
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC(sp2)–H selenylation of substituted benzo[4,5]imidazo[2,1-b]thiazoles using phenyliodine(III)bis(trifluoroacetate) as a mediator†
Prasanjit Ghosh *a,
Gautam Chhetria,
Anirban Mandala,
Yu Chenbc,
William H. Hershbc and
Sajal Das*a
*a,
Gautam Chhetria,
Anirban Mandala,
Yu Chenbc,
William H. Hershbc and
Sajal Das*a
aDepartment of Chemistry, University of North Bengal, Darjeeling-734013, India. E-mail: prasanjitmeister@yahoo.com; sajal.das@hotmail.com; sajaldas@nbu.ac.in
bDepartment of Chemistry and Biochemistry, Queens College and the Graduate Centre of City University of New York, Flushing, New York 11367-1597, USA. E-mail: yu.chen1@qc.cuny.edu
cPhD Program in Chemistry, The Graduate Center of the City University of New York, 365 Fifth Ave., New York, 10016, USA
First published on 2nd February 2024
Abstract
Herein, an expeditious metal-free regioselective C–H selenylation of substituted benzo[4,5]imidazo[2,1-b]thiazole derivatives was devised to synthesize structurally orchestrated selenoethers with good to excellent yields. This PIFA [bis(trifluoroacetoxy)iodobenzene]-mediated protocol operates under mild conditions and offers broad functional group tolerance. In-depth mechanistic investigation supports the involvement of radical pathways. Furthermore, the synthetic utility of this methodology is portrayed through gram-scale synthesis.
Introduction
Benzoimidazo[2,1-b]thiazoles, an important class of fused heterobicyclic scaffolds and their congeners, constitute a coveted field of research among a myriad of mainstream organic scaffolds. These frameworks adorn the core structures of several bioactive compounds and natural products with promising pharmacological activities, such as antibacterial,1 antidiabetic,2 antitumor,3 anti-inflammatory,4 anticardiovascular,5 antitubercular,6 antineurodegenerative7 and immunosuppressive activities.8Some drug candidates having an imidazo[2,1-b]thiazole core unit are shown in Fig. 1. In recent years, several approaches for benzoimidazo[2,1-b]thiazole synthesis have been well documented in the literature and have received immense attention from medicinal and synthetic chemists owing to their fascinating structural architecture.
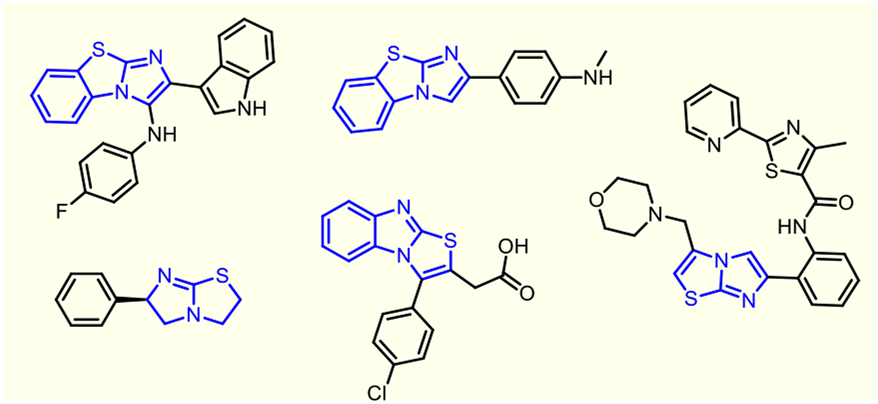 | ||
| Fig. 1 Representative drug candidates containing the imidazo[2,1-b]thiazole core unit. | ||
However, their derivatizations are still in a nascent stage.9 Consequently, devising novel synthetic strategies to access this high-value scaffold and fabricating it via selective functionalization is a vast and intriguing part of research. Moreover, the presence of Se functionality in organic molecules often favourably alters the pharmacokinetics and physicochemical properties of parent compounds on account of hydrogen bond acceptor and electron-donor properties, which play pivotal roles in the drug discovery field, as well as in catalysis, agrochemistry, fluorescence probe imaging, and materials science.10 Ebselen, the first organoselenium compound, is recognized as a quintessential antioxidant and neuroprotective agent.11 A few representative molecules are outlined in Fig. 2.
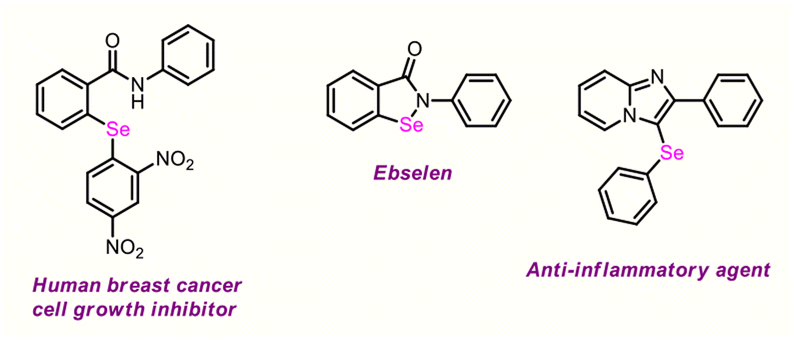 | ||
| Fig. 2 Bioactive diaryl selenide and aryl–selenyl–heteroaryl scaffolds. | ||
However, the supply of selenium-rich organic scaffolds from natural sources is scarce. Thus, the production of organoselenium compounds largely relies on man-made protocols, and devising straightforward synthetic methods for such purposes is a highly sought-after area of research. So far, several approaches have been developed, including cross-coupling reactions12 and transition metal-catalyzed C(sp2)–H chalcogenylation,13 for these purposes. Moreover, these procedures possess major pitfalls such as the requirement of transition metal catalysts, toxic reagents, and harsh reaction conditions. Nevertheless, chemists are particularly interested in new reactivity paradigms to construct organoselenium derivatives under environment-friendly conditions. In this regard, Du and coworkers exploited the reactivity of PhICl2 for the preparation of α-chalcogenylenamines under ambient conditions.14a Later, Du and Zhao disclosed a facile route for the synthesis of 3-chalocogenyl chromones through regioselective cyclization of alkynyl aryl ketones induced by PIFA [phenyliodine(III)bis(trifluoroacetate)].14b Recently, we accomplished the regioselective insertion of a thio/seleno ether at the C-3 position of biologically potent 4-quinolone and 4H-pyrido-[1,2-a]-pyrimidin-4-ones via metal-free C(sp2)–H bond functionalization.15 Moreover, various reports are well documented in the literature on the direct C–Se functionalization of imidazo[2,1-b]thiazoles and imidazo[1,2-a]pyridines.16 Recently, Yan and Wang et al. demonstrated the PIFA-induced regioselective C(sp2)–H chalcogenylation of benzo[d]imidazo[5,1-b]thiazoles.17 Encouraged by the above propitious outcomes and our continuous interest in the organochalcogenide chemistry of heterocyclic motifs, herein, for the first time, we propose a transitional metal-free C(sp2)–H selenylation of substituted benzo[4,5]imidazo[2,1-b]thiazoles with diselenides under ambient conditions using PIFA as an inducer (Fig. 3). Notably, our strategy provides direct access to structurally enriched C–Se coupled products of substituted benzo[4,5]imidazo[2,1-b]thiazole derivatives with good to excellent yields.
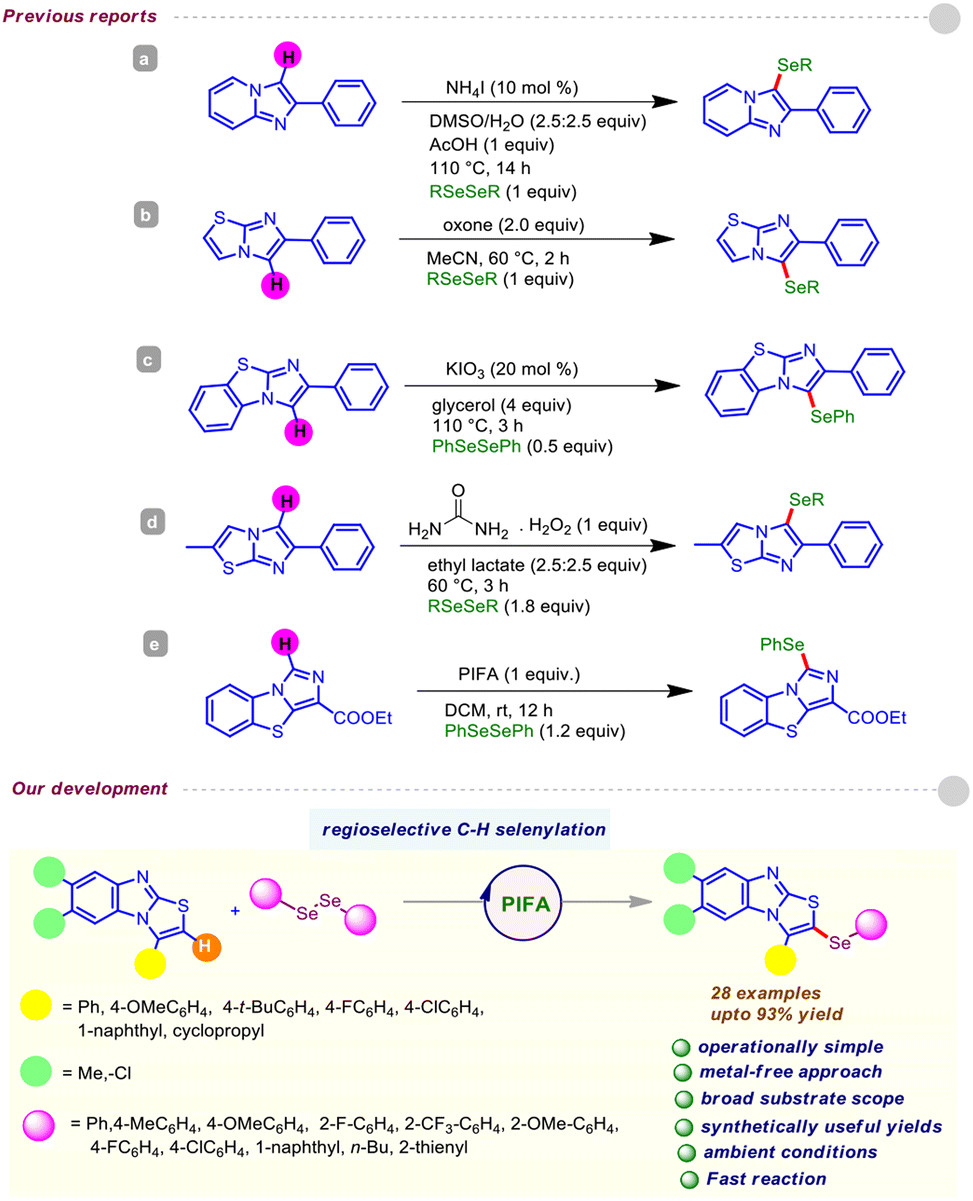 | ||
| Fig. 3 PIFA-mediated regioselective C–H selenylation of substituted benzo[4,5]imidazo[2,1-b]thiazoles. | ||
Results and discussion
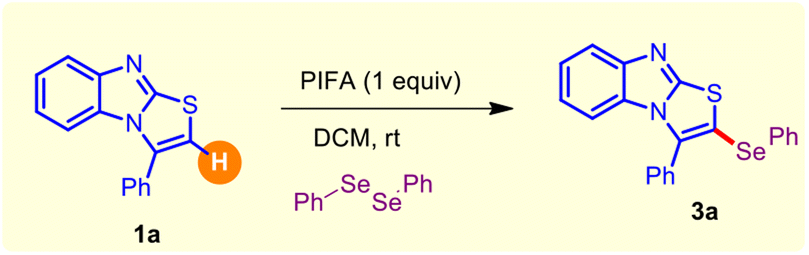
To envision this hypothesis, a judicial evaluation of different reaction parameters was performed employing 3-phenyl-benzo[4,5]imidazo[2,1-b]thiazole 1a and diphenyl diselenide 2a (as arylselenylating agent) as benchmark coupling partners, and a detailed summary is depicted in Table 1. Delightfully, we observed the regioselective formation of the target product 3-phenyl-2-(phenylselanyl)benzo[4,5]imidazo[2,1-b]thiazole 3a in high yield in the presence of the bench-stable oxidant phenyliodine(III)bis(trifluoroacetate) (PIFA) at room temperature under ambient condition (Table 1; entry 1). Of note, the reaction was finished within 40 min, with complete consumption of the starting material 1a (Table 1; entry 1). The screening of other alternative oxidants, such as PIDA/PhICl2 and PhIO, provided low to moderate yields of 3a, whereas TBHP/TBPB/DTBP and K2S2O8 were found to be ineffective for this transformation (Table 1; entries 2–3). Especially, the solvent played a decisive role in this regioselective C–H selenylation reaction. Switching the reaction solvent from DCM to other polar solvents, like MeCN/DCE and DMF, furnished the desired coupling product 3a in 35–70% yield (Table 1; entry 4). Unfortunately, the reaction was completely halted in the presence of DMSO and EtOH with absolute recovery of the starting material 1a (Table 1; entry 4). Moreover, examining other solvents, such as THF/1,4-dioxane and toluene, gave inferior outcomes even after 24 h (Table 1; entry 5). Reaction temperature enhancement did not significantly alter the yield of the anticipated product 3a (Table 1; entry 6). Next, we varied the stoichiometric amount of the oxidant (PIFA), although no improvement of the yield of 3a was noticed (Table 1; entries 7–8). The control experiment clearly showed that the reaction required PIFA (Table 1; entry 9). Performing a similar reaction under inert conditions (N2 atmosphere) also delivered an acceptable yield of the desired product 3a (Table 1; entry 10). Finally, the overall studies signified that 1 equiv. PIFA as an oxidant in the presence of DCM as solvent at room temperature was optimal to facilitate the regioselective C–H selenylation, rendering the target scaffold 3a in excellent yield.
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yieldb (%) |
| a Reaction conditions: 1a (0.25 mmol), 2a (1.5 equiv., 0.375 mmol), PIFA (1 equiv.), DCM (4 ml) at rt under air for 60 min.b Isolated yields.c Reaction continued up to 24 h. | ||
| 1 | None | 89 |
| 2c | PIDA/PhICl2/PhIO instead of PIFA | 25/41/32 |
| 3c | TBHP/TBPB/DTBP/K2S2O8 instead of PIFA | NR |
| 4c | MeCN/DCE/DMF/DMSO/EtOH instead of DCM | 70/51/35/trace/NR |
| 5c | THF/1,4-dioxane/toluene instead of DCM | 35/41/25 |
| 6 | At 40 °C | 85 |
| 7 | 0.5 equiv. of PIFA | 65 |
| 8 | 0.8 equiv. of PIFA | 78 |
| 9 | Without PIFA | NR |
| 10 | Under inert atmosphere (N2) | 75 |
Having determined the suitable reaction conditions, we next investigated the scope and generalizability of the procedure with an array of electronically and sterically diversified organodiselenides with 1a as the standard substrate. As summarized in Table 2, a broad range of organodiselenides were amenable to couple with 3-phenyl-benzo[4,5]imidazo[2,1-b]thiazole 1a, offering the desired C–H selenylated derivatives 3a–3i with reasonable to high efficiency. Diaryldiselenides encompassing electron-donating substituents, like methyl and alkoxy (–OMe), gave the intended derivatives 3b–3c in uniformly high yields. Synthetically modifiable common halo functionalities (–F, –Cl) displayed a negligible impact on the reaction efficiency (Table 2; entries 3d–3e). Notably, electron-withdrawing substituents bearing diaryldiselenides delivered the corresponding selenylated benzo[4,5]imidazo[2,1-b]thiazoles in slightly high yields in comparison to electron-releasing functional groups, which might be attributed to the stable intermediates (see the mechanism). In addition, this protocol permitted the usage of the ortho-substituent (–F) containing organodiselenide, although 1,2-bis(2-methoxyphenyl)diselane turned out to be futile in this PIFA-induced regioselective chalcogenation (Table 2; entries 3f–3g). Substrates with bulkier aryl frameworks like naphthyl also exhibited smooth reactivity, affording the desired product 3h in 89% yield, although heterocyclic diselenides (such as 1,2-di(thiophen-2-yl)diselane) showed a detrimental result under the standard reaction conditions (Table 2; entry 3i).

Next, the adaptability of this PIFA-mediated C–Se coupling was explored with structurally orchestrated benzo[4,5]imidazo[2,1-b]thiazole derivatives (Table 3). The reaction scope was broad, and 3-aryl-benzo[4,5]imidazo[2,1-b]thiazoles having electron-rich and electron-deficient groups boded well under the current standard conditions.
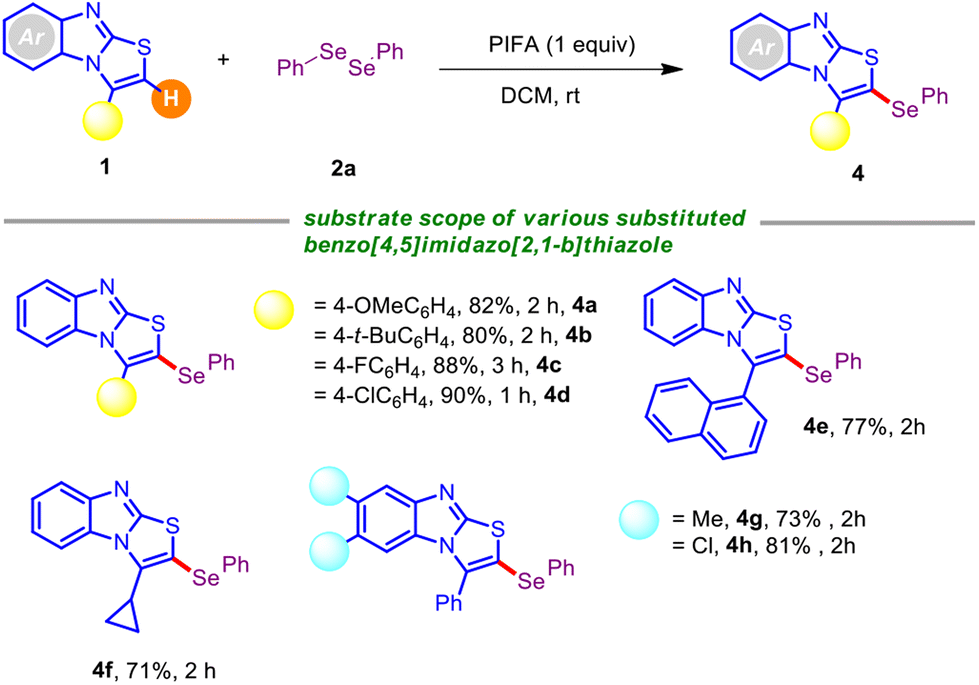
Fortunately, various functional groups (–OMe, –t-Bu, –F, and –Cl) at the periphery of the thiazole unit did not impede the progress of the reaction, providing the respective mono-selenylated scaffolds (4a–4d) with high yields. The protocol also worked efficiently with the naphthyl-embedded thiazole framework, rendering 4e in serviceable yield. Fortunately, benzo[4,5]imidazo[2,1-b]thiazole with an alkyl analog at the C-3 position posed no problem, and offered the corresponding selenylated derivative 4f in decent yield. It is noteworthy that the alkyl (–Me) and halo (–Cl) substitution at the arene backbone of benzo[4,5]imidazo[2,1-b]thiazole derivative caused a small variation in the reaction efficiency, forging 4g and 4h in 73% and 81% yields, respectively.
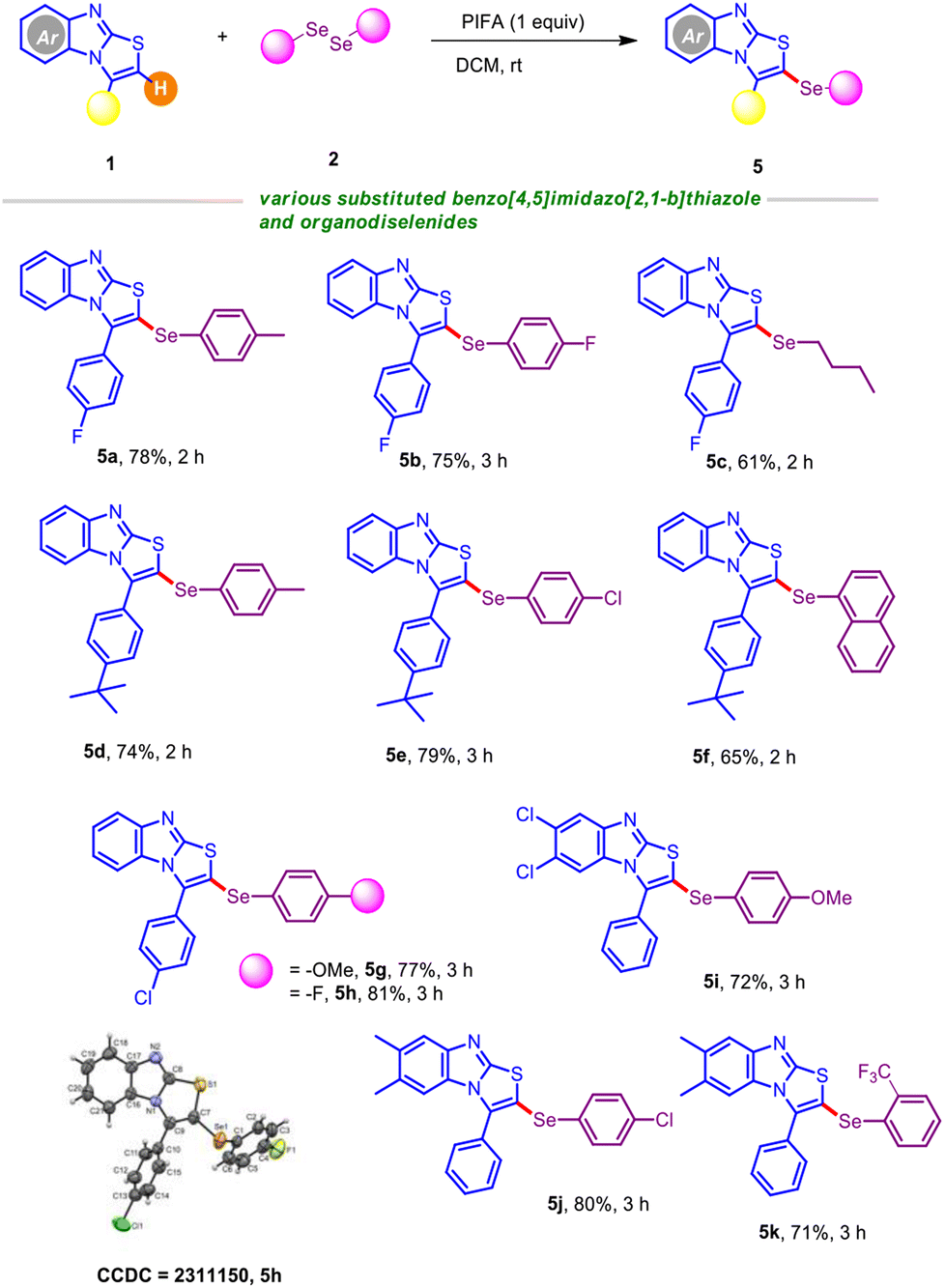
We next turned our attention to explore the breadth of substrate compatibility of different benzo[4,5]imidazo[2,1-b]thiazoles with diverse organodiselenides, as outlined in Table 4. Satisfyingly, 3-(4-fluorophenyl)benzo[4,5]imidazo[2,1-b]thiazole was smoothly engaged with organodiselenides bearing methyl and fluoro substituents, furnishing the target scaffolds 5a–5b in good yields. Moreover, participation of the less reactive organodiselenide (alkyl congener) was especially significant and afforded 5c in 61% yield. The protocol also functioned well with 3-(4-(tert-butyl)phenyl)benzo[4,5]imidazo[2,1-b]thiazole in the presence of a broad spectrum of diaryldiselenides (Table 4; entries 5d–5f). For instance, the sterically hindered naphthyl diselenide gave 5f in reasonable yield. To our delight, C–H selenylation of 3-(4-chlorophenyl)benzo[4,5]imidazo[2,1-b]thiazole was also tested with 1,2-bis(4-methoxyphenyl)diselane and 1,2-bis(4-chlorophenyl)diselane, which led to the C–Se coupled products 5g and 5h in high yields. Moreover, we attempted to crystallize the selenylated product 5h; X-ray analysis unambiguously confirmed the regioselective insertion of the ArSe unit of the scaffold. Gratifyingly, 6,7-dichloro-3-(p-tolyl)benzo[4,5]imidazo[2,1-b]thiazole was found to be suitable when coupled with 1,2-bis(4-methoxyphenyl)diselane, furnishing the desired product 5i in 72% yield. Furthermore, we judiciously incorporated the ortho-bromo and ortho-trifluoromethyl substituted selane in the presence of 6,7-dimethyl-3-(p-tolyl)benzo[4,5]imidazo[2,1-b]thiazole and no noticeable electronic effects were observed, providing comparable yields of C–Se coupled products (Table 4; entries 5j–5k).
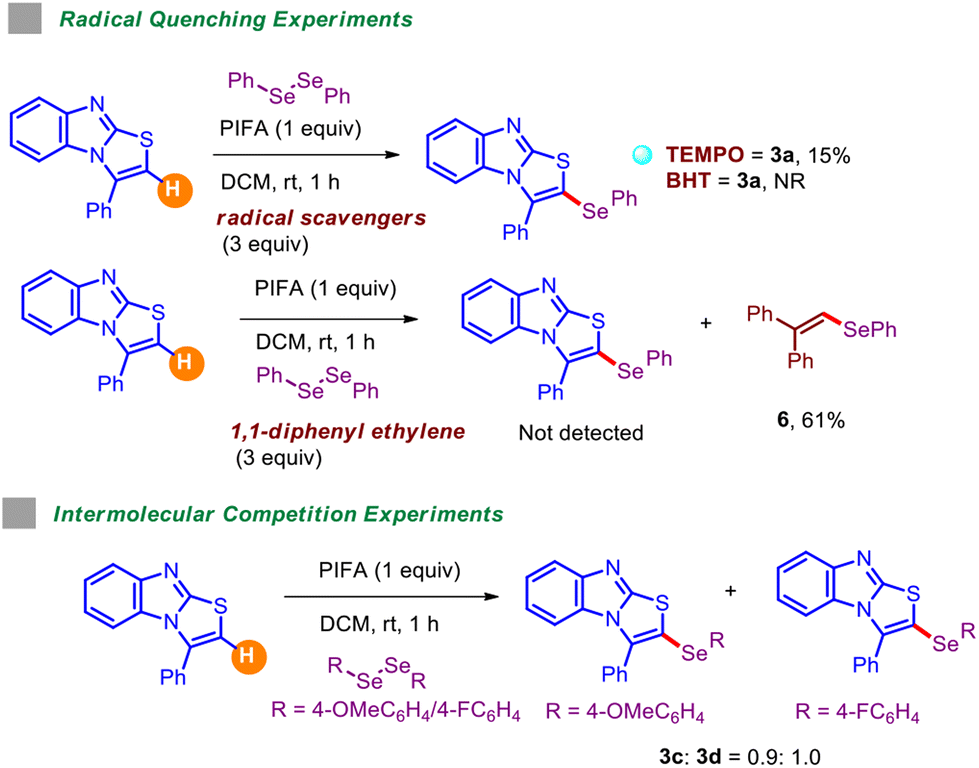
Several radical trapping experiments carried out in the presence of excess amounts of radical scavengers, including TEMPO, BHT (butylated hydroxy toluene), and 1,1-diphenylethylene (Table 5), reported detrimental outcomes, signifying the involvement of the radical mechanism. Intermolecular competition experiments were also performed by coupling 1,2-bis(4-methoxyphenyl)diselane and 1,2-bis(4-fluorophenyl)diselane with 1a in one-pot fashion under standard conditions. These outcomes suggested that electronically deficient diselenides react at a slightly higher rate than electron-rich ones.
In light of the control experiments and prior literature precedents,14,15 a plausible mechanism for the PIFA-mediated regioselective C–H selenylation is delineated in Scheme 1. At the onset, PIFA reacts with diphenyldiselenide and produces Int-A, which rapidly undergoes homolytic cleavage and results in the formation of a phenyl selenide radical Int-B and radical Int-C. Subsequently, an electrophilic attack from the C-3 position of the 3-phenylbenzo[4,5]imidazo[2,1-b]thiazole moiety 1a on the phenyl selenide radical Int-B results in the generation of another intermediate 1ab. The driving force for this electrophilic attack may be attributed to forming a stable benzylic radical. Then, the oxidation of Int-1ab by Int-C delivers an ionic intermediate Int-1ac, which can be stabilized to Int-1ac' by resonance. Finally, deprotonation of Int-1ac furnishes the desired C–Se coupled product 3a.
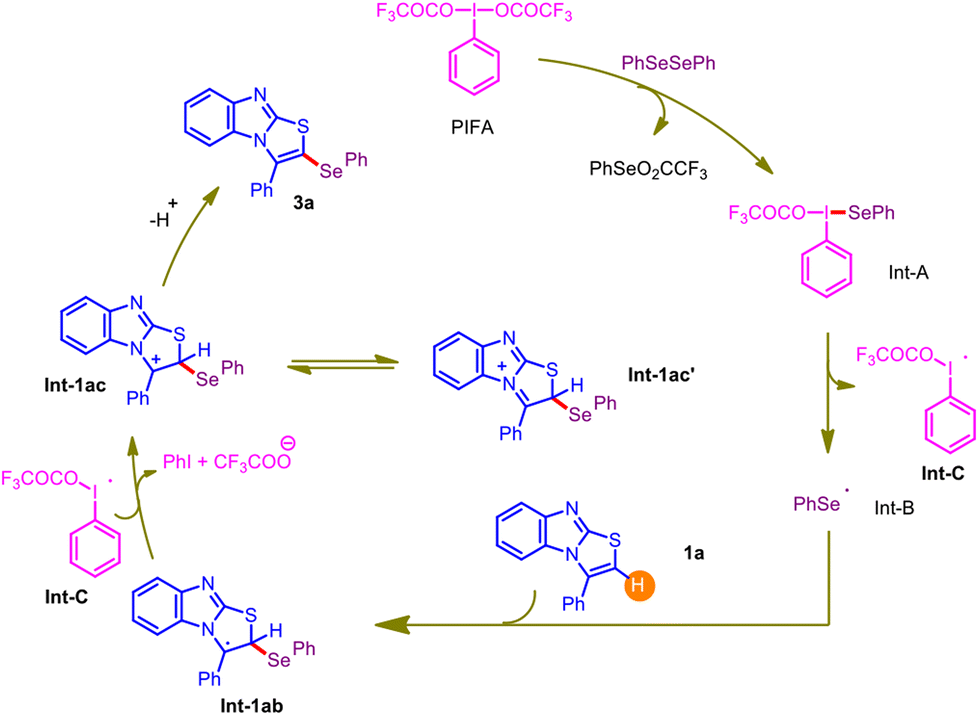 | ||
| Scheme 1 The possible mechanistic route (radical pathway). | ||
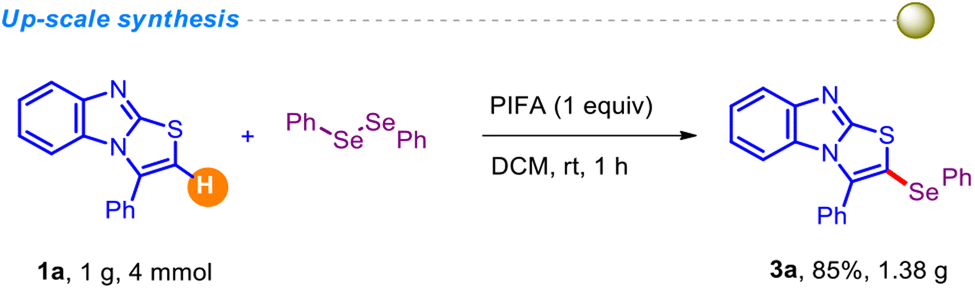
To assess the robustness of the current protocol, we conducted a relatively large-scale experiment and obtained the anticipated product 3a without significant loss in yield (Scheme 2).
 | ||
| Scheme 2 Application. | ||
Conclusion
In summary, we have unveiled the first example of the PIFA-induced regioselective C–H selenylation of substituted benzo[4,5]imidazo[2,1-b]thiazole derivatives with readily available organodiselenides under ambient conditions. The reaction scope was broad, encompassing both electron-donating and electron-withdrawing functionalities, and structurally diversified C–H chalcogenated derivatives were obtained in good to excellent yields. Moreover, the synthetic potentiality of this strategy was showcased through upscale synthesis without perturbing the reaction efficiency. The protocol offers a novel synthetic route due to its operational-simplicity, transition-metal free approach and mild reaction conditions. Initial mechanistic experiments showed that the reaction proceeds via a radical pathway. Due to the widespread application of organoselenium compounds in drug synthesis, these studies can unlatch a new avenue in pharmaceutical and medicinal chemistry. Further development of regioselective C–H chalcogenation and functionalization of substituted benzo[4,5]imidazo[2,1-b]thiazoles are underway in our laboratory, and will be reported in due course.Experimental
General consideration
Unless stated otherwise, all reagents (such as various PIFA, DCM and solvents) were used as received from commercial suppliers. Organodiselenides were prepared by the reported procedure.18 NMR spectra were recorded on a 400 MHz spectrometer at 298 K with calibration done on the basis of the solvent residual peak. Products were purified using column chromatography on silica gel (60–120 mesh). Ethyl acetate and petroleum ether (60–80 °C) were used as eluents. Progress of the reaction was monitored using silica gel TLC.Preparation of various substituted benzo[4,5]imidazo[2,1-b]thiazoles
All of the starting compounds are prepared by the following literature reports.9Preparation of various selenylated derivatives of substituted benzo[4,5]imidazo[2,1-b]thiazoles derivatives (3a–3i/4a–4h/5a–5k)
Initially, various substituted benzo[4,5]imidazo[2,1-b]thiazoles (0.25 mmol), organodiselenides (1.5 equiv., 0.375 mmol) and PIFA (1 equiv., 108 mg) were taken in dichloromethane (4 ml) in a 25 ml round-bottomed flask. Afterwards, the reaction mixture was stirred at room temperature. Completion of the reaction was monitored through TLC, and the reaction mixture was diluted with (3 × 10 ml) DCM and an added requisite amount of water. The organic layer was extracted and dried over anhydrous Na2SO4, and concentrated under reduced pressure. The crude product was then purified by column chromatography using 20–30% ethyl acetate/petroleum ether as the eluent.Experimental procedure for the radical trapping experiment
Initially, 3-phenyl-benzo[4,5]imidazo[2,1-b]thiazoles (0.25 mmol, 62.5 mg), diphenyl diselenide (0.375 mmol, 117 mg), PIFA (1 equiv., 108 mg) and radical scavengers (TEMPO/BHT/1,1-diphenylethylene, 3 equiv.) were taken in dichloromethane (4 ml) in a 25 ml round-bottomed flask. Afterwards, the reaction mixture was stirred at room temperature for 60 min. Completion of the reaction was monitored through TLC, and the reaction mixture was diluted with (3 × 10 ml) DCM and an added requisite amount of water. The organic layer was extracted and dried over anhydrous Na2SO4, and concentrated under reduced pressure. The crude product was then purified by column chromatography using 20–30% ethyl acetate/petroleum ether as the eluent.Experimental procedure for gram-scale synthesis (3a)
Initially, 3-phenyl-benzo[4,5]imidazo[2,1-b]thiazoles (4 mmol, 1.0 g), diphenyl diselenide (6 mmol, 1.88 g) and PIFA (1 equiv., 1.72 g) were taken in dichloromethane (6 ml) in a 15 ml reaction vial. Afterwards, the reaction mixture was stirred at room temperature for 1 h. Then, the product was extracted with dichloromethane (3 × 20 mL). The organic layer was dried over anhydrous Na2SO4, and concentrated under reduced pressure. The crude product was then purified by column chromatography using 20–30% ethyl acetate/petroleum ether solution, and the desired product 3a was isolated in 85% (1.38 g) yield.Physical characteristics and spectral data of compounds
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 111.6, 119.2, 120.6, 123.6, 125.4, 125.7, 127.6, 129.5, 129.8, 130.1, 130.9, 131.1, 138.1, 148.1, 153.7, 157.3. HRMS (ESI-TOF) m/z: [M + H]+ C25H23N2SSe calcd 463.0747; found 463.0732.
111.6, 119.2, 120.6, 123.6, 125.4, 125.7, 127.6, 129.5, 129.8, 130.1, 130.9, 131.1, 138.1, 148.1, 153.7, 157.3. HRMS (ESI-TOF) m/z: [M + H]+ C25H23N2SSe calcd 463.0747; found 463.0732.HRMS (ESI-TOF) m/z: [M + H]+ C22H15Cl2N2OSSe calcd 504.9447; found 504.9428.
Author contributions
All authors conceived of the study design and experiments conducted herein. P. G., G. C. and A. M. analyzed the spectral data. All authors contributed to the writing of this manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. M. gratefully acknowledges UGC for his fellowship. We also acknowledge DST-FIST for providing infrastructure facilities.Notes and references
- M. Palkar, M. Noolvi, R. Sankangoud and V. Maddi, Arch. Pharm. Chem. Life Sci., 2010, 343, 353–359 CrossRef CAS PubMed.
- C. B. Vu, J. E. Bemis, J. S. Disch, P. Y. Ng, J. J. Nunes, J. C. Milne, D. P. Carney, A. V. Lynch, J. J. Smith, S. Lavu, P. D. Lambert, D. J. Gagne, M. R. Jirousek, S. Schenk, J. M. Olefsky and R. B. Perni, J. Med. Chem., 2009, 52, 1275–1283 CrossRef CAS PubMed.
- (a) A. Andreani, S. Burnelli, M. Granaiola, A. Leoni, A. Locatelli, R. Morigi, M. Rambaldi, L. Varoli, N. Calonghi, C. Cappadone, M. Voltattorni, M. Zini, C. Stefanelli, L. Masotti and R. H. Shoemaker, J. Med. Chem., 2008, 51, 7508–7513 CrossRef CAS PubMed; (b) J. H. Park, M. I. El-Gamal, Y. S. Lee and G. H. Oh, Eur. J. Med. Chem., 2011, 46, 5769–5777 CrossRef CAS PubMed; (c) T. U. Mayer, T. M. Kapoor, S. J. Haggarty, R. W. King, S. L. Schreiber and T. J. Mitchison, Science, 1999, 286, 971–974 CrossRef CAS PubMed.
- B. Tozkoparan, M. Ertan, P. Kelicen and R. Demirdamar, Farmaco, 1999, 54, 588–593 CrossRef CAS PubMed.
- A. Locatelli, S. Cosconati, M. Micucci, A. Leoni, L. Marinelli, A. Bedini, P. Loan, S. M. Spampinato, E. Novellino, A. Chiarini and R. Budriesi, J. Med. Chem., 2013, 56, 3866–3877 CrossRef CAS PubMed.
- G. C. Moraski, L. D. Markley, M. Chang, S. Cho, S. G. Franzblau, C. H. Hwang, H. Boshoff and M. J. Miller, Bioorg. Med. Chem., 2012, 20, 2214–2220 CrossRef CAS PubMed.
- N. Pietrancosta, A. Moumen, R. Dono, P. Lingor, V. Planchamp, F. Lamballe, M. Bahr, J. L. Kraus and F. Maina, J. Med. Chem., 2006, 49, 3645–3652 CrossRef CAS PubMed.
- T. Mase, H. Arima, K. Tomioka, T. Yamada and K. Murase, J. Med. Chem., 1986, 29, 386–394 CrossRef CAS PubMed.
- (a) S. Jana, A. Chakraborty, V. Z. Shirinian and A. Hajra, Adv. Synth. Catal., 2018, 360, 2402–2408 CrossRef CAS; (b) S. Ambethkar, M. Vellimalai, V. Padmini and N. Bhuvanesh, New J. Chem., 2017, 41, 75–80 RSC; (c) G. Shen, B. Yang, X. Huang, Y. Hou, H. Gao, J. Cui, C. Cui and T. Zhang, J. Org. Chem., 2017, 82, 3798–3805 CrossRef CAS PubMed.
- (a) T. Ando, T. S. Kwon, A. Kitagawa, T. Tanemura, S. Kondo, H. Kunisada and Y. Yuki, Macromol. Chem. Phys., 1996, 197, 2803–2810 CrossRef CAS; (b) P. K. Khanna and B. K. Das, Mater. Lett., 2004, 58, 1030–1034 CrossRef CAS; (c) T. E. Frizon, D. S. Rampon, H. Gallardo, A. A. Merlo, P. H. Schneider, O. E. D. Rodrigues and A. L. Braga, Liq. Cryst., 2012, 39, 769–777 CrossRef CAS; (d) A. Kumar, G. K. Rao, F. Saleem and A. K. Singh, Dalton Trans., 2012, 41, 11949–11977 RSC; (e) F. Frankel, M. Priven, E. Richard, C. Schweinshault, O. Tongo, A. Webster, E. Barth, K. Slejzer and S. Edelstein, Int. J. Food Prop., 2016, 19, 537–548 CrossRef CAS; (f) A. Ivanova and P. Arsenyan, Coord. Chem. Rev., 2018, 370, 55–68 CrossRef CAS.
- (a) C. W. Nogueira and J. B. T. Rocha, J. Braz. Chem. Soc., 2010, 21, 2055–2071 CrossRef CAS; (b) A. Muller, E. Cadenas, P. Graf and H. Sies, Biochem. Pharmacol., 1984, 33, 3235–3239 CrossRef CAS PubMed; (c) D. A. Dawson, H. Masayasu, D. I. Graham and I. M. Macrae, Neurosci. Lett., 1995, 185, 65–69 CrossRef CAS PubMed; (d) I. Saito, T. Asano, K. Sano, K. Takakura, H. Abe, T. Yoshimoto, H. Kikuchi, T. Ohta and S. Ishibashi, Neurosurgery, 1998, 42, 269–277 CrossRef CAS PubMed.
- (a) G. Mugesh and H. B. Singh, Acc. Chem. Res., 2002, 35, 226–236 CrossRef CAS PubMed; (b) D. Ma, Q. Geng, H. Zhang and Y. Jiang, Angew. Chem., Int. Ed., 2010, 49, 1291–1294 CrossRef CAS PubMed; (c) Z. Qiao, J. Wei and X. Jiang, Org. Lett., 2014, 16, 1212–1215 CrossRef CAS PubMed; (d) Y. Li, M. Wang and X. Jiang, ACS Catal., 2017, 7, 7587–7592 CrossRef CAS; (e) Y. Fang, T. Rogge, L. Ackermann, S.-Y. Wang and S.-J. Ji, Nat. Commun., 2018, 9, 2240 CrossRef PubMed; (f) Y. -C. Shieh, K. Du, R. S. Basha, Y. J. Xue, B. H. Shih, L. Li and C. F. Lee, J. Org. Chem., 2019, 84, 6223–6231 CrossRef CAS PubMed; (g) R. Sikari, S. Sinha, S. Das, A. Saha, G. Chakraborty, R. Mondal and N. D. Paul, J. Org. Chem., 2019, 84, 4072–4085 CrossRef CAS PubMed.
- (a) F.-L. Yang and S.-K. Tian, Angew. Chem., 2013, 125, 5029–5032 (Angew. Chem., Int. Ed., 2013, 52, 4929–4932) CrossRef; (b) P. Saravanan and P. Anbarasan, Org. Lett., 2014, 16, 848–851 CrossRef CAS PubMed; (c) B. Duan, Y. Wu, Y. Gao, L. Ying, J. Tang, S. Hu, Q. Zhao and Z. Song, Chem. Commun., 2022, 58, 11555–11558 RSC; (d) Y. Cao, J. Liu, F. Liu, L. Jiang and W. Yi, Org. Chem. Front., 2019, 8, 825–829 RSC; (e) W. Ma, N. Kaplaneris, X. Fang, L. Gu, R. Mei and L. Ackermann, Org. Chem. Front., 2020, 7, 1022–1060 RSC; (f) T. Zhang, G. Deng, H. Li, B. Liu, Q. Tan and B. H. Xu, Org. Lett., 2018, 20, 5439–5443 CrossRef CAS PubMed; (g) H. Wang, Y. Li, Q. Lu, M. Yu, X. Bai, S. Wang, H. Cong, H. Zhang and A. Lei, ACS Catal., 2019, 9, 1888–1894 CrossRef CAS.
- (a) Z. Shang, Q. Chen, L. Xing, Y. Zhang, L. Wait and Y. Du, Adv. Synth. Catal., 2019, 361, 4932–4936 Search PubMed; (b) Z. Ai, J. Xiao, Y. Li, B. Guo, Y. Du and K. Zhao, Org. Chem. Front., 2020, 7, 3935–3940 RSC.
- (a) P. Ghosh, A. K. Nandi, G. Chhetri and S. Das, J. Org. Chem., 2018, 83, 12411–12419 CrossRef CAS PubMed; (b) P. Ghosh, G. Chhetri, E. Perl and S. Das, Adv. Synth. Catal., 2021, 363, 2148–2156 CrossRef CAS.
- (a) L. Betttanin, S. Saba, C. V. Doerner, M. S. Franco, M. Godoi, J. Rafique and A. L. Braga, Tetrahedron, 2018, 74, 3971–3980 CrossRef; (b) I. Rodrigues, A. M. Barcellos, A. L. Belladona, J. A. Roehrs, R. Cargnelutti, D. Alves, G. Perin and R. F. Schumacher, Tetrahedron, 2018, 74, 4242–4246 CrossRef CAS; (c) J. Rafique, S. Saba, M. S. Franco, L. Bettanin, A. R. Schneider, L. T. Silva and A. L. Braga, Chem.–Eur. J., 2017, 24, 4173 CrossRef PubMed; (d) C. A. O. Moraes, R. B. C. Santos, M. F. O. Cavalcante, J. S. Guilhermi, M. A. Ali, G. V. Botteselle, T. E. A. Frizon, M. I. A. Shah, L. M. Liao, A. Beatriz, S. Saba and J. Rafique, ACS Omega, 2023, 8, 39535–39545 CrossRef CAS PubMed.
- M. Liu, K. Yan, J. Wen, N. Zhang, X. Chen, X. Li and X. Wang, Asian. J. Org. Chem., 2022, 11, e202200381 CrossRef CAS.
- D. Singh, A. M. Deobald, L. R. S. Camargo, G. Tabarelli, O. E. D. Rodrigues and A. L. Braga, Org. Lett., 2010, 12, 3288–3291 CrossRef CAS PubMed.
- J.-D. Fang, X.-B. Yan, W.-J. Lin, Y.-C. Zhao and X.-Y. Liu, Org. Lett., 2019, 21, 7635–7638 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2311150. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra00057a |
| This journal is © The Royal Society of Chemistry 2024 |