
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlycerol carbonate synthesis via transesterification of enriched glycerol and dimethyl carbonate using a Li-incorporated MCM-41 framework
Jakkrapong
Jitjamnong
a,
Parinya
Khongprom
 ab,
Thanate
Ratanawilai
c and
Sukritthira
Ratanawilai
*a
ab,
Thanate
Ratanawilai
c and
Sukritthira
Ratanawilai
*a
aDepartment of Chemical Engineering, Faculty of Engineering, Prince of Songkla University, Hat Yai, Songkhla 90110, Thailand. E-mail: sukritthira.r@psu.ac.th
bAir Pollution and Health Effect Research Center, Prince of Songkla University, Songkhla, 90110, Thailand
cDepartment of Industrial and Manufacturing Engineering, Faculty of Engineering, Prince of Songkla University, Hat Yai, Songkhla 90110, Thailand
First published on 19th February 2024
Abstract
Waste crude glycerol was successfully enriched and utilized as an inexpensive source for producing value-added chemicals, such as glycerol carbonate (GC) – a valuable compound with extensive industrial applications. The Li/MCM-41 heterogeneous catalyst was synthesized and used for the transesterification of enriched glycerol and dimethyl carbonate (DMC) to produce GC. The catalyst's physicochemical properties were characterized using thermogravimetric, Hammett indicator, inductively coupled plasma-optical emission spectroscopy, nitrogen adsorption–desorption, X-ray diffractometry, scanning electron microscopy, and Fourier-transform infrared spectroscopy analyses. Reaction conditions were optimized using response surface methodology and analysis of variance, yielding an accurate quadratic model to predict the GC yield under different transesterification variables. The results revealed that 5%Li/MCM-41 served as the optimal catalyst, achieving the highest TOF of 4.72 h−1. The DMC: enriched glycerol molar ratio had the greatest impact on the GC yield, with an R2 = 0.9743 and adjusted R2 = 0.9502. The optimal GC yield (58.77%) with a final purity of 78% was attained at a 5.15 wt% catalyst loading relative to the initial amount of enriched glycerol, DMC: enriched glycerol molar ratio of 4.24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and a reaction temperature of 86 °C for 165 min. The 5%Li/MCM-41 heterogeneous catalyst could be reused for four cycles with a decreased GC yield from 58.77% to 45.72%. Thus, the Li/MCM-41 catalyst demonstrated a remarkable efficiency and potential as a heterogeneous catalyst for synthesizing GC. This method not only contributes to environmental sustainability by making use of a byproduct from biodiesel production but also aligns with the principles of a circular economy.
:1, and a reaction temperature of 86 °C for 165 min. The 5%Li/MCM-41 heterogeneous catalyst could be reused for four cycles with a decreased GC yield from 58.77% to 45.72%. Thus, the Li/MCM-41 catalyst demonstrated a remarkable efficiency and potential as a heterogeneous catalyst for synthesizing GC. This method not only contributes to environmental sustainability by making use of a byproduct from biodiesel production but also aligns with the principles of a circular economy.
1. Introduction
The world is grappling with the predicament of global warming, resulting principally from the combustion of fossil fuels, such as petroleum, natural gas, and coal, that result in the net release and accumulation of more greenhouse gases (GHGs), hydrocarbons, and particulate matter in the atmosphere that contribute to climate change.1,2 Responding to these concerns, in common with many other nations, the Royal Thai government has recently unveiled plans for its Bio-Circular-Green Economic Model aimed at fostering sustainable and inclusive growth and curtailing GHG emissions. Capitalizing on Thailand's strengths, this government policy has established a renewable energy target, particularly for biodiesel, aspiring to reach 30% of the total final energy consumption by 2036.3 Biodiesel emerges as a fitting solution to these challenges, being an eco-friendly biofuel capable of substantially mitigating GHG emissions while ensuring an energy supply for future generations.4 The clamor for biodiesel as a renewable biofuel has resulted in a marked global escalation in its production, with global biodiesel production projected to attain 23.57 billion liters by 2025.5Crude glycerol, a prominent by-product of biodiesel production from alcohol transesterification with vegetable oils, animal fats, algae, and recycled restaurant grease, constitutes approximately 10 wt% of total biodiesel production and is commonly discarded as industrial waste.6–8 By 2025, the cumulative volume of crude glycerol is estimated to surpass an impressive 2.4 billion liters, presenting an enormous reservoir brimming with potential for refinement and conversion into value-added products. Consequently, the development of sustainable techniques for harnessing this cost-effective organic raw material becomes imperative.5 From an economic standpoint, this glut of crude glycerol has reduced its value and so it becomes crucial to efficiently utilize crude glycerol via its conversion to other value-added chemical products, thereby curtailing the level of waste generated during the biodiesel manufacturing process and fostering the economically and environmentally sustainable advancement of bio-refineries.
Crude glycerol derived from the biodiesel production process is typically comprised of approximately 50% or less glycerol, accompanied by a substantial quantity of methanol, water, fatty acid salts (soap), fatty acid methyl esters (FAMEs), free fatty acids (FFAs), and ash content.5 The presence of ash is attributed to catalyst residues and impurities originating from the oil during chemical reactions. The specific impurities found in waste crude glycerol are contingent upon the feedstock (natural oil sources and alcohol) utilized during the biodiesel production. Crude glycerol can be refined and distilled to yield pure glycerol. The general enrichment process for crude glycerol is comprised of three primary steps. Firstly, the removal of metals and soap occurs through a precipitation process during acidification, leading to the formation of metal salts and conversion of soap into FFAs. In cases where a base catalyst is utilized in the transesterification reactions, acid treatment is employed, whereas in reactions involving acid catalysts, alkaline treatment is applied. The excess alcohol in the crude glycerol mixture is then evaporated, refining the glycerol stream and enhancing the glycerol purity to around 95% (w/w).9,10 A glycerol purity of approximately 93.7% was obtained from crude glycerol through a process of acidification, solvent extraction, and purification from crude glycerol, but byproducts, such as water, glycerides, and ash, were also obtained.11 Similarly, a glycerol purity of roughly 93.34% was obtained using sulphuric acid (H2SO4) acidification, sodium hydroxide (NaOH) neutralization, solvent extraction, and enrichment, resulting in low levels of contaminants.12 The sequential saponification, acidification, neutralization, solvent extraction, and purification of crude glycerol led to glycerol with a 97.5% purity and minimal residual FFA, water, and ash contents.13
Glycerol serves as a prominent renewable raw material within the chemical industry, boasting a broad spectrum of applications as a raw material and an active solvent. Notably, glycerol carbonate (GC) has emerged as a substance of significant interest, particularly within the pharmaceutical and beauty sectors, where its value is greatly augmented by achieving high levels of purity.14,15 Furthermore, glycerol plays a pivotal role in various other domains. It finds extensive usage as an electrolyte in lithium (Li) and Li-ion batteries, as well as in applications like gas separation membranes, solvents, detergent compositions, chemical intermediates, polymers, and in constructing eco-composites.15–19
Synthesis of GC can be achieved through various chemical routes, including the transesterification reaction of glycerol with dimethyl carbonate (DMC),1,4,17,18 carbon monoxide (CO),20,21 carbon dioxide (CO2),22,23 phosgene, or urea.24–26 However, several challenges and limitations are associated with these routes. For CO, its toxicity, environmental pollution, catalyst poisoning, and explosive nature restrict its suitability as a reagent. The use of CO2 suffers from difficulties in breaking the carbon–oxygen double bond in CO2, resulting in low GC yields and relatively high production costs. The use of phosgene as a reagent poses hazards due to its high toxicity, corrosiveness, and pollution potential, making this route highly constrained. The reaction with urea necessitates expensive equipment due to the requirement for vacuum conditions for the continuous removal of the formed ammonia in order to shift the thermodynamic equilibrium. Consequently, this route yields low GC yields.27 A promising pathway involves the transesterification reaction of glycerol with DMC using either homogeneous or heterogeneous catalysts. This pathway is advantageous due to its mild reaction conditions and the ease of product separation. However, homogeneous catalysts pose challenges in terms of catalyst separation, purification, and reusability. Therefore, the utilization of heterogeneous catalysts shows more promise in driving more economical GC production, primarily due to their potential for reusability.
Catalysts tailored for the transesterification reaction are primarily comprised of inorganic bases with alkali or alkaline earth metals as the active species. The catalyst's surface contains basic sites responsible for initiating the transesterification process. Catalysts functionalized with metals, such as potassium (K),28,29 sodium (Na),30 Li,6,29 magnesium (Mg),28,29 strontium (Sr),28 and zirconium,22 exhibit an enhanced performance in GC production.
Mobil Composition of Matter no. 41 (MCM-41) has garnered considerable attention in the field of materials science. MCM-41 stands out for its uniform hexagonal straight channel structure, controlled mesoporous pore size (ranging from 1.5 to 20 nm), substantial pore volume (exceeding 0.6 cm3 g−1), high surface area (approximately 700–1500 m2 g−1), and exceptional hydrothermal, chemical, and mechanical stability. Consequently, MCM-41 finds wide-ranging applications, particularly in catalytic conversions involving large molecules. Nonetheless, siliceous MCM-41 lacks a sufficient number of basic sites. To address this, the catalytic potential of siliceous materials can be readily modified by introducing heteroatoms into the silicate framework, thereby enhancing their catalytic prowess within confined spaces.31 For instance, Li impregnation onto mesoporous MCM-41 enhanced its catalytic activity. They achieved a glycerol conversion level of 99% with a GC yield of 93% when maintaining a glycerol: DMC molar ratio of 3:1, a catalyst loading of 13 wt%, and a temperature of 90 °C for 3 h.6 Cobalt oxide (Co3O4) supported on MCM-41 as a catalyst for the synthesis of GC via transesterification of glycerol and DMC gave a glycerol conversion level of 98.7% and GC yield of 94.1% under the optimized conditions of a DMC: glycerol molar ratio of 3:1, a catalyst dosage of 6 wt%, and a reaction time of 2 h at 90 °C.32 The efficacy of several active metals, such as Li, K, and barium (Ba), doped onto ash was shown for catalyzing GC synthesis via transesterification.29 In that study the active metals were doped at varying weight percentages using a straightforward wet impregnation method. Likewise, evaluation of the impact of K, Sr, and Mg on the catalytic stability of red mud in GC synthesis revealed that a 30 wt% K loading and calcination at 800 °C resulted in the most favorable catalytic performance due to the maximal surface concentration of active K2O.28
The synthesis process of glycerol carbonate is known for its high energy consumption and significant costs, necessitating optimization through the application of appropriate statistical tools. Specifically, the identification of optimal reaction parameters to maximize glycerol carbonate yield is crucial. Surprisingly, the utilization of response surface methodology (RSM) via central composite design (CCD), a statistical tool, has not been documented in existing research to the best of our knowledge. Hence, the implementation of RSM becomes imperative for optimizing the reaction factors and achieving a higher yield of glycerol carbonate. RSM techniques offer the advantage of identifying the true optimum point while accounting for interactions between factors, all while requiring fewer experimental runs. This approach has proven successful in numerous coagulation studies. Therefore, employing RSM in the optimization of glycerol carbonate synthesis can not only enhance efficiency but also provide valuable insights into the intricate relationships among reaction parameters, ultimately contributing to improved yields and cost-effectiveness.33–36
In the present work, crude glycerol was first enriched and then the enriched glycerol was used as a starting reactant for GC synthesis. The active species were incorporated onto the MCM-41 framework via impregnation. The research focused on investigating the impact of different types of active species (Li, Na, K, and Ba) loaded onto the MCM-41 support, with the goal of identifying the variant that produced the highest GC yield. Subsequently, the chosen catalyst was employed to optimize the transesterification reaction conditions via varying the catalyst loading (2–6 wt% relative to the initial amount of enriched glycerol), DMC: enriched glycerol molar ratio (1–5:1), reaction temperature (70–90 °C), and reaction time (60–180 min) using RSM based on a CCD. The reusability of the catalyst was also evaluated.
2. Materials and methods
2.1 Materials
The crude glycerol was supplied from the specialized research and development center for alternative energy from palm oil and oil crops, Prince of Songkla University, Thailand. Cetyl trimethylammonium bromide (CTAB; ≥98%) as the directing agent, tetraethyl orthosilicate (TEOS; ≥99%) as the silica source, and ammonium hydroxide solution as the mineralizing agent, all used in the synthesis of the MCM-41 solid catalyst support, were provided from KemAus Co. Ltd, Sigma-Aldrich Co. Ltd, and Macron Fine Chemicals Co. Ltd, respectively. Lithium nitrate (LiNO3; ≥98%) and barium nitrate [Ba(NO3)2; ≥99%], sodium nitrate (NaNO3; ≥99%); and potassium nitrate (KNO3; ≥99%), used as the Li, Ba, Na, and K precursors, respectively, were purchased from HiMedia Laboratories Pvt. Ltd, Loba Chemie Pvt. Ltd, and Kemaus Co. Ltd, respectively. Absolute ethanol (≥99.9%) and H2SO4 (≥98%) were acquired from Anhui Fulltime Specialized Solvent & Reagent Co. Ltd, China and Qrec Co. Ltd, New Zealand, while NaOH (99%) as a neutralizing agent was obtained from Pine Chemical Co. Ltd Hexane (C6H14; analytical reagent grade) as a accelerate agent for precipitation of salts was purchased from Macron Fine Chemicals. The DMC (C3H6O3; ≥99%) employed as a starting material for transesterification was obtained from Thermo Fisher Scientific Co. Ltd, China. The GC standard (C4H6O4; ≥90%) used for calibration in the gas chromatography was purchased from Tokyo Chemical Industry Co. Ltd, Japan while, iso-propanal [(CH3)2CHOH; 99.5%] and glycerol (≥99.5%) were supplied from KemAus Co. Ltd, Australia. Other chemical reagents, including bromothymol blue, phenolphthalein, 2,4-dinitroaniline, and 4-nitroaniline were used as indicators. Bromothymol blue and phenolphthalein were obtained from KemAus Co. Ltd, and Ajx Finechem Pty Ltd, while 2,4-dinitroaniline, and 4-nitroaniline were from Sigma-Aldrich Co. Ltd, USA. Benzoic acid powder (analytical reagent grade; C7H6O2) applied as a standard reagent for titration, was obtained from KemAus Co. Ltd All the chemicals in this study were used without purification. Distillated water was used to prepare chemical solutions.2.2 Crude glycerol enrichment
Initially, the crude glycerol, which had a dark brown hue (Fig. 1), was gently heated on a hot plate at 60 °C for 30 min. Subsequently, acidification was employed to expedite the precipitation of salts and FFA present in the crude glycerol. Acidification was performed by the addition of 1 M H2SO4 with stirring at 350 rpm until the pH was reduced to approximately 2–4. The resultant mixture was then transferred to a separator funnel for gravity-assisted sedimentation and allowed to stand for 48 h. Following this period, the three distinct layers that formed were separated – an upper layer of matter organic non-glycerol (MONG), a lower phase of inorganic salts, and a middle glycerol layer (Fig. 1a). The glycerol-rich phase was harvested, neutralized to pH 7.0 using 5 M NaOH, and left for 2 h before being filtered to remove the precipitated salt. Subsequently, hexane was added to the glycerol in an equal volume to accelerate salt precipitation. After stirring this solution for 30 min, it was transferred to a separatory funnel and left to separate into two distinct phases over a 2 h period. The lower phase containing crystallized salts was removed, while the upper glycerol-rich phase was retained. A 1.2 μm glass microfiber filter grade GF/C was employed to remove any remaining inorganic salts. To address the dark brown color of the obtained glycerol, activated carbon was introduced, and the mixture was stirred at 350 rpm for 3 h and then filtered to remove the activated carbon from the solution, yielding the enriched glycerol. | ||
| Fig. 1 (a) Three distinct layers of crude glycerol purification, (b) crude glycerol (c) enriched glycerol before activated carbon treatment, and (d) purified glycerol. | ||
2.3 Physical characterization of the crude and enriched glycerol
The water content of both the crude and enriched glycerol samples was determined using a Karl Fischer Coulometer (Mettler Toledo DL39 model). In this procedure, 5 mL of the sample was introduced using a syringe. The weight of the sample was measured by recording the syringe's weight before and after injection. Subsequently, the titration process was initiated.For pH measurement, 1.00 ± 0.1 g of the sample was dissolved in 50 mL of deionized water and the pH value was assessed at ambient temperature using a digital pH meter (pH 150 Eutech model), which had been calibrated using buffer solutions of pH 4 and 7.37,38
The viscosity of the prepared samples was analyzed using a digital viscometer (LVDV-I Prime Model, Brookfield, USA) at 25 ± 0.5 °C.37,38
Ash content of the crude and enriched glycerol was measured by burning 1 g of sample in a muffle furnace at 750 °C for 3 h following ISO 2098-1972.37,38 The ash content was then calculated from eqn (1):
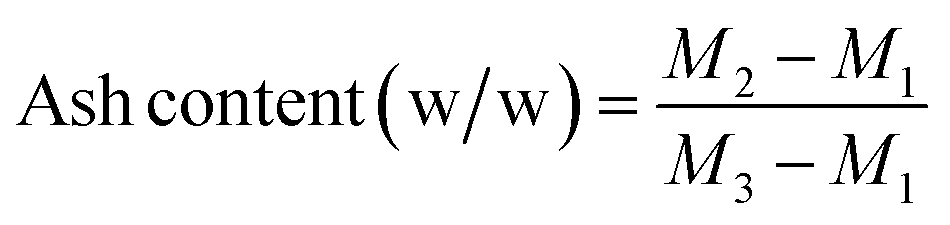 | (1) |
The enriched glycerol content was determined using the titration method. Initially, the sample (0.5 g) was dissolved in distilled water to achieve a solution volume of 50 mL in a 500 mL Erlenmeyer flask. Approximately 5–6 droplets of bromothymol blue were added to the solution and the pH was adjusted to 2–4 by the addition of H2SO4 (1 M). Subsequently, the solution was neutralized (turns yellow) by the addition of NaOH (0.05 M). Distilled water was utilized as a blank sample for reference. Following this, 50 mL of sodium metaperiodate was introduced, and the resulting mixture was allowed to age in darkness for 30 min. Next, 10 mL of a 1:1 (w/w) ratio mixture of ethylene glycol: distilled water was added and kept in darkness for 20 min,39,40 and then 300 mL of distilled water was added to this solution. Finally, NaOH (0.125 M) was slowly added drop by drop until the solution color changed from yellow to blue. The enriched glycerol content was then calculated using eqn (2):
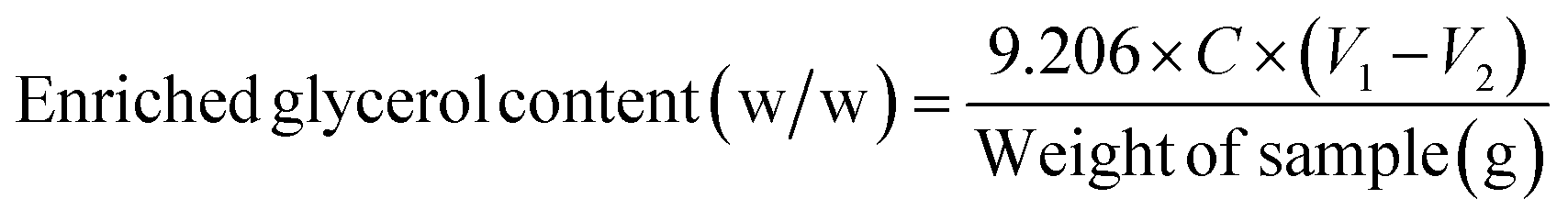 | (2) |
2.4 Catalyst preparation
The mesoporous silica MCM-41 support was synthesized using CTAB and TEOS as surface directing agents.6 In this synthesis, 4.8 g of CTAB was added to 240 g of distilled water under continuous stirring. Next, 16 mL of concentrated ammonium hydroxide solution was incorporated into the mixture solution and stirred for 5 min. To this solution, 40 mL of TEOS was gradually introduced and stirred at ambient temperature for 12 h, during which time the white material gradually precipitated. This was then filtered and thoroughly washed multiple times using ethanol and distilled water until the eluate had a pH of approximately 7. Subsequently, the white powder was dried in a hot air oven at 105 °C overnight and then calcined in a muffle furnace at 550 °C for 5 h at a heating rate of 5 °C min−1. This process yielded the MCM-41 support, which was stored in a desiccator for subsequent use.A series of MCM-41 supports doped with various metal types (Li, Na, K, and Ba) and metal loadings were prepared through incipient wetness impregnation. For instance, in the preparation of 5%Li/MCM-41, approximately 0.0805 g of the LiNO3 precursor was dissolved in 7.5 mL of distilled water and used to impregnate 1.5 g of dried MCM-41 support at ambient temperature for 24 h. The Li-impregnated MCM-41 sample was then dried at 105 °C overnight. Prior to the activity test in the transesterification reaction, the catalysts underwent calcination in a muffle furnace at 450 °C for 3 h at a heating rate of 5 °C min−1. A similar procedure was followed for the different metal types and metal loading levels (2–6 wt% relative to the initial amount of enriched glycerol), with the synthesized catalysts being designated as n%M/MCM-41, where n% represents the wt% of the loaded metal relative to the amount of metal and MCM-41 and M designates the type of metal.
2.5 Catalyst characterization
The crystalline structure of the prepared catalysts was identified using a wide-angle X-ray diffractometry (XRD; Panalytical brand, Empyrean model, Netherlands). The XRD patterns were obtained utilizing a 2.2 kW Cu anode that generated CuK-alpha radiation (1.5405 Å) as the radiation source. The X-ray tube was operated at a voltage of 40 kV and a current of 30 mA. The scanning was conducted over a 2θ range spanning from 10° to 90°, with a scan speed of 5° per min and a scan step of 0.02. The XRD profiles were compared to standard reference files.The functional groups present in the samples were analyzed through Fourier-transform infrared spectroscopy (FTIR; Bruker Vertex70, Germany). The 1 mg of catalyst powder was mixed, pelleted with 99 mg of KBr powder, and compressed into a disk form. The FTIR spectrum was recorded across a wavenumber range of 4000–400 cm−1 with a resolution of 4 cm−1 in absorbance mode, and 32 scans were performed.
The morphology of the prepared catalyst was visualized using scanning electron microscopy (SEM; Apreo Model), with an accelerating voltage of 2.0 kV. Each sample was positioned on a stub, sputtered, and then placed in the SEM machine's sample holder. Images were acquired at a magnification of 50000×.
The thermal stability and decomposition properties of both the synthesized MCM-41 support and 5%Li/MCM-41 were evaluated using thermal gravimetric analysis (TGA; PerkinElmer, TGA8000 model, USA). Approximately 4–10 mg of the prepared sample was loaded into a platinum pan, and the analysis was conducted under a nitrogen (N2) atmosphere at a heating rate of 10 °C per minute, spanning a temperature range of 30 to 900 °C.
The surface area and pore size distribution of the prepared catalyst were characterized using N2 adsorption–desorption surface area analysis (ASAP2060 model, Micromeritics brand, USA). Prior to the test, any adsorbed water and volatiles on the sample surface were removed by heating the sample to 250 °C overnight under vacuum conditions. The Brunauer–Emmett–Teller (BET) method was used to determine the surface area (SBET) and the total pore volume (Vp), which was measured up to a relative pressure (P/P0) of 0.99.
The actual metal loading levels on the MCM-41 catalyst were characterized using inductively coupled plasma-optical emission spectrometry (ICP-OES; PerkinElmer AVIO 500, USA). The prepared catalysts were digested in an aqua regia solution and subsequently diluted prior to characterization. The method was calibrated using known concentration standards, and the actual concentration values were then calculated from the standard curve.
The basic strength of the solid catalyst was determined using the Hammett indicator method using the following indicators: bromothymol blue (H_ = 7.2), phenolphthalein (H_ = 9.3), 2,4-dinitroaniline (H_ = 15.0), and 4-nitroaniline (H_ = 18.4). Approximately 0.025 g of the catalyst was placed in a beaker and 5 mL of the selected indicator was added and thoroughly mixed for 2 h. The total basicity of the prepared catalyst was then determined via Hammett indicator titration. Initially, 0.1 g of the catalyst was mixed with 4 mL of methanol, and then 0.2 mL of phenolphthalein indicator was added. The titration was performed using benzoic acid (0.1 M) as a titrant, gradually added until the color transitioned from pink to colorless.
2.6 Experimental design
The optimization of GC synthesis using the 5%Li/MCM-41 catalyst was conducted through RSM using a CCD in the Design Expert version 13 (trial) software (Stat-Ease Inc., Minneapolis, MN, USA). The experimental design varied four variables consist of catalyst loading, DMC: enriched glycerol molar ratio, reaction temperature, and reaction time. Each independent variable was explored at five levels (−2, −1, 0, +1, +2), as detailed in Table 1. A total of 30 experimental runs were conducted, comprised of 16 factorial points, eight axial point trials (two trials per variable), and six replication points situated at the central point. The repetition of central points was carried out to assess experimental error and data reproducibility. The experimental data were subjected to analysis using a second-order polynomial quadratic equation, depicted in eqn (3), for the purpose of predicting the GC yield.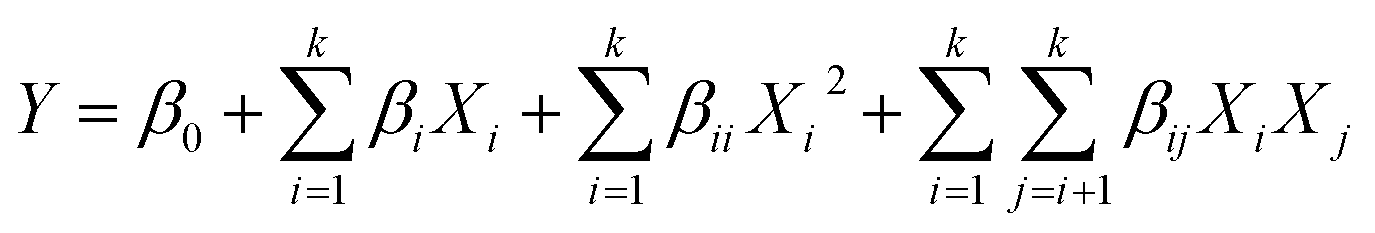 | (3) |
| Factor | Unit | Symbols | Coded factor levels | ||||
|---|---|---|---|---|---|---|---|
| −2 | −1 | 0 | +1 | +2 | |||
| Catalyst loading | wt% relative to the initial amount of enriched glycerol | A | 2 | 3 | 4 | 5 | 6 |
| DMC: enriched glycerol molar ratio | — | B | 1:1 |
2:1 |
3:1 |
4:1 |
5:1 |
| Reaction temperature | °C | C | 70 | 75 | 80 | 85 | 90 |
| Reaction time | min | D | 60 | 90 | 120 | 150 | 180 |
2.7 Synthesis of GC
The transesterification of enriched glycerol and DMC to GC was carried out in a 100 mL three-necked round-bottom flask equipped with a reflux condenser. The setup was placed in an oil bath to ensure the desired temperature on a hotplate and stirrer. Typically, 10 g of enriched glycerol was introduced into the above flask on a magnetic stirring hotplate set to the designated temperature (70, 75, 80, 85, or 90 °C). The appropriate quantity of DMC (to give DMC: enriched glycerol molar ratios of 1, 2, 3, 4, or 5) was then added and left until the desired temperature was attained. Subsequently, the predetermined catalyst loading (2–6 wt% relative to the initial amount of enriched glycerol) was incorporated into the flask to initiate the reaction, which was maintained for the specified duration (60, 90, 120, 150, or 180 min). Following the transesterification reaction, the flask was removed and allowed to cool. The catalyst was then separated from the solution through filtration. The remaining DMC and methanol byproduct were removed via vacuum distillation retaining the GC product for analysis.Upon completion of the reaction, the 5%Li/MCM-41 catalyst was recovered by filtration and washed with ethanol two to four times to eliminate adhered products like enriched glycerol and GC. Subsequently, the catalyst was dried in an oven at 105 °C overnight, preparing it for further utilization in subsequent experimental runs.
2.8 Analysis of the GC yield and purity
The identification of the obtained GC was carried out using gas chromatography (6890 series) equipped with a flame ionization detector (Hewlett Packard, USA). An HP-5MS column was employed, measuring 30 m in length with an internal diameter of 0.32 mm and a thickness of 0.25 μm. The carrier gas was helium at a flow rate of 79 mL min−1, while air and hydrogen served as the combustion gases. The injector temperature was set at 250 °C with a split ratio of 20:1, and the detector temperature was maintained at 270 °C. The initial oven temperature was 45 °C, increased at 10 °C min−1 up to 100 °C, and subsequently raised to 250 °C at 30 °C min−1 and then held at 250 °C for a total runtime of 27 min. To quantify the GC and enriched glycerol content, a calibration curve was prepared using different concentrations of isopropanol. Considering the enriched glycerol as the limiting reactant, the enriched glycerol conversion level, GC yield, GC selectivity, and turn over frequency of catalyst (TOF) were evaluated using eqn (4)–(7) as follows: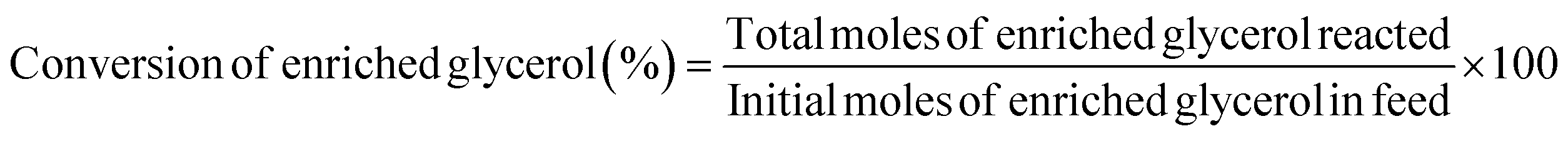 | (4) |
 | (5) |
 | (6) |
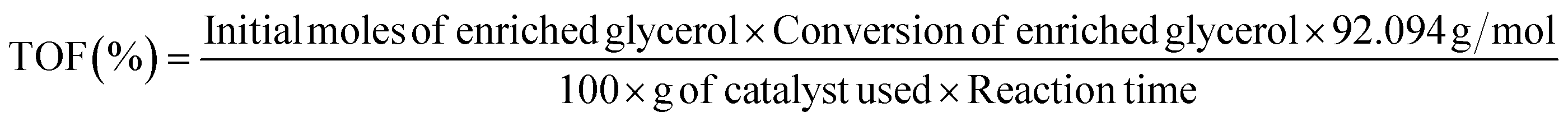 | (7) |
3. Results and discussion
3.1 Physical properties of the crude and enriched glycerol
The compositions of the commercial, crude, and enriched glycerol are presented in Table 2. The obtained crude glycerol exhibited a viscosity at 25 °C of 355.65 cP, which was lower than that of its enriched glycerol counterpart (823.30 cP). This difference in viscosity reflects their differing compositions.37 Although the crude glycerol contained 39.63 wt% glycerol, along with 7.26 wt% moisture, and 5.77 wt% ash, it had a higher content of MONG at 47.34 wt%. The ash content was attributed to salts from the unreacted catalyst in the transesterification process, resulting from the saponification of FFA. The water content in biodiesel could be attributed to the rinsing process that was carried out to remove residual catalyst during biodiesel production and its formation during the acid-base neutralization reaction.12,13 Some FFAs were released as dissolved soap, while others were either dissolved or suspended in the glycerol solution. In the subsequent neutralization step, these FFAs and FAMEs reacted with the residual catalyst to form soap, which remained in the glycerol residue.12| Component | Commercial glycerol | Crude glycerol | Enriched glycerol | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Ref. 30 | Ref. 37 | Ref. 38 | Ref. 37 | This work | Ref. 30 | Ref. 37 | Ref. 38 | This work | |
| a Measured by titration. b Measured by Karl Fischer. c Calculated by subtracting the total of the amount of glycerol, water, and ash. | |||||||||
| Glycerol (wt%)a | 99.9 | 99.98 | 98 min | 35.60 | 39.63 | 96.0 | 98.20 | 98.67 | 98.44 ± 0.11 |
| Water content (wt%)b | 0.01 | 0.01 | 2.0 max | 9.38 | 7.26 | 1.30 | 0.63 | 0.42 | 0.54 ± 0.01 |
| Ash content (wt%) | 0.0 | 0.002 | — | 4.73 | 5.77 ± 0.21 | 1.04 | 0.39 | 0.08 | 0.02 ± 0.03 |
| MONG (wt%)c | 0.0 | 0.001 | 1.0 max | 50.29 | 47.34 | 1.09 | 0.78 | 0.83 | 1.00 |
| Density (g cm−3) | 1.27 | — | — | — | 1.07 ± 0.03 | 1.26 | — | 1.26 ± 0.01 | |
| pH | 6.97 | 7 | — | 9.6 | 10.32 ± 0.01 | 6.98 | 7.08 | 7.1 | 6.95 ± 0.02 |
| Viscosity (cp at 25 °C) | — | — | — | — | 355.65 ± 2.22 | — | — | 823.30 ± 0.58 | |
| Color | Clear | Clear | Clear | Dark brown | Dark brown | Clear | Clear | Clear | Clear |
The combined process of acidification, neutralization, and phase separation increased the glycerol content 2.4-fold from the initial 39.63 wt% of crude glycerol to 98.44 wt% in the enriched glycerol, with an elimination of the FFA, FAME, and MONG from the crude glycerol. However, the formation of water and salt during the neutralization reaction slightly decreased the glycerol purity.11 According to Kongjao et al., acidification to a pH of 1 increases the glycerol content and removes most residues, but increasing the pH during the acidification stage led to a significantly higher glycerol yield and reduced amounts of inorganic salt and FFA.12 The glycerol obtained in this study before the activated carbon treatment appeared as a yellow liquid (Fig. 1c). Finally, activated carbon was utilized to remove the color, and also eliminated residual fatty acids and other impurities with molecular sizes smaller than the pore sizes of activated carbon,11 to yield the clear enriched glycerol.
The reported enriched glycerol yield in this study was the average of over 30 batches, with some variation in individual yields being noted. Overall, the enrichment process resulted in a 62.7% glycerol yield with a 98.44% glycerol purity, as compared to the previously reported values of 98.20 wt%41 and 96.0 wt%.38
The crude glycerol often contains a considerable amount of water, which lowers its viscosity. However, during enrichment the water is separated from the glycerol, leading to a more concentrated glycerol solution with an increased viscosity.
The pH of the crude glycerol was 10.32, which was higher than that of the enriched glycerol at 6.95, due to the presence of residues, such as the alkaline catalyst, alcohol, and FAMEs, which can elevate the pH.37 These impurities, primarily composed of MONG, can result from the transesterification process used in the biodiesel production. Previously, a pH of around 9.6 was reported for crude glycerol due to the significant amount of MONG and associated impurities present in the sample.41,42
The crude glycerol had a lower density (1.079 g cm−3) than the enriched glycerol (1.26 g cm−3), which was again due to the extraction of residual wastes during the enrichment steps, including methanol, fatty acids, catalyst residues, water, and other compounds originally derived from the oil used.43 The density of the enriched glycerol closely matched that of the commercially available pure glycerol, indicating the success of the enrichment process in producing a high-quality product.
3.2 Catalyst characterization
The wide-angle XRD profiles of MCM-41 and various Li/MCM-41 catalysts are illustrated in Fig. 2. The 2θ value at 21.97° in all the catalysts represented the broad peak characteristic of amorphous silica. Following the incorporation of the Li precursor at 2–6 wt% relative to the initial amount of enriched glycerol onto the MCM-41 framework and subsequent calcination at 450 °C for 3 h, the XRD profiles of the catalysts showed no distinct peaks of LiNO3 and/or Li2O. Typically, the calcination step is employed to convert the LiNO3 phase into the Li2O phase. However, the absence of the Li2O peak could be attributed to the complete dispersion of the Li-active species within the pores of the MCM-41 framework. Additionally, Li is lighter than silica, resulting in a weak diffraction that could be overshadowed by the higher scattering from the silica of the MCM-41 support.6,16,44–46 The limitation in instrumental detection hindered the recognition of small-sized lithium molecules.47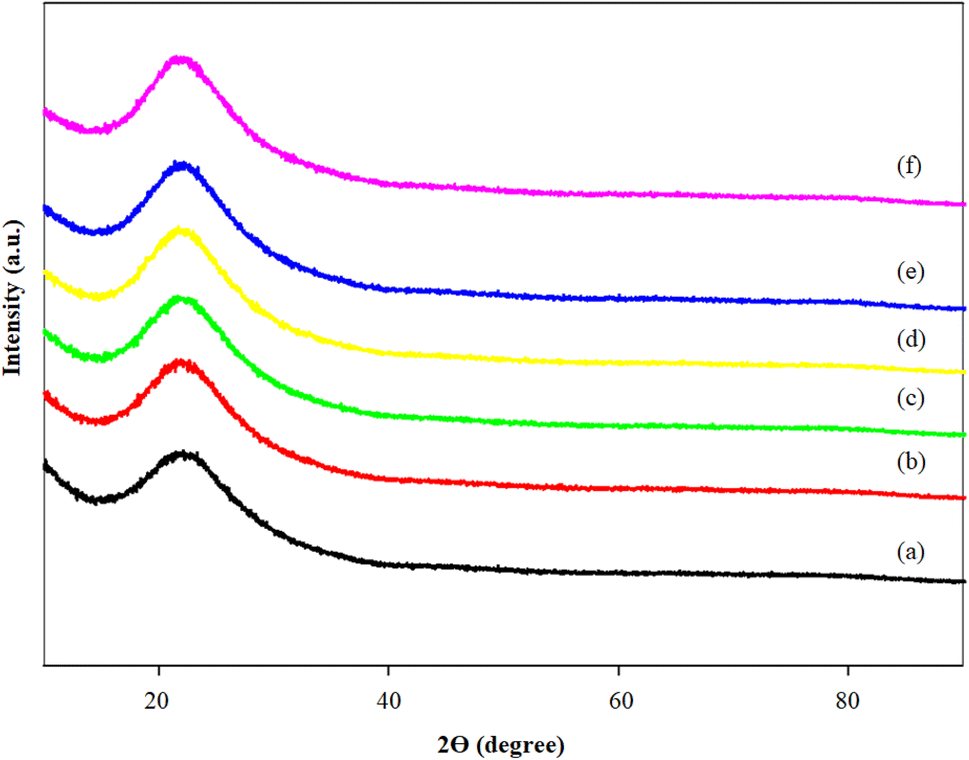 | ||
| Fig. 2 X-ray diffraction profile of (a) MCM-41; (b) 2%Li/MCM-41; (c) 3%Li/MCM-41; (d) 4%Li/MCM-41; (e) 5%Li/MCM-41; and (f) 6%Li/MCM-41. | ||
The FTIR profiles of MCM-41 and the 5%Li/MCM-41 catalyst were analyzed to investigate the stretching vibrational modes of various functional groups (Fig. 3). Across all catalyst spectra, the broad vibrational peaks observed at 3446.0–3447.2 and 1632.7–1636.0 cm−1 can be attributed to the hydroxyl groups linked with the silanol groups (Si–OH), arising from physically adsorbed moisture molecules on the catalyst surface.47 The vibrational peaks at 2918.6–2919.7 and 2850.5–2851.1 cm−1 are associated with C–H vibrations of the encapsulated surfactant molecules.36 The weak peak observed at 1465.7–1468.1 cm−1 is indicative of –CH2 groups.48 Peaks observed at 1088.8–1091.4 and 802.7–805.7 cm−1 are attributed to asymmetric stretching and bending of Si–O–Si bonds within the MCM-41 framework.49 The band at 464.4–467.6 cm−1 can be attributed to the vibration of Si–O bonds, indicating the mesoporous silica nature.48 Moreover, after Li doping onto the MCM-41 framework, a little bump peak appeared at 1383.6 cm−1 appeared, which was attributed to the N–O group of the LiNO3 precursor due to the presence of the metal oxide within the catalyst.6
 | ||
| Fig. 3 FTIR profiles of (a) MCM-41 and (b) 5%Li/MCM-41. | ||
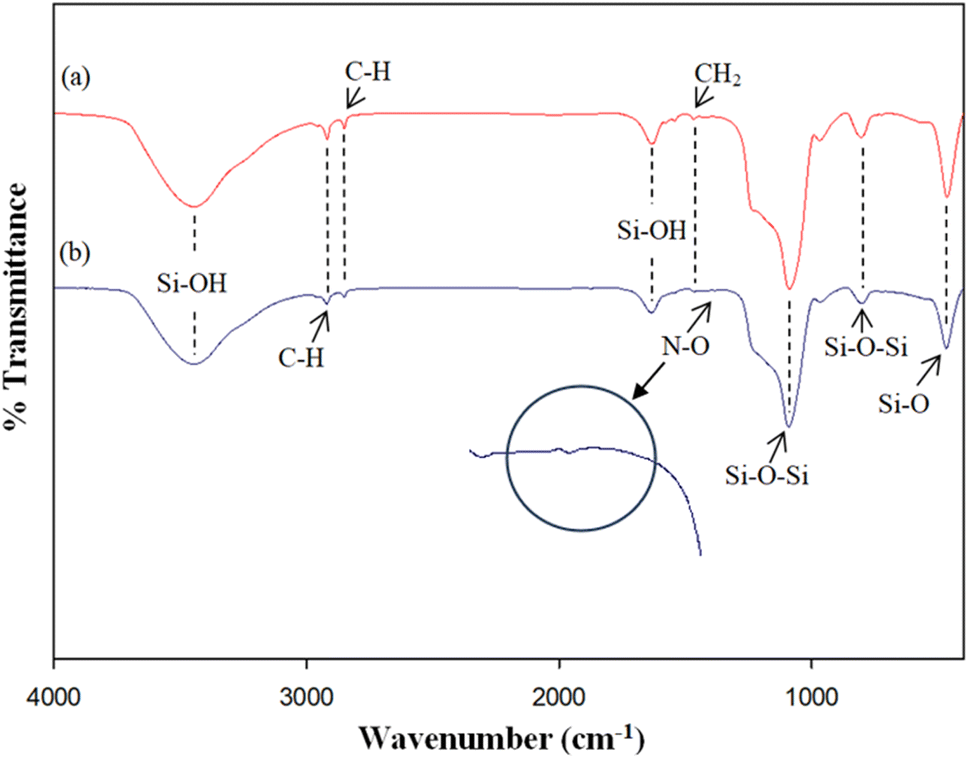
The surface morphological characteristics of prepared catalyst at a magnification of 50000× are illustrated in Fig. 4. The morphological images of prepared catalyst shown the spherical particles.
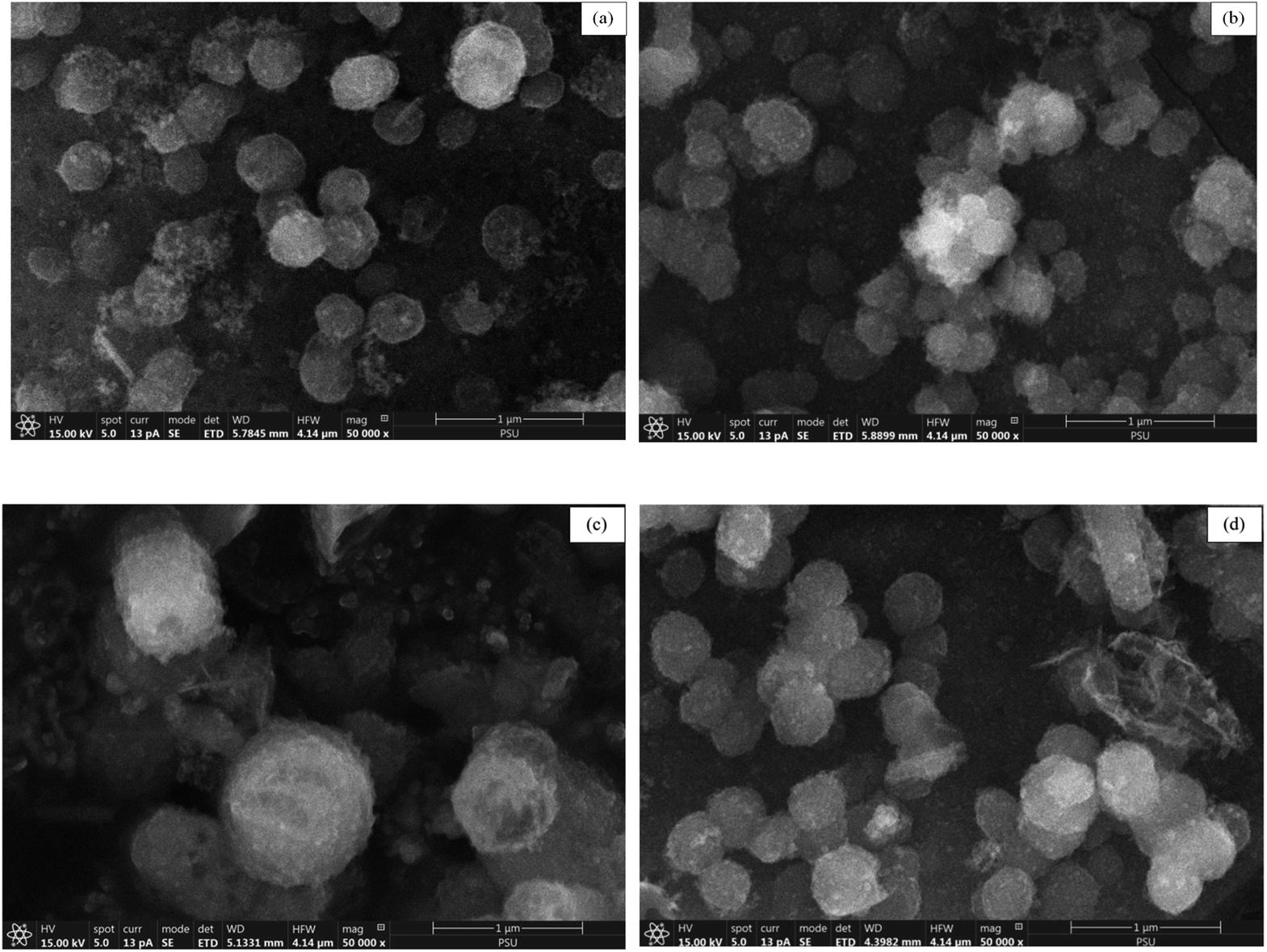 | ||
| Fig. 4 SEM images at 50000× magnification of (A) MCM-41, (B) 5%Li/MCM-41, (C) 5%Li/MCM-41 (4th reused), and (D) 5 wt% of LiNO3 doped on 5%Li/MCM-41 (4th reused). | ||
The thermal behavior of MCM-41 and 5%Li/MCM-41 was analyzed using TGA (Fig. 5). The TGA profile of the MCM-41 support showed a two-stage thermal decomposition with the first stage up to 193.67 °C accounting for an approximately 2.55% mass loss, which was due to the desorption of adsorbed moisture from the pores of the MCM-41 framework. The second stage, above 193.67 °C, accounted for 1.59% mass loss and was due to the decomposition of organic impurities within the MCM-41 pores. The TGA profile of the 5%Li/MCM-41 catalyst also exhibited a significant stage at above 400–530 °C resulted in an approximately 1.35% mass loss, and might be attributed to the decomposition of organic impurities with the pores of this sample together with the salt decomposition.
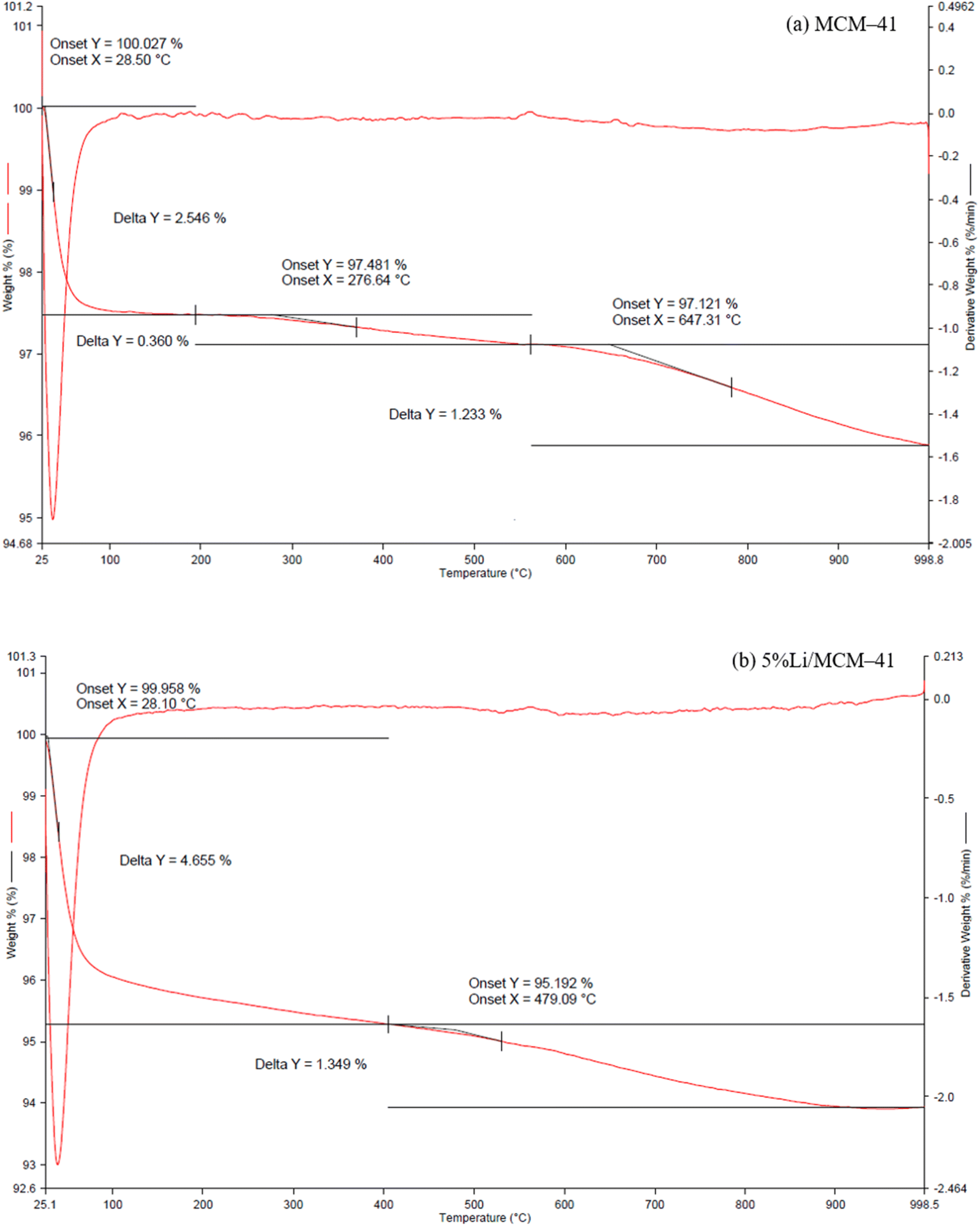 | ||
| Fig. 5 TGA profile of (a) MCM-41 and (b) 5%Li/MCM-41. | ||
The SBET, average pore diameter (dp), and Vp play essential roles in the activity of MCM-41 and 5%Li/MCM-41 catalysts. The N2 adsorption–desorption isotherms were characterized using liquid N2 at −195.85 °C (Fig. 6). Following the IUPAC nomenclature standards, the isotherms of the prepared catalysts were classified as type IV isotherms with H1 hysteresis loops6 (Fig. 6a and b). Both MCM-41 and 5%Li/MCM-41 isotherms exhibited very narrow loops confined within a relative pressure range (P/P0) of 0.5–1.0. The MCM-41 catalyst had a SBET of 642.05 m2 g−1, a Vp of 0.685 cm3 g−1, and an average dp of 4.27 nm, which is indicative of a well-defined mesoporous structure. It was previously shown that mesoporous catalysts exhibited high catalytic properties in the transesterification of enriched glycerol and DMC into GC, which was attributed to their ability to accommodate active metal species that enhance this catalytic activity.50
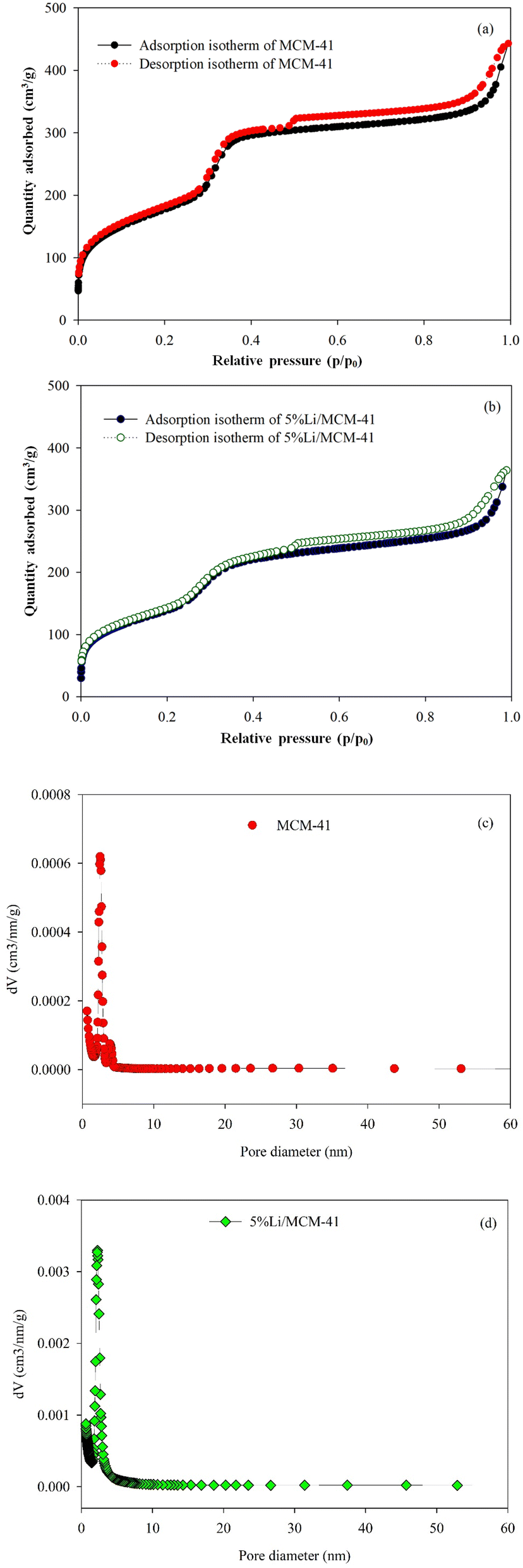 | ||
| Fig. 6 Nitrogen adsorption–desorption isotherm of (a) MCM-41; (b) 5%Li/MCM-41 and pore size distribution of (c) MCM-41; (d) 5%Li/MCM-41 and pore size. | ||
After incorporation of Li onto the MCM-41 framework, the SBET and Vp were slightly decreased to 504.12 m2 g−1 and 0.563 cm3 g−1, respectively, which is likely to be due to the incorporation of Li active metal onto the MCM-41 framework, whilst the dispersion of Li species within the mesoporous structure facilitates the transesterification reaction. However, the incorporation of the Li increased the average dp of 5%Li/MCM-41 to 4.47 nm, likely to be due to formation of metal oxide bond on the wall of MCM-41 framework.6
3.3 Screening of the catalysts
The mesoporous structure of MCM-41 support has found application with various catalytically active species, owing to its high surface areas and pore volumes that augment its potential for new functionalization. Addressing this, the catalytic capability of siliceous materials can be readily altered by introducing heteroatoms into the silicate framework. The impregnation of Li onto mesoporous MCM-41 significantly enhanced its catalytic activity. The GC yield and glycerol conversion level obtained with the MCM-41 support and the different active metals supported on MCM-41 for the transesterification of enriched glycerol and DCM were investigated with a catalyst loading of 5 wt% relative to the initial amount of enriched glycerol, a DMC: enriched glycerol molar ratio of 4:1, and a reaction temperature of 85 °C for 150 min. As expected, the MCM-41 support exhibited the lowest GC yield (Table 3). That the MCM-41 framework does not possess adequate catalytic activity for this transesterification reaction may reflect the low basic strength of the material (H_ ≤ 7.2) and the relatively low concentration of basic sites (1.11 mmol g−1).
| Catalyst | Basicity (mmol g−1) | Basic strength (H_) | GC yield (%) |
|---|---|---|---|
|
a Reaction conditions: catalyst loading of 5 wt%, DMC to enriched glycerol molar ratio of 4:1, reaction temperature of 85 °C, and reaction time of 150 min.
|
|||
| MCM-41 | 1.11 | H_ ≤ 7.2 | 2.96 |
| 2%Li/MCM-41 | 1.45 | 7.2 ≤ H_ ≤ 9.8 | 17.24 |
| 3%Li/MCM-41 | 2.03 | 7.2 ≤ H_ ≤ 9.8 | 22.25 |
| 4%Li/MCM-41 | 2.49 | 7.2 ≤ H_ ≤ 9.8 | 29.30 |
| 5%Li/MCM-41 | 4.70 | 9.8 ≤ H_ ≤ 15 | 58.77 |
| 6%Li/MCM-41 | 5.05 | 9.8 ≤ H_ ≤15 | 47.27 |
Incorporation of an active metal (Li, Na, K, or Ba) at a 5 wt% metal loading into the MCM-41 framework, improved the transesterification reaction by introducing essential basic sites for GC synthesis. The resulting GC yields for 5%Li/MCM-41, 5%Na/MCM-41, 5%K/MCM-41, and 5%Ba/MCM-41 were 57.18%, 25.11%, 42.33%, and 30.87%, respectively (Fig. 7). Notably, the 5%Li/MCM-41 catalyst exhibited the highest catalytic activity (in terms of GC yield), which can be attributed to the strong ion size effect of Li.6 Consequently, the Li/MCM-41 catalyst emerged as a promising candidate for this transesterification reaction.
 | ||
| Fig. 7 Effect of types of catalyst for GC synthesis (reaction conditions: catalyst loading of 5 wt%, DMC to enriched glycerol molar ratio of 4:1, reaction temperature of 85 °C, and reaction time of 150 min). | ||
To explore the effect of the Li metal loading level on the MCM-41 framework, Li loading levels from 2 wt% to 6 wt% were examined (Fig. 8). As the Li metal loading increased from 2 to 5 wt%, the GC yield and enriched glycerol conversion rates were increased from 17.24% to 57.18% and from 18.94% to 57.35%, respectively. These findings imply that the incorporation of Li species into the catalysts enhanced their basicity, as seen in Table 3. These improved basicity properties are likely key factors contributing to the catalysts' enhanced performance compared to the MCM-41 framework. However, at a Li loading level of 6 wt%, there was a slight decrease in the GC yield to 47.27%. The exploration of catalyst performance involved TOF calculation. Elevating the active metal loading from 2 wt% to 5 wt% resulted in a TOF increase from 1.52 h−1 to 4.72 h−1. However, a subsequent increase to 6 wt% led to a TOF decrease due to the formation of a multilayer dispersion of Li metal on the MCM-41 support. This would lead to pore blockage or agglomeration of Li metal within the MCM-41 framework.6 Based on these observations, it was inferred that an optimal the highest GC yield and the 5%Li/MCM-41 catalyst was selected for determining the optimal conditions for GC synthesis.
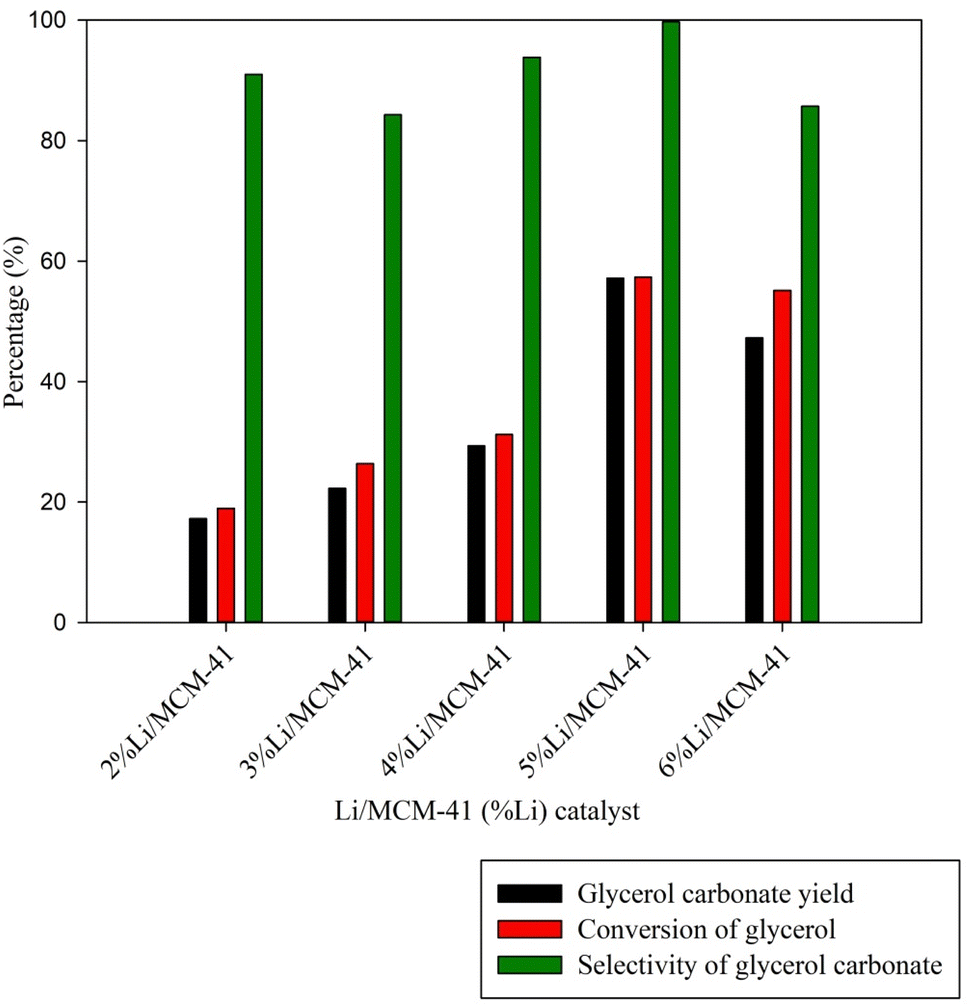 | ||
| Fig. 8 Effect of catalyst loading level for GC synthesis (reaction conditions: catalyst loading of 5 wt%, DMC to enriched glycerol molar ratio of 4:1, reaction temperature of 85 °C, and reaction time of 150 min). | ||
3.4 Optimization of GC synthesis by RSM
The incorporation of RSM and CCD into this study facilitated a more systematic and comprehensive optimization of the GC synthesis and allowed for identification of the optimal reaction conditions based on the experimental data (Table 4). CCD is particularly useful when there is a suspicion or prior knowledge that quadratic effects play a significant role in the response. CCD offers flexibility in choosing the number of factorial points, making it adaptable to different experimental scenarios. This flexibility can be crucial when dealing with a varying number of factors. By employing a quadratic model to assess the effects of different factors on the GC yield, the study managed to pinpoint the most influential factors on the reaction and subsequently optimize the reaction conditions accordingly. Model selection was guided by the p-value of the models, and the corresponding p-values for each model can be found in Table 5. From the available information, it was evident that the quadratic model was optimal (p-value below 0.05) and was a suitable choice for capturing the relationship between the variables and the response in the study.| Standard run | Catalyst loading (wt%) | DMC: enriched glycerol molar ratio | Reaction temperature (°C) | Reaction time (min) | GC yield (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Coded | Real | Coded | Real | Coded | Real | Coded | Real | Observed | Predicted | |
| 1 | −1 | 3 | +1 | 4:1 |
−1 | 75 | −1 | 90 | 36.77 | 36.71 |
| 2 | +1 | 5 | +1 | 4:1 |
−1 | 75 | −1 | 90 | 43.62 | 44.25 |
| 3 | +1 | 5 | −1 | 2:1 |
−1 | 75 | +1 | 150 | 44.15 | 43.08 |
| 4 | +1 | 5 | −1 | 2:1 |
−1 | 75 | −1 | 90 | 42.08 | 43.00 |
| 5 | 0 | 4 | 0 | 3:1 |
0 | 80 | +2 | 180 | 53.75 | 54.28 |
| 6 | +1 | 5 | −1 | 2:1 |
+1 | 85 | −1 | 90 | 37.78 | 38.72 |
| 7 | 0 | 4 | 0 | 3:1 |
0 | 80 | 0 | 120 | 54.90 | 55.31 |
| 8 | +1 | 5 | +1 | 4:1 |
−1 | 75 | +1 | 150 | 46.82 | 46.92 |
| 9 | −1 | 3 | −1 | 2:1 |
−1 | 75 | −1 | 90 | 46.34 | 44.32 |
| 10 | −1 | 3 | −1 | 2:1 |
+1 | 85 | −1 | 90 | 41.09 | 40.53 |
| 11 | 0 | 4 | 0 | 3:1 |
+2 | 90 | 0 | 120 | 48.77 | 47.72 |
| 12 | 0 | 4 | 0 | 3:1 |
0 | 80 | −2 | 60 | 48.16 | 47.65 |
| 13 | −1 | 3 | −1 | 2:1 |
−1 | 75 | +1 | 150 | 45.94 | 45.82 |
| 14 | −1 | 3 | +1 | 4:1 |
−1 | 75 | +1 | 150 | 41.31 | 40.81 |
| 15 | 0 | 4 | 0 | 3:1 |
0 | 80 | 0 | 120 | 55.64 | 55.31 |
| 16 | 0 | 4 | 0 | 3:1 |
0 | 80 | 0 | 120 | 55.75 | 55.31 |
| 17 | 0 | 4 | 0 | 3:1 |
0 | 80 | 0 | 120 | 56.46 | 55.31 |
| 18 | +1 | 5 | +1 | 4:1 |
+1 | 85 | +1 | 150 | 57.18 | 59.65 |
| 19 | 0 | 4 | 0 | 3:1 |
−2 | 70 | 0 | 120 | 37.72 | 38.78 |
| 20 | −1 | 3 | +1 | 4:1 |
+1 | 85 | +1 | 150 | 55.40 | 54.03 |
| 21 | +1 | 5 | +1 | 4:1 |
+1 | 85 | −1 | 90 | 54.86 | 54.52 |
| 22 | +2 | 6 | 0 | 3:1 |
0 | 80 | 0 | 120 | 51.33 | 49.70 |
| 23 | 0 | 4 | +2 | 5:1 |
0 | 80 | 0 | 120 | 46.25 | 45.02 |
| 24 | 0 | 4 | 0 | 3:1 |
0 | 80 | 0 | 120 | 55.61 | 55.31 |
| 25 | +1 | 5 | −1 | 2:1 |
+1 | 85 | +1 | 150 | 41.64 | 41.25 |
| 26 | −2 | 2 | 0 | 3:1 |
0 | 80 | 0 | 120 | 43.76 | 45.40 |
| 27 | −1 | 3 | +1 | 4:1 |
+1 | 85 | −1 | 90 | 45.96 | 47.48 |
| 28 | −1 | 3 | −1 | 2:1 |
+1 | 85 | +1 | 150 | 44.67 | 44.49 |
| 29 | 0 | 4 | 0 | 3:1 |
0 | 80 | 0 | 120 | 53.51 | 55.31 |
| 30 | 0 | 4 | −2 | 1:1 |
0 | 80 | 0 | 120 | 32.99 | 34.23 |
| Source | Sum of squares | Degree of freedom | Mean square | F-value | p-value | Remark |
|---|---|---|---|---|---|---|
| Mean vs. total | 67233.21 | 1 | 67233.21 | |||
| Linear vs. mean | 388.30 | 4 | 97.08 | 2.52 | 0.0666 | |
| 2FI vs. linear | 305.25 | 6 | 50.87 | 1.47 | 0.2417 | |
| Quadratic vs. 2FI | 623.56 | 4 | 155.89 | 67.17 | <0.0001 | Suggested |
| Cubic vs. quadratic | 28.88 | 8 | 3.61 | 4.26 | 0.0358 | Aliased |
| Residual | 5.93 | 7 | 0.8470 | |||
| Total | 68585.14 | 30 | 2286.17 |
The ANOVA statistical analysis was used to reveal the significant process parameters (Table 6). The p-values of this model were less than 0.0001, indicating the model terms were significant. The coefficient of the linear terms (A, B, C, and D) interactive terms (AB and BC), and quadratic terms (A2, B2, C2, and D2) were significant. The F-value of model is 40.54, indicating that it is statistically significant. This demonstrates that the model has a 0.01% chance of producing an F-value this large due to noise. Moreover, the lack of fit F-value is 2.90, implying that it is not significant model. There is only a 12.60% chance due to noise. The coefficient of determination (R2) explained the variability for the GC yield was 97.43%. Furthermore, the predicted determination coefficient (predicted R2) and adjusted determination coefficient (adjusted R2) were observed to be 86.81% and 95.02%, respectively, representing that the predicted and actual values were well-fitting, and indicating a statistically significant correlation between the GC yield and the four variable factors that affect it. The predicted versus actual values plot on the GC yield is illustrated in Fig. 9. A linear relationship between the actual and predicted values means was observed, indicating that the predictive model captured the underlying patterns in the data and is able to accurately predict the GC yield based on the input variables. The predicted equation for the GC yield is displayed in eqn (8):
| GC yield (%) = 55.31 + 1.07A + 2.70B + 2.24C + 1.66D + 2.21AB − 0.1231AC − 0.3569AD + 3.64BC + 0.6494BD + 0.6119CD − 1.94A2 − 3.92B2 − 3.02C2 − 1.09D2 | (8) |
| Source | Sum of squares | Degree of freedom | Mean square | F-value | p-value | Remark |
|---|---|---|---|---|---|---|
| Model | 1317.11 | 14 | 94.08 | 40.54 | <0.0001 | Significant |
| A | 27.71 | 1 | 27.71 | 11.94 | 0.0035 | Significant |
| B | 174.69 | 1 | 174.69 | 75.28 | <0.0001 | Significant |
| C | 119.93 | 1 | 119.93 | 51.68 | <0.0001 | Significant |
| D | 65.97 | 1 | 65.97 | 28.43 | <0.0001 | Significant |
| AB | 78.46 | 1 | 78.46 | 33.81 | <0.0001 | Significant |
| AC | 0.2426 | 1 | 0.2426 | 0.1045 | 0.7509 | |
| AD | 2.04 | 1 | 2.04 | 0.8781 | 0.3636 | |
| BC | 211.78 | 1 | 211.78 | 91.26 | <0.0001 | Significant |
| BD | 6.75 | 1 | 6.75 | 2.91 | 0.1088 | |
| CD | 5.99 | 1 | 5.99 | 2.58 | 0.1290 | |
| A 2 | 103.24 | 1 | 103.24 | 44.49 | <0.0001 | Significant |
| B 2 | 214.77 | 1 | 214.77 | 181.74 | <0.0001 | Significant |
| C 2 | 249.35 | 1 | 249.35 | 107.45 | <0.0001 | Significant |
| D 2 | 32.44 | 1 | 32.44 | 13.98 | 0.0020 | Significant |
| Residual | 34.81 | 15 | 2.32 | |||
| Lack of fit | 29.69 | 10 | 2.97 | 2.90 | 0.1260 | Not significant |
| Pure error | 5.12 | 5 | 1.02 | |||
| Cor total | 1351.92 | 29 | ||||
| R 2 = 0.9743, adj R2 = 0.9502 |
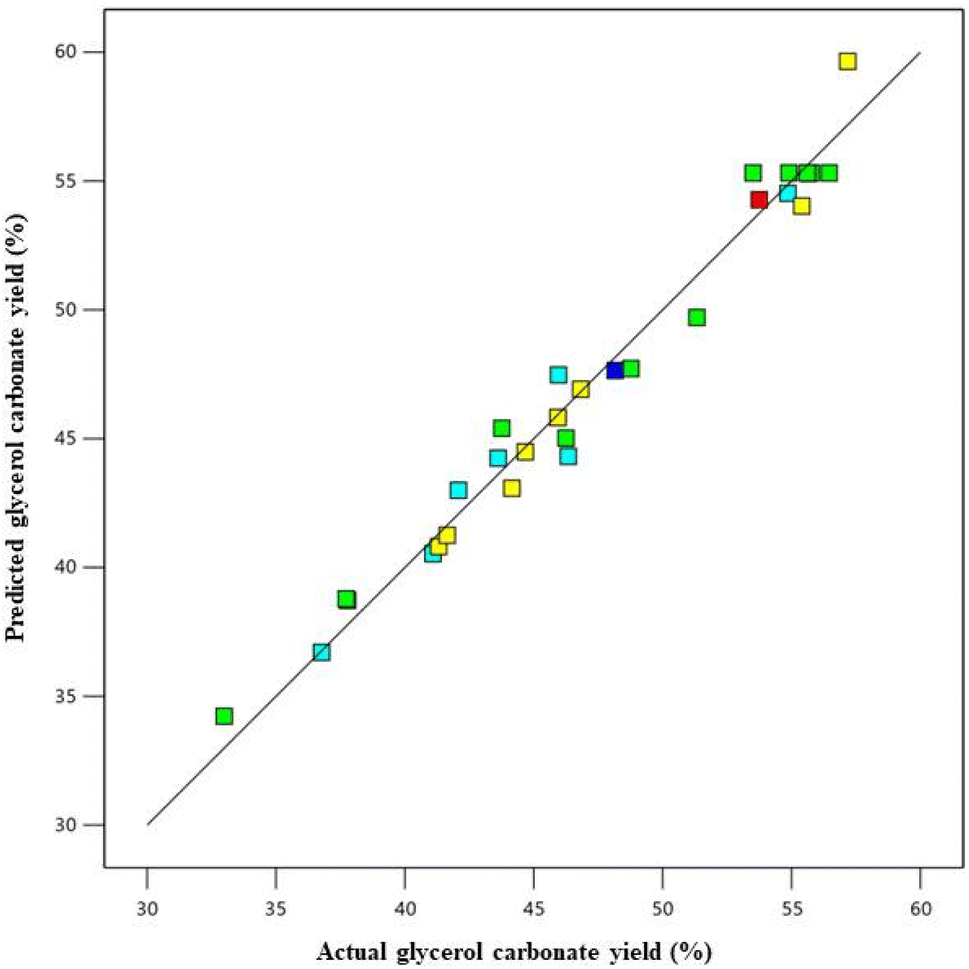 | ||
| Fig. 9 Predicted vs. actual data plot of GC yield. | ||
The combined influence of the four variables on the GC yield is visualized in Fig. 10. This was achieved by constructing perturbation graphs, which enabled a comparative analysis of the effects of the process variables at specific points within the design range.51 The four parameters under investigation were factor A (catalyst loading level: 4 wt% relative to the initial amount of enriched glycerol), factor B (DMC: enriched glycerol molar ratio: 3:1), factor C (reaction temperature: 80 °C), and factor D (reaction time: 180 min). These variables were identified as the controlling factors in determining the GC yield. In a perturbation plot, the steepness of the slope generally signifies the magnitude of a factor's impact on the yield, with a steeper slope indicating a more substantial effect compared to a flatter slope. As per the findings of the study, the perturbation diagram unveiled an inflection point in relation to the DMC: enriched glycerol molar ratio. This indicated that this particular parameter exerted a more pronounced influence on the GC yield compared to the other parameters. Following this, the sequence of impact was observed to be as follows: DMC: enriched glycerol molar ratio, reaction temperature, reaction time, and catalyst loading level.
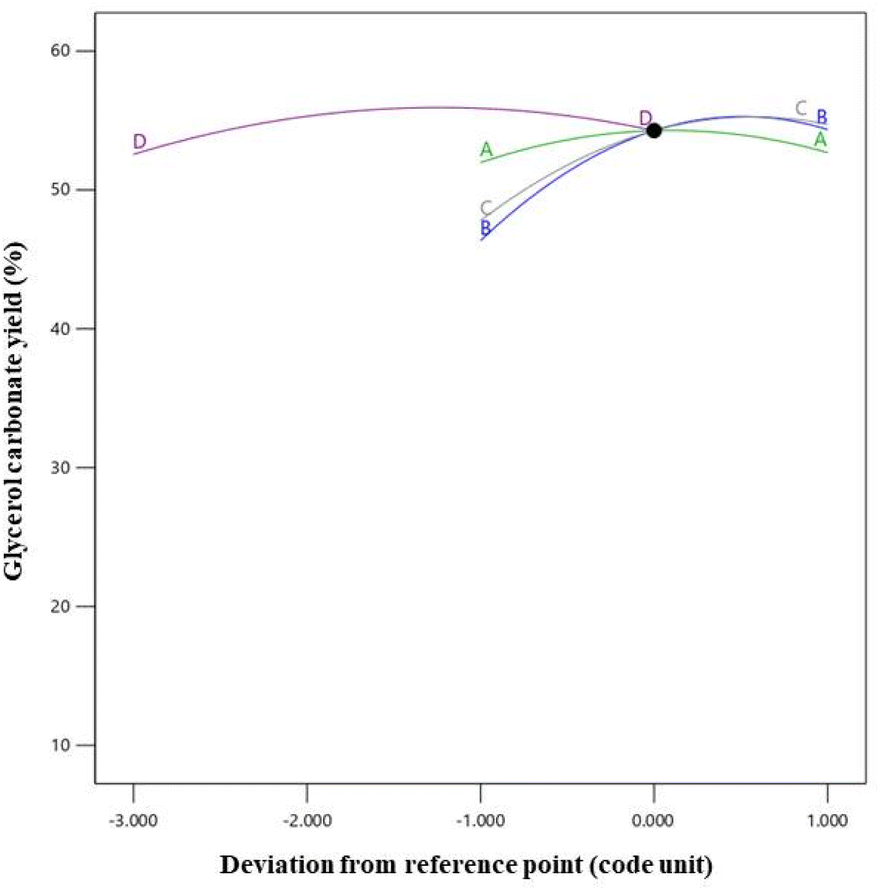 | ||
| Fig. 10 Perturbation plot including catalyst loading (A), DMC to enriched glycerol molar ratio (B), reaction temperature (C), and reaction time (D). | ||
3.5 Interaction effects of the parameters on the GC yield via RSM approach
The optimum conditions were determined using the Design Expert software, which included a catalyst loading level of 5.15 wt% relative to the initial amount of enriched glycerol, a DMC: enriched glycerol molar ratio of 4.24:1, a reaction temperature of 86 °C, and a reaction time of 165 min. When performed at this optimum condition, the reaction gave a GC yield of 58.77%, which is similar to the predicted value (Table 7). Thus, the predicted optimum conditions for the reaction, as determined by Design Expert software, can be used to produce GC with a reasonable degree of accuracy.
| Predicted GC yield (%) | Experimental GC yield (%) | Error (%) |
|---|---|---|
|
a Reaction conditions: catalyst loading of 5.15 wt%, DMC: enriched glycerol molar ratio of 4.24:1, reaction temperature of 86 °C, and reaction time of 165 min.
|
||
| 59.75 | 58.77 | 1.64 |
The three-dimensional (3D) surface diagrams (Fig. 11) offer a comprehensive visualization of how the operational factors interact with each other. Varying the catalyst loading level (A) from 2–6 wt% relative to the initial amount of enriched glycerol and the DMC: enriched glycerol molar ratio (B) from 1:1 to 5:1 on the GC yield, with a constant reaction temperature (80 °C) and reaction time (180 min), is shown in Fig. 11a–c. In chemical processes, the catalyst is pivotal, expediting product formation and lowering the reaction's activation energy. From the 3D surface diagrams, it is evident that increasing the catalyst loading from 2 wt% to 4 wt% (relative to the initial amount of enriched glycerol) led to a corresponding rise in the GC yield from 45.40% to 55.31%. This increase in yield can be attributed to the greater number of Li active sites present, which play a vital role in facilitating the transesterification reaction.19 The lower catalyst concentrations often resulted in a diminished GC yield due to the scarcity of active sites. Hence, the catalyst's activity is closely linked to its active sites during the reaction. Conversely, when the catalyst loading level was elevated to 6 wt% (relative to the initial amount of enriched glycerol), it potentially led to pore blockage and increased mass transfer resistance between the reactants and catalyst phases, resulting in a decline in the GC yield.4,30
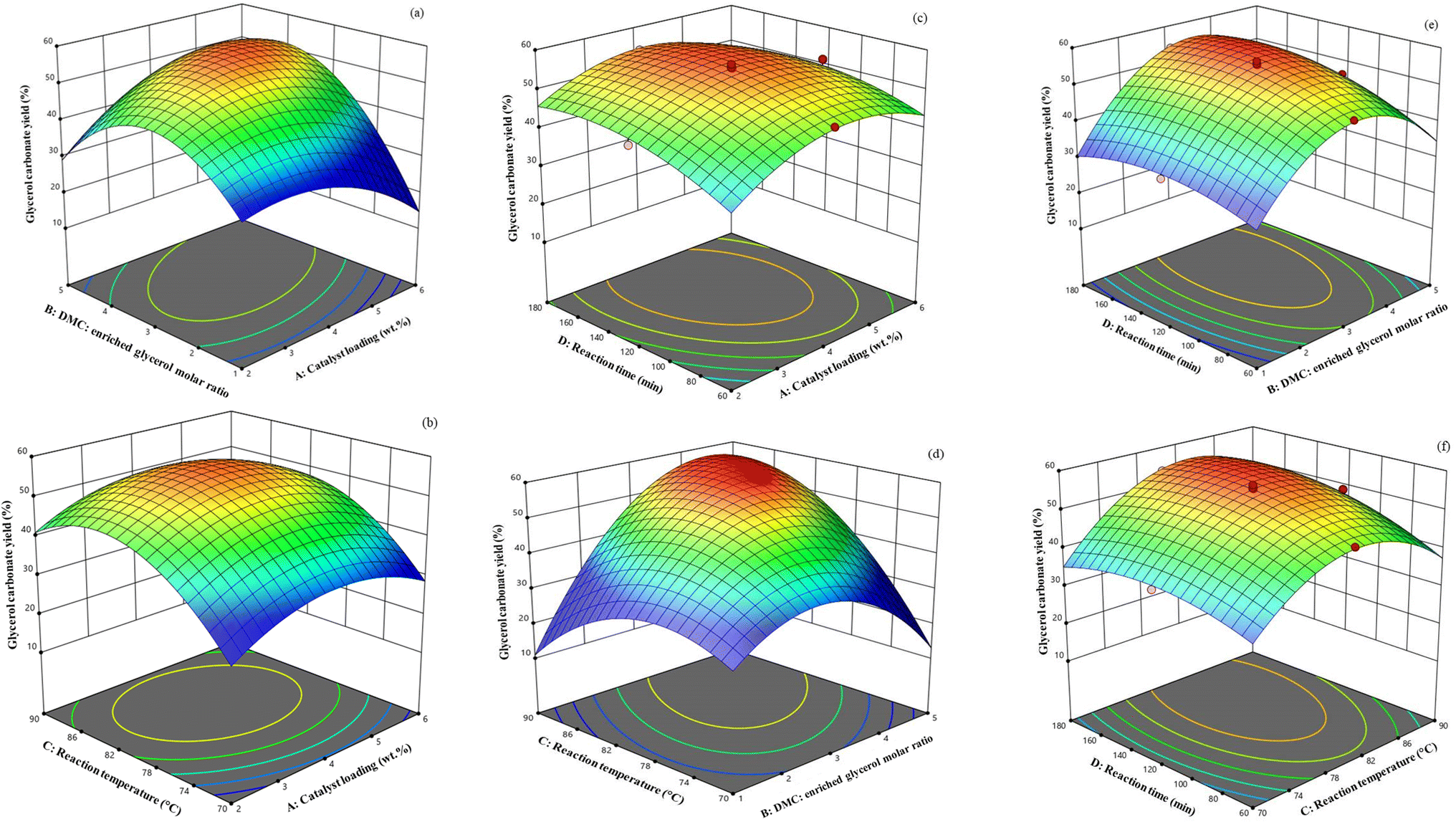 | ||
| Fig. 11 Interaction effects of (a) catalyst loading and DMC to enriched glycerol molar ratio, (b) catalyst loading and reaction temperature, (c) catalyst loading and reaction time, (d) DMC to enriched glycerol molar ratio and reaction temperature, (e) DMC to enriched glycerol molar ratio and reaction time, and (f) reaction temperature and reaction time on GC yield. | ||
The molar ratio of DMC to enriched glycerol has proven to be crucial for achieving the highest yield in the transesterification reaction of glycerol. The influence of the DMC to enriched glycerol molar ratio (1:1 to 5:1) on the GC yield (under a constant reaction temperature of 80 °C, catalyst loading level of 4 wt% (relative to the initial amount of enriched glycerol), and reaction time of 180 min) is illustrated in Fig. 11a, d and e. The GC yield was predicted to be 34.20% at a DMC: enriched glycerol molar ratio of 1:1 and markedly increased when raising the DMC: enriched glycerol molar ratio to 3:1. This is because an excess of DMC is necessary to facilitate the reaction in the forward direction.48 However, increasing the molar ratio to 5:1 did not enhance the GC yield; instead, it decreased to 45.02% owing to the poor miscibility of reactants at high concentrations.4
The effect of the reaction temperature (70–90 °C) and catalyst loading on the reaction (with a fixed DMC: enriched glycerol molar ratio of 3:1 and reaction time of 180 min) is illustrated in Fig. 11b. The GC yield increased noticeably when increasing the temperature from 70 °C to 80 °C. This effect is presumably attributed to the temperature rise enhancing the dispersibility and contact between the reactants. Additionally, the reduced viscosity of the reaction mixture facilitates improved mixing between the enriched glycerol and DMC, ultimately resulting in an increased reaction rate.53 However, further increasing the temperature to 90 °C reduced the GC yield (Fig. 11b, d and f), suggesting that the decarboxylation reaction occurs at high temperatures, leading to the formation of glycidol.52 Another contributing factor is the loss of DMC through evaporation, especially given that the boiling temperature of DMC is close to 90 °C. High temperatures can have a negative impact on chemical reactions, such as promoting unwanted side reactions (e.g., decarboxylation), and so decrease the effectiveness of catalysts and reduce their ability to facilitate the desired reactions.4
The reaction time was evaluated within the range of 60–180 min while keeping the other three variables constant (Fig. 11c, e and f). The GC yield increased gradually as the reaction time increased, and reached a maximum of 55.31% at 120 min. However, the yield then slowly decreased to 54.28% at 180 min, which could be attributed to the occurrence of side reactions involving the products, such as the formation of glycidol from GC. After a certain time period, the reaction progresses towards glycidol selectivity.53
3.6 Reaction mechanism
The reaction mechanism of the purified glycerol and DMC as reactants via transesterification process using 5%Li/MCM-41 as a catalyst to produce glycerol carbonate was represented in Fig. 12. The reaction pathway was exactly described using Langmuir–Hinshelwood–Hougen–Watson model.34,47 The overall mechanism of the 5%Li/MCM-41 heterogeneous catalyst can be explained as follows: first, purified glycerol and DMC from the bulk fluid diffuse into the external active sites of 5%Li/MCM-41 catalyst surface. Second, the purified glycerol and DMC are diffused from the pore mouth into the pores of the 5%Li/MCM-41 catalyst. The third step is the adsorption of purified glycerol and DMC onto the 5%Li/MCM-41 surface. Then, the reaction is carried out on the 5%Li/MCM-41 surface where purified glycerol was donated H+ ion on the basic sites while DMC was produced positive carbonyl carbon. The –OH group of purified glycerol was activated with carbonyl carbon of DMC. The cyclization was occurred to produce glycerol carbonate molecule. Next, the glycerol carbonate and methanol were desorbed from the active sites 5%Li/MCM-41 surface. Subsequently, the glycerol carbonate and methanol are diffused from the interior of the catalyst to the pore mouth of the 5%Li/MCM-41 surface. Finally, the glycerol carbonate and methanol were transferred into the bulk phase from the 5%Li/MCM-41 catalyst surface. These reactions generate glycerol carbonate as the main product and methanol as a byproduct.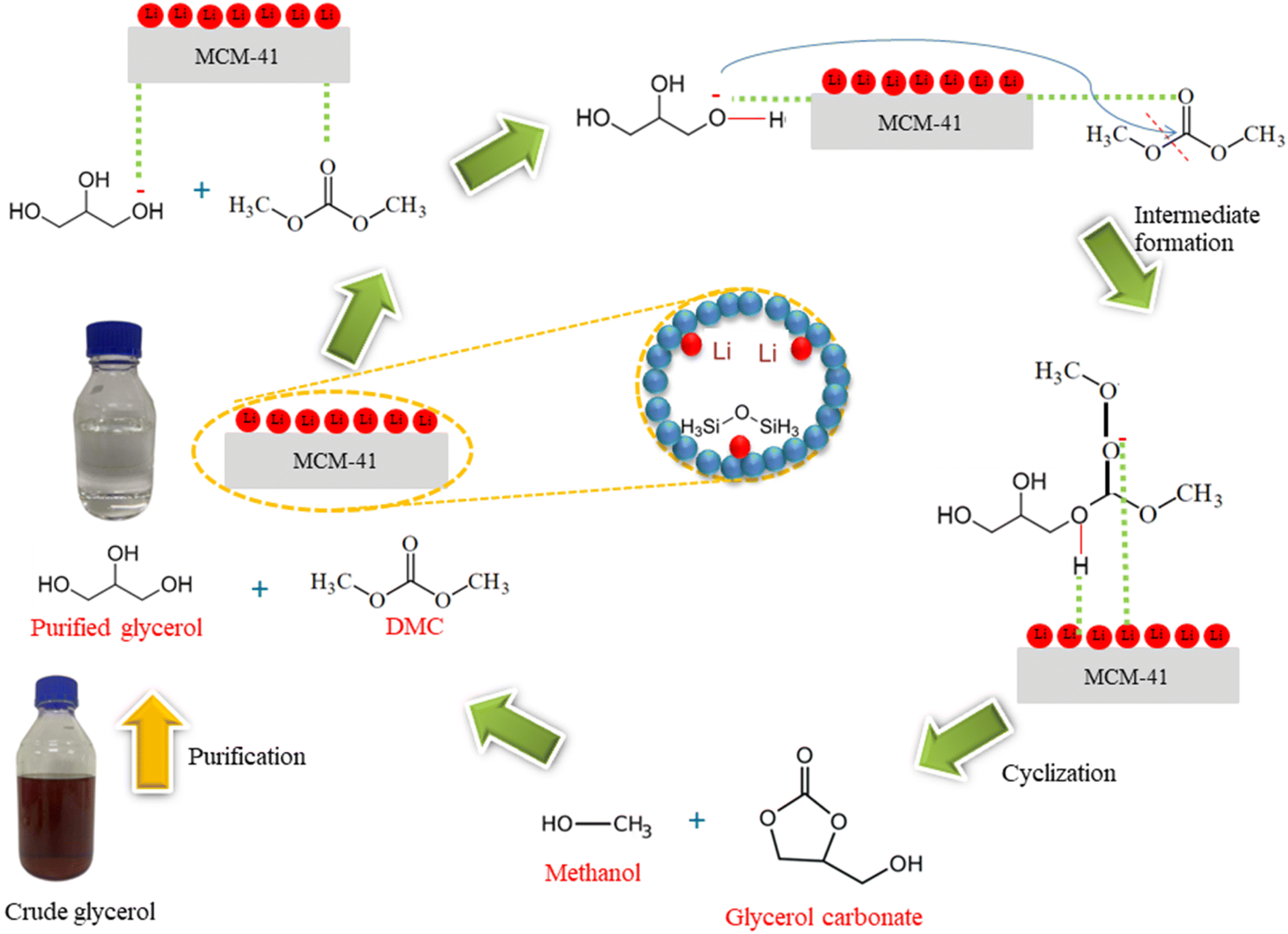 | ||
| Fig. 12 Possible reaction mechanism of enriched glycerol and DMC on 5%Li/MCM-41 catalyst for glycerol carbonate synthesis via transesterification. | ||
3.7 Reusability of the catalyst
The reusability of the 5%Li/MCM-41 catalysts and the 5 wt% (relative to the initial amount of enriched glycerol) of LiNO3 doped on 5%Li/MCM-41 (4th reused) were evaluated under the optimized reaction condition (DMC: enriched glycerol molar ratio of 4.24:1, catalyst loading of 5.15 wt% (relative to the initial amount of enriched glycerol), reaction temperature of 86 °C, and reaction time of 165 min). The GC yield decreased from 58.77% (first run) to 45.72% after the fourth run (Fig. 13). However, the glycerol carbonate yield for 5 wt% of LiNO3 doped on 5%Li/MCM-41 (4th reused) was 59.23%.
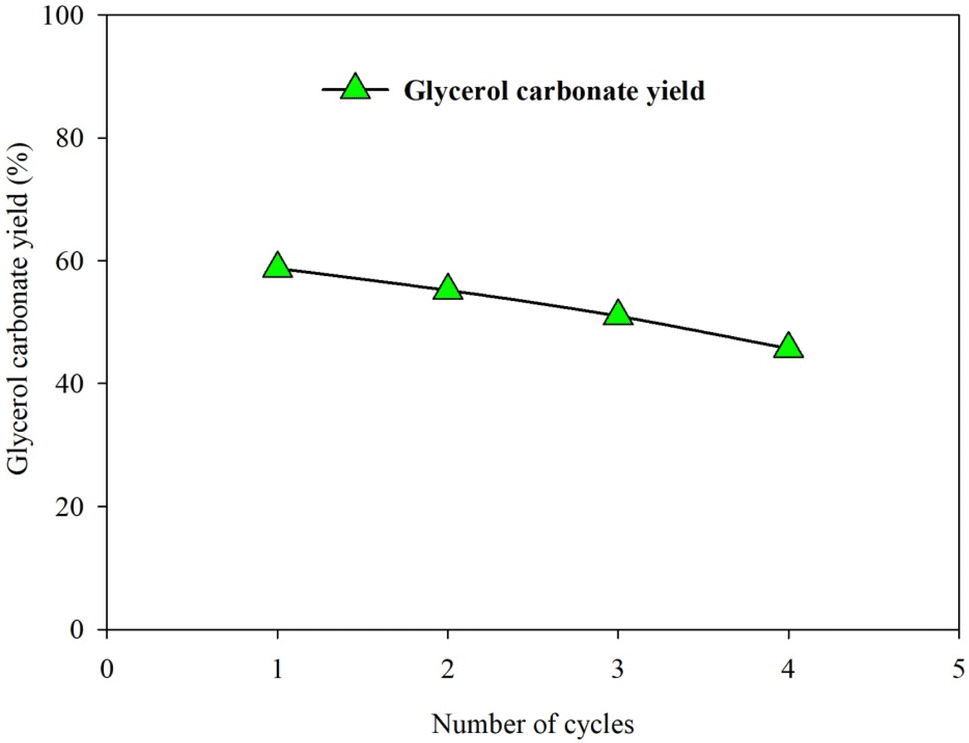 | ||
| Fig. 13 Reusability of 5%Li/MCM-41 catalyst in the GC synthesis from enriched glycerol and DMC. | ||
The ICP-OES analysis revealed that the concentration of Li active species of the 5%Li/MCM-41 was decreased from 3440.00 ± 20 ppm (first run) to 490.30 ± 11.9 ppm after the fourth run (Fig. 14). Thus, the reduced activity of the catalyst over four runs was likely to be due to leaching of the Li active species.
 | ||
| Fig. 14 ICP-OES analysis of fresh and 4th regenerated of 5%Li/MCM-41 catalyst. | ||
The basicity (Table 8) was markedly declined from 4.70 mmol g−1 (fresh catalyst) to 2.82 mmol g−1 after the fourth cycle. This decline suggests a potential gradual reduction in active basic sites, likely due to the leaching of Li during successive applications. The basic strength (Table 8) consistently fell within the range of 9.8 ≤ H_ ≤ 15 for both the fresh and secondarily reused catalysts. However, for the third and fourth cycles, it decreased to 7.2 ≤ H_ ≤ 9.8. This implies that while the number of basic sites may decrease upon reuse, the remaining sites retain a relatively stable strength. A noteworthy finding is the impact of LiNO3 doping on the fourth reused catalyst, resulting in a substantial increase in basicity to 7.62 mmol g−1. This suggests that LiNO3 addition has a rejuvenating effect on the catalyst's basic sites, potentially enhancing their activity. The basic strength remained within the range of 9.8 ≤ H_ ≤ 15, indicating that the added LiNO3 contributes to stronger basic sites.
| Catalyst | Basicity (mmol g−1) | Basic strength (H_) |
|---|---|---|
|
a Reaction conditions: catalyst loading of 5.15 wt%, DMC to enriched glycerol molar ratio of 4.24:1, reaction temperature of 86 °C, and reaction time of 165 min.
|
||
| 5%Li/MCM-41 (fresh) | 4.70 | 9.8 ≤ H_ ≤ 15 |
| 5%Li/MCM-41 (2nd reused) | 4.27 | 9.8 ≤ H_ ≤ 15 |
| 5%Li/MCM-41 (3rd reused) | 3.73 | 7.2 ≤ H_ ≤ 9.8 |
| 5%Li/MCM-41 (4th reused) | 2.82 | 7.2 ≤ H_ ≤ 9.8 |
| 5 wt% of LiNO3 doped on 5% Li/MCM-41 (4th reused) | 7.62 | 9.8 ≤ H_ ≤ 15 |
The surface morphological characteristics of 5%Li/MCM-41 (4th reused) and 5 wt% of LiNO3 doped on 5%Li/MCM-41 (4th reused) at a magnification of 50000× is exhibited in Fig. 4c and d. The morphological images consisted of spherical particles.
3.8 Comparison of various heterogeneous catalysts for GC synthesis
The GC yield obtained with the 5%Li/MCM-41 catalyst in the transesterification of enriched glycerol and DMC is compared to that in other studies44,54–57 in Table 9. The 5%Li/MCM-41 catalyst with enriched glycerol and DMC as reactants gave a high GC yield (58.77%), glycerol conversion level (59.05%), and GC purity (78%). Song et al.44 employed a LiNO3 heterogeneous catalyst for glycerol carbonate production through transesterification using commercial glycerol and DMC as reactants, yielding a glycerol carbonate yield of 15.16% and glycerol conversion of 15.81% under moderate reaction conditions. In contrast, by utilizing the 5%Li/MCM-41 catalyst with purified glycerol and DMC as reactants, a higher glycerol carbonate yield of 58.77% and glycerol conversion of 59.05% was achieved, all while employing lower catalyst loading, reaction temperature, and reaction time. The obtained glycerol carbonate displayed a high purity level of 78% and was colorless, albeit containing traces of purified glycerol. Similarly, Kumar et al.54 utilized a 3Ca–La catalyst, achieving a higher glycerol carbonate yield of 74.0% and glycerol conversion of 94.0%. However, this required a higher DMC to glycerol molar ratio, catalyst loading, and reaction temperature compared to the 5%Li/MCM-41 catalyst. In another study by Parameswaram et al.,56 a Mg/Zr/Sr solid catalyst was employed under similar reaction conditions to Kumar et al.,54 yielding a lower glycerol carbonate yield of 56.0% compared to the 5%Li/MCM-41 catalyst. Likewise, Liu et al.55 utilized hydrotalcites-Ni catalyst, requiring a high catalyst loading (10 wt%) and elevated reaction temperature (100 °C) for a glycerol carbonate yield of 55.0%. Among the prepared catalysts, certain formulations demonstrated notable advantages in their application for GC synthesis. These catalysts exhibited good performance, could be reused for subsequent runs after regeneration, and displayed environmental friendliness. Notably, this study stands out by employing crude glycerol as the reactant, which had undergone a simple enrichment process to obtain the enriched glycerol. This approach not only contributes to environmental sustainability by utilizing a byproduct of biodiesel production but also aligns with the principles of a circular economy. Moreover, the biodegradable nature of glycerol carbonate further enhances its environmental advantages over non-biodegradable alternatives. This strategy presents a cost-effective route, harnessing the value of crude glycerol waste from biodiesel production to yield a value-added product.| Feed glycerol | Catalyst | DMC: enriched glycerol (molar ratio) | Catalyst loading (wt% of enriched glycerol) | Temperature (°C) | Time (min) | GC yield (%) | Glycerol conversion (%) | Selectivity (%) | Reference |
|---|---|---|---|---|---|---|---|---|---|
| a Note: C-glycerol is commercial glycerol. | |||||||||
| C-glycerol | LiNO3 | 2:1 |
5 | 95 | 240 | 15.16 | 15.81 | 95.89 | 40 |
| C-glycerol | 3Ca–La | 5:1 |
10.8 | 90 | 90 | 74.0 | 94.0 | 78.72 | 51 |
| C-glycerol | Hydrotalcites-Ni | 3:1 |
10 | 100 | 120 | 55.0 | Not reported | — | 52 |
| C-glycerol | 3:1:1 Mg/Zr/Sr |
5:1 |
15 | 90 | 90 | 56.0 | 96.0 | 58.33 | 53 |
| C-glycerol | MgO | 5:1 |
15 | 75 | 90 | 12.1 | 12.4 | 97.58 | 54 |
| Enriched glycerol | 5%Li/MCM-41 | 4.24:1 |
5.15 | 86 | 165 | 58.77 | 59.05 | 99.53 | This study |
4. Conclusions
In this study, waste crude glycerol was effectively enriched and then utilized in the synthesis of GC, resulting in a 62.7% glycerol yield and a 98.44% glycerol purity. Among the tested catalysts, the 5%Li/MCM-41 catalyst displayed the highest GC yield (57.18%), followed by the 5%K/MCM-41 (42.33%), 5%Ba/MCM-41 (30.87%), and 5%Na/MCM-41 (25.11%) catalysts, in that order. Based on this assessment, the 5%Li/MCM-41 catalyst was chosen to determine the optimal conditions for GC synthesis. The optimized reaction parameters were identified as a DMC: enriched glycerol molar ratio of 4.24:1, a catalyst loading of 5.15 wt% (relative to the initial amount of enriched glycerol), and a reaction temperature and time of 86 °C and a 165 min, respectively. Under these established conditions, a notable GC yield of 58.77% was attained, underscoring the viability of this process for industrial-scale GC production. Furthermore, the catalyst demonstrated favorable reusability for up to four cycles, enhancing the cost-effectiveness and practicality of the process for industrial applications. These catalysts displayed excellent performance, could be reused after regeneration, and demonstrated environmental friendliness. Noteworthy is the unique aspect of this study, utilizing crude glycerol as the reactant after a simple enrichment process to obtain enriched glycerol.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by National Science, Research and Innovation Fund (NSRF), Thailand and Prince of Songkla University (Grant no. ENG6505084c) and Faculty of Engineering, Graduate School, Prince of Songkla University for PhD Program, Thailand.References
- J. Zhu, D. Chen, Z. Wang, Q. Wu, Z. Yin and Z. Wei, Chem. Eng. Sci., 2022, 258, 117760 CrossRef CAS.
- S. Jaiswal, S. Sahani and Y. C. Shar, J. Environ. Chem. Eng., 2022, 10(3), 107485 CrossRef CAS.
- A. Na Rungsi, A. Luengnaruemitchai, N. Chollacoop, S. Y. Chen, T. Mochizuki, H. Takagi and Y. Yoshimura, Fuel, 2023, 331, 125919 CrossRef CAS.
- G. Pradhan, S. Jaiswal and Y. C. Sharma, Mol. Catal., 2022, 526, 112332 CrossRef CAS.
- S. Nomanbhay, M. Y. Ong, K. W. Chew, P. L. Show, M. K. Lam and W. H. Chen, Energies, 2020, 13(6), 1–23 CrossRef.
- S. Arora, V. Gosu, U. K. A. Kumar and V. Subbaramaiah, J. Cleaner Prod., 2021, 295, 126437 CrossRef CAS.
- J. M. López, F. J. P. Flores, E. C. Rosales, E. O. Muñoz, S. Hernández-Anzaldo, H. V. Lima and Y. R. Ortega, Chem. Eng. J. Adv., 2022, 10, 100257 CrossRef.
- S. Brahma, B. Nath, B. Basumatary, B. Das, P. Saikia, K. Patir and S. Basumatary, Chem. Eng. J. Adv., 2022, 10, 100284 CrossRef CAS.
- L. R. Kumar, S. K. Yellapu, R. D. Tyagi and X. Zhang, Bioresour. Technol., 2019, 293, 122155 CrossRef CAS PubMed.
- C. Ningaraju, K. V. Yatish, P. R. Mithun, M. Sakar and B. R. Geetha, J. Cleaner Prod., 2022, 363, 132448 CrossRef.
- C. G. Chol, R. Dhabhai, A. K. Dalai and M. Reaney, Fuel Process. Technol., 2018, 178, 78–87 CrossRef CAS.
- S. Kongjao, S. Damronglerd and M. Hunsom, Korean J. Chem. Eng., 2010, 27(3), 944–949 CrossRef CAS.
- R. Dhabhai, E. Ahmadifeijani, A. K. Dalai and M. Reaney, Sep. Purif. Technol., 2016, 168, 101–106 CrossRef CAS.
- V. P. Indran, N. A. S. Zuhaimi, M. A. Deraman, G. P. Maniam, M. M. Yusoff, T. Y. Hin and M. H. Ab. Rahim, RSC Adv., 2014, 48, 25257–25267 RSC.
- J. R. Ochoa-Gómez, O. Gómez-Jiménez-Aberasturi, C. Ramírez-López and M. Belsué, Org. Process Res. Dev., 2012, 16(3), 389–399 CrossRef.
- S. Jaiswal, G. Pradhan and Y. C. Sharma, J. Taiwan Inst. Chem. Eng., 2021, 128, 388–399 CrossRef CAS.
- A. Das, D. Shi, G. Halder and S. L. Rokhum, Fuel, 2022, 330, 125511 CrossRef CAS.
- J. Keogh, G. Deshmukh and H. Manyar, Fuel, 2022, 310, 122484 CrossRef CAS.
- G. Pradhan and Y. C. Sharma, J. Cleaner Prod., 2021, 315, 127860 CrossRef CAS.
- J. Hu, J. Li, Y. Gu, Z. Guan, W. Mo, Y. Ni, T. Li and G. Li, Appl. Catal., A, 2010, 386, 188–193 CrossRef CAS.
- T. Mizuno, T. Nakai and M. Mihara, Heteroat. Chem., 2010, 21(7), 541–545 CrossRef CAS.
- J. Liu, Y. Li, H. Liu and D. He, Biomass Bioenergy, 2018, 118, 74–83 CrossRef CAS.
- C. Hu, M. Yoshida, H. C. Chen, S. Tsunekawa, Y. F. Lin and J. H. Huang, Chem. Eng. Sci., 2021, 235, 116451 CrossRef CAS.
- L. Wang, Y. Ma, Y. Wang, S. Liu and Y. Deng, Catal. Commun., 2011, 12(15), 1458–1462 CrossRef CAS.
- K. Jagadeeswaraiah, C. R. Kumar, P. S. S. Prasad, S. Loridant and N. Lingaiah, Appl. Catal., A, 2014, 469, 165–172 CrossRef CAS.
- G. P. Fernandes and G. D. Yadav, Catal. Today, 2018, 309, 153–160 CrossRef CAS.
- P. H. P. S. Guedes, R. F. Luz, R. M. Cavalcante and A. F. Young, Biomass Bioenergy, 2023, 168, 106659 CrossRef CAS.
- B. Das and K. Mohanty, Ind. Eng. Chem. Res., 2019, 58(35), 15803–15817 CrossRef CAS.
- G. Pradhan, S. Jaiswal and Y. C. Sharma, Environ. Technol. Innovation, 2021, 23, 101568 CrossRef CAS.
- S. Jaiswal and Y. C. Sharma, Waste Manage., 2023, 156, 148–158 CrossRef CAS PubMed.
- Y. Huang, G. Zhang and Q. Zhang, ACS Omega, 2021, 6(5), 3875–3883 CrossRef CAS PubMed.
- S. Arora, V. Gosu, V. Subbaramaiah and T. C. Zhang, Can. J. Chem. Eng., 2022, 100(8), 1868–1883 CrossRef CAS.
- H. S. Kusuma, A. Ansori, S. Wibowo, D. S. Bhuana and M. Mahfud, Korean Chem. Eng. Res., 2018, 56(4), 435–440 CAS.
- H. S. Kusuma, A. N. Amenaghawon, H. Darmokoesoemo, Y. A. B. Neolaka, B. A. Widyaningrum, C. L. Anyalewechi and P. I. Orukpe, Environ. Technol. Innovation, 2021, 24, 102005 CrossRef CAS.
- H. S. Kusuma, A. N. Amenaghawon, H. Darmokoesoemo, Y. A. B. Neolaka, B. A. Widyaningrum, S. U. Onowise and C. L. Anyalewechi, Ind. Crops Prod., 2022, 186, 115194 CrossRef.
- D. Moentamaria, Z. Irfin, F. Maya and H. S. Kusuma, Egypt. J. Chem., 2023, 66, 199–205 Search PubMed.
- S. Hu, X. Luo, C. Wan and Y. Li, J. Agric. Food Chem., 2012, 60(23), 5915–5921 CrossRef CAS.
- M. Nanda, Z. Yuan, W. Qin, M. Poirier and X. Chunbao, Austin Chem. Eng., 2014, 1(1), 1–7 Search PubMed.
- M. L. Pisarello, B. O. Dalla Costa, N. S. Veizaga and C. A. Querini, Ind. Eng. Chem. Res., 2010, 49(19), 8935–8941 CrossRef CAS.
- M. L. Pisarello, M. A. Maquirriain and C. A. Querini, Energy Fuels, 2018, 32, 8431–8437 CrossRef CAS.
- A. A. Abdul Raman, H. W. Tan and A. Buthiyappan, Front. Chem., 2019, 7, 1–9 CrossRef.
- M. Yuliana, L. Trisna, F. Sari and V. B. Lunardi, J. Environ. Chem. Eng., 2021, 9(3), 105239 CrossRef CAS.
- F. D. Pitt, A. M. Domingos and A. A. C. Barros, S. Afr. J. Chem. Eng., 2019, 29, 42–51 Search PubMed.
- X. Song, Y. Wu, F. Cai, D. Pan and G. Xiao, Appl. Catal., A, 2017, 532, 77–85 CrossRef CAS.
- Y. Li, J. Liu and D. He, Appl. Catal., A, 2018, 564, 234–242 CrossRef CAS.
- R. Malhotra and A. Ali, Renewable Energy, 2018, 119, 32–44 CrossRef CAS.
- S. Arora, V. Gosu, V. Subbaramaiah and B. H. Hameed, J. Environ. Chem. Eng., 2021, 9(5), 105999 CrossRef CAS.
- P. Sikarwar, U. K. A. Kumar, V. Gosu and V. Subbaramaiah, J. Environ. Chem. Eng., 2018, 6, 1736–1744 CrossRef CAS.
- M. Palaniappan, D. Selvaraj, S. Kandasamy, Y. H. Kahng, M. Narayanan, R. Rajendran and R. Rangappan, Environ. Res., 2022, 215, 114325 CrossRef CAS PubMed.
- Y. Wang, C. Liu, J. Sun, R. Yang and W. Dong, Sci. China: Chem., 2015, 58(4), 708–715 CrossRef CAS.
- B. Maleki, B. Singh, H. Eamaeili, Y. K. Venkatesh, S. S. A. Talesh and S. Seetharaman, Ind. Crops Prod., 2023, 193, 116261 CrossRef CAS.
- G. P. Deshmukh and G. D. Yadav, Mol. Catal., 2021, 515, 111934 CrossRef CAS.
- A. Chotchuang, P. Kunsuk, A. Phanpitakkul, S. Chanklang, M. Chareonpanich and A. Seubsai, Catal. Today, 2022, 388–389, 351–359 CrossRef CAS.
- P. Kumar, P. With, V. C. Srivastava, R. Gläser and I. M. Mishra, Ind. Eng. Chem. Res., 2015, 54(50), 12543–12552 CrossRef CAS.
- P. Liu, M. Derchi and E. J. M. Hensen, Appl. Catal., B, 2014, 144, 135–143 CrossRef CAS.
- G. Parameswaram, M. Srinivas, B. Hari Babu, P. S. Sai Prasad and N. Lingaiah, Catal. Sci. Technol., 2013, 3(12), 3242–3249 RSC.
- J. R. Ochoa-Gómez, O. Gómez-Jiménez-Aberasturi, B. Maestro-Madurga, A. Pesquera-Rodríguez, C. Ramírez-López, L. Lorenzo-Ibarreta, J. Torrecilla-Soria and M. C. Villarán-Velasco, Appl. Catal., A, 2009, 366(2), 315–324 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |