DOI:
10.1039/D4RA00470A
(Paper)
RSC Adv., 2024,
14, 11877-11884
First-principles study of the mechanical and thermodynamic properties of aluminium-doped magnesium alloys†
Received
18th January 2024
, Accepted 27th March 2024
First published on 15th April 2024
Abstract
Magnesium–aluminum (Mg–Al) alloys are widely used in aerospace, automobile and medical equipment owing to their advantages of easy casting, high strength-to-mass ratio and good biocompatibility. The structural, mechanical, electronic and thermodynamic properties of MgxAly alloys (x + y = 16, x = 1, 2,…, 15) with varying Al-doping contents were studied using the first-principles method. In this work, the structures of MgxAly alloys were constructed by replacing Mg atoms in a supercell with Al atoms. The lattice parameters of the Al-doped MgxAly alloys decrease with an increasing Al content because of the smaller atomic size of Al than that of Mg. The calculated formation energies show that Mg11Al5, Mg5Al3 and Mg9Al7 have prominent structural stability. The analyses of the mechanical properties reveal that the doping of Al improves the ductility of MgxAly alloys. The elastic moduli increase with an increasing Al content, and Mg9Al7 has a notable ability to resist deformation, while Mg11Al5 and Mg5Al3 have better plasticity. The calculated results of their electronic properties reveal that Mg11Al5, Mg5Al3 and Mg9Al7 are good conductors without magnetism. Furthermore, CDD analyses show that the inner layer charges of Al atoms migrated to the outer layer, and the charges of Mg atoms accumulated significantly in the outer region of Al atoms. The Debye temperature of Mg9Al7 is higher than that of Mg11Al5 and Mg5Al3, indicating that it has better thermodynamic stability. Our findings would be helpful for the design of Mg–Al alloys with excellent mechanical and thermodynamic performances.
1. Introduction
Magnesium (Mg) based alloys have the advantages of easy casting, high strength-to-mass ratio and good biocompatibility, which are widely used in fields such as the aircraft industry, automotive production and medical equipment.1–5 Among all Mg-based alloys, magnesium–aluminum (Mg–Al) alloys hold an important and special position.6–8 Firstly, the addition of Al could improve the flow ability, hot cracking resistance and mechanical properties of Mg alloys without significantly increasing their density.9 The density of Mg–Al alloys ranges from 1.74 to 2.7 g cm−3, with a maximum Young's modulus of 78.4 GPa, which has attracted widespread attention in the field of mechanical manufacturing.10 Secondly, the density and mechanical properties of Mg–Al alloys are close to that of human cortical bones, and they have good biocompatibility and biodegradability.11,12 These advantages make them extremely suitable as repair materials for human bone injuries. Due to the special applications of Mg–Al alloys, their mechanical, thermodynamic and electronic properties have been extensively studied through experiments.13–17 For example, Hsu et al.17 prepared several Mg–Al alloy electrodes with different Mg/Al mass ratios using powder metallurgy. They found that the Mg–Al alloy with 75 wt% has the highest efficiency in water electrolysis experiments, considering its anticorrosion property and material costs. Although many studies have been conducted on the properties of Mg–Al alloys, there are still significant challenges in exploring and preparing potential Mg–Al alloys with practical value because of the limitations in experimental equipment and technology. With the development of computer technology and scientific computing software, simulating the properties of materials at the atomic and electron level has become popular. Computational simulation is of great significance for the discovery of new materials and the improvement of material performance through specific designs. Doping other elements or clusters into materials is one of the efficient methods for discovering new high-performance materials, and many valuable works have been done in this regard.18–23 For example, Abbasi et al.20 studied the effects of elemental doping on the structural and electronic properties of stanene. Their results show that the influence of different doping elements on the electronic properties of the structure is significant, which provides a good idea for the design of electronic and optoelectronic devices in the future.
Many factors can affect the mechanical and thermodynamic properties of metal alloys, such as mass ratio, phase composition and grain boundaries.24–27 Among these factors, the mass ratio of elements in alloys is fundamental and has a significant impact on their performance.24,25 For Mg–Al alloys, some studies on mass ratios have also been reported.28–30 Using the atomic substitution method, Zhou et al.28 obtained the supercell of Mg15Al and studied the mechanical and electronic properties. The calculated B/G ratio of pure Mg and Mg15Al are 1.92 and 1.8, respectively, indicating that both structures are ductile. However, computational studies on discovering potential structures of Mg–Al alloys through doping procedures are still very rare. In this work, we aim to screen out potential structures with the lowest energy and stable state from massive initial Mg–Al supercells. Firstly, a large number of Mg–Al supercells with different Mg/Al ratios and substitution sites were constructed using the atomic substitution method. Then, potential stable structures were determined according to formation energies and the convex hull diagram. For the selected structures, we studied their structural features, electronic properties, and mechanical and thermodynamic properties based on density functional theory (DFT) schemes.31 Our work will provide guidance for designing new Mg–Al alloys and give a further understanding of their structural performance relationships.
2. Methods
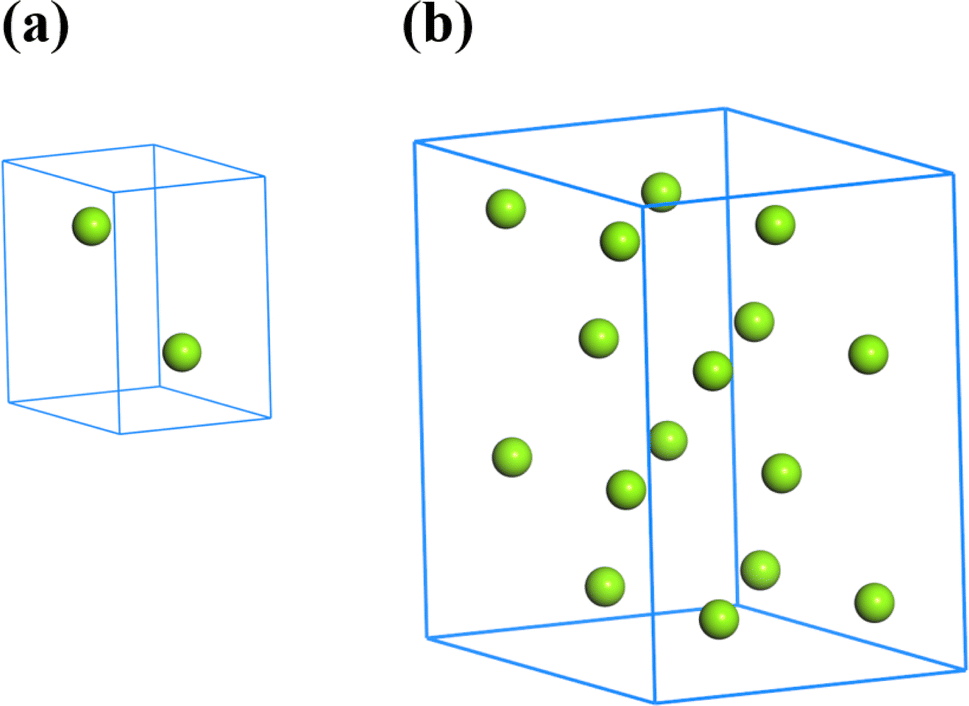
The single crystal of Mg belongs to a hexagonal close-packed structure (space group P63/mmc). As shown in Fig. 1a, each unit cell of Mg contains 2 atoms. The lattice parameters are a = b = 3.2094 Å and c = 5.2105 Å, which were determined by Batchelder and Raeuchle.32 A 2 × 2 × 2 supercell with lattice parameters a = b = 6.4188 Å and c = 10.421 Å was constructed with a total of 16 Mg atoms, as shown in Fig. 1b. The site of each Mg atom can be doped by an Al atom, assuming that the lattice parameters have no change. To study the effect of the Mg/Al ratio on the mechanical and thermodynamic properties of Mg–Al alloys, structures of 15 proportions were constructed according to the formula MgxAly (x + y = 16, x = 1, 2,…, 15). For each Mg/Al ratio, the number of the potential structures is Cy15, without considering the structural symmetry. Then, optimization calculations were performed on all structures to determine the structure with the lowest energy for each Mg/Al atomic ratio. To further evaluate and determine the stable structures, we calculated the formation energies and convex hull diagram of MgxAly structures with different Mg/Al ratios.
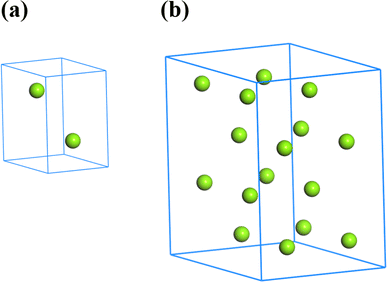 |
| | Fig. 1 The unit cell and supercell of Mg. | |
In this work, all of the calculations were performed using the Vienna ab initio simulation package (VASP).33,34 The generalized gradient approximation (GGA) using the Perdew–Burke–Ernzerhof (PBE) function35 was used to describe the exchange and correlation interactions, and the projector-augmented wave (PAW)36,37 method was also used in this work. The kinetic-energy cutoff was set to 500 eV for the plane-wave basis set. All lattice parameters and atomic coordinates were fully relaxed until a force tolerance of 0.01 eV Å−1 is reached. The Monkhorst–Pack k-point grids38 used for all MgxAly structures are 8 × 8 × 12. The selected parameters ensure that the accuracy of the total energy is converged to within 1.0 meV per atom. The elastic constants were calculated by the stress–strain method.39 Phonon calculations were carried out using the direct supercell method as implemented in the PHONOPY program40 to determine the dynamical stability of MgxAly structures.
3. Results and discussion
3.1 Lattice parameters and formation energies
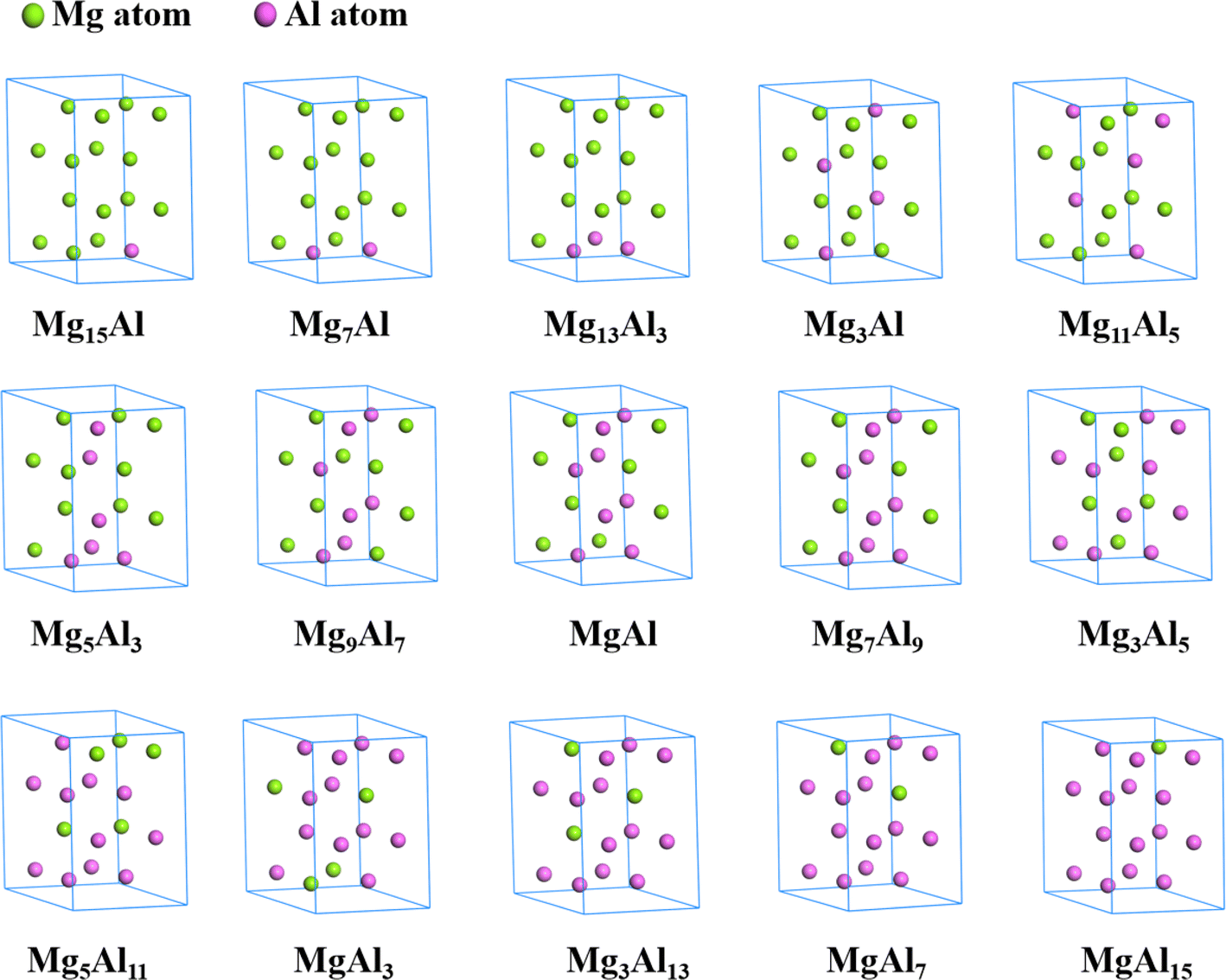
All optimized structures of MgxAly with the lowest energy were displayed by the Materials Studio software, as shown in Fig. 2. It is worth noting that the atom number of MgxAly structures discussed in this paper are all 16, and in order to unify writing standards, x and y are both the simplest ratios. For example, Mg14Al2 is abbreviated as Mg7Al. The lattice constants, volume and density for Mg16, fcc Al and MgxAly structures are listed in Table 1. When the number of doped Al atoms is less than 5, the structures with the lowest energy under each Mg/Al ratio show a bias of Al atom aggregation. The results reveal that the doping of Al atoms causes the change of MgxAly structures, and the values of a, c and c/a fluctuate and show a decreasing trend in general with increasing Al atoms. Due to the smaller atomic radii of Al compared with Mg, the volume of MgxAly decreases with increasing Al content. In addition, Al has a heavier atomic mass, which leads to an increase in density.
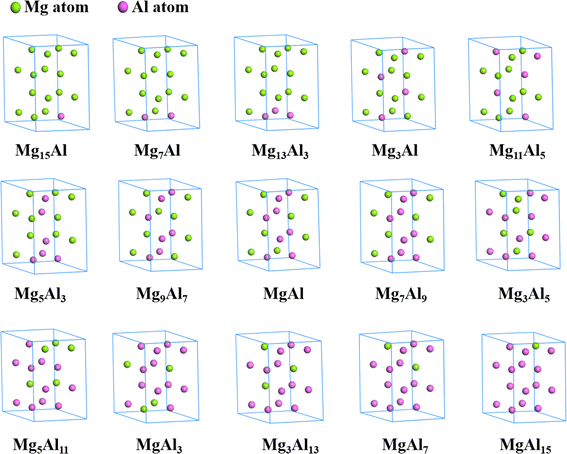 |
| | Fig. 2 The structures of MgxAly with the lowest energy. | |
Table 1 The lattice constants, volume and density of Mg16, fcc Al and MgxAly alloys
| Structures |
a (Å) |
c (Å) |
c/a |
V (Å3) |
ρ (g cm−3) |
| Mg16 |
6.430 |
10.39 |
1.616 |
365.4 |
1.745 |
| Mg15Al |
6.349 |
10.28 |
1.619 |
358.7 |
1.792 |
| Mg7Al |
6.339 |
10.24 |
1.615 |
351.6 |
1.842 |
| Mg13Al3 |
6.233 |
10.29 |
1.651 |
346.0 |
1.886 |
| Mg3Al |
6.194 |
10.03 |
1.619 |
333.6 |
1.971 |
| Mg11Al5 |
6.164 |
10.02 |
1.626 |
327.7 |
2.022 |
| Mg5Al3 |
6.172 |
9.936 |
1.610 |
321.9 |
2.074 |
| Mg9Al7 |
6.102 |
9.772 |
1.601 |
314.4 |
2.140 |
| MgAl |
6.056 |
9.652 |
1.594 |
308.8 |
2.194 |
| Mg7Al9 |
5.998 |
9.581 |
1.597 |
302.4 |
2.257 |
| Mg3Al5 |
6.043 |
9.412 |
1.558 |
300.0 |
2.292 |
| Mg5Al11 |
6.050 |
9.403 |
1.554 |
295.9 |
2.341 |
| MgAl3 |
6.019 |
9.291 |
1.544 |
290.7 |
2.400 |
| Mg3Al13 |
6.013 |
9.140 |
1.520 |
285.9 |
2.458 |
| MgAl7 |
5.955 |
9.142 |
1.535 |
281.2 |
2.516 |
| MgAl15 |
5.893 |
9.165 |
1.555 |
275.7 |
2.584 |
| Al |
4.044 |
|
|
66.17 |
2.712 |
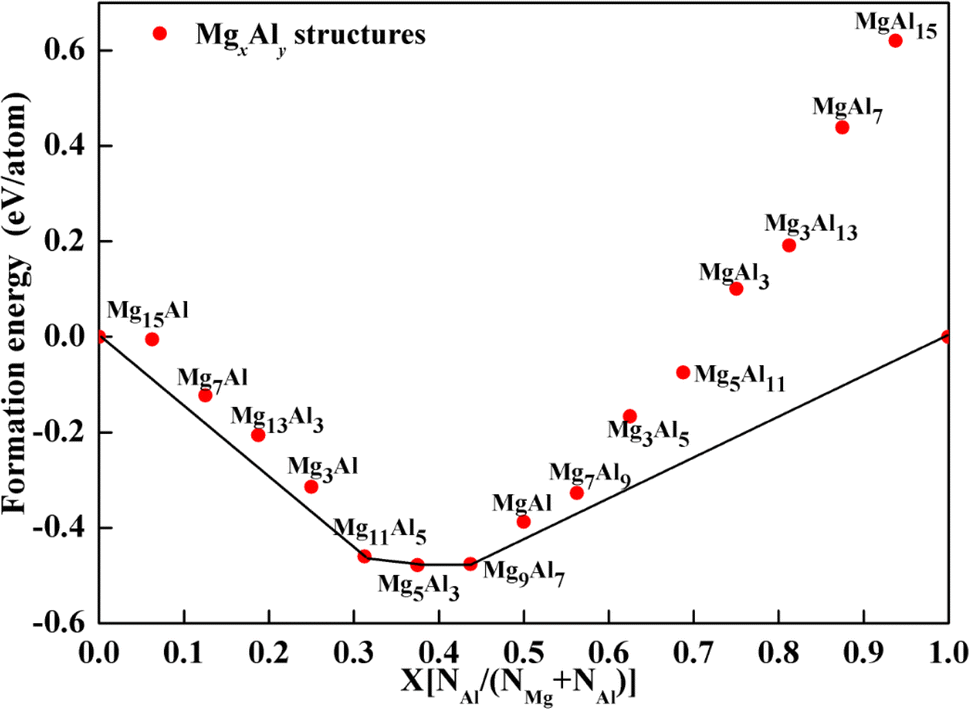
To preliminarily evaluate the stability of the structures, formation energies were calculated and a convex hull diagram was plotted. The formation energy of the MgxAly structure at 0 K is defined as follows,
| | |
Ef = (E(MgxAly) − xE(Mg) − yE(Al))/(x + y)
| (1) |
where
E(Mg
xAl
y) refers to the total energy of the Mg
xAl
y structure in the ground state, and
E(Mg) and
E(Al) represent the energy per atom of hcp Mg and fcc Al, respectively.
41 Fig. 3 shows the calculated formation energies and convex hull of Mg
xAl
y structures with different Mg/Al ratios. As depicted in
Fig. 3, when the number of Al atoms substituted in the Mg
xAl
y structures is greater than 11, the formation energies are positive, indicating that those structures are unstable. Due to the fact that structures with formation energies above the convex hull are meta-stable,
42 Mg
11Al1
5, Mg
5Al
3 and Mg
9Al
7 were confirmed to be the most stable structures and adopted for further study.
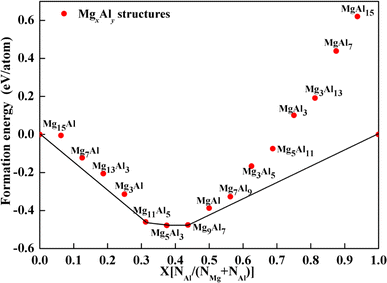 |
| | Fig. 3 Calculated formation energy of MgxAly alloys as a function of Al-doping contents. | |
3.2 Elastic properties
In addition to the formation energy, another index of structural stability is mechanical stability, which can be studied by elastic constants. We calculated the elastic constants of Mg16, fcc Al and MgxAly structures, as shown in Table 2. For Mg16, the value of C11 in this work is 2 GPa smaller than that of Wazzan et al.44 measured through experiments. The values of C12 and C13 are also very close to the results obtained by Wazzan et al. with the largest discrepancy being 3.83%. However, the C33 and C44 values of Mg16 are closer to the results obtained by Slutsky et al.45 through experiments with the largest discrepancy of 3.16%. For fcc Al, the values of C11 and C12 in this work are very close to those obtained by Le Page et al.39 by ab initio calculations with the largest discrepancy of 1.94%, while the value of C44 is 3.1 GPa smaller than theirs. Considering the differences in experimental and computational methods, our calculated elastic constants Cij have a good agreement with their results. Due to the addition of Al, the elastic constants of MgxAly alloys increase significantly, and the elastic constants of Mg11Al5, Mg5Al3 and Mg9Al7 increase with increasing Al content. Born's stability criteria47 is also usually used to determine the mechanical stability of materials. According to Born, any applied strain should increase the energy of the stable ground-state solid. The eigenvalues of the stiffness tensor and its matrix are positive. Therefore, the elastic constants of crystals should satisfy certain conditions according to their symmetry. The formulas of mechanical stability criteria for hcp Mg16, fcc Al and MgxAly structures are given in ESI.† From our calculated elastic constants, we confirm that Mg16, fcc Al, Mg11Al5, Mg5Al3 and Mg9Al7 are mechanically stable.
Table 2 The elastic constants (GPa) of Mg16, fcc Al and MgxAly alloys
| Structures |
C11 |
C12 |
C13 |
C33 |
C44 |
| Ref. 43. Ref. 44. Ref. 45. Ref. 39. Ref. 46. Present work. |
| Mga |
62.4 |
23.5 |
22.0 |
58.9 |
16.3 |
| Mgb |
59.5 |
26.1 |
21.8 |
61.6 |
16.4 |
| Mgc |
63.5 |
25.9 |
21.7 |
66.5 |
18.4 |
| Mg16f |
57.5 |
27.1 |
21.0 |
68.6 |
18.1 |
| Mg11Al5 |
70.7 |
37.5 |
30.5 |
79.4 |
12.9 |
| Mg5Al3 |
73.7 |
38.5 |
31.6 |
80.7 |
14.5 |
| Mg9Al7 |
76.9 |
44.6 |
31.4 |
88.3 |
21.6 |
| Ald |
114.0 |
62.0 |
|
|
32.0 |
| Ale |
120.0 |
61.0 |
|
|
28.0 |
| Alf |
115.2 |
63.2 |
|
|
28.9 |
Based on elastic constants, we also calculated the bulk modulus B, shear modulus G and Young's modulus E of Mg16, fcc Al and MgxAly structures, as shown in Table 3. The elastic moduli and Poisson's ratio v were obtained using the Voigt–Reuss–Hill averaging scheme,48–50
| | |
v = (3B − 2G)/[2(3B + G)]
| (5) |
where
BR and
BV represent the bulk moduli with the Reuss and Voigt averaging scheme, respectively.
GR and
GV represent the shear moduli with the Reuss and Voigt averaging scheme, respectively. The formulas for calculating
BR,
BV,
GR and
GV through
Cij are also given in ESI.
† For Mg
16, the value of Young's modulus
E is 45.6 GPa, which is 0.9 GPa smaller than the value calculated by Velikokhatnyi
et al.43 by first-principles calculations and 1.1 GPa larger than the result measured by Wazzan
et al.44 through experiments. The bulk modulus
B of Mg
16 is 35.7 GPa, which is very close to the value calculated by Velikokhatnyi
et al. However, the discrepancy is 2.52%, compared with the result from Wazzan
et al.. In addition, the shear modulus
G of Mg
16 is 17.7 GPa, which differs from the values of Velikokhatnyi
et al. and Wazzan
et al. by 0.4 and 0.5 GPa, respectively. In general, our results are very close to the values obtained by others through experiments and calculations, indicating that the method used in this work to calculate elastic properties is appropriate. For Mg
xAl
y, the elastic moduli increase with increasing Al content. It should be noted that
E and
G of Mg
11Al
5 and Mg
5Al
3 have similar values as Mg
16, while the bulk moduli
B are larger. The doping of Al atoms changes the structure of the Mg supercell, and the results reveal that the effect of Al doping on the volume compression is greater than that of shear and tension. The elastic moduli of Mg
9Al
7 are significantly higher than those of Mg
11Al
5 and Mg
5Al
3, which is mainly because the Al atoms in Mg
9Al
7 supercell are more concentrated.
B/
G value can be used to characterize the ductility and brittleness of materials. According to the Pugh criterion,
51 B/
G values greater than 1.75 indicate ductility, while a value less than 1.75 characterizes brittleness. Our calculated
B/
G values show that Mg
xAl
y alloys have better ductility than pure Mg. In addition, the elastic moduli of Mg
11Al
5, Mg
5Al
3 and Mg
9Al
7 increase with increasing Al content, while the
B/
G values show an opposite trend. The results indicate that Mg
9Al
7 has the best ability to resist deformation, while Mg
11Al
5 and Mg
5Al
3 have better plasticity.
Table 3 The elastic moduli (GPa) of Mg16, fcc Al and MgxAly alloys
| Structures |
E |
B |
G |
v |
B/G |
AU |
Hv |
| Ref. 43. Ref. 44. Present work. |
| Mga |
46.5 |
35.4 |
18.1 |
0.28 |
1.96 |
|
|
| Mgb |
44.5 |
36.6 |
17.2 |
0.29 |
2.06 |
|
|
| Mg16c |
45.6 |
35.7 |
17.7 |
0.29 |
2.02 |
0.12 |
3.2 |
| Mg11Al5 |
43.6 |
46.4 |
16.2 |
0.34 |
2.86 |
0.27 |
2.0 |
| Mg5Al3 |
46.6 |
47.9 |
17.4 |
0.34 |
2.75 |
0.19 |
2.2 |
| Mg9Al7 |
53.9 |
50.8 |
20.4 |
0.32 |
2.49 |
0.24 |
2.8 |
| Al |
74.6 |
80.5 |
27.7 |
0.35 |
2.91 |
0.01 |
2.9 |
The elastic anisotropy of these structures was also studied according to a universal index of AU = 5GV/GR + BV/BR − 6, which is related to the occurrence of micro-cracks in materials.52 The departure of AU from zero (for isotropic crystal) defines the extent of crystal anisotropy and accounts for both the shear and the bulk contributions.53 As shown in Table 3, the elastic anisotropy index shows that Mg16 and MgxAly structures have considerable elastic anisotropy compared with fcc Al. The AU value of Mg11Al5 is 0.27, indicating that the anisotropy is the most obvious. In addition, the hardness Hv of these three structures was evaluated according to the empirical correlation,54
| | |
Hv = 0.92(G/B)1.137G0.708
| (6) |
where
G and
B represent the shear modulus and bulk modulus, respectively. Without considering the intercept term, the hardness formula obtained by Chen
et al.54 eliminates the possibility of unrealistic negative hardness. For Mg
xAl
y, the hardness of Mg
9Al
7 is larger than that of Mg
11Al
5 and Mg
5Al
3, as shown in
Table 3. Hardness can be regarded as the comprehensive resistance of chemical bonds to indentation, indicating that the interaction between atoms was enhanced with increasing Al atoms.
3.3 Electronic properties
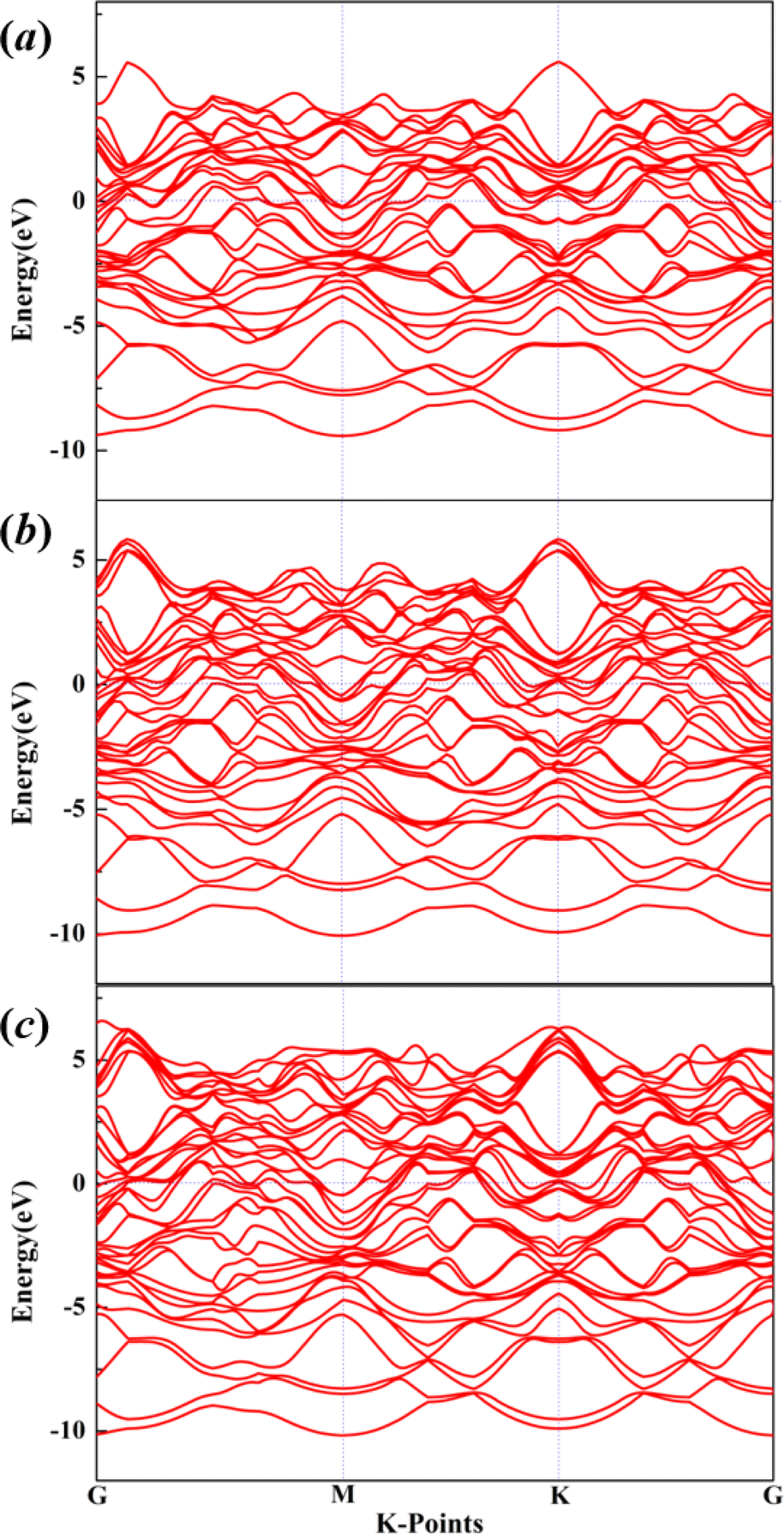
The electronic properties are of great significance for the development and application of new materials. In this work, we also employed the HSE06 hybrid functional55 to calculate the electronic band structures of Mg11Al5 (a), Mg5Al3 (b) and Mg9Al7 (c) with the Fermi level shifted to zero, as shown in Fig. 4. The results reveal that the valence bands and conduction bands of these structures overlap at the Fermi level, resulting in a band gap value of 0. As is well known, conductivity occurs in one or more partially filled energy bands in metals, which possess both valence and conduction band properties. Therefore, our results also confirm that Mg–Al alloy is a good conductor. In addition, we also calculated the magnetization of the structures considering spin polarization. The calculated magnetization values of Mg11Al5, Mg5Al3 and Mg9Al7 are 0.0029, 0.0028 and 0.0027 A m−1, respectively, which are much lower than the magnetization values of magnetic materials,56–58 indicating that these structures are in a non-magnetic state.
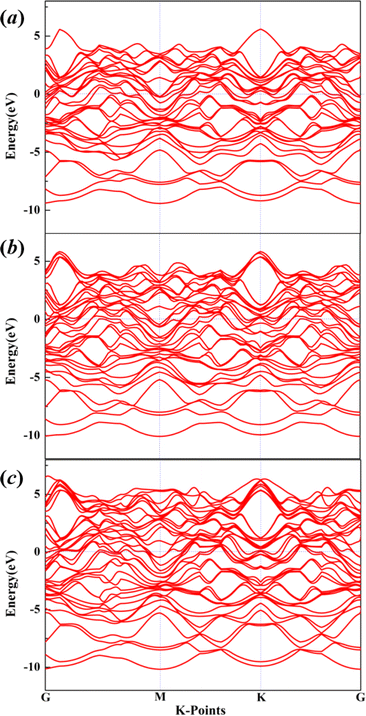 |
| | Fig. 4 The electronic band structures of Mg11Al5 (a), Mg5Al3 (b) and Mg9Al7 (c) with the Fermi level shifted to zero. | |
To discuss the electronic structure of the system further, we also calculated the charge density difference (CDD) for Mg11Al5, Mg5Al3 and Mg9Al7. The CDD is one of the important ways to investigate electronic structure, which could help us understand the charge transfer induced by the doping of Al atoms. Usually, the charge density of the total system is obtained by subtracting the density of two discrete segments that make it,
| | |
Δρ = ρ(Mg+Al) − ρ(Mg) − ρ(Al)
| (7) |
where
ρ(Mg+Al) denotes the total charge density of the Mg
xAl
y structure.
ρ(Mg) and
ρ(Al) represent the charge densities of Mg and Al fragments taken from the Mg
xAl
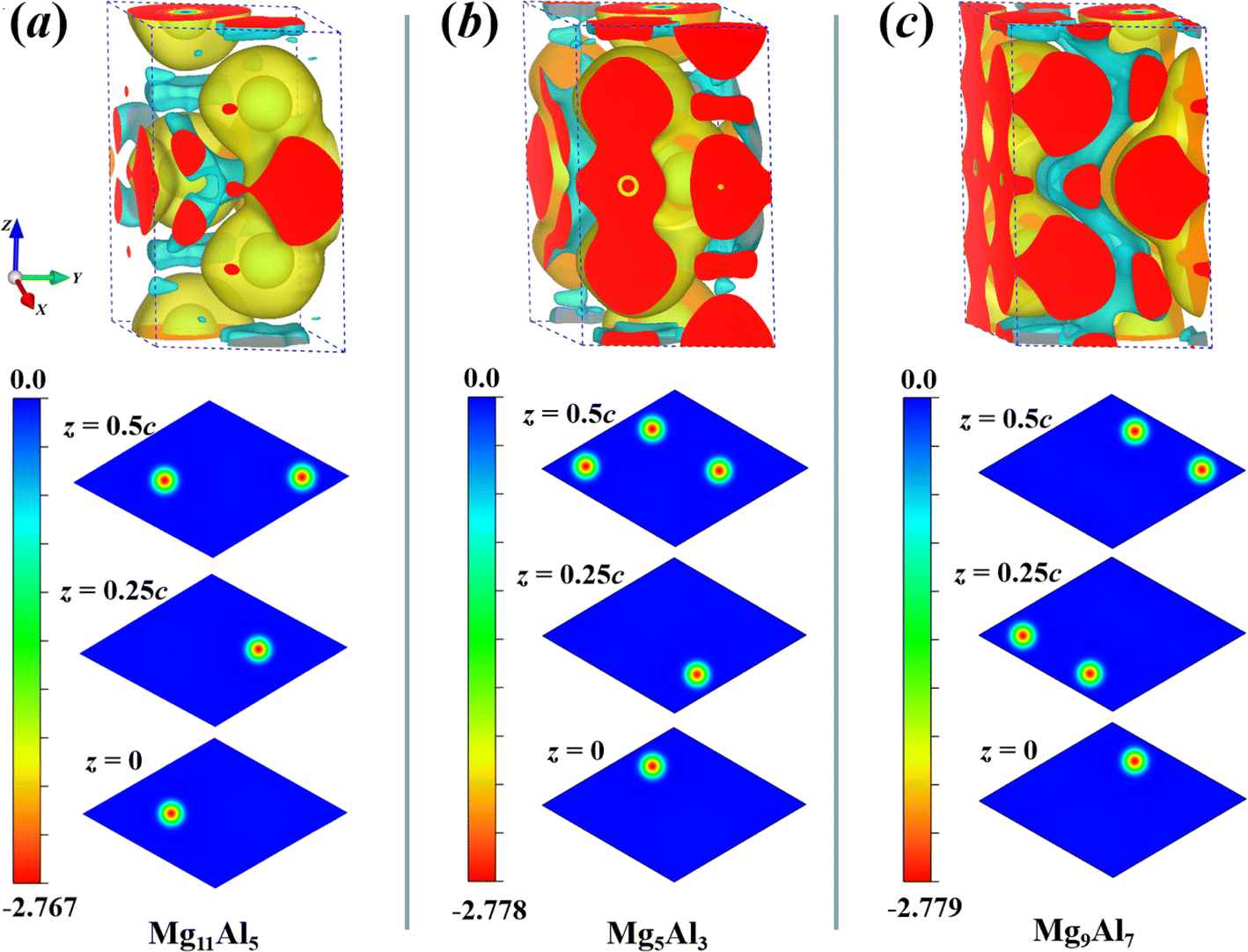
y supercell, respectively. As shown in
Fig. 5, the yellow and cyan regions represent the positive (electron accumulation) and negative (electron depletion) values, respectively. For Mg
11Al
5, Mg
5Al
3 and Mg
9Al
7, their CDD exhibits similar charge transfer behavior. The inner layer charges of Al atoms migrated to the outer layer, and the charges of Mg atoms also accumulated significantly in the outer region of Al atoms. Besides, the increase in the charge density at the middle of Al–Al domains reveals the formation of covalent bonds between these atoms. To observe the charge transfer of Al atoms at different sites in the
Z-direction, two-dimensional cross-sectional color maps at different
z-positions are also provided. As can be seen from
Fig. 5, the charge transfer of Al atoms at different sites is similar; that is, they all migrate outward from the atomic center, with a maximum charge transfer value of −2.779
e Bohr
−3.
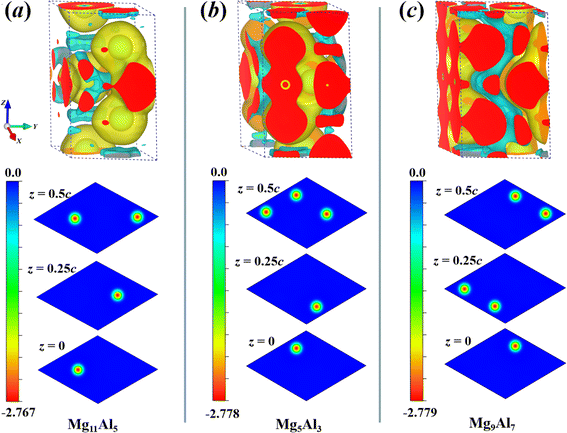 |
| | Fig. 5 The charge density difference (CDD) of Mg11Al5 (a), Mg5Al3 (b) and Mg9Al7 (c) with the isosurface plot level of 0.002 obtained using VESTA software.59 The yellow and cyan regions represent positive and negative values, respectively. Correspondingly, two-dimensional cross-sectional color maps at different z-positions are also provided. | |
3.4 Thermodynamic properties
The Debye temperature (θD) is closely related to the elastic constants, melting temperature and specific heat of materials, which can be used to characterize the strength of covalent bonds.60 In this work, Debye temperature was calculated from the average sound velocity (vm) as a function of longitudinal sound velocity (vl) and shear sound velocity (vt).60,61
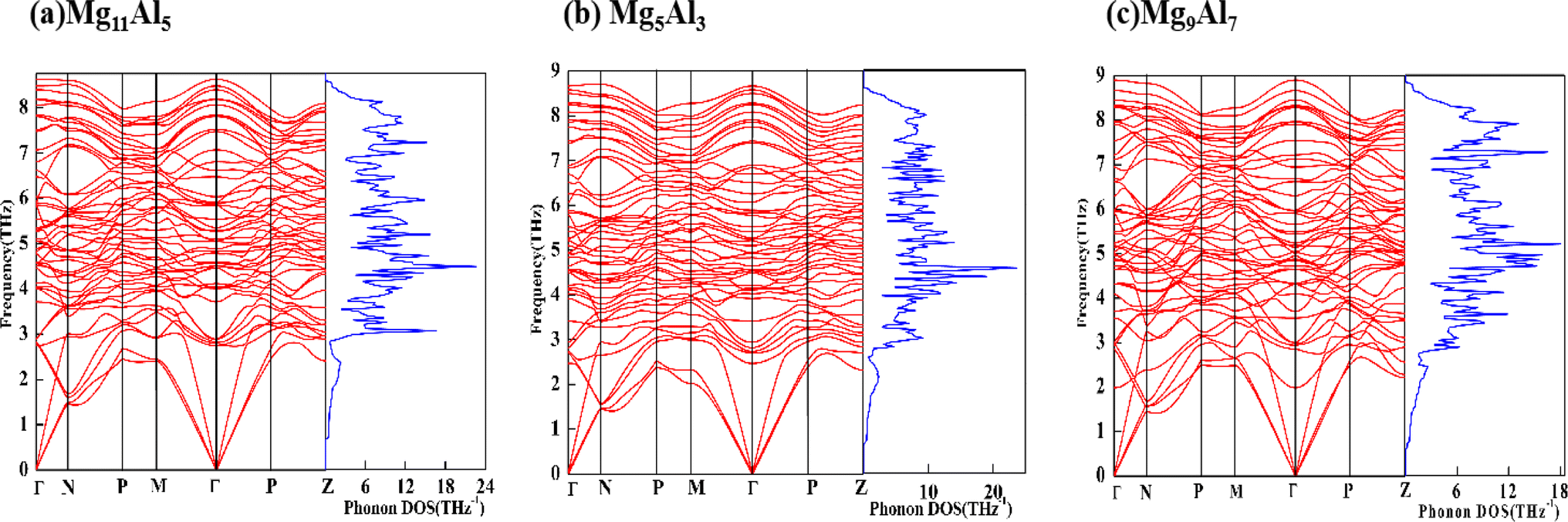
The calculated θD, vl, vt and vm of Mg11Al5, Mg5Al3 and Mg9Al7 are listed in Table 4. The θD value increases with increasing Al content, and the value for Mg9Al7 increases by 7.19% compared with Mg5Al3. The results reveal that the covalent bond in Mg9Al7 is stronger than that of other phases. Hence, the mechanical and thermal stability of Mg9Al7 is the best among these three MgxAly structures, which agrees with the earlier calculated elastic properties. In addition, we further studied the phonon spectrum of the structures, which is another key criterion to characterize the stability of materials.62 The absence of virtual phonon modes means that the system is dynamically stable. As shown in Fig. 6, the phonon modes for Mg11Al5, Mg5Al3 and Mg9Al7 in the first Brillouin zone are all real, which is consistent with their negative formation energies and their elastic constants that satisfy Born's criteria. The corresponding phonon density of states (PDOS) was also given in order to visualize the distribution of phonon modes better. Many flat phonon bands were observed, giving rise to sharp peaks in the PDOS plots.
Table 4 Theoretically calculated thermal properties of MgxAly alloys, including longitudinal sound velocity (vl), shear sound velocity (vt), average sound velocity (vm) and Debye temperature (θD)
| Structures |
vl (m s−1) |
vt (m s−1) |
vm (m s−1) |
θD (K) |
| Mg11Al5 |
5800.02 |
2831.13 |
3180.35 |
345.99 |
| Mg5Al3 |
5854.61 |
2896.26 |
3251.07 |
355.80 |
| Mg9Al7 |
6035.30 |
3086.31 |
3457.53 |
381.38 |
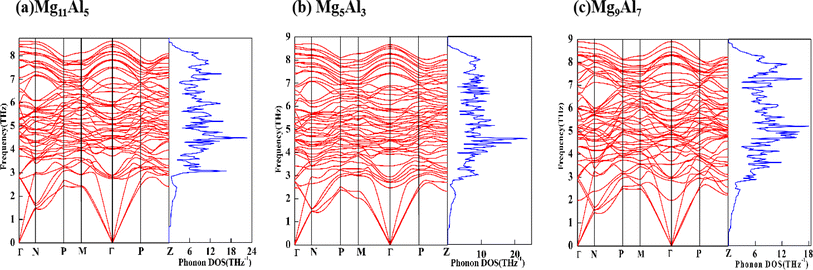 |
| | Fig. 6 Phonon spectrum and PHDOS of Mg11Al5, Mg5Al3 and Mg9Al7. | |
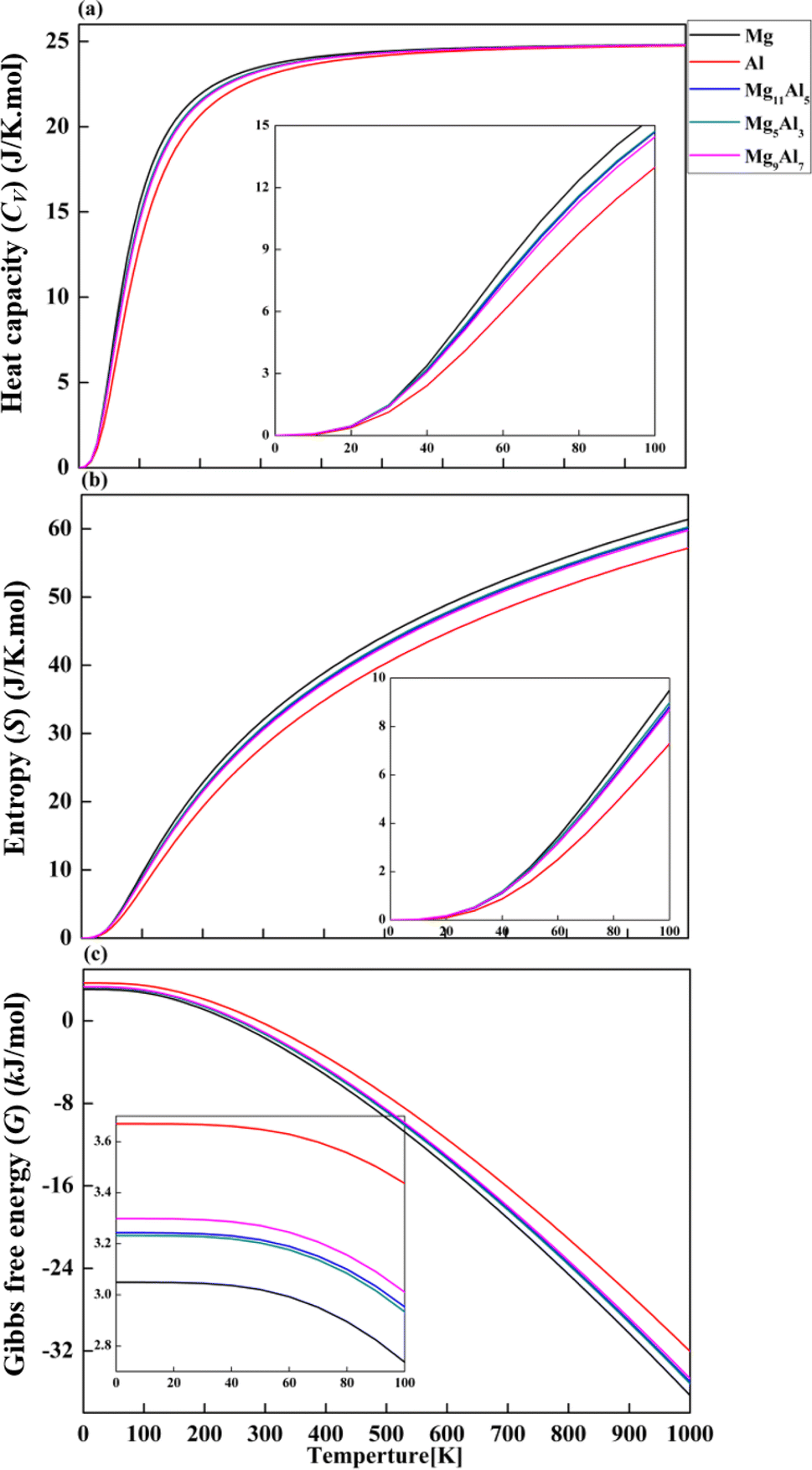
Based on phonon properties, we studied the effect of temperature on heat capacity (Cv), entropy (S) and Gibbs free energy (G) of pure Mg, Al, Mg11Al5, Mg5Al3 and Mg9Al7. The thermodynamic properties were calculated by PHONOPY software under 0 GPa.40 As shown in Fig. 7a, the heat capacity Cv increases with increasing temperature and tends to achieve a balance at higher temperatures. In the initial stage (<200 K), Mg and Al show the fastest and slowest increasing speed in Cv with temperature, respectively. The Cv curves of Mg11Al5, Mg5Al3 and Mg9Al7 are between that of Mg and Al and have similar responses to temperature. At about 200 K, the increasing speed of Cv starts to slow down with the change in temperature. After 500 K, Cv changes slightly with temperature and tends to achieve the maximum value of 25 J K−1 mol−1. The entropy S increases gradually with increasing temperature, as depicted in Fig. 7b. The entropy of Mg is greater than that of Al in the whole temperature range. The entropy–temperature curves of Mg11Al5, Mg5Al3 and Mg9Al7 follow the same pattern and have values between that of Mg and Al. From Fig. 7c, it is found that the Gibbs free energy G gradually decreases with increasing temperature. In the whole range of temperature, the values of G obey the following sequence: Al > Mg9Al7 > Mg11Al5 > Mg5Al3 > Mg.
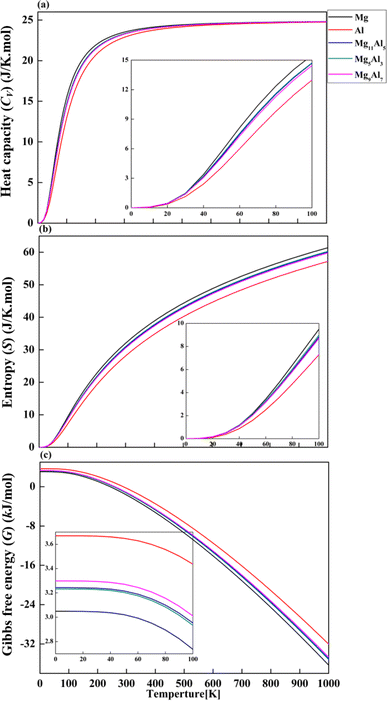 |
| | Fig. 7 Thermodynamic properties of (a) heat capacity (CV), (b) entropy (S) and (c) Gibbs free energy (G) for Mg16, Al and MgxAly alloys. | |
4. Conclusion
In this work, the structures of MgxAly alloys (x + y = 16, x = 1, 2,…, 15) were constructed by atomic substitution method. Structural optimization calculations were performed to determine structures with the lowest energy at each Mg/Al ratio. Their formation energies, elastic moduli, electronic properties and thermodynamic properties were studied using DFT calculations. The lattice parameters of MgxAly alloys decrease with increasing Al content, which is mainly due to the smaller atomic size of the Al atom compared with that of Mg. According to the convex hull diagram, Mg11Al5, Mg5Al3 and Mg9Al7 have the best structural stability. The calculated bulk modulus B, shear modulus G, Young's modulus E and Poisson ratio v show that Mg11Al5, Mg5Al3 and Mg9Al7 have better ductility than pure Mg. Mg9Al7 has the largest values of elastic moduli, indicating that it has a better ability to resist deformation. The calculated results of their electronic properties reveal that Mg11Al5, Mg5Al3 and Mg9Al7 are good conductors without magnetism. Furthermore, the CDD analyses show that the inner layer charges of Al atoms migrated to the outer layer, and the charges of Mg atoms also accumulated significantly in the outer region of Al atoms. The Debye temperature of Mg9Al7 is significantly higher than that of Mg11Al5 and Mg5Al3, revealing better thermodynamic stability. In terms of mechanical and thermodynamic properties, Mg9Al7 is the most promising structure for application. Our computational results would be helpful for the design of Mg–Al alloys and give a further understanding of structure–property relationships.
Author contributions
Wenjie Zhu: data curation, investigation, and writing-original draft, methodology; Xingtao Ma: writing-review and editing, validation; Yarui Wang: writing-review and editing, validation; Chaoyong Wang: writing-review and editing; Wei Li: writing-review and editing. The descriptions are accurate and agreed upon by all authors.
Data availability
The raw/processed data required to reproduce these findings cannot be shared at this time as the data also forms part of an ongoing study.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was supported by the Natural Science Foundation of Henan Province, the Foundation of Henan Educational Committee (Grant No. 232300420362).
References
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- C. Potzies and K. U. Kainer, Adv. Eng. Mater., 2004, 6, 281–289 CrossRef CAS.
- Z. B. Sajuri, T. Umehara, Y. Miyashita and Y. Mutoh, Adv. Eng. Mater., 2003, 5, 910–916 CrossRef.
- B. Mordike and T. Ebert, Mater. Sci. Eng., A, 2001, 302, 37–45 CrossRef.
- M. K. Kulekci, Int. J. Adv. Des. Manuf. Technol., 2007, 39, 851–865 CrossRef.
- N. C. Goel, J. R. Cahoon and B. Mikkelsen, Metall. Mater. Trans. A, 1989, 20, 197–203 CrossRef.
- R. J. Nimal, M. Sivakumar, S. G. Raj, S. A. Vendan and G. Esakkimuthu, Mater. Today: Proc., 2018, 5, 5886–5890 CAS.
- K. Park, J. Park and H. Kwon, J. Alloys Compd., 2018, 739, 311–318 CrossRef CAS.
- F. Wang, S. Sun, B. Yu, F. Zhang, P. Mao and Z. Liu, Trans. Nonferrous Met. Soc. China, 2016, 26, 203–212 CrossRef CAS.
- B. Zheng, L. Zhao, X. B. Hu, S. J. Dong and H. Li, Physica B, 2019, 560, 255–260 CrossRef CAS.
- F. Peng, H. Li, D. H. Wang, P. Tian, Y. X. Tian, G. Y. Yuan, D. M. Xu and X. Y. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 35033–35044 CrossRef CAS PubMed.
- J. Zou, Z. M. Shi, H. W. Xu and X. L. Li, BioMed Res. Int., 2017, 2017, 5763173 Search PubMed.
- S. W. Choi, Y. C. Kim, C. S. Kang, J. M. Jung and S. K. Hong, Adv. Mater. Res., 2013, 813, 427–430 CAS.
- Z. H. Wang, X. L. Zhang, S. B. Li, K. Liu and W. B. Du, Mater. Sci. Forum, 2014, 788, 98–102 Search PubMed.
- S. Maity, A. Sinha and S. Bera, Nano-Struct. Nano-Objects, 2018, 16, 63–68 CrossRef CAS.
- Z. Jiang, Y. Zhuang, Y. Li and S. Li, J. Iron Steel Res. Int., 2013, 20, 6–10 CrossRef CAS.
- W. N. Hsu, T. S. Shih and M. Y. Lin, Adv. Mater. Sci. Eng., 2014, 2014, 594984 Search PubMed.
- L. W. Huang, O. Elkedim and R. Hamzaoui, J. Alloys Compd., 2011, 509, S328–S333 CrossRef CAS.
- Q. Mahmood, G. Murtaza, R. Ahmad, T. Hussain and I. G. Will, Curr. Appl. Phys., 2016, 16, 361–370 CrossRef.
- A. Abbasi and J. J. Sardroodi, Appl. Surf. Sci., 2018, 456, 290–301 CrossRef CAS.
- A. Abbasi, RSC Adv., 2019, 9, 16069–16082 RSC.
- A. Abbasi, Phys. E, 2019, 108, 34–43 CrossRef CAS.
- A. Abbasi, J. Inorg. Organomet. Polym., 2019, 29, 1895–1915 CrossRef CAS.
- J. Li, G. Huang, X. Mi, L. Peng, H. Xie and Y. Kang, Materials, 2019, 12, 2855 CrossRef CAS PubMed.
- W. Cheng, L. Tian, Y. Bai, S. Ma and H. Wang, J. Mater. Res., 2017, 32, 2315–2324 CrossRef CAS.
- G. Rastelli and E. Cappelluti, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 184305 CrossRef.
- Y. Hu and T. J. Rupert, J. Mater. Sci., 2019, 54, 3975–3993 CrossRef CAS.
- L. Zhou, K. Su, Y. Wang, Q. Zeng and Y. Li, J. Alloys Compd., 2014, 596, 63–68 CrossRef CAS.
- C. Wang, P. Han, L. Zhang, C. L. Zhang, X. Yan and B. S. Xu, J. Alloys Compd., 2009, 482, 540–543 CrossRef CAS.
- P. Volovitch, M. Serdechnova and K. Ogle, Corrosion, 2012, 68, 557–570 CrossRef CAS.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- F. W. Von Batchelder and R. F. Raeuchle, Phys. Rev., 1957, 105, 59–61 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed.
- M. Methfessel and A. T. Paxton, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 3616–3621 CrossRef CAS PubMed.
- Y. Le Page and P. Saxe, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 104104 CrossRef.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- Y. Q. Wu and M. F. Yan, Physica B, 2010, 405, 2700–2705 CrossRef CAS.
- R. Arroyave, A. van de Walle and Z. K. Liu, Acta Mater., 2006, 54, 473–482 CrossRef CAS.
- O. I. Velikokhatnyi and P. N. Kumta, Mater. Sci. Eng., B, 2018, 230, 20–23 CrossRef CAS.
- A. R. Wazzan and L. B. Robinson, Phys. Rev., 1967, 155, 586–594 CrossRef CAS.
- L. J. Slutsky and C. W. Garland, Phys. Rev., 1957, 107, 972–976 CrossRef CAS.
- G. Simmons and H. Wang, Single Crystal Elastic Constants and Calculated Aggregate Properties, Cambridge, 1971 Search PubMed.
- M. Born, Proc. Cambridge Philos. Soc., 1940, 36, 160 CrossRef CAS.
- W. Voigt, Lehrbuch der Kristallpysik, Leipzig, Berlin, 1928 Search PubMed.
- A. Reuss, Z. Angew. Math. Mech., 1929, 9, 49–58 CrossRef CAS.
- R. Hill, Proc. Phys. Soc., London, Sect. A, 1952, 65, 349–355 CrossRef.
- S. F. Pugh, Relations between the elastic moduli and the plastic properties of polycrystalline pure metals, Nabu Press, 1954, vol. 45, pp. 823–843 Search PubMed.
- P. Shao, L. P. Ding, D. B. Luo, J. T. Cai, C. Lu and X. F. Huang, J. Alloys Compd., 2017, 695, 3024–3029 CrossRef CAS.
- S. I. Ranganathan and M. Ostoja-Starzewski, Phys. Rev. Lett., 2008, 101, 055504 CrossRef PubMed.
- X. Q. Chen, H. Y. Niu, D. Z. Li and Y. Y. Li, Intermetallics, 2011, 19, 1275–1281 CrossRef CAS.
- J. Heyd, J. E. Peralta, G. E. Scuseria and R. L. Martin, J. Chem. Phys., 2005, 123, 174101 CrossRef PubMed.
- N. Shao, J. Li, S. L. Che, J. W. Zheng, L. Qiao, Y. Ying, J. Yu and W. C. Li, J. Magn. Magn. Mater., 2024, 594, 171476 CrossRef CAS.
- X. Q. Wu, P. Y. Xu, Y. F. Duan, C. Hu and G. P. Li, Int. J. Min. Sci. Technol., 2012, 22, 825–830 CrossRef CAS.
- T. Akune, H. Abe, N. Sakamoto and Y. Matsumoto, Physica C, 2003, 388, 169–170 CrossRef.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- O. L. Anderson, J. Phys. Chem. Solids, 1963, 24, 909–917 CrossRef CAS.
- P. Wachter, M. Filzmoser and J. Rebizant, Physica B, 2001, 293, 199–223 CrossRef CAS.
- G. Grimvall, B. Magyari-Köpe, V. Ozoliņš and K. A. Persson, Rev. Mod. Phys., 2012, 84, 945–986 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The mechanical stability criteria and formulas of elastic moduli are listed. Except for Mg11Al5, Mg5Al3 and Mg9Al7, the elastic constants, elastic moduli and thermal properties of MgxAly are also given in Tables S1, S2 and S3, respectively. See DOI: https://doi.org/10.1039/d4ra00470a |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Yarui Wang*a,
Chaoyong Wanga and
Wei Liab
a,
Yarui Wang*a,
Chaoyong Wanga and
Wei Liab