
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiological activity of lyophilized chitosan scaffolds with inclusion of chitosan and zinc oxide nanoparticles†
Jorge Eliecer Viloria Angaritaa,
Daniel Insuastyb,
Juan David Rodríguez M.c,
Jorge Iván Castrod,
Carlos Humberto Valencia-Llanoe,
Paula A. Zapataf,
Johannes Delgado-Ospinag,
Diana Paola Navia-Porrasg,
Alberto Albis h and
Carlos David Grande-Tovar*a
h and
Carlos David Grande-Tovar*a
aGrupo de Investigación de Fotoquímica y Fotobiología, Universidad del Atlántico, Carrera 30 Número 8-49, Puerto Colombia 081008, Colombia. E-mail: carlosgrande@mail.uniatlantico.edu.co; Tel: +57-5-3599-484
bDepartamento de Química y Biología, División de Ciencias Básicas, Universidad del Norte, Km 5 Vía Puerto Colombia, Barranquilla 081007, Colombia
cPrograma de Medicina, Facultad de Ciencias de la Salud, Universidad Libre, Km 5 Vía Puerto Colombia, Barranquilla 081007, Colombia
dTribology, Polymers, Powder Metallurgy and Solid Waste Transformations Research Group, Universidad del Valle, Calle 13 No. 100-00, Cali 76001, Colombia. E-mail: Jorge.castro@correounivalle.edu.co
eGrupo Biomateriales Dentales, Escuela de Odontología, Universidad del Valle, Calle 4B # 36-00, Cali 76001, Colombia
fGrupo de Polímeros, Facultad de Química y Biología, Universidad de Santiago de Chile, Santiago 9170020, Chile
gGrupo de Investigación Biotecnología, Facultad de Ingeniería, Universidad de San Buenaventura Cali, Carrera 122 # 6-65, Cali 76001, Colombia
hGrupo de Investigación en Bioprocesos, Universidad del Atlántico, Facultad de Ingeniería, Carrera 30 Número 8-49, Puerto Colombia 081008, Colombia. E-mail: albertoalbis@uniatlantico.edu.co
First published on 25th April 2024
Abstract
The constant demand for biocompatible and non-invasive materials for regenerative medicine in accidents and various diseases has driven the development of innovative biomaterials that promote biomedical applications. In this context, using sol–gel and ionotropic gelation methods, zinc oxide nanoparticles (NPs-ZnO) and chitosan nanoparticles (NPs-CS) were synthesized with sizes of 20.0 nm and 11.98 nm, respectively. These nanoparticles were incorporated into chitosan scaffolds through the freeze-drying method, generating a porous morphology with small (<100 μm), medium (100–200 μm), and large (200–450 μm) pore sizes. Moreover, the four formulations showed preliminary bioactivity after hydrolytic degradation, facilitating the formation of a hydroxyapatite (HA) layer on the scaffold surface, as evidenced by the presence of Ca (4%) and P (5.1%) during hydrolytic degradation. The scaffolds exhibited average antibacterial activity of F1 = 92.93%, F2 = 99.90%, F3 = 74.10%, and F4 = 88.72% against four bacterial strains: K. pneumoniae, E. cloacae, S. enterica, and S. aureus. In vivo, evaluation confirmed the biocompatibility of the functionalized scaffolds, where F2 showed accelerated resorption attributed to the NPs-ZnO. At the same time, F3 exhibited controlled degradation with NPs-CS acting as initiation points for degradation. On the other hand, F4 combined NPs-CS and NPs-ZnO, resulting in progressive degradation, reduced inflammation, and an organized extracellular matrix. All the results presented expand the boundaries in tissue engineering and regenerative medicine by highlighting the crucial role of nanoparticles in optimizing scaffold properties.
Introduction
According to the World Health Organization (WHO), for every 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people worldwide who suffer severe injuries compromising body tissues, 8% die during the treatment stages. At the same time, many do not achieve satisfactory outcomes due to a lack of biocompatibility and availability.1 This highlights the need for research in the field of tissue engineering, aiming to develop materials that enable the restoration, maintenance, or enhancement of tissue functionality.2
000 people worldwide who suffer severe injuries compromising body tissues, 8% die during the treatment stages. At the same time, many do not achieve satisfactory outcomes due to a lack of biocompatibility and availability.1 This highlights the need for research in the field of tissue engineering, aiming to develop materials that enable the restoration, maintenance, or enhancement of tissue functionality.2
Generally, biopolymers have been used due to the stimulating interaction with different cells, which causes the absence of an immune response.3 However, these biopolymers have poor mechanical properties, and their poor water barrier results in a low recovery rate and a low synthesis rate.4 On the other hand, synthetic polymers possess good mechanical properties but have more immune responses related to non-stimulation of cell adhesion and proliferation.5 In this sense, several researchers have focused on generating nanocomposites where these polymers are present to overcome each polymer's barriers. Among the synthetic polymers are polyvinyl alcohol (PVA), polylactic acid (PLA), and polycaprolactone (PCL), among others. At the same time, among biopolymers, the most used is chitosan (CS) because it stimulates good adhesion and cell proliferation.6 Additionally, the addition of plasticizing agents such as essential oils or inorganic agents with biocompatible properties and antimicrobial activity such as zinc oxide nanoparticles (NPs-ZnO) is emerging as a promising alternative to the mechanical, thermal, or biological challenges of the scaffold to be created.7
CS is a natural polymer derived from modifying chitin, the primary constituent of crustacean shells. It is a binary compound of glucosamine linked to N-acetylglucosamine, widely recognized for biomedical applications.8 As mentioned above, CS has wide applications in the biomedical field, especially in subdermal or bone tissue engineering, due to its low immune response and excellent biodegradation.9 Additionally, this material has been evaluated with different materials such as nanofillers, polymers, and ceramics with the condition that these materials have good biocompatibility and cell proliferation properties to overcome the barriers of CS, such as its low solubility and poor mechanical resistance in biological fluids.10–13
For this reason, there is a need to synthesize and functionalize nanostructured compounds to enhance these scaffolds' strength and mechanical compatibility. Incorporating these particles alters the properties of the scaffolds at the nanoscale level, providing the durability and resistance necessary to optimize their ability to maintain and stimulate tissue regeneration. Specifically, chitosan nanoparticles (NPs-CS)14 and zinc oxide nanoparticles (NPs-ZnO)15 are prominent examples in nanotechnology applications. NPs-CS demonstrate excellent biocompatibility in targeted drug delivery and tissue engineering.16 On the other hand, NPs-ZnO, with their antimicrobial properties, high chemical and thermal stability, and low toxicity, contribute to improving the hygiene and durability of the materials they constitute.17
Different research groups have developed CS-based scaffolds incorporating different molecules such as PCL, collagen, or carbonaceous nanomaterials and found that these types of scaffolds effectively promote the generation of subdermal tissues.18,19 Additionally, the antimicrobial activity of CS is essential because it does not induce an immune response around the scaffold. Studies on the incorporation of metal nanoparticles have shown that these materials can reduce the minimum inhibitory concentration, in other words, increasing their biocompatibility in the medium of administration in the human body, either through a scaffold or a drug.20 Ruan and co-workers developed a scaffold based on CS-ZnO and NPs-Se using the freeze-drying method, where they showed antimicrobial activity against S. aureus and E. coli bacteria; in addition, a significant improvement of CS-ZnO/NPs-Se scaffold in comparison with CS was observed in planimetric and histopathological indices, causing a total wound closure.21
Ramzan and co-workers reported the synthesis of CS, sodium alginate, and elastin-based scaffolds with NPs-ZnO, where they exhibited fluid absorption capacity for cell attachment and growth as well as demonstrated high activity against E. coli and S. aureus bacteria, making it a promising and effective candidate for the treatment of chronic wounds and skin regeneration.22 Tayebi and co-workers reported the synthesis of PCL/CS films incorporated with NPs-CS, demonstrating that adding NPs-CS improved biocompatibility and cell adhesion.23
Despite the higher number of investigations in tissue engineering using CS and metal nanoparticles, there have been no investigations involving NPs-CS, NPs-ZnO, and CS where biocompatibility under in vivo and in vitro conditions has been evaluated. In this work, we assessed the preliminary biocompatibility of four CS/NPs-CS/NPs-ZnO composite formulations based on subdermal implantations in vivo of Wistar rats and in vitro studies with Baby Hamster Kidney fibroblast cells (BHK-21).
Materials and methods
Materials
In this research, zinc chloride (ZnCl2, 98% purity), sodium hydroxide (NaOH, 98% purity) purchased from Lobachemie, isopropyl alcohol (2-propanol; 99.8% purity), sodium tripolyphosphate (TPP, 85% purity), low molecular weight chitosan (1 × 106 g mol−1) with a degree of deacetylation of 75%, and a viscosity of 494 cP acquired from Sigma-Aldrich® and glacial acetic acid (CH3COOH; 100% purity, diluted to 1% v/v) purchased from Supelco® were used.Experimental
Synthesis of NPs-CS
The synthesis of NPs-CS was carried out according to the reported methodology, using sodium tripolyphosphate (TPP) as the cross-linking agent.24 Two solutions were prepared: the first one contained 1% v/v acetic acid, to which 2 g of low molecular weight chitosan was added using an analytical balance RADWAG AS 220.R2 (RADWAD, Poland). This mixture was stirred for 24 hours at room temperature to dissolve the chitosan completely. Subsequently, the pH was adjusted to 5.2 using a 1 N NaOH solution. The second solution was prepared by adding 96 mg of TPP to 100 mL of ultrapure water obtained from a Direct-Q® 3UV distiller (Millipore, Darmstadt, Germany) with a water resistivity of 18.2 Megaohms (MΩ). This solution was stirred until completely dissolved and then dropwise added to the first solution while stirring for 30 minutes. The final solution was centrifuged for 20 minutes at 3200 rpm using an SL 8R centrifuge (Thermo Scientific, Dreieich, Germany) to form pellets. These pellets were separated from the supernatant and placed in an LC-FD-06H lyophilizer (Müller Scientific, Zhengzhou, China), where they were frozen at −50 °C for 8 hours. Subsequently, the lyophilization (freeze-drying) process was initiated by applying a vacuum using a 2XZ-4 rotary vane vacuum pump (East vacuum, Beijing, China) until reaching 5.5 Pa, which removed all the solvent from the solution over 72 hours.25Synthesis of NPs-ZnO
The nanoparticles were synthesized according to the procedure reported by Becheri.26 To do this, two solutions were prepared. The first solution was made with 5.5 g of ZnCl2 in a beaker containing 200 mL of distilled water. The second solution consisted of 5 M NaOH. Once the solutions were prepared, small volumes of 5 M NaOH were taken with a disposable syringe and added dropwise to the first solution. The mixture was then constantly stirred on a Scilogex hot plate (SCI550-2, EU) at 900 rpm, maintaining the temperature at 90 °C for 10 minutes. Subsequently, the obtained suspension was collected by vacuum filtration and washed with distilled water to reduce the concentration of sodium chloride (NaCl). Next, the nanoparticles were dispersed in a Branson Bransonic® CPX Digital Bath 2800 ultrasonic bath (Bransonic, Danbury, USA) for 12 minutes at a frequency of 40 kilohertz (kHz) with 230 V power and mixed with 2-propanol for 10 minutes at 20 °C. Additionally, the ZnO-NPs were centrifuged at 5000 rpm for 15 minutes to wash with 2-propanol thrice. Finally, the nanoparticles were calcined in a Nabertherm LHT 02/18 furnace (Lilienthal, Bremen, Germany) at 250 °C for five hours.27Synthesis of chitosan scaffolds with inclusion of NPs-CS and NPs-ZnO
The scaffolds were synthesized following Mohandes and colleagues' modified protocol.28 First, a 2% v/v glacial acetic acid solution was prepared, which was used to dissolve the chitosan from the four proposed formulations (Table 1). The nanoparticles in each formulation were dispersed in distilled water using ultrasound for 30 minutes at a frequency of 40 kHz with a power of 230 V. After achieving completely dispersed and homogeneous solutions, the solvent was removed by freeze-drying at −50 °C and 5.5 Pa over 96 hours.29| Compounds | Formulations | |||
|---|---|---|---|---|
| F1 | F2 | F3 | F4 | |
| Chitosan (mg) | 2000 | 1900 | 1900 | 1900 |
| Zinc oxide nanoparticles (mg) | — | 100 | — | 50 |
| Chitosan nanoparticles (mg) | — | — | 100 | 50 |
| Acetic acid (mL) | 100 | 100 | 100 | 100 |
Characterization of chitosan NPs-CS and NPs-ZnO
The polydispersity index (PDI) was calculated using the average diameter of the nanoparticles with eqn (1):
 | (1) |
Characterization of lyophilized scaffolds (F1, F2, F3, F4)
Hydrolytic degradation of the scaffolds
The hydrolytic degradation of the scaffolds was determined using samples of 1.0 by 1.5 cm immersed in Roswell Park Memorial Institute (RPMI) culture medium containing 2% penicillin–streptomycin (P/S) and 10% Fetal Bovine Serum (FBS), with a volume of 25 mL for each formulation.1 The samples were kept agitated in an orbital shaker (SK-O180-2; Dlab, Beijing, China) at 37 °C in an incubator (Memmert, Schwabach, Germany) for 1 to 16 days. The mass of the four formulations was assessed each day. During this period, the pH of the medium was also recorded using an Accumet™ AB150 pH meter (Fisherbrand, Ottawa, Canada).
 | (2) |
Before immersion in the test solution, the sample was weighed (W0), and at the end of the test, the samples were removed from the medium and superficially dried with a dry cloth to be weighed (Ww). Subsequently, they were dried in the incubator at 37 °C for 2 hours and weighed again (Wd). The scaffolds' weight loss (Wl) was determined using eqn (2).36 Finally, scanning electron microscopy (SEM) images were obtained after 1 and 16 days of sample immersion in a culture medium (RPMI) with 10% Fetal Bovine Serum (FBS) using a JEOL JSM 6490 LV scanning electron microscope coupled with an energy-dispersive X-ray spectrometer (EDS) (Akishima, Tokyo, Japan) to verify the presence of calcium and phosphorus as indicators of hydroxyapatite formation.
Mechanical properties of the scaffolds
The compression test was performed using a SHIMADZU EZ-LZ universal testing machine (Shimadzu, Tokyo, Japan) and a 500 N load cell. The samples were tested at a 10 mm min−1 speed, and at least five samples per formulation were used. The samples were cylinders with a height of 10 mm and a diameter of 25 mm.37In vitro biocompatibility analysis
Similarly, a stock dispersion of NPs-ZnO was prepared at a concentration of 20 mM by taking 8.1 mg of NPs-ZnO and dispersing them in 5 mL of distilled water. The NPs-ZnO were dispersed using vortex agitation followed by 2 minutes of ultrasound. Four successive dilutions were prepared (10, 5, 2.5, 1.25 mM).
Enterobacteriaceae (Salmonella enterica ATCC 53648, Staphylococcus aureus ATCC 55804, Klebsiella pneumoniae, Enterobacter cloacae sub cloacae, Enterobacter asburiae, Enterobacter hormaechei, and Enterobacter ludwigii) previously identified molecularly (unpublished data) from the culture collection of the Universidad de San Buenaventura Cali, were selected for analysis. These strains were cultured in Brain Heart Infusion (BHI) broth for 24 hours and subsequently subjected to a washing process. The washing was carried out through three cycles of vortex agitation for one minute, followed by centrifugation at 6000 rpm for five minutes in a sterile 0.85% saline solution. From the resulting solution, dilutions were made to adjust the concentration to 106 colony-forming units per milliliter (CFU mL−1) using optical density (OD600), following the McFarland scale previously established with Tryptic Soy Broth (TSB).38
In a 96-well polystyrene microplate (BRANDplates), 100 μL of the enterobacteria and 100 μL of the NPs solution were inoculated and incubated at 37 °C for 24 hours. The minimum inhibitory concentration (MIC) value was determined as the lowest concentration at which no turbidity or bacterial growth was observed. To confirm biological activity, 20 μL of a 0.2% triphenyl tetrazolium chloride (TTC) solution was added and incubated at 37 °C for two hours. A red color indicated biological activity.39
The minimum bactericidal concentration (MBC) value was determined from the wells that showed no visible growth of the microorganism (lack of turbidity and no reaction with TTC). Ten μL from the well were taken and plated on a nutrient agar plate; subsequently, it was incubated at 37 °C for 24 hours to confirm the microorganism's death. The tests were performed in triplicate. To evaluate the ability of nanoparticles to inhibit biofilm formation on polystyrene microplates, the procedure of Rossi et al.40 was followed without modification.
Once the incubation time was completed, the tubes from each treatment were vortexed for one minute, followed by serial dilutions in 0.85% saline solution and plating 100 μL on nutrient agar plates. The plates were then incubated at 37 °C for 48 hours. The viable cell count was performed using the plate counting technique. Each test was conducted in triplicate, and the results were reported as the percentage of material inhibition.
Preliminary analysis of the bioactivity of the scaffolds
To assess the scaffolds' in vitro bioactivity, images were obtained using scanning electron microscopy (SEM) after 1 and 16 days of sample immersion in a culture medium (RPMI) with 10% Fetal Bovine Serum (FBS). A JSM 6490 LV model scanning electron microscope from JEOL coupled with an energy-dispersive X-ray spectrometer (EDS) (Akishima, Tokyo, Japan) verified the presence of calcium and phosphorus as indicators of hydroxyapatite formation.In vitro cytotoxicity assays
To estimate the cellular viability of the scaffolds, segments of 10 mm diameter and 1.5 mm thickness were taken from each formulation and sterilized using ultraviolet light in a Purifier™ Cell Logic™ biological safety cabinet (Madrid, Spain) for 60 minutes. Subsequently, they were immersed in Roswell Park Memorial Institute (RPMI) medium containing 2% penicillin–streptomycin (P/S) and 10% fetal bovine serum (FBS), with a volume of 5 mL for each formulation. These segments were incubated in an OrbiCult™ IBS orbital shaker (IBS-NR, Changi South Street, Singapore) at a temperature of 37 °C and a speed of 200 rpm for periods of four, fifteen, and twenty-one days. BHK-21 cells were seeded in a 96-well plate at a density of 1000 cells per well with a 5% CO2 atmosphere to evaluate cell viability. Subsequently, the samples were exposed to the extracts for 24 hours, using twelve replicated wells for each formulation and twenty-four for the negative control. The latter consists solely of RPMI culture medium, aiming to prevent any negative impact on the cells and allow for an accurate comparison of cellular viability. Finally, a 0.5% solution of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma Aldrich, St. Louis, MO, USA) in RPMI 1640 medium without fetal bovine serum was added to the plate. After a 3 hour incubation at 37 °C, cell viability was determined by the optical density of the wells at 570 nm using a FLUOstar Omega microplate reader (BMG LABTECH, Allmendgrün, Germany).42 The percentage viability was calculated using eqn (3):
 | (3) |
Using the optical density measurements of the sample and the control.
In vivo viability analysis of the scaffolds
The experiment followed the guidelines of the UNE 10993-6 standard, which refers to the biological evaluation of medical devices, specifically about testing local effects after their implementation. In this case, three biomodels were used: three-month-old male Wistar rats with an average weight of 280 g, provided by the Intermediate Preclinical Research and Livestock Laboratory (LABBIO) of the Universidad del Valle. The biomodels were randomly selected by the LABBIO laboratory staff from the animals in the laboratory that met the age, sex, and weight requirements.
The sample size of three biomodels was determined following the recommendations of ISO 10993-6, which establishes three as the minimum number of animals to obtain results.44 This also allowed Russell and Burch to adopt the “Reduction” principle.45 The study followed the guidelines established in the “Animal Research: Reporting of In Vivo Experiments” (ARRIVE) guidelines.46 No complications were recorded during or after surgery, and no biomodels died during the research. Still, it is also taken into account that the vivariums for biomodels handle a very high degree of consanguinity to try to ensure that the sample is representative of the population of biomodels and retains a lot of similarities. In the genotype, it is due to genetic closeness, and in the phenotype, it is due to the controlled environment of the laboratory.
The ethical approval and supervision of this work were overseen by the Animal Experimentation Ethics Committee of Universidad del Valle, Cali, Colombia, through Resolution No. CEAS 006-022.
After making incisions in the biomodels, several blocks were inserted to evaluate initial biocompatibility. For this purpose, four surgical pockets were created by making four independent incisions of five millimeters in length and total thickness on the right side of the midline, using hemostatic forceps to a depth of fifteen millimeters.47
Fragments measuring one centimeter by two centimeters were cut from the four available types of scaffolds, each with an average weight of 0.0175 grams. Subsequently, they were implanted into the pockets and sutured with a simple stitch using an absorbable suture (ETHICON, Johnson and Johnson, New Jersey, USA). Then, gentamicin was topically applied (Gentamicin 1%, Procaps, Cali, Valle del Cauca, Colombia), and for postoperative medication, Diclofenac 75 mg was administered (La Sante, Bogotá, Colombia).
The biological models were sedated by administering Ketamine (70 mg kg−1, Laboratorio Blaskov, Bogotá, Colombia) and Xylazine (30 mg kg−1, Laboratorios ERMA, Celta, Colombia). After a 30 day implantation period, the biological models were euthanized by intraperitoneal injection of an excess Ketamine/Xylazine solution, and the samples were retrieved. Subsequently, a visual observation of the areas where the skin implants with hair had been made was conducted, followed by hair removal to obtain the samples.
Histological analysis
The samples were fixed for 48 hours in buffered formalin. Subsequently, they were processed using Autotechnicon Tissue Processor™ equipment (Leica Microsystems, Mannheim, Germany). The obtained samples were embedded in paraffin blocks using the Histoplast™ equipment (Thermo Fisher Scientific, Waltham, MS, USA), and 6-micron sections were made with the Leica RM2125 RTS microtome (Leica Microsystems, Mannheim, Germany). All sections underwent Hematoxylin and Eosin (H&E) staining, Masson's Trichrome (MT), and Gomori's Trichrome (GT). Histological images were captured using a Leica DM750 optical microscope and a Leica DFC 295 camera. Images were processed with Leica Application Suite version 4.12.0 software (Leica Microsystems, Mannheim, Germany).Statistical analysis
All cell viability and hydrolytic degradation test results were expressed as the mean of at least three replicates ± standard deviation. In the cell viability analysis, significant differences were determined using ANOVA with a 95% confidence interval using The Unscrambler X software. For the results of mechanical and antimicrobial tests, Minitab 19 software was used to analyze variance for treatments using ANOVA, with a significance level of 0.05.Results and discussion
FT-IR analysis of chitosan, NPs-CS, and NPs-ZnO
The band between 3400 cm−1 and 3300 cm−1, as evidenced in Fig. S1A and present† in both spectra, is characteristic of the symmetric and asymmetric tensions of the N–H bonds of the primary amine. The width of the band at 3400 cm−1 for the chitosan spectrum is related to the stretching of the –OH groups of chitosan, which results from the O–H stretching vibration associated with intra, inter, and free molecule-bound hydroxyl groups. Similarly, the small band at 2871 cm−1 corresponds to the C–H vibration.48The bands at 1648 cm−1 and 1570 cm−1 in chitosan are attributed to the bending of the –NH bonds of the primary amine and the amide; however, in the NPs, these bands shift to 1637 cm−1 and 1544 cm−1. This could be due to the interactions between the phosphate groups of TPP and the ammonium ions of chitosan, depending on the degree of cross-linking.48 The bands at 1374 cm−1 and 1405 cm−1 for NPs-CS and CS are due to the scissoring bending of C–C bonds, while the band at 1026 cm−1 corresponds to C–O vibrations, and at 660 cm−1 indicates vibrations produced by the C–O–C bonds of chitosan. The band at 798 cm−1 in NPs-CS can be attributed to the P–O vibration and the P–O–P bond.49
Size of NPs-CS by DLS
After synthesizing the NPs-CS, particle size was analyzed using the DLS method. The polydispersity index (PDI) and the average diameter were calculated using eqn (1), resulting in a value of 0.02. Based on DLS measurements, the particle size distribution appears as a bell-shaped curve, maintaining a moderate amplitude, indicating a homogeneous size distribution. Most of the NPs-CS were found in the 150 to 200 nm range, with the predominant size being the peak maximum of 169.9 nm (Fig. S1B†),50 suggesting that the NPs-CS are monodisperse. The low polydispersity of the NPs-CS is related to the ionotropic synthesis method of nanoparticles, as this method allows for control over the shape and size of the NPs.TEM analysis of NPs-CS
The predominant morphology of the nanoparticles revealed a spherical nature. However, agglomeration formation could be identified, as evidenced in Fig. S1C.† This phenomenon is expected within the methodology developed for synthesizing these chitosan-based nanoparticles.Upon meticulous examination of the dimensions of the NPs-CS, the use of ImageJ software facilitated a comprehensive quantitative analysis. The size distribution histogram, as shown in Fig. S1D,† provides an accurate visual representation of the dimensional variability of these nanoparticles. While an average size of 11.98 nm ± 3.98 is recorded, it is essential to highlight that the dimensions of the NPs exhibit inherent variability. This variability could be attributed to fluctuations in synthesis conditions or particle formation heterogeneity during the process. Such dimensional heterogeneity could have significant implications, especially when considering the specific application of these nanoparticles. For instance, in biomedical applications, the size and uniformity of nanoparticles can influence their biodistribution, drug-loading capacity, and interaction with specific cells or tissues.51
FTIR analysis of NPs-ZnO
In Fig. S2A,† the band at 3499 cm−1 is evident, corresponding to the symmetric stretching of the hydroxyl (O–H) groups, primarily attributed to dissociative adsorption on the irregular edges of the ZnO NPs surface.52In the fingerprint region, a broad band at 905 cm−1 is observed, attributed to the stretching vibration of the O–Zn–O lattice. Additionally, two bands appear at 720 cm−1 and 563 cm−1, arising from the stretching and deformation of the Zn–O bonds.53
Meanwhile, the band at 1042 cm−1 signifies the vibration of the absorbed carbonate lattice.54
Size of NPs-ZnO by DLS
The particle size was measured using the DLS method, and the polydispersity index (PDI) was calculated using eqn (1) with the average diameter of the nanoparticles. The result was 0.03, indicating that the nanoparticles are monodisperse. The particle size distribution is a bell-shaped curve with moderate amplitude, suggesting a homogeneous size distribution. However, most NPs-ZnO were in the 360 to 490 nm range, with the predominant size at the peak maximum of 420.2 nm (Fig. S2B†).Specifically, the size dispersions of the NPs-ZnO are attributed to their thermodynamic metastability due to their high surface area. This means that they do not exhibit the Ostwald ripening phenomenon since there is no evidence of an increase in the dispersion of the NPs-ZnO due to low energies during nanoparticle preparation.55
TEM analysis of NPs-ZnO
The TEM analysis of the NPs-ZnO, as illustrated in Fig. S2C,† reveals distinctive morphological characteristics of these nanoparticles. Notably, these NPs-ZnO predominantly appear in a spherical and monodisperse shape. This uniformity in shape and dispersion initially suggests a well-controlled synthesis process. However, it is crucial to highlight the presence of aggregates observed within the sample. As cited in previous research, these aggregates may arise due to interactions between nanoparticles, primarily due to collision phenomena amplified by their interaction with water.56 A histogram was generated with data from 300 nanoparticles using ImageJ software (Fig. S2D†), showing an average size of 20 nm ± 7.The variability in size, reflected in the standard deviation of ±7, could have significant implications for the functional properties of these nanoparticles. For instance, specific size and dispersion can influence their chemical reactivity, their ability to disperse in different mediums, or their interaction with biological systems.
Analysis of scaffolds incorporated with NPs-CS and NPs-ZnO
 | ||
| Fig. 1 FTIR of the different scaffolds based on NPs-ZnO and NPs-CS. F1, 2000 mg CS/100 mL acetic acid; F2, 1900 mg CS/100 mg NPs-ZnO/100 mL acetic acid; F3, 1900 mg CS/100 mg NPs-CS/100 mL acetic acid; F4, 1900 mg CS/50 mg NPs-ZnO/50 mg NPs-CS/100 mL acetic acid. | ||
The bands at 1374 and 1405 cm−1 in chitosan and NPs-CS are attributed to C–C bonds. Similarly, the bands in the 1070–1026 cm−1 region indicate vibrations produced by the C–O–C bonds of chitosan. The bands around 650 and 560 cm−1 are typically associated with the structure's C–N vibration of glucosamine (N-acetylglucosamine).
Interestingly, changes in the bands are observed as nanoparticles are incorporated. In the case of F2, the suppression of bands at 2880, 1635, and 650 cm−1 is noticeable, which can be explained as a shielding effect between the bands of the NPs-ZnO and those of the scaffold. On the other hand, with the inclusion of only NPs-CS into F3, an increase in the intensity of the scaffold bands can be observed, as NPs-CS and the chitosan-based scaffold share the exact chemical nature.
However, in the last formulation (F4) that contains both nanoparticles, a slight decrease in the band's intensity at 1645 cm−1 is observed. Additionally, the bands between 650 cm−1 and 560 cm−1 intensify due to the combined presence of both nanoparticles.
Determination of structural ordering by XRD
The X-ray diffraction (XRD) analysis provided insights into the structural changes of chitosan scaffolds upon incorporating NPs-CS and NPs-ZnO, as shown in Fig. 2. The diffractogram of formulation F1 (2% CS) revealed two characteristic peaks of the chitosan structure, located at 2θ values of 21.3° (111) and 29.3° (211), along with another peak present in F3 (1.9% CS:0.1% NPs-CS) at 11.3° (111). This profile arises from interactions between hydrogen bonds formed by the amino, amide, and alcohol functional groups present in the molecule.57 However, there is an apparent emergence of prominent peaks at 2θ values of 46.6° (321), 47.5° (321), 56.6° (322), and 63.8° (422). These can be attributed to the cross-linking generated during the fabrication process of the chitosan scaffold.
 | ||
| Fig. 2 Diffractogram of the different scaffolds based on NPs-ZnO and NPs-CS. F1, 2000 mg CS/100 mL acetic acid; F2, 1900 mg CS/100 mg NPs-ZnO/100 mL acetic acid; F3, 1900 mg CS/100 mg NPs-CS/100 mL acetic acid; F4, 1900 mg CS/50 mg NPs-ZnO/50 mg NPs-CS/100 mL acetic acid. | ||
In the diffraction spectrum of F2 (1.9% CS:0.1% NPs-ZnO), a decrease in the intensity of the 2θ peaks at 29.5° (111), 38.6° (210), 42.3° (211), 46.3° (211), 47.7° (220), 59.7° (311), 63.6° (320), 71.8° (400), and 76.3° (411) is observed. This decrease can be associated with the distortion of the crystalline lattice due to the incorporation of NPs-ZnO into the polymeric matrix. This results in overlapping the NPs-ZnO peaks (represented by black dots in the F2 diffractogram) and the formation of imperfections and crystalline distortions with those of the chitosan scaffold.58 This leads to peaks at 2θ of 42.3°, 47.7°, 56.6°, and 59.6° and an increase in the peak at 38.6°.
An inverse relationship between the interplanar distance (d-spacing) and the angle θ can also be established. The greater the spacing between the atomic planes, the smaller the diffraction angle θ. Therefore, the decrease in intensity is also associated with a slight leftward shift of the diffraction peaks of the material.59
Regarding formulation F3 (1.9% CS:0.1% NPs-CS), the diffraction pattern significantly changes. A peak at 2θ of 11.3° (111) is observed, and the loss of other previously present peaks is noted. This is due to the presence of the NPs-CS, whose chemical nature is similar to the scaffold, and due to their size, they are incorporated into the planes of the crystalline lattice. This results in a loss of material crystallinity, making it more amorphous,60 altering the planes in the 2θ peaks of 21.4 (311), 29.4 (420), 42.3 (540), 47.8 (551), 56.6 (653).
In the latest formulation, F4 (1.9% CS:0.05% NPs-ZnO:0.05% NPs-CS), the presence of both NPs-CS and NPs-ZnO significantly enhances the material's crystallinity. Well-defined peaks, a reduction in the interplanar distance, an increase in the intensity of the prominent peaks, and a slight shift towards more considerable angles evidence this. This shift is due to the rise in the interatomic distance within the material's crystalline lattice, caused by the introduction of the nanoparticles, resulting in a longer wavelength being diffracted within the material.61
TGA and DSC analysis
The primary objective of thermogravimetric analysis (TGA) is to evaluate a material's response to increasing temperature, focusing on determining its thermal stability. In Fig. 3 and 4, the thermogravimetric curves of the scaffolds in their various formulations are presented. In the case of chitosan, its thermal degradation occurs in three distinct stages. Still, from the exclusive perspective of polymer decomposition, the most significant is observed in the temperature range from 268 °C to 312 °C.62,63 | ||
| Fig. 3 Thermogram for the different scaffolds based on NPs-ZnO and NPs-CS. F1, 2000 mg CS/100 mL acetic acid; F2, 1900 mg CS/100 mg NPs-ZnO/100 mL acetic acid; F3, 1900 mg CS/100 mg NPs-CS/100 mL acetic acid; F4, 1900 mg CS/50 mg NPs-ZnO/50 mg NPs-CS/100 mL acetic acid. | ||
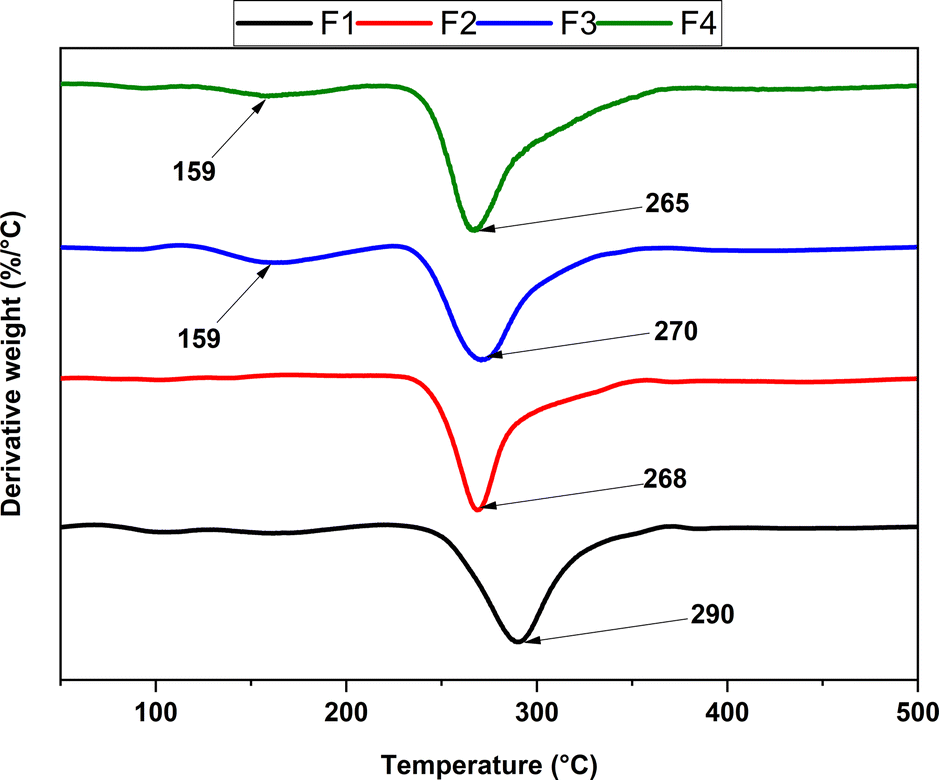 | ||
| Fig. 4 Derivative curves for the different scaffolds based on NPs-ZnO and NPs-CS. F1, 2000 mg CS/100 mL acetic acid; F2, 1900 mg CS/100 mg NPs-ZnO/100 mL acetic acid; F3, 1900 mg CS/100 mg NPs-CS/100 mL acetic acid; F4, 1900 mg CS/50 mg NPs-ZnO/50 mg NPs-CS/100 mL acetic acid. | ||
The thermogravimetric analysis reveals two main stages in the thermal degradation profile of chitosan scaffolds: NPs-CS and NPs-ZnO. However, only one primary phase is evident in two proposed formulations, specifically F1 and F2. For F1, a thermal degradation peak is observed, indicating the decomposition of chitosan marked by polymer depolymerization and the release of volatile compounds. This event has a Tmax of 290 °C, resulting in a mass loss of 43%. This process originates due to the depolymerization of chitosan structures and degradation of pyranose rings through water loss and deamination, followed by ring opening. In the case of F2, a single thermal degradation peak is also observed at approximately 268 °C. This peak suggests a primary thermal degradation reaction in this formulation, influenced by NPs-ZnO. This could lead to degradation at a lower temperature by facilitating bond breakage in chitosan, resulting in thermal degradation at a lower temperature. Moreover, this interaction might lead to the release of volatile compounds at lower temperatures compared to the F1 formulation.64
On the other hand, both F3 and F4 display two thermal degradation peaks recorded at approximately 159 °C, with mass losses of 6.8% and 4.6%, respectively. This behavior indicates the release of water firmly bound to chitosan molecules in this phase. Additionally, F3 and F4 show a second thermal degradation peak, with values around 270 °C and 265 °C, and mass losses of 36.34% and 42.9%, respectively, associated with polymer decomposition. Incorporating NPs-CS in these formulations impacts the complexity of the thermal reactions, resulting in comparable thermal stability. The thermogravimetric analysis suggests that scaffolds made from chitosan, including NPs-CS and NPs-ZnO, maintain their thermal stability up to approximately 230–235 °C in a nitrogen atmosphere.65
Chitosan, a semi-crystalline polysaccharide with a significant amorphous fraction, exhibits physical properties that undergo noticeable changes during the glass transition temperature, a crucial parameter of the amorphous state.66 differential scanning calorimetry (DSC) was used to clarify the thermal properties of the four scaffold formulations proposed for the research development, as illustrated in Fig. 5.
 | ||
| Fig. 5 DSC curves for the different scaffolds based on NPs-ZnO and NPs-CS. F1, 2000 mg CS/100 mL acetic acid; F2, 1900 mg CS/100 mg NPs-ZnO/100 mL acetic acid; F3, 1900 mg CS/100 mg NPs-CS/100 mL acetic acid; F4, 1900 mg CS/50 mg NPs-ZnO/50 mg NPs-CS/100 mL acetic acid. | ||
Incorporating nanoparticles (NPs) into the polymeric matrix subtly alters the glass transition temperature. In formulation two (F2 CS 1.9%:NPs-ZnO 0.1%), a slight reduction in transition temperature is evident, indicative of surface interactions between the NPs-ZnO and chitosan. This interaction, influenced by the low polydispersity of the NPs-ZnO according to DLS analysis, affects the molecular mobility of the polymeric matrix, impacting its ability to transition from a glassy state to a more elastic one. Compared to formulation four (F4 CS 1.9%:NPs-ZnO 0.05%:NPs-CS 0.05%), a different impact on thermal properties is observed due to interactions between nanoparticles and chitosan, which can alter the polymer's molecular structure.67 In formulations one (F1 CS 2%) and three (F3 1.9% CS:NPs-CS 0.1%), a similar glass transition temperature was evident, suggesting thermal stability. This highlights that the NPs-CS do not significantly affect the glass transition temperature.68
SEM analysis of scaffolds
Fig. 6 presents representative images obtained through scanning electron microscopy (SEM) of highly porous three-dimensional scaffolds designed for biomedical applications. These scaffolds have three pore categories: small, medium, and large. The irregular morphology, with sheet-like pores, suggests a flat and extended geometry rather than a spherical one across all four formulations. | ||
| Fig. 6 Morphology of CS/PVA/TTEO films by SEM images of the different scaffolds based on NPs-ZnO and NPs-CS. F1, (a), 2000 mg CS/100 mL acetic acid; F2, (b), 1900 mg CS/100 mg NPs-ZnO/100 mL acetic acid; F3, (c), 1900 mg CS/100 mg NPs-CS/100 mL acetic acid; F4, (d), 1900 mg CS/50 mg NPs-ZnO/50 mg NPs-CS/100 mL acetic acid. | ||
The large, irregular, and uniformly interconnected pores result from the lyophilization method, enabling discrete control over pore diameter. Scaffolds were manufactured with small (<100 μm), medium (100–200 μm), and large (200–450 μm) pore sizes.69 The interconnections among the large pores allow massive transport and cell migration throughout the scaffold. The fabrication protocol was optimized to ensure consistent scaffold properties, particularly in interconnectivity, facilitating efficient cell migration and massive transfer between different pore sizes. Through comprehensive SEM analysis, it was observed that the interconnection and pore connectivity remain consistent across all pore sizes of the four formulations examined. At higher magnifications, the scaffolds exhibit a uniform laminar surface; this aspect adds to the polymeric nature of chitosan, providing essential mechanical properties such as flexibility and strength. The amino groups allow positive interaction by forming specific bonds with cell membrane molecules. This set of characteristics operates synergistically, which is crucial in facilitating fundamental processes such as cell adhesion, proliferation, and protein secretion. This composition and morphology are particularly beneficial in the context of tissue engineering applications, where the scaffolds' ability to promote positive cellular interactions is essential for the success of tissue regeneration and the functionality of biomimetic constructs.70
Analysis of the hydrolytic degradation of functionalized scaffolds
Fig. 7 displays functionalized scaffolds' weight loss (%) after 16 days of agitation in a simulated biological fluid. Generally, increased scaffold stability is observed, manifested as a decrease in weight loss. When analyzing daily degradation rates, it is evident that after six days of immersion, F2 (31%) and F4 (34%) show lower degradation rates compared to F1 (46%) and F3 (41%). Additionally, a difference is observed at 16 days, where a weight loss of F1 (64%) and F3 (75%) is higher than that of F2 (46%) and F4 (49%).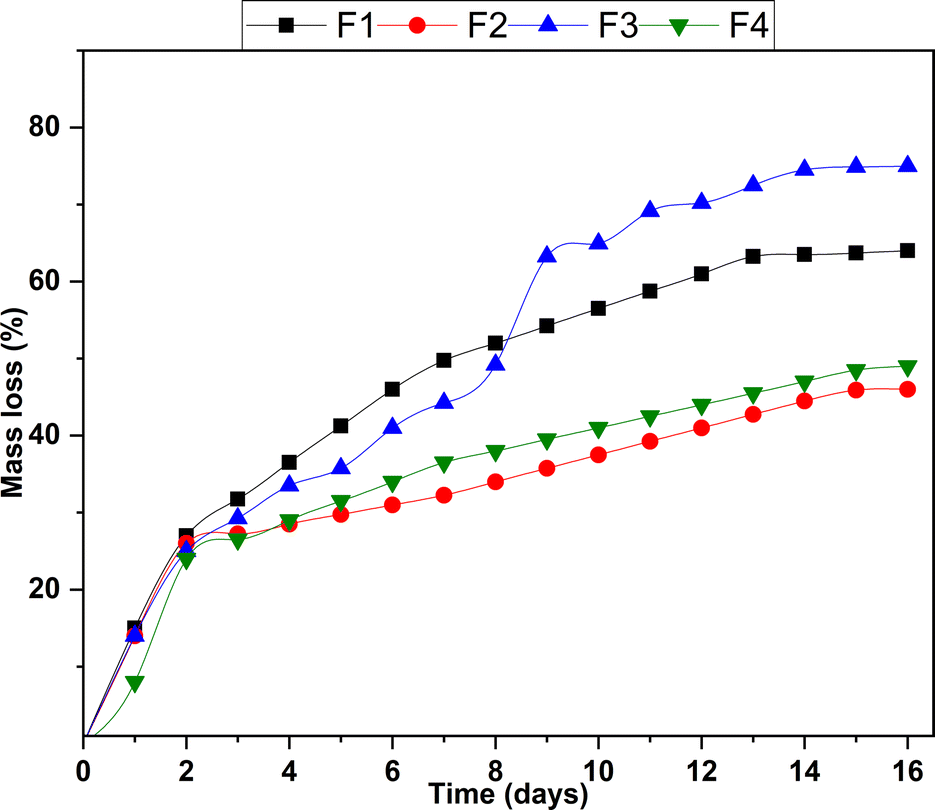 | ||
| Fig. 7 Diagram of the hydrolytic degradation of functionalized scaffolds. | ||
F2 and F4 stand out as the most stable formulations, exhibiting lower degradation rates and reduced levels of total degradation compared to F1 and F3. The NPs-ZnO present in F2 and F4 seem to have a stabilizing effect on these formulations. This phenomenon could be related to the unique physical and chemical properties of the NPs-ZnO. The presence of these nanoparticles moderately interferes with hydrolysis processes by establishing a physicochemical barrier with chitosan, slowing down degradation. F2 and F4 are more stable formulations suitable for applications with a more controlled and sustained degradation over time. On the other hand, F1 and F3, with higher degradation rates, could be helpful in applications where rapid or complete release is desired.
Mechanical properties of the scaffolds
In tissue engineering, there is a demand for biomaterials with specific mechanical and thermal properties tailored to the environment in which they will remain until complete degradation. Chitosan scaffolds, made from this natural polymer derived from chitin, have emerged as a promising option.71The results of the mechanical properties under compression of the functionalized chitosan scaffolds in the four formulations (Table 2) reveal a significant decrease in the scaffolds' deformation stress as NPs-ZnO is incorporated and an increase with the inclusion of NPs-CS. Among them, scaffold F3 exhibits the best mechanical properties with minimal deformation (higher Young's modulus), followed by F4, F2, and F1, which has the highest deformation percentage since it does not contain any of the nanoparticles used in its composition.
| Formulation | M. elastic (MPa) | Max. tension (MPa) | Max. deformation (%) |
|---|---|---|---|
| F1 | 0.04458 ± 0.02 | 0.49205 ± 0.05 | 269.534 ± 42.2 |
| F2 | 0.02232 ± 0.005 | 0.35722 ± 0.07 | 256.946 ± 63.2 |
| F3 | 0.05028 ± 0.09 | 0.45118 ± 0.012 | 163.586 ± 18 |
| F4 | 0.03268 ± 0.02 | 0.40198 ± 0.06 | 232.572 ± 102 |
These results are consistent with the interpretation of the pore size analysis calculated from the SEM images for each of the nanoparticles, where their homogeneous dispersion in the polymeric matrix can be observed. On the other hand, the increase in Young's modulus was less than expected, perhaps because the diameter of the NPs was between 70–110 nm. The smaller the diameter of the NPs, the higher their Young's modulus, and therefore, it would be expected that reinforcing the polymeric matrix with the NPs would result in a higher Young's modulus.72
On the other hand, a linear regression analysis was performed for each of the formulations in the three tests conducted. Overall, low R2 values were obtained in the tests for Young's modulus (elastic), maximum compression stress (tension), and maximum deformation, where only 27.38%, 26.31%, and 31.86% of the data adequately fit the linear model, respectively. However, an analysis of variance was carried out using one-way ANOVA with a 95% confidence level (5α = 0.05) to verify the proposed linear model.
The null hypothesis is accepted for the Young's modulus test with a critical F = 2.01, greater than the experimental F = 0.153, indicating no significant differences in the variation in response to each formulation. Similarly, the null hypothesis is accepted for the maximum compression stress test with a critical F = 1.90, more significant than the experimental F = 0.170, and for the maximum deformation test with a critical F = 2.49, more significant than the experimental F = 0.097. By accepting the null hypothesis, it can be stated that there is no considerable variation in the response of the proposed linear model. There is no linear relationship between the percentage of nanoparticles incorporated into the polymeric matrix and the values of the three tests obtained; therefore, the mechanical properties cannot be modulated according to the concentration of nanoparticles incorporated, even though differences are noted among the four formulations.
In vitro biocompatibility and antibacterial capacity analysis
 | ||
| Fig. 8 SEM-EDS images and percentage weight results of the scaffolds after 16 days of immersion in RPMI with 10% SFB: (a) micrograph of F1 (2% CS), (b) weight percentage of analyzed elements for F1; (c) micrograph of F2 (1.9% CS:0.1% NPs-ZnO), (d) weight percentage of analyzed elements for F2; (e) micrograph of F3 (1.9% CS:0.1% NPs-CS), (f) weight percentage of analyzed elements for F3; (g) micrograph of F4 (1.9% CS:0.05% NPs-ZnO:0.05% NPs-CS), (h) weight percentage of analyzed elements for F4. | ||
The predominant elements identified in formulations F2, F3, and F4 are carbon (C), oxygen (O), and sodium (Na). In contrast, in the F1 formulation, its significant constituents are carbon (C), oxygen (O), and calcium (Ca). The presence of these elements is attributed to chitosan and its bioactivity, which induces hydroxyapatite formation on its surface, as well as inorganic salts such as sodium, calcium, silicon, chlorine, and potassium inherent to the RPMI medium.
Notably, the only scaffolds with phosphorus (P) deposits are formulations F2 and F4, which contain NPs-ZnO. The presence of these NPs may promote the formation of hydroxyapatite (Ca5(PO4)3(OH)).73 Incorporating components capable of interacting or binding to biological tissues in the scaffolds enhances bioactivity. This, in turn, promotes osteoconduction (the process of bone tissue growth), osteointegration (firm attachment of scaffolds to surrounding bone tissue), osteoinduction (stimulation of immature cells' differentiation into osteogenic cells), and angiogenesis (increased development of blood vessels).74
Evaluation of the antimicrobial capacity and biofilm formation of NPs-CS and NPs-ZnO
Different Enterobacteriaceae strains were used because antimicrobial-resistant Gram-negative pathogens have been identified as an urgent threat due to their multiple resistance mechanisms.75 In Latin America, the prevalence of Enterobacterales with antibiotic resistance increased from 22% in 2015 to 32% in 2020.76 Additionally, Gram-negative bacteria are responsible for infection in the skin and soft tissue wounds, which is a possible application of nanoparticles. This study will also demonstrate a possible application of nanoparticles and the fact that in vivo studies can be carried out with specifically resistant strains. The ability to reduce and inhibit bacterial growth of NPs-CS and NPs-ZnO was analyzed by determining the minimum inhibitory concentration (MIC) value. This was established as the lowest concentration at which no turbidity or bacterial growth was observed against the biofilm formation of common Gram-negative and Gram-positive strains after 24 hours of incubation at 37 °C. Tables 3 and 4 summarize the results obtained for NPs-CS and NPs-ZnO, respectively.| Microorganism | CMI | CMB | Inhibition of biofilm formation (%) |
|---|---|---|---|
| a Results are expressed as means ± standard deviations. Different letters in the same column indicate significant differences (P < 0.05). | |||
| S. aureus ATCC 55804 | 0.625 | >10 | 0.0 ± 1.3 a |
| S. enterica ATCC 53648 | 0.625 | >10 | 89.3 ± 3.6 b |
| K. pneumoniae | 0.625 | >10 | 96.6 ± 2.8 c |
| E. cloacae sub cloacae | 0.625 | >10 | 97.3 ± 2.3 c |
| E. asburiae | 1.25 | >10 | 94.1 ± 4.5 c |
| E. hormaechei | 0.625 | >10 | 98.0 ± 2.0 c |
| E. ludwigii | 0.625 | >10 | 88.1 ± 2.3 b |
| Microorganism | CMI | CMB | Inhibition of biofilm formationa (%) |
|---|---|---|---|
| a Results are expressed as means ± standard deviations. Different letters in the same column indicate significant differences (P < 0.05). | |||
| S. aureus ATCC 55804 | 2.5 | >20 | 0.0 ± 1.5 a |
| S. enterica ATCC 53648 | >20 | >20 | 0.0 ± 1.5 a |
| K. pneumoniae | >20 | >20 | 30.1 ± 1.8 b |
| E. cloacae sub cloacae | >20 | >20 | 52.5 ± 2.4 c |
| E. hormaechei | >20 | >20 | 0.0 ± 5.1 a |
| E. asburiae | >20 | >20 | 0.0 ± 3.6 a |
| E. hormaechei | >20 | >20 | 28.5 ± 1.5 b |
| E. ludwigii | >20 | >20 | 0.0 ± 6.9 a |
The inhibition by NPs-CS against biofilm development in Gram-negative strains can be explained by the increased permeabilization of the membrane and neutralization of the lipopolysaccharides that comprise it.38 NPs-CS exhibit electrostatic interactions between the positively charged chitosan chains and the phosphoryl groups present in the bacterial membrane phospholipids, leading to gaps in the membrane and facilitating the release of intracellular material.77 Conversely, S. aureus, a Gram-positive bacterium, lacks an outer membrane, a distinctive feature of Gram-negative bacteria.39 This absence of an outer membrane could influence the observed lack of response or inhibition against NPs-CS.
Specifically, Gram-negative bacteria often develop resistance to oxidative stress during the growth phase by inducing a sigma factor known as RpoS.78 The presence of this factor may explain the low anti-biofilm inhibition of NPs-ZnO, as the presence of these nanoparticles has a high capacity to generate reactive oxygen species.
Scaffolds antimicrobial capacity
The formulations F1, F2, and F4 of the functionalized scaffolds exhibited high rates of antimicrobial activity against each of the four strains used. These results are consistent with the inherent antibacterial activity of chitosan. However, it is noteworthy that the efficacy against the E. cloacae strain was 100% for all formulations (Table 5).| Strain | F1 | F2 | F3 | F4 |
|---|---|---|---|---|
| a Results are expressed as means ± standard deviations. Different letters in the same row indicate significant differences (P < 0.05). | ||||
| K. pneumoniae | 100 ± 0.0 b | 99.94 ± 0.10 b | 96.50 ± 0.90 a | 99.972 ± 0.05 b |
| E. cloacae | 100 ± 0.0 a | 100 ± 0.0 a | 100 ± 0.0 a | 100 ± 0.0 a |
| S. enterica ATCC 53648 | 81.17 ± 16.36 b | 99.84 ± 0.14 c | 0.0 ± 0.0 a | 83.82 ± 8.88 b |
| S. aureus ATCC 55804 | 100 ± 0.0 a | 100 ± 0.0 a | 99.84 ± 0.21 a | 84.01 ± 16.04 a |
However, formulation F3 was less effective against the multidrug-resistant strain of S. enterica Typhimurium (ATCC® 53648™). Likely, the presence of antigens on the surface of this bacterial strain may have triggered adaptive response mechanisms to the solution enriched with the scaffold's properties. Indeed, the percentages of antimicrobial activity in the other formulations are lower, except for formulation F2, which only incorporates NPs-ZnO.
The mechanisms by which different types of nanoparticles can control or kill bacteria are unclear and may be explained by additive effects. For ZnO-based NPs, several pathways have been proposed, such as the toxicity of the Zn2+ ion from the dissolution of ZnO NPs, reactive oxygen species (ROS) generated from the photocatalytic process, and bacterial membrane dysfunction.79 For chitosan NPs, it is considered that the interaction of the amino group with the negative charges of the cell wall of microorganisms generates the lysis of these structures, which leads to the loss of protein compounds and other intracellular constituents. Furthermore, its chelating property allows it to selectively bind to metals in the external structures of microorganisms, thus inhibiting the production of toxins or enzymes due to the chitosan-DNA interaction that alters messenger RNA synthesis.80
Overall, the fact that scaffolds, especially those with incorporated NPs-ZnO, exhibit high antimicrobial activity against Gram-negative and Gram-positive bacterial species demonstrates their effectiveness against protective barriers within their cellular structure, indicating a broad spectrum against mixed infections and their potential uses as therapeutic materials.40 Moreover, their efficacy against multidrug-resistant bacteria supports the reduction of aggressive bacteria in hospital settings and even minimizes antibiotic use, significantly enhancing patient safety.41
In vitro cytotoxicity assays of the scaffolds
To understand the interactions between the scaffolds functionalized with the four formulations, three cell viability assays were conducted using Baby Hamster Kidney Fibroblasts (BHK-21) as the cell line in an RPMI culture medium containing the formulations for 4, 15, and 21 days. The results revealed significant differences in cell viability percentages among the formulations (Fig. 9). | ||
| Fig. 9 Cell viability of the four formulations (n = 3). | ||
Formulations F1 (2% CS) and F3 (1.9% CS:0.1% NPs-CS), which contained only chitosan as the base compound, exhibited a viability level greater than 96%, suggesting a favorable response in the cytocompatibility of BHK-21 cells to these formulations.
However, for formulations F2 (1.9% CS:0.1% NPs-ZnO) and F4 (1.9% CS:0.05% NPs-ZnO:0.05% NPs-CS), low cell viability values were recorded, suggesting that the presence of NPs-ZnO may be toxic to BHK-21 cells at these concentrations. These findings emphasize the importance of understanding the potential effects of components in functionalized scaffolds, especially when using sensitive cell lines.81,82
Fig. 9 displays the cell viability percentage results for the assays conducted at 4, 15, and 21 days. When comparing the means of the three formulations, no significant differences were found between the daily tests. However, there were considerable variations among the four formulations.
Analysis of the in vivo viability of the scaffolds
 | ||
| Fig. 10 The dorsal skin surface of a Wistar rat after 30 days of healing. (A) Dorsal area with hair; (B) dorsal area with trichotomy; (C) internal skin surface. IZ: implantation zone. | ||
The hair recovery on the skin of the biomodels after 30 days following the surgical procedure indicates successful skin healing. Hair regeneration is a positive sign that the skin is returning to its normal state and that biological functions are returning to normality. For this reason, it is used for the evaluation of dermatological treatments.83,84
Histological analysis
The formulation F1, which consists of 2% CS, reveals that after 30 days of implantation, healing progresses through a foreign body reaction, evidenced by the formation of a thin fibrous capsule surrounding the implantation site (Fig. 11A). Using Masson's trichrome and Gomori staining techniques, the capsule is identified to comprise type I collagen fibers, depicted in blue (Fig. 11A), and type III collagen fibers, illustrated in green (Fig. 11B). | ||
| Fig. 11 Subdermal tissue implanted with scaffolds at 30 days. (A) Sample F1, MT technique, image at 4× magnification. (B) Sample F1, GT technique, image at 4× magnification. (C) Sample F1, MT technique, image at 100× magnification. (D) Sample F2, HE technique, image at 4× magnification. (E) Sample F2, GT technique, image at 40× magnification. (F) Sample F2, HE technique, image at 100× magnification. (G) Sample F3, TG technique, image at 4× magnification. (H) Sample F3, GT technique, image at 100× magnification. (I) Sample F3, HE technique, image at 40× magnification. (J) Sample F4, HE technique, image at 4× magnification. (K) Sample F4, MT technique, image at 40× magnification. (L) Sample F4, HE technique, image at 100× magnification. D: Dermis. M: muscle. IZ: implantation zone. FC: fibrous capsule. Yellow arrow: material particles. Purple arrow: type III collagen fibers. Blue arrow: type I collagen fibers. Black arrow: inflammatory cells. Red circle: area of histological interest. Red star: inflammatory infiltrate. BV: blood vessel. | ||
The scaffold fragment obtained from formulation F1 is composed of a fibrillar structure. The interfibrillar spaces are filled with a type III collagen matrix (Fig. 11B) and numerous inflammatory cells (Fig. 11C). The scaffold's fibrillar structure shows minimal evidence of reabsorption/degradation.
The formulation F2 (1.9% CS/0.1% NPs-ZnO) represents a modification of the initial formulation, incorporating NPs-ZnO, which stimulated a more pronounced inflammatory response. Furthermore, the presence of a fibrous capsule persists, accompanied by an inflammatory process progressing centripetally from the implantation zone's periphery towards the defect's center (Fig. 11D). The inflammatory infiltrate is substantial, and as it progresses, it incorporates small fragments of the material for phagocytosis, as evidenced by the red circle in Fig. 11D. The advancing zone of the inflammatory process consists of a connective tissue matrix with numerous cells, as depicted in Fig. 11E. The presence of multinucleated cells in the interfibrillar spaces of the scaffold is also notable, as seen in the red circles in Fig. 11F. Unlike formulation, F1, the fibers composing the scaffold appear more irregular due to the effect of phagocytic cells, which can be observed in Fig. 11F.
In formulation F3 (1.9% CS:0.1% NPs-CS), the NPs-ZnO were replaced by NPs-CS; this alteration manifested in the scar response. At 30 days post-implantation, an inflammatory response was not as pronounced as in formulation F2. However, a thin fibrous capsule remains evident (Fig. 11G), along with a centripetal absorption pattern surrounding the scaffold fragment. Within this absorption zone, collagen fibers are discernible. Fig. 11H shows some type III collagen fibers, along blood vessels and inflammatory cells, are visible. In Fig. 11I, captured at a 40× magnification, the progression and absorption of the scaffold fragment by the inflammatory infiltrate can be distinctly observed.
In formulation F4 (1.9% CS:0.05% NPs-ZnO:0.05% NPs-CS), chitosan incorporates both NPs-ZnO and NPs-CS. However, as depicted in Fig. 11J, the pattern of centripetal absorption persists. A fragile fibrous capsule is evident, and within the absorption zone, a fibrillar matrix can be observed where scaffold fragments are incorporated (Fig. 11K). In Fig. 11L, numerous phagocytic cells are visible within the scaffold's interfibrillar spaces.
The scaffolds studied in this research were synthesized from chitosan, a product known for its significant biological properties such as biocompatibility, biodegradability/resorption, and the ability to interact with various cells and extracellular matrix.85 The histological results for formulation 1 are consistent with the healing of a biocompatible material implanted subdermally. The presence of a fibrous capsule is considered a normal response to implanted materials and serves as a control mechanism by the immune system to ensure that the healing process continues while the material is being reabsorbed/degraded.86
The capsule observed at 30 days is very small and appears to delineate the reabsorption zone (Fig. 11A). In formulation F2, there is a faster reabsorption and degradation of the material, possibly caused by increased hydrophilicity. This can be explained by the presence of ZnO NPs, which are also rich in –OH groups on their surface besides being biocompatible.87
In formulation F3, the incorporation of NPs-CS results in more controlled degradation and resorption of the scaffold because NPs-CS might act as initiation sites for degradation, affecting the rate at which enzymes, other agents, and biological factors such as lymphocytes, macrophages, and fibroblasts can access to break down the material and deposit the temporary matrix of connective tissue.88
In formulation, F4, both NPs-CS and NPs-ZnO were incorporated into chitosan. Their presence reduced the degradation rate of CS by providing a larger surface area, more initiation sites for degradation and reabsorption, and more sites for interaction with cells. Thus, the material's progressive degradation and reabsorption process occurred, accompanied by a moderate inflammatory infiltrate and the formation of an organized extracellular matrix rich in type I and III collagens in the reabsorption zone.
Overall, the porous chitosan matrices studied in this research induced a foreign body reaction-type inflammatory response characterized by a fragile fibrous capsule and a reabsorption process of the polymeric structure. This reabsorption was characterized by simultaneous degradation of the structure along with the deposition of connective tissue, similar to what has been reported by Popryaduhin, Yukina, Suslov, et al.89
Chitosan freeze-dried porous scaffolds have demonstrated biocompatibility in different regeneration tissue therapies. For example, improving fibroblast cell adhesion, proliferation, and mechanical reinforcement for hard tissues with pre-loaded hydroxyapatite and bioglass nanoparticles benefits hard tissue regeneration.90
On the other hand, chitosan-based nanocomposites have been used for wound healing due to their excellent antimicrobial, stimuli-responsiveness, and easy handling. Besides, incorporating several nanoparticles, including silver, iron, copper, and zinc hydroxyapatite, as fillers for wound dressing applications has demonstrated an improvement in the antimicrobial and regeneration capacity, indicating the potential of these nanocomposite scaffolds for tissue regenerative therapies.91–94 However, some metallic nanoparticles are concerned about their safety, limiting their use. Therefore, the results presented here incorporating NPs-CS for soft tissue regeneration in chitosan scaffolds are promising since they avoid using toxic nanoparticles while retaining mechanical, thermal, and antimicrobial reinforcement.
Conclusions
The research on chitosan scaffolds prepared by freeze-drying, incorporating NPs-CS and NPs-ZnO, has provided fundamental insights for their application in tissue regeneration. The size of the nanoparticles was effectively controlled, exhibiting monodisperse and homogeneous characteristics. These inclusions significantly impacted the mechanical properties of the scaffolds; for instance, NPs-ZnO reduced deformation, whereas NPs-CS enhanced Young's modulus, with formulation F3 standing out. Thermogravimetric and hydrolytic degradation analyses revealed distinct profiles across formulations, suggesting improved stability with NPs-ZnO. FTIR and XRD techniques confirmed chemical interactions and structural changes, validating the efficacy of freeze-drying in functionalization.Regarding antimicrobial properties, NPs-CS effectively inhibited Gram-negative bacteria but showed limitations against S. aureus. NPs-ZnO exhibited low inhibition due to specific responses of Gram-negative bacteria to oxidative stress. The scaffolds F1, F2, and F4 demonstrated overall high antimicrobial activity, but F3 showed constraints against a multidrug-resistant strain of S. enterica Typhimurium. The histological analysis reflected biocompatibility, showing controlled immunological responses and potential acceleration in degradation with NPs-ZnO. In summary, this study expands the boundaries in tissue engineering and regenerative medicine by highlighting the crucial role of nanoparticles in optimizing scaffold properties.
Author contributions
Formal analysis, Carlos David Grande-Tovar; investigation, Carlos David Grande-Tovar, Jorge Iván Castro, Jorge Eliecer Viloria Angarita and Carlos Humberto Valencia-Llano; methodology, Carlos David Grande-Tovar, Jorge Eliecer Viloria Angarita, Diana Paola Navia-Porras, Johannes Delgado-Ospina, Paula A. Zapata, and Carlos Humberto Valencia-Llano; project administration, Carlos David Grande-Tovar; resources, Juan David Rodríguez M, Daniel Insuasty and Alberto Albis; validation, Carlos David Grande-Tovar and Carlos Humberto Valencia-Llano; writing – original draft, Jorge Eliecer Viloria Angarita, Carlos David Grande-Tovar and Carlos Humberto Valencia-Llano; writing – review & editing, Carlos David Grande-Tovar and Jorge Eliecer Viloria Angarita.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors want to acknowledge Fondecyt Regular under Regular Project 1220093.References
- M. M. Islam, M. Shahruzzaman, S. Biswas, M. Nurus Sakib and T. U. Rashid, Bioact. Mater., 2020, 5, 164–183 Search PubMed.
- K. G. Nathan, K. Genasan and T. Kamarul, Mar. Drugs, 2023, 21, 1–18 CrossRef PubMed.
- S. Saravanan, R. S. Leena and N. Selvamurugan, Int. J. Biol. Macromol., 2016, 93, 1354–1365 CrossRef CAS PubMed.
- N. B. Shelke, R. James and C. T. Laurencin, Polym. Adv. Technol., 2014, 25, 448–460 CrossRef CAS.
- B. Dhandayuthapani, Y. Yoshida, T. Maekawa and D. S. Kumar, Int. J. Polym. Sci., 2011, 2011, 1–19 CrossRef.
- S. P. Soundarya, A. H. Menon, S. V. Chandran and N. Selvamurugan, Int. J. Biol. Macromol., 2018, 119, 1228–1239 CrossRef PubMed.
- S. Shankar, L.-F. Wang and J.-W. Rhim, Mater. Sci. Eng., C, 2018, 93, 289–298 CrossRef CAS PubMed.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS.
- L. L. Fernandes, C. X. Resende, D. S. Tavares, G. A. Soares, L. O. Castro and J. M. Granjeiro, Polimeros, 2011, 21, 1–6 CrossRef CAS.
- S. Ruiz, J. A. Tamayo, J. Delgado Ospina, D. P. Navia Porras, M. E. Valencia Zapata, J. H. Mina Hernandez, C. H. Valencia, F. Zuluaga and C. D. Grande Tovar, Biomolecules, 2019, 9, 109 CrossRef CAS PubMed.
- A. Ali and S. Ahmed, Int. J. Biol. Macromol., 2018, 109, 273–286 CrossRef CAS PubMed.
- S. Saravanan, R. S. Leena and N. Selvamurugan, Int. J. Biol. Macromol., 2016, 93, 1354–1365 CrossRef CAS PubMed.
- R. Jayakumar, R. Ramachandran, V. V. Divyarani, K. P. Chennazhi, H. Tamura and S. V. Nair, Int. J. Biol. Macromol., 2011, 48, 336–344 CrossRef CAS PubMed.
- S. Kunjachan, S. Jose and T. Lammers, Asian J. Pharm., 2010, 4, 148 CrossRef CAS.
- E. Y. Shaba, J. O. Jacob, J. O. Tijani and M. A. T. Suleiman, Appl. Water Sci., 2021, 11, 48 CrossRef CAS.
- B. R. Rizeq, N. N. Younes, K. Rasool and G. K. Nasrallah, Int. J. Mol. Sci., 2019, 20, 5776 CrossRef CAS PubMed.
- D. Mutukwa, R. T. Taziwa and L. Khotseng, Nanomaterials, 2022, 12, 4469 CrossRef CAS PubMed.
- C. D. Grande Tovar, J. I. Castro, C. H. Valencia, D. P. Navia Porras, J. H. Mina Hernández, M. E. Valencia, J. D. Velásquez and M. N. Chaur, Biomolecules, 2019, 9, 684 CrossRef PubMed.
- J. I. Castro, C. H. Valencia-Llano, M. E. Valencia Zapata, Y. J. Restrepo, J. H. Mina Hernandez, D. P. Navia-Porras, Y. Valencia, C. Valencia and C. D. Grande-Tovar, Polymers, 2021, 13, 3753 CrossRef CAS PubMed.
- A. Haider, S. Khan, D. N. Iqbal, M. Shrahili, S. Haider, K. Mohammad, A. Mohammad, M. Rizwan, Q. Kanwal and G. Mustafa, Eur. Polym. J., 2024, 112983 CrossRef CAS.
- Q. Ruan, L. Yuan, S. Gao, X. Ji, W. Shao, J. Ma and D. Jiang, Int. Wound J., 2023, 20, 1819–1831 CrossRef PubMed.
- A. Ramzan, A. Mehmood, R. Ashfaq, A. Andleeb, H. Butt, S. Zulfiqar, M. Nasir, A. Hasan, K. Khalid and M. Yar, Int. J. Biol. Macromol., 2023, 233, 123519 CrossRef CAS PubMed.
- T. Tayebi, A. Baradaran-Rafii, A. Hajifathali, A. Rahimpour, H. Zali, A. Shaabani and H. Niknejad, Sci. Rep., 2021, 11, 7060 CrossRef CAS PubMed.
- P. Calvo, C. Remunan-Lopez, J. L. Vila-Jato and M. J. Alonso, J. Appl. Polym. Sci., 1997, 63, 125–132 CrossRef CAS.
- F. Goycoolea, Monografías de la Real Academia Nacional de Farmacia, 2009 Search PubMed.
- A. Becheri, M. Dürr, P. Lo Nostro and P. Baglioni, J. Nanopart. Res., 2008, 10, 679–689 CrossRef CAS.
- C. D. Grande-Tovar, J. I. Castro, C. H. Valencia Llano, D. L. Tenorio, M. Saavedra, P. A. Zapata and M. N. Chaur, Pharmaceutics, 2022, 15, 43 CrossRef PubMed.
- F. Mohandes and M. Salavati-Niasari, RSC Adv., 2014, 4, 25993–26001 RSC.
- C. Valencia, C. H. Valencia, F. Zuluaga, M. E. Valencia, J. H. Mina and C. D. Grande-Tovar, Molecules, 2018, 23, 2651 CrossRef PubMed.
- E. R. Carmona, T. Plaza, G. Recio-Sánchez and J. Parodi, Rev. Invest. Vet. Perú, 2018, 29, 1195–1202 CrossRef.
- D. Asmat-Campos, D. Delfín-Narciso, L. Juárez-Cortijo, R. Nazario, R. Rengifo and E. Asmat, Proc. LACCEI Int. Multi-Conference Eng. Educ. Technol., 2019, 2019-July, 24–26 Search PubMed.
- I. Ijaz, A. Bukhari, E. Gilani, A. Nazir, H. Zain, R. Saeed, S. Hussain, T. Hussain, A. Bukhari, Y. Naseer and R. Aftab, Environ. Nanotechnol., Monit. Manage., 2022, 18, 100704 CAS.
- J. Carvajal De La Osa, A. Pacheco Leal, Y. N. Artíles, C. R. Figueroa Hernández, R. Gómez González, J. Luis, V. Santos, Y. S. Riverio, A. Valdés Martínez, T. R. Moliner, J. Prohías Martínez and Y. D. Roller, Rev. Soc. Cubana Ing., 2021, XII, 279 Search PubMed.
- M. Singh, D. Vadher, V. Dixit and C. Jariwala, Mater. Today: Proc., 2022, 48, 690–692 CAS.
- C. Fonseca, A. Ochoa, M. T. Ulloa, E. Alvarez, D. Canales and P. A. Zapata, Mater. Sci. Eng., C, 2015, 57, 314–320 CrossRef CAS PubMed.
- F. Maraver Eyzaguirre, I. Corvillo, L. Aguilera and F. Armijo, Bol. Soc. Esp. Hidrol. Med., 2005, 20, 43–47 Search PubMed.
- S. Sánchez-González, N. Diban and A. Urtiaga, Membranes, 2018, 8, 12 CrossRef PubMed.
- C. Rossi, F. Maggio, C. Chaves-López, L. Valbonetti, M. Berrettoni, A. Paparella and A. Serio, J. Appl. Microbiol., 2022, 132, 1866–1876 CrossRef CAS PubMed.
- C. Rossi, C. Chaves-López, S. S. Možina, C. Di Mattia, S. Scuota, I. Luzzi, T. Jenič, A. Paparella and A. Serio, Lebensm.-Wiss. Technol., 2019, 111, 16–22 CrossRef CAS.
- C. Rossi, A. Serio, C. Chaves-López, F. Anniballi, B. Auricchio, E. Goffredo, B. T. Cenci-Goga, F. Lista, S. Fillo and A. Paparella, Food Control, 2018, 86, 241–248 CrossRef CAS.
- Z.-C. Xing, W.-P. Chae, J.-Y. Baek, M.-J. Choi, Y. Jung and I.-K. Kang, Biomacromolecules, 2010, 11, 1248–1253 CrossRef CAS PubMed.
- I. Mitra, S. Mukherjee, P. B. Reddy Venkata, S. Dasgupta, C. K. Jagadeesh Bose, S. Mukherjee, W. Linert and S. C. Moi, RSC Adv., 2016, 6, 76600–76613 RSC.
- Á. M. Villegas, E. Naranjo and D. M. Gómez, In Vivo, 2008, 16, 38–44 Search PubMed.
- AEN/CTN, Evaluación biológica de los productos sanitarios Parte: Parte 6: Ensayos relativos a los efectos locales después de la implantación, 1995, vol. 51 Search PubMed.
- R. C. Hubrecht and E. Carter, Animals, 2019, 9, 754 CrossRef PubMed.
- N. P. Du Sert, V. Hurst, A. Ahluwalia, S. Alam, M. T. Avey, M. Baker, W. J. Browne, A. Clark, I. C. Cuthill, U. Dirnagl, M. Emerson, P. Garner, S. T. Holgate, D. W. Howells, N. A. Karp, S. E. Lazic, K. Lidster, C. J. MacCallum, M. Macleod, E. J. Pearl, O. H. Petersen, F. Rawle, P. Reynolds, K. Rooney, E. S. Sena, S. D. Silberberg, T. Steckler and H. Würbel, PLoS Biol., 2020, 18, 1–12 Search PubMed.
- Biological evaluation of medical devices - Part 6: Tests for local effects after implantation, UNE-EN ISO 10993-6:2017, 2017 Search PubMed.
- C. Pan, J. Qian, C. Zhao, H. Yang, X. Zhao and H. Guo, Carbohydr. Polym., 2020, 241, 116349 CrossRef CAS PubMed.
- A. Anitha, V. V. Divya Rani, R. Krishna, V. Sreeja, N. Selvamurugan, S. V. Nair, H. Tamura and R. Jayakumar, Carbohydr. Polym., 2009, 78, 672–677 CrossRef CAS.
- J. Vera, W. Herrera, E. Hermosilla, M. Díaz, J. Parada, A. B. Seabra, G. Tortella, H. Pesenti, G. Ciudad and O. Rubilar, Antioxidants, 2023, 12, 784 CrossRef CAS PubMed.
- A. Patricia, H. Barros, M. Teresa, A. Morantes, M. Ignacio Castro Hoyos, L. José and M. Ospino, Rev. EIA, 2016, 12, E75–E83 Search PubMed.
- H. Noei, H. Qiu, Y. Wang, E. Löffler, C. Wöll and M. Muhler, Phys. Chem. Chem. Phys., 2008, 10, 7092–7097 RSC.
- S. Haq, M. B. Ali, A. Mezni, A. Hedfi, W. Rehman, G. Sarwar, Z. Ul-abdin, S. U. Din, F. U. Rehman, S. A. Abbasi and A. L. Lone, Dig. J. Nanomater. Biostruct., 2022, 17, 499–505 CrossRef.
- N. M. Shamhari, B. S. Wee, S. F. Chin and K. Y. Kok, Acta Chim. Slov., 2018, 65, 578–585 CrossRef CAS PubMed.
- R. Zanella, Mundo Nano. Rev. Interdiscip. en Nanociencia y Nanotecnología, 2012, 5, 69–81 Search PubMed.
- D. Zhou and A. A. Keller, Water Res., 2010, 44, 2948–2956 CrossRef CAS PubMed.
- J. Kumirska, M. Czerwicka, Z. Kaczyński, A. Bychowska, K. Brzozowski, J. Thöming and P. Stepnowski, Mar. Drugs, 2010, 8, 1567–1636 CrossRef CAS PubMed.
- A. Guedri, M. Zaabat, B. Boudine and A. Hafdallah, J. Inorg. Organomet. Polym. Mater., 2020, 30, 4884–4894 CrossRef CAS.
- A. Moulahi, Egypt. J. Chem., 2021, 64, 6147–6154 Search PubMed.
- M. Achehboune, M. Khenfouch, I. Boukhoubza, L. Leontie, C. Doroftei, A. Carlescu, G. Bulai, B. Mothudi, I. Zorkani and A. Jorio, Mater. Today: Proc., 2021, 53, 319–323 Search PubMed.
- J. Cai, Y. Li, H. Wang, C. Ma, J. Luo and P. Zhang, NDT E Int., 2021, 121, 102461 CrossRef CAS.
- Y. C. Cárdena-Pérez, R. Vera-Graziano, E. d. J. Muñoz-Prieto and E. Y. Gómez-Pachón, Ing. Compet., 2017, 19, 134 Search PubMed.
- C. Qiao, X. Ma, X. Wang and L. Liu, Lebensm.-Wiss. Technol., 2021, 135, 109984 CrossRef CAS.
- A. Z. Bazmandeh, E. Mirzaei, M. Fadaie, S. Shirian and Y. Ghasemi, Int. J. Biol. Macromol., 2020, 162, 359–373 CrossRef CAS PubMed.
- I. Corazzari, R. Nisticò, F. Turci, M. G. Faga, F. Franzoso, S. Tabasso and G. Magnacca, Polym. Degrad. Stab., 2015, 112, 1–9 CrossRef CAS.
- J. Lamarra, S. Rivero, L. Damonte and A. Pinotti, Adv. Mater. Sci. Eng., 2018, 3965783 Search PubMed.
- E. Prokhorov, J. G. Luna-Bárcenas, J. B. González-Campos and I. C. Sanchez, Mol. Cryst. Liq. Cryst., 2011, 536, 24 CAS /[256]–32/[264]..
- P. Jithendra, A. M. Rajam, T. Kalaivani, A. B. Mandal and C. Rose, ACS Appl. Mater. Interfaces, 2013, 5, 7291–7298 CrossRef CAS PubMed.
- M. Haghbin, J. Esmaeilzadeh and S. Kahrobaee, Macromol. Res., 2020, 28, 1232–1240 CrossRef CAS.
- R. Zhang and P. X. Ma, J. Biomed. Mater. Res., 1999, 45, 285–293 CrossRef CAS PubMed.
- X. Dong, Q. Cheng, Y. Long, C. Xu, H. Fang, Y. Chen and H. Dai, Polym. Degrad. Stab., 2020, 181, 109322 CrossRef CAS.
- Y. Tian, H. Zhang and Z. Zhang, Composites, Part A, 2017, 98, 1–8 CrossRef CAS.
- M. E. Álvarez-Monsiváis, B. R. Cruz-Ortiz, I. Olivas-Armendáriz, A. Sáenz-Galindo, S. G. Solís-Rosales and E. M. Múzquiz-Ramos, Afinidad, 2022, 79, 61–69 Search PubMed.
- G. Turnbull, J. Clarke, F. Picard, P. Riches, L. Jia, F. Han, B. Li and W. Shu, Bioact. Mater., 2018, 3, 278–314 Search PubMed.
- E. V. Lemos-Luengas, S. Rentería-Valoyes, D. M. A. Muñoz, C. K. G. Gonzalez, G. Guerrón-Gómez and J. A. Ramos-Castaneda, Diagn. Microbiol. Infect. Dis., 2024, 109, 116235 CrossRef CAS PubMed.
- M. G. Wise, J. A. Karlowsky, E. V Lemos-Luengas, R. R. Valdez and D. F. Sahm, Braz. J. Infect. Dis., 2023, 27, 102759 CrossRef CAS PubMed.
- Y. C. Chung and C. Y. Chen, Bioresour. Technol., 2008, 99, 2806–2814 CrossRef CAS PubMed.
- P. Stoodley, K. Sauer, D. G. Davies and J. W. Costerton, Annu. Rev. Microbiol., 2002, 56, 187–209 CrossRef CAS PubMed.
- A. Karami, Z. Xie, J. Zhang, M. S. Kabir, P. Munroe, S. Kidd and H. Zhang, Mater. Sci. Eng., C, 2020, 107, 110220 CrossRef CAS PubMed.
- G. Ayala Valencia, Sci. Agroaliment., 2015, 2, 32–38 Search PubMed.
- R. Mohammadinejad, M. A. Moosavi, S. Tavakol, D. Ö. Vardar, A. Hosseini, M. Rahmati, L. Dini, S. Hussain, A. Mandegary and D. J. Klionsky, Autophagy, 2019, 15, 4–33 CrossRef CAS PubMed.
- S. Singh, Toxicol. Mech. Methods, 2019, 29, 300–311 CrossRef CAS PubMed.
- Z. H. Yang, L. J. Gu, D. L. Zhang, Z. Li, J. J. Li, M. R. Lee, C. Y. Wang, Z. Wang, J. H. Cho and C. Sung, Asian-Australas. J. Anim. Sci., 2012, 25, 708–716 CrossRef PubMed.
- M. S. Orasan, I. I. Roman, A. Coneac, A. Muresan and R. I. Orasan, Clujul Med., 2016, 89, 327–334 Search PubMed.
- A. D. Gholap, S. Rojekar, H. S. Kapare, N. Vishwakarma, S. Raikwar, A. Garkal, T. A. Mehta, H. Jadhav, M. K. Prajapat and U. Annapure, Carbohydr. Polym., 2023, 323, 121394 CrossRef PubMed.
- M. R. Major, V. W. Wong, E. R. Nelson, M. T. Longaker and G. C. Gurtner, Plast. Reconstr. Surg., 2015, 135, 1489–1498 CrossRef CAS PubMed.
- S. Rashki, E. A. Dawi, M. R. Zilaei, H. Safardoust-Hojaghan, M. Ghanbari, A. Ryadh, H. A. Lafta, A. Khaledi and M. Salavati-Niasari, Int. J. Biol. Macromol., 2023, 253, 127583 CrossRef CAS PubMed.
- D. D. Zhao, S. Yu, B. Sun, S. Gao, S. Guo and K. Zhao, Polymers, 2018, 10, 462 CrossRef PubMed.
- P. V. Popryaduhin, G. Y. Yukina, D. N. Suslov, I. P. Dobrovolskaya, E. M. Ivankova and V. E. Yudin, Tsitologiia, 2016, 58, 701–777 Search PubMed.
- P. Kumar, M. Saini, B. S. Dehiya, A. Umar, A. Sindhu, H. Mohammed, Y. Al-Hadeethi and Z. Guo, Int. J. Biol. Macromol., 2020, 149, 1–10 CrossRef CAS PubMed.
- A. Mohandas, B. S. Anisha, K. P. Chennazhi and R. Jayakumar, Colloids Surf., B, 2015, 127, 105–113 CrossRef CAS PubMed.
- B. Lu, F. Lu, Y. Zou, J. Liu, B. Rong, Z. Li, F. Dai, D. Wu and G. Lan, Carbohydr. Polym., 2017, 173, 556–565 CrossRef CAS PubMed.
- X. Huang, X. Bao, Z. Wang and Q. Hu, RSC Adv., 2017, 7, 34655–34663 RSC.
- W. Shao, J. Wu, S. Wang, M. Huang, X. Liu and R. Zhang, Carbohydr. Polym., 2017, 157, 1963–1970 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00371c |
| This journal is © The Royal Society of Chemistry 2024 |