
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLithium aluminate flotation by pH- and light-switchable collectors based on the natural product punicine†
Ali Zgheiba,
Sophie Ackera,
Maximilian Hans Fischera,
Jan C. Namysloa,
Franziska Strubeb,
Martin Rudolphb,
Ursula E. A. Fittschenc,
Annett Wollmannd,
Alfred P. Weberd,
Martin Niegere and
Andreas Schmidt *a
*a
aClausthal University of Technology, Institute of Organic Chemistry, Leibnizstrasse 6, D-38678 Clausthal-Zellerfeld, Germany. E-mail: schmidt@ioc.tu-clausthal.de
bHelmholtz-Zentrum Dresden-Rossendorf, Helmholtz Institute Freiberg for Resource Technology, Chemnitzer Str. 40, D-09599 Freiberg, Germany
cClausthal University of Technology, Institute of Inorganic and Analytical Chemistry, Arnold-Sommerfeld-Str. 4, D-38678 Clausthal-Zellerfeld, Germany
dClausthal University of Technology, Institute of Particle Technology, Leibnizstrasse 19, D-38678 Clausthal-Zellerfeld, Germany
eUniversity of Helsinki, Department of Chemistry, P. O. Box 55, FIN-00014 Helsinki, Finland
First published on 20th March 2024
Abstract
Derivatives of the natural product punicine [1-(2′,5′-dihydroxyphenyl)pyridinium chloride] were developed as switchable collectors for the flotation of lithium-containing engineered artifical minerals (EnAMs). These EnAMs are e.g. formed by pyrometallurgical processing of end-of-life lithium-ion batteries. Depending on the pH value and the lighting conditions, punicines exist in water as cations, two different electrostatically neutral mesomeric betaines, anionic tripoles, radical cations or radical anions. The radical species form by photochemically induced disproportionation reactions. We prepared punicine derivatives introducing alkyl chains in the pyridinium moiety (4-methyl, 4-ethyl, 4-octyl and 4-undecanyl) to install hydrophobic groups and examined the recovery rates of the flotation of lithium aluminate (LiAlO2). We varied the lighting conditions (darkness, daylight, LED irradiation at λ = 390–400 nm) and the pH value, the collector's and frother's concentration, and the flotation time. With our collectors, recovery rates of lithium aluminate up to 90% were accomplished when the flotation was conducted in Hallimond tubes exposed to daylight at pH 11 in water.
Introduction
Modern technologies cause a considerably increasing demand for raw materials. Thus, the technology-critical element lithium plays a crucial role for energy storage in lithium-ion batteries especially caused by the current development of electromobility. Apart from batteries, lithium is also applied in the optoelectronic1 and nuclear2 industries, in ceramics,3 glass,4 lubricants,5 and refrigeration technologies.6 Although electric vehicles fortunately have already avoided 53 Mt of carbon dioxide in 2019,7 the increasing use of lithium-ion batteries is problematic from different viewpoints.8 Thus, assuming a 36% annual increase of electric vehicles, as predicted by the International Energy Agency,7 370 kt of lithium will be required in 2030. Lithium, however, is produced mainly by two countries, Australia (ca. 55%) and Chile (ca. 23%)9 and is considered as critical raw material for the EU's raw material supply.10 Hence, the effective recovery of lithium from waste such as end-of-life lithium-ion batteries becomes crucial.11 The pyrometallurgical processing is one of the most important routes to recycle lithium-ion batteries. Whereas the key elements cobalt, nickel and copper form alloys during this process, the ignoble element lithium is usually found in the slag phase.12 Intense research in mineral processing technologies is currently directed toward the formation of lithium-rich artificial minerals in the slags, their separation, and the generation of concentrates for subsequent hydrometallurgical processing. Lithium aluminate (LiAlO2) is a promising engineered artificial mineral (EnAM) with respect to this recycling route. It forms from Li2O–Al2O3–SiO2–CaO-mixtures, and the influence of other elements such as manganese or magnesium is currently being investigated.13 The most important and versatile mineral processing technique to separate valuable minerals from undesired gangue materials is flotation.14 This technique exploits the difference in surface hydrophobicity of fine particles, which are obtained by crushing and grinding of raw minerals to separate valuable components from interlocking particles. An aqueous suspension is conditioned with a selection of reagents such as collectors, frothers, depressants, activators, and regulators before air is blown into the dispersion. Whereas hydrophilic particles remain suspended in the pulp, hydrophobic particles collide with and attach to the rising air bubbles forming a froth which can be separated. The aforementioned reagents influence the interfacial properties of particles and bubbles and thus affect their interactions. The overall success of the flotation is i.a. governed by an interplay between these interactions and hydrodynamic parameters.15 The interaction of the valuable mineral and the bubble can be controlled by the use of collectors, that attach selectively to said particles, rendering them hydrophobic and thus enhance the probability of attachment to the gas bubble. The goal of intense efforts is often directed toward the maximization of the adsorption of the collectors on the surfaces to enhance selectively the hydrophobicity of the particles and improve particle-bubble attractive interactions.16 Sodium oleate has been described as a very simple surfactant-type collector for the flotation of lithium aluminate,17 and other collectors have also been developed to improve the economic potential of the process.18 As example, it has been shown that lithium aluminate can be separated from gangue materials such as silicates by flotation.19Our studies aimed at the development of switchable collectors, whose properties can reversibly be adjusted by external parameters such as pH value and the lighting conditions during the flotation. Based on our previous experiences, we designed and synthesized collectors based on the natural product punicine and studied the application of these punicine derivatives in flotation. We chose the EnAM lithium aluminate (LiAlO2) as valuable and gehlenite (Ca2Al[AlSiO7]) as gangue material for our experiments.
The alkaloid punicine was discovered in its protonated form 1 in the leaves of Punica granatum20 (pomegranates) and characterized later.21 At neutral pH, it belongs to the substance class of heterocyclic mesomeric betaines, because it can exclusively be represented by canonical formulae in which the positive and negative charges are delocalized within the common π-electron system22 (Scheme 1). Under these conditons, punicine exists as a mixture of two different tautomers, 1+/−A and 1+/−B which are in equilibrium. These tautomers possess different chemical and physical properties, because 1+/−A is a conjugated and 1+/−B is a cross-conjugated mesomeric betaine. In bases, deprotonation of the mesomeric betaines 1+/−A and 1+/−B occurs so that an anionic tripole 1− is formed which delocalizes two negative and one positive charge within its common π-electron system.23 The most outstanding property of punicines, however, is their switchability by light. On irradiation, punicines form radical species such as radical cations 1+ and radical anions 1− depending on the conditions.24
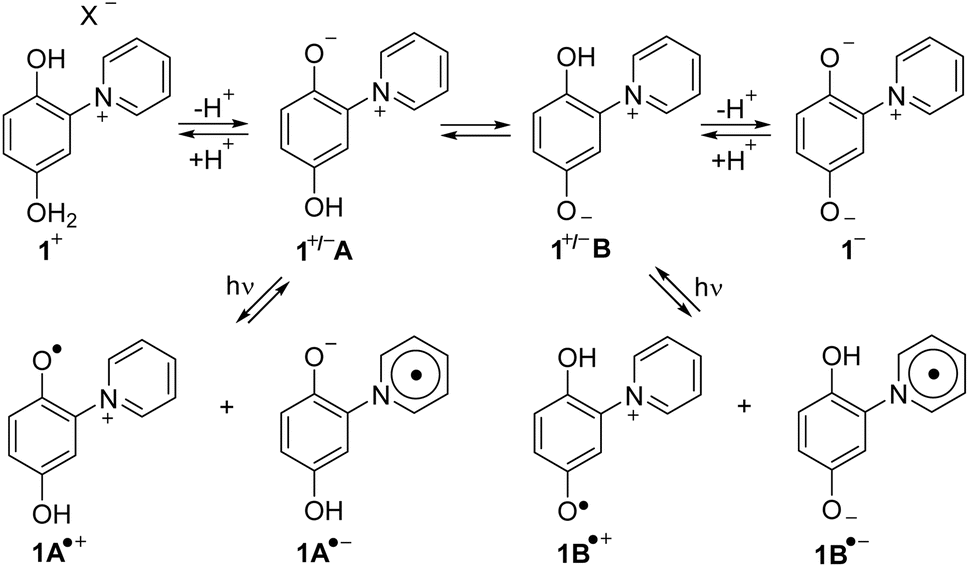 | ||
| Scheme 1 Structure and properties of the natural product punicine. | ||
Correspondingly, punicine gives an intense signal in ESR spectroscopy25,26 at g = 2.003927 (W band, 94.2 GHz) depending the conditions, which shows a hyperfine triplet (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) attributable to the 14N nucleus. The radical ions of punicine and of its polymer-functionalized derivatives have also been measured and calculated.27 Due to these properties, derivatives of punicine and its derivatives can be used in coupled photocatalytic electron-transfers,26 as partial structures of photoresponsive28,29 and redox-active materials,30 and as substituents for non-symmetric bis(thienyl)ethene (BTE) based molecular switches.31 Both tautomers 1+/−A and 1+/−B are π-donors as well as π-acceptors, as they possess π-electron-rich phenolate and π-electron-poor pyridinium rings, respectively. In addition, they have σ-donating groups in common (Fig. 1).24 The σ-, π- and chemical properties of the two tautomers 1+/−A and 1+/−B, however, are different. As example, electron-deficient boron compounds such as B(C6F5)3 selectively form adducts to the 2-olate group of tautomer 1+/−A,32 and cycloadditions with diethyl diazodicarboxylate (DEAD) as electron-deficient dienophile also start from this position.25 On tautomerization, the olate and hydroxyl group change their roles as hydrogen bond donors and acceptors.
:1:1) attributable to the 14N nucleus. The radical ions of punicine and of its polymer-functionalized derivatives have also been measured and calculated.27 Due to these properties, derivatives of punicine and its derivatives can be used in coupled photocatalytic electron-transfers,26 as partial structures of photoresponsive28,29 and redox-active materials,30 and as substituents for non-symmetric bis(thienyl)ethene (BTE) based molecular switches.31 Both tautomers 1+/−A and 1+/−B are π-donors as well as π-acceptors, as they possess π-electron-rich phenolate and π-electron-poor pyridinium rings, respectively. In addition, they have σ-donating groups in common (Fig. 1).24 The σ-, π- and chemical properties of the two tautomers 1+/−A and 1+/−B, however, are different. As example, electron-deficient boron compounds such as B(C6F5)3 selectively form adducts to the 2-olate group of tautomer 1+/−A,32 and cycloadditions with diethyl diazodicarboxylate (DEAD) as electron-deficient dienophile also start from this position.25 On tautomerization, the olate and hydroxyl group change their roles as hydrogen bond donors and acceptors.
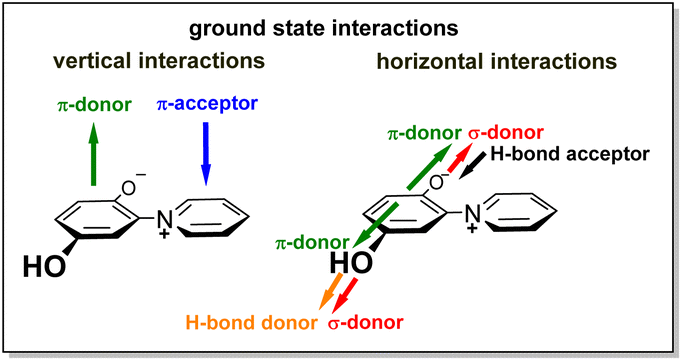 | ||
| Fig. 1 Intermolecular interactions of the zwitterionic punicine. | ||
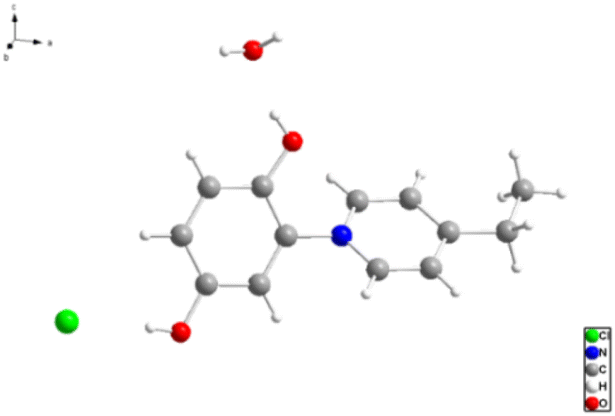 | ||
| Fig. 2 Crystal structure of 1-(2′,5′-dihydroxyphenyl)-4-ethylpyri-dinium chloride 3 (“C2-punicine”). Displacement parameters are drawn at 50% probability level. | ||
Hence, it was concluded that on changing the pH value or the lighting conditions, the intermolecular interactions toward inorganic particles also change, because the overall-charge of the molecule, the charge distribution, and the vertical as well as the horizontal interactions change. We chose four punicine derivatives which possess a methyl, an ethyl, an octanyl and an undecanyl chain in the 4-position of the pyridinium ring, respectively, to gradually enhance the hydrophobicity in this part of the molecule. In continuation of our interest in the chemistry of mesomeric betaines,33 switchable molecules,34 the intersection of organic chemistry and recycling,35 and in view of previous results concerning switchable metal-ion punicine interactions in solution,41 we report here on the applicability of punicines as switchable collectors for the single mineral flotation of lithium aluminate and flotations in the presence of its gangue material gehlenite.
Results and discussion
Syntheses
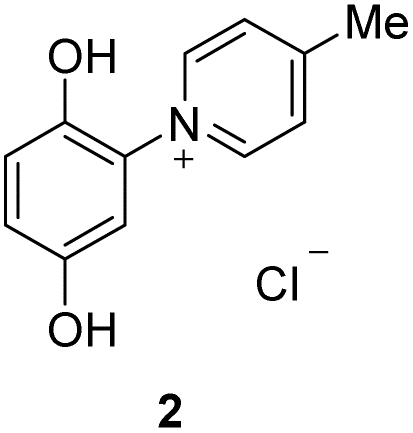
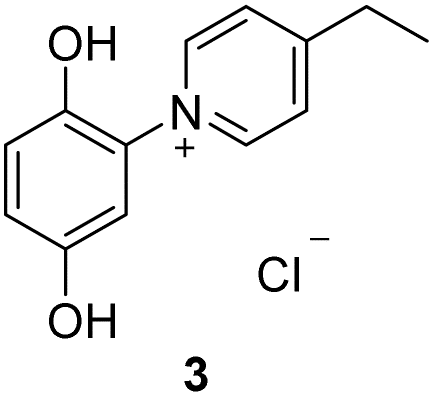
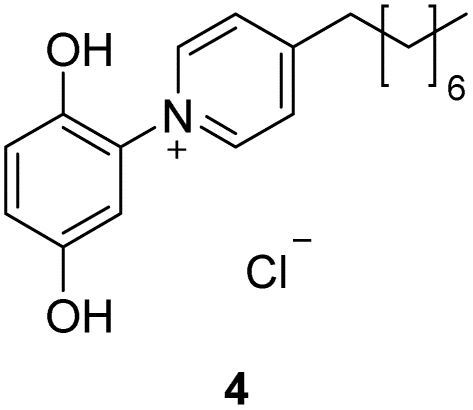
The target punicines were prepared as follows. Reaction of p-benzoquinone with 4-methylpyridine, 4-ethylpyridine, 4-octylpyridine, 4-undecylpyridine, respectively, in the presence of glacial acetic acid followed by stirring overnight (Scheme 2) gave the desired C1-, C2-, C8- and C11-punicine derivatives 2, 3, 4 and 5, where Cn stands for the chain length in the 4-position of the pyridinium moiety. The starting materials 4-octyl- and 4-undecanyl pyridine were synthesized according to literature36 from 4-methylpyridine by a sequence of deprotonation and alkylation. | ||
| Scheme 2 Synthesis of the 4-alkylated punicine derivatives 2, 3, 4 and 5 with chain lengths of 1, 2, 8, and 11 C-atoms (“C1-, C2-, C8-, C11-punicines”). | ||
We were able to grow a single crystal of C2-punicine 3 possessing an ethyl group in the 4-position from hydrochloric acid, from which a single crystal X-ray analysis was performed. In the single crystal, the pyridinium ring is twisted by 133.84(10)° from planarity. Hydrogen bonds are formed between the OH-groups of the punicine and its chloride anion as well as to one molecule of water of crystallization.
For further analysis, UV-Vis spectra of the punicines were measured in aqueous solutions. As example, C11-punicine 5 shows absorption maxima at λmax = 222.5 nm and λmax = 254.5 nm under neutral conditions, as well as shoulders at approximately λmax = 351 nm (Fig. 3). After the addition of 2 μl of 0.1 M Britton-Robinson buffer to the solution of C11-punicine 5, these maxima shift only slightly. On addition of 2 μl of 0.1 M aqueous sodium hydroxide solution a shoulder at λmax = 338 nm is formed, in addition to two absorption maxima at λmax = 224.5 nm and 254.5 nm. The existence of different absorption bands is indicative of the formation of the dianionic (pH 13), betainic (pH 7) and protonated (pH 2) tautomer of C11-punicine 5, which has been similarly observed for previously reported punicine derivatives.21 The spectra of the C1-, C2-, and C8-punicines 2, 3 and 4 are shown in the ESI.†
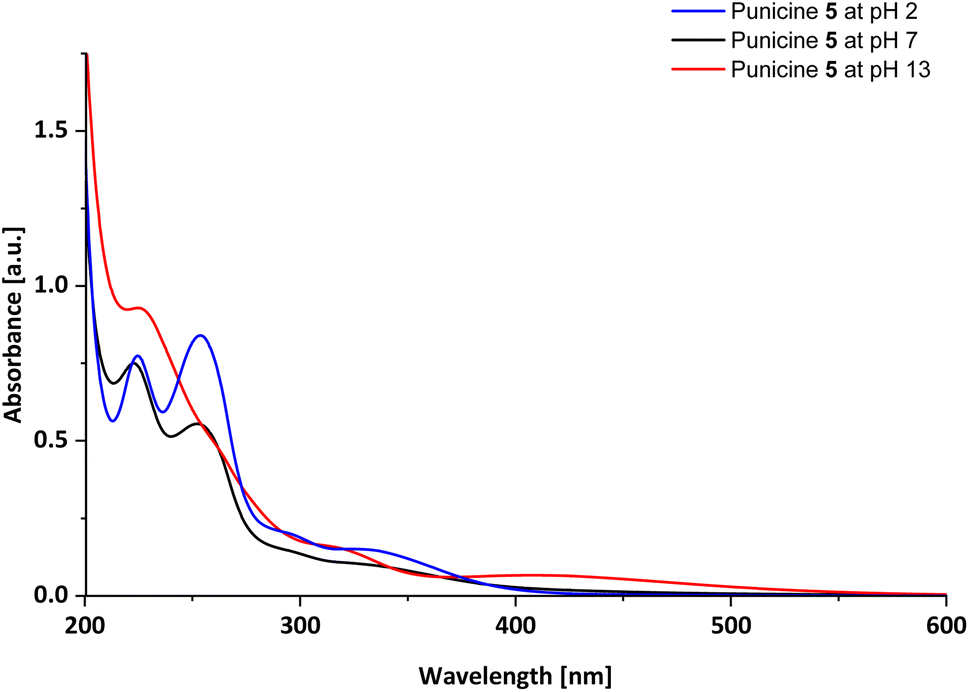 | ||
| Fig. 3 UV-Vis spectrum of C11-punicine 5. | ||
DFT-calculations
The frontier orbital profiles of the different states of C2-punicine 3 were calculated (B3LYP/6-31G*) to gain knowledge about the potential π-donor, π-acceptor, and σ-donor capacities. Fig. 4 shows the highest occupied molecular orbitals (HOMO) of its cationic, zwitterionic, and anionic forms. The HOMO of the cation 3+ is exclusively located in the hydroquinone moiety, whereas considerable atomic orbital coefficients of the HOMO can be seen in the pyridinium ring of the conjugated tautomer 3+/−A. As expected, these are smaller in the cross-conjugated tautomer 3+/−B. On betaine formation, the HOMO energy shifts from −9.13 eV (3+) to −4.24 (3+/−A) and −3.61 (3+/−B) which are therefore much stronger π-bases than the punicine in protonated form. The anionic punicine 3− is an even stronger π-base, as its HOMO energy was calculated to be 0.34 eV. As expected, the LUMO of the cation is exclusively located in the pyridinium ring, indicative of π-acceptor properties. Additional coefficients can be calculated in the hydroquinone rings of the betaines, respectively. These are more pronounced in the conjugated tautomer 3+/−A than in its cross-conjugated analog 3+/−B. The LUMO energy of the cation shifts from −5.85 eV to 1.85 eV which are indicative of considerably reduced π-acceptor properties in the betaines and the anion.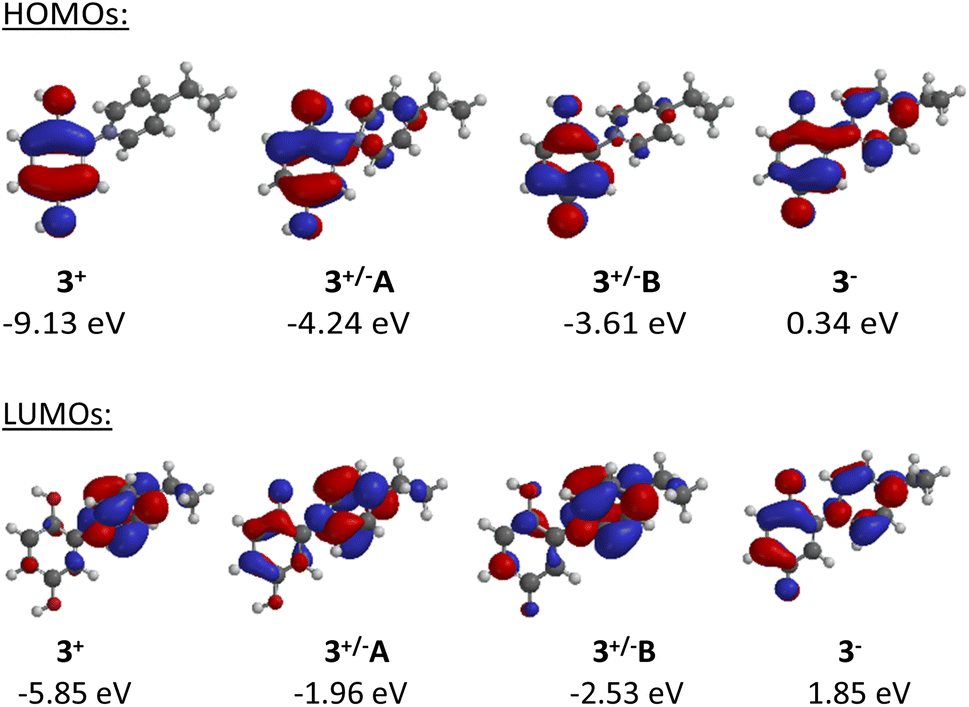 | ||
| Fig. 4 Highest occupied (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) of the different states of C2-punicine 3. | ||
Wettability
Prior to the performance of flotation experiments, the apparent contact angles were measured with a bubble in aqueous environment on particle beds. The results show low contact angles for the pure LiAlO2 of 14.3° ± 3.6° and for gehlenite of 14.2° ± 0.8°, indicating a hydrophilic sample surface. The contact angle of the pure LiAlO2 shows a higher confidence interval caused by bubbles on the sample surface which specifically occur on LiAlO2. Conditioning the samples with the C2-, C8- and C11-punicines 3–5 possessing a longer aliphatic residue, regardless of the type used, slightly increased the contact angle up to 15.1–18.6 ± 2.26° in comparison to those conditioned with C1-punicine 2 which has a methyl group in the γ-position (cf. ESI†). Furthermore, it could be observed that samples containing LiAlO2, especially the conditioned LiAlO2 with punicines 3–5, tend to cause pinning effects of the bubble (cf. ESI†). The results show that the longer the aliphatic residues the more homogenous pinning effects and increasing contact angles could be observed, however, given the large confidence intervals no significant trend can be derived. Furthermore, obtaining contact angles from particles is rather challenging, as a proper contact angle analysis requires a flat, smooth and homogeneous surface. The process used in this study has its limitations, as the contact angle is affected by the roughness of the particle bed, porosity and irregularity of shapes, as well as the appearance of dirt particles or the formation of bubbles.40Single mineral flotation
Lithium aluminate, which serves as model for a lithium EnAM, was purchased from Alfa Aesar. The gehlenite which we used was of natural origin. It has a purity of around 67% and is further composed of 20–24% garnet including iron-rich grossular, as well as impurities of 7% calcite and 1% quartz, as evidenced by XRD measurements (cf. ESI†). The quantitative XRD values have an error between 1–2%. ICP-OES measurements confirmed the varying composition of gehlenite. According to the molecular formula Ca2Al[AlSiO7], the theoretical content is 19.7% of aluminum, 29.2% of calcium and 10.2% of silicon. In the present material, 10.3% of aluminum, 25.6% of calcium and 10.6% of silicon were found as well as small amounts of the elements iron (<5%) and magnesium (<5%) (cf. ESI†). The lithium aluminate was also characterized via XRD and it was found that besides LiAlO2 (59%) also LiAl5O8 (14%) and LiAl2(OH)7·xH2O (27%) were present. Furthermore, particle size distributions were measured. Table 1 summarizes the results. The x50 value for the particle size of the lithium aluminate particles was 15 μm and 90% of the particles were smaller than 43 μm. The gehlenite particle size value for x50 is 25 μm and 90% of the particles are below 73 μm. The particle size distribution ranged between 1 μm to 80 μm.| Material | X16 | X50 | X90 |
|---|---|---|---|
| Lithium aluminate | 7 μm | 15 μm | 43 μm |
| Gehlenite | 11 μm | 25 μm | 73 μm |
Microflotation experiments in Hallimond tubes were done in the presence of the C1-, C2-, C8- and C11-punicine derivatives 2, 3, 4 and 5 as collectors and pine oil as frother, respectively, under variation of the collector's concentration, the pH value (pH 2 and pH 11) as well as the lighting conditions. Thus, we floated in daylight (>3000 lux), under LED UV irradiation (λ = 390–400 nm, 4500 lux), and in the dark (<40 lux). Stock solutions of the individual collectors with a concentration of 1 wt% in H2O or H2O/ethanol (for punicines 4, 5) were prepared. To assure that the addition of ethanol does not interfere with flotation results, a blank test was performed, which confirmed no effect of this co-solvent. All measurements were repeated 5 to 6 times. We determined the average values and the confidence intervals. Table 2 shows the molar concentrations of the collectors in the Hallimond tube that have been deployed. In the experiments, samples of 2.00 g of lithium aluminate were subjected to flotation, and the recovery yields were calculated from the mass ratios of concentrate and the residing pulp. For specifications of the light sources including emission spectra, see the ESI.†
| Collector | |||
|---|---|---|---|
| Conc. [mol l−1] | 1 × 10−5 | 2 × 10−5 | 1 × 10−4 |
| Vol. added [μl] | 60 | 120 | 600 |
| C1 | C2 | C8 | C11 |
 |
 |
 |
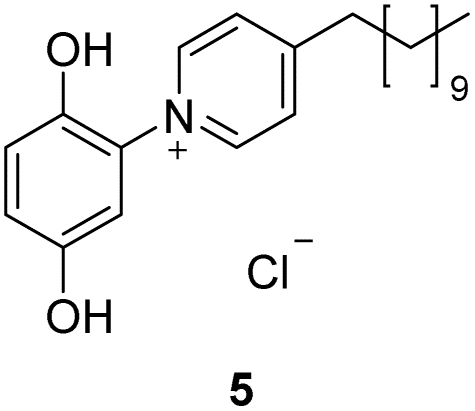 |
We first tested the performance of the C1-, C2-, C8- and C11-punicines 2–5 in the flotations of lithium aluminate under identical conditions. As Fig. 5 shows, the flotation of lithium aluminate without any collectors and frother (“blank”) has an average recovery rate of 17.82 ± 3.70%. Upon the addition of the punicine derivates, the recovery rate increased gradually and reached its maximum value of 49.89 ± 9.09% using C11-punicine 5. Obviously, the chain length of the substituent at C4 of the pyridinium ring influences the flotation's results considerably, as a longer chain length gives better recovery rates. C2-punicine 3 gives 9%, and C8-punicine 5 gives 32% more yield, compared to the blank test (Fig. 5).
 | ||
| Fig. 5 Comparison of the recovery rates in the presence of the collectors 2–5 under identical conditions, i.e. 2.00 g of LiAlO2, 1 min of mixing (500 rpm) with 25 ml of distilled water, 60 μl (1 × 10−5 M) of the corresponding collector, 1 min of mixing, 30 μl of frother, and another 1 min of mixing. The flotation was done for 3 min at an air flow rate 32 ml min−1 using 250 ml distilled water in daylight at pH 11. | ||
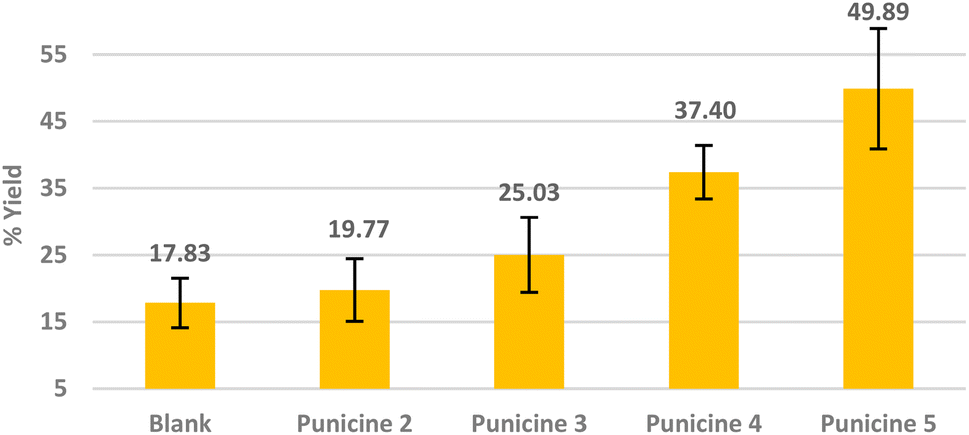
As punicine is a switchable collector, we next performed flotations of lithium aluminate under different lighting conditions, i.e. in daylight, in UV light and in the dark. Flotations in UV light (390–400 nm) were performed in a closed box with four LEDs in the middle of the four walls in which the Hallimond tube was installed. Fig. 6 shows three blank probes with samples of pure lithium aluminate without collector and frother, and the performance of C11-punicine 5 as collector. No changes were observed under the different light conditions in the absence of C11-punicine 5, so that the light does not influence the lithium aluminate itself during the flotation. Applying C11-punicine 5 as collector, a significant effect of the lighting conditions can be measured. Thus, the average yield increases from 44.13 ± 7.54% in daylight to 66.55 ± 2.75% under UV irradiation. Flotation in the dark gave a recovery rate of 61.89 ± 4.58%.
 | ||
| Fig. 6 Flotation of lithium aluminate using C11-punicine 5 as a collector under irradiation in daylight (yellow), in the dark (black), and under UV-light (blue). The conditioning was as following: 120 μl collector (2 × 10−5 M), 1 min mixing, 30 μl frother, 1 min mixing, stirring speed 500 rpm, air flow rate 32 ml min−1 using 250 ml distilled water and flotation time 3 min at pH 11. | ||
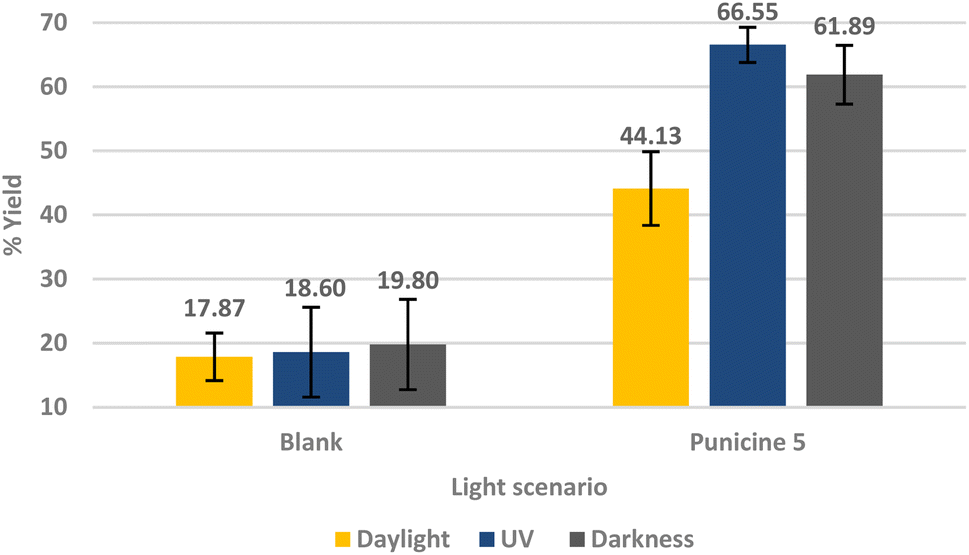
In a control experiment, sodium oleate was used as collector and the lighting conditions were changed as before. Fig. 7 shows that the recovery rate of lithium aluminate does not change on variation of the lighting conditions in the presence of this classical collector.
 | ||
| Fig. 7 Flotation of lithium aluminate using sodium oleate 1% wt as a collector. The conditioning was as following: 60 μl collector (1 × 10−5 M), 1 min mixing, 30 μl frother, 1 min mixing, stirring speed 500 rpm, air flow rate 32 ml min−1 using 250 ml distilled water and flotation time 3 min. | ||
Punicines not only change their properties in dependence on the light but also on variation of the pH. Therefore, we performed flotations under acidic conditions (pH 2) to compare the results with flotations at pH 11, which is the natural pH value of lithium aluminate in aqueous suspension. pH 2 was adjusted by addition of 11 ml of 0.1 M NaOH to 40 ml of 0.1 M Britton-Robinson buffer. Fig. 8 shows a comparison between the natural product punicine from Punica granatum without any substituent in the γ-position (carbon chain length = 0), and the C1-, C2-, C8- and C11-punicines 2, 3, 4, and 5 as collectors in the flotation of lithium aluminate at pH 2 under variation of the lighting conditions. The optimum in this series of flotations was reached with C11-punicine 5 at pH 2 under UV irradiation (54.49 ± 2.67%). At pH 11, this punicine floated 66.54 ± 2.75% of lithium aluminate in the dark. The lowest recovery yields were obtained in daylight in all cases. Additional details are given in the ESI.†
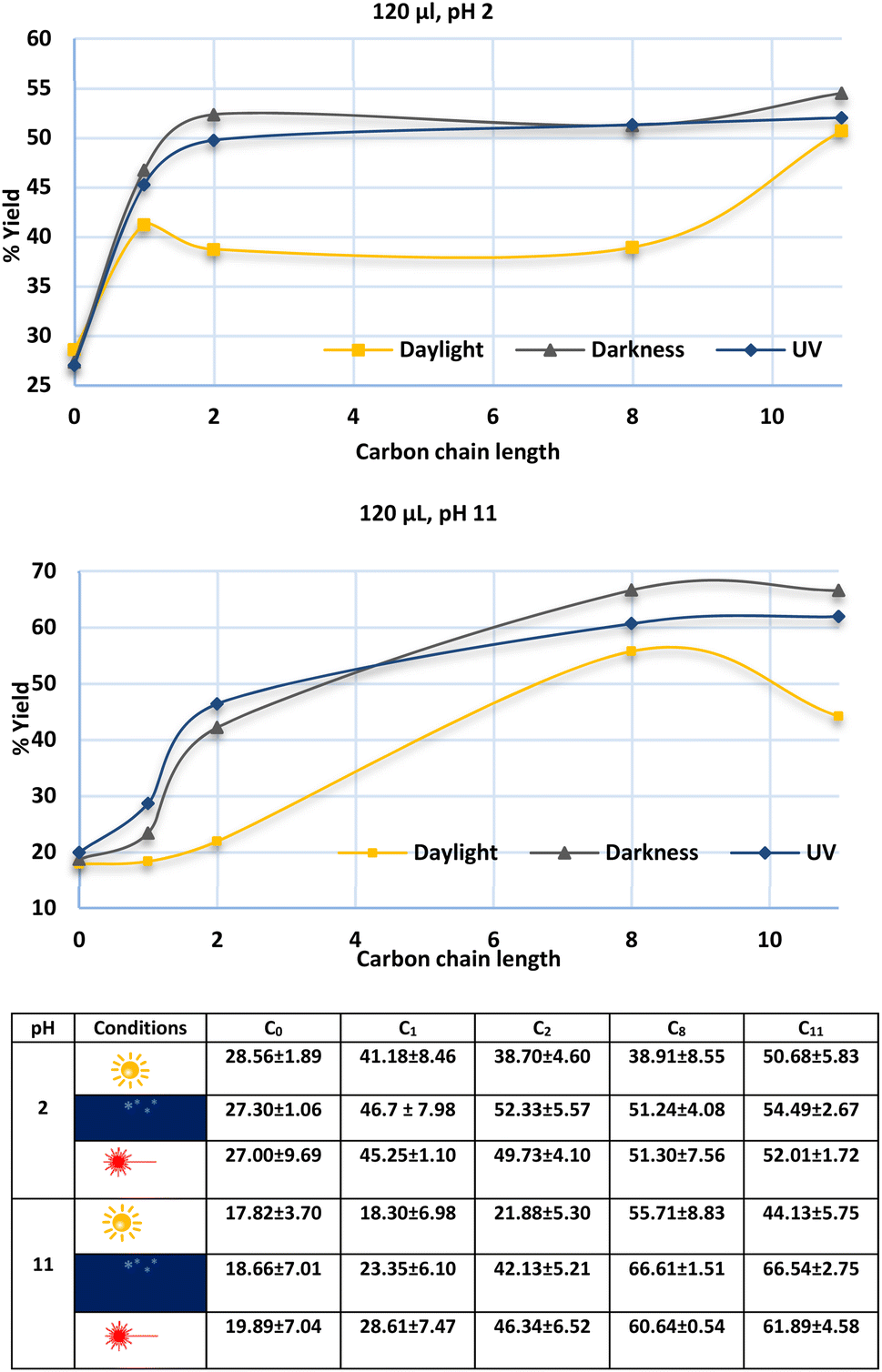 | ||
| Fig. 8 Flotation of lithium aluminate using different punicine derivatives as collector at pH 2 (above) and pH 11 (below), and under different lighting conditions. The conditioning here was as following: 120 μl collector, 1 min mixing, 30 μl frother, 1 min mixing, stirring speed 500 rpm, air flow rate 32 ml min−1 using 250 ml distilled water and flotation time 3 min. The confidence interval is 95%. Symbols that indicate the lighting conditions are explained in Table 3. | ||
In order to test the effect of increasing the collector's concentration and flotation time on the recovery rate of lithium aluminate under different light conditions, the flotation was performed using 600 μl of C8- and C11-punicine 4 and 5 which correspond to 101.96 μmol l−1 and 103.32 μmol l−1 or 61.2 g t−1 LiAlO2 and 70.8 g t−1 LiAlO2, respectively. We floated for 5 min and under the 3 different lighting conditions at pH 11. Lithium aluminate was recovered in 86.2 ± 1.34% and 85.05 ± 7.08% yield by C8- and C11-punicine 4 and 5, respectively, under UV-irradiation under these conditions (Fig. 9), whereas flotations in daylight gave considerably lower yields.
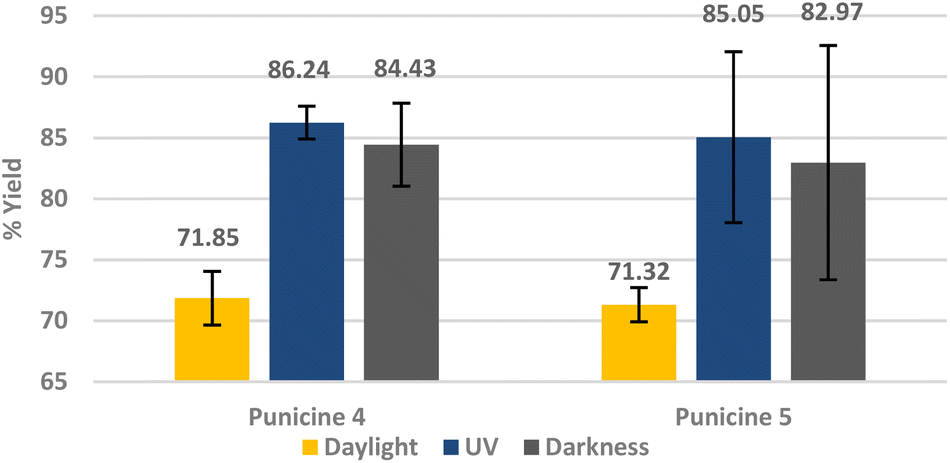 | ||
| Fig. 9 Flotation of lithium aluminate using punicines 4 and 5 as collector at pH 11, and under different lighting conditions. The conditioning was as follows: 600 μl collector (1 × 10−4 M), 1 min mixing, 30 μl frother, 1 min mixing, stirring speed 500 rpm, air flow rate 32 ml min−1 using 250 ml distilled water and flotation time 5 min. | ||
A series of flotation experiments applying an air flow of 170 cm3 min−1 were also conducted. These results are summarized in Table 3. We enlarged the scope of parameters for these studies, enhanced collector concentrations (conditions B, C) and flotation times (conditions A, B: 10 min; condition C: 15 min). Not surprisingly, the recovery rates of lithium aluminate are much higher under these more vigorous conditions. As an example, at pH 2 C1-punicine 2 as collector increased the yield by 156% in comparison to the conditions shown in Fig. 8. At pH 11 similarly increased yields were obtained in comparison to those shown in Fig. 8.
| a Flotation conditions: A: FT = 10 min, 600 μl collector = 101.24 (2) and 95.59 (3) μmol l−1, 1.2 × 10−2 g l−1 frother. B: FT = 10 min, 900 μl collector = 151.86 (2) μmol l−1, 1.2 × 10−2 g l−1 frother. C: FT = 15 min, 900 μl collector = 151.86 (2) μmol l−1, 1.8 × 10−2 g l−1 frother. Values of the recovery rate are average values of 3–6 individual measurements, respectively, and the confidence interval is 95%. |
|---|
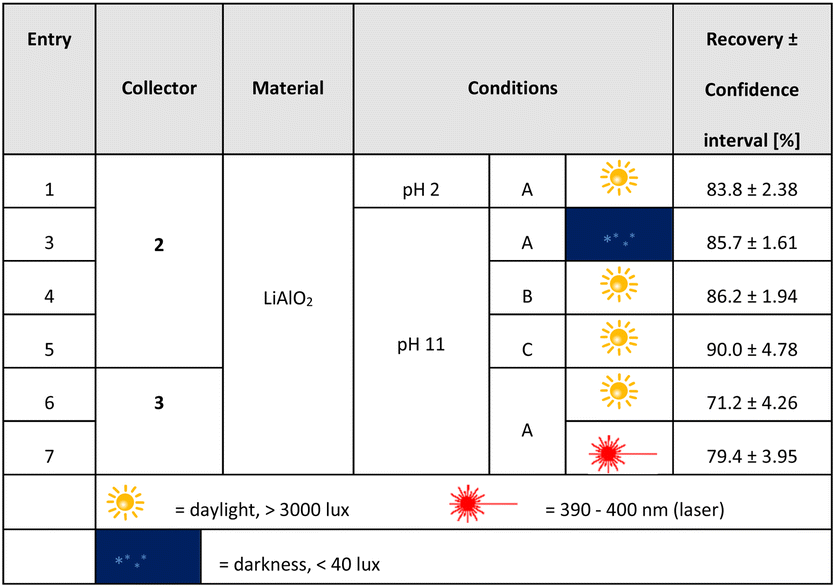 |
Fig. 10 shows the results of flotations of lithium aluminate and of gehlenite under identical conditions in the presence of C11-punicine 5 as collecor. Only 15.29 ± 2.00% recovery of gehlenite was obtained under the conditions applied. This difference in the recovery of lithium aluminate and gehlenite serves as a promising result for a selective separation.
 | ||
| Fig. 10 Single mineral flotation of lithium aluminate and gehlenite using C11-punicine 5 as collector at pH 11 for lithium aluminate and pH 5.8 for gehlenite in daylight. The conditioning was as follows: 60 μl collector, 1 min mixing, 30 μl frother, 1 min mixing, stirring speed 500 rpm, air flow rate 32 cm3 min−1 using 250 ml distilled water and flotation time 3 min. | ||
Mixed mineral flotation
Based on these results a 1:1 mass fraction mixture of lithium aluminate and gehlenite was tested using C11-punicine 5 as collector. Three different flotation conditions were tested: pH 11 in daylight, pH 11 under UV-irradiation, and pH 2 in daylight to take advantage of the light switchability of the punicine. To evaluate the recovery of lithium aluminate and gehlenite, quantitative XRD analysis of the concentrates and tailings were acquired and the results are displayed in Fig. 11. From this it can be noticed that there is a clear trend in the separation, since for all investigated flotation conditions higher fractions of lithium aluminate were recovered in the concentrate, while higher fractions of gehlenite were recovered in the tailings. In combination with the mass balance of the flotation, the recovery of each lithium aluminate fraction and gehlenite fraction was calculated. The results are plotted in a Fuerstenau upgrading plot (Fig. 12), which shows the recovery of lithium aluminate against the recovery of gehlenite. The red solid line represents the splitting curve where no enrichment occurs. The blank indeed shows a higher enrichment of the lithium aluminate even without collector due to its finer particle size and lower density (lithium aluminate 2.62 kg m−3, gehlenite 3.02 kg m−3)39 which influences the settling velocity and thus the entrainment behavior,42 since the entrainment increases with decreasing particle size and decreasing density.43
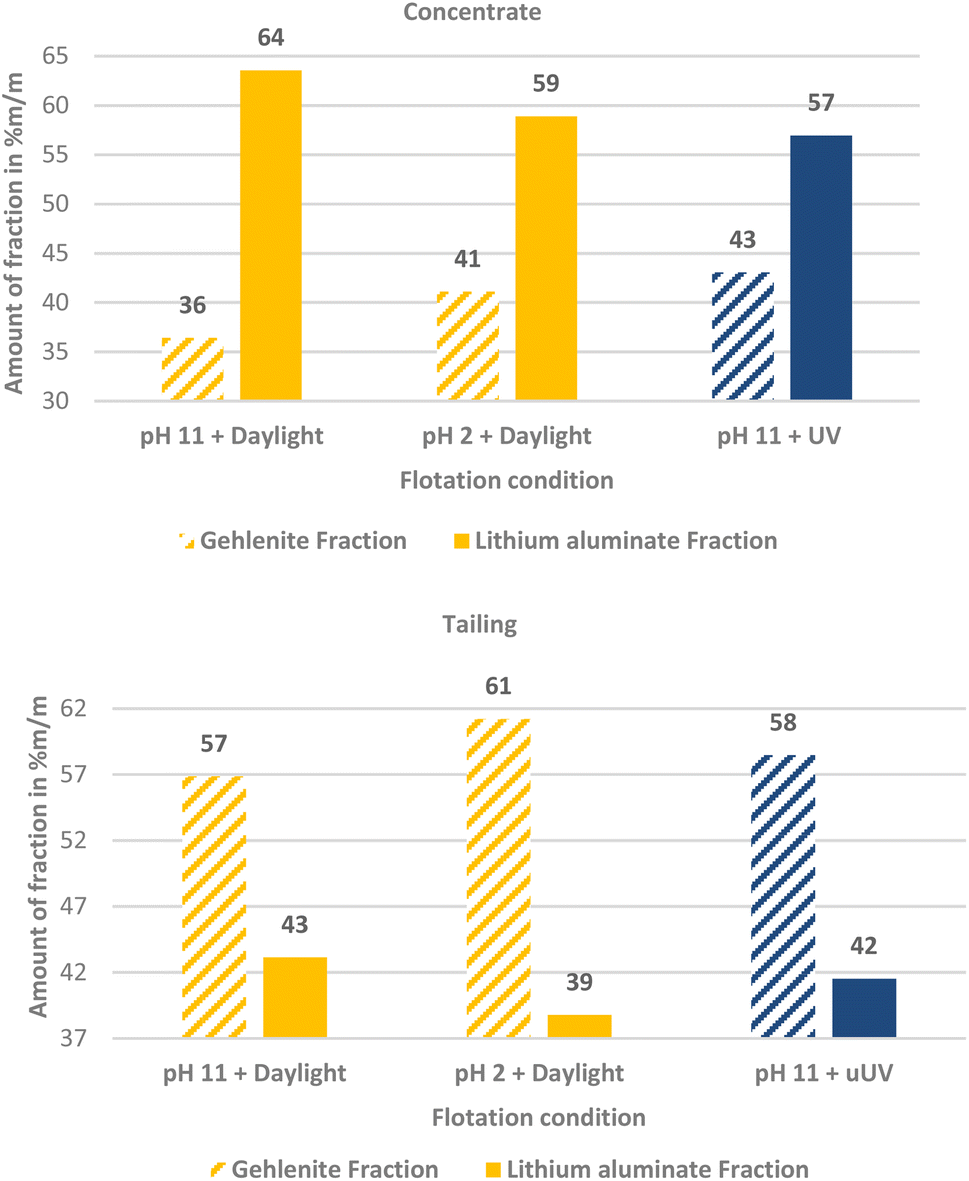 | ||
| Fig. 11 The mass fraction % of lithium aluminate and gehlenite present in the concentrate and the tailing of their mixed micro flotation obtained from quantitative XRD measurements. | ||
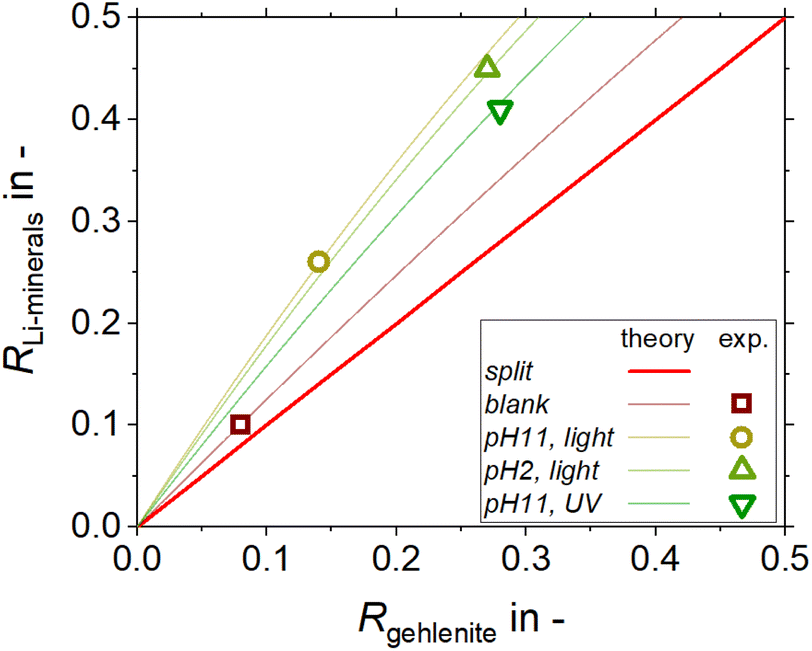 | ||
| Fig. 12 Fuerstenau upgrading diagram for the mixed mineral micro flotation tests with 2.00 g of a 1:1 mixture of LiAlO2 and gehlenite, 1 × 10−5 M C11-punicine 5 (C11), 1.2 × 10−3 g l−1 pine oil, conditioning time 3 min, flotation for 3 min with 32 cm3 min−1 air flow and 250 ml of distilled water. | ||
Analyses of the particle size distribution of the flotation feed and of the products, i.e. concentrate and tailings, further supports this, as a higher fraction of fines is found in the concentrates (see Fig. 13). The same behavior in terms of particle size distribution is observed for the tests where C11-punicine 5 is used, as a higher amount of fines is reporting to the concentrate. Furthermore, with the addition of C11-punicine 5 the slope of the calculated theoretical curves (added to guide the eye) increases, and so does the selectivity. This is most probably due to greater bubble-particle attachment induced by the punicine, resulting in higher recoveries of the lithium aluminate fraction. It can be observed that the condition at pH 11 and under ambient light resulted in the highest selectivity.
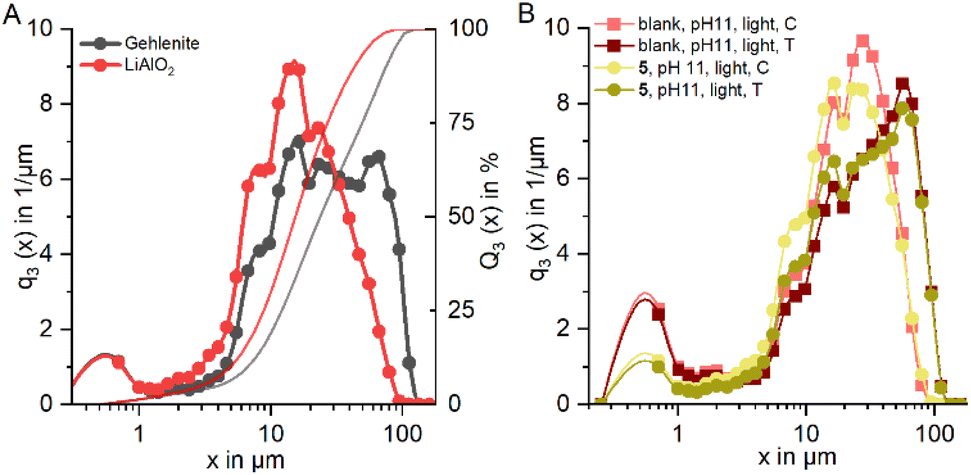 | ||
| Fig. 13 (A) sum and density distribution of gehlenite (black) and lithium aluminate (red); (B) density distribution of blank, pH 11, concentrate and tailing, as well as result of binary flotation with C11-punicine 5 at pH 11 and light exposure. | ||
Zeta potential
To gain more insight into the particle–collector interactions, zeta potential measurements for the mineral lithium aluminate used in this study was carried out over a pH range of 2, 5 and 11. It turned out that the mineral does not provide stable values at certain pH values. In order to visualize the effect of punicines on the zeta potential of lithium aluminate at different pH ranges, similar experiments were done, in which the C8- and C11-punicines 4 and 5 were added. The result is shown in Fig. 14. First, at extreme acidic condition, the zeta potential of pure lithium aluminate was ranging between 2.23 mV and 5.83 mV. Upon the addition of C8-punicine 4, the zeta potential increased significantly to +26.8 mV, which could indicate the assembly of C8-punicine 4 on the surface of lithium aluminate or a reaction with LiAl2(OH)7 on its surface by nucleophilic replacement of OH groups to the punicine, thus increasing its zeta potential. Upon the addition of C11-punicine 5, the zeta potential of lithium aluminate stayed approximately in the same range (2.88 mV). The results could indicate that lithium aluminate interacts more strongly with C8-punicine 4 in acidic medium which could also be noticed in the flotation results, in which the recovery rate of lithium aluminate using C8-punicine 4 in the acidic medium was higher than that using C11-punicine 5. At pH 5, the zeta potential of lithium aluminate was −31.5 mV, it has been increased in both C8-punicine 4 and C11-punicine 5 (−26.5 mV and −23 mV respectively). While in an extreme basic condition, the zeta potential of lithium aluminate has been decreased from −35.2 mV to −46.8 mV and −43.8 mV upon the addition of the punicines 4 and 5, respectively. This confirms the interaction between punicine and lithium aluminate, in which the punicine at basic condition has a negative charge and upon its accumulation at the surface of lithium aluminate, it leads to the further decrease of its zeta potential.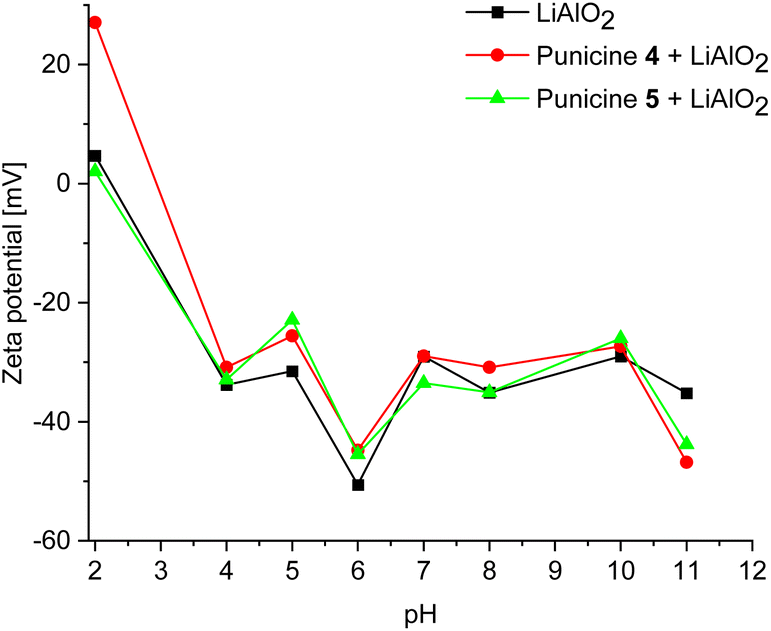 | ||
| Fig. 14 Zeta potential of pure lithium aluminate, lithium aluminate with 2.4 μl of 42.5 mmol l−1 punicine 4 and lithium aluminate with 2.4 μl of 42.5 mmol l−1 punicine 5 in distilled water and under different pH values. | ||
In order to test the effect of concentration of the punicine on the zeta potential of lithium aluminate, a series of zeta potential measurements were done, in which the volume of the added C8-punicine 4 and the C11-punicine 5 were increased from 2.4 μl to 4.8 μl and 7.2 μl at the 3 different pH ranges. Both punicine induce a decrease in the zeta potential of lithium aluminate at pH 11 and pH 5 as their concentration increases. At pH 2 the zeta potential slightly increased upon increasing the volume added of the C11-punicine 5. In C8-punicine 4, upon increasing the volume added from 2.4 μl to 7.2 μl, an increase in the zeta potential of lithium aluminate is observed (27.2 mV). A further increase in punicine concentration increases the zeta potential to 27 mV.
Table 4 summarizes the zeta potential of lithium aluminate affected by C0-, C1- and C2-punicines 1, 2 and 3 under different pH values. A comparison of the values with each other shows that an increase in the concentration of the respective collector leads to an increase in the measured zeta potential. The measurements with 1 × 10−4 M for collector 2 and 3 lead to a decrease of the zeta potential.
| Collector | Amount of collector [mol l−1] | pH value | Zeta potential [mV] |
|---|---|---|---|
| 1 | 1 × 10−5 | 10.50 | −18.43 |
| 2 | 1 × 10−5 | 9.02 | −17.65 |
| 1 × 10−4 | 8.66 | −3.73 | |
| 5 × 10−4 | 10.2 | −12.37 | |
| 3 | 1 × 10−5 | 8.32 | −15.63 |
| 1 × 10−4 | 8.98 | −5.75 | |
| 1.5 × 10−4 | 10.1 | −13.55 |
Conclusion
In summary, first switchable collectors based on punicines for the recovery of the engineered artificial mineral lithium aluminate are presented. With increasing chain length of the alkyl residue of the punicines the recovery rate in micro flotations also increases. Apart from these structural variations, the properties can reversibly be tuned by the lighting conditions and the pH value. Thus, selectivities can be adjusted. Recovery rates up to 90 ± 4.78% were achieved in single mineral flotation experiments of lithium aluminate. In mixed mineral flotations of lithium aluminate and its gangue material gehlenite, separations of both minerals were achieved.Experimental section
General
All chemicals used within this study were purchased from commercial suppliers and used as received. The measurements of 1H and 13C spectra have been performed on Bruker Avance (400 MHz) and Avance III (600 MHz) NMR spectrometer. The multiplicities are given with the following abbreviations: s = singlet, bs = broad singlet, d = doublet, dd = doublet of a doublet, t = triplet, tt = triplet of a triplet, q = quartet, quint = quintet, sext = sextet, m = multiplet. IR-spectroscopy has been performed by the FT-IR-spectrometer alpha-T from Bruker with a platinum-ATR-module. The spectra consider a range of 400 to 4000 cm−1. The high-resolution mass spectra were measured with an Impact II mass spectrometer from Bruker. The samples for the IR experiments were mixed with the respective mineral by suspension in methanol and then dried. The mixing ratio was 50:50.
The chemical analysis of gehlenite was performed with a Varian model Vista MPX and with an evaluation program from Varian. The sample was digested in two steps. In the first step, aqua regia was used, followed by diluted HF (0.8%) in the second. The value shown is the average of three measurements. Lithium aluminate (LiAlO2) was bought from Alfa Aesar. Gehlenite was provided by the Institute of Mineral and Waste Processing, Recycling and Circular Economy Systems, Clausthal-Zellerfeld.
Crystal structure determination of 1-(2,5-dihydroxyphenyl)-4-ethylpyridin-1-ium chloride water solvate (3)
The single-crystal X-ray diffraction study was carried out on a Bruker D8 Venture diffractometer with Photon II detector at 173(2) K using Mo-Kα radiation (λ = 0.71073 Å). Dual space methods (SHELXT)44 were used for structure solution and refinement was carried out using SHELXL-2014 (full-matrix least-squares on F2). Hydrogen atoms were localized by difference electron density determination and refined using a riding model (H(O) free). A semi-empirical absorption correction was applied.3: Yellow crystals, C13H14NO2+ Cl−·H2O, Mr = 269.72, crystal size 0.35 × 0.20 × 0.15 mm, monoclinic, space group P21/n (no. 14), a = 11.9361(3) Å, b = 7.3938(2) Å, c = 14.4601(4) Å, β = 93.592(1)°, V = 1273.64(6) Å3, Z = 4, ρ = 1.407 Mg m−1−3, μ(Mo-Kα) = 0.30 mm−1, F(000) = 568, T = 173(2) K, 2θmax = 55.2°, 31472 reflections, of which 2944 were independent (Rint = 0.054), 175 parameters, 6 restraints, R1 = 0.030 (for 2801 I > 2σ (I)), wR2 = 0.083 (all data), S = 1.05, largest diff. peak/hole = 0.28/−0.19 e Å−3.
Flotation experiments
The flotation experiments have been performed in a Hallimond tube (Fig. 15) with a volume of 250 ml. Lithium aluminate and gehlenite were tested as the single minerals (2.00 g for each measurement). The collector concentrations are in a range of 7.87 μmol l−1 to 151.86 μmol l−1 and for the frother (pine oil) between 1.2 × 10−3 g l−1 and 1.8 × 10−2 g l−1. In addition to the concentrations and the flotation time, the pH-value was varied. The air flows were adjusted to 32 cm3 min−1 and 170 cm3 min−1. The continuous stirring speed of the magnetic stirrer was 500 rpm. The conditioning time was three times one minute for each addition step. The flotation time varied between three and ten minutes as indicated in the tables. In addition to the concentrations and the flotation time were also varied the pH-value and the light scenario from daylight (>5000 lux) to halogen element (400 W, >8550 lumen) or laser (390–400 nm) and to darkness (<40 lux).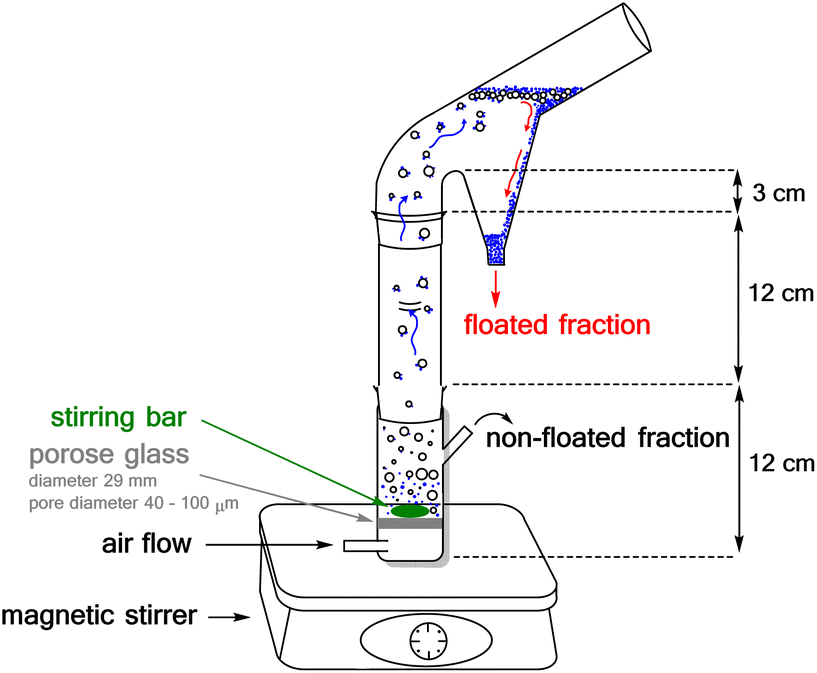 | ||
| Fig. 15 Pictorial representation of the Hallimond tube used. | ||
The Hallimond tubes used (250 ml) possess a medium-pore fritted glass through which the air can flow into the apparatus. A magnetic stirrer, adjusted to 500 rpm, ensures continuous mixing. The flotation tailings are collected separately in the upper area during flotation. In the first step, the mineral was conditioned in distilled water or buffer (one minute). The buffer consisted of 0.04 M boric acid, 0.04 M phosphoric acid, and 0.04 M acetic acid, and the required pH was achieved by the addition of 0.1 M NaOH. The buffer was used for measurements at a pH of 2. Then the collector was added and, after another minute, the frother. The remaining volume was filled up to 250 ml and floated for 3 or 10 minutes.
The light tests were carried out in a closed box in which the Hallimond tube was installed. The box measures 60 × 60 × 60 cm and can be opened on two sides with hinges. In the middle of each of the four walls there is a aluminium heat sink from Fischer (thermal resistance: 0.7–1.65 K W−1, dimensions: 200 × 200 × 25 mm), on which four LEDs from Avonec (Premium 3W LED, 390–400 nm, colour: UV-A, max. power: 750 mA, operating voltage: 3.5–4.5 V) were attached, which can be operated via a power supply unit (APC-16-700, input: 100–240 V, output: +24 V, max. 700 mA, class 2 power supply). For flotation experiments under irradiation by the halogen element, a spotlight possessing a halogen element R7s, 220–240 V, 400 W, 8550 lumen, 2700–3300 Kelvin was positioned at a distance of 50 cm from the Hallimond tube. The light sources for all experiments were switched on at the beginning of the conditioning in all experiments.
Zeta potential measurements
The measurements were conducted with a Zetasizer Nano from Malvern. The respective collector was added to the solution. The measurements have been performed with the supernatant. Lithium aluminate samples were exclusively measured at the natural pH value.Particle size measurements
The particle size distribution of mixed samples and single mineral samples of LiAlO2 and gehlenite was determined with dry measurement via the HELOS laser diffractometer from Sympatec, Germany, at an air pressure of 4 bar.UV measurements
The UV-Vis spectra were measured with a Jasco V-650 spectrophotometer and were evaluated by the program Spectra Manager. Precision cuvettes made of quartz glass from Hellma were used: Type No. 110-QS, light path 10 mm with a volume of 3 ml. The measurements were performed in distilled water. A stock solution of the individual collectors with a concentration of 1% by mass in distilled water was prepared. A volume of 2.5 μl of the stock solution was placed into the cuvette, filled up with distilled water and was measured. 10 μl of a 0.1 M HCl or 0.1 M NaOH solution were added for the different pH values.Wettability analysis
The contact angles of conditioned and untreated LiAlO2 and gehlenite materials were conducted with a OCA-50 Pro optical contour analysis (dataphysics, Germany). Therefore, powder of the sample material with a particle size smaller than 80 μm was fixed on a tab (PELCO Tabs TM 6 mm OD from TED PELLA, INC) by pressing with a spatula and two microscopic glass slides. For the measurement a 7 μl air bubble (2.5 mm diameter) was injected into 10 mM KCl solution with a syringe and deposited on the sample surface (Fig. 2). The sample surface was previously checked with a light microscope (Zeiss AXI0) on tab coverage and homogeneity of particles. For each sample at least 3 measurements were done. The contact angle was afterwards determined with the help of ImageJ software (version 1.54f).XRD measurements
The XRD samples were prepared according to the following conditions: concentration of 10.33 μmol l−1 of collector 5, 1.2 × 10−3 g l−1 of pine oil as frother, a conditioning time of 3 minutes with punicine-C11. The flotation was performed for 3 minutes at air flow rate of 32 cm3 min−1 using 250 ml distilled water in a Hallimond tube. For the XRD measurments a PANalytical Empyrean (Malvern, UK) X-ray diffractometer with a Co X-ray source, a voltage of 35 kV and a current of 35 mA was used. Prior to the measurement the samples were split into 2.5 ml sample (<400 μm) was ground in ethanol with zirconium-oxide grinding media in a McCrone mill (Retsch, Germany), ideally producing particles with a narrow size distribution below 1 μm and an intact crystallographic structure. Afterwards the sample was dried overnight and the dried sample homogenized with small steel balls (4 mm diameter) by using the mixer mill MM 400 from Retsch. The diffractogram and quantitative phase determination were performed via Rietveld refinement using Profex 4.1.0 software. The results need to be considered with an error between 1–2%.Preparation of 1-(2′,5′-dihydroxyphenyl)-4-methylpyridinium chloride (2)
To a suspension of p-benzoquinone (5.40 g, 50 mmol) in 35 ml of concentrated glacial acid were added 4-methylpyridine (4.90 ml, 50 mmol). The mixture was stirred overnight at room temperature and was then treated with an excess of concentrated hydrochloric acid. The resulting solid was filtered off and alternately washed with chloroform and ethylacetate. Recrystallization from water revealed 7.913 g (66%) of a yellow solid. 1H NMR (400 MHz, DMSO-d6): δ = 10.43 (bs, 1H), 9.75 (bs, 1H), 9.00–8.98 (m, 2H), 8.09–8.07 (m, 2H), 7.11 (d, JH–H = 8.9 Hz, 1H), 7.03 (d, JH–H = 2.8 Hz, 1H), 6.96 (dd, JH–H = 8.9, 2.8 Hz, 1H) ppm. Data are in agreement to those reported earlier.Preparation of 1-(2′,5′-dihydroxyphenyl)-4-ethylpyridinium chloride (3)
To a suspension of p-benzoquinone (2.20 g, 20 mmol) in 20 ml of concentrated glacial acid were added 4-ethylpyridine (2.3 ml, 20 mmol). The mixture was stirred for 3 d at room temperature and was then treated with an excess of concentrated hydrochloric acid. The resulting solid was filtered off and alternately washed with chloroform and ethylacetate. Recrystallization from water revealed 1.702 g (34%) of a yellow solid; decomposition 119 °C, 258 °C, 277 °C (DSC). 1H NMR (400 MHz, DMSO-d6): δ = 10.38 (bs, 1H), 9.70 (bs, 1H), 9.03–9.01 (m, 2H), 8.12–8.11 (m, 2H), 7.10 (d, JH–H = 8.9 Hz, 1H), 7.03 (d, JH–H = 2.9 Hz, 1H), 6.96 (dd, JH–H = 8.9, 2.9 Hz, 1H), 3.01 (q, JH–H = 7.6 Hz, 1H), 1.32 (t, JH–H = 7.6 Hz, 1H) ppm. 13C NMR (100 MHz, DMSO-d6): δ = 165.0 (*, 1C), 150.3 (*, 1C), 145.6 (+, 2C), 142.7 (*, 1C), 129.7 (*, 1C), 126.9 (+, 2C), 119.1 (+, 1C), 118.0 (+, 1C), 112.6 (+, 1C), 28.2 (−, 1C), 13.4 (+, 1C) ppm; IR (ATR): ν = 3397, 3330, 3208, 3038, 1631, 1507, 1457, 1380, 1341, 1282, 1203, 1179, 1131, 849, 822, 787, 716, 692, 662, 628, 585, 549, 477, 464 cm−1. HR-ESI-MS: calculated for [C13H14NO2]+: 216.1019. Found: 216.1034.Preparation of 4-octylpyridine
Diisopropylamine (1.41 ml, 10.03 mmol) were dissolved in anhydrous THF (60 ml) and cooled to 0 °C under N2 protective gas atmosphere. N-butyllithium (3.50 ml, 10.03 mmol, 2.8 M in cyclohexane) were then added dropwise and the mixture was stirred for 30 minutes. The reaction solution was cooled to −50 °C. 4-Picoline (0.93 ml, 9.55 mmol) were then added dropwise and the mixture was stirred at −50 °C for further 60 minutes. 1-Bromoheptane (1.50 ml, 9.55 mmol) were added dropwise to the orange solution and the mixture was stirred at −50 °C for a further 60 minutes and then warmed to room temperature. After 16 h the reaction was quenched with saturated NH4Cl solution, the organic phase was separated off and the aqueous part was extracted twice with diethyl ether. The combined organic phases were then washed with water and subsequently with NaCl solution, dried over Na2SO4 and the solvent was removed under reduced pressure. 1.45 g of a brown liquid are obtained (79%).1H NMR (400 MHz, DMSO-d6): δ = 8.43 (d, JH–H = 6.0 Hz, 2H), 7.20 (d, JH–H = 6.0 Hz, 2H), 2.57 (t, JH–H = 7.5 Hz, 2H), 1.56 (t, JH–H = 7.5 Hz, 2H), 1.24 (m, 10H), 0.84 (t, 3H) ppm. Data are in agreement with the literature.37
Preparation of 4-undecylpyridine
Diisopropylamine (3.18 ml, 22.55 mmol) were dissolved in anhydrous THF (60 ml) and cooled to 0 °C under N2 protective gas atmosphere. N-butyllithium (8.05 ml, 22.55 mmol, 2.8 M in cyclohexane) were then added dropwise and the mixture was stirred for 30 minutes. The reaction solution was cooled to −50 °C. 4-Picoline (2.09 ml, 21.48 mmol) were then added dropwise and the mixture was stirred at −50 °C for further 60 minutes. 1-Iododecane (4.57 ml, 21.48 mmol) were added dropwise to the orange solution and the mixture was stirred at −50 °C for a further 60 minutes and then warmed to room temperature. After 16 h the reaction was quenched with saturated NH4Cl solution, the organic phase was separated off and the aqueous part was extracted twice with diethyl ether. The combined organic phases were then washed with water and subsequently with NaCl solution, dried over Na2SO4 and the solvent was removed under reduced pressure. 4.58 g of a brown liquid are obtained (91%). 1H NMR (400 MHz, DMSO-d6): δ = 8.48 (d, JH–H = 5.9 Hz, 2H), 7.10 (d, JH–H = 5.9 Hz, 2H), 2.60 (t, JH–H = 7.6 Hz, 2H), 1.62 (quint., JH–H = 7.6 Hz, 2H), 1.34–1.24 (m, 16H), 0.88 (t, 3H) ppm. Data are in agreement with the literature.38Preparation of 1-(2,5-dihydroxyphenyl)-4-octylpyridinium chloride (4)
To a suspension of p-benzoquinone (0.61 g, 5.64 mmol) in 10 ml of concentrated glacial acetic acid were added 4-octylpyridine (1 g, 5.64 mmol) dropwise. The mixture was stirred for 12 h at room temperature and was then treated with concentrated hydrochloric acid. The mixture was then stirred for 2 more hours. Subsequently water added to precipitate the product, which was filtered off and washed with diethylether, which yields 0.499 g of a yellow crystalline solid (23%), decomposition at 119 °C. 1H-NMR (600 MHz, DMSO-d6) δ = 10.40 (s, 1H), 9.72 (s, 1H), 9.00 (d, JH–H = 6.9 Hz, 2H), 8.10 (d, JH–H = 6.9 Hz, 2H), 7.10 (d, JH–H = 8.9 Hz, 1H), 7.03 (d, JH–H = 2.9 Hz, 1H), 6.97 (dd, JH–H = 8.9, 2.9 Hz, 1H), 2.95 (t, JH–H = 7.6 Hz, 2H), 1.71 (quint., JH–H = 7.6 Hz, 2H), 1.37–1.22 (m, 10H), 0.86 (t, 3H) ppm. 13C-NMR (150 MHz, DMSO-d6) δ = 164.3 (*, 1C), 150.8 (*, 1C), 145.9 (+, 2C), 143.1 (*, 1C), 130.2 (*, 1C), 127.8 (+, 2C), 119.6 (+, 1C), 118.5 (+, 1C), 113.1 (+, 1C), 35.4 (−, 1C), 31.7 (−, 1C), 22.6; 29.0; 29.1; 29.2; 29.6 (−, 5C), 14.4 (+, 1C) ppm. IR (ATR): ν = 3369, 3117, 3070, 3042, 2956, 2916, 2849, 1640, 1627, 1507, 1456, 1333, 1272, 1252, 1202, 1124, 829, 816, 794, 721, 628, 614, 594, 556, 532, 483, 469 cm−1. HR-ESI-MS: calculated for [C19H26NO2]+: 300.1958 found: 300.1955 (Δ = 0.0003).Preparation of 1-(2,5-dihydroxyphenyl)-4-undecylpyridinium chloride (5)
To a suspension of p-benzoquinone (0.46 g, 4.28 mmol) in 15 ml of concentrated glacial acetic acid were added 4-undecylpyridine (1 g, 4.28 mmol) dropwise. The mixture was stirred for 12 h at room temperature and was then treated with concentrated hydrochloric acid, the mixture was stirred for a further 2 hours. The water was further added to the solution until solid precipitates, which was filtered off and washed with diethylether, whereupon 0.671 g of a yellow crystalline solid are obtained; decomposition at 136 °C. 1H-NMR (600 MHz, DMSO-d6) δ = 10.37 (s, 1H), 9.70 (s, 1H), 9.00 (d, JH–H = 6.6 Hz, 2H), 8.10 (d, JH–H = 6.6 Hz, 2H), 7.09 (d, JH–H = 8.8 Hz, 1H), 7.03 (d, JH–H = 2.8 Hz, 1H), 6.96 (dd, JH–H = 8.8, 2.8 Hz, 1H), 2.96 (t, JH–H = 7.6 Hz, 2H), 1.71 (quint., JH–H = 7.6 Hz, 2H), 1.33–1.25 (m, 16H), 0.85 (t, 3H) ppm. 13C-NMR (150 MHz, DMSO-d6) δ = 163.3 (*, 1C), 150.3 (*, 1C), 145.5 (+, 2C), 142.6 (*, 1C), 129.7 (*, 1C), 127.3 (+, 2C), 119.1 (+, 1C), 118.0 (+, 1C), 112.6 (+, 1C), 34.9 (−, 1C), 31.30 (−, 1C), 28.6; 28.7; 28.7; 28.9; 29.0; 29.1 (−, 6C), 22.1(−, 1C), 13.9(+, 1C) ppm. IR (ATR): ν = 3369, 3117, 3070, 3042, 2956, 2916, 2849, 1640, 1627, 1507, 1456, 1333, 1272, 1252, 1202, 1124, 829, 816, 794, 721, 628, 614, 594, 556, 532, 483, 469 cm−1. HR-ESI-MS: calculated for [C22H32NO2]+: 342.2428 found: 342.2432 (Δ = 0.0004).Author contributions
Ali Zgheib, Sophie Acker and Maximilian Hans Fischer synthesized the collectors and Ali and Sophie performed the flotation experiments. Max and Ali measured the zeta potentials and Jan C. Namyslo performed the calculations. Martin Nieger made the single structure X-ray analysis. Franziska Strube and Martin Rudolph performed particle size measurements and examined the samples of mixed mineral flotation concerning composition and recovery. Martin Rudolph, Ursula Fittschen, Alfred Weber/Annett Wollmann and Andreas Schmidt are group leaders of DFG projects within the DFG Priority Program SPP 2315, respectively. The project of Andreas Schmidt is dealing with switchable collectors for the flotation of engineered artifical minerals (EnAM).Conflicts of interest
There are no conflicts to declare.Acknowledgements
Birgit Wawrzinek and Florian Ahrend-Vaiana are gratefully acknowledged for measuring the NMR-spectra. Maike Weigert is gratefully acknowledged for ESI-MS spectra. Maike Gamenik and Sarah Tuchtfeld are gratefully acknowledged for measuring the zeta potentials for the minerals. Doreen Ebert from Helmholtz Institute for Resource Technology in Freiberg is gratefully acknowledged for performing the quantitative XRD analysis and the data evaluation with Rietveld refinement. The DFG is gratefully acknowleged for funding of this project Schm1371/18-1 as well as RU 2184/2-1 within the DFG Priority Program SPP 2315.Notes and references
- Y. Cao, S. L. Tan, E. J. H. Cheung, S. Y. Siew, C. J. Li, Y. Liu, C. S. Tan, M. Lal, G. Y. Chen, K. Dogheche, P. Yang, S. Pennycock, A. T. S. Wee, S. Chua, E. Dogheche, T. Venkatesan and A. Danner, Adv. Mater., 2021, 33, 2101128 CrossRef CAS PubMed.
- (a) E. Stefanelli, M. Puccini, A. Pesetti, R. Lo Frano and D. Aquaro, Nucl. Mater. Energy, 2022, 30, 101131 CrossRef CAS; (b) L. J. Mao, P. R. Zhang, H. Q. Ju, X. L. Zhou, Z. X. Xue, C. M. Wang, J. H. Sun, Y. Z. Jia, F. Shao, X. W. Zou, B. Li and Y. Jing, J. Mol. Liq., 2022, 367, 120357 CrossRef CAS.
- (a) Y. Di, G. Chen, L. L. Zhang, Z. Chen, R. Zhang, M. I. Asghar, S. J. Geng and P. D. Lund, J. Power Sources, 2021, 503, 120070 Search PubMed; (b) C. Venkateswaran, H. Sreemoolanadhan and R. Vaish, Int. Mater. Rev., 2022, 67, 620–657 CrossRef CAS; (c) S. Bahel, R. Singh, G. Kaur and S. B. Narang, Ferroelectrics, 2016, 502, 49–56 CrossRef CAS.
- (a) V. N. Sigaev, A. S. Lipat'ev, S. S. Fedotov, S. V. Lotarev, A. S. Naumov and D. M. Shevyakina, Glass Ceram., 2022, 79, 45–47 CrossRef CAS; (b) S. J. Huang, W. Z. Wang, H. Jiang, H. F. Zhao and Y. P. Ma, Materials, 2022, 15, 4555 CrossRef CAS PubMed.
- (a) F. M. Yeh, V. Volli, L. W. Bin, P. H. Tung and C. M. Shu, Appl. Therm. Eng., 2019, 150, 1328–1336 CrossRef CAS; (b) Z. H. Song, Y. M. Liang, M. J. Fan, F. Zhou and W. M. Liu, Tribol. Int., 2014, 70, 136–141 CrossRef CAS.
- (a) W. Rivera, G. Moreno-Quintanar, C. O. Rivera, R. Best and F. Martinez, Sol. Energy, 2011, 85, 38–45 CrossRef CAS; (b) K. Sedighi, M. Farhadi and M. Liaghi, Proc. Inst. Mech. Eng., Part C, 2007, 221, 1345–1351 CrossRef CAS.
- IEA, Global EV Outlook 2020, 2020, https://www.iea.org/reports/global-ev-outlook-2020.
- C. Scheller, S. Blömeke, M. Nippraschk, K. Schmidt, M. Mennenga, T. S. Spengler, C. Herrmann and D. Goldmann, Procedia CIRP, 2021, 98, 464–469 CrossRef.
- B. W. Jaskula, Lithium, available online, https://pubs.usgs.gov/periodicals/mcs2020/mcs2020-lithium.pdf, accessed on 29 November 2020.
- European Commission, Study on the EU's List of Critical Raw Materials – Final Report, 2020 Search PubMed.
- H. Qiu, C. Peschel, M. Winter, S. Nowak, J. Köthe and D. Goldmann, Metals, 2022, 12, 677 CrossRef CAS.
- (a) T. Schirmer, H. Qiu, H. Li, D. Goldmann and M. Fischlschweiger, Metals, 2020, 10, 1633 CrossRef CAS; (b) T. Schirmer, H. Qiu, D. Goldmann, C. Stallmeister and B. Friedrich, Minerals, 2022, 12, 310 CrossRef CAS.
- A. Wittkowski, T. Schirmer, H. Qiu, D. Goldmann and U. E. A. Fittschen, Metals, 2021, 11, 188 CrossRef CAS.
- (a) B. A. Wills and T. Napier-Munn, Mineral Processing Technology, Elsevier, Amsterdam, 2006, p. 267 Search PubMed; (b) R. M. G. Lima, P. R. G. Brandao and A. E. C. Peres, Miner. Eng., 2005, 18, 267–273 CrossRef CAS; (c) X. Wenig, G. Mei and T. Zhao, Sep. Purif. Technol., 2013, 103, 187–194 CrossRef; (d) A. V. Nguyen and H. J. Schulze, Colloidal Science of Flotation, Marcel Dekker Inc, New York, 2004 Search PubMed; (e) M. Ejtemaei, M. Gharabaghi and M. Irannajad, Adv. Colloid Interface Sci., 2014, 206, 68–78 CrossRef CAS PubMed; (f) S. Aghazadeh, S. K. Mousavinezhad and M. Gharabaghi, Adv. Colloid Interface Sci., 2015, 225, 203–217 CrossRef CAS PubMed; (g) Z. J. Wu, X. M. Wang, H. N. Liu, H. F. Zhang and J. D. Miller, Adv. Colloid Interface Sci., 2016, 235, 190–200 CrossRef CAS PubMed.
- Y. Xing, M. Xu, X. Gui, Y. Cao, B. Babel, M. Rudolph, S. Weber, M. Kappl and H.-J. Butt, Adv. Colloid Interface Sci., 2018, 256, 373–392 CrossRef CAS PubMed.
- (a) T. N. Hunter, R. J. Pugh, G. V. Franks and G. J. Jameson, Adv. Colloid Interface Sci., 2008, 137, 57–81 CrossRef CAS PubMed; (b) H. Polat and D. Erdogan, J. Hazard. Mater., 2007, 148, 267–273 CrossRef CAS PubMed; (c) R. M. Rahman, S. Ata and G. J. Jameson, Int. J. Miner. Process., 2012, 106, 70–77 CrossRef; (d) D. Zamboulis, S. Pataroudi, A. Zouboulis and K. Matis, Desalination, 2004, 162, 159–168 CrossRef CAS.
- L. Filippov, S. Farrokhpay, L. Lyo and I. Filippova, Minerals, 2019, 9, 372 CrossRef CAS.
- (a) J. Tian, L. Xu, H. Wu, S. Fang, W. Deng, T. Peng, W. Sun and Y. Hu, J. Cleaner Prod., 2018, 174, 625–633 CrossRef CAS; (b) H. Wu, J. Tian, L. Xu, S. Fang, Z. Zhang and R. Chi, Miner. Eng., 2018, 127, 42–47 CrossRef CAS.
- (a) A. Haas, T. Elwert, D. Goldmann and T. Schirmer, in Proceedings of the European Mineral Processing & Recycling Congress, ed. GDMB Verlag GmbH, GDMB, Clausthal-Zellerfeld, Germany, 2018, ISBN 978-3-940276-84-1 Search PubMed; (b) T. Elwert, K. Strauss, T. Schirmer and D. Goldmann, Recycling von Li-Ionen-Traktionsbatterien; Recycling und Rohstoffe, Neuruppin, Germany, 2012, pp. 679–690 Search PubMed; (c) T. Elwert, K. Strauss, T. Schirmer and D. Goldmann, World Metall.--Erzmet., 2012, 65, 163–171 CAS.
- M. A. Nawwar, S. A. Hussein and I. Merfort, Phytochemistry, 1994, 37, 1175 CrossRef CAS.
- A. Schmidt, T. Mordhorst and M. Nieger, Nat. Prod. Res., 2005, 19, 541–546 CrossRef CAS PubMed.
- W. D. Ollis, S. P. Stanforth and C. A. Ramsden, Tetrahedron, 1985, 41, 2239–2329 CrossRef CAS.
- A. Schmidt and T. Mordhorst, ARKIVOC, 2003, XIV, 233–245 Search PubMed.
- M. Albrecht, M. Gjikaj and A. Schmidt, Tetrahedron, 2010, 66, 7149–7154 CrossRef CAS.
- A. Schmidt, M. Topp, T. Mordhorst and O. Schneider, Tetrahedron, 2007, 63, 1842–1848 CrossRef CAS.
- A. Schmidt, T. Mordhorst, H. Fleischhauer and G. Jeschke, ARKIVOC, 2005, X, 150–164 Search PubMed.
- A. Schmidt, M. Albrecht, T. Mordhorst, M. Topp and G. Jeschke, J. Mater. Chem., 2007, 17, 2793–2800 RSC.
- A. Schmidt and M. Albrecht, Z. Naturforsch., 2008, 63b, 465–472 CrossRef.
- M. Albrecht, M. Yulikov, T. Kohn, G. Jeschke, J. Adams and A. Schmidt, J. Mater. Chem., 2010, 20, 3025–3034 RSC.
- M. Albrecht, O. Schneider and A. Schmidt, Org. Biomol. Chem., 2009, 7, 1445–1453 RSC.
- S. Nagorny, F. Lederle, V. Udachin, T. Weingartz, E. G. Hübner, S. Dahle, W. Maus-Friedrichs, J. Adams and A. Schmidt, Eur. J. Org Chem., 2021, 3178–3189 CrossRef CAS.
- C. F. Otto, M. Liu, C. Herzberger, J. C. Namyslo, M. Nieger, E. G. Hübner, F. Lederle, T. Freese and A. Schmidt, Tetrahedron, 2020, 76, 131627 CrossRef CAS.
- S. Mummel, F. Lederle, E. Hübner, J. C. Namyslo, M. Nieger and A. Schmidt, Angew. Chem., Int. Ed., 2021, 60, 18882–18887 CrossRef CAS PubMed.
- (a) S. Nagorny, T. Weingartz, J. C. Namyslo, J. Adams and A. Schmidt, Eur. J. Org Chem., 2022, e202200996 Search PubMed; (b) S. Nagorny, F. Lederle, V. Udachin, T. Weingartz, E. G. Hübner, S. Dahle, W. Maus-Friedrichs, J. Adams and A. Schmidt, Eur. J. Org Chem., 2021, 3178–3189 CrossRef CAS.
- S. Acker, J. C. Namyslo, M. Rudolph, F. Strube, U. E. A. Fittschen, H. Qiu, D. Goldmann and A. Schmidt, RSC Adv., 2023, 13, 6593–6605 RSC.
- R. Hulst, I. Muizebelt, P. Oosting, C. Van Der Pol, A. Wagenaar, J. Smisterova, E. Bulten, C. Driessen, D. Hoekstra and J. B. F. N. Engberts, Eur. J. Org. Chem., 2004, 4, 835–849 CrossRef.
- C. M. Hendy, G. C. Smith, Z. Xu, T. Lian and N. T. Jui, J. Am. Chem. Soc., 2021, 143, 8987–8992 CrossRef CAS PubMed.
- R. Hulst, I. Muizebelt, P. Oosting, C. van der Pol, A. Wagenaar, J. Šmisterová, E. Bulten, C. Driessen, D. Hoekstra and J. B. F. N. Engberts, Eur. J. Org Chem., 2004, 835–849 CrossRef CAS.
- J. B. Ferguson and A. F. Buddington, Am. J. Sci., 1920, 50, 138 Search PubMed.
- A. Vanderbruggen, J. Sygusch, M. Rudolph and R. Serna-Guerrero Rodrigo, Colloids Surf., 2021, 626, 127111 CrossRef CAS.
- A. Schmidt, C. F. Otto, J. C. Namyslo, A. Fischer and K. Filip, Bull. Entomol. Res., 2021, 10, 601–620 Search PubMed.
- F. Maggi, J. Geophys. Res. Oceans, 2013, 118, 2118–2132 CrossRef.
- P. G. Smith and L. J. Warren, Miner. Process. Extr. Metall. Rev., 1989, 5(1–4), 123–145 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., 2015, A71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2291748 (3) contains the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra00116h |
| This journal is © The Royal Society of Chemistry 2024 |