
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereochemical insights into β-amino-N-acylhydrazones and their impact on DPP-4 inhibition†
Eduardo Reinaa,
Lucas Silva Franco ab,
Teiliane Rodrigues Carneiroa,
Eliezer J. Barreiroab and
Lidia Moreira Lima*ab
ab,
Teiliane Rodrigues Carneiroa,
Eliezer J. Barreiroab and
Lidia Moreira Lima*ab
aInstituto Nacional de Ciência e Tecnologia de Fármacos e Medicamentos (INCT-INOFAR), Laboratório de Avaliação e Síntese de Substâncias Bioativas (LASSBio®), Universidade Federal do Rio de Janeiro (UFRJ), CCS, Cidade Universitária, Rio de Janeiro-RJ, Brazil. E-mail: lidialima@ufrj.br
bPós-graduação em Farmacologia e Química Medicinal, Instituto de Ciências Biomédicas, Universidade Federal do Rio de Janeiro, Rio de Janeiro-RJ, Brazil
First published on 22nd February 2024
Abstract
Dipeptidyl peptidase IV (DPP-4) is a key enzyme that regulates several important biological processes and it is better known to be targeted by gliptins as a modern validated approach for the management of type 2 diabetes mellitus (T2DM). However, new generations of DPP-4 inhibitors capable of controlling inflammatory processes associated with chronic complications of T2DM are still needed. In this scenario, we report here the design by molecular modelling of new β-amino-N-acylhydrazones, their racemic synthesis, chiral resolution, determination of physicochemical properties and their DPP4 inhibitory potency. Theoretical and experimental approaches allowed us to propose a preliminary SAR, as well as to identify LASSBio-2124 (6) as a new lead for DPP-4 inhibition, with good physicochemical properties, favourable eudismic ratio, scalable synthesis and anti-diabetes effect in a proof-of-concept model. These findings represent an interesting starting point for the development of a new generation of DPP-4 inhibitors, useful in the treatment of T2DM and comorbidities.
Introduction
Dipeptidyl peptidase-4 (DPP-IV/DPP-4/CD26, EC3.4.14.5) is a 766 amino acid serine protease belonging to the α,β-hydrolase (family S9B), sequentially related to the prolyl oligopeptidases (POPs). DPP-4 specifically cleaves X-proline (X-pro) or X-alanine (X-ala) dipeptides from the free N-terminal sequence of peptides.1,2 Almost all peptides bearing an X-pro or X-ala sequence are potential substrates for DPP-4, including cytokines, chemokines, growth factors and several peptides such as pituitary adenylate cyclase-activating polypeptide, peptide YY, substance P, neuropeptide Y and glucagon-like peptide-1 (GLP-1).2–4 DPP-4 mediates limited proteolysis of these peptides resulting in their activation, activity modulation or initiation of degradation process. Thus, it has been linked to the regulation of blood pressure, immune system, and glucose control.3,4In diabetes, DPP-4 is of particular importance as it is the enzyme involved in the GLP-1 metabolism. After secretion, GLP-1 is rapidly hydrolysed and inactivated by DPP-4 action (GLP-1 half-life = 2–4 minutes), making its use as a natural antidiabetic drug impossible.3,4 Therefore, the development of DPP-4 inhibitors has emerged as a central strategy to circumvent this limitation and DPP-4 is a validated target to develop new drugs to treat type 2 diabetes mellitus (T2DM).5 Although it is expected that DPP-4 inhibitors may prevent or delay the onset of chronic complications of diabetes, due to their ability to minimise the damage promoted by glucotoxicity process,6 it should be considered that many patients that are diagnosed with T2DM already show significant damage to the macro- and microcirculation.7 These damages, mainly due to local inflammation and oxidative stress, are not reversed or controlled by DPP-4 inhibitors.
In continuity with a line of research aimed at discovering new candidates for antidiabetic drugs, we conjecture the possibility of combining in a same molecule the structural features that confer the ability to inhibit DPP-4 and to modulate TNF-α production, a key player in the development of sub-chronic inflammation. This approach seeks to broaden the therapeutic spectrum of conventional DPP-4 inhibitors, enabling them to control the hyperglycaemia and to prevent or to regulate the chronic complications of diabetes.
Therefore, we proposed molecular hybridisation between and sitagliptin (Januvia®) (1), the first DDP-4 inhibitor approved by the FDA for the treatment of T2DM8 and the prototype discovered by our group, LASSBio-1773 (2), which was described as a hypoglycemic agent able to reduce thermal hyperalgesia and mechanical allodynia in a murine model of STZ-induced diabetic neuropathic pain9–11 (Fig. 1). In addition, LASSBio-1773 (2) has revealed important ability to modulate the production of TNF-α, an important cytokine involved in subchronical and chronical inflammatory stages.9–11
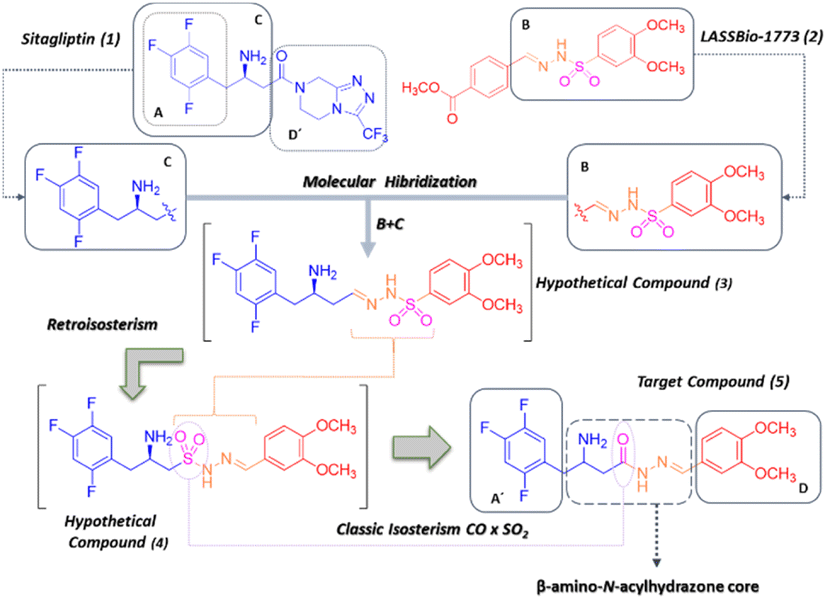 | ||
| Fig. 1 Design concept of the new the β-amino-N-acylhydrazone derivative (5) proposed as a DPP-4 inhibitor by molecular hybridisation between sitagliptin (1) and LASSBio-1773 (2). | ||
Our conceptual design was guided by the need to retain the structural requirements described for enzyme inhibition,5,8,12,13 such as: (1) a hydrophobic region (described for sitagliptin by subunit A, Fig. 1); (2) a primary or secondary amine group (which appears to be responsible for the selectivity towards prolyl endopeptidases, described for sitagliptin by subunit C, Fig. 1); and (3) a spacer or a linker connecting both the hydrophobic group and the amine group to a second aromatic subunit (D, Fig. 1), which has been proposed as the region of the molecule promoting selectivity over other exo-amino(di)peptidases.
One of the most notable features of the resulting hybrid compound (5) is the presence of the β-amino-N-acylhydrazone core (Fig. 1), which possess particular stereochemical properties in terms of absolute configuration and conformation (Fig. 2). With this assumption in mind, a key objective was to investigate the impact of selected five variables of the β-amino-N-acylhydrazone core, described in Fig. 2, on the DDP-4 enzymatic activity. We first carried out a molecular modelling study on DPP-4 enzyme to design the target compounds that represented the five variables under investigation (Fig. 2). Next, the designed compounds were synthesized and tested against DPP-4 enzyme in vitro. Our findings allowed us to propose a preliminary SAR model that may be useful in the design of new DPP-4 inhibitors with anti-inflammatory activity. In addition, our results suggest that the combination of the anti-inflammatory activity with inhibition of the DPP-4 enzyme could be explored to improve the current profile of DPP-4 inhibitors.
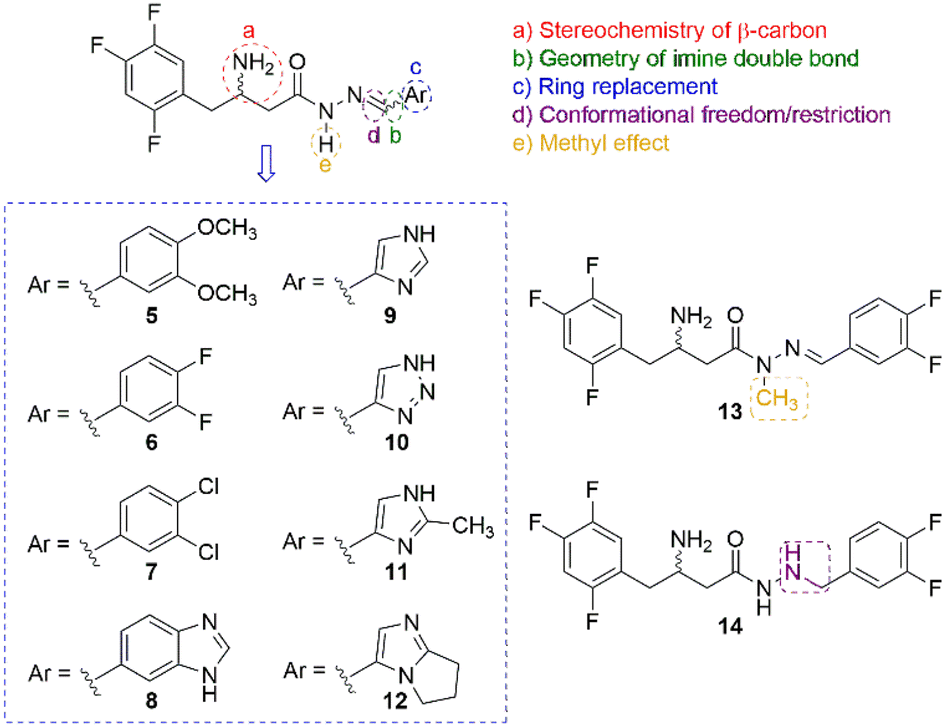 | ||
| Fig. 2 Selected variables considered in the study of the β-amino-N-acylhydrazone core. | ||
Taken together, these results are also important beyond their implications for T2DM therapies, considering that DPP-4 is a pharmacological target that remains of considerable ongoing interest, with several studies suggesting that DPP-4i could be used for other important indications such as cardiometabolic diseases,14,15 atherosclerosis,16 Alzheimer's disease17–20 and viral pathologies, including COVID-19.21,22
Results and discussion
Design concept
The starting point of our design concept was the application of molecular hybridisation between the C-subunit of sitagliptin (1) and the B-subunit of LASSBio-1773 (2) (Fig. 1). Subsequently, the design of compound (4) was envisioned through the application of a retroisosterism strategy.23 Considering several evidence of the isosteric relationship between sulfonylhydrazone (RSO2NHN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHR) and N-acylhydrazone (RCONHNCHR) functions,23,24 based primarily on the classical isosterism of carbonyl (CO) versus sulfonyl (SO2) group, we designed compound (5) as the desired hybrid compound. It is noteworthy that the N-acylhydrazone framework has been reported as a privileged structure for the design of several bioactive compounds, including anti-inflammatory agents acting by different mechanism of action.25 Thus, the proposed replacement of the SO2 group by the CO group could potentially preserve the anti-inflammatory profile of the original prototype LASSBio-1773 (2). The structural analysis of the designed compound (5) allowed us to identify five variables, two related to the absolute configuration and three related to conformational changes, that may influence its theoretical degree of affinity by DPP-4. First, the stereochemistry nature of the carbon bearing the amine group, given that the eudysmic ratio for the two enantiomers of sitagliptin (1) is well known, with the R-enantiomer being the eutomer with an IC50 = 20 nM and the S-enantiomer being the distomer with IC50 = 440 nM.26 Second, the geometry of the imine double bond since E or Z isomers will exhibit remarkably different orientations of the D-subunit in the pocket 2 of the DPP-4 enzyme (ESI Fig. S1†). Third, ring replacement of the D-subunit with other aromatic groups, which were designed as a congeneric series by replacing the 3,4-dimethoxyphenyl group in compound 4 (D-subunit, Fig. 2) with other related aromatic rings, resulting in compounds (5–12). Fourth, we decided to investigate the impact of increasing conformational freedom of the β-amino-N-acylhydrazone core on DDP-4 inhibition by designing compound (13) via reduction of the imine double bond. Finally, we designed compound (14) by methylation of the sp3 nitrogen of the β-amino-N-acylhydrazone core (Fig. 2) to evaluate the impact of the methylation effect in the NAH framework, a strategy that has been reviewed.27 To evaluate these five variables, initially we first performed a molecular docking study of the designed compounds (Fig. 2) using a DPP-4 co-crystallized with sitagliptin (1), as follows.
CHR) and N-acylhydrazone (RCONHNCHR) functions,23,24 based primarily on the classical isosterism of carbonyl (CO) versus sulfonyl (SO2) group, we designed compound (5) as the desired hybrid compound. It is noteworthy that the N-acylhydrazone framework has been reported as a privileged structure for the design of several bioactive compounds, including anti-inflammatory agents acting by different mechanism of action.25 Thus, the proposed replacement of the SO2 group by the CO group could potentially preserve the anti-inflammatory profile of the original prototype LASSBio-1773 (2). The structural analysis of the designed compound (5) allowed us to identify five variables, two related to the absolute configuration and three related to conformational changes, that may influence its theoretical degree of affinity by DPP-4. First, the stereochemistry nature of the carbon bearing the amine group, given that the eudysmic ratio for the two enantiomers of sitagliptin (1) is well known, with the R-enantiomer being the eutomer with an IC50 = 20 nM and the S-enantiomer being the distomer with IC50 = 440 nM.26 Second, the geometry of the imine double bond since E or Z isomers will exhibit remarkably different orientations of the D-subunit in the pocket 2 of the DPP-4 enzyme (ESI Fig. S1†). Third, ring replacement of the D-subunit with other aromatic groups, which were designed as a congeneric series by replacing the 3,4-dimethoxyphenyl group in compound 4 (D-subunit, Fig. 2) with other related aromatic rings, resulting in compounds (5–12). Fourth, we decided to investigate the impact of increasing conformational freedom of the β-amino-N-acylhydrazone core on DDP-4 inhibition by designing compound (13) via reduction of the imine double bond. Finally, we designed compound (14) by methylation of the sp3 nitrogen of the β-amino-N-acylhydrazone core (Fig. 2) to evaluate the impact of the methylation effect in the NAH framework, a strategy that has been reviewed.27 To evaluate these five variables, initially we first performed a molecular docking study of the designed compounds (Fig. 2) using a DPP-4 co-crystallized with sitagliptin (1), as follows.
Molecular modelling studies
Molecular docking studies of the newly designed compounds (5–14) were performed using the co-crystallographic structure of DPP-4 with sitagliptin (1), available in Protein Data Bank (PDB) with code 1 × 70.26 All protein structures were obtained from human DPP-4 enzyme and showed a reasonable resolution (<2.85 Å). After validation by re-docking approach, the ChemPLP score function (without water) showed the best RMSD value (0.387 Å) (ESI Fig. S3†) and was therefore selected to perform the docking studies with the designed compounds (5–14).| Compound | Score (R) | Score (S) |
|---|---|---|
| Sitagliptin (1) | 88.02 | 72.01 |
| (5) | 83.9 | 75.1 |
| (6) | 86.9 | 79.3 |
| (7) | 84.6 | 82.3 |
| (8) | 80.2 | 77.1 |
| (9) | 79.7 | 70.2 |
| (10) | 74.2 | 67.9 |
| (11) | 76.4 | 71.2 |
| (12) | 80.3 | 75.8 |
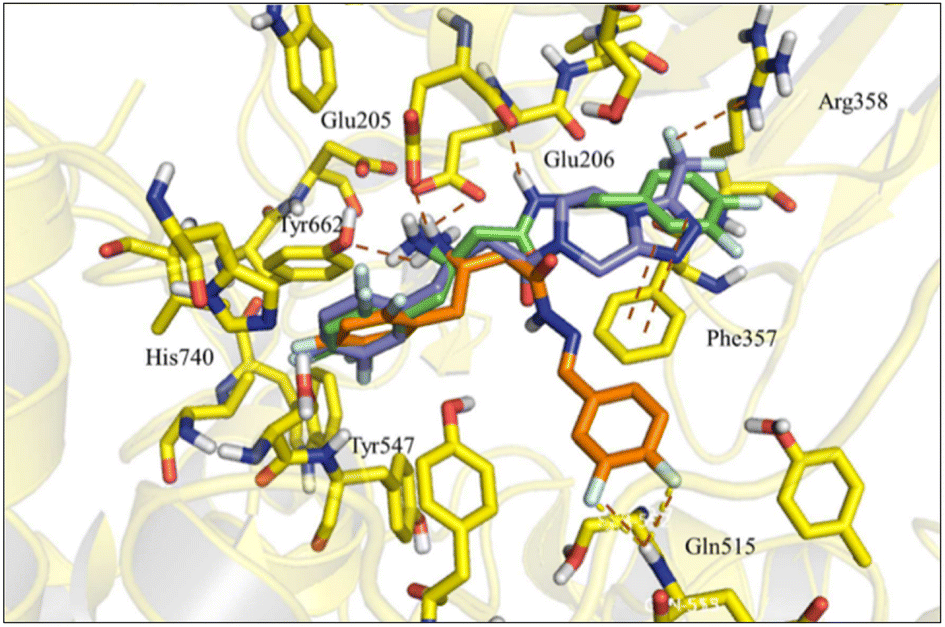 | ||
| Fig. 3 Superposition of compounds (6-R) (blue), (6-S) (orange) and sitagliptin (1-R) (green) inside DPP-4 active site. | ||
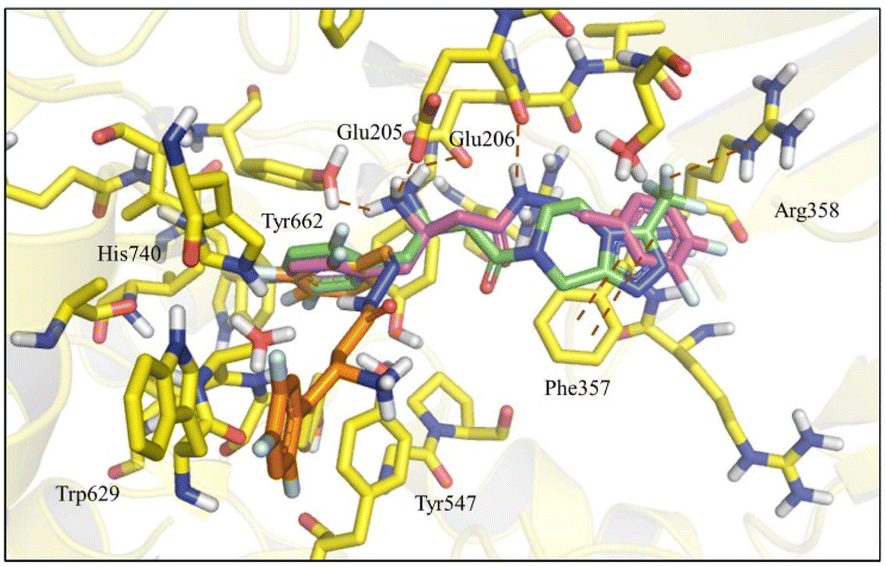 | ||
| Fig. 4 Superposition of compounds (6-R-E) (purple), (6-S-Z) (orange) and R-sitagliptin (1-R) (green) at the DPP-4 active site. | ||
Nevertheless, a higher score was found for compound (6-R), which showed a similar binding mode to R-sitagliptin (1-R). The attempt to replace the 3,4-dimethoxyphenyl by a benzimidazole or imidazole ring or by a pyrrolo[1,2-a]imidazole nucleus has resulted in a decrease in the score value, which was even more pronounced when 1,2,3-triazol or 2-methyl imidazole systems were docked (ESI Fig. S5†). With this information in mind, our next step was to study how conformational changes introduced directly into the β-amino-N-acylhydrazone framework would affect the binding to DPP-4. First, the increase in conformational freedom by reducing the β-amino-N-acylhydrazone core was investigated, and second, the impact of the methylation effect by substitution of the amidic hydrogen was investigated.26
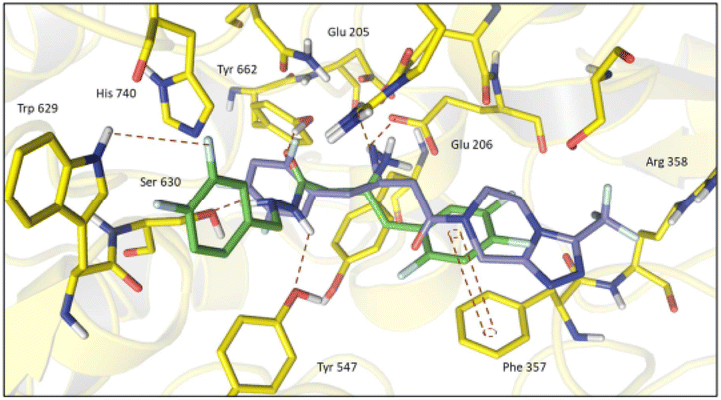 | ||
| Fig. 5 Superposition of compounds (13-R) (green) and R-sitagliptin (1-R) (purple) inside DPP-4 active site. | ||
Synthesis and structural characterization
To confirm the modelling results, we proposed to synthesise compounds that could represent each variable of interest (Fig. 2). In terms of the stereochemistry of the β-carbon atom linked to the amino group, we proposed to obtain (6-R), its enantiomer (6-S) and its racemate (6-R,S). The geometry Z was predicted as detrimental in all the compounds analysed and, in addition, it has been established that obtaining of Z diastereoisomers NAH is thermodynamically unfavourable, so we expected and verified the E-geometry of all the β-amino-N-acylhydrazones synthesised. Regarding the third variable, the conformational changes promoted by the D-subunit substitution, it was observed two possible orientations, one that could be represented by the synthesis of (6-R), and the second one, that could be represented by compounds (5) and (7), which have presented high scores and an adequate drug-like profile (ESI Table S4†). On the other hand, compound (9) was selected as a “negative control”, as it was predicted to be a weak ligand to the DPP-4 binding site and to have unsatisfactory drug-like properties (ESI Table S4†). Finally, as previously discussed, the proposed change in the conformational nature of the β-amino-N-acylhydrazone framework by N-methylation and imine double bond reduction, was predicted to be favourable for compound (14) and unfavourable for compound (13). Therefore, both compounds were selected for synthesis and evaluation against DPP-4 enzyme.To obtain the target compounds designed and selected by the docking studies two synthetic approaches were proposed. The first, exploring a synthetic route to obtain the target compounds as racemic mixtures, as depicted in Scheme 1A. The second approach was based on the synthesis of the enantiomerically pure compounds from the chiral resolution of the β-amino ester intermediate (20) (Scheme 1B).
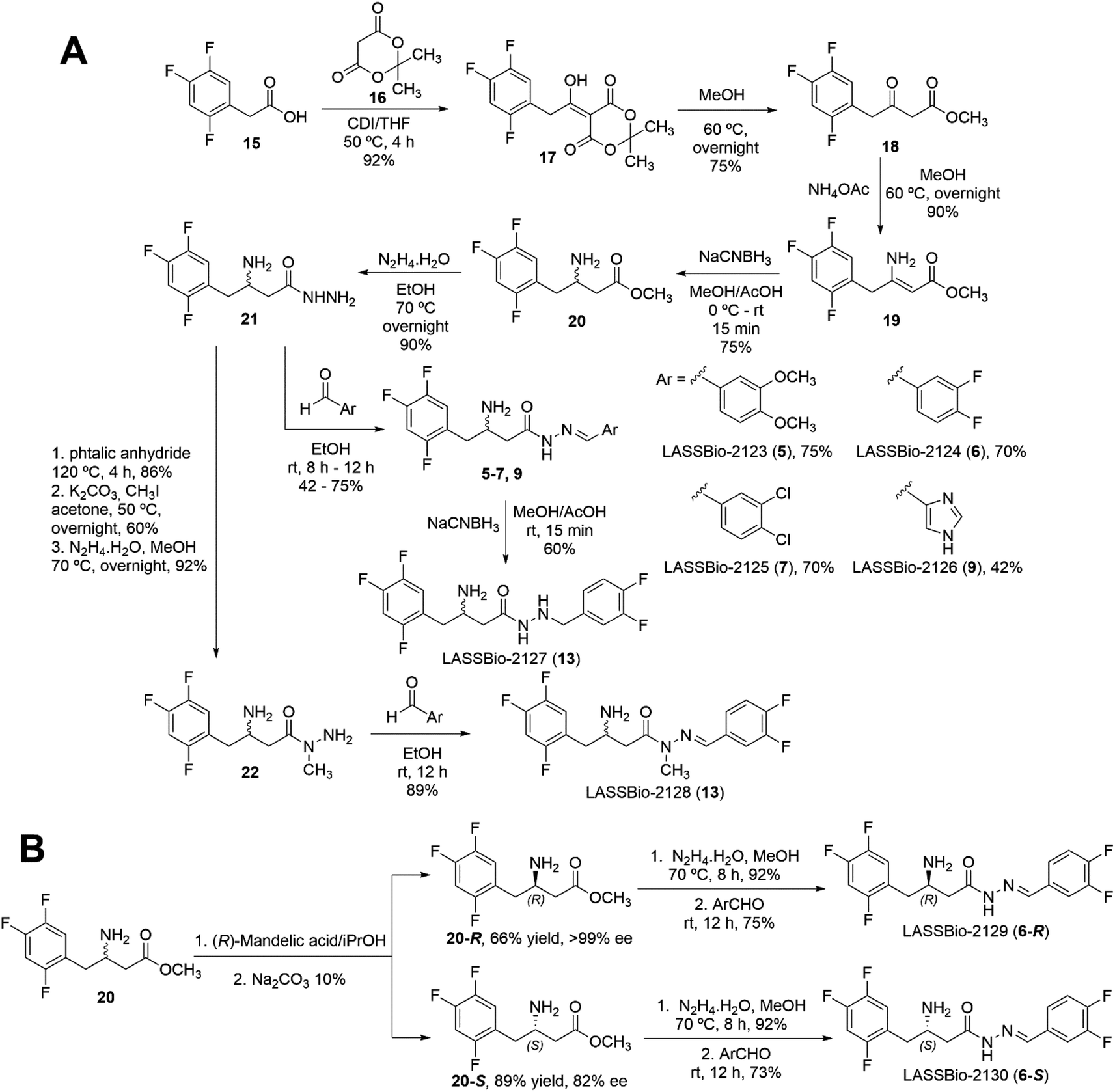 | ||
| Scheme 1 (A) Synthesis of compounds (5–7), (9), (13–14) as racemic mixtures. (B) Methodology for the chiral resolution of β-aminoester (20) and its application in the synthesis of the target compounds (6-R) and (6-S). | ||
The racemic synthesis of the selected compounds started with the synthesis of Meldrum's adduct (17), which was prepared by the reaction of 2,4,5-trifluorophenylacetic acid (15) with Meldrum acid (16) in the presence of CDI under inert atmosphere (Scheme 1).29 Treatment of Meldrum's adduct (17) with MeOH under reflux in inert atmosphere yielded the β-keto ester (18), which was subjected to reductive amination in two steps. First, the β-enaminoester (19) was formed by treating the β-keto ester (18) with ammonium acetate in methanol under inert atmosphere (Scheme 1B). The β-enamine ester was then reduced with sodium cyanoborohydride (NaCNBH3), with pH adjusted to 4–5 with glacial AcOH. This reaction was carried out in MeOH under inert atmosphere, affording the β-amino ester (20) (Scheme 1). Next, the hydrazide intermediate (21) was synthetized by the treating of β-amino ester (20) with excess of hydrazine hydrate in ethanol under reflux.30 This reaction led to the desired hydrazide (21) in 90% of yield (Scheme 1). The racemic β-aminohydrazide (21), obtained in five linear steps in 15% of overall yield, was outlined as the key intermediate for the synthesis of target β-amino-N-acylhydrazone derivatives, designed as new DPP-4 inhibitors. With this intermediate in hand, we were able to obtain target compounds (5) (LASSBio-2123), (6) (LASSBio-2124), (7) (LASSBio-2125), and (9) (LASSBio-2126) by condensation reaction between the β-aminohydrazide intermediate (21) and the corresponding aldehydes dissolved in ethanol, at room temperature (Scheme 1).
Although the synthesised β-amino-N-acylhydrazones have showed a high degree of purity as assessed by high performance liquid chromatography (HPLC), signal duplication was observed in the 1H and 13C NMR spectra (ESI Table S5†). After a combination of different dynamic NMR and molecular modelling studies we proposed that the β-amino-N-acylhydrazones might exist in solution as a mixture of s-cis and s-trans conformers (Fig. 6). It is interesting to note that a similar conformational behaviour has been reported for some N-acylhydrazones by Lopes and colleagues.31–34
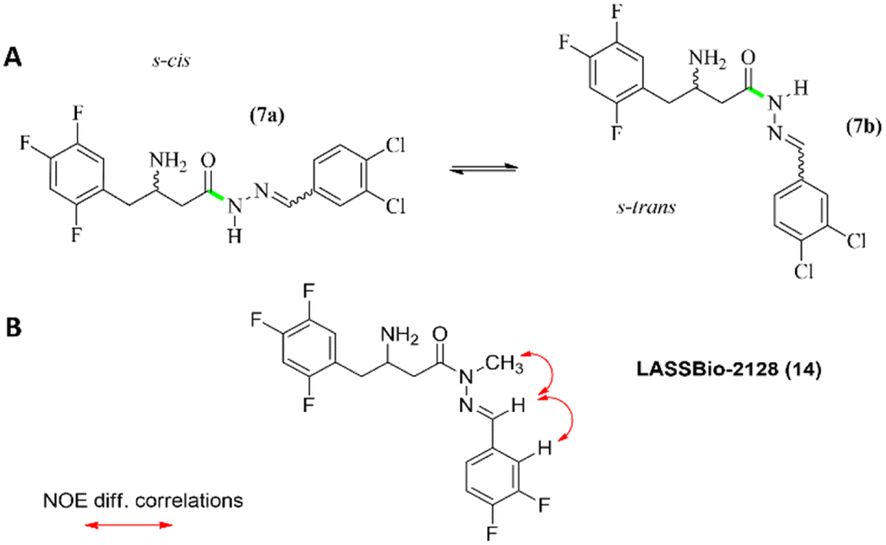 | ||
| Fig. 6 (A) Conformers s-cis and s-trans of compound (7). (B) NOE correlations found for compound (14). | ||
Regarding the geometry of the imine double bond, several pieces of evidence led us to propose an E configuration, firstly because this is the energetically favoured isomer,31–34 and secondly because the chemical shift of the imine hydrogen at δH 8.15 in the NMR spectra performed at 90 °C is expected for this geometry, compared to the chemical shift for a Z configuration, which is expected at δH ≈ 7.00.31,32 The E-geometry of the imine double was later confirmed by NOE studies on the target compound LASSBio-2128 (14), the direct analogue of LASSBio-2144 (6) (ESI Fig. S9†), as it will be described later.
The hydrazide target compound (13), designed as a non-conformationally restricted analogue of LASSBio-2124 (6), was synthesised from compound (6) by exploring the reduction of the imine double bond, using sodium cyanoborohydride in acid medium (Scheme 1).
The strategy used to obtain compound (14) (LASSBio-2128), the N-methyl analogue of LASSBio-2124 (6), was based on the protection of the two primary amine groups of the β-amine hydrazide (21) with phthalic anhydride, followed by methylation of the phthalimide intermediate product, its deprotection and subsequent condensation of the N-methyl-hydrazide with 3,4-diflurobenzaldehyde (Scheme 1). The desired LASSBio-2128 (14) was obtained in nine linear steps, in 15% of overall yield.
It is worth mentioning that no signal duplication was observed in the NMR spectra for LASSBio-2128 (14), which shows a different pattern when compared to the non-methylated β-amino-N-acylhydrazone (i.e., LASSBio-2124, 6). This fact can be explained considering that the N-methylation of the amide nitrogen of the β-amino-N-acylhydrazone scaffold causes a conformational restriction of the amide bond, preventing its rotation. Consequently, it reduces the existence of conformers and abolishes the duplication of the signals in the NMR spectra. These results also corroborate with the hypothesis of the existence of amide s-cis/s-trans conformers, which was previously mentioned in an attempt to explain the duplication of signals in the NMR spectra of LASSBio-2123 to LASSBio-2126 (5–7, 9).
Furthermore, to confirm the relative configuration of LASSBio-2128 (14), NOEdiff experiments were performed. As depicted in Fig. S9 (ESI),† the experiments have revealed the spatial correlation between the irradiated hydrogens of the methyl group with the hydrogen of the imine function. These data allowed to confirm an E configuration of the imine double bond, as previously proposed, through the analysis of the chemical shifts of the amide and imine hydrogens of β-amino-N-acylhydrazone derivatives.
The second part of the synthetic plan was based on efforts to implement a synthetic methodology to separately obtain both S and R-enantiomers of the selected β-amino-N-acylhydrazone derivatives. We decided to explore the chiral resolution of the β-amino ester intermediate (20), followed by the reactions of hydrazinolysis and condensation with the selected aldehydes (Scheme 1B). We started with the formation of diastereomeric salts by treating the (R,S)-β-amino ester (20) with (R)-mandelic acid in isopropanol to furnish the mixture of the diastereoisomers, which were separated and purified by recrystallization. Separate treatment with Na2CO3 10% solution afforded the free amine (20-R) with an enantiomeric excess greater than 99% and the amine (20-S) with 82% of enantiomeric excess (ESI Section 4†).
It should be noted that an absolute stereochemistry of R was assigned to the intermediate (20-R) (HPLC retention time: 28.7 min), and consequently S for the intermediate (20-S) (HPLC retention time: 23.4 min). A detailed explanation of the determination of the absolute stereochemistry can be found in the ESI (Section 4.2).†
Once accomplished the resolution and determination of the absolute stereochemistry of the β-amino esters (20-R) and (20-S), the next step was the synthesis of the hydrazides (21-R) and (21-S), which was carried out by hydrazinolysis of the corresponding intermediates (20-R and 20-S) (Scheme 1). Finally, each of the enantiopure hydrazides (21-R and 21-S) were reacted with 3,4-difluorobenzaldehyde to give the target compounds LASSBio-2129 (6-R) and LASSBio-2130 (6-R).
With all the target compounds in hand, we decided to evaluate their key physicochemical properties, such as aqueous solubility, pKa, and chemical stability.
Psychochemical properties assessment
Aqueous solubility at physiological pH (7.4) was measured by spectrophotometric assay for the target compounds LASSBio-2123 (5), LASSBio-2124 (6), LASSBio-2125 (7) and LASSBio-2126 (9) (ESI Section 5†). As shown in Table 2, all compounds exhibited aqueous solubility values greater than 24 mg mL−1 (≥66 μM). The high aqueous solubility of the free amines (5–7, 9) (LASSBio-2123–LASSBio-2125), even considering the lipophilic properties of the aromatic subunit linked to the imine moiety, was attributed to the presence of the primary amine and its pKa value, which should contribute to the ionization of the molecules at pH = 7.4. To confirm that, the experimental determination of dissociation constants of compounds LASSBio-2123 to LASSBio-2126 (5–7, 9) was recognised as an important goal.| Compound | Score value | R2 |
|---|---|---|
| LASSBio-2123 (5) | 51.5 (105) | 0.9999 |
| LASSBio-2124 (6) | 24.6 (66) | 0.9999 |
| LASSBio-2125 (7) | 33.2 (82.4) | 0.9999 |
| LASSBio-2126 (9) | >48.8 (>150.1) | 0.9954 |
The dissociation constant, usually expressed as negative the logarithm (pKa), indicates the degree of ionisation of acidic/basic compounds and is an important parameter influencing physicochemical properties such as lipophilicity, solubility, and permeability. In addition, the pKa value is useful to assess the pharmacodynamic behaviour in the biophase of a given drug with ionisable groups. Thus, considering the presence of a β-amino group in the structure of our target compounds, and taking into account the pharmacophoric nature of this group, which makes key interactions with Glu205/206 in the active site of DPP-4,26 the experimental dissociation constant values for compounds 5–7 and 9 was determined (ESI Section 6†). The experimental pKa values for the target β-amino-N-acylhydrazone range from 7.51 to 8.05, indicating that at physiologic pH (7.4) compounds 5–7 and 9 exist in a mixture of dissociated and non-dissociated forms (probably with percentages close to the 50–60%), with the equilibrium tending towards the dissociated form. These data may explain the high aqueous solubility of these compounds and suggest that, as with sitagliptin (1), an ionic bridge-type interaction may be involved in the recognition of the β-amino group with the Glu residues in the active site of DPP-4. For compound LASSBio-2126 (8), the pKa value of 4.43 indicates that the imidazole subunit is fully protonated at pH 7.4, which would explain its higher water solubility.
Finally, we considered studying the chemical stability of compounds (5–7, 9) in an aqueous media with a pH system that could mimic the pH of the enzymatic test (pH 7.4) (ESI Section 7†), with the aim of verifying that these compounds maintain their physical and chemical properties unchanged to reach its target. In addition, we extended our chemical stability study to pH = 2.0 to see how these compounds behave in the acidic conditions of the stomach. The results indicated that in the neutral medium (pH 7.4), which mimics pH environment of the test, all the compounds were stable during the time analysed from 0 to 240 min, with less than 10% of degradation (Fig. 7). However, in acidic buffer (mimicking gastric pH) the compounds showed a degree of instability. LASSBio-2123 (5) and LASSBio-2124 (6) were degraded by about 20% in the first 30 min, increasing to 30% degradation from 60 min to the end of the experiments (after 240 min). LASSBio-2123 (5) showed slower degradation kinetics, with 30% of decomposition at 120 min and 50% of recovery after 240 min. Taken together, we have observed a lower stability of the target compounds in PBS pH = 2.0 compared to pH = 7.4. However, the instability was time dependent and did not exceed 30% in the first 2 hours.
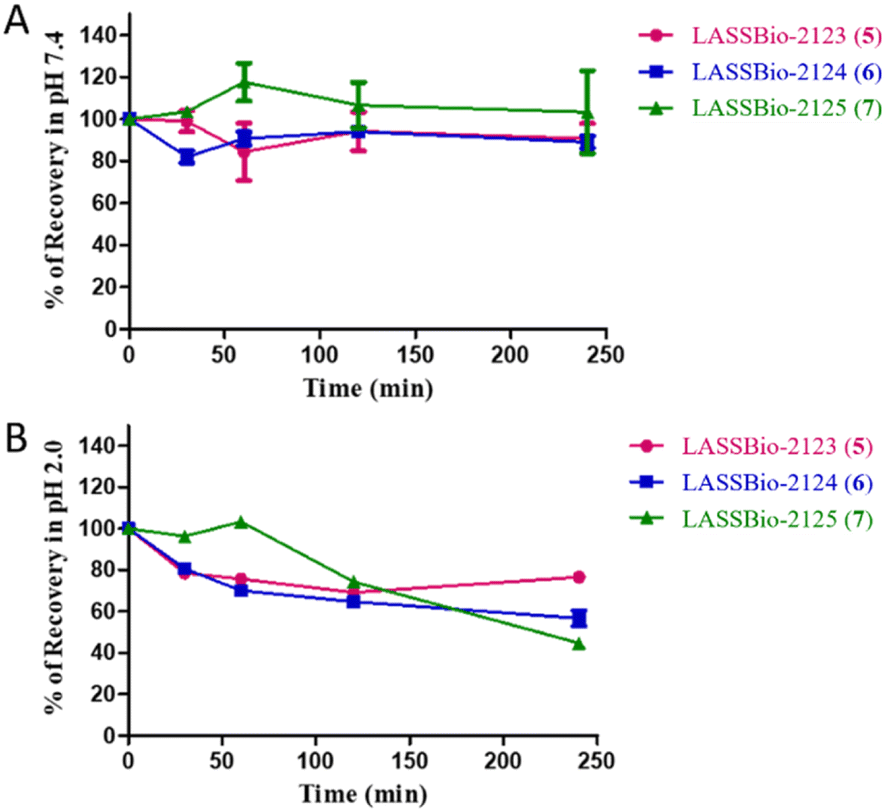 | ||
| Fig. 7 Chemical stability of LASSBio-2123 (5), LASSBio-2124 (6) and LASSBio-2125 (7) (A) results at pH = 7.4. (B) Results at pH = 2.0. The experiments were conducted in triplicate, and the values shown represent the average of the experiments. | ||
These results indicate that our target compounds have adequate aqueous solubility and chemical stability under the test conditions. Additionally, a pKa values of 7.7–8.05 led us to propose that these compounds could be protonated at the primary amine in the biophase. This is important considering that Glu266/265 are key pharmacophoric points capable of making ionic interactions with the primary amine of the tested compounds.
In vitro evaluation
After the synthesis and determination of key physicochemical parameters (solubility, pKa and chemical stability) compounds 5 (LASSBio-2123), 6 (LASSBio-2124), 7 (LASSBio-2125), 9 (LASSBio-2126), 13 (LASSBio-2127), 14 LASSBio-2128, 6-R (LASSBio-2129) and 6-S (LASSBio-2130) were evaluated for their inhibition profile against human recombinant DPP-4 enzyme (Table 3).‡| Compound | Inhibition of DPP-4 IC50 (μM)a | R2 |
|---|---|---|
| a The IC50 values displayed are the mean of three experiments in μM.b N.A. = no active. | ||
| (R)-Sitagliptin (1-R) | 0.092 | 0.9991 |
| LASSBio-1773 (2) | N.A.b | — |
| LASSBio-2123 (5) | 34.3 | 0.9964 |
| LASSBio-2124 (6) | 10.6 | 0.9835 |
| LASSBio-2125 (7) | 30.8 | 0.9972 |
| LASSBio-2126 (9) | 24.0 | 0.9768 |
| LASSBio-2127 (13) | 2.93 | 0.9911 |
| LASSBio-2128 (14) | 74.67 | 0.9184 |
| LASSBio-2129 (6-R) | 5.08 | 0.9994 |
| LASSBio-2130 (6-S) | 44.74 | 0.9970 |
We started the biological evaluation by validating the DPP-4 assay kit with (R)-sitagliptin (1).§ (R)-sitagliptin (1) showed inhibitory activity with an IC50 of 92 nM. This value was considered comparable to that reported in the literature (IC50 = 19 nM).26 The difference was expected since the conditions of the literature's experiment (buffers and equipment) were not the same as those used in this DPP-4 assay. However, considering that both values (IC50 = 92 nM vs. IC50 = 19 nM) are in the same order of magnitude, we have considered the kit satisfactorily validated.
Next, we used the DPP-4 kit to investigate the concentration–response relationship and determine the IC50 values of compounds previously mentioned. As expected, LASSBio-1773 (2) was inactive. On the other hand, the new hybrid compounds LASSBio-2123 (5), LASSBio-2124 (6), LASSBio-2125 (7), LASSBio-2126 (9) showed activity in the same range of magnitude (IC50 = 10 to 74 μM), with LASSBio-2124 (6) being most potent (Table 3). This result is consistent with the docking study (described above), which showed both R and S enantiomers of LASSBio-2124 (6) with higher score values (Table 1).
To analyse the effect of the stereochemistry of the β-amino group in the DPP-4 inhibition profile, docking studies projected the R-enantiomer of LASSBio-2124 (6) as being able to reproduce the interactions of (R)-sitagliptin in the active site of DPP-4. The comparison of LASSBio-2124 (racemate), LASSBio-2129 (R-isomer), LASSBio-2130 (S-isomer) is shown in Table 3. LASSBio-2129 (6-R) showed a potency of 5.08 μM, while its antipode, LASSBio-2130 (6-S), showed an IC50 = 44,74 μM. An almost nine-fold difference in the comparative potency of these compounds suggests that the R-enantiomer (LASSBio-2129, 6-R) is the eutomer, and the S-enantiomer (LASSBio-2130, 6-S) is the distomer (eudysmic ratio (ER): 8.8), whereas the ER described in literature for sitagliptin (1) is 22. The difference in potency of the enantiomers LASSBio-2129 (6-R) and LASSBio-2130 (6-S) was anticipated by docking studies, as previously discussed in the molecular modelling section. Surprisingly, the pharmacological results also demonstrated that there was no satisfactory gain in potency when we compared the inhibitory profile of the racemic mixture, LASSBio-2124 (6), with the R-eutomer, LASSBio-2129 (6-R).
With regard to the geometry of the imine double bond, as suggested by molecular modelling studies, the β-amino-N-acylhydrazones with an E-geometry LASSBio-2123 (5), LASSBio-2124 (6), LASSBio-2125 (7) and LASSBio-2126 (9), tested as a racemic mixture, were able to inhibit the DPP-4 enzyme with IC50 values of 34.3, 10.6, 30.8 and 24.0 μM, respectively. These data clearly indicate that although the target compounds were active against human DPP-4, they showed lower potency when compared to (R)-sitagliptin (Table 3).
On the other hand, changes in the D-subunit did not result in a significant gain in potency and considering that no statistical difference was observed for the potency of compounds LASSBio-2123 (5), LASSBio-2125 (7) and LASSBio-2126 (9) (Table 3), we can assume that the stereoelectronic effects of the substituents linked to the imine subunit had little contribution to the inhibition of DPP-4. This assumption contradicts the docking results, discussed in the molecular modelling section. However, another important point to consider about the activity of these β-amino-N-acylhydrazones, is related to the duplicated signals obtained in the NMR spectra of the β-amino-N-acylhydrazones synthesised for this study. This behaviour was attributed to a mixture of two predominant conformations, s-cis and s-trans, even in polar solvents such as DMSO. This raises the possibility that these compounds, in the biophase, could be present in two different conformations with different affinities for DPP-4. In this respect, it is interesting to note that the s-trans conformation (Fig. 6B) proposed for LASSBio-2125 (7) resemble that projected for LASSBio-2128 (14) (Fig. 6B), which is considered inactive in the DPP-4 assay. Conformationally restricted analogues will be required to address this issue and should be included in new optimization campaigns.
The increase in conformational freedom obtained through by reducing of the imine double bond, has resulted in a more potent compound (IC50 = 2.93 μM) than LASSBio-2124 (6) (IC50 = 10.6 μM) and its enantiomers LASSBio-2129 (6-R) and LASSBio-2130 (6-R). The N-methyl analogue LASSBio-2128 (14) induced a strong decrease in the DPP-4 inhibition (IC50 = 74.8 μM). These results are consistent with the predicted activity suggested by the docking approach (see molecular modelling section).
Based on the described pharmacological results and considering the structural variables proposed at the beginning of the project, we have proposed a preliminary structure–activity relationship (Fig. 8). In summary, the 2,4,5-trifluorophenyl moiety has been highlighted as a relevant pharmacophoric point capable of making hydrophobic interactions, probably involving Phe357 (S2-pocket) or amino acid residues at the S1-pocket. The stereochemistry of the β-amino group was confirmed as important for the inhibitor potency, e.g., LASSBio-2124 (6) has displayed an eudysmic ratio of 8.81, favouring the R configuration. Alkylation at the sp3 nitrogen of the β-amino-N-acylhydrazones core was detrimental to activity. The conformational freedom introduced by the reduction of the imine double bond was favourable for DPP-4 inhibition. Substitution at the imine subunit did not play an important role in the inhibitory potency.
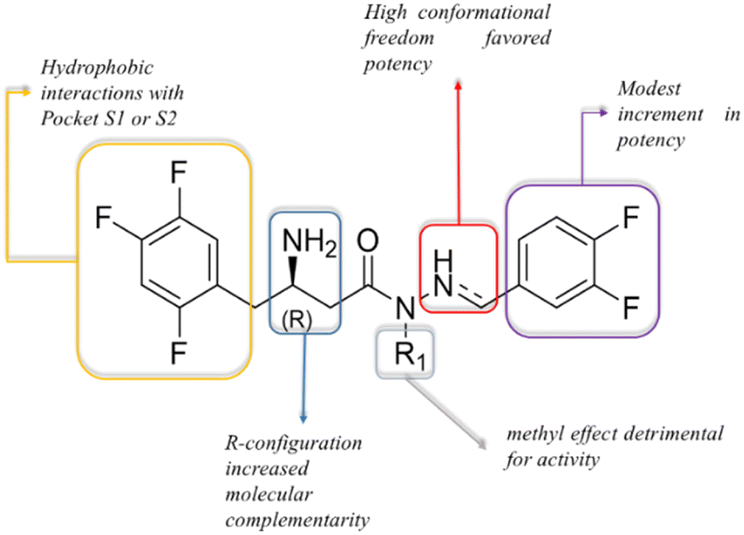 | ||
| Fig. 8 Preliminary structure–activity relationships for LASSBio-2123 to LASSBio-2130 (5–7, 9, 13, 14, 6-R, 6-S) as DPP-4 inhibitors. | ||
Having confirmed the ability of the designed target compounds to inhibit DDP4, the next challenge was to investigate whether such an inhibitory action would indeed imply an anti-diabetic effect in vivo. Therefore, we have selected LASSBio-2124 (6) to start the in vivo analysis. The criteria for this selection were because LASSBio-2124 (6), even in the racemic form, has shown a comparable potency to its R-enantiomer LASSBio-2129 (6-R). Moreover, considering the experimental methodologies developed in this work, the racemic approach has proven to be more scalable, when compared to the resolution of the β-amino ester intermediate, followed by the synthesis of the pure enantiomer.
Two grams of LASSBio-2124 (6) were synthesised and tested in in vivo assays via oral administration, using a murine model of T2DM induced by a combination of high fat diet and streptozotocin. As demonstrated by Alves and coworkers, like (R)-sitagliptin, LASSBio-2124 (6) showed antihyperglycemic effects and improved cardiac and renal dysfunction, which are commonly observed disturbances in T2DM associated with metabolic syndrome.35
Conclusions
In conclusion, a new kind of DPP-4 inhibitors were designed bearing a new β-amino-N-acylhydrazone framework, obtained by applying molecular hybridization between sitagliptin (1) and LASSBio-1773 (2) prototype. We evaluated five key variables of the β-amino-N-acylhydrazone core and their impact on DPP-4 inhibition. The stereochemistry of the β-amino group was important for activity, as well as the E geometry of the imine doubled bond, but the substituent on this region had little impact on potency and could be explored for optimization. On the other hand, an increase in conformational freedom obtained by reduction of the imine double bond, had a positive effect, resulting in LASSBio-2127 (13), with IC50 = 2.9 μM. Finally, the conformational change induced by the introduction of the N-methyl group had a detrimental effect on DPP-4 inhibition, as illustrated by LASSBio-2128 (14) (IC50 = 74.7 μM). In addition, these studies led us to identify LASSBio-2124 (6) as an authentic lead with good physicochemical properties, good DPP4 inhibition potency and in vivo antidiabetic activity, which can be synthesized in its racemic or pure enantiomeric form and used in future lead-optimization campaigns.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Instituto Nacional de Ciência e Tecnologia de Fármacos e Medicamentos (INCT-INOFAR).Notes and references
- L. Emami, Z. Faghih, A. Sakhteman, Z. Rezaei, Z. Faghih, F. Salehi and S. Khabnadideh, New J. Chem., 2020, 44, 19515–19531 RSC.
- E. E. Mulvihill and D. J. Drucker, Endocr. Rev., 2014, 35, 992–1019 CrossRef CAS PubMed.
- J. Fremaux, C. Venin, L. Mauran, R. Zimmer, F. Koensgen, D. Rognan, S. Bitsi, M. A. Lucey, B. Jones, A. Tomas, G. Guichard and S. R. Goudreau, Chem. Sci., 2019, 10, 9872–9879 RSC.
- C. F. Deacon, Nat. Rev. Endocrinol., 2020, 16, 642–653 CrossRef CAS PubMed.
- L. Juillerat-Jeanneret, J. Med. Chem., 2014, 57, 2197–2212 CrossRef CAS PubMed.
- S. Costes, G. Bertrand and M. A. Ravier, Int. J. Mol. Sci., 2021, 22, 5303 CrossRef CAS PubMed.
- D. Kannenkeril, A. Bosch, J. Harazny, M. Karg, S. Jung, C. Ott and R. E. Schmieder, Cardiovasc. Diabetol., 2018, 17, 128 CrossRef CAS PubMed.
- B. D. Patel and M. D. Ghate, Eur. J. Med. Chem., 2014, 74, 574–605 CrossRef CAS PubMed.
- L. M. Lima, G. Zapata-Sudo, I. K. da Costa Nunes, J. Segundo Chaves de Araujo, J. da Silva, M. Manhães Trachez, T. Fernandes da Silva, F. P. da Costa, R. Sudo and E. Barreiro, Drug Des., Dev. Ther., 2016, 10, 2869–2879 CrossRef PubMed.
- G. Zapata-Sudo, L. M. Lima, S. L. Pereira, M. M. Trachez, F. P. da Costa, B. J. Souza, C. E. S. Monteiro, N. C. Romeiro, E. D. D'Andrea, R. T. Sudo and E. J. Barreiro, Curr. Top. Med. Chem., 2012, 12, 2037–2048 CrossRef CAS PubMed.
- L. M. Lima, M. M. Trachez, J. S. C. Araújo, J. S. Silva, D. N. Amaral, R. T. Sudo, E. J. Barreiro and G. Zapata-Sudo, J. Diabetes Metab., 2014, 5, 392 Search PubMed.
- S. Kumar, A. Mittal and A. Mittal, Bioorg. Med. Chem., 2021, 46, 116354 CrossRef CAS PubMed.
- K. Tomovic, B. S. Ilic and A. Smelcerovic, J. Med. Chem., 2021, 64, 9639–9648 CrossRef CAS PubMed.
- J. Zhong, A. Maiseyeu, S. N. Davis and S. Rajagopalan, Circ. Res., 2015, 116, 1491–1504 CrossRef CAS PubMed.
- Q. Gong, S. Rajagopalan and J. Zhong, Int. J. Cardiol., 2015, 197, 170–179 CrossRef PubMed.
- H. Liu, L. Guo, J. Xing, P. Li, H. Sang, X. Hu, Y. Du, L. Zhao, R. Song and H. Gu, Eur. J. Pharmacol., 2020, 875, 173037 CrossRef CAS PubMed.
- E. Angelopoulou and C. Piperi, Ann. Transl. Med., 2018, 6, 255 CrossRef PubMed.
- Y. Chen, J. Zhang, B. Zhang and C.-X. Gong, Curr. Top. Med. Chem., 2015, 16, 485–492 CrossRef PubMed.
- N. S. S. Chalichem, P. S. S. Sai Kiran and D. Basavan, J. Drug Targeting, 2018, 26, 670–675 CrossRef CAS PubMed.
- Y. Ohyagi, K. Miyoshi and N. Nakamura, Therapeutic Strategies for Alzheimer's Disease in the View of Diabetes Mellitus, in Diabetes Mellitus, Advances in Experimental Medicine and Biology, ed. Y. Nakabeppu and T. Ninomiya, Springer, Singapore, 2019, vol. 1128, pp. 227–248 Search PubMed.
- R. Strollo and P. Pozzilli, Diabetes/Metab. Res. Rev., 2020, 36(8), e3330 CrossRef CAS PubMed.
- S. B. Solerte, A. Di Sabatino, M. Galli and P. Fiorina, Acta Diabetol., 2020, 57, 779–783 CrossRef CAS PubMed.
- L. M. Lima and E. J. Barreiro, in Comprehensive Medicinal Chemistry III, Elsevier, New York, 2017, vol. 1–8, pp. 186–210 Search PubMed.
- L. Lima and E. Barreiro, Curr. Med. Chem., 2005, 12, 23–49 CrossRef CAS PubMed.
- S. Thota, D. A. Rodrigues, P. de S. M. Pinheiro, L. M. Lima, C. A. M. Fraga and E. J. Barreiro, Bioorg. Med. Chem. Lett., 2018, 28, 2797–2806 CrossRef CAS PubMed.
- D. Kim, L. Wang, M. Beconi, G. J. Eiermann, M. H. Fisher, H. He, G. J. Hickey, J. E. Kowalchick, B. Leiting, K. Lyons, F. Marsilio, M. E. McCann, R. A. Patel, A. Petrov, G. Scapin, S. B. Patel, R. S. Roy, J. K. Wu, M. J. Wyvratt, B. B. Zhang, L. Zhu, N. A. Thornberry and A. E. Weber, J. Med. Chem., 2005, 48, 141–151 CrossRef CAS PubMed.
- E. J. Barreiro, A. E. Kümmerle and C. A. M. Fraga, Chem. Rev., 2011, 111, 5215–5246 CrossRef CAS PubMed.
- M. Eckhardt, E. Langkopf, M. Mark, M. Tadayyon, L. Thomas, H. Nar, W. Pfrengle, B. Guth, R. Lotz, P. Sieger, H. Fuchs and F. Himmelsbach, J. Med. Chem., 2007, 50, 6450–6453 CrossRef CAS PubMed.
- P. Sun, Y. Chen, and G. YU, Process for Preparing R-Beta-Amino Phenylbutyric Acid Derivatives, US Pat., US2011/0130587 A1, 2011 Search PubMed.
- A. E. Kümmerle, M. Schmitt, S. V. S. Cardozo, C. Lugnier, P. Villa, A. B. Lopes, N. C. Romeiro, H. Justiniano, M. A. Martins, C. A. M. Fraga, J.-J. Bourguignon and E. J. Barreiro, J. Med. Chem., 2012, 55, 7525–7545 CrossRef PubMed.
- A. Lopes, E. Miguez, A. Kümmerle, V. Rumjanek, C. Fraga and E. Barreiro, Molecules, 2013, 18, 11683–11704 CrossRef PubMed.
- G. Palla, G. Predieri, P. Domiano, C. Vignali and W. Turner, Tetrahedron, 1986, 42, 3649–3654 CrossRef CAS.
- P. Kumar, K. Kadyan, M. Duhan, J. Sindhu, V. Singh and B. S. Saharan, Chem. Cent. J., 2017, 11, 115 CrossRef PubMed.
- T. F. Silva, W. Bispo Júnior, M. S. Alexandre-Moreira, F. N. Costa, C. Monteiro, F. Furlan Ferreira, R. C. R. Barroso, F. Noël, R. T. Sudo, G. Zapata-Sudo, L. M. Lima and E. Barreiro, Molecules, 2015, 20, 3067–3088 CrossRef PubMed.
- B. E. O. Alves, A. K. N. de Alencar, L. E. R. Gamba, M. M. Trachez, J. S. da Silva, J. S. C. Araújo, T. L. Montagnoli, L. V. P. Mendes, P. M. Pimentel-Coelho, V. do M. N. Cunha, R. Mendez-Otero, G. M. M. Oliveira, L. M. Lima, E. J. Barreiro, R. T. Sudo and G. Zapata-Sudo, Pharmacol. Rep., 2019, 71, 1190–1200 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00450g |
| ‡ For the evaluation of the inhibitory activity of the compounds designed as DPP-4 inhibitors, we have acquired the DPP-4 inhibitor screening assay kit from cayman chemicals. The assay is based on a fluorometric test for screening and to establish the concentration–response curve of potential DPP-4-inhibitors (Cayman Chemicals, 2108). |
| § We tested the (R)-sitagliptin (7) (free base) in concentrations ranging from 0.001 to 10 μM, to establish the half maximal inhibitory concentration (IC50), and then compare the calculated IC50 value with that previous described in the literature. |
| This journal is © The Royal Society of Chemistry 2024 |