
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Glycerol-derived organic carbonates: environmentally friendly plasticizers for PLA†
Hyeon Jeong Seo‡
 ,
Yeong Hyun Seo‡,
Sang Uk Park,
Hyun Ju Lee,
Mi Ryu Lee,
Jun Hyeong Park,
Woo Yeon Cho,
Pyung Cheon Lee* and
Bun Yeoul Lee*
,
Yeong Hyun Seo‡,
Sang Uk Park,
Hyun Ju Lee,
Mi Ryu Lee,
Jun Hyeong Park,
Woo Yeon Cho,
Pyung Cheon Lee* and
Bun Yeoul Lee*
Department of Molecular Science and Technology, Ajou University, Suwon 16499, South Korea. E-mail: bunyeoul@ajou.ac.kr; pclee@ajou.ac.kr; Fax: +82-31-219-2394; Tel: +82-31-219-1844
First published on 5th February 2024
Abstract
Polylactic acid (PLA) stands as a promising material, sourced from renewables and exhibiting biodegradability—albeit under stringent industrial composting settings. A primary challenge impeding PLA's broad applications is its inherent brittleness, as it fractures with minimal elongation despite its commendable tensile strength. A well-established remedy involves blending PLA with plasticizers. In this study, a range of organic carbonates—namely, 4-ethoxycarbonyloximethyl-[1,3]dioxolan-2-one (1), 4-methoxycarbonyloximethyl-[1,3]dioxolan-2-one (2), glycerol carbonate (3), and glycerol 1-acetate 2,3-carbonate (4)—were synthesized on a preparative scale (∼100 g), using renewable glycerol and CO2-derived diethyl carbonate (DEC) or dimethyl carbonate (DMC). Significantly, 1–4 exhibited biodegradability under ambient conditions within a week, ascertained through soil exposure at 25 °C—outpacing the degradation of comparative cellulose. Further investigations revealed 1's efficacy as a PLA plasticizer. Compatibility with PLA, up to 30 phr (parts per hundred resin), was verified using an array of techniques, including DSC, DMA, SEM, and rotational rheometry. The resulting blends showcased enhanced ductility, evident from tensile property measurements. Notably, the novel plasticizer 1 displayed an advantage over conventional acetyltributylcitrate (ATBC) in terms of morphological stability. Slow crystallization, observed in PLA/ATBC blends over time at room temperature, was absent in PLA/1 blends, preserving amorphous domain dimensions and mitigating plasticizer migration—confirmed through DMA assessments of aged and unaged specimens. Nevertheless, biodegradation assessments of the blends revealed that the biodegradable organic carbonate plasticizers did not augment PLA's biodegradation. The PLA in the blends remained mostly unchanged under ambient soil conditions of 25 °C over a 6 month period. This work underscores the potential of organic carbonates as both eco-friendly plasticizers for PLA and as biodegradable compounds, contributing to the development of environmentally conscious polymer systems.
Introduction
Organic carbonates are valuable compounds with diverse applications,1 including benign solvents,2,3 lithium-ion battery electrolytes,4 fuel additives,5,6 and polymer synthesis.7,8 Traditionally, organic carbonates were prepared using highly toxic phosgene. However, currently, a more attractive method involves their production from CO2.9–11 One established process for synthesizing organic carbonates involves the coupling reaction of CO2 with ethylene oxide or propylene oxide, yielding ethylene carbonate or propylene carbonate (Scheme 1).12 These compounds can then be further converted to DMC or DEC while simultaneously producing anhydrous-grade ethylene glycol or propylene glycol.13–15 DMC or DEC can also serve as starting materials for the production of other organic carbonates through transcarbonation reactions with various alcohols.16,17 Additionally, they can be used as raw materials for the production of conventional bisphenol-A-based aromatic polycarbonates as well as biodegradable aliphatic polycarbonates.18–21 The production of organic carbonates and aliphatic polycarbonates holds great promise in the field of CO2 capture and utilization (CCU) technology.22–28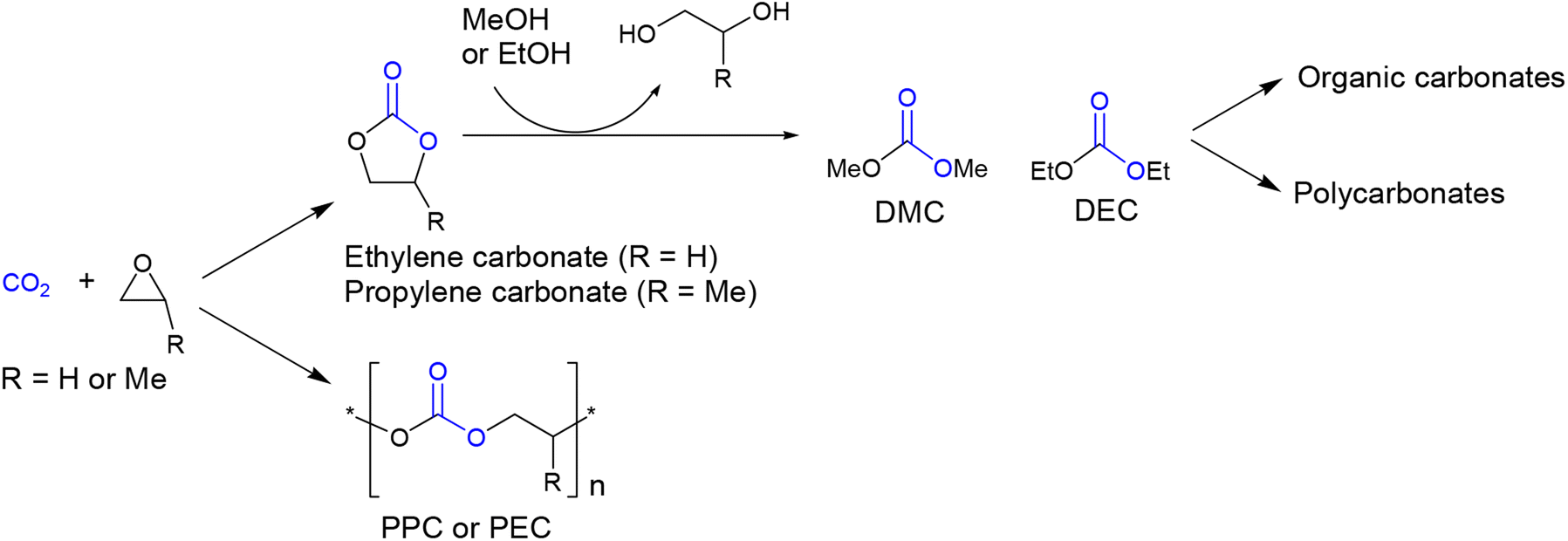 | ||
| Scheme 1 Exploiting CO2 for the synthesis of organic carbonates and polycarbonates. | ||
On the other hands, there has been recently a significant increase in interest regarding biodegradable polymers, particularly those manufactured using renewable resources.29,30 PLA serves as a typical example, but its annual production remains insignificant, amounting to only several hundred kilotons, when compared to the total polymer market size (∼300 megatons per year). One obstacle to the market expansion of PLA could be its non-biodegradability under ambient conditions, such as soil, fresh water, marine environments, and home composting, although it does biodegrade in industrial composting sites operating under harsh artificial conditions.31 Another challenge is its inherent brittleness.32 While PLA exhibits high tensile strength (approximately 60 MPa), its strain at break is low (around 5%). To address this issue, blending PLA with low molar mass organic compounds, commonly referred to as plasticizers, has been explored. Various ester compounds derived from citric acid,33,34 levulinic acid,35–37 tartaric acid,38 adipic acid,39,40 phthalic acid,41 glycerol,42–44 glucose,45 isosorbide,46 and lactones (e.g., oligolactide)47,48 as well as some ether compounds such as poly(ethylene glycol)49 and epoxidized fatty oil have been attempted as plasticizers for PLA to impart ductile properties and overcome brittleness.50–53 When selecting a plasticizer for PLA, compatibility is a crucial criterion, along with other factors such as non-volatility (boiling temperature > 300 °C), non-toxicity, non-fuming, and odorlessness. Ideally, the plasticizer itself should be biodegradable and manufactured using renewable resources, further enhancing its desirability. In this study, we demonstrate the efficacy of organic carbonates derived from CO2 and renewable glycerol as PLA plasticizers. Our findings reveal that these organic carbonates remain intact during the blending process at high temperatures, while exhibiting rapid biodegradation under ambient soil conditions.
Results and discussion
Preparation of glycerol based organic carbonates
4-Ethoxycarbonyloximethyl-[1,3]dioxolan-2-one (1) is an appealing compound that can potentially be synthesized from CO2, glycerol, and ethanol. Glycerol and ethanol are representative renewable carbon sources, with the former being a byproduct of biodiesel production and the latter obtained through fermentation of sugars.54 However, its synthesis has been rarely reported. A century ago, it was reported to be prepared from glycerol using the toxic ethyl chloroformate (EtOC(O)Cl) and a stoichiometric amount of Na metal.55 The yield was very low (15%), and its boiling temperature was reported to be 304–306 °C. In our study, we successfully synthesized it on a 90 g-scale with a high yield (81%) using DEC (Scheme 2a). However, it's worth noting that the reaction and process conditions, as discussed below, still posed substantial challenges when it comes to cost-effective large-scale production. The product is formed through transcarbonation reactions between glycerol and DEC, catalyzed by lithium alkoxide (1.0 mol% per glycerol). It should be noted that the transcarbonation reaction is a reversible process, and to shift the equilibrium towards the product, the generated byproduct EtOH needs to be continuously removed from the reaction mixture. To facilitate this, we designed a reactor employing the Dean–Stark apparatus, which effectively removes the generated EtOH during the reaction (Scheme 2c).16 EtOH is distilled from the reaction mixture along with the carrier solvent cyclohexane, and both compounds are condensed in the Dean–Stark trap filled with two phases of cyclohexane and water. EtOH diffuses into the lower water phase while cyclohexane returns back into the reactor. Despite our efforts to minimize the undesired formation of diglycerol tricarbonate by using an excess of DEC (7 eq per glycerol), a small amount of diglycerol tricarbonate formation might have been unavoidable. However, we were able to eliminate this side product through vacuum distillation, resulting in the desired product 1 containing 1.5 mol% glycerol carbonate (Fig. S1†) with a yield of 81%, which was not quantitative but still satisfactory.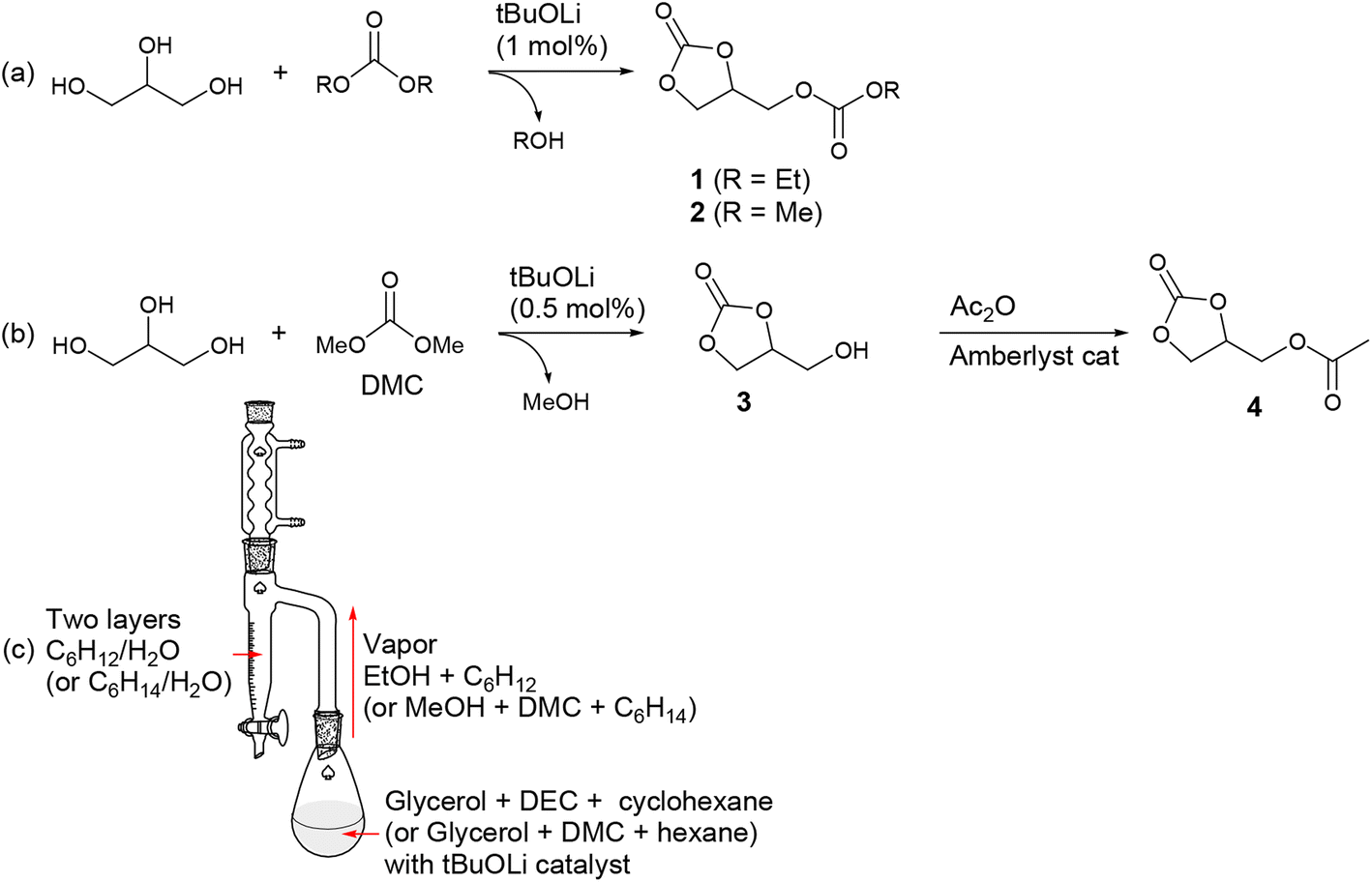 | ||
| Scheme 2 (a) and (b) Synthetic routes and (c) experimental setup for glycerol-based organic carbonates. | ||
Using the developed reactor, we successfully prepared high-purity 4-methoxycarbonyloximethyl-[1,3]dioxolan-2-one (2) on a 60 g-scale (Fig. S2†). During the reaction, a side product, diglycerol tricarbonate, could also be generated, which reduced the isolated yield to 64%. Product 2 precipitated from the reaction mixture and was isolated by filtration, leaving the side product in the solution phase. In this case, the reactant DMC (boiling point, 90 °C) was distilled from the reaction mixture, forming an azeotrope with the byproduct MeOH. However, selective separation of MeOH was achieved in the Dean–Stark trap, while DMC, along with the carrier solvent hexane, returned back into the reactor. It is important to use anhydrous grades of reactants (glycerol, DEC, and DMC) for successful synthesis. Otherwise, glycerol may not be completely converted to the desired products 1 and 2, with some portion persistently remaining as the intermediate glycerol carbonate (3). Water can act as a source of catalyst poison, transforming the active lithium alkoxide (ROLi) catalyst into inactive lithium alkyl carbonate species (ROCO2Li). A preparative-scale synthesis of 2 using glycerol, DMC (10 eq), and K2CO3 (10 mol%) as the catalyst has also been reported. In that study, byproduct MeOH was continuously removed through azeotropic distillation with DMC, which was fed in excess, using a Vigreux column.56 We encountered a significant challenge when attempting to reproduce the synthesis of 1 and 2. In some instances, glycerol was not completely converted into the desired products 1 and 2, a portion persistently remaining as the intermediate glycerol carbonate (3) even after extended reaction times. Eventually, we found that using anhydrous-grade reactants (glycerol, DEC, and DMC) was crucial for achieving reproducibility. We suspect that water may serve as a source of catalyst poisoning, potentially transforming the active lithium alkoxide (ROLi) catalyst into inactive lithium alkyl carbonate species (ROCO2Li).
Extensive efforts were made to synthesize glycerol carbonate (3) due to its potential applications in polyurethane synthesis and as a fuel additive.5,57 Various heterogeneous and homogeneous catalysts have been tested for the transcarbonation reaction of glycerol and DMC to produce 3.58 For example, a reaction condition was established to achieve a 98% yield of 3 with 99% conversion. This was accomplished by reacting glycerol with 4 eq DMC for 2.5 h at refluxing temperature, utilizing a 10 mol% triethylamine catalyst.56 In this study, a 150 g-scale synthesis of 3 was carried out by reacting glycerol with 3 eq DMC using a much smaller amount (0.5 mol%) of a simple lithium alkoxide catalyst, ultimately yielding 99% isolated yield (Scheme 2b). The catalyst was easily removed by filtration, followed by a reaction with the ion exchange resin Amberlyst®. In this particular case, it was unnecessary to remove the generated MeOH during the reaction, as the formation of the 5-membered cyclic carbonate from DMC is an irreversible process and thermodynamically favorable. Some side product 2 (6.5 mol%) and residual reactant glycerol (5.0 mol%) persisted in the reaction mixture along with the desired product. However, during the distillation process, which was performed to remove the byproduct MeOH and excess DMC particularly in the presence of the lithium alkoxide catalyst, most of these impurities were converted to the desired product, resulting in the presence of 2.3 mol% of glycerol and 1.3 mol% of 2 (Fig. S3†). The synthesized glycerol carbonate was then transformed into glycerol 1-acetate 2,3-carbonate (4) by treating it with 1.2 eq acetic anhydride in the presence of an Amberlyst® catalyst under neat conditions (Fig. S4†).
Blending the organic carbonates with PLA
Previous report indicate that propylene carbonate is miscible with PLA and can function as a plasticizer for PLA.59 However, its boiling temperature of 242 °C is not sufficiently high for practical use as a plasticizer. During storage under ambient conditions, we observed a slow escape of propylene carbonate from the polymer matrix. Specifically, initially transparent specimens, fabricated using a blend containing 20 phr of propylene carbonate, became hazy after being stored at room temperature for one month. Furthermore, their ductile properties deteriorated significantly, leading to a notable decrease in elongation at break, which went from 310 ± 190% to 35 ± 3%. The miscibility of polymers with organic compounds is highly dependent on their structure. When attempting to blend PLA with other organic carbonates such as ethylene carbonate and 3, phase separation was observed. To our delight, we found that 1 exhibited good miscibility with PLA, as demonstrated by a significant decrease in the glass transition temperature (Tg) upon blending (Table 1). The Tg values gradually decreased from neat PLA value of 60 °C to 46, 34, 24, and 14 °C as the amount of 1 increased (10, 20, 30, and 40 phr, respectively). Notably, this decrease displayed a linear trend empirically (Fig. 1). To elaborate further, the Tg values of the blends conformed well to the Fox equation: 1/Tg-blend = wPLA/Tg-PLA + w1/Tg-1, where wPLA and w1 represent the weight fractions of PLA and 1, respectively. The calculated Tg values, estimated using this equation with the measured Tg values of neat PLA and 1 (333 and 220 K, respectively; Fig. S5†), closely matched the values determined from the DSC curves (318, 307, 298, and 290 K, compared to the measured values of 319, 307, 297, and 287 K, respectively). Under the same amount of plasticizer, the Tg values of the PLA/1 blends were similar to those of the PLA/ATBC blend (34 vs. 30 °C at 20 phr; Fig. S5†) as well as the blend of oligolactide EtO[C(O)CH(Me)O]4.5C(O)CH3 (46 vs. 45 °C at 10 phr).33,47 This similarity is noteworthy, considering that the Tg value of ATBC is significantly lower than that of 1 (−84 °C versus −53 °C). The Tg curve was fairly distinct for the PLA blends containing 10–30 phr of 1, while the curve was rather blurred for the blend containing 40 phr of 1 (denoted as PLA/140 phr). PLA/ATBC blends displayed a relatively more blurred transition curve than the PLA/1 blends under the same amount of plasticizer. 2 and 4 also exhibited miscibility with PLA, showing a similar linear dependency of the Tg values on the amounts of 2 and 4 (Fig. S7 and S8†).| Plasticizer (phr) | Tg (°C) | Tcc (°C); ΔH (J g−1) | Tm (°C); ΔH (J g−1) | tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ peak Tg (°C) δ peak Tg (°C) |
E′′ peak Tg (°C) | E′ at 25 °C (MPa) | E′′ at 25 °C (MPa) | Tensile strength (MPa) | Strain at break (%) |
|---|---|---|---|---|---|---|---|---|---|
| PLA | 60 | — | 148–156; 0.2 | 65 | 59 | 1600 | 17 | 60 ± 2 | 3 ± 0.1 |
| 1 (10) | 46 | 103–125; 12 | 140–153; 12 | 54 | 48 | 1500 | 27 | 50 ± 4 | 3 ± 1 |
| 1 (15) | 37 | 96–121; 16 | 132–148; 17 | 48 | 42 | 1100 | 34 | 41 ± 4 | 76 ± 43 |
| 1 (20) | 34 | 93–118; 18 | 131–147; 14 | 43 | 33 | 1300 | 190 | 26 ± 4 | 260 ± 30 |
| 1 (30) | 24 | 87–120; 12 | 124–142; 14 | 36 | 26 | 710 | 310 | 30 ± 2 | 470 ± 40 |
| 1 (40) | 14 | 82–111; 10 | 118–138; 12 | 28 | 17 | 270 | 140 | 21 ± 3 | 150 ± 20 |
| 2 (10) | 43 | 100–121; 17 | 136–151; 17 | 58 | 52 | 1600 | 38 | 42 ± 3 | 4 ± 2 |
| 2 (20) | 31 | 94–110; 19 | 129–145; 16 | 45 | 36 | 1400 | 180 | 22 ± 3 | 440 ± 160 |
| 2 (30) | 24 | 90–110; 14 | 125–143; 13 | 37 | 27 | 820 | 310 | 14 ± 6 | 540 ± 100 |
| 2 (40) | 15 | 87–111; 7 | 123–138; 8 | 49, 30 | 23 | 580 | 260 | — | — |
| 4 (10) | 49 | 107–140; 20 | 140–156; 18 | 54 | 49 | 1500 | 35 | 21 ± 7 | 31 ± 13 |
| 4 (15) | 41 | 96–116; 30 | 143–151; 27 | 39 | 30 | 680 | 150 | 17 ± 0.4 | 180 ± 6 |
| 4 (20) | 35 | 94–122; 28 | 142–149; 24 | 43 | 34 | 1300 | 170 | 9 ± 1 | 110 ± 20 |
| 4 (30) | 25 | 88–110; 23 | 123–145; 20 | 36 | — | 320 | 49 | — | — |
| 4 (40) | 21 | 83–103; 26 | 134–143; 23 | 33 | — | 360 | 60 | — | — |
| ATBC (10) | 46 | 98–125; 25 | 139–155; 25 | 52 | 46 | 2000 | 43 | 52 ± 3 | 5 ± 3 |
| ATBC (15) | 37 | 95–119; 30 | 135–154; 26 | 47 | 38 | 1500 | 100 | 26 ± 0.4 | 220 ± 14 |
| ATBC (20) | 30 | 87–100; 28 | 141–152; 27 | 42 | 32 | 840 | 120 | 27 ± 1 | 300 ± 57 |
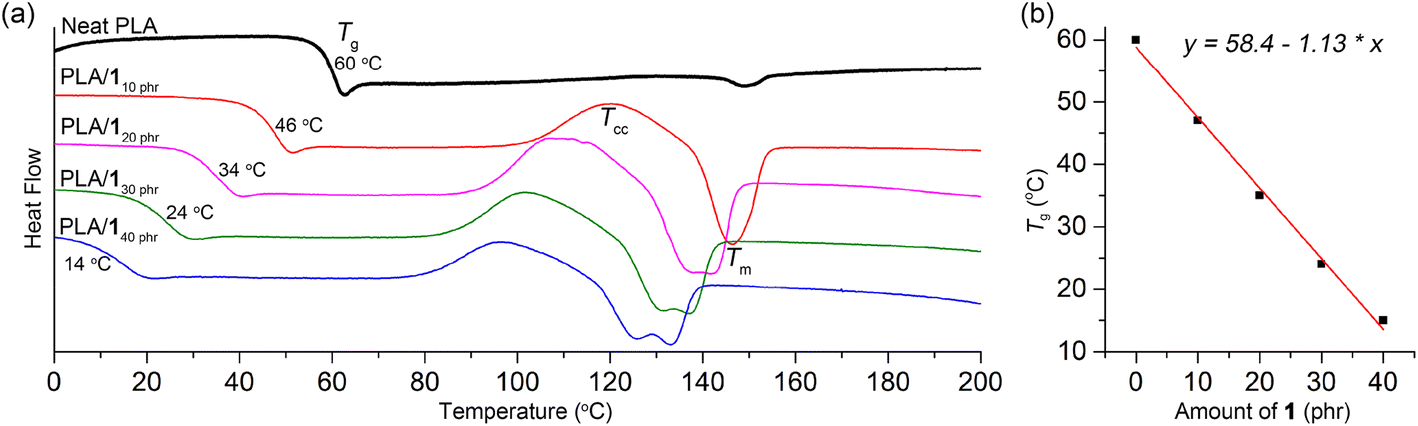 | ||
| Fig. 1 (a) Second-heating DSC curves of PLA/1 blends compared with neat PLA. (b) Linear correlation between the content of 1 and Tg of the blends. | ||
Plasticizer 1 facilitated the crystallization of PLA, and the cold crystallization temperature (Tcc) signals were detected in the second heating DSC traces of the blends (Fig. 1a).39,60,61 In contrast, the Tcc signal was negligibly observed in neat PLA (Natureworks 2003D),48,60 even though some literature reports have indicated its presence.32,34,49,50 As the amount of 1 increased (10, 20, 30, and 40 phr), the Tcc signals gradually shifted to lower temperatures (103–125, 93–118, 87–120, and 82–111 °C, respectively), while the heat of crystallization remained almost constant (ΔH, 12–18 J g−1). This shift can be attributed to the increased chain mobility at higher plasticizer content or the plasticizer's role as a nucleating agent.61 Due to the occurrence of cold crystallization, the melting temperature (Tm) signals were observed in the blended PLAs with the same intensity as the Tcc signals, whereas the Tm signal was very weak in neat PLA. The Tm signals also gradually shifted to lower temperatures compared to neat PLA (148–156 °C), reaching 140–153, 131–147, 124–142, and 118–138 °C with increasing amounts of 1 (10, 20, 30, and 40 phr, respectively). This shift is a common behavior observed in plasticized semicrystalline polymers, and it can be attributed to the presence of a less perfect crystalline phase resulting from higher levels of plasticizer content.33,60 For the blend containing 10 and 15 phr of 1, a single melting signal was observed, while two melting signals were observed for blends containing 20, 30, and 40 phr of 1. This behavior is commonly observed in PLA blends and is attributed to cold crystallization. The higher temperature signal corresponds to the α-form lamellar crystals, whereas the lower temperature signals correspond to the less perfect α′-form crystals.62,63 Similar trends were also observed for PLA/2 and PLA/4 blends in terms of Tcc and Tm (Table 1 and Fig. S7 and S8†).
The homogeneity of plasticizer distribution was further demonstrated through dynamic mechanical analysis (DMA), a highly sensitive technique for detecting phase separation in blends. The blends containing 10, 20, and 30 phr exhibited fairly narrow and unimodal loss factor (tanδ) curves, indicating an even distribution of 1 within the PLA matrix (Fig. 2a). However, PLA/140 phr showed a broad and weak, yet still distinguishable tanδ signal. In cases of uneven distribution, wide signals with fluctuating multiple peaks or shoulders are typically observed.36 For well-known plasticizers, such as triethyl citrate and ATBC, prominent tanδ curves were observed up to 17.5 phr content but became very broad at 25 phr or higher content.64 The peaks of tanδ signals, also known as α-relaxation transition temperature (Tα), indicated the Tg values, which gradually decreased with increasing amounts of plasticizer 1, as was observed in DSC studies. The Tα values were found to be approximately 10 °C higher than the Tg values measured using DSC.
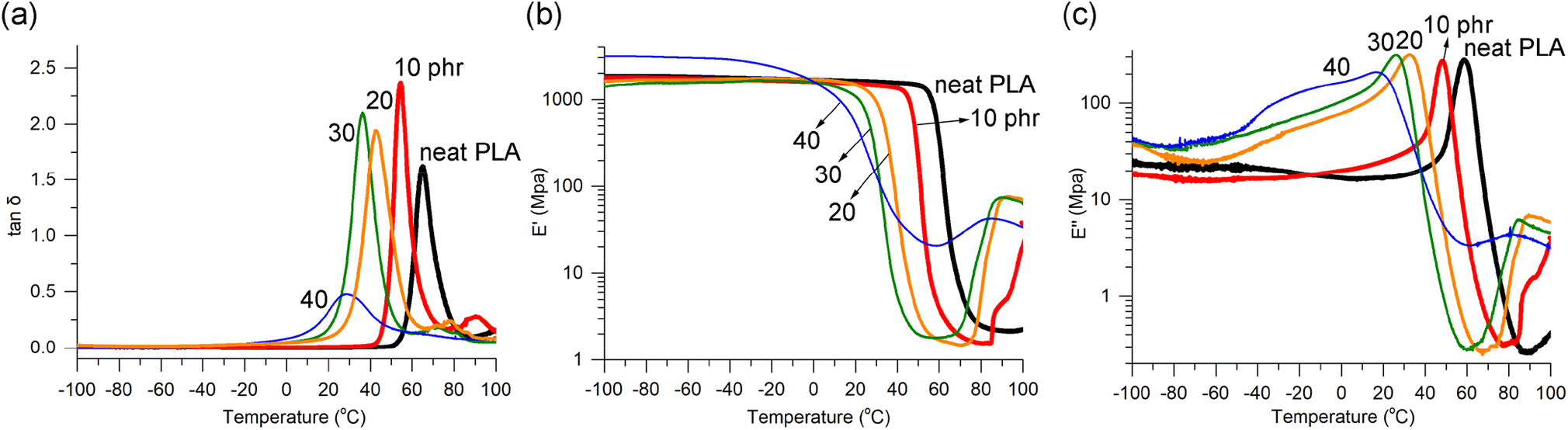 | ||
| Fig. 2 Temperature dependence of (a) tanδ, (b) storage modulus (E′), (c) loss modulus (E′′) curves from DMA runs illustrated for PLA/1 blends compared to neat PLA. | ||
The storage modulus (E′) curves of the blends containing up to 30 phr of 1 exhibited similar features to neat PLA. They showed a plateau line in the glassy state, followed by a sharp drop due to glass transition, and a subsequent increase due to cold crystallization (Fig. 2b). The temperature at which the E′ drop occurred (i.e., Tg) gradually decreased, consistent with the findings from DSC studies. The levels of the plateau lines (i.e., E′ values) of the blends remained almost unchanged after blending with 1 (10, 20, and 30 phr), which is in contrast to the findings reported for blends with acetylated oligolactide. In those blends, the E′ value decreased from 2.9 GPa in neat PLA to 0.1–1.3 GPa in the blends.47 The shape of the E′ curve for PLA/140 phr deviated somewhat from the features observed in neat PLA and the blends containing 10, 20, and 30 phr. It did not exhibit the sharp drop observed in the other cases, which aligns with the broad Tg signal observed in DSC and the broad and weak tanδ signal. Similar deviations were reported to be observed in the blends with ATBC when the plasticizer contents were 25 phr or higher, although the E′ curve feature looked normal up to 20 phr contents of ATBC (Fig. S9†).64 However, a substantial reduction of the E′ value at the glassy plateau region was observed for PLA/ATBC20 phr, from 1.8 to 1.5 GPa in neat PLA to 0.6–0.9 GPa.
The loss modulus (E′′) curves of the blends containing 10, 20, and 30 phr exhibited the same features as neat PLA, showing distinct peaks that allowed for determining the Tg values, which were almost identical to the values obtained from DSC (Fig. 2c). In cases where miscibility between PLA and the plasticizer is poor, resulting in phase separation, signals related to the plasticizer are typically observed, especially at low-temperature regions.34,44 However, such signals were absent in the blends with 1. The shape of the E′′ curve for PLA/140 phr also deviated somewhat from the typical features. The DMA runs of PLA/2 blends showed similar features in tanδ, E′, and E′′ lines compared to PLA/1 blends, with the exception of the observation of two tanδ signals in PLA/240 phr (Fig. S10†). The DMA curves for the blends containing 20 phr or lower amounts of plasticizer 4 exhibited the expected normal features. However, significant deviations from the normal features were observed when the amounts of plasticizer were high at 30 phr and 40 phr (Fig. S11†). In the tanδ curve, shoulder signals were observed around −40 °C, in addition to the main peak signals at 36 and 33 °C, respectively. In the E′ curve, a gradual decrease was observed instead of the typical sharp drop around Tg, and the E′′ curve exhibited non-linearity in the low-temperature region below 0 °C.
PLA/110 phr demonstrated brittleness similar to neat PLA. There was no significant improvement in elongation at break compared to neat PLA (3%), while the tensile strength marginally decreased (60 vs. 50 MPa) and Young's modulus slightly increased (2.1 vs. 2.4 GPa). These results suggest that the plasticizing effect was negligible at the small amount of 1 (10 phr). However, when the amount of 1 increased to 15 phr, the tensile curve exhibited ductile behavior. It showed an elastic region with a Young's modulus of 1.6 GPa, an upper yield point with an ultimate tensile strength of 41 ± 4 MPa, a lower yield point with a substantial rapid decrease in stress to 17 ± 2 MPa, strain hardening, and eventual breakage (Fig. 3). The elongation at break values were substantially increased compared to neat PLA, although they varied significantly depending on the specimens (76 ± 43%). PLA/120 phr displayed similar characteristics to PLA/115 phr, but with considerably lower and non-uniform yield strengths (13 ± 7 MPa). The strain hardening effect was prominent, and elongation at break values were consistently high and uniform (260 ± 30%) with fairly high ultimate tensile strengths (26 ± 4 MPa). Finally, PLA/130 phr exhibited an elastomer-like tensile curve, without distinct yield points, but showing a simple strain hardening line from the origin to the breaking points. The tensile strength and strain at break were enhanced to 30 ± 2 MPa and 470 ± 40%, respectively, compared to PLA/120 phr.
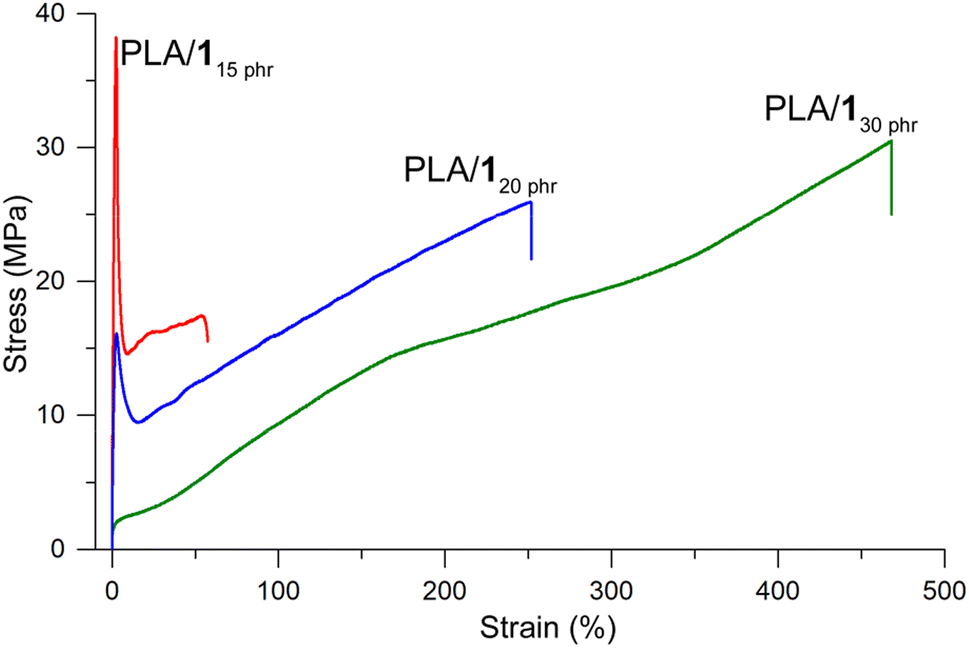 | ||
| Fig. 3 Stress–strain behavior for PLA/1 blends. | ||
PLA/210 phr did not exhibit ductile properties, whereas PLA/220 phr and PLA/230 phr displayed ductile behavior with noticeable strains at break, albeit with notable fluctuations (440 ± 160 and 540 ± 100%, respectively; Fig. S12†). The significant fluctuations in strain at break indicated an uneven distribution of plasticizer 2 within the material. PLA/120 phr showed a distinct yield point, although it was non-uniform. In contrast, PLA/220 phr did not exhibit such a distinct yield point and instead showed a simple elastomer-like tensile curve, with a slightly reduced ultimate tensile strength (22 ± 3 MPa) compared to PLA/120 phr (26 ± 4 MPa). PLA/230 phr exhibited a tensile behavior similar to PLA/220 phr, but with non-uniform and reduced ultimate tensile strength (14 ± 6 MPa) and strain (540 ± 100%) values. In accordance with the results in the DMA studies, PLA/430 phr and PLA/440 phr exhibited very weak mechanical strength, breaking at an early stage of low stress and strain. In contrast, blends with 10, 15, and 20 phr of 4 displayed ductile curves (Fig. S13†). However, the ultimate tensile strengths of these blends were substantially lower than those observed for the blends with 1. Specifically, the yield strength (ultimate tensile strength) of PLA/410 phr was non-uniform and low at 21 ± 7 MPa compared to PLA/115 phr (41 ± 4 MPa). The tensile curve of PLA/415 phr displayed a similar trend to that of PLA/120 phr, but with an inferior tensile strength at break and less strain hardening effect (17 ± 0.4 vs. 26 ± 4 MPa).
In the case of the representative plasticizer ATBC, a similar negligible plasticizing effect was observed at a low amount of 10 phr.33 However, PLA/ATBC15 phr also exhibited ductile properties (Fig. S14†), although its yield strength was substantially weaker compared to PLA/115 phr (18 ± 6 vs. 41 ± 4 MPa), while the elongation at break was higher (220 ± 14 vs. 76 ± 43%). Contrary to the reported limitations of ATBC's ability to uniformly plasticize PLA at 25 phr or higher content,64 as indicated by the observation of blurred tanδ curves, our findings revealed the presence of ductile properties with high tensile strengths and elongations at break in both PLA/ATBC30 phr and PLA/ATBC40 phr. Specifically, PLA/ATBC30 phr exhibited tensile strengths of 27 ± 2 MPa and elongations at break of 290 ± 37%, while PLA/ATBC40 phr displayed tensile strengths of 26 ± 6 MPa and elongations at break of 464 ± 53%.
The miscibility of plasticizers with PLA was assessed through scanning electron microscopy (SEM) images (Fig. 4). Blends containing 30 phr or less of 1 exhibited smooth fracture surfaces, indicating good miscibility without phase separation. The observed SEM images aligned with the tensile properties that demonstrated ductile behavior with consistent strains at break (260 ± 30 and 470 ± 40%, respectively) and notable tensile strengths (26 ± 4 and 30 ± 2 MPa, respectively) for PLA/120 phr and PLA/130 phr. However, PLA/140 phr displayed rough and irregular surfaces, suggesting phase separation. Due to phase separation, weak mechanical properties were observed in the PLA/140 phr blend. Conversely, SEM images revealed that 2 is unsuitable as a plasticizer due to its crystalline nature with a melting temperature of 80 °C. In fact, solid compounds are rarely employed as plasticizers. SEM images showed the presence of small dot-like particles (possibly crystals of 2) even in blends containing as little as 10 phr of 2 and exhibited highly rough surfaces in PLA/220 phr (Fig. S15†). The uneven distribution of plasticizer resulted in fluctuating tensile properties for PLA/220 phr and PLA/230 phr. While both materials exhibited ductile behavior with relatively high average strains at break, they also displayed significant fluctuations in the strain at break (440 ± 160 and 540 ± 100%, respectively). Additionally, over time, plasticizer 2 migrated from the polymer matrix, forming solid particles on the blend's surface (“sweat-out” phenomenon). Blends containing 20 phr or less of 4 displayed smooth surfaces, while those with 30 phr or higher of 4 exhibited rough and irregular surfaces, indicative of phase separation (Fig. S16†). Notably, the detrimental “sweat-out” phenomenon was absent in samples showing smooth surfaces (i.e., blends containing 30 phr or less of 1 and 20 phr or less of 4).
 | ||
| Fig. 4 SEM images of the tensile fracture surfaces of PLA blends with 1, 2, and 4. | ||
In gel permeation chromatography (GPC) studies, notable chain scissions were observed in the PLA chains during the blending process with 4. This led to a significant reduction in the weight-average molecular weight (Mw). More specifically, the initial Mw of pristine PLA stood at 260 kDa with a dispersity (Ð) of 2.2. However, after undergoing identical thermal treatment without the addition of plasticizers, this value decreased to 180 kDa (Ð = 2.2). It's important to note that this reduction in Mw was consistently observed, resulting in Mw value of either 180 kDa or 170 kDa (Ð = 2.2), even when the thermal treatment was conducted under a N2 atmosphere with eliminating the possibility of water inclusion. This phenomenon, which is well-documented in the case of PLA, can be attributed to chain scission rather than hydrolysis.65
In the case of PLA/420 phr, the Mw value experienced a substantial decrease to 44 kDa (Ð = 2.0) (Fig. 5). The significant decrease in the Tg value from 60 to 35 °C can primarily be attributed to the plasticizer effect rather than the reduction in molecular weight. The Tg of PLA with a molecular weight (Mw) of 44 kDa and a dispersity of 2.0 is estimated to be 58 °C using the modified Flory–Fox equation (Tg = Tg∞ − 52/(Mn × Mw)0.5 = 60 − 52/(22 × 44)0.5).66 This estimate is in close agreement with the reported Tg value of 55 °C found in the literature.67 Importantly, both of these values are not significantly lower than the Tg of high molecular weight PLA, which stands at 60 °C. This reduction in molecular weight contributed to the material's weak tensile properties, with a tensile strength of 9 MPa and a strain at break of 110%. Typically, commercial PLA grades are primarily synthesized through ring-opening polymerization of lactide using a tin(II) catalyst.68 These catalyst residues might catalyze transesterification reactions between PLA and 4, which possesses an ester bond, consequently leading to molecular weight reduction. Interestingly, ATBC, another plasticizer with ester linkages, did not exhibit such molecular weight reduction observed in the blending with 4.47 In the case of PLA/ATBC20 phr, it maintained a Mw value of 200 kDa (Ð = 2.2). The ester bonds in ATBC may possess steric hindrance, which could impede transesterification reactions. In contrast, the ester bonds in 4 are more openly configured, making them highly susceptible to transesterification reactions.
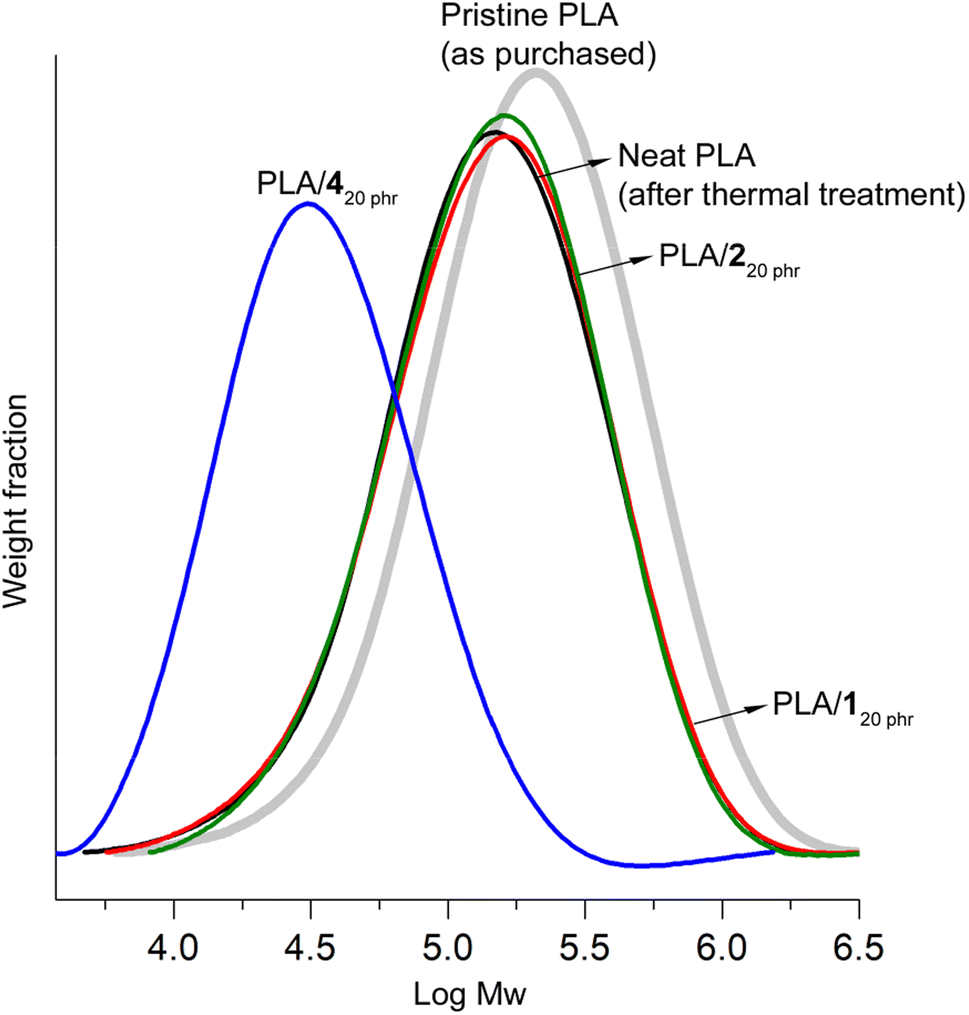 | ||
| Fig. 5 GPC curves of pristine purchased PLA, neat PLA post thermal treatment under blending conditions, PLA/120 phr, PLA/220 phr, and PLA/420 phr. | ||
Blending with 1 and 2, characterized by carbonate linkages exclusively, did not result in any discernible molecular weight reduction. The Mw values for PLA/120 phr and PLA/220 phr [190 kDa (Ð = 2.2) and 190 kDa (Ð = 2.1), respectively] closely paralleled that of neat PLA. The decrease in molecular weight during the blending process is a widely acknowledged concern, particularly evident when employing plasticizers containing alcohol groups.38 In literatures, a common method involved mixing PLA and plasticizers in chloroform at room temperature, followed by complete solvent evaporation to create blends. These blends were subsequently processed using a twin-screw extruder, typically operated at optimally minimized temperatures (e.g., 130–150 °C) for a brief duration (e.g., 20 min).47 In contrast, our research adopted a more practical blending approach that eliminated the use of a solvent and instead employed relatively rigorous conditions. We subjected the materials to a prolonged thermal treatment (2 h at 170 °C for melting) and an additional 30 min of stirring to ensure thorough blending and to assess the robustness of the blends. It is worth mentioning that even under these extremely rigorous blending conditions, the PLA/1 and PLA/2 blends, which were formulated with plasticizers consisting solely of carbonate bonds, remained structurally intact. This stands in stark contrast to the significant reduction in molecular weight observed during the formulation with 4, which possesses an ester bond. Generally, carbonate linkages exhibit greater resilience than ester linkages, whether in terms of hydrolysis or transesterification (transcarbonation) reactions.
The thermal stability of PLA/1 blends was evaluated using thermal gravimetry analysis (TGA) in combination with DSC. The temperatures corresponding to 5 wt% weight loss (Td 5 wt%) were found to be 277, 253, 244, and 244 °C for blends containing 10, 15, 20, and 30 phr of 1, respectively, and 206 °C for pure 1. These values were substantially lower than neat PLA (338 °C) (Fig. S17†) and were slightly higher than the acetyl-end-capped oligolactide plasticizer EtO[C(O)CH(Me)O]4.5C(O)Me (210–245 °C) reported previously.47 It is worth noting that the processing temperatures for PLA typically range from 185 to 250 °C, depending on the melting temperature (Tm) of PLA, which ranges from 130 to 230 °C. The weight loss observed in PLA/1 blends was minimal, 0.56–0.76 and 1.0–1.5 wt%, at plausible processing temperatures of PLA/1 blends, e.g., 185 and 200 °C, though the weight losses observed in pure 1 were significantly higher, reaching 2.4 and 4.0% at the same temperatures, respectively. In the DSC curves, two prominent overlapping signals were observed, peaking at 340–350 °C and 367–371 °C, along with small PLA Tm signals peaking at 152, 147, 143, and 139 °C, which gradually decreased with increasing amounts of plasticizer (Fig. S18†). These two major signals were reasonably attributed to the evaporation of 1 and lactide (formed by depolymerization in the sample holder), with reported boiling points of 305 and 286 °C, respectively. The onset temperatures of these signals were 270 °C or higher, indicating that they were sufficiently higher than the plausible processing temperatures of the blends, thereby alleviating concerns about the destruction or evaporation of 1 during thermal processing (Fig. 6).
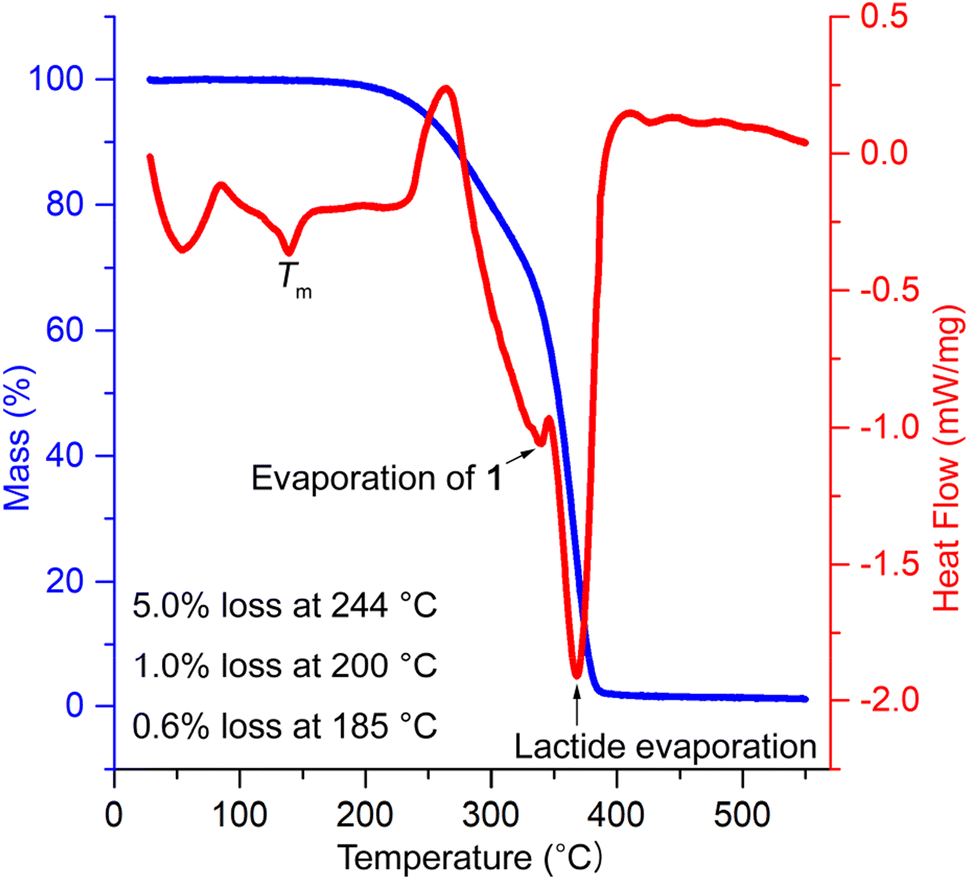 | ||
| Fig. 6 TGA/DSC analysis of PLA/130 phr blend. | ||
The complex viscosities (η*) obtained from rotational rheometer measurements at 170 °C showed significant reductions upon blending, and the viscosity gradually decreased with increasing 1 content (Fig. 7a). For instance, at a low frequency of 0.1 rad s−1, the η* values for PLA/110 phr, PLA/120 phr, and PLA/130 phr were 1120, 660, and 450 Pa s, respectively. In contrast, neat PLA exhibited a substantially higher η* value of 6450 Pa s, comparable to that of another well-known biodegradable polyester, poly(butylene adipate-co-terephthalate) (PBAT), with Mw = 230 kDa (5400 Pa s). Both neat PLA and the blends displayed similar features in the η*-angular frequency curves, showing Newtonian plateau behavior at low frequencies and shear-thinning behavior at high frequencies.61 With an increase in plasticizer content, the Newtonian plateau lines extended to higher frequencies. In contrast, PBAT exhibited shear-thinning behavior across the entire frequency range, likely due to the broad dispersity of the commercial grade PBAT sample (Mw/Mn = 4.4). Such shear-thinning behavior is beneficial in polymer processing techniques like blow molding and blown film extrusion.69 Certain PLA blends, such as the one containing 25 wt% glyceryl triacetate, were reported not to exhibit any shear-thinning behavior.43 PLA/110 phr, PLA/120 phr, and PLA/130 phr blends, as well as PBAT, displayed a viscous nature throughout the measured frequency range (i.e., loss modulus G′′ > storage modulus G′) (Fig. S19†). In contrast, neat PLA exhibited a transition from viscous to elastic behavior at a critical frequency of 126 rad s−1.50 The Cole–Cole plot, representing the relationship between the real (η′) and imaginary (η′′) parts of η*, revealed semicircular lines for PLA/110 phr, PLA/120 phr, and PLA/130 phr blends, indicating good compatibility and phase homogeneity between PLA and 1 even in the melt state (Fig. 7b).40
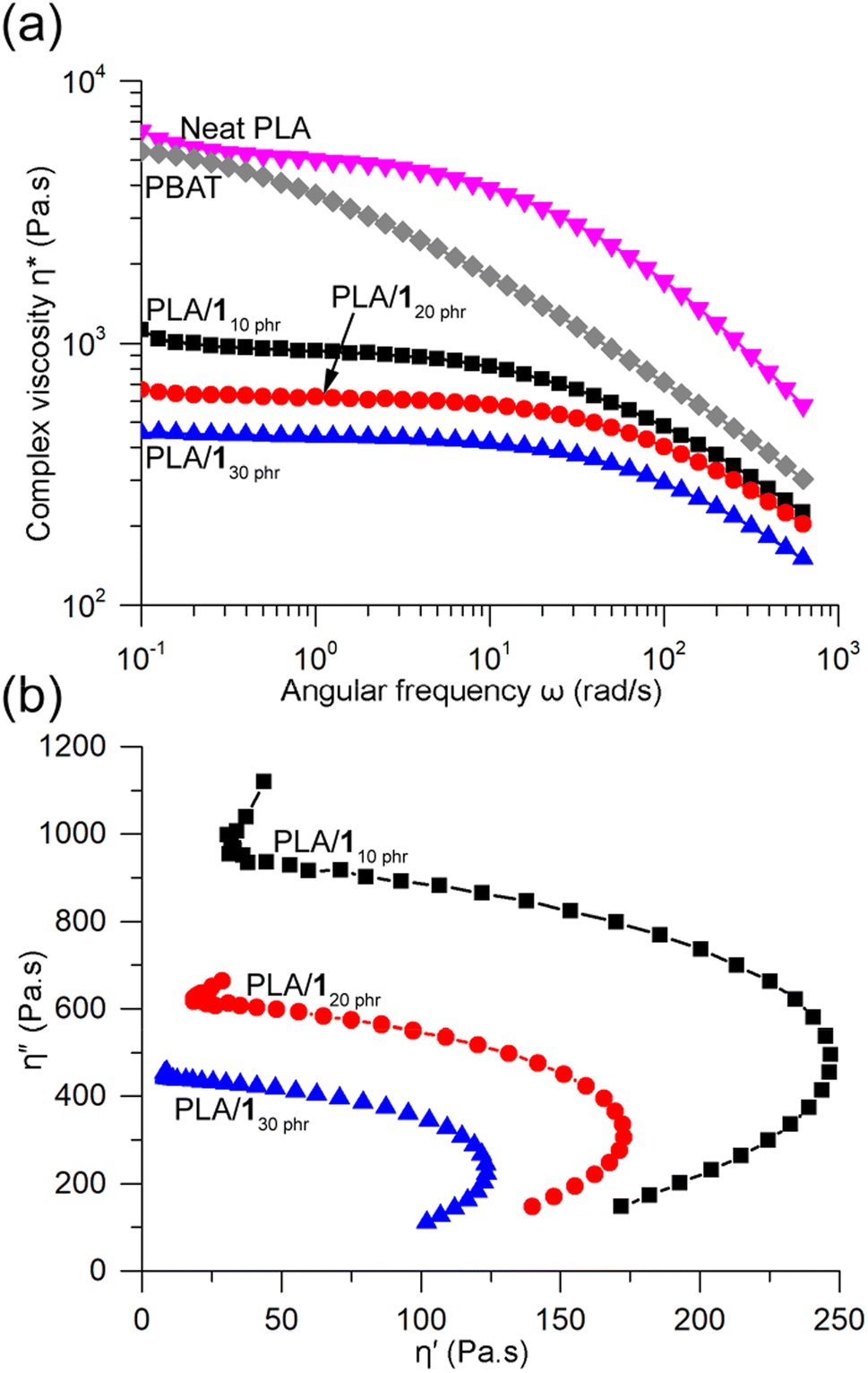 | ||
| Fig. 7 (a) Dynamic viscosities of PLA/110 phr, PLA/120 phr, and PLA/130 phr compared to neat PLA and PBAT measured using rotational rheometer at 170 °C. (b) Cole–Cole plots of PLA/110 phr, PLA/120 phr, and PLA/130 phr. | ||
ATBC is widely recognized as one of the most efficient plasticizers for PLA due to its biobased origin, biodegradability, biocompatibility, and approval for food contact applications.70 However, concerns have been raised about the morphological stability of PLA when blended with tributyl citrate (TBC).34 The inclusion of plasticizer TBC leads to the crystallization of PLA chains even at room temperature, resulting in a reduction in the size of amorphous domains. Consequently, this phenomenon leads to a gradual opaqueness and deformation of the specimens over time. Remarkably, after aging for 6 weeks at room temperature, a previously transparent PLA/ATBC20 phr specimen underwent a discernible transformation, becoming hazy. DMA revealed a significant weakening of the tanδ peak and pronounced alterations in the E′ and E′′ curves when compared to the unaged specimen (Fig. 8a). The decline in E′ and E′′ values across the Tg region was less abrupt and less dramatic than that observed in the unaged sample, implying a substantial crystallization process occurring during the 6 week storage period at room temperature. Crystallization due to aging was also evident in the wide-angle X-ray diffraction (WAXD) patterns. Initially, for PLA/ATBC20 phr, only broad signals were present at 2θ = 10–25°. Nevertheless, following the aging process, a distinct and sharp signal at 2θ = 16.5° emerged (Fig. S20a†), aligning with a prominent signal observed in PLA crystals. This signal can be attributed to the reflection from the (110/200) plane of the α′ or α crystal form of PLA.61
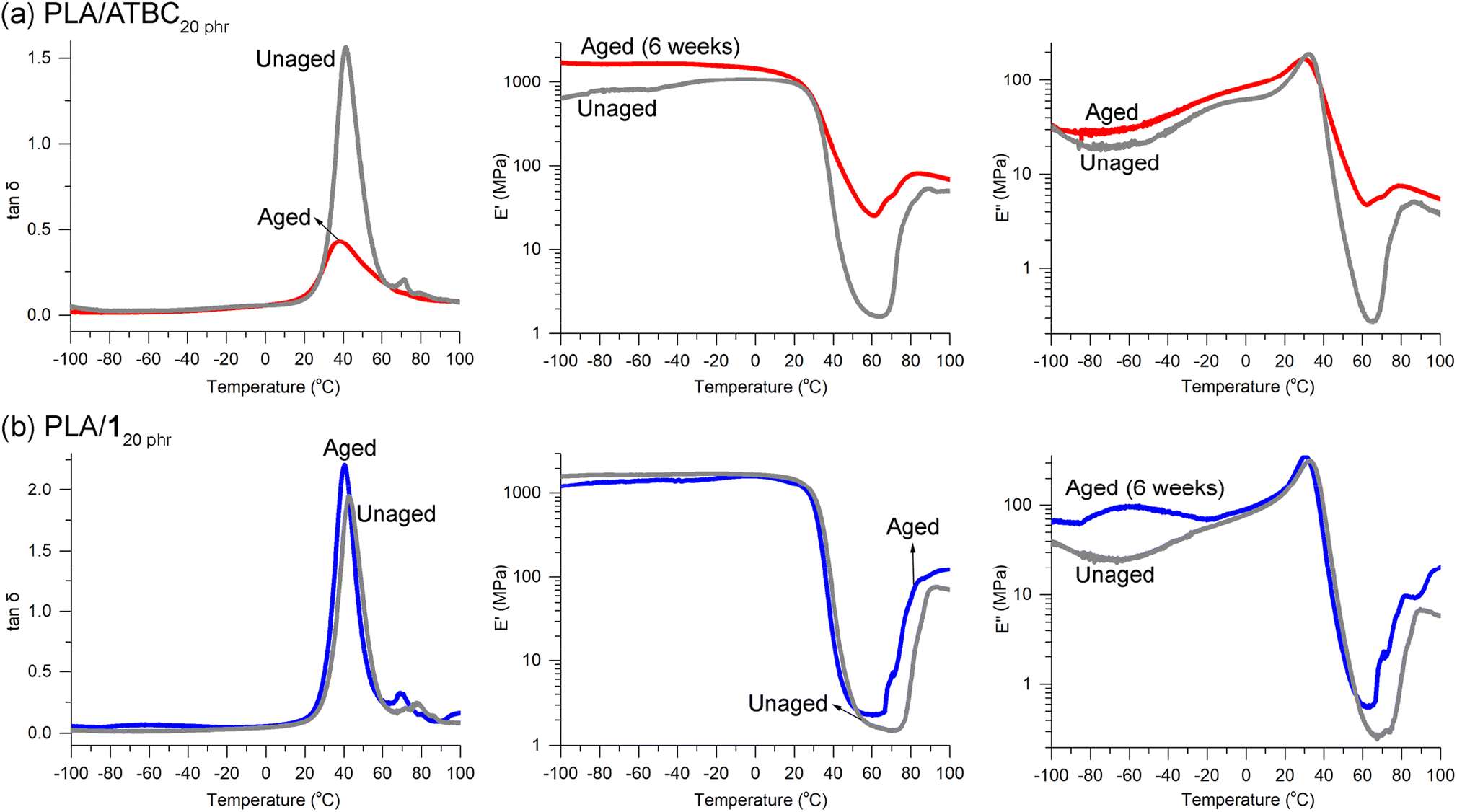 | ||
| Fig. 8 DMA curves for unaged and aged (6 weeks) specimens of (a) PLA/ATBC20 phr and (b) PLA/120 phr. | ||
Crystallization in the specimens has been previously reported to result in a degradation of the tensile properties of PLA.71 In our observations, the tensile strength of neat PLA was found to decrease from 60 ± 2 to 50 ± 9 MPa when the specimens were annealed at 110 °C for 1 h to induce crystallization. This degradation in properties was similarly noted in the case of PLA/120 phr. Notably, both the tensile strength and elongation at break decreased from 26 ± 4 to 21 ± 2 MPa and from 260 ± 30 to 10 ± 2%, respectively, after annealing under the same conditions. Encouragingly, the DMA curves of the PLA/120 phr specimen remained remarkably unchanged (Fig. 8b), and the specimen maintained its transparency throughout the 6 week aging period. This outcome suggests the absence of crystallization in the presence of plasticizer 1. Furthermore, in the WAXD curves of both unaged and aged specimens of PLA/120 phr, only broad signals were detected (Fig. S20b†), in contrast to appearance of the clear and sharp signal in the aged PLA/ATBC20 phr. This absence of a sharp and distinct signal suggests that crystallization did not take place during the aging process.
Biodegradability studies
Prior to investigating the biodegradabilities of PLA/130 phr, PLA/230 phr, and PLA/430 phr blends, we first examined the biodegradabilities of organic carbonates 1, 2, and 4, individually, using the standard method and soil conditions in an ECHO® respirometer set at 25 °C and 50–55% water content [ISO 17556 (2019)]. The evolved CO2 amount was continuously monitored under an air flow for 1, 2, and 4, compared to microcrystalline cellulose. Surprisingly, all tested organic carbonates exhibited very rapid biodegradability in soil at 25 °C, with almost complete consumption within a week, showing higher CO2-evolution rates than microcrystalline cellulose (Fig. 9a). The total evolved CO2 amounts in 2 weeks corresponded to 71, 69, and 68% of the CO2 amounts estimated for the full conversions of the carbons in 1, 2, and 4 to CO2 gas, respectively. It is worth noting that even in cases of complete biodegradation, not all carbon may evolve into CO2; a fraction could be assimilated into microbial biomass. Similarly, only 68% of the estimated CO2 was detected in the case of microcrystalline cellulose, suggesting that a portion of the carbon might have been assimilated into microbial biomass rather than being fully evolved into CO2. Glycerol carbonate 3 and propylene carbonate also exhibited similar rapid biodegradability (Fig. S21†). After unexpectedly observing rapid biodegradability of the organic carbonates, we assessed the stability of 2 in water, as it is fairly soluble, by monitoring its 1H NMR spectra in D2O over a period of 2 months (Fig. S22†). In 10 days, most of the signals corresponding to 2 remained intact, with the appearance of a set of small intensity signals assigned to glycerol carbonate 3 (5 mol%). The intensity of the signals corresponding to 3 increased over time without the formation of other signals, reaching 11 mol% in 30 days and 18 mol% in 60 days. The observed stability of organic carbonates in water implies that their rapid biodegradability is attributable to the collective effects of various soil constituents, including microbial communities.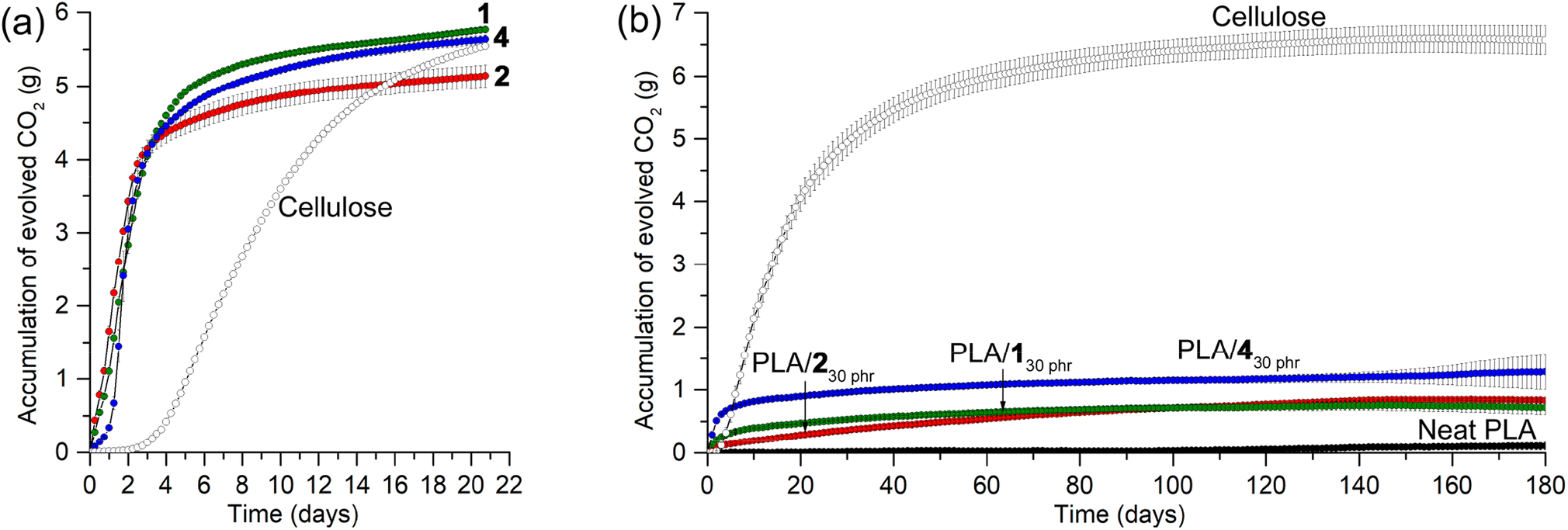 | ||
| Fig. 9 Biodegradability assessments of (a) organic carbonates and (b) their blends monitored by measuring evolved CO2 over time in a respirometer set to 25 °C and 50–55% water content with continuous air flow. | ||
PLA is non-biodegradable under ambient conditions in soil, and the CO2 evolution was minimal in the respirometer conditions at 25 °C and 50–55% water content, even after 180 days (1.1% of the estimated CO2 evolution; Fig. 9b). The PLA/130 phr, PLA/230 phr, and PLA/430 phr blends exhibited slightly higher rates of CO2 evolution that neat PLA, but this could be attributed to the biodegradation of the plasticizers rather than substantial PLA degradation. The total evolved CO2 amounts in 180 days corresponded to 39, 48, and 68% of those estimated for the complete conversion of the carbons in plasticizers 1, 2, and 4 to CO2 gas, respectively. After incubation in the respirometer for 180 days, PLAs in soils were extracted with CH2Cl2, and pure PLA was isolated by filtration of the CH2Cl2 solution through a short pad of silica gel, as confirmed by the analysis of 1H NMR spectra. The Mw values were 200 kDa (Ð = 2.1), 95 kDa (Ð = 2.0), and 130 kDa (Ð = 2.1) for PLAs extracted from soils incubated with pristine PLA, PLA/130 phr, and PLA/230 phr, respectively. The Mw values were only marginally reduced, indicating that PLAs, both individually and in blends, remained largely intact. This observation is consistent with PLA's known resistance to degradation in ambient conditions, whereas it is completely degraded under harsh industrial composting conditions with higher temperature of 60 °C and abundant microbiomes.
Experimental
General remarks
Reagents of glycerol, DEC, acetic anhydride, lithium tert-butoxide, Amberlyst® 15 in hydrogen form (4.7 mmol-H+/g), cyclohexane, hexane, and anhydrous grade DMC were procured from Merck. ATBC was acquired from TCI (Tokyo Chemical Industry) and used as received, with precautions taken to prevent moisture exposure. PLA (Grade 2003D) was supplied by NatureWorks; prior to use, it was dried for 24 h in a vacuum oven at 70 °C. 1H NMR (600 MHz) and 13C NMR (150 MHz) spectra were recorded using a JEOL ECZ600 spectrometer. Thermal transition temperatures (Tg, Tcc, and Tm) along with enthalpy of melting (ΔH) were determined via a DSC 200F3 Maia® differential scanning calorimeter (DSC) from NETZSCH. Measurements were taken during the second heating cycle at a rate of 10 °C min−1. DMA was performed using a TA Instruments TA Q800 dynamic mechanical analyzer. Single cantilever mode was employed, heating from −100 to 100 °C at a constant rate of 3 °C min−1. The deformation amplitude was set at 15 μm, and frequency at 1 Hz. Tensile tests followed the KSM527-2 type 5A standard test method, utilizing a Qmesys QM100T universal testing machine. Tests were conducted at a drawing rate of 50 mm min−1 with a gauge length of 25 mm. Morphological analysis was carried out using a field emission SEM (JSM-7900F, JEOL) at an accelerating voltage of 2.0–3.0 kV. GPC data were acquired at 40 °C, using chloroform as an eluent and Shodex GPC HK-400 series with polystyrene standards. TGA/DSC experiments were performed using a NETZSCH model STA449F3 Jupiter® within the temperature range of 35–700 °C. The heating rate was set at 10 °C min−1 under a N2 flow rate of 50 mL min−1. Dynamic shear melt rheological data were obtained using a TA Instruments DHR10 rotational rheometer at an angular frequency of 0.1–628 rad s−1 and a strain of 1% at 170 °C.4-Ethoxycarbonyloximethyl-[1,3]dioxolan-2-one (1)
Glycerol (50.0 g, 0.543 mol), DEC (449 g, 3.80 mol), tBuOLi (0.435 g, 5.43 mmol, 1.0 mol%), and cyclohexane (70 mL) were combined in a one-necked flask. The flask was fitted with a Dean–Stark apparatus, with the trap containing two layers of distilled water (400 mL) and cyclohexane (130 mL), connected to a manifold equipped with vacuum and N2 gas lines (Scheme 2c). After purging with N2 gas, the reaction mixture was placed in an oil bath set at 120–125 °C and stirred for 21 hours. Throughout the reaction, the resulting byproduct EtOH was continuously removed via the Dean–Stark apparatus. The volume of the aqueous phase gradually increased due to the absorption of the generated EtOH, and a portion of the aqueous phase was periodically discarded to prevent its flow into the reactor. To remove the base catalyst, ion exchange resin (Amberlyst® 15 hydrogen form, 2.3 g, 11 mmol-H+), pre-washed with propylene carbonate and subsequently with DEC, was added. The solution was then filtered to eliminate the ion exchange resin. Excess DEC was removed by vacuum distillation under full vacuum at an oil bath temperature of 60 °C, followed by collecting the product as the oil bath temperature was raised to 150 °C (86.9 g, 81% yield). Although small impurity signals (primarily 1.5 mol% glycerol carbonate) were observed in the 1H and 13C NMR spectra of the collected portion (Fig. S1†), it was used without further purification for blending studies. 1H NMR (600 MHz, (CD3)2CO): δ 5.14 (m, H), 4.69 (dd, J = 9.0, 8.4 Hz, H), 4.47 (dd, J = 12.6, 3.0 Hz, H), 4.42 (dd, J = 9.0, 6.6 Hz, H), 4.39 (dd, J = 12.6, 4.8 Hz, H), 4.18 (q, J = 7.8 Hz, 2H), 1.26 (t, J = 7.8 Hz, CH3, 2H) ppm. 13C NMR (150 MHz, (CD3)2CO): 154.8, 74.4, 66.7, 66.0, 64.4, 13.8 ppm.4-Methoxycarbonyloximethyl-[1,3]dioxolan-2-one (2)
The compound 2 was synthesized using the same procedure and conditions as 1. Glycerol (50.0 g, 0.543 mol), DMC (342 g, 3.80 mol) as a replacement for DEC, tBuOLi (0.435 mg, 5.43 mmol, 1.0 mol%), hexane (200 mL), and water (400 mL) were utilized in the reaction. The reaction was conducted at a bath temperature of 85 °C for 28 hours. Upon completion of the reaction, while the reaction solution was still hot, ion exchange resin (Amberlyst® 15 hydrogen form, 2.3 g, 11 mmol-H+) was added, followed by filtration after a 1 hour neutralization reaction. To remove some of the volatiles (hexane and DMC), the solution was subjected to rotary evaporation, resulting in a 180 g solution from which crystalline solids precipitated upon storage at ambient temperature for 12 hours (46.0 g, 48%). The purity of the isolated solids as 2 was confirmed by 1H and 13C NMR spectra (Fig. S2†). Additional yield of the product was obtained as second crops when the filtrate was stored overnight at ambient temperature after reducing its mass to 65 g using a rotary evaporator (14.9 g, 16%, total yield 64%). 1H NMR (600 MHz, (CD3)2CO): δ 5.13 (m, H), 4.69 (dd, J = 9.0, 8.4 Hz, H), 4.48 (dd, J = 12.6, 3.0 Hz, H), 4.42 (dd, J = 9.0, 6.6 Hz, H), 4.40 (dd, J = 12.6, 4.2 Hz, H), 3.77 (s, CH3, 3H) ppm. 13C NMR (150 MHz, (CD3)2CO): 155.9, 154.7, 74.5, 67.1, 66.2, 55.1 ppm.Glycerol carbonate (3)
Glycerol (156 g, 1.69 mol), DMC (458 g, 5.08 mol), and tBuOLi (0.678 g, 8.47 mmol, 0.5 mol%) were combined in a one-neck flask (1 L). After purging with N2 gas, the reaction mixture was stirred at 80 °C for 1.67 hours. Analysis by 1H NMR of a sample confirmed the formation of the desired product in good yield. However, there was still a remaining amount of glycerol (6.5 mol%) as well as some side product 2 (5.0 mol%). Nonetheless, byproduct MeOH and excess DMC were removed by vacuum distillation, which was performed at a bath temperature of 30–35 °C with the receiver cooled by a dry ice/acetone bath. Encouragingly, most of the remaining glycerol and 2 were converted to the desired product during the distillation process, leaving behind 1.3 mol% of glycerol (Fig. S3†). Finally, ion exchange resin (Amberlyst® 15 hydrogen form, 3.6 g, 17 mmol-H+) was added to the resulting product to remove the base catalyst. After stirring for 1 hour at ambient temperature, the resin was filtered off, yielding a colorless oil (198 g, 99% yield), which was used without further purification for blending studies. 1H NMR (600 MHz, (CD3)2CO): δ 4.87 (m, H), 4.57 (dd, J = 8.4, 7.8 Hz, H), 4.43 (t, J = 5.4 Hz, –OH), 4.41 (dd, J = 8.4, 6.0 Hz, H), 3.88 (ddd, J = 12.6, 4.8, 3.0 Hz, H), 3.71 (ddd, J = 12.6, 6.0, 3.6 Hz, H), 3.77 (s, CH3, 3H) ppm. 13C NMR (150 MHz, (CD3)2CO): 155.7, 77.4, 66.2, 61.7 ppm.Glycerol 1-acetate 2,3-carbonate (4)
Glycerol carbonate (35.7 g, 302 mmol), acetic anhydride (37.0 g, 363 mmol), and ion exchange resin (Amberlyst® 15 hydrogen form, 0.30 g, 1.4 mmol-H+) were added to a one-neck flask. The reaction mixture was then purged with N2 gas and stirred at 60 °C for 48 hours. After cooling, the ion exchange resin was removed by filtration, and the volatiles were eliminated using a rotary evaporator, giving a colorless oil (41.4 g, 86%). Analysis of the 1H NMR spectrum revealed that the isolated product was contaminated with 3.4 mol% of triacetyl glycerol (Fig. S4†). 1H NMR (600 MHz, (CD3)2CO): δ 5.10 (m, H), 4.67 (dd, J = 9.0, 8.4 Hz, H), 4.39 (dd, J = 9.0, 6.0 Hz, H), 4.38 (dd, J = 12.6, 3.0 Hz, H), 4.31 (dd, J = 12.6, 4.2 Hz, H), 2.06 (s, CH3, 3H) ppm. 13C NMR (150 MHz, (CD3)2CO): 170.4, 155.1, 74.7, 66.4, 63.7, 20.1 ppm.Polymer blending
With a restricted amount of in-house prepared plasticizer, we designed and fabricated a compact reactor (25 mL capacity) equipped with a mechanical stirrer for blending (Fig. S23†). PLA samples (18 g) were combined with the plasticizer (3.6 g, 20 phr) under a N2 atmosphere within the custom-made reactor and, subsequently, the reactor was immersed in an oil bath at 170 °C for a duration of 2 h, ensuring complete melting of the PLA (Tm, 148–156 °C). Following the melting process, stirring was performed at 170 °C utilizing a custom-made screw-type blade, for a period of 30 min to ensure thorough blending. Upon completion of the blending procedure, the reactor was opened while still hot, and the blended polymers were extracted from the reactor using long-nose pliers. To preserve the quality and prevent moisture contact from the surrounding air, the obtained blended polymers were vacuum-sealed within polyethylene film using a countertop vacuum sealer (FoodSaver, FM5460-071).Biodegradable studies
To emulate the natural biodegradation conditions for PLAs, soil samples were collected from three geographically distinct sites: Mt. Gwanggyosan, Suwon, South Korea (37°20.1360 N 127°1.1640′ E); Ajou University's field campus in Suwon (37°17.1210 N 127°2.6710 E), South Korea; and flower beds in Hwaseong, South Korea (37°10.8030 N 126°58.8720 E), in compliance with ISO 17556 (2019). The collected soil was air-dried naturally at room temperature for 24 hours, homogenized, and then sieved through a 2 mm mesh using an AS 200 Control vibrating sieve (Retsch, Germany). The 2400 g portions of sieved soil were supplemented with mineral nutrients (0.2 g KH2PO4, 0.1 g MgSO4, 0.4 g NaNO3, 0.4 g CO(NH2)2, and 0.4 g NH4Cl per kg of soil) and then equitably combined. PLA blends (neat PLA, PLA/130 phr, PLA/230 phr, and PLA/430 phr) were mechanically ground and sieved through a 2 mm mesh using a Type ZM 200 Ultra Centrifugal Mill (Retsch, Nordrhein-Westfalen). Microcrystalline cellulose (20 μm, Sigma-Aldrich, St. Louis, MO, USA) served as a positive control for biodegradability assays. In each reactor, 15 g of PLA blends or microcrystalline cellulose was uniformly blended with 800 g of soil and then aerobically incubated in a reactor (triplicates) at 25 °C in the dark using a 12-channel ECHO® respirometer (ECHO Instruments, Slovenske Konjice, Slovenia). A blank soil sample (800 g) was also incubated to normalize CO2 evolution. Real-time monitoring of CO2 evolutions was enabled by an integrated near-infrared (NIR) detector in the ECHO® Respiratory system, with a consistent air flow rate of 200 mL min−1 regulated by an integrated mass flow controller. Likewise, 15 g of organic carbonate plasticizers 1, 2, or 4 were each mixed with 800 g of soil and incubated under identical conditions.Conclusions
4-Ethoxycarbonyloximethyl-[1,3]dioxolan-2-one (1), synthesized using renewable glycerol and CO2-derived DEC (CO2 content in 1, 46 wt%), proves to be a highly effective plasticizer for PLA. The plasticization effect was evident through the gradual reduction in Tg as the concentration of plasticizer increased, following a linear relationship represented by the equation “Tg (°C) = 58.4 − 1.13 × [amount of 1 (phr)]”. DMA revealed PLA/1 blends exhibiting narrow and unimodal tanδ curves, along with distinctive E′ and E′′ curves, confirming excellent miscibility of PLA with up to 30 phr of 1. This was further supported by SEM images. The blending process endowed the PLA with ductile properties, leading to a remarkable increase in strain at break, up to 470% for PLA/130 phr, however, inevitably with some reduction in tensile strength (25–40 MPa). Chain scission was not observed during the melt blending process at 170 °C, as indicated by GPC studies. TGA displayed minimal weight loss (∼1%) at a plausible processing temperature of 200 °C. The dynamic viscosity curves demonstrated that while the blends retained similar features to neat PLA, the complex viscosity was significantly reduced, highlighting their improved processability. The Cole–Cole plot suggested phase homogeneity in the melt state. Significantly, 1 outperformed conventional ATBC plasticizer in terms of morphological stability. Unlike PLA/ATBC blends, PLA/1 blends exhibited no cold crystallization over time at room temperature. Furthermore, all synthesized organic carbonates exhibited rapid biodegradability under ambient soil conditions at 25 °C within a week. However, even over an extended 6 month period under the same ambient conditions, the PLA in the blends remained predominantly undegraded.
Author contributions
Hyeon Jeong Seo and Yeong Hyun Seo conducted the blending and collection of analysis data. Sang Uk Park and Hyun Ju Lee were responsible for preparing the plasticizers. Mi Ryu Lee and Jun Hyeong Park managed the data collection and analysis. Woo Yeon Cho conducted the biodegradation studies. Bun Yeoul Lee and Pyung Cheon Lee supervised the project and offered invaluable suggestions. All the authors read, edited, and approved the final manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was supported by the Carbon to X Program of Ministry of Science and ICT (grant number 2020M3H7A1098281) and by Korea Institute of Marine Science & Technology Promotion (KIMST) funded by the Ministry of Oceans and Fisheries (20220258).References
- R. H. Heyn, Chapter 7 – Organic carbonates, in Carbon Dioxide Utilisation, ed. Styring P., Quadrelli E. A. and Armstrong K., Elsevier, Amsterdam, 2015, pp. 97–113 Search PubMed.
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Organic Carbonates as Solvents in Synthesis and Catalysis, Chem. Rev., 2010, 110(8), 4554–4581 CrossRef PubMed.
- R. Porcar, P. Lozano, M. I. Burguete, E. Garcia-Verdugo and S. V. Luis, Dimethyl carbonate as a non-innocent benign solvent for the multistep continuous flow synthesis of amino alcohols, React. Chem. Eng., 2018, 3(4), 572–578 RSC.
- A. L. Michan, B. S. Parimalam, M. Leskes, R. N. Kerber, T. Yoon, C. P. Grey and B. L. Lucht, Fluoroethylene Carbonate and Vinylene Carbonate Reduction: Understanding Lithium-Ion Battery Electrolyte Additives and Solid Electrolyte Interphase Formation, Chem. Mater., 2016, 28(22), 8149–8159 CrossRef CAS.
- M. Szőri, B. R. Giri, Z. Wang, A. E. Dawood, B. Viskolcz and A. Farooq, Glycerol carbonate as a fuel additive for a sustainable future, Sustainable Energy Fuels, 2018, 2(10), 2171–2178 RSC.
- M. A. Pacheco and C. L. Marshall, Review of Dimethyl Carbonate (DMC) Manufacture and Its Characteristics as a Fuel Additive, Energy Fuels, 1997, 11(1), 2–29 CrossRef CAS.
- N. von Seggern, T. Schindler and S. Naumann, Dual Catalytic Ring-Opening Polymerization of Ethylene Carbonate for the Preparation of Degradable PEG, Biomacromolecules, 2020, 21(7), 2661–2669 CrossRef CAS PubMed.
- Z. Abdel Baki, H. Dib and T. Sahin, Overview: Polycarbonates via Ring-Opening Polymerization, Differences between Six- and Five-Membered Cyclic Carbonates: Inspiration for Green Alternatives, Polymers, 2022, 14(10), 2031 CrossRef CAS PubMed.
- D. Shi, S. Heyte, M. Capron and S. Paul, Catalytic processes for the direct synthesis of dimethyl carbonate from CO2 and methanol: a review, Green Chem., 2022, 24(3), 1067–1089 RSC.
- A. Raza, M. Ikram, S. Guo, A. Baiker and G. Li, Green Synthesis of Dimethyl Carbonate from CO2 and Methanol: New Strategies and Industrial Perspective, Adv. Sustainable Syst., 2022, 6(8), 2200087 CrossRef CAS.
- A. Brege, B. Grignard, R. Méreau, C. Detrembleur, C. Jerome and T. Tassaing, En Route to CO2-Based (a)Cyclic Carbonates and Polycarbonates from Alcohols Substrates by Direct and Indirect Approaches, Catalysts, 2022, 12(2), 124 CrossRef CAS.
- T. Weidlich and B. Kamenická, Utilization of CO2-Available Organocatalysts for Reactions with Industrially Important Epoxides, Catalysts, 2022, 12(3), 298 CrossRef CAS.
- L. F. S. Souza, P. R. R. Ferreira, J. L. de Medeiros, R. M. B. Alves and O. Q. F. Araújo, Production of DMC from CO2 via Indirect Route: Technical–Economical–Environmental Assessment and Analysis, ACS Sustain. Chem. Eng., 2014, 2(1), 62–69 CrossRef CAS.
- J.-Q. Wang, J. Sun, C.-Y. Shi, W.-G. Cheng, X.-P. Zhang and S.-J. Zhang, Synthesis of dimethyl carbonate from CO2 and ethylene oxide catalyzed by K2CO3-based binary salts in the presence of H2O, Green Chem., 2011, 13(11), 3213–3217 RSC.
- S.-H. Pyo, J. H. Park, T.-S. Chang and R. Hatti-Kaul, Dimethyl carbonate as a green chemical, Curr. Opin. Green Sustainable Chem., 2017, 5, 61–66 CrossRef.
- J. G. Kim, J. Y. Jeon, J. Chun, C. S. Kim, P. C. Lee and B. Y. Lee, Efficient synthesis of organic carbonates and poly(1,4-butylene carbonate-co-terephthalate)s, J. Appl. Polym. Sci., 2017, 134(24), 44951 CrossRef.
- X. Bi, N. Yao, X. Meng, M. Gou and P. Zhao, MnCO3-Catalyzed Transesterification of Alcohols with Dimethyl Carbonate Under Mild Conditions, Catal. Lett., 2021, 151(2), 454–462 CrossRef CAS.
- S. Fukuoka, M. Tojo, H. Hachiya, M. Aminaka and K. Hasegawa, Green and sustainable chemistry in practice: development and industrialization of a novel process for polycarbonate production from CO 2 without using phosgene, Polym. J., 2007, 39(2), 91–114 CrossRef CAS.
- J. H. Park, J. Y. Jeon, J. J. Lee, Y. Jang, J. K. Varghese and B. Y. Lee, Preparation of High-Molecular-Weight Aliphatic Polycarbonates by Condensation Polymerization of Diols and Dimethyl Carbonate, Macromolecules, 2013, 46(9), 3301–3308 CrossRef CAS.
- S. Y. Park, J. Chun, J. Y. Jeon, P. C. Lee, Y. Hwang, B. G. Song, R. Ramos, C. Y. Ryu and B. Y. Lee, Branched poly(1,4-butylene carbonate-co-terephthalate)s: LDPE-like semicrystalline thermoplastics, J. Polym. Sci., Part A: Polym. Chem., 2015, 53(7), 914–923 CrossRef CAS.
- W. Zhu, X. Huang, C. Li, Y. Xiao, D. Zhang and G. Guan, High-molecular-weight aliphatic polycarbonates by melt polycondensation of dimethyl carbonate and aliphatic diols: synthesis and characterization, Polym. Int., 2011, 60(7), 1060–1067 CrossRef CAS.
- C. Ngassam Tounzoua, B. Grignard and C. Detrembleur, Exovinylene Cyclic Carbonates: Multifaceted CO2-Based Building Blocks for Modern Chemistry and Polymer Science, Angew. Chem., Int. Ed., 2022, 61(22), e202116066 CrossRef CAS PubMed.
- P. Tyagi, D. Singh, N. Malik, S. Kumar and R. Singh Malik, Metal catalyst for CO2 capture and conversion into cyclic carbonate: progress and challenges, Mater. Today, 2023, 65, 133–165 CrossRef CAS.
- J. Wu, Q. Guo, H. Hong, R. Xie and N. Zhu, The tandem reaction of propargylamine/propargyl alcohol with CO2: reaction mechanism, catalyst activity and product diversity, J. CO2 Util., 2022, 65, 102192 CrossRef CAS.
- S. Bang, J. Y. Jang, Y.-J. Ko, S. M. Lee, H. J. Kim and S. U. Son, Hydroboration of Hollow Microporous Organic Polymers: A Promising Postsynthetic Modification Method for Functional Materials, ACS Macro Lett., 2022, 11(8), 1034–1040 CrossRef CAS PubMed.
- C. Seo, S. E. Kim, H. Kim and H.-Y. Jang, CO2 Fixation by Dual-Function Cu(triNHC) Catalysts as a Route to Carbonates and Carbamates via α-Alkylidene Carbonates, ACS Sustain. Chem. Eng., 2022, 10(17), 5643–5650 CrossRef CAS.
- Y. H. Seo, Y. B. Hyun, H. J. Lee, J. W. Baek, H. C. Lee, J. H. Lee, J. Lee and B. Y. Lee, Preparation of double-metal cyanide catalysts with H3Co(CN)6 for propylene oxide homo- and CO2-copolymerization, J. CO2 Util., 2021, 53, 101755 CrossRef CAS.
- C. A. L. Lidston, S. M. Severson, B. A. Abel and G. W. Coates, Multifunctional Catalysts for Ring-Opening Copolymerizations, ACS Catal., 2022, 12(18), 11037–11070 CrossRef CAS.
- H. J. Lee, W. Y. Cho, H. C. Lee, Y. H. Seo, J. W. Baek, P. C. Lee and B. Y. Lee, Rapid Biodegradable Ionic Aggregates of Polyesters Constructed with Fertilizer Ingredients, J. Am. Chem. Soc., 2022, 144(35), 15911–15915 CrossRef CAS PubMed.
- P. Skoczinski, L. Krause, A. Raschka, L. Dammer and M. Carus, Chapter One – Current status and future development of plastics: solutions for a circular economy and limitations of environmental degradation, in Methods in Enzymology, ed. Weber G., Bornscheuer U. T. and Wei R., Academic Press, 2021, vol. 648, pp. 1–26 Search PubMed.
- T. Rheinberger, J. Wolfs, A. Paneth, H. Gojzewski, P. Paneth and F. R. Wurm, RNA-Inspired and Accelerated Degradation of Polylactide in Seawater, J. Am. Chem. Soc., 2021, 143(40), 16673–16681 CrossRef CAS PubMed.
- Y. Park and J. Lee, Comparison of recently developed toughening strategies for polylactic acid blends, J. Ind. Eng. Chem., 2023, 125, 50–57 CrossRef CAS.
- L. V. Labrecque, R. A. Kumar, V. Davé, R. A. Gross and S. P. McCarthy, Citrate esters as plasticizers for poly(lactic acid), J. Appl. Polym. Sci., 1997, 66(8), 1507–1513 CrossRef CAS.
- N. Ljungberg and B. Wesslén, Preparation and Properties of Plasticized Poly(lactic acid) Films, Biomacromolecules, 2005, 6(3), 1789–1796 CrossRef CAS PubMed.
- W. Xuan, M. Hakkarainen and K. Odelius, Levulinic Acid as a Versatile Building Block for Plasticizer Design, ACS Sustain. Chem. Eng., 2019, 7(14), 12552–12562 CAS.
- W. Xuan, K. Odelius and M. Hakkarainen, Tailoring Oligomeric Plasticizers for Polylactide through Structural Control, ACS Omega, 2022, 7(16), 14305–14316 CrossRef CAS PubMed.
- W. Xuan, K. Odelius and M. Hakkarainen, Dual-Functioning Antibacterial Eugenol-Derived Plasticizers for Polylactide, Biomolecules, 2020, 10(7), 1077 CrossRef CAS PubMed.
- K. Zawada, A. Plichta, D. Jańczewski, H. Hajmowicz, Z. Florjańczyk, M. Stępień, A. Sobiecka and L. Synoradzki, Esters of Tartaric Acid, A New Class of Potential “Double Green” Plasticizers, ACS Sustain. Chem. Eng., 2017, 5(7), 5999–6007 CrossRef CAS.
- J. Anakabe, A. M. Zaldua Huici, A. Eceiza, A. Arbelaiz and L. Avérous, Combined effect of nucleating agent and plasticizer on the crystallization behaviour of polylactide, Polym. Bull., 2017, 74(12), 4857–4886 CrossRef CAS.
- Y. Hao, H. Yang, H. Zhang, G. Zhang, Y. Bai, G. Gao and L. Dong, Diethylene glycol monobutyl ether adipate as a novel plasticizer for biodegradable polylactide, Polym. Bull., 2016, 73(11), 3143–3161 CrossRef CAS.
- S.-L. Yang, Z.-H. Wu, B. Meng and W. Yang, The effects of dioctyl phthalate plasticization on the morphology and thermal, mechanical, and rheological properties of chemical crosslinked polylactide, J. Polym. Sci., Part B: Polym. Phys., 2009, 47(12), 1136–1145 CrossRef CAS.
- N. Ljungberg and B. Wesslén, The effects of plasticizers on the dynamic mechanical and thermal properties of poly(lactic acid), J. Appl. Polym. Sci., 2002, 86(5), 1227–1234 CrossRef CAS.
- T. Wan, Y. Lin and Y. Tu, Plasticizing effect of glyceryl tribenzoate, dipropylene glycol dibenzoate, and glyceryl triacetate on poly(lactic acid), Polym. Eng. Sci., 2016, 56(12), 1399–1406 CrossRef CAS.
- G. Coativy, M. Misra and A. K. Mohanty, Microwave Synthesis and Melt Blending of Glycerol Based Toughening Agent with Poly(lactic acid), ACS Sustain. Chem. Eng., 2016, 4(4), 2142–2149 CrossRef CAS.
- X. Yang and M. Hakkarainen, Migration resistant glucose esters as bioplasticizers for polylactide, J. Appl. Polym. Sci., 2015, 132(18), 41928 CrossRef.
- Y. Yang, Z. Xiong, L. Zhang, Z. Tang, R. Zhang and J. Zhu, Isosorbide dioctoate as a “green” plasticizer for poly(lactic acid), Mater. Des., 2016, 91, 262–268 CrossRef CAS.
- H. Jeong, J. S. Yuk, H. Lee, S. Kang, H. Park, S. H. Park and J. Shin, Lactide-derived ester oligomers for highly compatible poly(lactide) plasticizer produced through an eco-friendly process: renewable resources, biodegradation, enhanced flexibility, and elastomeric performance, Green Chem., 2021, 23(19), 7549–7565 RSC.
- N. Burgos, D. Tolaguera, S. Fiori and A. Jiménez, Synthesis and Characterization of Lactic Acid Oligomers: Evaluation of Performance as Poly(Lactic Acid) Plasticizers, J. Polym. Environ., 2014, 22(2), 227–235 CrossRef CAS.
- D. Li, Y. Jiang, S. Lv, X. Liu, J. Gu, Q. Chen and Y. Zhang, Preparation of plasticized poly (lactic acid) and its influence on the properties of composite materials, PLoS One, 2018, 13(3), e0193520 CrossRef PubMed.
- R. N. Darie-Niţă, C. Vasile, A. Irimia, R. Lipşa and M. Râpă, Evaluation of some eco-friendly plasticizers for PLA films processing, J. Appl. Polym. Sci., 2016, 133(13), 43223 CrossRef.
- A. Sustaita-Rodríguez, A. Vega-Rios, A. Bugarin, V. H. Ramos-Sánchez, A. A. Camacho-Dávila, B. Rocha-Gutiérrez and D. Chávez-Flores, Chemoenzymatic Epoxidation of Highly Unsaturated Fatty Acid Methyl Ester and Its Application as Poly(lactic acid) Plasticizer, ACS Sustain. Chem. Eng., 2021, 9(50), 17016–17024 CrossRef.
- A. Carbonell-Verdu, M. D. Samper, D. Garcia-Garcia, L. Sanchez-Nacher and R. Balart, Plasticization effect of epoxidized cottonseed oil (ECSO) on poly(lactic acid), Ind. Crops Prod., 2017, 104, 278–286 CrossRef CAS.
- R. Turco, R. Tesser, M. E. Cucciolito, M. Fagnano, L. Ottaiano, S. Mallardo, M. Malinconico, G. Santagata and M. Di Serio, Cynara cardunculus Biomass Recovery: An Eco-Sustainable, Nonedible Resource of Vegetable Oil for the Production of Poly(lactic acid) Bioplasticizers, ACS Sustain. Chem. Eng., 2019, 7(4), 4069–4077 CrossRef CAS.
- T. J. Tse, D. J. Wiens and M. J. T. Reaney, Production of Bioethanol—A Review of Factors Affecting Ethanol Yield, Fermentation, 2021, 7(4), 268 CrossRef CAS.
- C. F. Allpress and W. Maw, CCXCIX.—The carbonates of ethylene glycol and related compounds, J. Chem. Soc., Trans., 1924, 125, 2259–2264 RSC.
- J. R. Ochoa-Gómez, O. Gómez-Jiménez-Aberasturi, C. Ramírez-López and B. Maestro-Madurga, Synthesis of glycerol 1,2-carbonate by transesterification of glycerol with dimethyl carbonate using triethylamine as a facile separable homogeneous catalyst, Green Chem., 2012, 14(12), 3368–3376 RSC.
- S. Kotanen, T. Laaksonen and E. Sarlin, Feasibility of polyamines and cyclic carbonate terminated prepolymers in polyurethane/polyhydroxyurethane synthesis, Mater. Today Commun., 2020, 23, 100863 CrossRef CAS.
- S. Sahani, S. N. Upadhyay and Y. C. Sharma, Critical Review on Production of Glycerol Carbonate from Byproduct Glycerol through Transesterification, Ind. Eng. Chem. Res., 2021, 60(1), 67–88 CrossRef CAS.
- G. Lee, H. Lee and Y. Kim, Thermal and Mechanical Properties of Poly(L-lactic Acid) Films Plasticized with Propylene Carbonate, Polymer (Korea), 2019, 43, 113–122 CrossRef CAS.
- H. Xiao, W. Lu and J.-T. Yeh, Effect of plasticizer on the crystallization behavior of poly(lactic acid), J. Appl. Polym. Sci., 2009, 113(1), 112–121 CrossRef CAS.
- H. Kang, Y. Li, M. Gong, Y. Guo, Z. Guo, Q. Fang and X. Li, An environmentally sustainable plasticizer toughened polylactide, RSC Adv., 2018, 8(21), 11643–11651 RSC.
- M. Hesami and A. Jalali-Arani, Cold crystallization behavior of poly(lactic acid) in its blend with acrylic rubber; the effect of acrylic rubber content, Polym. Int., 2017, 66(11), 1564–1571 CrossRef CAS.
- S. Saeidlou, M. A. Huneault, H. Li and C. B. Park, Poly(lactic acid) crystallization, Prog. Polym. Sci., 2012, 37(12), 1657–1677 CrossRef CAS.
- M. Maiza, M. T. Benaniba, G. Quintard and V. Massardier-Nageotte, Biobased additive plasticizing Polylactic acid (PLA), Polímeros, 2015, 25, 581–590 CrossRef CAS.
- S. Farah, D. G. Anderson and R. Langer, Physical and mechanical properties of PLA, and their functions in widespread applications - A comprehensive review, Adv. Drug Delivery Rev., 2016, 107, 367–392 CrossRef CAS PubMed.
- R. Zhang, F. Du, K. Jariyavidyanont, E. Zhuravlev, C. Schick and R. Androsch, Glass transition temperature of poly(d,l-lactic acid) of different molar mass, Thermochim. Acta, 2022, 718, 179387 CrossRef CAS.
- G. Li, M. Zhao, F. Xu, B. Yang, X. Li, X. Meng, L. Teng, F. Sun and Y. Li, Synthesis and Biological Application of Polylactic Acid, Molecules, 2020, 25(21), 5023 CrossRef CAS PubMed.
- H. R. Kricheldorf and S. M. Weidner, Syntheses of polylactides by means of tin catalysts, Polym. Chem., 2022, 13(12), 1618–1647 RSC.
- H. J. Lee, J. W. Baek, T. J. Kim, H. S. Park, S. H. Moon, K. L. Park, S. M. Bae, J. Park and B. Y. Lee, Synthesis of Long-Chain Branched Polyolefins by Coordinative Chain Transfer Polymerization, Macromolecules, 2019, 52(23), 9311–9320 CrossRef CAS.
- J. Tian, Z. Cao, S. Qian, Y. Xia, J. Zhang, Y. Kong, K. Sheng, Y. Zhang, Y. Wan and J. Takahashi, Improving tensile strength and impact toughness of plasticized poly(lactic acid) biocomposites by incorporating nanofibrillated cellulose, Nanotechnol. Rev., 2022, 11(1), 2469–2482 CrossRef CAS.
- B. Ma, X. Wang, Y. He, Z. Dong, X. Zhang, X. Chen and T. Liu, Effect of poly(lactic acid) crystallization on its mechanical and heat resistance performances, Polymer, 2021, 212, 123280 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08922c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |