion and extraction of Na+ ions.58
 | ||
| Fig. 2 XRD spectra (a), Raman spectra (b), FTIR spectra (c), TG curves (d), XPS spectra (e), Fe2p high-resolution spectra (f) of NaFeHCF samples. | ||
According to the principle of electrical neutrality, the emergence of a sodium-rich phase results in a lower average oxidation state of transition metal elements in PBAs materials, which signifies a reduction in the average oxidation state of iron.59 Due to the sensitivity of the cyanide stretching vibration modes to the surrounding chemical environment, Raman spectroscopy can be employed to identify the average oxidation state of iron ions. Typically, the Raman peaks associated with cyanide ligands coordinated to Fe2+ ions appear at lower wavenumbers than those bound to Fe3+ ions.60 The Raman peaks observed around 2131 and 2094 cm−1 are generated by the vibrations of FeIII–CN and FeII–CN, respectively, in the Prussian blue structure.61 Fig. 2(b) presents the Raman spectroscopy results for five groups of Fe–PBAs samples. It can be observed that the peaks of the samples synthesized with the assistance of a Y-tube have shifted towards lower wavenumbers compared to C-PB-0.3. Among these, Y-PB-15 exhibits the most significant shift towards lower wavenumbers, indicating a reduction in the average oxidation state of the iron ions. The FTIR spectroscopy results shown in Fig. 2(c) reveal that all five sample groups exhibit absorption peaks for the O–H bond at 3590 and 3620 cm−1 and for the H–O–H bond at 1617 cm−1. These peaks are attributed to the crystal water in the NaFeHCF. The Y-PB-0.3 sample exhibits lower peak intensities related to crystal water vibrations compared to C-PB-0.3, indicating that Y-PB-0.3 contains less crystal water. The absorption peaks located at 2060 cm−1 and 594 cm−1 arise from the –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N and Fe–CN bonds in the NaFeHCF crystal framework. The larger peak areas at 2060 cm−1 and 594 cm−1 for Y-PB-15 suggest a higher content of corresponding chemical bonds in the crystal, implying fewer [Fe(CN)6]4− vacancies in the crystal structure.62,63
N and Fe–CN bonds in the NaFeHCF crystal framework. The larger peak areas at 2060 cm−1 and 594 cm−1 for Y-PB-15 suggest a higher content of corresponding chemical bonds in the crystal, implying fewer [Fe(CN)6]4− vacancies in the crystal structure.62,63
TG analysis was performed to determine the water content in the samples, with the results presented in Fig. 2(d). Between 30 and 100 °C, the loss of water corresponds to the desorption of physically adsorbed water, while the loss between 100 and 200 °C is associated with the removal of interstitial water.11 Both types of water are located on the material's surface without chemical bonds to NaFeHCF, facilitating their removal. Coordinated water, on the other hand, is covalently bonded to Fe atoms in the crystal framework, making it difficult to remove.9 After 200 °C, coordinated water begins to be removed, and as the temperature continues to rise, weight loss plateaus occur between 250 and 270 °C, indicating the nearly complete removal of coordinated water.58,64,65 Subsequently, as the temperature increases further, NaFeHCF undergoes thermal decomposition.60,61 Based on the TG result, the calculated crystal water (from 100 °C to 250–270 °C) contents for C-PB-0.3, Y-PB-0.3, Y-PB-15, Y-PB-30, and Y-PB-41 are 15.15%, 14.39%, 13.59%, 13.88%, and 14.59%, respectively. The use of a Y-tube during the synthesis process has led to a reduction in the water content of the crystals, and an appropriate increase in feed flow rate results in a further decrease in water content. Y-PB-15 exhibits the lowest water content, suggesting that this crystal has the fewest defects and the highest crystallinity. ICP-OES and elemental analysis were employed to further assess the chemical composition of the samples, and the results are presented in Table 1. Compared to C-PB-0.3, Y-PB series samples have a higher sodium content and fewer vacancy defects, aligning with the previously discussed characterization outcomes. After increasing the flow rate, the sodium content initially rises and then decreases, while vacancy defects show a corresponding decrease followed by an increase. This suggests that when the flow rate reaches 15 mL min−1, the synthesized sample exhibits the most favorable crystal structure.
| Sample | Na (wt%) | Fe (wt%) | C (wt%) | N (wt%) | H2O (wt%) | Molecular formula |
|---|---|---|---|---|---|---|
| C-PB-0.3 | 9.12 | 33.68 | 19.33 | 21.72 | 15.15 | Na1.18Fe[Fe(CN)6]0.80□0.20·2.49H2O |
| Y-PB-0.3 | 10.23 | 33.35 | 19.83 | 22.66 | 14.39 | Na1.38Fe[Fe(CN)6]0.85□0.15·2.46H2O |
| Y-PB-15 | 11.12 | 32.87 | 20.14 | 21.88 | 13.59 | Na1.56Fe[Fe(CN)6]0.90□0.10·2.42H2O |
| Y-PB-30 | 10.34 | 33.69 | 19.94 | 22.31 | 13.88 | Na1.37Fe[Fe(CN)6]0.84□0.16·2.34H2O |
| Y-PB-41 | 9.56 | 33.72 | 19.42 | 22.12 | 14.59 | Na1.24Fe[Fe(CN)6]0.81□0.19·2.41H2O |
The XPS survey spectrum of the five NaFeHCF samples depicted in Fig. 2(e) reveals the presence of characteristic peaks at binding energies of 285.4, 397.9, 532.5, 709.3, and 1072.4 eV, which correspond to the C 1s, N 1s, O 1s, Fe 2p, and Na 1s orbital peaks, respectively.66,67 Fig. 2(f) displays the high-resolution Fe 2p spectra for these samples. By quantifying the peak areas of FeII and FeIII in the XPS spectra, the FeII/FeIII ratio in Fe–PBAs can be determined. The peak positions and the ratios of the peak areas for FeII and FeIII are documented in Table 2. It is apparent that in comparison to C-PB-0.3, the FeII/FeIII peak area ratio in Y-PB-0.3 is elevated, which signifies an increased proportion of FeII, thus suggesting a lower average oxidation state of iron ions in Y-PB-0.3. Furthermore, as the flow rate increases, both the peak area ratio and the proportion of FeII incrementally rise, indicating a progressive reduction in the average oxidation state of the iron ions.
| Sample | FeII2p3/2 peak position (eV) | FeII2p1/2 peak position (eV) | FeIII2p3/2 peak position (eV) | FeIII2p1/2 peak position (eV) | Peak area ratio of FeII to FeIII | Proportion of FeII (%) |
|---|---|---|---|---|---|---|
| C-PB-0.3 | 708.63 | 721.47 | 710.88 | 724.52 | 1.62 | 61.77 |
| Y-PB-0.3 | 708.62 | 721.46 | 710.87 | 724.58 | 1.95 | 66.09 |
| Y-PB-15 | 708.65 | 721.47 | 711.16 | 724.12 | 2.21 | 68.84 |
| Y-PB-30 | 708.67 | 721.49 | 711.06 | 724.35 | 2.31 | 69.81 |
| Y-PB-41 | 708.65 | 721.48 | 711.11 | 724.42 | 2.84 | 73.96 |
The morphologies of the various samples were examined using, and the findings are depicted in Fig. 3(a)–(e). It is discernible that the samples consist of uniformly distributed, quasi-cubical particles with an average particle size in the range of 1–2 μm. According to the particle size distribution measurements presented in Fig. 3(a′)–(e′), the average particle size for C-PB-0.3, Y-PB-0.3, Y-PB-15, Y-PB-30, and Y-PB-41 are 1.76 ± 0.40 μm, 1.20 ± 0.19 μm, 1.37 ± 0.20 μm, 1.47 ± 0.41 μm and 1.92 ± 0.52 μm, respectively. Notably, the particles in the Y-PB-0.3 sample exhibit a reduced size and an increased degree of agglomeration compared to those in C-PB-0.3. The introduction of the Y-tube in the synthesis process leads to a reduction in particle size and a narrowing of the particle size distribution. Moreover, as the flow rate increases, there is a trend towards larger particle sizes and a broader particle size distribution. Fig. 3(f) and (g) showcase the TEM images of the samples, where the cubic particles exhibit sharp edges, corroborating the observations made in the SEM images. Fig. 3(k)–(o) display the Energy-Dispersive X-ray Spectroscopy (EDS) maps for the Y-PB-15 sample, indicating a relatively uniform distribution of Na, Fe, N, and C elements within the product. This suggests that the synthesized product has a good level of uniformity.
 | ||
| Fig. 3 SEM images of C-PB-0.3 (a), Y-PB-0.3 (b), Y-PB-15 (c), Y-PB-30 (d), and Y-PB-41 (e); particle size distribution graph of C-PB-0.3 (a′), Y-PB-0.3 (b′), Y-PB-15 (c′), Y-PB- 30 (d′), and Y-PB-41 (e′); TEM images of C-PB-0.3 (f), Y-PB-0.3 (g), Y-PB-15 (h), Y-PB-30 (i), and Y-PB-41 (j); EDS mapping results of Y-PB-15 (k–o). | ||
Fig. 4(a) illustrates the rate capability of each electrode at varying current densities. It is evident that, across all the tested rates, Y-PB-0.3 demonstrates a markedly higher specific capacity compared to C-PB-0.3. With the incorporation of the Y-tube, the electrodes' rate capability initially improves with increasing flow rate but subsequently declines. Notably, Y-PB-15 stands out, maintaining a specific capacity of 71.9 mA h g−1 even at the demanding rate of 10C. This trend may be attributed to an excessive increase in particle size due to high flow velocities, which is detrimental to the delivery of high-rate capacities. Fig. 4(b) depicts the cycling stability of each electrode cycled at 0.5C. The application of the Y-tube technique results in enhanced initial capacities, while cycling performance remains relatively unchanged. As the flow rate is escalated, there is a noticeable increase in initial capacity, with Y-PB-15 reaching an initial capacity of 99.2 mA h g−1. After 200 cycles, it retains 69.8% of its initial capacity. The enhanced performance is likely due to the Y-tube assistance reducing the grain size, which in turn increases the specific surface area and the number of reactive sites, ultimately boosting the material's capacity. A higher flow rate can intensify turbulence and mixing within the Y-tube, leading to a more homogeneous reactant mixture. However, if the flow is too rapid, it can impede crystal growth and introduce additional defects, which may negatively impact the cycling stability of the material. Fig. 4(c)–(g) depict charge–discharge curves of each electrode at different cycle numbers. It can be observed that discharge curves at various cycle numbers exhibit two distinct discharge plateaus around 3.3 V and 3 V. These correspond to the discharge of low-spin Fe and high-spin Fe in two different chemical environments within the Fe–PBAs structure.68,69 With an increase in cycle numbers, the discharge plateaus of each sample progressively shorten. The capacity shows a slow decay in the initial 10 cycles, followed by a faster decay. However, for Y-PB-15, the discharge plateau shows no significant change even after 100 cycles, indicating its excellent cycling performance. SEM images of various samples after cycling tests (Fig. S1, ESI†) indicate that after 200 cycles, the surfaces of the electrodes across the samples show varying extents of cracking and pore emergence. The particle morphology of each sample remains well-preserved, predominantly exhibiting cubic shapes, without significant structural collapse or volume changes. Y-PB-15 stands out with its minimal cracking and pore presence on the electrode surfaces, coupled with a denser and more consistent appearance, signifying superior structural integrity.
 | ||
| Fig. 4 Rate performance (a), cyclic performance at 0.5C (b) of each sample. Charge and discharge curves at 0.1C (first turn) and 0.5C (2nd, 10th, and 100th) for C-PB-0.3 (c), Y-PB-0.3 (d), Y-PB-15 (e), Y-PB-30 (f), and Y-PB-41 (g). | ||
Fig. 5 presents the CV profiles alongside the corresponding differential capacity (dQ/dV) plots for the electrodes. As depicted in Fig. 5(c), Y-PB-0.3 features redox peaks at 3.059/2.823 V and a peak at 2.613 V, attributable to the high-spin Fe3+/Fe2+ transitions (HS Fe–N, 8c) within the CN ligands bound to nitrogen atoms. Additionally, the redox couple observed at 3.469/3.303 V is associated with the low-spin Fe3+/Fe2+ (LS Fe–C, 8c) states in CN ligands coordinated to carbon atoms.70 In comparison, C-PB-0.3 shown in Fig. 5(a) displays a diminishing reduction peak at 2.614 V with subsequent cycles, which suggests a depletion in sodium content within the C-PB-0.3 lattice, leading to compromised cycling stability. Both C-PB-0.3 and Y-PB-0.3 samples exhibit a third oxidation peak above 4.0 V, which markedly diminishes in the second cycle, indicative of Na+ deintercalation from the 24d sites and collateral reactions between the electrolyte and water. Moving to Fig. 5(e), (g), and (i), an escalation in flow rates sees Y-PB-15, Y-PB-30, and Y-PB-41 samples showcasing more pronounced redox peaks associated with high-spin Fe3+/Fe2+ states, suggesting that reduced grain size enhances electron/ion diffusion kinetics and activates the electrochemical functionality of the high-spin Fe3+/Fe2+ system. Additionally, these samples reveal a novel redox feature around 4 V, signifying the Na+ deintercalation/intercalation at the 24d sites, due to the sodium-enriched monoclinic phase crystal structures of Y-PB-15, Y-PB-30, and Y-PB-41, which facilitate the accommodation of Na+ ions into the 24d positions.71,72 The potential separation (ΔE) between the oxidation and reduction peaks in the CV curves is intimately connected to the material's crystallographic stability and the battery's polarization, and the elevated values of ΔE are indicative of heightened polarization.73 The ΔE metrics for the high-spin and low-spin Fe atoms involved in Fe3+/Fe2+ electrode reactions are 0.301 and 0.232, 0.236 and 0.166, 0.100 and 0.110, 0.128 and 0.142, and 0.106 and 0.133 V for the C-PB-0.3, Y-PB-0.3, Y-PB-15, Y-PB-30, and Y-PB-41 samples in respective order. Significantly, Y-PB-15 exhibits the least degree of polarization, hinting at superior reversibility in its electrode reactions. As shown in Fig. 5(b), (d), (f), (h), and (j), the dQ/dV curves of the five electrodes exhibit peaks corresponding to the redox peaks in the CV curves. It is clearly visible that the Fe3+/Fe2+ electrode reaction peaks for high-spin Fe atoms and low-spin Fe atoms remain consistent. The second reduction peak associated with high-spin Fe atoms in C-PB-0.3 disappears after the first cycle, which similarly confirms that Y-PB-0.3 has a higher sodium content compared to C-PB-0.3. With the increase in flow rate, the peak intensity of the redox peaks associated with low-spin Fe atoms in the sample continuously increases. This indicates the activation of more low-spin Fe3+/Fe2+, thereby exhibiting a larger capacity.74
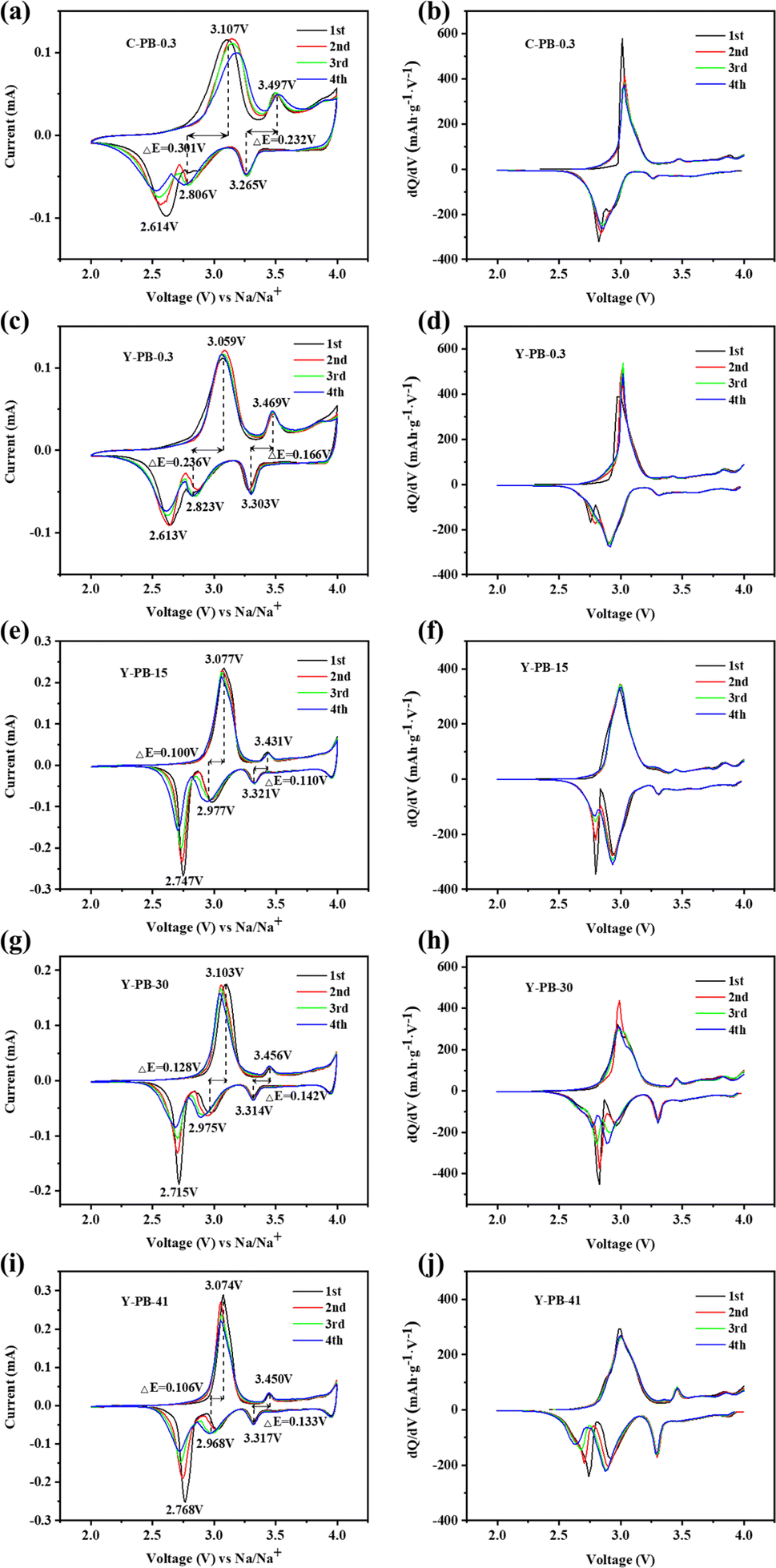 | ||
| Fig. 5 CV curves (a, c, e, g, i) and dQ/dV curves (b, d, f, h, j) for NaFeHCF samples. | ||
EIS measurements were conducted on the test cells before charge and discharge to obtain dynamic parameters such as charge transfer resistance (Rct) and solution resistance (Rs). As shown in Fig. 6(a), compared with C-PB-0.3, Y-PB-0.3 has a smaller semicircle in the mid-to-high frequency region, and the solution impedance Re does not change much, but the charge transfer impedance Rct is significantly reduced. After increasing the flow rate, Re and Rct show a trend of first decreasing and then increasing. The Zview software was utilized for curve fitting, and the corresponding equivalent circuit is provided in the inset of Fig. 6(a). Table 3 reveals that Y-PB-15 has smaller Re and Rct values, which are 5.23 Ω and 1989 Ω, respectively. This is attributed to the assistance of the Y-tube and the appropriate flow rate, which facilitate the electrode–electrolyte interface's transition to a balanced state that remains stable throughout subsequent cycles. σ is defined as the slope of the straight line between Zre and ω−1/2 in the low-frequency range, as shown in Fig. 6(b). The σ value of Y-PB-0.3 is significantly smaller than that of C-PB-0.3. After increasing the flow rate, the σ value also shows a trend of first decreasing and then increasing, with Y-PB-15 having the smallest σ value. A smaller σ value correlates with an enhanced diffusion rate of sodium ions, enabling more efficient utilization of the material and contributing to the stability of the battery.75 This relationship between the σ value and ion diffusion underpins the superior cycling performance observed in Y-PB-15.
 | ||
| Fig. 6 (a) Nyquist plots of the impedance spectra for each sample (inset depicts the equivalent circuit model); (b) fitted curves of the real part of impedance (Zre) as a function of ω−1/2 in the low-frequency range. | ||
| Sample | Re (Ω) | Rct (Ω) | σ |
|---|---|---|---|
| C-PB-0.3 | 2.032 | 3428 | 125.28 |
| Y-PB-0.3 | 6.177 | 2405 | 107.18 |
| Y-PB-15 | 5.23 | 1989 | 59.69 |
| Y-PB-30 | 17.49 | 3560 | 69.20 |
| Y-PB-41 | 10.93 | 4823 | 97.97 |
4. Conclusion
In this study, we employed a Y-tube assisted coprecipitation technique to fabricate NaFeHCF. The Y-tube offers an excellent micro-mixing environment that compensates for the inadequate micro-mixing effects encountered in conventional reactors, which is beneficial for synthesizing high-quality materials characterized by small particle sizes and a narrow size distribution. Moreover, by suitably increasing the reactant flow rate, we can enhance the turbulence and disturbance within the Y-tube, ensuring a more complete mixing of the reactants. Optimal conditions were achieved at a flow rate of 15 mL min−1, resulting in a sample that exhibited the optimal sodium content, minimal vacancy defects, and the least amount of crystal water, thereby delivering the best overall electrochemical performance. This sample demonstrated a capacity of 71.9 mA h g−1 at a high discharge rate of 10C, and maintained a capacity retention rate of 69.8% after 200 cycles at 0.5C. Nonetheless, an excessively high fluid velocity can impede crystal growth and introduce an excess of defects, leading to diminished cycling performance. The Y-tube assisted coprecipitation method used in this study offers high cost-effectiveness, simplicity in operation, and the capacity for continuous production. Moreover, it is beneficial in producing products with reduced vacancy defects and crystal water content, leading to excellent sodium storage performance. The findings offer novel insights into the efficient fabrication of high-performance PBAs cathode materials for potential applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22109107), Natural Science Foundation of Sichuan Province (No. 23NSFSC1450), and Sichuan Science and Technology Project (No. 2021YFH0131).References
- J. Xu, X. Cai, S. Cai, Y. Shao, C. Hu, S. Lu and S. Ding, Energy Environ. Mater., 2022, 6, e12450 CrossRef.
- A. Ramesh, A. Tripathi and P. Balaya, Int. J. Appl. Ceram. Technol., 2022, 19, 913–923 CrossRef CAS.
- S. Baharak, H. Hayley, P. Saurabh, N. Long Hoang Bao, Z. Minghao and M. Ying Shirley, MRS Energy Sustain., 2022, 9, 183–197 CrossRef.
- X. Wang, S. Roy, Q. Shi, Y. Li, Y. Zhao and J. Zhang, J. Mater. Chem. A, 2021, 9, 1938–1969 RSC.
- M. He, R. Davis, D. Chartouni, M. Johnson, M. Abplanalp, P. Troendle and R. P. Suetterlin, J. Power Sources, 2022, 548, 232036 CrossRef CAS.
- D. S. Baji, S. Nair and D. Santhanagopalan, Sustainable Energy Fuels, 2022, 6, 1719–1726 RSC.
- P. Gupta, S. Pushpakanth, M. A. Haider and S. Basu, ACS Omega, 2022, 7, 5605–5614 CrossRef CAS PubMed.
- H. Zhang, Y. Gao, X. H. Liu, L. F. Zhou, J. Y. Li, Y. Xiao, J. Peng, J. Z. Wang and S. L. Chou, Adv. Energy Mater., 2023, 13, 2300149 CrossRef CAS.
- T. Yuan, Y. Chen, X. Gao, R. Xu, Z. Zhang, X. Chen and L. Cui, Small Methods, 2023, 2301372 CrossRef PubMed.
- J. Peng, W. Zhang, Q. N. Liu, J. Z. Wang, S. L. Chou, H. K. Liu and S. X. Dou, Adv. Mater., 2022, 34, 2108384 CrossRef CAS.
- B. X. Xie, B. Y. Sun, T. Y. Gao, Y. L. Ma, G. P. Yin and P. J. Zuo, Coord. Chem. Rev., 2022, 460, 214478 CrossRef CAS.
- D. O. Ojwang, M. Svensson, C. Njel, R. Mogensen, A. S. Menon, T. Ericsson, L. Häggström, J. Maibach and W. R. Brant, ACS Appl. Mater. Interfaces, 2021, 13, 10054–10063 CrossRef CAS.
- L. Yang, Q. Liu, M. Wan, J. Peng, Y. Luo, H. Zhang, J. Ren, L. Xue and W. Zhang, J. Power Sources, 2020, 448, 227421 CrossRef CAS.
- C. M. Xu, J. Peng, X. H. Liu, W. H. Lai, X. X. He, Z. Yang, J. Z. Wang, Y. Qiao, L. Li and S. L. Chou, Small Methods, 2022, 6, 2200404 CrossRef CAS PubMed.
- Y. P. Wang, B. P. Hou, X. R. Cao, S. Q. Wu and Z. Z. Zhu, J. Electrochem. Soc., 2022, 169, 010525 CrossRef CAS.
- Y. Gao, Y. Huang, H. Pan, L. Ji, L. Wang, Y. Tang, Y. Zhu, M. Yan, G. Sun, W. Ni and Y. Jiang, J. Alloys Compd., 2023, 950, 169886 CrossRef CAS.
- J. E. Kang, T. N. Vo, S.-k. Ahn, S.-W. Lee and I. T. Kim, J. Alloys Compd., 2023, 939, 168773 CrossRef CAS.
- Q. Wang, J. Li, H. Jin, S. Xin and H. Gao, InfoMat, 2022, 4, e12311 CrossRef CAS.
- D. Yang, J. Xu, X. Z. Liao, H. Wang, Y. S. He and Z. F. Ma, Chem. Commun., 2022, 58, 13661 RSC.
- M. Jiang, L. Ren, Z. Hou, W. Hua, D. Lei, Y. Cao, Y. Zhang and J. G. Wang, J. Power Sources, 2023, 554, 232334 CrossRef CAS.
- Q. Y. Li, C. Xu, Y. R. Liang, Z. Yang, N. Lege, J. Peng, L. Chen, W. H. Lai, Y. X. Wang, Z. Tao, M. Liu and S. Chou, ACS Appl. Mater. Interfaces, 2022, 14, 47747–47757 CrossRef CAS PubMed.
- S. Fan, Y. Liu, Y. Gao, Y. Liu, Y. Qiao, L. Li and S. L. Chou, SusMat, 2023, 3, 749–780 CrossRef CAS.
- J. Peng, J. Huang, Y. Gao, Y. Qiao, H. Dong, Y. Liu, L. Li, J. Wang, S. Dou and S. Chou, Small, 2023, 19, 2300435 CrossRef CAS PubMed.
- Z. Y. Chen, X. Y. Fu, L. L. Zhang, B. Yan and X. L. Yang, ACS Appl. Mater. Interfaces, 2022, 14, 5506–5513 CrossRef CAS PubMed.
- H. Zhang, J. Peng, L. Li, Y. A. Zhao, Y. Gao, J. Z. Wang, Y. L. Cao, S. X. Dou and S. L. Chou, Adv. Funct. Mater., 2023, 33, 2210725 CrossRef CAS.
- Y. M. Xi and Y. C. Lu, Powder Technol., 2023, 423, 118500 CrossRef CAS.
- Y. M. Xi and Y. C. Lu, ACS Appl. Mater. Interfaces, 2022, 14, 39022–39030 CrossRef CAS PubMed.
- Z. Y. Chen, X. Y. Fu, L. L. Zhang, B. Yan and X. L. Yang, ACS Appl. Mater. Interfaces, 2022, 14, 5506–5513 CrossRef CAS PubMed.
- C. Wei, X. Y. Fu, L. L. Zhang, J. Liu, P. P. Sun, L. Gao, K. J. Chang and X. L. Yang, Chem. Eng. J., 2021, 421, 127760 CrossRef CAS.
- Y. Liu, Y. Qiao, W. X. Zhang, Z. Li, X. Ji, L. Miao, L. X. Yuan, X. L. Hu and Y. H. Huang, Nano Energy, 2015, 12, 386–393 CrossRef CAS.
- Y. Liu, D. D. He, Y. J. Cheng, L. Li, Z. S. Lu, R. Liang, Y. Y. Fan, Y. Qiao and S. L. Chou, Small, 2020, 16, 1906946 CrossRef CAS.
- A. J. Zhou, Z. M. Xu, H. C. Gao, L. G. Xue, J. Z. Li and J. B. Goodenough, Small, 2019, 15, 1902420 CrossRef CAS PubMed.
- J. Song, L. Wang, Y. Lu, J. Liu, B. Guo, P. Xiao, J. J. Lee, X. Q. Yang, G. Henkelman and J. B. Goodenough, J. Am. Chem. Soc., 2015, 137, 2658–2664 CrossRef CAS PubMed.
- W. L. Wang, Y. Gang, J. Peng, Z. Hu, Z. C. Yan, W. H. Lai, Y. F. Zhu, D. Appadoo, M. Ye, Y. L. Cao, Q. F. Gu, H. K. Liu, S. X. Dou and S. L. Chou, Adv. Funct. Mater., 2022, 32, 2111727 CrossRef CAS.
- Y. Tang, W. X. Zhang, L. H. Xue, X. L. Ding, T. Wang, X. X. Liu, J. Liu, X. C. Li and Y. H. Huang, J. Mater. Chem. A, 2016, 4, 6036–6041 RSC.
- Z. Y. Chen, L. L. Zhang, X. Y. Fu, B. Yan and X. L. Yang, ACS Appl. Mater. Interfaces, 2022, 14, 43308–43318 CrossRef CAS PubMed.
- T. Y. Pan, Ruqia, C. Y. Wu, C. S. Ni, S. Gull, A. Haider and H. Y. Chen, Electrochim. Acta, 2022, 427, 140778 CrossRef CAS.
- M. J. Piernas-Munoz, E. Castillo-Martinez, J. L. Gomez-Camer and T. Rojo, Electrochim. Acta, 2016, 200, 123–130 CrossRef CAS.
- X. M. Zhang, Z. Xu, J. Xie, Y. Lu, S. Y. Liu, X. W. Xu, J. Tu, B. Xu and X. B. Zhao, J. Energy Storage, 2024, 80, 110263 CrossRef.
- C. X. Yan, A. Zhao, F. P. Zhong, X. M. Feng, W. H. Chen, J. F. Qian, X. P. Ai, H. X. Yang and Y. L. Cao, Electrochim. Acta, 2020, 332, 135533 CrossRef CAS.
- Y. Xu, M. Y. Ou, Y. Liu, J. Xu, X. P. Sun, C. Fang, Q. Li, J. T. Han and Y. H. Huang, Nano Energy, 2020, 67, 104250 CrossRef CAS.
- Y. You, X. L. Wu, Y. X. Yin and Y. G. Guo, Energy Environ. Sci., 2014, 7, 1643–1647 RSC.
- W. J. Li, S. L. Chou, J. Z. Wang, Y. M. Kang, J. L. Wang, Y. Liu, Q. F. Gu, H. K. Liu and S. X. Dou, Chem. Mater., 2015, 27, 1997–2003 CrossRef CAS.
- S. A. F. S. M. Fadzil, H. J. Woo, A. D. Azzahari, T. Winie and M. Z. Kufian, Mater. Today Chem., 2023, 30, 101540 CrossRef.
- J. W. Hu, H. W. Tao, M. L. Chen, Z. C. Zhang, S. L. Cao, Y. Shen, K. Jiang and M. Zhou, ACS Appl. Mater. Interfaces, 2022, 14, 12234–12242 CrossRef CAS.
- Y. C. Chen, H. J. Woo, S. A. F. S. M. Fadzil, W. N. Tan, F. Wang and A. K. M. Arof, ACS Appl. Nano Mater., 2022, 5, 4833–4840 CrossRef CAS.
- W. Wang, Y. Gang, Z. Hu, Z. Yan, W. Li, Y. Li, Q. F. Gu, Z. Wang, S. L. Chou, H. K. Liu and S. X. Dou, Nat. Commun., 2020, 11, 980 CrossRef CAS PubMed.
- X. Y. Wu, C. H. Wu, C. X. Wei, L. Hu, J. F. Qian, Y. L. Cao, X. P. Ai, J. L. Wang and H. X. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 5393–5399 CrossRef CAS PubMed.
- H. Jeong, S. H. Ahn and C. Jo, Chem. Eng. J., 2023, 465, 142834 CrossRef CAS.
- Y. Xi and Y. Lu, Chem. Eng. J., 2021, 405, 126688 CrossRef CAS.
- Y. Xi and Y. Lu, Cryst. Growth Des., 2021, 21, 1086–1092 CrossRef CAS.
- R. S. Abiev, Y. S. Kudryashova, A. V. Zdravkov and N. Y. Fedorenko, Inorganics, 2023, 11, 49 CrossRef CAS.
- P. Zhu, Y. Wang, J. Li and Y. Jin, ACS Appl. Energy Mater., 2023, 6, 6141–6150 CrossRef.
- E. Mosheva and I. Krasnyakov, Exp. Therm. Fluid Sci., 2024, 151, 111100 CrossRef CAS.
- X. Wan, J. Li, N. Li, J. Zhang, Y. Gu, G. Chen and S. Ju, Materials, 2023, 16, 2217 CrossRef CAS PubMed.
- H. Helisaz, M. Babaei and A. Sadeghi, Chem. Eng. Sci., 2018, 191, 358–368 CrossRef CAS.
- P. Wang, Y. Li, D. Zhu, F. Gong, S. Fang, Y. Zhang and S. Sun, Dalton Trans., 2022, 51, 9622–9626 RSC.
- W. Wang, Y. Gang, J. Peng, Z. Hu, Z. Yan, W. Lai, Y. Zhu, D. Appadoo, M. Ye, Y. Cao, Q. F. Gu, H. K. Liu, S. X. Dou and S. L. Chou, Adv. Funct. Mater., 2022, 32, 2111727 CrossRef CAS.
- W. Geng, Z. Zhang, Z. Yang, H. Tang and G. He, Chem. Commun., 2022, 58, 4472–4475 RSC.
- M. Yang, K. Wang, Q. Liu, S. Cao, J. Wang and Y. Liu, J. Alloys Compd., 2024, 973, 172899 CrossRef CAS.
- Y. Luo, J. Peng and Y. Yan, RSC Adv., 2021, 11, 31827–31833 RSC.
- Y. Li, Q. Dang, W. Chen, L. Tang and M. Hu, J. Inorg. Organomet. Polym. Mater., 2021, 31, 1877–1893 CrossRef CAS.
- W. C. Chen, S. J. Li, H. Y. Xu, S. H. Xu and G. T. Fei, ChemPhysChem, 2024, e202300960 CrossRef CAS PubMed.
- W.-J. Li, C. Han, G. Cheng, S.-L. Chou, H.-K. Liu and S.-X. Dou, Small, 2019, 15, 1900470 CrossRef PubMed.
- X. Liu, H. Gong, C. Han, Y. Cao, Y. Li and J. Sun, Energy Storage Mater., 2023, 57, 118–124 CrossRef.
- M. Fayaz, W. Lai, J. Li, W. Chen, X. Luo, Z. Wang, Y. Chen, D. Li, S. M. Abbas and Y. Chen, Mater. Res. Bull., 2024, 170, 112593 CrossRef CAS.
- X. Dong, H. Wang, J. Wang, Q. Wang, H. Wang, W. Hao and F. Lu, Molecules, 2023, 28, 7267 CrossRef CAS PubMed.
- Y. Shan, G. Zhang, W. Yin, H. Pang and Q. Xu, Bull. Chem. Soc. Jpn., 2022, 95, 230–260 CrossRef CAS.
- K. Nakamoto, R. Sakamoto, Y. Sawada, M. Ito and S. Okada, Small Methods, 2019, 3, 1970010 CrossRef.
- H. Hayashi, Front. Phys., 2022, 10, 214 Search PubMed.
- V. D. Ivanov, Ionics, 2020, 26, 531–547 CrossRef CAS.
- S. Kjeldgaard, I. Dugulan, A. Mamakhel, M. Wagemaker, B. B. Iversen and A. Bentien, R. Soc. Open Sci., 2021, 8, 201779 CrossRef CAS PubMed.
- Q. Li, T. Li, C. Shao and W. Liu, Prog. Chem., 2023, 35, 1053–1064 CAS.
- Y. Luo, L. Yang, Q. Liu and Y. Yan, R. Soc. Open Sci., 2021, 8, 211092 CrossRef CAS PubMed.
- Y. Li, W. X. He, X. Y. Zheng, S. L. Yu, H. T. Li, H. Y. Li, R. Zhang and Y. Wang, J. Inorg. Mater., 2019, 34, 365–372 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00762j |
| This journal is © The Royal Society of Chemistry 2024 |