
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring Cassia mimosoïdes as a promising natural source of steroids with potent anti-cancer, urease inhibition, and antimicrobial properties†
Robert Viani Kepdieu Tchebouab,
Umar Farooq*bc,
Rémy Bertrand Teponno*a,
Tanveer A. Wani e,
Léon Azefack Tapondjoua,
Azhar Rasoold,
Rizwana Sarwarb,
Aneela Khushalb,
Syed Majid Bukharib,
Seema Zargarf,
Hong-Guang Xuc and
Sara Khan*b
e,
Léon Azefack Tapondjoua,
Azhar Rasoold,
Rizwana Sarwarb,
Aneela Khushalb,
Syed Majid Bukharib,
Seema Zargarf,
Hong-Guang Xuc and
Sara Khan*b
aResearch Unit of Environmental and Applied Chemistry, Department of Chemistry, Faculty of Science, University of Dschang, Box 67, Dschang, Cameroon. E-mail: remyteponno@gmail.com
bDepartment of Chemistry, COMSATS University Islamabad, Abbottabad 22010 KPK, Pakistan. E-mail: umarf@cuiatd.edu.pk; sarakhancuiatd.edu.pk
cBeijing National Laboratory for Molecular Sciences, State Key Laboratory of Molecular Reaction Dynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
dDepartment of Zoology, GC University, Faisalabad, Pakistan
eDepartment of Pharmaceutical Chemistry, College of Pharmacy, King Saud University, POBox 2457, Riyadh 11451, Saudi Arabia
fDepartment of Biochemistry College of Science, King Saud University, PO Box 22452, Riyadh 11451, Saudi Arabia
First published on 18th March 2024
Abstract
The genus Cassia is a rich source of physiologically active secondary metabolites, including a novel compound named 21-methylene-24-ethylidene lophenol, alongside 15 known compounds. These compounds were characterized using different spectroscopic techniques. They exhibited promising antimicrobial activity, particularly against bacteria causing gastrointestinal infections. Compound 1 showed strong anti-bacterial activity against H. pylori and S. aur with MIC values of 0.28 and 0.12 μg mL−1 respectively. The study investigated their impact on H. pylori, a contributor to ulcer development, by inhibiting the urease enzyme. Inhibiting urease can reduce H. pylori's pathogenic potential, evident from the fact that the compounds evaluated toward urease enzyme showed higher inhibitory activity (1.024 ± 0.43 <IC50 > 6.678±0.11 μM) compared to standard thiourea (IC50 = 18.61 ± 0.11 μM). Molecular docking studies confirmed their inhibitory action, with compound 7 notably outperforming thiourea in inhibiting urease (−6.95 kcal mol−1 vs. −3.13 kcal mol−1). Additionally, these compounds showed positive effects on liver functioning, which H. pylori can impair. Compound 9 shows the best response against human HepG2 liver cancer cell lines i.e., % viability is 14.47% ± 0.69 and IC50 is 7.8 μM ± 0.21. These compounds hold potential as lead compounds for addressing gastrointestinal and liver disorders caused by H. pylori.
1. Introduction
Various medicinal plants have been used for years in daily life to treat diseases around the world. The interest in medicinal plants reflects the recognition of the validity of many traditional claims regarding the value of natural products in health care. Cassia mimosoides L. (Caesalpiniaceae), is distributed in various countries including Cameroon, Luzon, Mindanao, India, China, Malaysia, and Australia. This plant is widely used by tribal people to treat various ailments including typhoid fever and other microbial infections.1,2 It is used in Uganda to treat pediatric cough.3 In northwestern Tanzania, the aerial parts of C. mimosoides are pounded and mixed with animal fat, applied topically or taken orally for fractures, cleaning of the uterus by pregnant women, and as an antibacterial. In addition, some South African diviners used these plants for their neurogenic properties.4 The roots are used to treat diarrhea, colic, dysentery, and stomach spasms.Parts of C. mimosoides are known to be an important source of secondary metabolites, including anthraquinones. Physcion, chrysophanol, 1,8-dihydroxy-6-methoxy-2-methyl anthraquinone, 1,8-dihydroxy-6-methoxy-3-methyl anthraquinone, and emodin have been reported from the aerial part.5 Luteolin, emodin, 1,3-benzenediol, oleanolic acid, (R)-artabotriol, α-L-rhamnose, β-sitosterol, and daucosterol were isolated from an ethanolic extract of C. mimosoides.6 In addition to phenolic compounds and their derivatives, phytochemical screening of the latter revealed the presence of alkaloids, steroids, saponins, carbohydrates, tannins, glycosides, proteins, and amino acids.7
Ureases are common metalloenzymes made by bacteria, fungi, and plants but not by animals. They quickly catalyze the hydrolysis of urea to produce ammonia and carbamate, followed by the urea's breakdown into a second ammonia molecule and carbon dioxide. By giving bacteria nitrogen in the form of ammonia for their growth, the enzyme plays a critical part in their pathogenicity. The pathogenicity of gastrointestinal disorders such as gastritis, duodenal, peptic ulcer, and gastric cancer is significantly influenced by Helicobacter pylori's ureolytic activity. Ammonia produced by ureases is responsible for human and animal cases of hepatic encephalopathy, hepatic coma, urolithiasis, pyelonephritis, and urinary catheter encrustation. In light of this, urease inhibitors have garnered a lot of interest as potential treatments for infections brought on and facilitated by ureolytic activity. Several urease inhibitors, such as fluorofamide, hydroxyureas, and hydroxamic acids have been reported in the past. Some of these inhibitors have, however, been banned from usage in vivo because of their instability or toxicity. Active metabolites found in plants are well known to help treat a variety of viral disorders. To address concerns about toxicity right away, a lot of emphasis has been paid to examining the unique biological features of phytochemicals extracted from food plants. It has been demonstrated that many medicinal plants, herbs, extracts, and isolated substances have anti-urease properties.
Staphylococcus aureus is an important nosocomial and community-acquired facultative intracellular pathogen. Staphylococcus aureus, notorious for causing bacterial infections in liver transplant recipients, often leads to intestinal symptoms.8 Meanwhile, Helicobacter pylori, a culprit behind peptic ulcers and gastritis, is suspected of playing a role in hepatitis, gallstones, and even hepatobiliary tumors. Helicobacter pylori's ureolytic activity is a game-changer. It significantly impacts gastrointestinal disorders like gastritis, peptic ulcers, and gastric cancer, due to the ammonia it produces. This ammonia also contributes to health problems such as hepatic encephalopathy, hepatic coma, and more.9,10
There is therefore a need to identify alternative, safe and less expensive inhibitors for the treatment of these various conditions that undermine the health of populations. In this intricate web of health challenges, the search for novel, affordable, and less toxic treatments is paramount. Medicinal plants like Cassia mimosoides, with their arsenal of compounds, hold the potential to revolutionize healthcare, addressing conditions that continue to undermine the well-being of populations. These isolated compounds from nature may just be the key to unlocking safer and more effective treatments for hepatic diseases and beyond.
2. Results and discussion
Fifteen compounds were extracted from the ethyl acetate and chloroform fractions of C. mimosoides. These compounds underwent purification through column chromatography and HPLC techniques. The resulting pure compounds were then analyzed using 1H and 13C NMR to determine their chemical structures. Characterization of isolated compounds. Compound 1, obtained as a white powder, exhibited in the SAS-RS-APCI m/Z 425.42 [M + H]+ (positive-ion mode) a molecular formula of C30H49O.All proton and carbon signals of 1 were assigned on the basis of 1H NMR, 1H–1H COSY, HSQC and HMBC experiments (Table 1). The 1H NMR spectrum of compound 1 (Table) exhibited in the up field region signals of two tertiary methyl resonating at δH 0.53 (Me-18) and 0.81 (Me-19), and four secondary methyl at δH 1.59 (3H; d; J = 6.8 Hz; Me-29), 0.98 (3H; d; J = 6.9 Hz; Me-27), 0.97 (3H; d; J = 6.5 Hz; Me-26) and at δH 0.99 (3H; d; J = 6.2 Hz; Me-30). Signals of two olefinic protons were also observed at δH 5.17 (1H; dd; J = 5.9; 2.3 Hz; H-7) and 5.09 (1H; d; J = 6.9 Hz; H-28). In addition, the signals of two methylene protons were observed in this spectrum at δH 4.72 (1H; s; H-21a) and 4.66 (1H; t; J = 1.7 Hz; H-21b). The 13C NMR spectrum of compound 1 combined to the HMBC spectrum showed a set of 30 carbons. The most interesting resonances were those of olefinic carbons depicted at δC 117.6 (C-6), 139.1 (C-8), 116.4 (C-28), 148.5 (C-24) and those of the methylene carbons at δC 105.8 (C-21), 156.8 (C-20). The backbone of compound 2 was identified as citrostadienol and all the 1H and 13C NMR spectral data were in good agreement with literature values (Rondet et al., 1999; Schaller, 2010; X. Zhang et al., 2006).11–13 Resonances of two methylene protons at δH 4.72 (1H; s; H-21a) and 4.66 (1H; t; J = 1.7 Hz; H-21b) giving HSQC correlations with the carbon at δC 105.8 (C-21) were also observed. The HMBC correlations observed from the protons at δH 0.97 (Me-26) and 1.59 (Me-29) to the carbons at δC 116.4 (C-28), and 148.5 (C-24) as well as the 1H–1H COSY correlation between the protons at δH 5.17 (H-7) and 2.12 (H-6) allowed us to locate the positions of the double bonds 8 (Fig. 1). Thus, the structure of 1 was established as 21-methylene-24-ethylidene lophenol, a previously unreported avenasterol-type phytosterols to which we gave the trivial name 21citrostadienol.
| Positions | δ 13C | δ 1H (mult, J) |
|---|---|---|
| 1 | 36.9 | 1.84 (m, 1H) |
| 1.81 (m, 1H) | ||
| 2 | 30.9 | 1.80 (m, 1H) |
| 1.46 (m, 1H) | ||
| 3 | 76.2 | 3.12 (m, 1H) |
| 4 | 40.2 | 1.30 (m, 1H) |
| 5 | 46.6 | 1.01 (m, 1H) |
| 6 | 26.6 | 2.13 (m, 1H) |
| 2.09 (m, 1H) | ||
| 7 | 117.6 | 5.17(dt, J = 5.9, 2.3 Hz, 1H) |
| 8 | 139.1 | — |
| 9 | 49.6 | 1.64 (m, 1H) |
| 10 | 34.8 | — |
| 11 | 21.3 | 1.55 (m, 1H) |
| 1.45 (m, 1H) | ||
| 12 | 39.4 | 2.02 (m, 1H) |
| 1.22 (m, 1H) | ||
| 13 | 43.3 | — |
| 14 | 54.9 | 1.80 (m, 1H) |
| 15 | 22.8 | 1.51 (m, 1H) |
| 1.25 (m, 1H) | ||
| 16 | 29.6 | 1.25 (m, 1H) |
| 17 | 55.9 | 1.22 (m, 1H) |
| 18 | 11.8 | 0.53 (s, 3H) |
| 19 | 14.1 | 0.83 (s, 3H) |
| 20 | 156.8 | — |
| 21 | 105.8 | 4.72 (s, 1H) |
| 4.66 (t, J = 1.7 Hz) | ||
| 22 | 29.6 | 1.25 (m, 1H) |
| 23 | 27.9 | 1.91 (m, 1H) |
| 1.27 (m, 1H) | ||
| 24 | 145.8 | — |
| 25 | 28.5 | 2.85 (m, 1H) |
| 26 | 21.0 | 0.97 (d, J = 6.5 Hz, 3H) |
| 27 | 20.9 | 0.98 (d, J = 6.9 Hz, 3H) |
| 28 | 116.4 | 5.09 (q, J = 6.9 Hz, 1H) |
| 29 | 12.7 | 1.59 (d, J = 6.8 Hz, 3H) |
| 30 | 15.1 | 0.99 (d, J = 6.2 Hz, 3H) |
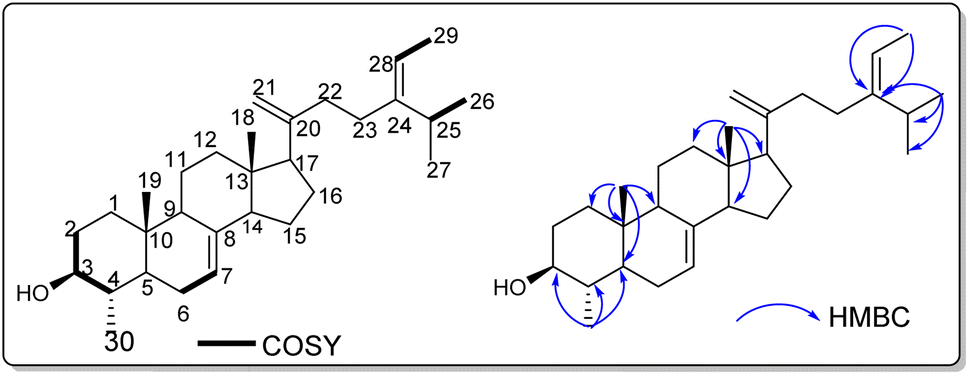 | ||
| Fig. 1 1H–1H COSY and HMBC correlations of compound 1. | ||
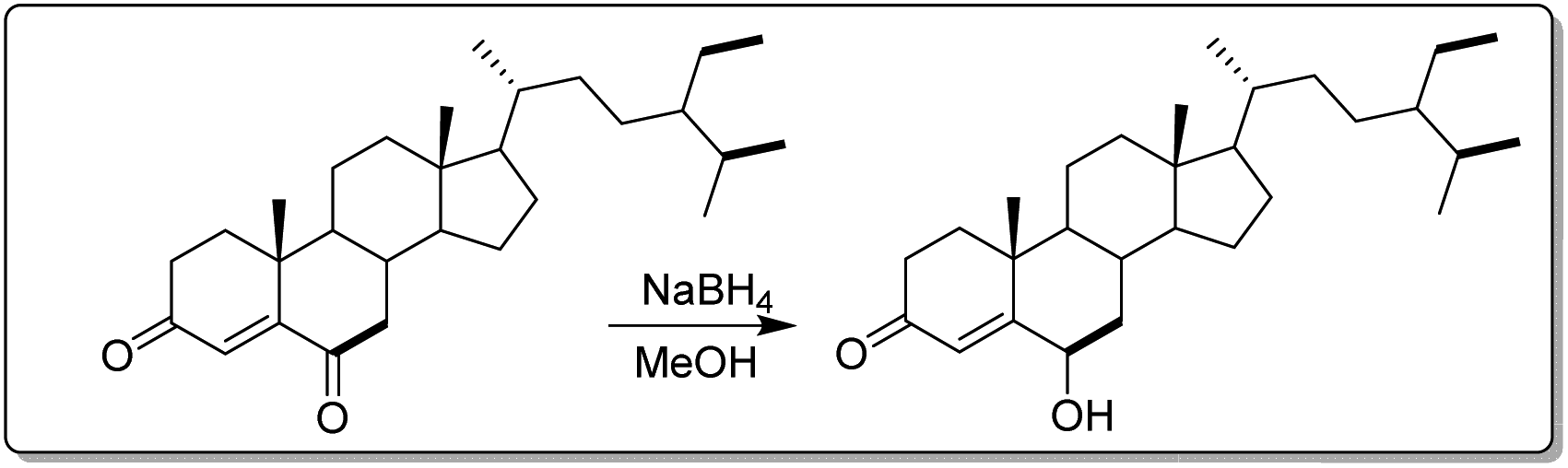 | ||
| Fig. 2 Synthetic scheme semi-derivative of stigmast-4-en-3,6-dione. | ||
2.1. Luteolin1
Yellow powder, 1H NMR (600 MHz, Acetone-d6) δ (ppm): 6.59 (1H, s, H-3), 12.99 (1H, s, OH-5), 6.26 (1H, d, J = 2.1 Hz, H-6), 6.53 (1H, d, J = 2.3 Hz, H-8), 7.47 (1H, dd, J = 8.4, 2.2 Hz, H-2′), 7.01 (1H, d, J = 8.4 Hz, H-3′), 7.50 (1H, d, J = 2.2 Hz, H-6′). 13C NMR (150 MHz, Acetone-d6) δ (ppm): 164.3 (C-2), 103.1 (C-3), 182.2 (C-4), 162.3 (C-5), 98.8 (C-6), 164.2 (C-7), 93.8 (C-8), 157.8 (C-9), 104.3 (C-10), 122.6 (C-1′), 119.1 (C-2′), 115.6 (C-3′), 149.4 (C-4′), 145.7 (C-5′), 113.0 (C-6′).2.2. 3′,4′,7-Trihydroxyflavone2
Yellow powder, 1H NMR (600 MHz, Acetone-d6) δ (ppm): 6.64 (1H, s, H-3), 7.62 (1H, d, J = 8.4 Hz, H-5), 6.79 (1H, dd, J = 8.4, 2.0 Hz, H-6), 6.83 (1H, d, J = 2.0 Hz, H-8), 7.35 (1H, dd, J = 8.3, 2.1 Hz, H-2′), 6.95 (1H, d, J = 8.2 Hz, H-3′), 7.58 (1H, d, J = 2.1 Hz, H-6′). 13C NMR (150 MHz, Acetone-d6) δ (ppm): 146.3 (C-2), 111.3 (C-3), 181.5 (C-4), 125.5 (C-5), 114.0 (C-6), 168.0 (C-7), 98.5 (C-8), 165.7 (C-9), 112.5 (C-10), 117.7 (C-1′), 124.7 (C-2′), 115.6 (C-3′), 147.3 (C-4′), 145.2 (C-5′), 124.5 (C-6′).2.3. Luteolin-5-methyl ether3
Yellow powder, 1H NMR (400 MHz, CD3OD) δ (ppm): 6.47 (1H, s, H-3), 6.40 (1H, d, J = 2.2 Hz, H-6), 6.53 (1H, d, J = 2.1 Hz, H-8), 7.33 (1H, dd, J = 8.9, 2.3 Hz, H-2′), 6.89 (1H, d, J = 8.9 Hz, H-3′), 7.30 (1H, d, J = 2.3 Hz, H-6′), 3.88 (3H, s, OCH3-5). 13C NMR (100 MHz, CD3OD) δ (ppm): 162.4 (C-2), 105.1 (C-3), 178.8 (C-4), 161.0 (C-5), 96.1 (C-6), 163.6 (C-7), 94.9 (C-8), 159.8 (C-9), 106.8 (C-10), 122.2 (C-1′), 118.5 (C-2′), 115.3 (C-3′), 149.1 (C-4′), 145.6 (C-5′), 112.5 (C-6′), 55.0 (OCH3-5).2.4. Apigenin-8-C-β-D-glucopyranoside1
Yellow powder, 1H NMR (400 MHz, DMSO-d6) δ (ppm): 6.77 (1H, s, H-3), 13.17 (1H, s, OH-5), 6.26 (1H, s, H-6), 8.02 (2H, d, J = 8.02, H-2′/H-6′), 6.88 (2H, d, J = 8.02 Hz, H-3′/H-5′), 4.67 (1H, dd, J = 9.8, 3.4 Hz, H-1′′), 3.82 (1H, dd, J = 9.8, 3.4 Hz, H-2′′), 3.25 (1H, m, H-3′′), 3.36 (1H, m, H-4′′), 3.23 (1H, m, H-5′′), 3.76 (1H, d, J = 12.1 Hz, H-6a′′), 3.52 (1H, d, J = 6.4 Hz, H-6b''). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 164.6 (C-2), 102.9 (C-3), 182.5 (C-4), 161.0 (C-5), 98.6 (C-6), 163.1 (C-7), 105.2 (C-8), 156.7 (C-9), 104.6 (C-10), 122.6 (C-1′), 129.4 (C-2'/C-6′), 116.2 (C-3′/C-5′), 73.9 (C-1′′), 71.2 (C-2′′), 79.1 (C-3′′), 70.9 (C-4′′), 82.3 (C-5′′), 61.7 (C-6′′).2.5. Luteolin-8-C-β-D-glucopyranoside (orientin)1
Yellow powder, 1H NMR (400 MHz, DMSO-d6) δ (ppm): 6.64 (1H, s, H-3), 13.17 (1H, s, OH-5), 6.25 (1H, s, H-6), 7.52 (1H, dd, J = 8.5, 2.2 Hz, H-2′), 6.87 (1H, d, J = 8.4 Hz, H-3′), 7.46 (1H, d, J = 2.2 Hz, H-6′), 4.67 (1H, dd, J = 9.8, 3.4 Hz, H-1′′), 3.83 (1H, m, H-2′′), 3.24 (1H, m, H-3′′), 3.36 (1H, m, H-4′′), 3.28 (1H, m, H-5′′), 3.77 (1H, m, H-6a′′), 3.53 (1H, m, H-6b′′). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 164.6 (C-2), 102.8 (C-3), 182.4 (C-4), 160.7 (C-5), 98.6 (C-6), 162.9 (C-7), 104.9 (C-8), 156.4 (C-9), 104.4 (C-10), 122.3 (C-1′), 119.8 (C-2′), 116.0 (C-3′), 150.1 (C-4′), 146.2 (C-5′), 114.4 (C-6′), 73.8 (C-1′′), 71.2 (C-2′′), 79.1 (C-3′′), 71.0 (C-4′′), 82.3 (C-5′′), 61.7 (C-6′′).2.6. Luteolin-6-C-β-D-glucopyranoside14
Yellow powder, 1H NMR (400 MHz, DMSO-d6) δ (ppm): 6.70 (1H, s, H-3), 13.58 (1H, s, OH-5), 6.50 (1H, s, H-8), 7.44 (1H, dd, J = 8.2, 2.3 Hz, H-2′), 6.87 (1H, d, J = 8.3 Hz, H-3′), 7.42 (1H, d, J = 2.3 Hz, H-6′), 4.60 (1H, d, J = 9.8 Hz, H-1′′), 4.07 (1H, d, J = 9.2 Hz, H-1′′), 3.21 (1H, m, H-3′′), 3.14 (1H, m, H-4′′), 3.18 (1H, m, H-5′′), 3.70 (1H, m, H-6a′′), 3.42 (1H, m, H-6b''). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 164.0 (C-2), 103.7 (C-3), 182.3 (C-4), 161.2 (C-5), 109.3 (C-6), 163.7 (C-7), 93.9 (C-8), 156.7 (C-9), 103.9 (C-10), 121.9 (C-1′), 116.4 (C-2′), 116.0 (C-3′), 150.1 (C-4′), 146.2 (C-5′), 114.4 (C-6′), 73.4 (C-1′′), 70.6 (C-2′′), 79.4 (C-3′′), 71.0 (C-4′′), 82.0 (C-5′′), 61.7 (C-6′′).2.7. Butin4
An orange powder, 1H NMR (600 MHz, Acetone-d6) δ (ppm): 5.40 (1H, dd, J = 12.8, 2.9 Hz, H-2), 3.02 (1H, dd, J = 16.7, 12.8 Hz, H-3a), 2.68 (1H, dd, J = 16.7, 3.0 Hz, H-3b), 7.73 (1H, d, J = 8.6 Hz, H-5), 6.58 (1H, dd, J = 8.6, 2.3 Hz, H-6), 6.43 (1H, d, J = 2.3 Hz, H-8), 7.05 (1H, d, J = 1.9 Hz, H-2′), 6.87 (1H, d, J = 8.1 Hz, H-5′), 6.89 (1H, dd, J = 8.2, 1.9 Hz, H-6′). 13C NMR (150 MHz, Acetone-d6) δ (ppm): 79.6 (C-2), 43.8 (C-3), 189.6 (C-4), 128.5 (C-5), 110.2 (C-6), 163.5 (C-7), 102.7 (C-8), 163.2 (C-9), 114.3 (C-10), 131.2 (C-1′), 113.7 (C-2′), 145.6 (C-3′), 145.6 (C-4′), 114.7 (C-5′), 118.2 (C-6′).2.8. Stigmast-4-en-3,6-dione5
White powder, 1H NMR (600 MHz, C5D5N) δ (ppm): 1.69 (1H, m, H-1a), 1.52 (1H, m, H-1b), 1.78 (1H, m, H-2a), 1.39 (1H, m, H-2b), 6.41 (1H, s, H-4), 2.67 (1H, dd, J = 16.1, 4.4 Hz, H-7a), 2.05 (1H, dd, J = 16.1, 12.3 Hz, H-7b), 1,78 (1H, m, H-8), 1.24 (1H, m, H-9), 1.48 (2H, m, H-11), 2.00 (1H, m, H-12a), 1,15 (1H, m, H-12b), 1.02 (1H, m, H-14), 1.51 (1H, m, H-15a), 1.31 (1H, m, H-15b), 1.83 (2H, m, H-16), 1.03 (1H, m, H-17), 0.68 (3H, s, H-18), 1.00 (3H, s, H-19), 1.38 (1H, m, H-20), 0.98 (3H, d, J = 6.3 Hz, H-21), 2.39 (1H, m, H-22a), 1,05 (1H, m, H-22b), 1.24 (2H, m, H-23), 1.49 (1H, m, H-24), 1.68 (1H, m, H-25), 0.88 (3H, d, J = 6.1 Hz, H-26), 0.85 (3H, d, J = 6.8 Hz, H-27), 1.31 (2H, m, H-28), 0.90 (3H, t, H-29). 13C NMR (150 MHz, C5D5N) δ (ppm): 35.1 (C-1), 33.9 (C-2), 198.9 (C-3), 125.3 (C-4), 160.8 (C-5), 201.6 (C-6), 46.5 (C-7), 39.1 (C-8), 50.4 (C-9), 34.0 (C-10), 20.7 (C-11), 39.5 (C-12), 42.4 (C-13), 55.8 (C-14), 23.1 (C-15), 28.1 (C-16), 56.2 (C-17), 11.7 (C-18), 16.9 (C-19), 36.0 (C-20), 18.6 (C-21), 33.8 (C-22), 26.1 (C-23), 45.8 (C-24), 29.2 (C-25), 19.3 (C-26), 18.9 (C-27), 23.1 (C-28), 11.9 (C-29).2.9. Stigmast-4-en-3β,6α-diol (Zhao et al.,15 2005)
White powder, 1H NMR (400 MHz, Acetone-d6) δ (ppm): 1.73 (1H, m, H-1a), 1.69 (1H, m, H-1b), 1.40 (1H, m, H-2a), 1.27 (1H, m, H-2b), 4.08 (1H, m, H-3), 5.74 (1H, d, J = 1.7 Hz, H-4), 4.11 (1H, m, H-6), 2.02 (2H, m, H-7), 1,30 (1H, m, H-8), 0.72 (1H, m, H-9), 1.50 (1H, m, H-11a), 1.38 (1H, m, H-11b), 2.05 (1H, m, H-12a), 1,16 (1H, m, H-12b), 1.17 (1H, m, H-14), 1.23 (2H, m, H-15), 1.69 (2H, m, H-16), 1.05 (1H, m, H-17), 1.05 (3H, s, H-18), 0.74 (3H, s, H-19), 1.42 (1H, m, H-20), 0.96 (3H, d, J = 6.4 Hz, H-21), 1.67 (1H, m, H-22a), 1,40 (1H, m, H-22b), 1.92 (2H, m, H-23), 0.98 (1H, m, H-24), 1.70 (1H, m, H-25), 0.88 (3H, m, H-26), 0.84 (3H, m, H-27), 1.30 (2H, m, H-28), 0.88 (3H, m, H-29). 13C NMR (100 MHz, Acetone-d6) δ (ppm): 36.4 (C-1), 34.3 (C-2), 67.4 (C-3), 121.5 (C-4), 147.5 (C-5), 67.0 (C-6), 42.5 (C-7), 29.5 (C-8), 54.5 (C-9), 37.4 (C-10), 20.8 (C-11), 39.7 (C-12), 42.4 (C-13), 56.1 (C-14), 25.8 (C-15), 24.0 (C-16), 56.0 (C-17), 19.2 (C-18), 11.4 (C-19), 36.0 (C-20), 18.2 (C-21), 33.7 (C-22), 28.0 (C-23), 45.8 (C-24), 29.1 (C-25), 19.2 (C-26), 18.4 (C-27), 22.8 (C-28), 11.3 (C-29).2.10. Emodin6
Orange powder, 1H NMR (600 MHz, Acetone-d6) δ (ppm): 12.10 (1H, s, OH-1), 7.15 (1H, s, H-2), 7.58 (1H, d, J = 1.6 Hz, H-4), 7.26 (1H, d, J = 2.4 Hz, H-5), 6.67 (1H, d, J = 2.4 Hz, H-7), 12.20 (1H, s, OH-8), 2.48 (3H, s, H-1′). 13C NMR (150 MHz, Acetone-d6) δ (ppm): 165.4 (C-1), 124.0 (C-2), 148.6 (C-3), 120.5 (C-4), 133.3 (C-4a), 108.9 (C-5), 162.3 (C-6), 107.9 (C-7), 165.9 (C-8), 109.1 (C-8a), 190.7 (C-9), 113.6 (C-9a), 181.4 (C-10), 135.6 (C-10a), 21.0 (C-1′).2.11. Chrysophanol7
Orange powder, 1H NMR (600 MHz, CDCl3) δ (ppm): 12.10 (1H, s, OH-1), 7.12 (1H, m, H-2), 7.67 (1H, d, J = 1.8 Hz, H-4), 7.84 (1H, dd, J = 7.5, 1.2 Hz, H-5), 7.69 (1H, d, J = 8.0 Hz, H-6), 7.31 (1H, dd, J = 8.5, 1.2 Hz, H-7), 12.03 (1H, s, OH-8), 2.49 (3H, s, H-1′). 13C NMR (150 MHz, CDCl3) δ (ppm): 162.4 (C-1), 124.5 (C-2), 149.3 (C-3), 121.3 (C-4), 133.2 (C-4a), 119.9 (C-5), 136.9 (C-6), 124.3 (C-7), 162.7 (C-8), 115.8 (C-8a), 192.5 (C-9), 113.7 (C-9a), 182.0 (C-10), 133.6 (C-10a), 22.2 (C-1′).2.12. Physcion6
Orange powder, 1H NMR (400 MHz, CDCl3) δ (ppm): 12.13 (1H, s, OH-1), 7.09 (1H, d, J = 1.5 Hz, H-2), 7.63 (1H, d, J = 1.7 Hz, H-4), 7.37 (1H, d, J = 2.6 Hz, H-5), 6.69 (1H, d, J = 2.5 Hz, H-7), 12.33 (1H, s, OH-8), 2.46 (3H, s, H-1′), 3.94 (3H, s, H-1′′). 13C NMR (100 MHz, CDCl3) δ (ppm): 162.5 (C-1), 124.5 (C-2), 148.4 (C-3), 121.3 (C-4), 133.2 (C-4a), 108.2 (C-5), 166.5 (C-6), 106.7 (C-7), 165.2 (C-8), 110.2 (C-8a), 190.8 (C-9), 113.7 (C-9a), 181.8 (C-10), 135.2 (C-10a), 22.1 (C-1′), 56.1 (C-1′′) (Fig. 3).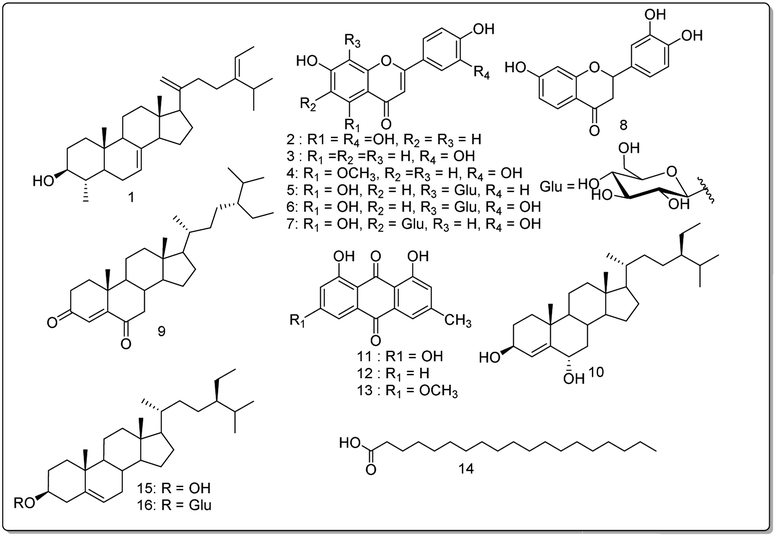 | ||
| Fig. 3 Structures of compounds (1–16) isolated from Cassia mimosoïdes. | ||
These compounds were tested for their antimicrobial activity, especially against bacteria that cause gastrointestinal infections. The study also evaluates their impact on H. pylori, a bacterium associated with ulcers, and identifies potential benefits through the inhibition of urease, a critical enzyme linked to ulcer development. Molecular docking analysis confirms the structure–activity relationship, demonstrating robust urease inhibition through various interactions, surpassing the standard thiourea. Furthermore, the compounds undergo testing on Hep G2 cells, indicating potential positive effects on liver function, given that H. pylori can disrupt liver function. These compounds offer targeted solutions for gastrointestinal and liver issues associated with H. pylori infection. These findings suggest that further optimization could lead to the development of these compounds as lead candidates for therapeutic purposes. The results follow as under.
2.13. In vitro analysis
| Sample | Alternaria alternata | Aspergillus fumigatus | Aspergillus niger | Penicillium citrii |
|---|---|---|---|---|
| Luteolin | 0.043 ± 0.002 | 0.01 ± 0.004 | 0.021 ± 0.001 | 0.05 ± 0.002 |
| 3′,4′,7-Trihydroxy flavone | 0.056 ± 0.001 | 0.013 ± 0.003 | 0.032 ± 0.005 | 0.016 ± 0.01 |
| Luteolin-5-methyl ether | 0.03 ± 0.006 | 0.04 ± 0.002 | 0.01 ± 0.006 | 0.06 ± 0.004 |
| Apigenin-8-C-β-D-glucopyranoside | 0.025 ± 0.007 | 0.01 ± 0.006 | 0.015 ± 0.001 | 0.01 ± 0.006 |
| Luteolin-8-C-β-D-glucopyranoside | 0.021 ± 0.005 | 0.014 ± 0.005 | 0.013 ± 0.006 | 0.015 ± 0.008 |
| Luteolin-6-C-β-D-glucopyranoside | 0.06 ± 0.007 | 0.25 ± 0.003 | 0.0072 ± 0.0003 | 0.022 ± 0.003 |
| Butin | 0.02 ± 0.006 | 0.017 ± 0.005 | 0.005 ± 0.0003 | 0.10 ± 0.003 |
| Stigmast-4-en-3,6-dione | 0.15 ± 0.005 | 0.125 ± 0.0015 | 0.02 ± 0.006 | 0.04 ± 0.003 |
| Stigmast-4-en-3β,6α-diol | 0.045 ± 0.003 | 0.01 ± 0.006 | 0.015 ± 0.005 | 0.20 ± 0.03 |
| Emodin | 0.01 ± 0.006 | 0.035 ± 0.005 | 0.04 ± 0.003 | 0.045 ± 0.005 |
| Chrysophanol | 0.012 ± 0.003 | 0.06 ± 0.001 | 0.03 ± 0.002 | 0.05 ± 0.001 |
| Physcion | 0.015 ± 0.005 | 0.035 ± 0.006 | 0.09 ± 0.009 | 0.03 ± 0.005 |
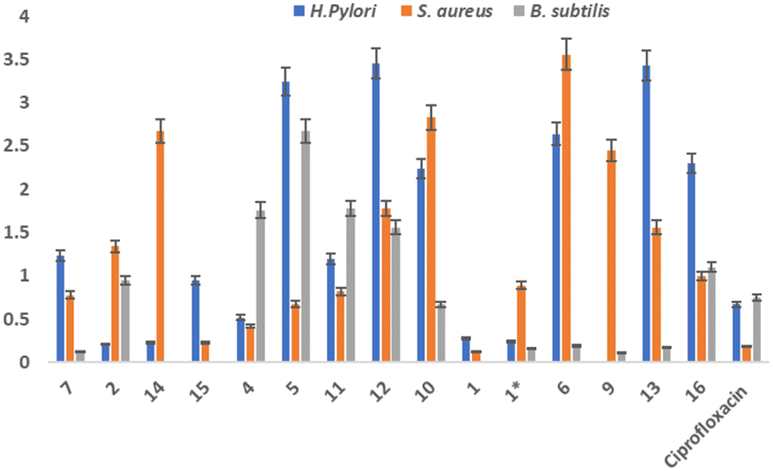 | ||
| Fig. 4 Antibacterial activity of isolated compounds against bacterial strains. H.pylori, B.subtilis and S.aureus. (1* = synthesized compound). | ||
| Compounds | % Inhibition | IC50 ± SEM (μM) |
|---|---|---|
| 7 | 81.57 | 1.22 ± 0.43 |
| 2 | 82.05 | 1.30 ± 0.28 |
| 14 | 80.42 | 1.95 ± 0.31 |
| 15 | 80.00 | 2.78 ± 0.24 |
| 4 | 82.73 | 3.87 ± 0.24 |
| 5 | 77.52 | 4.73 ± 0.22 |
| 11 | 72.89 | 5.20 ± 0.14 |
| 12 | 78.21 | 5.92 ± 0.25 |
| 10 | 76.57 | 6.68 ± 0.11 |
| 1 | 82.32 | 1.02 ± 0.35 |
| Synthesized compound | 69 | 8.03 ± 0.19 |
| 6 | 71.52 | 5.03 ± 0.12 |
| 9 | 13.42 | — |
| 13 | 63.47 | 6.24 ± 0.30 |
| 16 | 19.47 | — |
| Standard (thiourea) | 86.73 | 18.61 ± 0.11 |
The enzyme kinetic analysis focused on the most potent inhibitor identified, compound 1, against urease. The kinetic assay involved preparing varying concentrations of both the test compound and the substrate (urea), spanning 0 μM, 5 μM, 10 μM, 15 μM, and 20 μM. The resulting data was utilized to construct a Lineweaver–Burk plot. The Lineweaver–Burk plot revealed a competitive mode of inhibition for compound 1 (Fig. 5). This observation suggests a scenario where both the substrate and compound 1 compete for the enzyme's active site. This competitive relationship signifies that compound 1 effectively occupies the enzyme's active pocket, which aligns with the outcomes of the molecular docking study that also indicated a robust binding of compound 1 within the enzyme's active site.
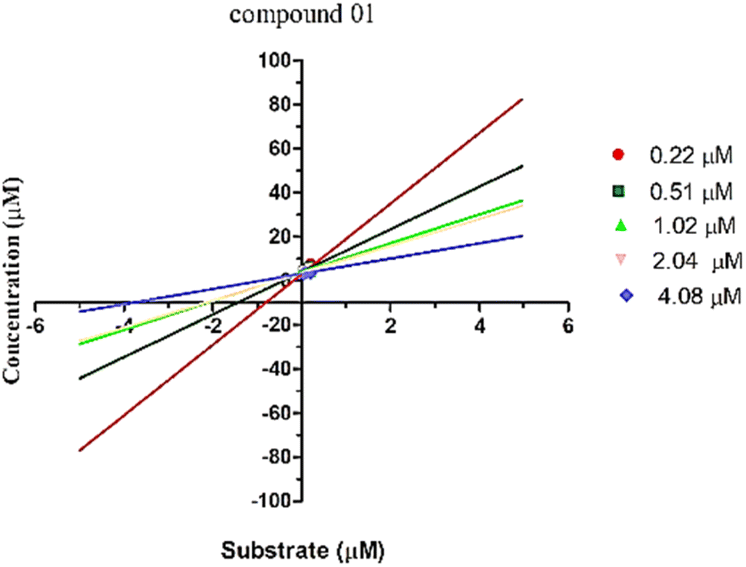 | ||
| Fig. 5 Lineweaver–Burk plot for the compound 1. | ||
Structure–activity relationship (SAR) analysis proves invaluable in discerning the structural attributes pivotal for the efficacy of the isolated compounds and their inhibitory potential against the target enzyme (Fig. 6). Examination of the inhibitory outcomes (as presented in Table 3) discloses that among the entire array of compounds studied, compounds 1, 2, and 7 exhibit the most potent inhibition, showcasing impressive IC50 values of 1.02 μM, 1.30 μM, and 1.22 μM, respectively. These compounds stand out due to the presence of olefinic, alkyl, and hydroxyl groups, which contribute to heightened lipophilicity and hydrophilicity. An in-depth SAR analysis of compound 1 reveals that the substitution of 5-isopropyl-2-methylhepta-1,5-diene at ring D (a five-membered ring) emerges as a crucial determinant of biological activity. This substitution notably enhances lipophilicity, thereby exerting a substantial impact on the observed inhibition. Additionally, hydroxyl and methyl moieties at the juncture of ring A, B, and ring C, D, along with those at ring A, play pivotal roles in the urease inhibition process.
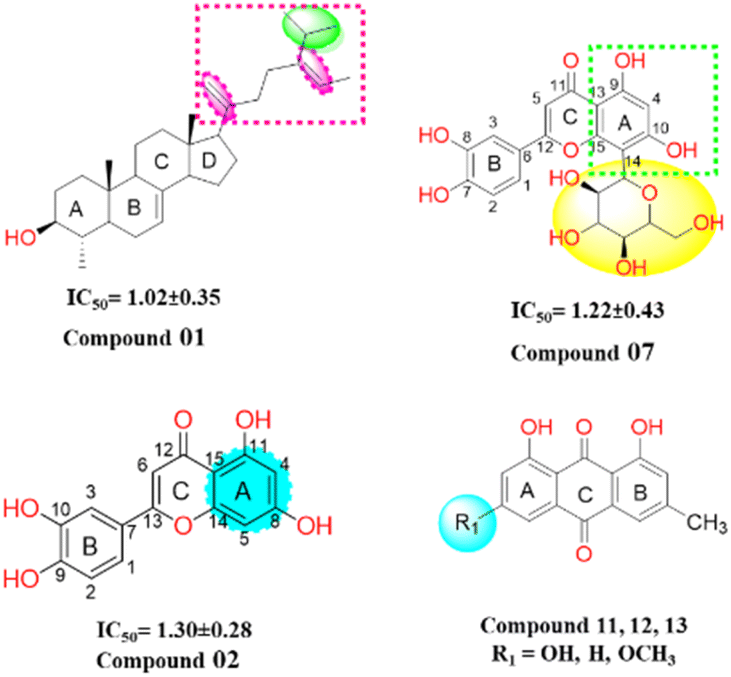 | ||
| Fig. 6 Structure of potent compounds with their IC50 -values. | ||
In compound 2, the introduction of hydroxyl groups at positions 8 and 11 in ring A, and positions 9 and 10 in ring B, leads to a notable enhancement in inhibitory activity against the urease enzyme. This effect is attributed to the electron-withdrawing nature of the hydroxyl group from the benzene moieties, which increases the compounds' hydrophilicity by facilitating hydrogen bonding interactions. Compound 7 similarly exhibits effective inhibition, displaying even greater potency compared to compound 2. This increased potency can be attributed to the substitution of the sugar moiety at position (C 14) of the flavonoid structure. Among the remaining compounds (14, 15, 4, 5, 11, 12, 10, 6, and 13), a range of good to moderate inhibition is observed, with corresponding IC50 values of 1.95, 2.78, 3.87, 4.73, 5.20, 5.92, 6.68, 5.03, and 6.24 μM, respectively. However, compounds 9 and 16 exhibit weaker inhibition against the urease enzyme, with inhibition percentages of 13.42% and 19.47%, respectively. Notably, in anthraquinones (compounds 11, 12, and 13), the presence of a hydroxyl or a methoxy group on the A ring diminishes their inhibitory potency. In the case of steroids, the inhibitory efficacy is contingent upon the steroid skeleton's nature and its specific substitutions. For instance, the substitution of a glucose unit for a hydrogen in sterol (compounds 15 and 16) significantly reduces its inhibitory potential against the urease enzyme. Furthermore, intriguing insights emerge from the reduction of the ketone function in compound 9 to an alcohol (compound 10), which substantially enhances its inhibitory activity. These findings are reinforced by in vitro docking studies, where all isolated compounds displayed strong binding energies in the active site, particularly the potent inhibitors. Moreover, the kinetic studies of the novel isolated compound affirmed a competitive mode of inhibition, adding further depth to our understanding of its inhibitory mechanism.
2.14 Molecular docking studies
We performed molecular docking study to evaluate the tendency of compounds identified from C. mimosides as potential inhibitor for urease enzyme. The protein (PDB ID: 3LA4) and ligands prepared as described in Materials and methods were docked into the active site of the urease enzyme. The result of this calculation is shown in Table 4, while the docking results are shown in Fig. 7, and S9.† The docking analysis results have provided valuable insights into the binding affinities and interactions between specific compounds and the 3LA4 protein. Among the compounds tested, 16, 6, 5, 1, and 7 demonstrated the highest binding affinities, with energy values of −7.43, −7.68, −8.31, −8.79, and −6.79 kcal mol−1, respectively. Conversely, compound 4 exhibited the weakest binding interaction, with a binding affinity of −7.84 kcal mol−1, indicating a lower likelihood of effective binding. The majority of protein–ligand complexes displayed the presence of hydrogen bonds, which play a pivotal role in stabilizing these interactions. However, some compounds, specifically 1, 8, 9, and 13, did not form hydrogen bonds with certain key amino acid residues, including Ala80, Asp295, Asp730, Glu34, Glu742, Gly714, Gly641, Ile148, Phe838, Ser421, and Tyr309. This absence of hydrogen bonds in these cases might influence the overall binding and stability of the complexes.| Compound name | Binding score (kcal mol−1) | RMSD (Å) |
|---|---|---|
| Luteolin-6-C-β-D-glucopyranoside (7) | −6.79 | 1.49 |
| Luteolin-8-C-β-D-glucopyranoside (6) | −7.68 | 1.80 |
| Apigenin-8-C-β-D-glucopyranoside (5) | −8.31 | 2.09 |
| Nonadecanoic acid (14) | −8.47 | 1.88 |
| Luteolin-5-methyl ether (4) | −7.84 | 1.53 |
| Luteolin (2) | −8.61 | 0.99 |
| β-Sitosterol 3-O-β-D-glucopyranoside (16) | −7.43 | 1.84 |
| Butin (8) | −7.38 | 1.67 |
| β-Sitosterol (15) | −6.97 | 1.37 |
| Stigmat-4-en-3,6-dione (9) | −6.19 | 1.26 |
| 21-Methylene-24-ethylidene lophenol (1) | −8.79 | 1.19 |
| Physine (13) | −5.84 | 1.45 |
| Emodin (11) | −5.59 | 1.84 |
| Chrysophanol (12) | −5.51 | 1.71 |
| 3′,4′,7-Trihydroxyflavone (3) | −8.49 | 1.32 |
| Stigmast-4-en-3β,6α-diol (10) | −7.05 | 1.72 |
| Reference (thiourea) | −3.13 | 2.15 |
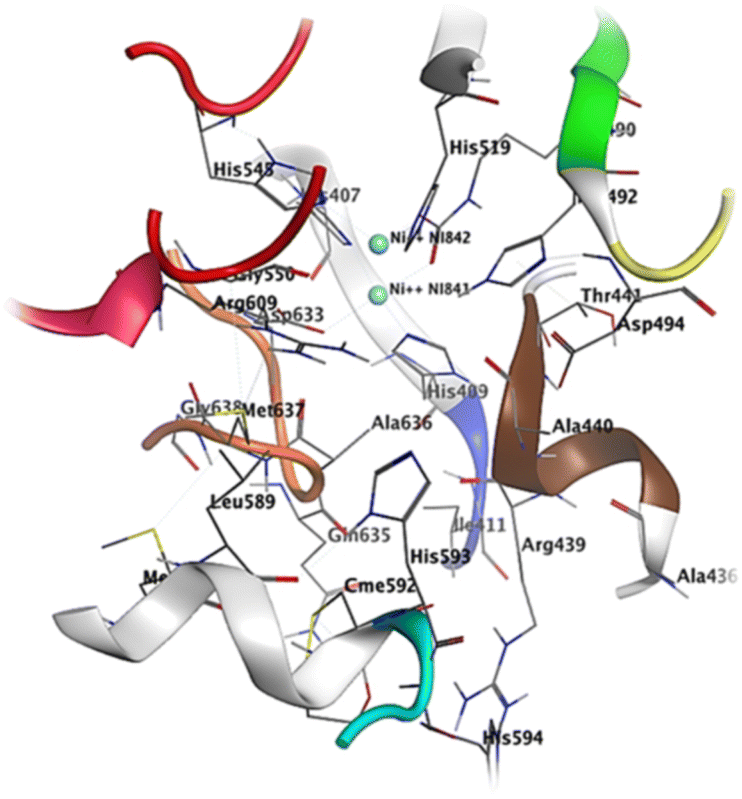 | ||
| Fig. 7 Active site residues including sheet S23 (residues 403–409, color coded silver) a small loop L30 (410–412, blue), helix H14 (435–441 color coded brown), loop L37 (444–454 color coded red), helix H20 (580–593 color coded green), loop L39 (594–598 color coded cyan), helix H21 (599–610 color coded pink) and a loop L41 (632–638 color coded orange). | ||
The crystal structure of urease reveals crucial residues that form the binding pocket, facilitating the retention of inhibitors within the active site. These include a Ni-metal atom, sheet S23 (residues 403–409), a compact loop L30 (410–412, shown in blue), helix H14 (435–441 in a brown color), loop L37 (444–454 in red), helix H20 (580–593 in green), loop L39 (594–598 in cyan), helix H21 (599–610 in pink), and a loop L41 (632–638 in orange). Compound 1 establishes a binding interaction with the urease active site, displaying a binding energy of −8.79 kcal mol−1. Notably, compound 1 possesses alkyl and olefinic moieties that enable interactions with the active pocket, leading to several pi–alkyl interactions observed with key residues in the active site, particularly with His549, Ala436, and Ala440. Compound 2 effectively binds to the urease active site, exhibiting a binding energy of −8.61 kcal mol−1. This interaction involves both nickel atoms within the active site, leading to the formation of a protein–inhibitor complex. Notably, conventional hydrogen bonds are established with amino acids Asp633, Gly550, Ala440, and H14. Furthermore, a π–alkyl interaction occurs between compound 2 and Met637, while a π–π interaction is observed between His519 and the compound. The binding of compound 7 to the urease active site is robust, displaying a binding energy of −6.79 kcal mol−1. This interaction centers around the active Ni-metal atom, facilitated by the hydroxyl group of the sugar moiety. Conventional hydrogen bonds are prominent, involving amino acid residues Arg436, Ala440, Arg609, and Glu493. Additionally, π–cation interactions are established with Met637, His593, and Asp494. Interestingly, a majority of the studied compounds exhibited enhanced accuracy in the docking protocol compared to the reference compound, with root-mean-square deviations (RSMDs) ranging from 0.63 to 1.79 Å. This indicates that the majority of the studied compounds had relatively precise docking predictions, reinforcing the reliability of the docking protocol for evaluating their binding interactions with the 3LA4 protein.
2.15 Drug similarity analyses and ADME studies
Similarity analyses with reference compounds and ADME predictions for the natural compounds studied were performed via the SwissADME web server.8 The results are presented in Tables 1S and 2S† In these analyses, no secondary metabolites studied had more than twelve hydrogen bond acceptors. However, compounds 5, 6 and 7 have the highest number of hydrogen bond acceptors with values between ten and eleven. On the other hand, although compounds 5, 6 and 7 obey the Lipinski rule while possessing more than five hydrogen bond donors. The molecular masses of the natural compounds studied are between 254.24 and 576.85 g mol−1.9,10 The drug character of a given molecule was predicted using topological polar surface area (TPSA).10 From these results, it can be seen that, the polar surface area of the majority of the studied compounds are less than 111.13 Å2.The water solubility of the studied secondary metabolites (ESOL, ALI and SILICOS-IT16) varies from poorly soluble to soluble. The high value of gastrointestinal absorption of compound 12 may therefore imply that it can cross the blood–brain barrier. We were also able to observe a variation in skin permeability −1.91 to −9.14 cm s−1.17 These results also show that all the molecules obeyed the rules of Lipinski (86.66%), Ghose (53.33%), Veber (73.33%), Egan (53.33%) and Muegge (46.66%), and Abbott's bioavailability scores18 were predicted at 0.55 for 80% of the molecules studied.
| Sample | % Viability |
|---|---|
| Chrysophanol (12) | 59.32 ± 2.20 |
| Physine (13) | 87.44 ± 2.55 |
| β-Sitosterol 3-O-β-D-glucopyranoside (16) | 96.40 ± 2.20 |
| Luteolin (2) | 82.46 ± 4.31 |
| β-Sitosterol (15) | 94.44 ± 4.42 |
| Stigmat-4-en-3,6-dione (9) | 31.13 ± 2.68 |
| Stigmast-4-en-3β,6α-diol (10) | 14.47 ± 0.68 |
| Emodin (11) | 83.89 ± 0.73 |
| Apigenin-8-C-β-D-glucopyranoside (5) | 94.67 ± 7.40 |
| Lutéolin-8-C-β-D-glucopyranoside (6) | 89.23 ± 2.50 |
| Luteolin-5-methyl ether (4) | 90.55 ± 2.15 |
| Luteolin-6-C-β-D-glucopyranoside (7) | 97.94 ± 3.53 |
3. Methodology
3.1. General experimental procedures
Acetone-d6, chloroform-d, metthanol-d4, and DMSO-d6 were used as the analytical solvent for 1H and 13C NMR spectra (Bruker AMX-400 MHz and a Bruker Avance III-600 MHz). Tetramethylsilane (TMS) was used as a reference to the solvent residue while coupling constants (J) and chemical shifts (δ) are expressed in Hz and parts per million units (ppm) respectively. Silica gel 60 (0.040–0.063 mm, Merck), 60 F254 (Merck), OSD, and Sephadex LH-20 gel were used in different chromatographic techniques (HPLC, column, and TLC). Sulfuric acid 10% and UV light (254 and 365 nm) were used to reveal different secondary metabolites.3.2. Plant material
The aerial part of C. mimosoïdes was collected in Dschang (5°27′0′′N, 10°4′0′′E), West Region of Cameroon, in October 2018 and identified at the National Herbarium of Cameroon (NHC), Yaounde, where a voucher specimen (No. 8521/HCN) was deposited.3.3. Extraction and isolation of the plant constituents
The aerial part of C. mimosoïdes was cut, dried and ground to give 6 kg of powder. 4 kg of this powder was extracted with ethanol (20 L) 3 × 24 h at room temperature followed by filtration. The filtrate obtained was concentrated under reduced pressure to give a crude extract (141 g). The extract was suspended in water and successively partitioned to ethyl acetate and butanol fractions. The ethyl acetate and n-butanol fractions were subjected to silica gel column chromatography using different solvent mixtures (n-hexane–ethyl acetate, ethyl acetate–methanol and ethyl acetate–methanol–water) in increasing polarity to give twelve subfractions (A-M). By repeating different chromatographic techniques (column and HPLC) of these twelve fractions, one new compound (1) and fifteen known secondary metabolites (2–16) were obtained (Fig. 1). It should be noted that all of these known compounds are reported herein for the first time from Cassia mimosoïdes.3.4. Synthesis of semi-synthetic derivative (10) from stigmast-4-en-3,6-dione
In a 50 mL flask, 20 mg (4.69 × 10–5 mol) of stigmast-4-en-3,6-dione (10) was dissolved in 6 mL of methanol and refluxed under stirring until complete dissolution and then allowed to cool (25 °C). While under continuous stirring, 2 mg sodium borohydride (NaBH4) previously dissolved in 3 mL distilled water was added drop-wise and the evolution of the reaction was monitored by TLC until complete disappearance of starting material (20 min) as illustrated in Fig. 2. The reaction medium was then transferred to a beaker (250 mL) containing 50 mL of acidified ice-cold distilled water (3 mL of concentrated HCl). After 20 minutes the formation of a precipitate was observed. The medium was then filtered to yield 14.8 mg or 73.3%.3.5. In vitro studies
| % Inhibition = 100 − (Abs(sample)/Abs(control)) × 100 |
4. Conclusions
All the compounds showed appreciable antimicrobial activity against selected strain of bacterial and fungal origin. Compound 5 and 6 showed moderate to good antifungal activity against all selected strains. The genus Cassia is a significant source of secondary metabolites that are physiologically active and come from several chemical classes. The current research discusses the spectroscopic elucidation of the structure and enzymatic activity in silico and in vitro of fifteen known compounds as well as a new unidentified avenasterol derivative called 21-methylene-24-ethylidene lophenol. The compound isolated from C. mimidosa holds significant potential as a drug candidate for treating illnesses associated with H. pylori infection. This potential arises from its demonstrated effectiveness in targeting the urease enzyme and its promising activity against the HEP-G2 cell line. The urease enzyme is a key factor in the survival of H. pylori within the stomach's acidic environment. Inhibiting urease activity can potentially impair the bacterium's ability to colonize and cause damage. The compound from C. mimidosa has shown strong inhibitory activity against the urease enzyme, suggesting its potential to disrupt H. pylori's survival mechanism. Compound 1 showed strong antibacterial activity against H. pylori and S. aur with MIC value of 0.28 and 0.12 respectively. The inhibitory activity of all the substances tested against urease was higher (1.224 ± 0.43 IC50 > 6.678 ± 0.11 M) than that of thiourea (IC50 = 18.61 ± 0.11 M). The compound's 10 and 9 positive effects on the HEP-G2 cell line further underscore their potential as a drug candidate. The HEP-G2 cell line is often used as a model to study the effects of potential drugs on cancer cells. The compound's activity against these cells indicates that it might have a broader impact on cellular processes, potentially offering therapeutic benefits beyond its anti-H. pylori effects. In conclusion, the compound isolated from C. mimidosa shows great promise as a multi-faceted drug candidate for treating illnesses associated with H. pylori infection. Its inhibition of urease enzyme activity suggests a targeted approach against H. pylori, while its positive effects on the HEP-G2 cell line hint at broader applications, possibly including anti-cancer properties. Further research and clinical studies are warranted to fully explore the compound's potential as a novel therapeutic agent.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge TWAS fellowship program for support in the completion of the project. The acknowledgement also extends to COMSATS University Islamabad, Abbottabad Campus for providing necessary facilities to conduct a portion of this research. A part of this research was funded by researcher supporting project number (RSP2024R357), King Saud University, Riyadh Saudi Arabia. The authors extend their thanks to Prof. Dr Xiaomeng Li from Northeast Normal University Chanchun, China for providing human liver cell lines.References
- D. T. D. Cuong, et al., Isolation and characterization of six flavonoids from the leaves of Sterculia foetida Linn, Vietnam J. Chem., 2019, 57(4), 438–442 CrossRef CAS.
- J. K. Dzotam, et al., In vitro antibacterial and antibiotic modifying activity of crude extract, fractions and 3′, 4′, 7-trihydroxyflavone from Myristica fragrans Houtt against MDR Gram-negative enteric bacteria, BMC Compl. Alternative Med., 2018, 18, 1–9 CrossRef PubMed.
- J. B. Harborne, C. A. Williams and K. L. Wilson, Flavonoids in leaves and inflorescences of Australian Cyperus species, Phytochemistry, 1982, 21(10), 2491–2507 CrossRef CAS.
- G. Tian, et al., Separation of flavonoids from the seeds of Vernonia anthelmintica Willd by high-speed counter-current chromatography, J. Chromatogr. A, 2004, 1049(1–2), 219–222 CrossRef CAS PubMed.
- M. Della Greca, P. Monaco and L. Previtera, Stigmasterols from Typha latifolia, J. Nat. Prod., 1990, 53(6), 1430–1435 CrossRef CAS.
- S. K. Ko, W. K. Whang and I. H. Kim, Anthraquinone and stilbene derivatives from the cultivated Korean Rhubarb Rhizomes, Arch. Pharmacal Res., 1995, 18, 282–288 CrossRef CAS.
- Prateeksha, M. A. Yusuf, B. N. Singh, S. Sudheer, R. N. Kharwar, S. Siddiqui, A. M. Abdel-Azeem, L. F. Fraceto, K. Dashora and V. K. Gupta, Chrysophanol: a natural anthraquinone with multifaceted biotherapeutic potential, Biomolecules, 2019, 9(2), 68 CrossRef CAS PubMed.
- W. L. DeLano, Pymol: an open-source molecular graphics tool, CCP4 Newsl. Protein Crystallogr, 2002, 40(1), 82–92 Search PubMed.
- F. Lovering, J. Bikker and C. Humblet, Escape from flatland: increasing saturation as an approach to improving clinical success, J. Med. Chem., 2009, 52(21), 6752–6756 CrossRef CAS PubMed.
- W. Wei, et al., Fsp3: a new parameter for drug-likeness, Drug Discovery Today, 2020, 25(10), 1839–1845 CrossRef CAS PubMed.
- X. Zhang, A. Cambrai, M. Miesch, S. Roussi, F. Raul, D. Aoude-Werner and E. Marchioni, Separation of ¢5- and ¢7-Phytosterols by Adsorption Chromatography and Semipreparative Reversed Phase High-Performance Liquid Chromatography for Quantitative Analysis of Phytosterols in Foods, J. Agric. Food Chem., 2006, 54, 1196–1202 CrossRef CAS PubMed.
- S. Rondet, M. Taton and A. Rahier, Identification, Characterization, and Partial Purification of 4α-Carboxysterol–C3-Dehydrogenase/ C4-Decarboxylase from Zea mays, Arch. Biochem. Biophys., 1999, 366, 249–260 CrossRef CAS PubMed.
- H. Schaller, Sterol and Steroid Biosynthesis and Metabolism in Plants and Microorganisms, Comprehensive Natural Products II: Chemistry and Biology, 2010, vol. 1, pp. 755–787 Search PubMed.
- N. Anantachoke, W. Kitphati, S. Mangmool and N. Bunyapraphatsara, Polyphenolic compounds and antioxidant activities of the leaves of Glochidion hypoleucum, Nat. Prod. Commun., 2015, 10(3), 479–482 CrossRef PubMed.
- C. C. Zhao, J. H. Shao, X. Li, J. Xu and P. Zhang, Antimicrobial constituents from fruits ofAilanthus altissima SWINGLE, Arch. Pharmacal Res., 2005, 28, 1147–1151 CrossRef CAS PubMed.
- J. Ali, et al., In silico prediction of aqueous solubility using simple QSPR models: the importance of phenol and phenol-like moieties, J. Chem. Inf. Model., 2012, 52(11), 2950–2957 CrossRef CAS PubMed.
- R. O. Potts and R. H. Guy, Predicting skin permeability, Pharm. Res., 1992, 9, 663–669 CrossRef CAS PubMed.
- Y. C. Martin, A bioavailability score, J. Med. Chem., 2005, 48(9), 3164–3170 CrossRef CAS PubMed.
- I. Khokhar, et al., Evaluation of antagonistic activity of soil bacteria against plant pathogenic fungi, Pak. J. Phytopathol., 2011, 23(2), 166–169 Search PubMed.
- R. Sarwar, et al., Evaluation of antioxidant, free radical scavenging, and antimicrobial activity of Quercus incana Roxb, Front. Pharmacol, 2015, 6, 277 Search PubMed.
- S. J. Teague, et al., The design of leadlike combinatorial libraries, Angew. Chem., Int. Ed., 1999, 38(24), 3743–3748 CrossRef CAS PubMed.
- M. Totir, et al., Macro-to-micro structural proteomics: native source proteins for high-throughput crystallization, PLoS One, 2012, 7(2), e32498 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08913d |
| This journal is © The Royal Society of Chemistry 2024 |