
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRole of the thiosugar ring in the inhibitory activity of salacinol, a potent natural α-glucosidase inhibitor†
Katsuki Takashima‡
 a,
Shinya Nakamura‡a,
Maiko Nagayamaa,
Shinsuke Marumotoc,
Fumihiro Ishikawaa,
Weijia Xied,
Isao Nakanishia,
Osamu Muraokab,
Toshio Morikawa*b and
Genzoh Tanabe*ab
a,
Shinya Nakamura‡a,
Maiko Nagayamaa,
Shinsuke Marumotoc,
Fumihiro Ishikawaa,
Weijia Xied,
Isao Nakanishia,
Osamu Muraokab,
Toshio Morikawa*b and
Genzoh Tanabe*ab
aFaculty of Pharmacy, Kindai University, 3-4-1 Kowakae, Higashi-osaka, Osaka 577-8502, Japan
bPharmaceutical Research and Technology Institute, Kindai University, 3-4-1 Kowakae, Higashi-osaka, Osaka 577-8502, Japan
cJoint Research Centre, Kindai University, 3-4-1 Kowakae, Higashi-osaka, Osaka 577-8502, Japan
dState Key Laboratory of Natural Medicines, Department of Medicinal Chemistry, China Pharmaceutical University, Nanjing 2100009, P. R. China
First published on 2nd February 2024
Abstract
Herein, ring-cleaved (24) and truncated (25) analogues of an azasugar, 1-deoxynojirimycin (23), exhibited inhibitory activity (Ki = 4–10 μM) equal to that of the parent compound (1, Ki = 14 μM). Based on this structure–activity relationship (SAR), four ring-cleaved (26a–26c and 27c) and three truncated (28a–28c) analogues of salacinol (1), a potent thiosugar-ring-containing α-glucosidase inhibitor, were synthesised. Bioassay results revealed that all the synthetics were inactive, indicating that the 5-membered thiosugar ring of 1 played an essential role in the potent activities of sulfonium-type inhibitors. The present findings are interesting and important in understanding the function of salacinol, considering that the observed inhibitory activity trend was contrary to the SAR observed in aza-compounds (23, 24, and 25) in a previous study, which suggested that the cyclic structure did not contribute to their strong inhibitory activity.
Introduction
The roots and stems of plants belonging to the Salacia genus, such as Salacia reticulata, S. oblonga, and S. chinensis, have traditionally been used in the Ayurvedic system for the treatment of diabetes. In 1997, a novel thiosugar-sulfonium sulphate inner salt, salacinol (1), was isolated from the extracts of S. reticulata roots and stems. Salacinol (1) was shown to be responsible for the antidiabetic properties of these extracts.1,2 The zwitterionic structure of 1 is unique in that it comprises a thiosugar sulfonium cation and a sulphate anion (within a 4-deoxy-D-erythritol-type side chain), as shown in Fig. 1.2 The mechanism of action of 1 involves the inhibition of α-glucosidase, and its inhibitory activity against rat α-glucosidases is as effective as those of clinically used diabetic medications such as voglibose and acarbose. In the following year, a side-chain-extended homologue, kotalanol3 (2), was isolated from Salacia extracts as an antidiabetic compound that also showed potent inhibitory activity against rat α-glucosidases.3 Moreover, after further research on the composition of Salacia extracts, a side-chain homologue, ponkoranol4 (3), as well as de-O-sulfonated versions of 1, 2, and 3 [neosalacinol5,6 (4), neokotalanol7,8 (5), and neoponkoranol9 (6), respectively] were isolated as active components with potent antidiabetic activities. Thereafter, these sulfonium salts (1–6) were also proven to effectively inhibit human maltase.10 Clinical trials involving patients with type 2 diabetes revealed that these extracts were effective in treating diabetes while minimizing serious acute toxicity or side effects.11–15 Based on this evidence, plants of the Salacia genus have attracted interest as therapeutic materials for type 2 diabetes and have garnered considerable attention as potential functional foods for individuals with diabetes and those with impaired glucose tolerance, who are at high risk of developing the disease in the future. | ||
| Fig. 1 A new class of natural α-glucosidase inhibitors (1–6). | ||
Due to their potent glucosidase inhibitory activities and unique structures, these natural products (1–6) have attracted much attention, and total syntheses of 1–6 have been achieved.16–18 Furthermore, structure–activity relationship (SAR) studies on these sulfonium salts have been ongoing since the early 2000s. Early-stage SAR studies mainly focused on the hydrogen bonding interactions between candidates and enzyme active sites; side-chain analogues (e.g., 7–16) and thiosugar analogues (e.g., 17–19) have been synthesised thus far (Fig. 2). An evaluation of their activities revealed the following findings regarding the structural features that contributed to their inhibitory activity. With respect to the side chain, (a) the sulphate anion is not always required for potent inhibition19 (e.g., 1 vs. 4); (b) the cooperative role of 2′S-OH with C4′-OH is critical for potent activity20 (1 vs. 12–14); (c) the R configuration at C4′ is crucial for the activity of inhibitors with a side chain longer than four carbons (e.g., 9 vs. 16);21–28 (d) if the side chain meets requirements (a), (b), and (c), the hydroxyl groups after C5′ are not necessarily required for potent activity29 (e.g., 2 vs. 7); and (e) loss of the oxygen atom at the 3′ position significantly reduces the inhibitory activity30 (e.g., 1, 4 vs. 15). On the other hand, regarding the thiosugar moiety, several related sulfonium salts have been synthesised to date. (f) Evaluation of their inhibitory activities against α-glucosidase suggests that hydroxyls in the D-arabino configuration of the 5-membered thiosugar, a characteristic shared with natural inhibitors 1–6, are considered favorable for potent activity31–33 (e.g., 1 vs. 17, 18, 19). Thus, structural determinants regarding hydrogen bonding interactions with the enzyme active site have been gradually revealed through SAR studies; however, potent inhibitors (superior to natural sulfonium salts 1–6) have not been produced in these SAR studies.
 | ||
| Fig. 2 A new class of natural α-glucosidase inhibitors (1–6) and synthesised analogues (7–19) in the early stage of SAR studies. | ||
In 2010, X-ray crystallographic studies34 on the complex of 1 and 2 with the N-terminal catalytic domain of human maltase-glucoamylase (hNtMGAM) showed that the OHs of the thiosugar (in addition to the 2′S-OH and C4′-OH of the side chains) closely interacted with the enzyme active site via hydrogen bonds. Furthermore, these studies suggested that the 3′-O-sulphate anion of 1 and 2 was constrained by the surrounding hydrophobic aromatic amino acid residues of the enzyme, which possibly caused negative binding interactions. Based on these findings, we synthesised a series of 3′-O-alkylated or aralkylated salacinol analogues (20 and 21) possessing a side chain in which the sulphate anion of 1 is substituted with a hydrophobic alkyl or benzyl moiety.35–37 Their inhibitory activities (IC50 = 0.13–1.7 μM) against rat intestinal maltase were significantly superior to those of 1 and 2 (IC50 = 5.2–7.2 μM). In silico docking studies of 20 and 21 with hNtMGAM identified additional structural determinants, with respect to the side chain. (g) van der Waals interactions between alkyl and/or aralkyl groups on the C3′ oxygen atom and the enzyme active site effectively enhance the α-glucosidase inhibitory activity.36,37 Furthermore, mimicking the structures of 20 and 21, Xie et al. recently prepared a derivative (22) with potent inhibitory activity comparable to those of 20 and 21 (ref. 38) (Fig. 3).
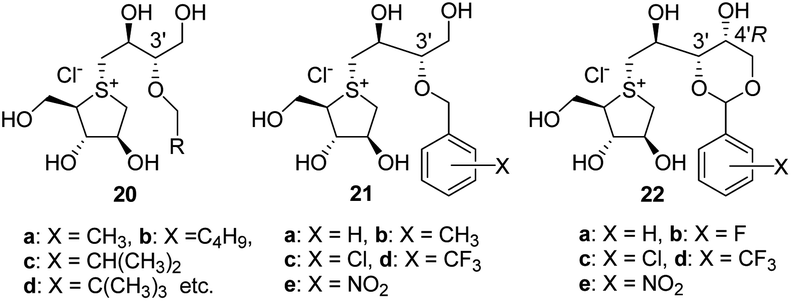 | ||
| Fig. 3 3′-O-Alkylated or benzylated analogues (20, 21 and 22). | ||
Thus, salacinol analogues that surpass the α-glucosidase inhibitory activity of the parent sulfonium salts have been developed, but all of them require a D-arabino-type thiosugar structure for inhibitory activity. While several synthetic methods have been reported,20,39–43 it remains difficult to acquire this thiosugar in large quantities for SAR studies due to the number of reaction steps required, limited yield of each step, and accessibility of the starting materials.
On the other hand, in 1994, Fowler et al. synthesised a ring-cleaved (24) and a truncated (25) analogues of the natural α-glucosidase inhibitor 1-deoxynojirimycin (23) and found that they exhibited inhibitory activity against yeast glucosidase comparable to that of 23 (23, Ki = 14.6 μM, 24, Ki = 10 μM 25, Ki = 4 μM, Fig. 4).44 This report demonstrated that OHs of acyclic candidates could successfully bind to enzyme active sites when their side chains possessed the same hydroxyl stereochemistry as those of the parent cyclic inhibitors. Existing SAR studies on salacinol (1) have not elucidated whether the thiosugar ring structure is necessary for inhibitory activity, and the role of this thiosugar ring remains unclear. Moreover, sulfonium salts [such as ring-cleaved salacinol analogues (26a–26c and 27c) and truncated salacinol analogues (28a–28c) newly synthesised in this study] are easier to synthesise than conventional salacinol analogues because they do not require the thiosugar moiety (Fig. 4). Therefore, we aimed to investigate whether potent inhibitory activity was retained in acyclic compounds 26–28.
 | ||
| Fig. 4 Acyclic analogues (24 and 25) of 1-deoxynojirimycin (23) and acyclic analogues (26, 27 and 28) of salacinol (1). | ||
Results and discussion
All compounds were synthesised as depicted in Scheme 1. First, the secondary hydroxyl of commercially available 1,3-di-O-benzylglycerol (29) was sulphonated with tosyl chloride to quantitatively give tosylate (30). The tosyloxy moiety of 30 was then treated with the sodium salt of 2-mercaptoethanol in DMF to displace to a sulpheny group, and the remaining primary hydroxyl of the corresponding sulphide (31) was protected as a benzyl ether to furnish tri-O-benzylated sulphide (32) in 84% yield via two steps. Next, according to the conventional method for S-alkylation of sulphides,45 32 was treated with a cyclic sulphate (33)19,46 to give the sulfonium salt (34) in 96% yield as an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric mixture. In the 13C NMR spectrum of 34, a downfield shift was observed with respect to the signals attributed to the C1′ methylene (at δC 41.7/42.1) and C2 methine (at δC 56.0/56.7) carbons, when compared to that of 32 [δC 31.3 (C1′ methylene) and 45.7 (C2 methine)]. A pair of signals, produced due to the introduction of an α-methylene carbon (C1′′) into the sulphur atom of 34, was also observed at δC 43.6/45.6, confirming that the sulfonium ion was generated successfully. In addition to the signals from these three carbons (C1′, C1′′, and C2), the remaining carbons (C1, C2′, C2′′, C3′′, and C4′′) were also detected as paired signals, thereby validating the diastereomeric structure of 34. Thereafter, the benzyl and acetonide moieties of 34 were simultaneously removed under acidic hydrogenation conditions to give 26a, which was transformed by acidic methanolysis into the corresponding sulfonium monomethyl sulphate (26b) in 92% yield. Finally, the CH3OSO3− anion of 26b was exchanged with the chloride anion via treatment with an ion exchange resin IRA-400 (Cl− form) to furnish a ring-cleaved analogue 26c in 93% yield. The positive mode ESI mass spectra of the sulfonium sulphate inner salt (26a) and its de-O-sulfonates (26b and 26c) displayed a peak at m/z 359 due to quasi-molecular ion [M + Na]+ and another at m/z 257 due to the sulfonium cation moiety ([M–OSO3Me]+ and [M–Cl]+). The 13C NMR spectroscopic properties of 26a, 26b, and 26c were similar, with the exception of signals attributed to the C-3′′ methine carbon of 26a (δC3′′ 83.3/83.4), which were highly downfield-shifted when compared to those of 26b and 26c (δC3′′ 75.4/75.5 and 75.4/75.6, respectively). These findings support the sulfonium structures depicted in Scheme 1. To synthesize the other ring-cleaved analogue (27c), optically pure epoxide (35) was prepared from isoascorbic acid according to the method previously reported by Abushanab et al.47 Then, 35 was subjected to regio-selective ring opening with a thiolate anion, which was generated in situ by reacting mercaptoethanol and potassium carbonate, to quantitatively give a chiral diol (36). The sulphur atom of 36 was alkylated in the presence of HBF4·Et2O with commercially available (S)-(−)-glucidol (37) to give the sulfonium salt (38) in an approximately 1:1 diastereomeric ratio. The BF4− ion of 38 was subsequently exchanged with the chloride anion in a manner similar to that used for the preparation of 26c to furnish the sulfonium chloride (39) in 98% yield. Finally, catalytic hydrogenolysis of 39 resulted in the formation of the target compound (27c, 89% yield). The positive mode ESI mass spectrum of 27c showed a peak at m/z 257 due to the sulfonium cation structure. The 13C NMR spectrum of 27c displayed three pairs of signals at δC1′′ 47.7/47.8, δC1′ 47.9/48.2, and δC1 48.2/48.6, which were attributed to three carbons α to the chiral sulfonium centre, validating that the mixture was composed of two diastereomeric isomers. The synthesis of a truncated salacinol analogue (28a) was more straightforward than those of the other two kinds of ring-cleaved analogues (26 and 27). Moreover, 28a was prepared in 87% yield (in three steps) from commercially available 2,2′-thiodiethanol (40), as depicted in Scheme 1. The acidic methanolysis of 28a gave 28b in 88% yield, and subsequent ion exchange of 28b furnished 28c in 87% yield. The 13C NMR spectra of all analogues synthesised in this manner were similar to each other, except that the signal attributed to the C-3′′ methine carbon in 28a (δC3′′ 83.2) was highly downfield-shifted in comparison to those in 28b and 28c (δC3′′ 74.4 and 75.5, respectively). Furthermore, the positive mode ESI mass spectra of 28a displayed a peak at m/z 329 due to the quasi-molecular ion [M + Na]+. Similarly, a peak at m/z 227 was observed in the positive mode ESI mass spectra of 28b and 28c, also consistent with the structure of both sulfonium cation moieties ([M–OSO3Me]+ and [M–Cl]+).
:1 diastereomeric mixture. In the 13C NMR spectrum of 34, a downfield shift was observed with respect to the signals attributed to the C1′ methylene (at δC 41.7/42.1) and C2 methine (at δC 56.0/56.7) carbons, when compared to that of 32 [δC 31.3 (C1′ methylene) and 45.7 (C2 methine)]. A pair of signals, produced due to the introduction of an α-methylene carbon (C1′′) into the sulphur atom of 34, was also observed at δC 43.6/45.6, confirming that the sulfonium ion was generated successfully. In addition to the signals from these three carbons (C1′, C1′′, and C2), the remaining carbons (C1, C2′, C2′′, C3′′, and C4′′) were also detected as paired signals, thereby validating the diastereomeric structure of 34. Thereafter, the benzyl and acetonide moieties of 34 were simultaneously removed under acidic hydrogenation conditions to give 26a, which was transformed by acidic methanolysis into the corresponding sulfonium monomethyl sulphate (26b) in 92% yield. Finally, the CH3OSO3− anion of 26b was exchanged with the chloride anion via treatment with an ion exchange resin IRA-400 (Cl− form) to furnish a ring-cleaved analogue 26c in 93% yield. The positive mode ESI mass spectra of the sulfonium sulphate inner salt (26a) and its de-O-sulfonates (26b and 26c) displayed a peak at m/z 359 due to quasi-molecular ion [M + Na]+ and another at m/z 257 due to the sulfonium cation moiety ([M–OSO3Me]+ and [M–Cl]+). The 13C NMR spectroscopic properties of 26a, 26b, and 26c were similar, with the exception of signals attributed to the C-3′′ methine carbon of 26a (δC3′′ 83.3/83.4), which were highly downfield-shifted when compared to those of 26b and 26c (δC3′′ 75.4/75.5 and 75.4/75.6, respectively). These findings support the sulfonium structures depicted in Scheme 1. To synthesize the other ring-cleaved analogue (27c), optically pure epoxide (35) was prepared from isoascorbic acid according to the method previously reported by Abushanab et al.47 Then, 35 was subjected to regio-selective ring opening with a thiolate anion, which was generated in situ by reacting mercaptoethanol and potassium carbonate, to quantitatively give a chiral diol (36). The sulphur atom of 36 was alkylated in the presence of HBF4·Et2O with commercially available (S)-(−)-glucidol (37) to give the sulfonium salt (38) in an approximately 1:1 diastereomeric ratio. The BF4− ion of 38 was subsequently exchanged with the chloride anion in a manner similar to that used for the preparation of 26c to furnish the sulfonium chloride (39) in 98% yield. Finally, catalytic hydrogenolysis of 39 resulted in the formation of the target compound (27c, 89% yield). The positive mode ESI mass spectrum of 27c showed a peak at m/z 257 due to the sulfonium cation structure. The 13C NMR spectrum of 27c displayed three pairs of signals at δC1′′ 47.7/47.8, δC1′ 47.9/48.2, and δC1 48.2/48.6, which were attributed to three carbons α to the chiral sulfonium centre, validating that the mixture was composed of two diastereomeric isomers. The synthesis of a truncated salacinol analogue (28a) was more straightforward than those of the other two kinds of ring-cleaved analogues (26 and 27). Moreover, 28a was prepared in 87% yield (in three steps) from commercially available 2,2′-thiodiethanol (40), as depicted in Scheme 1. The acidic methanolysis of 28a gave 28b in 88% yield, and subsequent ion exchange of 28b furnished 28c in 87% yield. The 13C NMR spectra of all analogues synthesised in this manner were similar to each other, except that the signal attributed to the C-3′′ methine carbon in 28a (δC3′′ 83.2) was highly downfield-shifted in comparison to those in 28b and 28c (δC3′′ 74.4 and 75.5, respectively). Furthermore, the positive mode ESI mass spectra of 28a displayed a peak at m/z 329 due to the quasi-molecular ion [M + Na]+. Similarly, a peak at m/z 227 was observed in the positive mode ESI mass spectra of 28b and 28c, also consistent with the structure of both sulfonium cation moieties ([M–OSO3Me]+ and [M–Cl]+).
 | ||
| Scheme 1 Reagents and conditions: (a) TsCl, Et3N, Me3N·HCl, CH2Cl2 (99%); (b) HSCH2CH2OH, NaH, DMF, 100 °C (93%); (c) BnBr, NaH, DMF, 0 °C (32: 90%, 41: 99%); (d) HFIP, K2CO3, reflux (34: 96%, 42: 95%); (e) H2, Pd–C, 10% aq. TFA, 1,4-dioxane, 50 °C (26a: 94%, 28a: 93%); (f) 5% methanolic HCl, (26b: rt, 92%, 28b: 50 °C, 88%); (g) IRA-400J (Cl− form), CH3OH, rt (26c: 93%, 28c: 87%, 39: 98%); (h) HSCH2CH2OH, K2CO3, C2H5OH, reflux (99%); (i) HBF4·(C2H5)2O, CH2Cl2, −78 °C (71%); (j) H2, Pd–C, CH3OH, rt (89%). | ||
The glycosidase inhibitory activities of the seven target compounds (26a, 26b, 26c, 27c, 28a, 28b, and 28c) were tested in vitro using enzymes obtained from rat small intestines.14 Their activities were compared with those of the parent compounds (1 and 4) to evaluate the effects of cleaving and truncating the thiosugar ring of 1 and 4. The results are summarized in Table 1. Although salacinol (1) and neosalacinol (4) effectively inhibited maltase, sucrase, and isomaltase (entries 8 and 9), the acyclic analogues 26b and 26c lacking the C2–C3 bond of the thiosugar moiety of 4 hardly inhibited these three enzymes even at a concentration of 100 μg mL−1 (26b, 272 μM, 26c, 342 μM, entries 2 and 3). The acyclic analogue 26a, which lacks the C2–C3 bond of the thiosugar moiety of 1, also exhibited poor inhibitory activity against these three enzymes at 100 μg mL−1 (301 μM), albeit performing slightly better than 26b and 26c (entry 1). The inhibitory activity of 27c, which lacks the C3–C4 bond of the thiosugar moiety of 4, was also negligible against the tested enzymes at 100 μg mL−1 (342 μM) (entry 4). Furthermore, the simplest acyclic analogues (28a, 28b, and 28c) lost considerable activity against all the enzymes regardless of the presence or absence of the sulphate anion moiety. These findings indicate that ring constraint of the sugar moiety is required for the potent inhibitory activity of sulfonium salts. Thus, clarifying factors that explain the substantial difference in the role of the ring structure between aza- and sulfonium-type inhibitors is significant for research and development in the field of glucosidase inhibitors.
| Entry | Compound | Maltase | Sucrase | Isomaltase |
|---|---|---|---|---|
| a The inhibitory activities of salacinol (1) and neosalacinol (4) were re-examined in this study to compare under the same experimental conditions as those of all synthetics (26, 27 and 28). Values in parentheses indicate inhibition (%) at 100 μg mL−1 (26a: 297 μM, 26b: 271 μM, 26c and 27c: 341 μM, 28a: 326 μM, 28b: 296 μM, 28c: 380 μM). | ||||
| 1 | 26a | >297 (8.3) | >297 (18.8) | >297 (17.3) |
| 2 | 26b | >271 (9.6) | >271 (3.2) | >271 (16.9) |
| 3 | 26c | >341 (3.4) | >341 (3.2) | >341 (5.9) |
| 4 | 27c | >341 (3.1) | >341 (4.4) | >341 (−2.3) |
| 5 | 28a | >326 (−0.2) | >326 (0.6) | >326 (−1.0) |
| 6 | 28b | >296 (0.5) | >296 (1.2) | >296 (5.3) |
| 7 | 28c | >380 (−1.1) | >380 (2.6) | >380 (−0.9) |
| 8 | Salacinol (1) | 5.2 | 1.3 | 1.1 |
| 9 | Neosalacinol (4) | 8.9 | 1.4 | 0.6 |
The α-glucosidase catalytic domains of rats are known to be homologous to those in humans. For example, the N-terminal catalytic domain of rat ntMGAM and that of human ntMGAM (hntMGAM) were reported to share 60% amino acid sequence similarity.48,49 Thus, it is not surprising that the inhibitory potencies of sulfonium salts such as 1 and 2 against rat maltase were comparable to those against human maltase.10 In contrast, although the full-length amino acid sequence of yeast α-glucosidase only shares 42% similarity with that of human maltase, the catalytic domain (in the range of 5 Å around the substrate binding site) of yeast α-glucosidase exhibits high similarity (83%) to that of human maltase.50 This explains why 1-deoxynojirimycin (23) is a potent inhibitor against both yeast α-glucosidase44 and human maltase.51 Thus, we conducted a docking study using hntMGAM and aimed to elucidate the structural factors involved in the differences between Fowler's results44 and ours.
Given that acyclic compounds occupy the active site of enzymes, they are generally regarded as less Gibbs-energetically favourable than their corresponding parent cyclic compounds. This is because the side chain of acyclic compounds can no longer rotate freely due to binding to the amino acid residue at the active site, leading to conformational restriction and significant entropic loss. Therefore, in general, acyclic compounds are often weaker inhibitors than their corresponding parent cyclic compounds. However, Fowler's report demonstrated that the OH of an acyclic inhibitor can successfully bind to the enzyme active site when their side chains possess the same hydroxyl stereochemistry as that of the parent cyclic inhibitors. However, they did not investigate the binding mode in detail. The present docking study revealed the key structural factors enabling the ring-cleaved analogue (24) to exhibit potent inhibitory activity equal to that of the parent cyclic compound, 1-deoxynojirimycin (23), at the enzyme active site. The superpositions of 23 and 24 in hntMGAM are displayed in Fig. 5A and B, respectively. As shown in Fig. 5A, 23 places four oxygen atoms on the C2, C3, C4, and C6 carbons in ideal positions to effectively form hydrogen bonding interactions (white double-headed dotted arrows) with the amino acid residues Asp327, Asp542, and His600 within a distance of 3.0 Å. This allows the enzyme to fit into the narrow pocket of the active site. Furthermore, the nitrogen atom of 23 is conveniently positioned 3.50 Å from a catalytic nucleophilic residue (Asp443) to form a salt bridge (yellow double-headed dotted arrow). Therefore, we speculated that 23 exhibited potent inhibitory activity due to the cooperative effect of these interactions. In contrast, 24 had to spread out its two side chains in the active site to avoid mutual intramolecular steric repulsion. Consequently, C3 and C4 of the side chains approached Asp542 and Asp327 at distances of 2.78 Å and 2.90 Å, respectively. These distances were much shorter than those of 23 (3.39 Å and 3.43 Å), presumably causing repulsive van der Waals interactions that diminished its inhibitory activity. In this repulsive conformation of side chains, the hydroxyl groups at the C2, C3, C4, and C6 positions of 24 stabilised the side chain via hydrogen bonding interactions with Asp327, Asp542, and His600, resulting in the nitrogen atom of 24 approaching Asp443 at a distance of 2.87 Å. This distance is approximately 0.6 Å smaller than that of 23 (3.50 Å), which would be expected to strengthen the salt bridge effect with Asp443 in 24 relative to that in 23. Therefore, this effect was considered to be an important interaction that counteracts the negative effects of the aforementioned van der Waals repulsion and entropic loss. On the other hand, the superpositioning of neosalacinol (4) in hntMGAM (Fig. 5D) indicated that the binding mode of thiosugar and side-chain hydroxyls with amino acid residues was largely similar to those previously reported for salacinol (1, Fig. 5C).37 Hence, we characterised the differences in binding mode between neosalacinol (4) and the corresponding ring-cleaved analogues (26c and 27c). Four hydrogen bonding interactions between the thiosugar hydroxyls of 4 and Asp327 and His600, as well as three hydrogen bonding interactions between the side-chain hydroxyls of 4 and Asp203, Arg526, and Asp524 (residues Asp203 and Arg526 are not shown), were detected in the narrow pocket. The distances between the oxygen atoms of all the hydroxyls and amino acid residues were calculated to be in the range of 2.6–3.3 Å, thus indicating that hydrogen bonding interactions were effectively induced between the hydroxyls and enzyme. The sulfonium ion centre of 4 was placed at an appropriate position, which allowed optimal contacts for electrostatic interactions with Asp443 via salt bridging (3.43 Å). Meanwhile, calculations of the binding mode of the acyclic analogues (26c and 27c) in the enzyme active site indicated that the C2 and C3 of 26c and the C3 of 27c approached the amino acid residues (Asp524 and/or Asp327) with a distance within 3.0 Å, which presumably caused van der Waals repulsion (Fig. 5E and F). Thus, the two side chains of 26c and 27c were in an environment similar to those of ring-cleaved 1-deoxynojirimycin (23). However, the additional side chain comprising the C1′–C4′ of 26c and 27c were anchored by hydrogen bonds to Asp203, Asp524, and Arg526, preventing the sulfonium cation from approaching Asp443. Thus, the stronger salt bridge observed for 24 (Fig. 5B) is unlikely to occur for 26c and 27c. The binding abilities of 26c and 27c to the enzyme were presumed to be significantly lower than those of 1 and 4 due to the following reasons: (1) hydroxyls within side chains have to retain hydrogen bonds with the enzyme in a conformation where van der Waals repulsion exists without the contribution of salt bridges and (2) when the hydrogen bond between side chains and the enzyme is formed, the loss of rotational flexibility of the side chains results in an entropic disadvantage. In consideration of the above findings, a computational analysis of the binding mode of the truncated salacinol analogues (28a–28c; lacking the C3-OH moiety that can bind Asp327 and His600 in the active site of hntMGAM) was not performed because it was apparent that their binding capacities to the active site are likely further reduced than those of 26c and 27c.
 | ||
| Fig. 5 Superpositions of 1-deoxynojirimycin (23): (A) acyclic analogue 24: (B) salacinol (1): (C) neosalacinol (4): (D) acyclic analogues 26c: (E) and 27c: (F) in the hNtMGAM active site. Double-headed dot arrows show hydrogen bonding (white) and salt bridge (yellow) residues with amino acid residues. Double-headed orange arrows show distances between side chain carbons and amino acid residues (Asp327 and Asp524). The numbers attached to arrows indicate the binding distance (Å). | ||
Conclusions
In this study, we synthesised acyclic analogues (26, 27, and 28) of natural sulfonium-type α-glucosidase inhibitors (1 and 4) based on the knowledge that acyclic analogues (24 and 25) of 1-deoxynojirimycin (23) retain strong α-glucosidase inhibitory activity. However, our results showed that all these sulfonium-type analogues were inactive against rat α-glucosidase. Our findings provide important insights into the structure–activity relationship (SAR) of 1 and reveal that the five-membered thiosugar ring structure is essential for salacinol-type inhibitors. This is interesting because the opposite was observed in a previous SAR study on aza-compounds (23, 24, and 25), suggesting that their cyclic structure does not contribute to inhibitory activity. Further SAR studies to identify novel analogues of these sulfonium-type inhibitors with stronger α-glucosidase inhibitory activity are currently in progress.Experimental section
Chemistry
IR spectra were measured on a FT-IR spectrophotometer. NMR spectra were recorded on a FT-NMR spectrometers (1H, 500, 600 or 800 MHz; 13C, 125, 150 or 200 MHz). Multiplicity was designated as s = singlet, d = doublet, t = triplet, quint = quintet, m = multiplet, br = broad. Chemical shifts (δ) and coupling constants (J) are given in ppm and Hz, respectively. Tetramethylsilane (TMS) was used as an internal standard for 1H NMR measurements in CDCl3, whereas 13C NMR measurements utilized the solvent signal (77.0 ppm) of CDCl3 for this purpose. When CD3OD and DMSO-d6 were used for the measurement of 1H and 13C NMR spectra, solvent signals [in CD3OD (δH 3.30 ppm and δC 49.0 ppm) and in DMSO-d6 (δH 2.49 ppm and δC 39.7 ppm)] were used as standards. Sodium 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) was used as an external standard in the measurement of 1H and 13C NMR spectra in D2O. 1D NMR peak assignments were confirmed by COSY and HSQC spectra. High-resolution mass spectra were recorded on an Orbitrap mass spectrometry (ESI). Optical rotations were determined with a digital polarimeter. All the organic extracts were dried over anhydrous Na2SO4 prior to evaporation. Column chromatography was performed over silica gel (45–106 μM).:EtOAc = 5:1) to give the title compound 30 (6.2 g, 14.5 mol, 99%) as a colourless oil.IR (neat): 3088, 3063, 6030, 2920, 2866, 1597, 1497, 1456, 1361, 1190, 1099, 918 cm−1. 1H NMR (600 MHz, CDCl3) δ: 2.39 (3H, s, C6H4CH3), 3.67 (4H, br d-like, J = ca. 5.6, CH2OBn), 4.42/4.46 (each 2H, d, J = 12.0, OCH2Ph), 4.76 (1H, br quint-like, J = 5.6, CHOTs), 7.24–7.18 (6H, m, arom.), 7.27–7.33 (6H, m, arom.), 7.78 (2H, d, J = 8.3, arom.). 13C NMR (150 MHz, CD3OD) δ: 21.6 (C6H4CH3), 68.7 (2C, CH2OBn), 73.3 (2C, OCH2Ph), 79.6 (CHOTs), 127.6 (4C)/127.7 (2C)/127.9 (2C)/128.3 (4C)/129.5 (2C) (d, arom.), 133.9/137.6 (2C)/144.5 (s, arom.). HRMS (ESI) m/z: [M + Na]+ calcd for C24H26O5NaS, 449.1393; found, 449.1384.
:EtOAc = 5:1) gave the title compound 31 (4.16 g, 12.59 mmol, 93%) as a colourless oil.IR (neat): 3429, 1605, 1454, 1361, 1280, 1207, 1118, 1072 cm−1. 1H NMR (500 MHz, CDCl3) δ: 2.79 (2H, t, J = 5.6, H-1′), 2.89 (1H, br t-like, J = ca. 5.5, OH), 3.08 (1H, quint, J = 6.2, H-2), 3.61/3.63/3.68/3.69 (each 1H, J = 6.2, H-1), 3.70–3.74 (1H, br m, H-2′), 4.54 (4H, s, OCH2Ph), 7.28–7.35 (10H, m, arom.). 13C NMR (125 MHz, CDCl3) δ: 35.6 (C-1′), 45.8 (C-2), 61.5 (C-2′), 71.2 (2C, C-1), 73.3 (OCH2Ph), 127.7 (2C)/127.7/128.4 (2C) (d, arom.), 137.9 (s, arom.). HRMS (ESI) m/z: [M + Na]+ calcd for C19H24O3NaS, 355.1344; found, 355.1338.
:EtOAc = 10:1) gave the title compound 32 (4.01 g, 9.48 mmol, 90%) as a colourless oil.IR (neat): 1605, 1497, 1454, 1361, 1308, 1288, 1261, 1204, 1103, 1026 cm−1. 1H NMR (800 MHz, CDCl3) δ: 2.83 (2H, t, J = 6.9, H-1′), 3.08 (1H, quint, J = 5.9, H-2), 3.63 (2H, t, J = 6.9, H-2′), 3.67/3.68 (each 2H, d, J = 5.9, H-1), 4.50/4.51 (each 2H, s, OCH2Ph), 7.26–7.33 (15H, arom.). 13C NMR (200 MHz, CDCl3) δ: 31.3 (C-1′), 45.7 (C-2), 70.2 (C-2′), 70.7 (C-1), 73.0/73.2 (OCH2Ph), 127.56/127.59/127.64/127.7/128.3/128.4 (d, arom.), 138.1/138.2 (s, arom.). HRMS (ESI) m/z: [M + Na]+ calcd for C26H30O3NaS, 445.1807; found, 445.1799.
:MeOH = 20:1) gave the title compound 34 (369 mg, 0.77 mmol, 96%) as a colourless oil consisting of ca. 1:1 diastereomeric isomers.1H NMR (800 MHz, CDCl3) δ: 1.26/1.30/1.35/1.40 [each 3H, s, (CH3)2C], 3.49–3.52 (0.5H, m, H-1′a), 3.74–3.99 (9.5H, m, H-1′b, H-1′a#, H-1′b#, H-1′′a, H-1′′a#, H-1a, H-1b, H-1c, H-1d, H-1a#, H-1b#, H-1c#, H-1d#, H-2′a, H-2′b, H-2′a#, H-2′b#, H-4′′b and H-4′′b#), 4.12 (0.5H, dd, J = 14.4, 4.0, H-1′′b), 4.15–4.27 (3.0H, m, H-1′′b#, H-2, H-4′′b, H-4′′b#, H-3′′ and H-3′′#), 4.35–4.56 (7.5H, H-2#, H-2′′, H-2′′# and PhCH2), 7.21–7.36 (15H, m, arom.). 13C NMR (200 MHz, CDCl3) δ: 19.1 (2C)/28.1/28.2 [(CH3)2C], 41.7/42.1 (C-1′), 43.6/45.6 (C-1′′), 56.0/56.7 (C-2), 62.38/62.42 (C-4′′), 63.9/64.6 (C-2′), 66.12/66.13/66.3/67.0 (C-1), 67.8/68.2 (C-2′′), 69.5/69.6 (C-3′′), 73.2/73.35/73.54/73.61/73.7 (2C) (OCH2Ph), 127.8/128.0/128.11/128.12/128.14/128.15/128.23/128.27/128.34/128.36/128.52/128.55/28.57/128.60/128.67/128.68 (d, arom.), 136.32/136.34/136.5 (2C)/136.71/136.75 (s, arom.). HRMS (ESI) m/z: [M + Na]+ calcd for C33H42O9NaS2, 669.2163; found, 669.2152.
:MeOH:H2O = 6:4:1) to give the title compound 26a (124 mg, 0.32 mmol, 94%) as a colourless oil consisting of ca. 1:1 diastereomeric isomers.1H NMR (800 MHz, D2O) δ: 3.72 (0.5H, dd, J = 9.9, H-1′′a), 3.77–3.82 (1.0H, m, H-1′a and H-1′a#), 3.91–3.97 (2.5H, m, H-1′b, H-1′b#, H-1′′a#, H-4′′a and H-4′′a#), 4.00 (0.5H, dd, J = 14.0, 3.0, H-1′′b#), 4.04 (0.5H, dd, J = 16.7, 3.9, 4′′b), 4.05 (0.5H, dd, J = 16.7, 3.9, 4′′b#), 4.11–4.23 (7.5H, m, H-1′′b, H-2′a, H-2′a#, H-2′b, H-2′b#, H-2, H-2#, H-1a, H-1b, H-1a# and H-1b#), 4.41 (0.5H, td, J = 6.9, 3.9, H-3′′), 4.43 (0.5H, td, 6.9. 3.9, H-3′′#), 4.46–4.49 (1.0H, m, H-2′′). 13C NMR (125 MHz, D2O) δ: 45.5/46.3 (C-1′), 46.4/46.7 (C-1′′), 59.2/59.3 (C-2′), 60.7/61.2 (C-2), 61.1/61.2/61.4 (C-1), 62.4 (C-4′′), 68.2/68.7 (C-2′′), 83.3/83.4 (C-3′′). HRMS (ESI) m/z: [M + Na]+ calcd for C9H20O9NaS2, 359.0441; found, 359.0437.
:MeOH = 5:1 → CHCl3:MeOH:H2O = 6:4:1) to give the title compound 26b (130 mg, 0.35 mol, 92%) as a colourless oil consisting of ca. 1:1 diastereomeric isomers.1H NMR (800 MHz, CD3OD) δ: 3.59–3.73 (4.5H, m, H-1′a, H-1′a#, H-1′′a, H-4′′a, H-4′′b, H-4′′a#, H-4′′b#, H-3′′, H-3′′#), 3.68 (3H, s, CH3OSO3), 3.51–3.85 (1H, m, H-1′b, H-1′′a#), 3.87–3.90 (1H, m, H-1′′b, H-1′′b#), 3.99–4.11 (8.5H, m, H-1′b#, H-2′a, H-2′b, H-2′a#, H-2′b#, H-2′′, H-2′′#, H-2, H-2#, H-1a, H-1b, H-1#a, H-1#b). 13C NMR (800 MHz, CD3OD) δ: 43.9/44.9 (C-1′), 45.0/45.3 (C-1′′), 55.2 (CH3OSO3), 58.13/58.14 (C-2′), 59.6/60.2 (C-2), 60.1/60.2 (C-1), 64.0/64.1 (C-4′′), 69.1/69.6 (C-2′′), 75.4/75.5 (C-3′′). HRMS (ESI) m/z: [M–CH3OSO3]+ calcd for C9H21O6S, 257.1053; found, 257.1051.
:MeOH:H2O = 6:4:1) to give the title compound gave 26c (70 mg, 0.24 mmol, 93%) as a colourless oil consisting of ca. 1:1 diastereomeric isomers.1H NMR (800 MHz, CD3OD) δ: 3.59–3.73 (4.5H, m, H-1′a, H-1′a#, H-1′′a, H-4′′a, H-4′′b, H-4′′a#, H-4′′b#, H-3′′, H-3′′#), 3.81–3.85 (1H, m, H-1′b, H-1′′a#), 3.87–3.91 (1H, m, H-1′′b, H-1′′b#), 3.99–4.11 (8.5H, m, H-1′b#, H-2′a, H-2′b, H-2′a#, H-2′b#, H-2′′, H-2′′#, H-2, H-2#, H-1a, H-1b, H-4#a, H-4#b). 13C NMR (800 MHz, CD3OD) δ: 43.9/45.0 (C-1′), 45.0/45.3 (C-1′′), 58.13/58.14 (C-2′), 59.6/60.1 (C-2), 60.2/60.3 (C-1), 64.0/64.1 (C-4′′), 69.1/69.6 (C-2′′), 75.4/75.6 (C-3′′). HRMS (ESI) m/z: [M–Cl]+ calcd for C9H21O6S, 257.1053; found, 257.1050.
:MeOH = 100:1) gave the title compound 36 (3.02 g, 8.70 mmol, 99%) as a colourless oil.1H NMR (800 MHz, CDCl3) δ: 2.64 (1H, dd, J = 14.0, 8.2, H-1′a), 2.71/2.72 (each 1H, t, J = 5.6, H-1a and H-1b), 2.86 (1H, dd, J = 14.0, 3.4, H-1′b), 3.62 (1H, dt, J = 6.2, 4.8, H-3′), 3.70 (1H, dd, J = 10.2, 4.8, H-4′a), 3.70/3.71 (1H, t, J = 5.6, H-2a and H-2b), 3.73 (1H, dd, J = 10.2, 4.8, H-4′b), 3.91 (ddd, J = 8.2, 6.2, 3.4, H-2′), 4.54/4.55 (each 1H, d, J = 12.0, OCH2Ph), 4.56/4.68 (each 1H, d, J = 11.6, OCH2Ph), 7.26–7.37 (10H, m, arom.). 13C NMR (200 MHz, CDCl3) δ: 35.7 (C-1′), 35.9 (C-1), 61.1 (C-2), 69.7 (C-4′), 71.4 (C-2′), 72.6/73.5 (OCH2Ph), 79.3 (C-3′), 127.7/127.8/127.9/128.4/128.5 (d, arom.), 137.7/138.0 (s, arom.). HRMS (ESI) m/z: [M + Na]+ calcd for C20H26O4NaS, 385.1444; found, 385.1423.
:MeOH = 15:1) to give the title compound 38 (294 mg, 0.56 mmol, 71%) as a colourless oil.δ: 3.15–3.81 (11H, m, H-1′a, H-1′b, H-1′a#, H-1′b#, H-1′′a, H-1′′b, H-1′′a#, H-1′′b#, H-1a, H-1b, H-1a#, H-1b#, H-3′a, H-3′b, H-3′a#, H-3′b#, H-3′′, H-3′′#, H-4′′a, H-4′′b, H-4′′a# and H-4′′b#), 3.93–4.00 (2.5H, m, H-2a, H-2b, H-2′, H-2a# and H-2b#), 4.11 (0.5H, tdd, J = 5.5, 4.8, 3.5, H-2′#), 4.26–4.29 (1H, m, H-2′′ and H-2′′#), 4.52/4.56/4.63/4.71 (each 1H, d, J = 11.5, OCH2Ph), 7.27–7.29 (2H, m, arom.), 7.32–7.36 (8H, m, arom.). 13C NMR (200 MHz, CD3OD) δ: 46.39/46.42 (C-1′′), 46.5/46.9 (C-1′), 47.1/47.3 (C-1), 57.8 (C-2), 65.8 (C-3′), 68.6/68.7 (C-2′′), 69.1/69.2 (C-2′), 69.51/69.53 (C-4′′), 73.7/73.8/74.5 (OCH2Ph), 81.9/82.0 (C-3′′), 128.82/128.83/128.87/128.89/129.00/129.02/129.23/129.24/129.44/129.46 (d, arom.), 139.42/139.46 (s, arom.). HRMS (ESI) m/z: [M–BF4]+ calcd for C23H33O6S, 437.1992; found, 437.1986.
:MeOH:H2O = 6:4:1) to give the title compound 39 (208 mg, 0.534 mmol, 98%) as a colourless oil.1H NMR (800 MHz, CD3OD) δ: 3.47–3.83 (11H, m, H-1′a, H-1′b, H-1′a#, H-1′b#, H-1′′a, H-1′′b, H-1′′a#, H-1′′b#, H-1a, H-1b, H-1a#, H-1b#, H-3′a, H-3′b, H-3′a#, H-3′b#, H-3′′, H-3′′#, H-4′′a, H-4′′b, H-4′′a# and H-4′′b#), 3.93–4.01 (2.5H, m, H-2a, H-2b, H-2′, H-2a# and H-2b#), 4.11 (0.5H, tdd, J = 5.5, 4.8, 3.5, H-2′#), 4.27–4.30 (1H, m, H-2′′ and H-2′′#), 4.52/4.56/4.63/4.71 (each 1H, d, J = 11.5, OCH2Ph), 7.27–7.29 (2H, m, arom.), 7.32–7.36 (8H, m, arom.). 13C NMR (200 MHz, CD3OD) δ: 46.36/46.42 (C-1′′), 46.6/47.0 (C-1′), 47.1/47.3 (C-1), 57.8 (C-2), 65.8 (C-3′), 68.6/68.7 (C-2′′), 69.1/69.2 (C-2′), 69.54/69.56 (C-4′′), 73.75/73.78/74.5 (OCH2Ph), 81.96/82.02 (C-3′′), 128.8/128.85/128.89/129.00/129.02/129.23/129.24/129.44/129.46/129.48 (d, arom.), 139.41/139.45 (s, arom.). HRMS (ESI) m/z: [M–Cl]+ calcd for C23H33O6S, 437.1992; found, 437.1987.
:MeOH:H2O = 6:4:1) to give the title compound 27c (83 mg, 0.28 mmol, 89%) as a colourless oil.1H NMR (800 MHz, D2O) δ: 3.66–3.83 (10H, m, H-1′a, H-1′a#, H-1′′a, H-1′′b, H-1′′#a, H-1′′#b, H-3′a, H-3′b, H-3′a#, H-3′b#, H-1a, H-1b, H-1a#, H-1b#, H-4′′a, H-4′′b, H-4′′a#, H-4′′b#, H-3′′ and H-3′′#), 3.86/3.88 (each 0.5H, dd, J = 14.0, 3.0, H-1′b and H-1′b#), 4.15 (2H, t, J = 5.5, H-2a, H-2b, H-2#a and H-2#b), 4.23 (1H, ddd, J = 9.3, 6.4, 3.0, H-2′′ and H-2′′#), 4.28–4.31 (1H, m, H-2′ and H-2′#). 13C NMR (200 MHz, D2O) δ: 47.7/47.8 (C-1′′), 47.9/48.2 (C-1′), 48.2/48.6 (C-1), 58.93/58.95 (C-2), 64.9 (C-3′), 66.8 (C-4′′), 69.87/69.93 (C-2′′), 69.97/70.00 (C-2′), 76.4 (C-3′′). HRMS (ESI) m/z: [M–Cl]+ calcd for C9H21O6S, 257.1053; found, 257.1051.
IR (neat): 2918, 2857, 1494, 1454, 1360, 1206, 1101 cm−1. 1H NMR (800 MHz, CDCl3) δ: 2.78 (4H, t, J = 7.0, SCH2CH2O), 3.64 (4H, t, J = 7.0, SCH2CH2O), 4.52 (4H, s, OCH2Ph), 7.28 (2H, td, J = 2.3, 5.7, arom.), 7.14–7.35 (8H, m, arom.). 13C NMR (200 MHz, CDCl3) δ: 32.1 (SCH2CH2O), 70.0 (SCH2CH2O), 73.0 (OCH2Ph), 127.6 (d, arom.), 127.7 (d, arom.), 128.4 (d, arom.), 138.1 (s, arom.). HRMS (ESI) m/z: [M + Na]+ calcd for C18H22O2NaS, 325.1232; found, 325.1233.
:MeOH = 30:1) to give the title compound 42 (501 mg, 1.17 mmol, 95%) as a white amorphous.Mp 127.0–127.5 °C. [α]24D + 13.8 (c = 1.83, CHCl3). IR (KBr): 1261, 1242, 1207, 1157, 1132, 1099, 1080, 1018 cm−1. 1H NMR (800 MHz, CDCl3) δ: 1.32/1.43 [each 3H, s, (CH3)2C], 3.62 (1H, ddd, J = 13.8, 5.5, 5.0, H-1a), 3.72 (1H, ddd, J = 13.8, 6.6, 4.8, H-1b), 3.76 (1H, ddd, J = 13.8, 5.6, 3.8, H-1c), 3.78 (1H, dd, J = 11.6, 9.6, H-4′a), 3.81–3.85 (3H, m, H-1d, H-2a, and H-2b), 3.86–3.92 (2H, m, H-2c and H-2d), 3.92 (1H, dd, J = 14.2, 3.8, H-1′a), 4.05 (1H, dd, J = 14.2, 3.8, H-1′b), 4.17 (1H, dd, J = 11.6, 5.8, H-4′b), 4.62 (1H, ddd, J = 9.6, 3.8, 3.8, H-2′), 4.40 (1H, td, J = 9.6, 5.8, H-3′), 4.48/4.51 (each 1H, d, J = 11.8, OCH2Ph), 4.49/4.52 (each 1H, d, J = 11.6, OCH2Ph), 7.26–7.37 (10H, m, arom.). 13C NMR (200 MHz, CDCl3) δ: 19.2/28.2 [(CH3)2C], 42.5/43.4 (C-1), 44.2 (C-1′), 62.4 (C-4′), 63.8/64.2 (C-2), 68.1 (C-3′) 69.5 (C-2′), 73.4 (OCH2Ph), 99.7 [(CH3)2C], 128.0/128.1/128.2/128.3/128.6/128.7 (d, arom.), 136.6/136.7 (s, arom.). HRMS (ESI) m/z: [M + Na]+ calcd for C25H34O8NaS2, 549.1587; found, 549.1577.
:MeOH = 5:1 → CHCl3:MeOH:H2O = 10:5:1) to give the title compound 28a (157 mg, 0.512 mmol, 93%) as a colourless oil.[α]24D + 23.4 (c = 1.11, CH3OH). IR (KBr): 3479, 1412, 1257, 1231, 1211, 1069, 1015 cm−1. 1H NMR (800 MHz, D2O) δ: 3.76–3.79 (4H, m, H-1), 3.79 (1H, dd, J = 14.0, 9.2, H-1′′a), 3.94 (1H, dd, J = 13.0, 3.2, H-4′′a), 3.97 (1H, dd, J = 14.0, 3.2, H-1′′b), 4.05 (1H, dd, J = 13.0, 3.2, H-4′′b), 4.16–4.18 (4H, m, H-2), 4.42 (1H, td, J = 7.2, 3.2, H-3′′), 4.49 (1H, ddd, J = 9.2, 7.2, 3.2, H-2′′). 13C NMR (200 MHz, D2O) δ: 47.1/47.5 (C-1), 47.9 (C-1′′), 58.9 (d, C-2), 62.4 (C-4′′), 68.2 (C-2′′), 83.2 (C-3′′). HRMS (ESI) m/z: [M + Na]+ calcd for C8H18O8NaS2, 329.0335; found, 329.0336.
:MeOH = 5:1 → CHCl3:MeOH:H2O = 6:4:1) to give the title compound 28b (68 mg, 0.20 mmol, 88%) as a colourless oil.[α]24D + 21.9 (c = 0.74, CH3OH). IR (neat): 3360, 1647, 1454, 1416, 1242, 1068 cm−1. 1H NMR (800 MHz, CD3OD) δ: 3.61–3.63 (2H, m, H-3′a and H-4′′a), 3.65 (2H, t-like, J = 5.5, H-1), 3.67–3.69 (3H, m, H-1 and H-4′′b), 3.68 (3H, s, CH3OSO3), 3.70 (1H, dd, J = 13.6, 9.2, H-1′′a), 3.79 (1H, dd, J = 13.6, 3.4, H-1′′b), 4.06–4.04 (4H, m, H-2), 4.10 (1H, ddd, J = 9.2, 6.2, 3.4, H-2′′). 13C NMR (200 MHz, CD3OD) δ: 45.6/46.2 (C-1), 46.6 (C-1′′), 55.1 (CH3OSO3), 57.8/57.8 (C-2), 64.0 (C-4′′), 69.1 (C-2′′), 74.4 (C-3′′). HRMS (ESI) m/z: [M–CH3OSO3]+ calcd for C8H19O5S, 227.0948; found, 227.0947.
:MeOH:H2O = 5:10:1) to give the title compound 28c (47 mg, 0.18 mmol, 87%) as a colourless oil. [α]24D + 24.6 (c = 0.59, CH3OH). IR (neat): 3360, 1647, 1454, 1416, 1242, 1068 cm−1. 1H NMR (800 MHz, CD3OD) δ: 3.61–3.64 (2H, m, H-3′′a and H-4′′a), 3.66 (2H, t-like, J = 5.5, H-1), 3.67–3.69 (3H, m, H-1cd and H-4′′b), 3.70 (1H, dd, J = 13.6, 8.7, H-1′′a), 3.79 (1H, dd, J = 13.6, 3.4, H-1′′b), 4.06–4.04 (4H, m, H-2), 4.11 (1H, ddd, J = 8.2, 6.2, 3.4, H-2′′). 13C NMR (200 MHz, CD3OD) δ: 45.6/46.2 (C-1), 46.6 (C-1′′), 57.8/57.8 (C-2), 64.0 (C-4′′), 69.1 (C-2′′), 75.5 (C-3′′). HRMS (ESI) m/z: [M–Cl]+ calcd for C8H19O5S, 227.0948; found, 227.0948.Inhibitory effects on rat intestinal α-glucosidases
The experiments were performed according to the method reported.51–53 Thus, rat small intestinal brush border membrane vesicles were prepared and their suspensions in 0.1 M maleate buffer (pH 6.0) were used as small intestinal α-glucosidases of maltase, sucrose, and isomaltase.53,54 A test sample was dissolved in dimethyl sulfoxide (DMSO), and the resulting solution was diluted with 0.1 M maleate buffer to prepare the test sample solution (concentration of DMSO 10%). A substrate solution in the maleate buffer (maltose 74 mM, sucrose 74 mM, isomaltase 7.4 mM, 50 μL), the test sample solution (25 μL), and the enzyme solution (25 μL) were mixed at 37 °C for 30 min, and then immediately heated by boiling water for 2 min to stop the reaction. The glucose concentrations were determined by a glucose-oxidase method. The final concentration of DMSO in the test solution was 2.5% and no influence of DMSO on the inhibitory activity was detected.Author contributions
KT, MN and SM performed chemistry and analytical experiments. FI and TM performed the biological experiments and analysed data. SN and IN performed molecular docking studies. GT supervised the study, obtained funding and wrote the paper. WX and OM revised the paper. The manuscript was reviewed and approved by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by a Grant-in-Aid for scientific research from the Japan Society for the Promotion of Science [JSPS (KAKENHI), 22K06541 (G. T.)], and a grant from the Mochida memorial foundation for medical and pharmaceutical research, 2021–2022 (K. T). The authors thank financial supports extended by the “Antiaging” Project for Private Universities (G. T. and T. M.).Notes and references
- M. Yoshikawa, T. Murakami, H. Shimada, H. Matsuda, J. Yamahara, G. Tanabe and O. Muraoka, Tetrahedron Lett., 1997, 38, 8367–8370 CrossRef CAS.
- M. Yoshikawa, T. Morikawa, H. Matsuda, G. Tanabe and O. Muraoka, Bioorg. Med. Chem., 2002, 10, 1547–1554 CrossRef CAS PubMed.
- M. Yoshikawa, T. Murakami, K. Yashiro and H. Matsuda, Chem. Pharm. Bull., 1998, 46, 1339–1340 CrossRef CAS PubMed.
- M. Yoshikawa, F. Xu, S. Nakamura, T. Wang, H. Matsuda, G. Tanabe and O. Muraoka, Heterocycles, 2008, 75, 1397–1405 CrossRef CAS.
- Y. Minami, C. Kuriyama, K. Ikeda, A. Kato, K. Takebayashi, I. Adachi and W. J. G. Fleet, Bioorg. Med. Chem., 2008, 16, 2734–2740 CrossRef CAS PubMed.
- G. Tanabe, W. Xie, A. Ogawa, C. Cao, T. Minematsu, M. Yoshikawa and O. Muraoka, Bioorg. Med. Chem. Lett., 2009, 19, 2195–2918 CrossRef CAS PubMed.
- S. Ozaki, H. Oe and S. Kitamura, J. Nat. Prod., 2008, 71, 981–984 CrossRef CAS PubMed.
- O. Muraoka, W. Xie, G. Tanabe, F. A. M. Amer, T. Minematsu and M. Yoshikawa, Tetrahedron Lett., 2008, 49, 7315–7317 CrossRef CAS.
- W. Xie, G. Tanabe, J. Akaki, T. Morikawa, K. Ninomiya, T. Minematsu, M. Yoshikawa, X. Wu and O. Muraoka, Bioorg. Med. Chem., 2011, 19, 2015–2022 CrossRef CAS PubMed.
- T. Morikawa, J. Akaki, K. Ninomiya, E. Kinouchi, G. Tanabe, Y. Pongpiryadacha, M. Yoshikawa and O. Muraoka, Nutrients, 2015, 7, 1480–1493 CrossRef CAS PubMed.
- M. Kobayashi, J. Akaki, K. Yamashita, T. Morikawa, K. Ninomiya, M. Yoshikawa and O. Muraoka, Jpn. Pharmacol. Ther., 2010, 38, 545–550 Search PubMed.
- J. A. Williams, Y. S. Choe, M. J. Noss, C. J. Baumgartner and V. A. Mustad, Am. J. Clin. Nutr., 2007, 86, 124–130 CrossRef CAS PubMed.
- M. H. S. Jayawardena, N. M. W. de Alwis, V. Hettigoda and D. J. S. Fernando, J. Ethnopharmacol., 2005, 97, 215–218 CrossRef CAS PubMed.
- O. Kajimoto, S. Kawamori, H. Shimoda, H. Kawahara, Y. Hirata and T. Takahashi, Nippon Eiyo · Shokuryo Gakkaishi, 2000, 53, 199–205 CrossRef.
- H. Shimoda, T. Fujimura, K. Makino, K. Yoshizima, K. Naitou, H. Ibota and Y. Minowa, Shokuhin Eiseigaku Zasshi, 1999, 40, 198–205 CrossRef.
- S. Mohan and B. M. Pinto, Carbohydr. Res., 2007, 342, 1551–1580 CrossRef CAS PubMed.
- W. Xie, G. Tanabe, J. Xu, X. Wu, T. Morikawa, M. Yoshikawa and O. Muraoka, Mini-Rev. Org. Chem., 2013, 10, 141–159 CrossRef CAS.
- L. Lue, X. Li and W. Xie, Chem.–Eur. J., 2019, 25, 13458–13471 CrossRef PubMed.
- G. Tanabe, K. Yoshikai, T. Hatanaka, M. Yamamoto, Y. Shao, T. Minematsu, O. Muraoka, T. Wang, H. Matsuda and M. Yoshikawa, Bioorg. Med. Chem., 2007, 15, 3926–3937 CrossRef CAS PubMed.
- O. Muraoka, K. Yoshikai, H. Takahashi, T. Minemastu, G. Lu, G. Tanabe, T. Wang, H. Matsuda and M. Yoshikawa, Bioorg. Med. Chem., 2006, 14, 500–509 CrossRef CAS PubMed.
- G. Tanabe, K. Matsuoka, M. Yoshinaga, W. Xie, N. Tsutsui, M. A. M. Amer, S. Nakamura, I. Nakanishi, X. Wu, M. Yoshikawa and O. Muraoka, Bioorg. Med. Chem., 2012, 24, 6321–6334 CrossRef PubMed.
- W. Xie, G. Tanabe, K. Matsuoka, M. A. M. Amer, T. Minematsu, X. Wu, M. Yoshikawa and O. Muraoka, Bioorg. Med. Chem., 2011, 24, 2252–2262 CrossRef PubMed.
- B. D. Johnston, H. H. Jensen and B. M. Pinto, J. Org. Chem., 2006, 71, 1111–1118 CrossRef CAS PubMed.
- R. Nasi, B. O. Patrick, L. Sim, D. R. Rose and M. Pinto, J. Org. Chem., 2008, 73, 6172–6181 CrossRef CAS PubMed.
- R. Nasi, L. Sim, D. R. Rose and B. M. Pinto, J. Org. Chem., 2007, 72, 180–186 CrossRef CAS PubMed.
- H. Liu, L. Sim, D. R. Rose and B. M. Pinto, J. Org. Chem., 2006, 71, 3007–3013 CrossRef CAS PubMed.
- H. Liu, R. Nasi, K. Jayakanthan, L. Sim, H. Heipel, D. R. Rose and B. M. Pinto, J. Org. Chem., 2007, 72, 6562–6572 CrossRef CAS PubMed.
- R. Eskandari, D. A. Kuntz, D. R. Rose and B. M. Pinto, Org. Lett., 2010, 12, 1632–1635 CrossRef CAS PubMed.
- K. Takashima, M. Sakano, E. Kinouchi, S. Nakamura, S. Marumoto, F. Ishikawa, K. Ninomiya, I. Nakanishi, T. Morikawa and G. Tanabe, Bioorg. Med. Chem. Lett., 2021, 33, 127751 CrossRef CAS PubMed.
- S. Nakamura, K. Takahira, G. Tanabe, T. Morikawa, M. Sakano, K. Ninomiya, M. Yoshikawa, O. Muraoka and I. Nakanishi, Bioorg. Med. Chem. Lett., 2010, 20, 4420–4423 CrossRef CAS PubMed.
- N. S. Kumar and B. M. Pinto, Carbohydr. Res., 2005, 340, 2612–2619 CrossRef CAS PubMed.
- A. Ghavami, B. D. Johnston, M. D. Maddess, S. M. Chinapoo, M. T. Jensen, B. Svensson and B. M. Pinto, Can. J. Chem., 2002, 80, 937–942 CrossRef CAS.
- P. Bagri, K. Chester, W. Khan and S. Ahmad, RSC Adv., 2017, 7, 28152–28185 RSC.
- L. Sim, K. Jayakanthan, S. Mohan, R. Nasi, B. D. Johnston, B. M. Pinto and D. R. Rose, Biochemistry, 2010, 49, 443–451 CrossRef CAS PubMed.
- G. Tanabe, T. Otani, W. Cong, T. Minematsu, K. Ninomiya, M. Yoshikawa and O. Muraoka, Bioorg. Med. Chem. Lett., 2011, 21, 3159–3162 CrossRef CAS PubMed.
- G. Tanabe, S. Nakamura, N. Tsutsui, G. Balakishan, W. Xie, S. Tsuchiya, J. Akaki, T. Morikawa, K. Ninomiya, I. Nakanishi, M. Yoshikawa and O. Muraoka, Chem. Commun., 2012, 48, 8646–8648 RSC.
- G. Tanabe, W. Xie, G. Balakishan, M. F. A. Amer, N. Tsutsui, H. Takemura, S. Nakamura, J. Akaki, K. Ninomiya, T. Morikawa, I. Nakanishi and O. Muraoka, Bioorg. Med. Chem., 2016, 24, 3705–3715 CrossRef CAS PubMed.
- L. Lu, J. Chen, W. Tao, Z. Wang, D. Liu, J. Zhou, X. Wu, H. Sun, W. Li, G. Tanabe, O. Muraoka, B. Zhao, L. Wu and W. Xie, J. Med. Chem., 2023, 66, 3484–3498 CrossRef CAS PubMed.
- H. Yuasa, T. Kajimoto and H. C. Wong, Tetrahedron Lett., 1994, 35, 8243 CrossRef CAS.
- Y. Yoshimura, K. Kitano, K. Yamada, H. Satoh, M. Watanabe, S. Miura, S. Sakata, T. Sasaki and A. Matsuda, J. Org. Chem., 1997, 62, 3140 CrossRef CAS PubMed.
- H. Satoh, Y. Yoshimura, S. Sakata, S. Miura, H. Machida and A. Matsuda, Bioorg. Med. Chem. Lett., 1998, 8, 989–992 CrossRef CAS PubMed.
- W. Chen, L. Sim, D. R. Rose and B. M. Pinto, Carbohydr. Res., 2007, 342, 1661–1667 CrossRef PubMed.
- T. S. Gunasundari and S. Chandrasekaran, Carbohydr. Res., 2013, 382, 30–35 CrossRef CAS PubMed.
- P. A. Fowler, A. H. Haines, R. J. K. Taylor, E. J. T. Chrystal and M. B. Gravestock, J. Chem. Soc., Perkin Trans. 1, 1994, 2229–2235 RSC.
- A. Ghavami, K. S. Sadalapure, B. D. Johnston, M. Lobera, B. B. Snider and M. Pinto, Synlett, 2003, 9, 1259–1262 Search PubMed.
- H. Yuasa, J. Takada and H. Hashimoto, Tetrahedron Lett., 2000, 41, 6615–6618 CrossRef CAS.
- E. Abushanab, P. Vemishetti, R. W. Leiby, H. K. Singh, A. B. Mikkilineni, D. C. J. Wu, R. Saibaba and R. P. Panzica, J. Org. Chem., 1988, 53, 2598–2602 CrossRef CAS.
- Maltase-glucoamylase Gene ID: (a) Entrez Gene 312272 [Rattus norvegicus (Norway rat)];; (b) Entrez Gene 8972 [Homo sapiens (human)]..
- S. Nakamura, K. Shimada, G. Tanabe, O. Muraoka and I. Nakanishi, Open J. Med. Chem., 2017, 7, 19–28 CAS.
- Glucosidase 2 subunit alpha, Gene ID: Entrez Gene 854708 [Saccharomyces cerevisiae S288C].
- D. Breitmeier, S. Gunther and H. A. Heymann, Arch. Biochem. Biophys., 1988, 346, 7–14 CrossRef PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- T. A. Halgren, J. Comput. Chem., 1996, 17, 490–519 CrossRef CAS.
- T. A. Halgren, J. Comput. Chem., 1996, 17, 520–552 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08485j |
| ‡ These authors contributed equally to this project. |
| This journal is © The Royal Society of Chemistry 2024 |