
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual-rotor strategy for organic cocrystals with enhanced near-infrared photothermal conversion†
Xinyi Wen‡
a,
Yixin Shao‡a,
Ye-Tao Chena,
Jiaxing He a,
Shun-Li Chen*a,
Li Dangab and
Ming-De Liab
a,
Shun-Li Chen*a,
Li Dangab and
Ming-De Liab
aCollege of Chemistry and Chemical Engineering, Key Laboratory for Preparation and Application of Ordered Structural Materials of Guangdong Province, Shantou University, Shantou 515063, China. E-mail: chenxl@stu.edu.cn
bChemistry and Chemical Engineering Guangdong Laboratory, Shantou 515031, China
First published on 2nd February 2024
Abstract
Organic cocrystal engineering provides a promising route to promote the near-infrared (NIR) light harvesting and photothermal conversion (PTC) abilities of small organic molecules through the rich noncovalent bond interactions of D/A units. Besides, the single-bond rotatable groups known as “rotors” are considered to be conducive to the nonradiative transitions of the excited states of organic molecules. Herein, we propose a single-/double-bond dual-rotor strategy to construct D–A cocrystals for NIR PTC application. The results reveal that the cocrystal exhibits an ultra-broadband absorption from 300 nm to 2000 nm profiting from the strong π–π stacking and charge transfer interactions, and the weakened p–π interaction. More importantly, the PTC efficiency of cocrystals at 1064 nm in the NIR-II region can be largely enhanced by modulating the number of rotor groups and the F-substituents of D/A units. As is revealed by fs-TA spectroscopy, the superior NIR PTC performance can be attributed to the nonradiative decays of excited states induced by the free rotation of the single-bond rotor (–CH3) from the donors and the inactive double-bond rotor (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(C
C(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N)2) being in the active form of [–C(CN)2] in the excited states from the acceptors. This prototype displays a promising route to extend the functionalization of small organic molecules based on organic cocrystal engineering.
N)2) being in the active form of [–C(CN)2] in the excited states from the acceptors. This prototype displays a promising route to extend the functionalization of small organic molecules based on organic cocrystal engineering.
Introduction
Due to the ability to directly convert UV-VIS-NIR light into thermal energy, photothermal conversion (PTC) materials have exciting prospects in a wide range of practical applications including photothermal imaging,1 photothermal therapy,2 and solar water evaporation.3 At present, the reported PTC materials mainly consist of metal nanomaterials,4 semiconductors,5 carbon-based materials,6 and organic polymers.7 The narrow absorption region far away from the NIR region makes small organic molecules seem not suitable for PTC application. However, since the first dibenzotetrathiafulvalene (DBTTF)-1,2,4,5-tetracyanobenzene (TCNB) cocrystal for NIR PTC and imaging was reported by Hu's group in 2018,1 organic donor–acceptor (D–A) cocrystal engineering provides a promising route to improve the NIR light harvesting and PTC abilities of small organic molecules based on the rich noncovalent interactions of D/A units.8It is revealed that strong D–A intermolecular interactions, e.g., π–π stacking, charge-transfer interaction, are conducive to the narrowing of the optical band gap, the red-shifting of the absorption band, and the exciton quenching of cocrystals.9 But the p–π interaction may have negative influence on these properties, which should be reduced or avoided in the rational design of cocrystals.10 From the perspective of molecular structure, a large conjugated skeleton is often favorable to the dense stacking and strong interactions of D/A units.8,9 The existence of single-bond rotatable groups (e.g. –CF3, –Ph, etc.) available in the ground states as the “rotors” can promote the possible nonradiative transitions of excited states of organic materials under light excitation.11,12 Our previous reports reveals that the inactive double-bond rigid moieties (C(CN)2) of conjugated quinoid molecules (e.g. tetracyanoethylene (TCNE), 7,7′,8,8′-tetracyanodimethyl-p-benzoquinone (TCNQ)) in the ground states can convert into active single-bond rotatable moieties (–C(CN)2) in the excited states, which can also serve as the rotors to strengthen the nonradiative transitions of excited states of cocrystals.13 It is very attracting that, when a certain conjugated skeleton of D/A units is satisfied, can the PTC properties of cocrystals be improved notably if we combine these two kinds of rotatable groups together?
To answer this significant question, we propose a single-/double-bond dual-rotor strategy to construct D–A cocrystals for the NIR PTC application. In detail, as described in Scheme 1, the anthracene-based donors provide the single-bond rotor (–CH3): e.g. 9-methylanthracene (MAT), 9,10-dimethylanthracene (DAT). The acceptors with different strengths of electron affinity provide the double-bond rotor (C(CN)2): e.g. TCNQ, 2,3,5,6-tetrafluoro-7,7′,8,8′-tetracyanodimethyl-p-benzoquinone (F4TCNQ). The polycyclic aromatic anthracene has a well-conjugated planar structure,14 which facilitates it to be a good donor candidate for cocrystals by connecting the electron-donating methyl units to endow MAT and DAT with a stronger electron-donating capacity. TCNQ and F4TCNQ have strong electron affinity and commonly serve as the acceptors for cocrystals.15 Three kinds of cocrystals, MAT–TCNQ (MTQ), DAT–TCNQ (DTQ) and DAT–F4TCNQ (DFQ), were synthesized by a conventional solution self-assembly method.
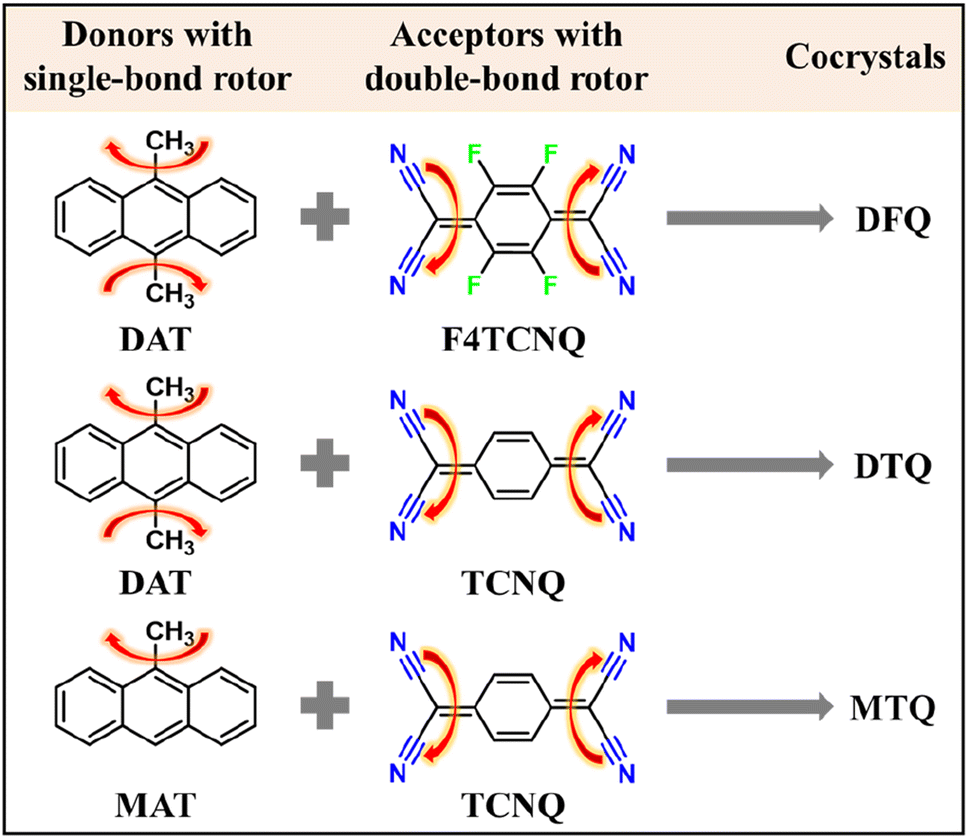 | ||
| Scheme 1 Dual-rotor strategy to construct D–A cocrystals in this work. | ||
It is exciting that the DFQ cocrystal exhibits an ultra-broadband absorption from 300 nm to 2000 nm, which is much broader than that of the anthracene-F4TCNQ cocrystal with only one double-bond rotor from our previous work.10 Under laser irradiation at 1064 nm in the NIR-II region, the DFQ cocrystal has a high PTC efficiency η of up to 73.6%, which is about 29.1% higher than that of the anthracene-F4TCNQ cocrystal.10 The MAT/DAT cocrystals have a board absorption band from 300 nm to 1100 nm/1300 nm and a PTC efficiency η of up to 48.6%/62.6%. Single crystal structural analysis, Fourier transform infrared absorption, electron paramagnetic resonance (EPR), ultrafast transient absorption and theoretical calculations are combined to explain these phenomena. The results unambiguously reveal that the DFQ cocrystal has the highest degree of charge transfer (DCT) in these cocrystals with the interaction between the electron-pushing methyl substituent and strong electron-attracting F4TCNQ. The J-aggregated stacking structure endows the DFQ cocrystal strong π–π interactions and weak p–π interactions, which affect its optical band gap and the width of ultra-broadband absorption, the free rotation of rotors and the relaxation dynamics of excited states, and the final photothermal performance.
Results and discussions
The DFQ/DTQ/MTQ cocrystals are synthesized via a simple solution volatilization method (Fig. 1a and S1a and b†). According to the crystal structures obtained by the XRD spectra (Fig. S2a–c†), all these three cocrystals display a mixed stacking mode (…D–A–D–A…), where the π–π and p–π interactions play important roles in the self-assembly stacking process. For the DFQ/DTQ/MTQ cocrystals, one π–π conjugated interaction between the benzene ring of donors and the six-membered ring of acceptors shows the distances of 3.36/3.47/3.53 Å (Fig. 1b and S1c and d†), while the p–π intermolecular interaction between the benzene ring of donors and the CC double bond of acceptors shows the distances of 3.38/3.47/3.45 Å (Fig. 1c and S1e and f†). Interestingly, the J-aggregated stacking structure makes the DFQ cocrystal only have one p–π noncovalent bond on one side of acceptors (Fig. 1c), while the DTQ/MTQ cocrystals have two p–π bonds. From the D–A interaction distances and bond number, it is revealed that the DFQ cocrystal has the strongest π–π interaction and the weakest p–π interaction compared to the DTQ/MTQ cocrystals. Such strong π–π interaction can enhance the red-shift absorption of cocrystals, while the weak p–π interaction facilitates the free rotation of the double-bond rotors (C(CN)2), thus favors the nonradiative electron leaps. As is clearly shown in Fig. S3,† the DFQ/DTQ/MTQ cocrystals exhibit a large red-shift up to ∼2000/1300/1100 nm compared to their D/A monomers. More significantly, the red-shift for the DFQ cocrystal is much larger than that of the DTQ/MTQ cocrystals (Fig. 1d), which unequivocally demonstrates the feasibility of extending the absorption range of cocrystals by modulating the intermolecular π–π interaction.
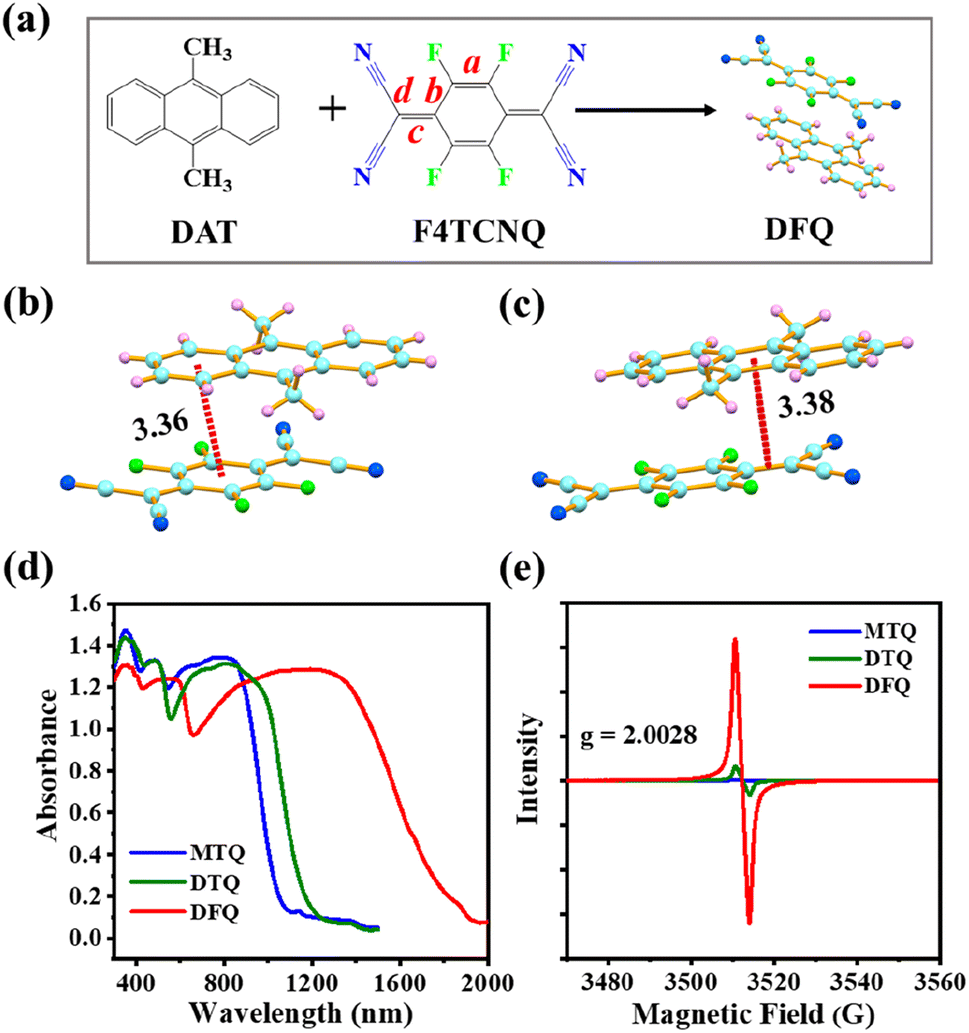 | ||
| Fig. 1 (a) DFQ cocrystal via the cocrystallization of the DAT/F4TCNQ monomers. The bond positions marked by a/b/c/d are adopted for the DCT calculation in subsequent analysis. (b) π–π and (c) p–π interactions between the DAT donor and F4TCNQ acceptor. (d) Diffuse reflection absorption spectra and (e) EPR spectra of the DFQ/DTQ/MTQ cocrystals. | ||
Density functional theory calculations reveal that the highest occupied molecular orbital (HOMO) of three cocrystals is close to the HOMO of donors, while the lowest unoccupied molecular orbital (LUMO) is close to the LUMO of acceptors (Fig. S4†), which is attributed to the intermolecular charge transfer interactions that allow the electron cloud to rearrange to form new molecular orbitals. Meanwhile, compared with the bandgaps of the precursors, the much narrower bandgaps of three cocrystals (Fig. S5†) are conducive to the red-shift absorption,16 which accords well with the characteristics of their absorption spectra shown in Fig. 1d.
It is well known that the degree of charge transfer (DCT) plays a crucial role in the red-shifted absorption of cocrystals.17 Then the EPR measurements are utilized to probe the relationship between the obtained radical concentration and photophysical properties of cocrystals. As shown in Fig. 1e and S6,† three cocrystals have distinct intensities of the EPR signals with the g-factor of ∼2.0028, which is close to the value (2.0032) for free electrons and indicates the presence of unpaired electrons and active radicals in all three cocrystals.18 Notably, the EPR signal of the DFQ cocrystal is much stronger than that of the DTQ/MTQ cocrystals. Namely, the DFQ cocrystal has the highest DCT and forms the strongest π-conjugation cocrystal system. The commonly used bond length parameters (Fig. 1a) from crystal cell structure19,20 are utilized to reconfirm the DCT of cocrystals. The DCT value of cocrystals can be calculated from the parameters αX = c/(b + d) with the equation as: DCT = (αCT − α0)/(α−1 − α0), where the subscripts CT, 0, and −1 refer to the cocrystal, neutral acceptor, and acceptor anion, respectively. The obtained DCT values are 0.2263/0.0871/0.0836 for the DFQ/DTQ/MTQ cocrystals. Furthermore, the Fourier transform infrared absorption,21 also supports the highest DCT of the DFQ cocrystal, which is revealed by the obvious red-shift natures of several characteristic peaks of the backbone vibrations, e.g. the b1uν18 mode of the CN stretching vibration, the stretching vibrations of the C–F and CC groups for the cocrystals compared to the D/A monomers (Fig. S7†). Based on the peak red-shift of the b1uν18 mode of the CN vibration,21 the calculated DCT values are ∼0.15/0.1/0.1 for the DFQ/DTQ/MTQ cocrystals. Compared to the TCNQ acceptor without F-substituents in the DTQ cocrystal, the F4TCNQ acceptor with four F-substituents on the skeleton structure largely improves the DCT in the DFQ cocrystal. The EPR signal of the DTQ cocrystal is higher than that of the MTQ cocrystal, which reveals that more methyl groups improve the electron-donating ability of the donors and further enhance the intermolecular charge transfer interaction.22 Thus, the DFQ cocrystal exhibits the broadest red-shift absorption (300–2000 nm) in the full solar spectrum (250–2500 nm). Meanwhile, the red-shift absorption (300–1300 nm) of the DTQ cocrystal is broader than that (300–1100 nm) of the MTQ cocrystal (Fig. 1d).
As is expected, three cocrystals constructed by dual-rotor strategy showcase excellent NIR PTC performance profiting from the intense π–π stacking and charge transfer interactions, the weakened p–π interaction, and the enhanced NIR light harvesting ability. Based on the tests through a homemade photothermal measuring system (Fig. S8†), the temperatures of the DFQ/DTQ/MTQ cocrystals are rapidly increased to 94.3/79.2/47.6 °C under the irradiation of the 1064 nm laser at 1.0 W cm−2, while the temperature of the blank quartz glass is virtually unchanged (Fig. 2a and S9†). From the cooling curves with detailed procedures shown in the ESI,†1 the obtained PTC efficiency η of the DFQ/DTQ/MTQ cocrystals are 73.6/62.6/48.6% (Fig. S10–S13†). Clearly, the DFQ cocrystal has the best NIR PTC performance, which is about 29.1% higher than that (∼57%) of the anthracene-F4TCNQ cocrystal reported in our previous work.10 Besides, the efficiency η of the DTQ cocrystal with the DAT donor modified by two methyl moieties is about 29.1% higher than that of the MTQ cocrystal with the MAT donor modified by only one methyl moiety.
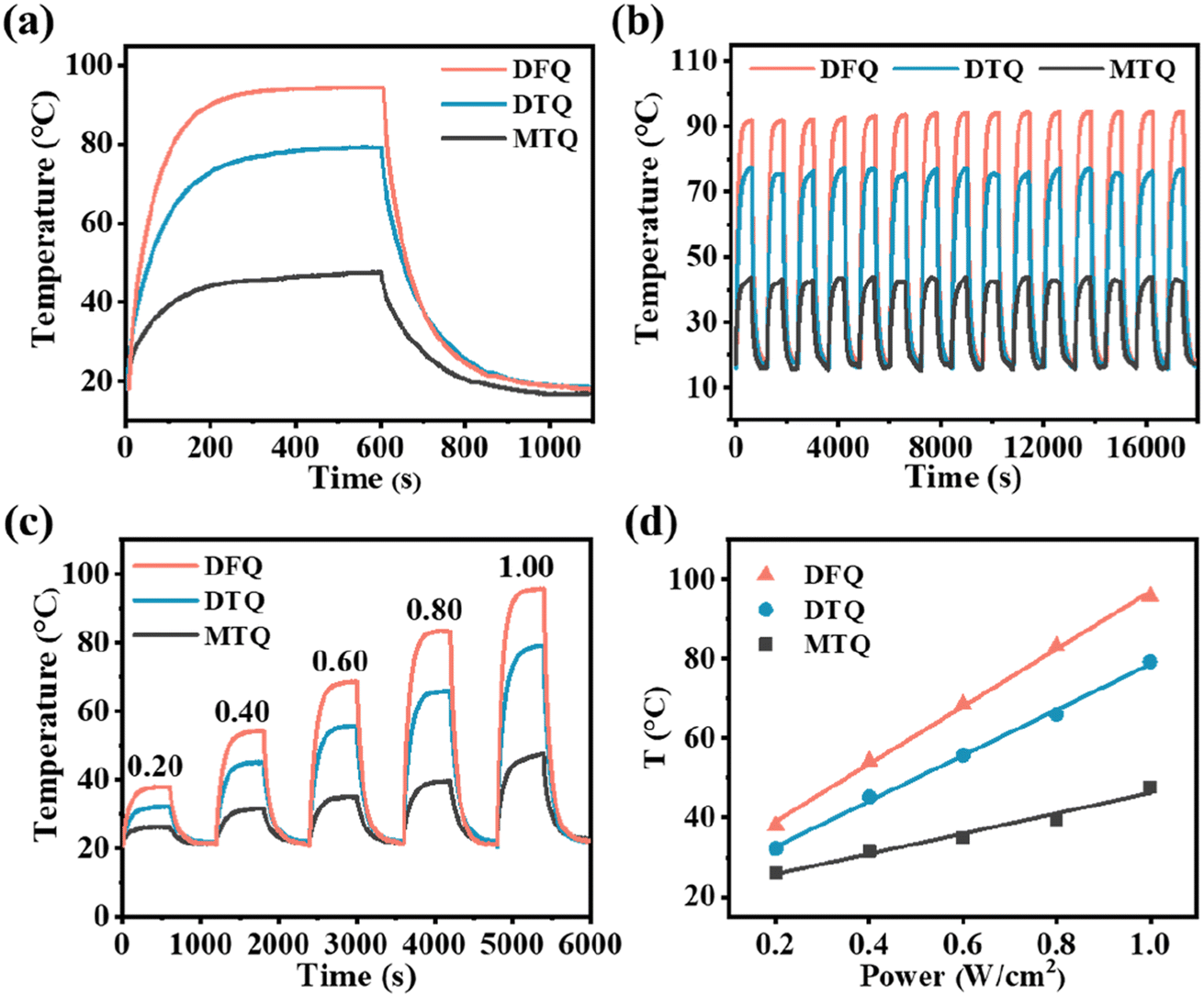 | ||
| Fig. 2 (a) Typical photothermal cyclic tests and (b) successive heating and cooling curves of three cocrystals under the 1064 nm laser at 1.0 W cm−2. (c) Photothermal cycling curves of three cocrystals under different laser powers. (d) The relationship between the temperature T of cocrystals and laser power. | ||
A successive 15-cycle (5 hours) heating-cooling experiments confirm the great photothermal stability of three cocrystals (Fig. 2b). Furthermore, the irradiation power dependence results from 0.2 W cm−2 to 1.0 W cm−2 display that the maximum temperature rising ΔT of three cocrystals increases proportionally with the laser irradiation power (Fig. 2c and d). That is, the photothermal behaviors of cocrystals can be flexibly controlled by fine adjustment of the excitation power. From the thermogravimetric analysis (TGA), the inflection point temperatures of weight loss are much higher than the maximum temperature rising ΔT of three cocrystals under laser irradiation (Fig. S14†). And the diffraction peaks of three cocrystals before and after laser irradiation are basically consistent with the simulations based on the powder X-ray diffraction (PXRD). Hence, these three cocrystals have an excellent photothermal stability (Fig. S15†).
The high PTC efficiency of three cocrystals indicate the active nonradiative decay processes of excited states. To gain deeper insights into the dissipation mechanism of cocrystals, femtosecond transient absorption (fs-TA) technique is employed to trace the excited-state dynamics of these cocrystals (Fig. 3a–c and S16–S18†).23–26 As shown in Fig. S16a and b to S18a and b,† the DFQ/DTQ/MTQ cocrystals display the excited-state absorption (ESA) bands peaked at around 330 nm under 400 nm-laser excitation. In particular, for the DFQ cocrystal, a significant ESA band of 330 nm is detected at 0.37 ps with the maximum intensity and then rapidly decreases. Similarly, the DTQ/MTQ cocrystals exhibit an ESA band of 330/329 nm at 0.52/0.36 ps with the maximum intensities. Meanwhile, under an 800 nm-NIR laser excitation, the fs-TA spectra of three cocrystals show similar kinetic processes (Fig. S16c and d to S18c and d†). The DFQ/DTQ/MTQ cocrystals display the ESA bands peaked at 324–336 nm under 800 nm-laser excitation. In detail, the DFQ/DTQ/MTQ cocrystals exhibit an ESA band of 336/324/328 nm at 0.41/0.52/0.92 ps with the maximum intensities.
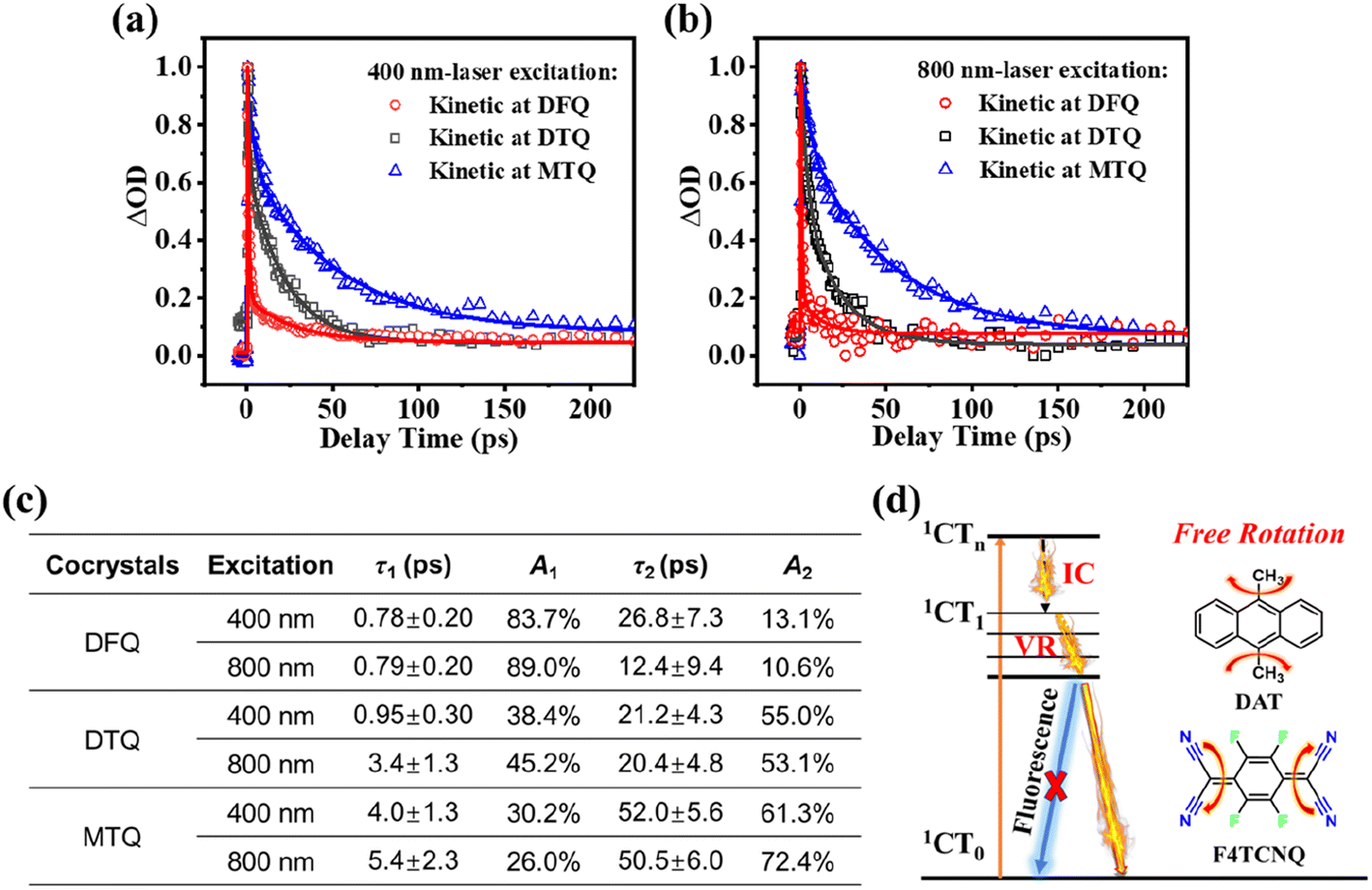 | ||
Fig. 3 The comparison of decay kinetics and the corresponding fitting curves of ESA in the three cocrystals excited at 400 nm (a) and 800 nm (b). (c) The dynamics parameters of the ESA evolution of three cocrystals obtained from fs-TA spectra excited at 400 nm/800 nm. (d) Jablonski diagram of excited-state evolution. The single-wavelength fitting of the decays of ESA was completed based on the equation: 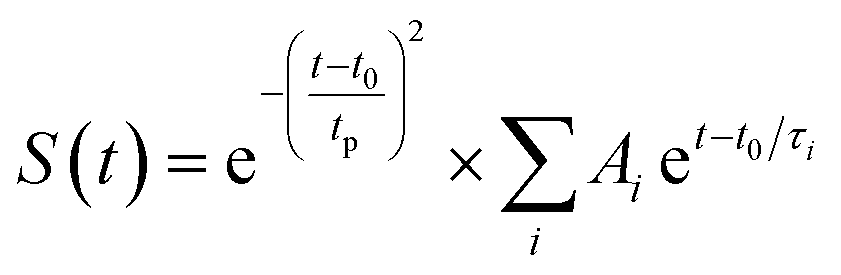 . Herein, the t is referred to the delay time, and t0 means the zero time. The tp, Ai and τi represent the instrument response value of ∼120 fs, the proportion (weight factor) and the lifetime of the ith process, respectively. Because the lifetime of the rise kinetics of ESA of cocrystals is close to the instrument response time, it is hard to resolve reliable information from this parameter and not discussed at here. . Herein, the t is referred to the delay time, and t0 means the zero time. The tp, Ai and τi represent the instrument response value of ∼120 fs, the proportion (weight factor) and the lifetime of the ith process, respectively. Because the lifetime of the rise kinetics of ESA of cocrystals is close to the instrument response time, it is hard to resolve reliable information from this parameter and not discussed at here. | ||
Because the dominant radiative or non-radiative decay process largely decides the photoluminescence emission or PTC property of cocrystals subject to light excitation,26,27 our emphasis focuses on the dominant ultrafast decay. As is clearly shown in Fig. 3a–c for the two excitation wavelengths (400 nm/800 nm), the first ultrafast decay with a lifetime of τ1 (0.78 ps/0.79 ps) and a dominant weight factor of A1 (83.7%/89.0%) for the DFQ with the highest PTC efficiency is obviously faster than those for the DTQ/MTQ cocrystals. If this ultrafast decay can be attributed to the nonradiative transition processes of ESA, this result conforms to the common rule well that the faster nonradiative charge recombination largely corresponds to the higher PTC efficiency.26,27
To exclude the possible radiative transition channels of excited states, we evaluate the luminescence emissions of three cocrystals. The results reveal that no emission was detected from these cocrystals compared to the strong emissions of the donors. Hence, the excited-state evolution is mainly dominated by the nonradiative decay processes. A Jablonski diagram of excited-state evolution is shown to illustrate the possible nonradiative relaxation channels of excited states responsible for the superior NIR PTC performance of cocrystals. Generally, the ultrafast decay of these ESA bands following the excitation can be attributed to the nonradiative decays of the excited charge transfer (CT) states from the CTn states to the CT1/CT0 states mainly through internal conversion (IC) and vibrational relaxation (VR). The first lifetime τ1 can be assigned to the IC transition from the higher excited states (1CTn) to the lower excited states (1CT1). And the second lifetime τ2 is assigned to the vibrational relaxation (VR) process from the excited states (1CT1) to the ground states (1CT0).1,10,13,23–26 From the perspective of D/A molecular structure, the detailed IC transition and VR can be produced easily by the free rotation of the single-bond rotor (–CH3) from the donors,11,12 and the double-bond rotor being in the form of [C(CN)2] in the ground states but being in the form of [–C(CN)2] in the excited states from the conjugated quinoid acceptors.13 These nonradiative decay processes together help the cocrystals efficiently convert the absorbed light into heat energy.
To better illustrate the validity of dual-motor strategy to improve the PTC ability of cocrystals, we made a more careful comparison of MTQ/DTQ/DFQ with the anthracene-TCNQ/F4TCNQ cocrystals10 in the ESI (Table S2†). From the perspective of the ultrafast decays of ESA, the common rule works well that the faster nonradiative charge recombination largely corresponds to the higher PTC efficiency, and the ultrafast decay of ESA often dominates the PTC property of cocrystals. Furthermore, different decay processes may have a nonlinear additive contribution to the final PTC property.26 From the perspective of D/A structure, the rotor mechanism may rely on the structural match and stacking distance of D/A units, which affect the possible free space for rotor rotation.
Conclusion
A single-/double-bond dual-rotor strategy is proposed to build organic cocrystals for the NIR PTC application. Profiting from the strong π–π stacking and charge transfer interactions, the weakened p–π interaction, and the enhanced NIR light harvesting ability, the DFQ cocrystal exhibits an ultra-broadband absorption from 300 nm to 2000 nm. Besides, the PTC efficiency at 1064 nm in the NIR-II region can be largely enhanced by modulating the number of rotor groups in the donors and the F-substituents on the skeleton structure of the acceptors. The internal mechanism responsible for the superior NIR PTC performance is uncovered utilizing fs-TA technique. The nonradiative decay processes of excited states including the internal conversion and vibrational relaxation together promote the cocrystals to efficiently convert the absorbed light into heat energy. From the perspective of D/A molecular structure, the detailed decays can be produced by the free rotation of the single-bond rotor (–CH3) from the donors and the double-bond rotor (C(CN)2) in the form of [–C(CN)2] in the excited states from the quinoid acceptors. This work proves the validity of dual-rotor strategy and provides a promising route to extend the functionalization of small organic molecules based on organic cocrystal engineering through the rich noncovalent bond interactions.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This project was financially supported by the National Natural Science Foundation of China (22273057, 21773151), the Guangdong Basic and Applied Basic Research Foundation (2023A1515012631), the Universities Joint Laboratory of Guangdong, Hong Kong and Macao (130/07422011), Shantou University Initial Funding (NTF21007).References
- Y. Wang, W. Zhu, W. Du, X. Liu, X. Zhang, H. Dong and W. Hu, Angew. Chem., Int. Ed., 2018, 57(15), 3963–3967 CrossRef CAS PubMed.
- C. Ou, W. Na, W. Ge, H. Huang, F. Gao, L. Zhong, Y. Zhao and X. Dong, Angew. Chem., Int. Ed., 2021, 60(15), 8157–8163 CrossRef CAS PubMed.
- S. Tian, Z. Huang, J. Tan, X. Cui, Y. Xiao, Y. Wan, X. Li, Q. Zhao, S. Li and C.-S. Lee, ACS Energy Lett., 2020, 5(8), 2698–2705 CrossRef CAS.
- M. Chen, Y. Wu, W. Song, Y. Mo, X. Lin, Q. He and B. Guo, Nanoscale, 2018, 10, 6186–6193 RSC.
- M. Zhang, K. Sun, Z. Zheng, H. Liu and X. Wang, Desalination, 2023, 550, 116380 CrossRef CAS.
- A. Guo, X. Ming, Y. Fu, G. Wang and X. Wang, ACS Appl. Mater. Interfaces, 2017, 9(35), 29958–29964 CrossRef CAS PubMed.
- J. Yang, J. Choi, D. Bang, E. Kim, E. Lim, H. Park, J. Suh, K. Lee, K. Yoo, E. Kim, Y.-M. Huh and S. Haam, Angew. Chem., Int. Ed., 2011, 50, 441–444 CrossRef CAS PubMed.
- L. Sun, W. Zhu, X. Zhang, L. Li, H. Dong and W. Hu, J. Am. Chem. Soc., 2021, 143(46), 19243–19256 CrossRef CAS PubMed.
- Y.-T. Chen, M.-P. Zhuo, X. Wen, W. Chen, K.-Q. Zhang and M.-D. Li, Adv. Sci., 2023, 10(11), 2206830 CrossRef CAS PubMed.
- Y.-T. Chen, X. Wen, J. He, Z. Li, S. Zhu, W. Chen, J. Yu, Y. Guo, S. Ni, S. Chen, L. Dang and M.-D. Li, ACS Appl. Mater. Interfaces, 2022, 14(25), 28781–28791 CrossRef CAS PubMed.
- D. Xi, M. Xiao, J. Cao, L. Zhao, N. Xu, S. Long, J. Fan, K. Shao, W. Sun, X. Yan and X. Peng, Adv. Mater., 2020, 32(11), 1907855 CrossRef CAS PubMed.
- C. Xu, R. Ye, H. Shen, J. W. Y. Lam, Z. Zhao and B.-Z. Tang, Angew. Chem., Int. Ed., 2022, 61(30), e202204604 CrossRef CAS PubMed.
- Y.-T. Chen, W. Chen, J. He, G. Zhang, X. Wen, S. Ran, Z. Deng, S. Zhu, H. Li, S. Ni, S. Chen, L. Dang and M.-D. Li, J. Phys. Chem. C, 2021, 125(46), 25462–25469 CrossRef CAS.
- Y.-X. Li, X.-F. Yang, Y. Cui, Y. Nie, F.-F. Wang and G.-X. Sun, J. Lumin., 2017, 187, 14–19 CrossRef CAS.
- A. N. Maity, B. Schwederski, B. Sarkar, S. Záliš, J. Fiedler, S. Kar, G. K. Lahiri, C. Duboc, M. Grunert, P. Gütlich and W. Kaim, Inorg. Chem., 2007, 46(18), 7312–7320 CrossRef CAS PubMed.
- S. Tian, H. Bai, S. Li, Y. Xiao, X. Cui, X. Li, J. Tan, Z. Huang, D. Shen, W. Liu, P. Wang, B.-Z. Tang and C.-S. Lee, Angew. Chem., Int. Ed., 2021, 60, 11758–11762 CrossRef CAS PubMed.
- S. K. Park, I. Cho, J. Gierschner, J. H. Kim, J. H. Kim, J. E. Kwon, O. K. Kwon, D. R. Whang, J.-H. Park, B.-K. An and S. Y. Park, Angew. Chem., Int. Ed., 2016, 55, 203–207 CrossRef CAS PubMed.
- Y. D. Zhao, J. Han, Y. Chen, Y. Su, Y. M. Cao, B. Wu, S. M. Yu, M.-D. Li, Z. Wang, M. Zheng, M.-P. Zhuo and L.-S. Liao, ACS Nano, 2022, 16, 15000–15007 CrossRef CAS PubMed.
- T. J. Kistenmacher, T. J. Emge, A. N. Bloch and D. O. Cowan, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38(4), 1193–1199 CrossRef.
- H. Jiang, P. Hu, J. Ye, K. K. Zhang, Y. Long, W. Hu and C. Kloc, J. Mater. Chem. C, 2018, 6(8), 1884–1902 RSC.
- P. Hu, K. Du, F. Wei, H. Jiang and C. Kloc, Cryst. Growth Des., 2016, 16(5), 3019–3027 CrossRef CAS.
- M.-P. Zhuo, Y. Yuan, Y. Su, S. Chen, Y.-T. Chen, Z.-Q. Feng, Y.-K. Qu, M.-D. Li, Y. Li, B.-W. Hu, X.-D. Wang and L.-S. Liao, Adv. Mater., 2022, 2107169 CrossRef CAS PubMed.
- W. Chen, S. Sun, G. Huang, S. Ni, L. Xu, L. Dang, D. L. Phillips and M.-D. Li, J. Phys. Chem. Lett., 2021, 12(24), 5796–5801 CrossRef CAS PubMed.
- X. Wen, Y.-T. Chen, J. He, B. Wang, X. Ye, Y. Guo, S. Ni, S. Chen, D. L. Phillips, L. Dang and M.-D. Li, Sol. RRL, 2023, 7(14), 2300262 CrossRef CAS.
- S.-L. Chen, M.-M. Zhang, J. Chen, X. Wen, W. Chen, J. Li, Y.-T. Chen, Y. Xiao, H. Liu, Q. Tan, T. Zhu, B. Ye, J. Yan, Y. Huang, J. Li, S. Ni, L. Dang and M.-D. Li, ChemSusChem, 2023, 16(14), e202300644 CrossRef CAS PubMed.
- M.-M. Zhang, S.-L. Chen, S. Huang, D. Zheng, H. Liang, B. Ye, J. Chen, X. Song, L. Liu, J. Li, W. Chen, S. Ji, L. Dang and M.-D. Li, J. Phys. Chem. Lett., 2024, 15, 68–75 CrossRef CAS PubMed.
- M.-M. Zhang, S.-L. Chen, A.-R. Bao, Y. Chen, H. Liang, S. Ji, J. Chen, B. Ye, Q. Yang, Y. Liu, J. Li, W. Chen, X. Huang, S. Ni, L. Dang and M.-D. Li, Angew. Chem., Int. Ed., 2024, e202318628 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The single-crystal data; UV-Vis-NIR absorption spectra; HOMO and LUMO molecular orbitals; HOMO and LUMO band diagram; electron paramagnetic resonance spectra; infrared vibrational spectra; photothermal conversion experiments; thermal stability tests; the photothermal conversion efficiency equation; femtosecond transient absorption spectra; the validity of dual-motor strategy (PDF). CCDC 2246243–2246245. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra00002a |
| ‡ X. Wen and Y. Shao contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2024 |